
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiphenylacetylene stabilised alkali-metal nickelates: synthesis, structure and catalytic applications†
Andryj M.
Borys
 and
Eva
Hevia
*
and
Eva
Hevia
*
Departement für Chemie, Biochemie und Pharmazie, Universität Bern, 3012 Bern, Switzerland. E-mail: eva.hevia@unibe.ch
First published on 11th January 2023
Abstract
Whilst low-valent nickelates have recently been proposed as intermediates in Ni-catalysed reactions involving polar organometallics, their isolation and characterisation is often challenging due to their high sensitivity and reactivity. Advancing the synthetic, spectroscopic and structural insights of these heterobimetallic systems, here we report a new family of alkyne supported alkali-metal nickelates of the formula Li4(solv)n(Ar)4Ni2{μ2:η2,η2-Ph–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ph} (where solv = Et2O, THF; Ar = Ph, o-Tol, naphthyl, 4-tBu-C6H4) which can be accessed through the combination of Ni(COD)2, Ph–CC–Ph and the relevant lithium aryl in a 2
C–Ph} (where solv = Et2O, THF; Ar = Ph, o-Tol, naphthyl, 4-tBu-C6H4) which can be accessed through the combination of Ni(COD)2, Ph–CC–Ph and the relevant lithium aryl in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4 ratio. Demonstrating the versatility of this approach, the sodium and potassium nickelates can also be accessed when using PhNa or via alkali-metal exchange with AMOtBu (AM = Na, K). When employing bulky or structurally constrained aryl-lithiums, mononickel complexes of the formula Li2(solv)n(Ar)2Ni{η2-Ph–CC–Ph} are instead obtained, highlighting the structural diversity of alkali-metal nickelates bearing alkyne ligands. Expanding the catalytic potential of these systems, their ability to promote the catalytic cyclotrimerisation of diphenylacetylene to hexaphenylbenzene was explored, with mononickel compounds bearing electron rich aryl-substituents displaying the best performance.
:1:4 ratio. Demonstrating the versatility of this approach, the sodium and potassium nickelates can also be accessed when using PhNa or via alkali-metal exchange with AMOtBu (AM = Na, K). When employing bulky or structurally constrained aryl-lithiums, mononickel complexes of the formula Li2(solv)n(Ar)2Ni{η2-Ph–CC–Ph} are instead obtained, highlighting the structural diversity of alkali-metal nickelates bearing alkyne ligands. Expanding the catalytic potential of these systems, their ability to promote the catalytic cyclotrimerisation of diphenylacetylene to hexaphenylbenzene was explored, with mononickel compounds bearing electron rich aryl-substituents displaying the best performance.
Introduction
The coordination of polar organometallics to Ni(0) was first investigated in the early 1970s,1,2 and succeeded the pioneering work of Wilke into ubiquitous Ni(0)-olefin complexes such as Ni(C2H4)3,3 Ni(ttt-CDT)4 and Ni(COD)2.5 It was observed that the treatment of these Lewis acidic Ni(0) complexes with simple polar organometallics can give rise to highly sensitive anionic nickelates in which the formally carbanionic centre now coordinates to Ni(0), often with displacement of an olefin ligand.2 Several examples of anionic nickelates were documented during these early studies, with a range of polar organometallics such as organolithiums,6–10 organomagnesiums,11 organoaluminium12 or hydrido-aluminate species.13–15 Typically, one or more olefins remain coordinated to Ni(0) to act as a π-accepting ligand to modulate the high electron density at the d10 metal centre.2 In the absence of olefins, through forced displacement with excess PhLi or PhNa, it was discovered that even N2 can coordinate in a side-on and bridging motif {μ2-η2:η2} between two Ni centres,16–18 hinting at the key role of π-accepting ligands for the stabilisation and isolation of low valent nickelates. For alkali-metal nickelates, examples of both charge-separated (Scheme 1a, I)6,9 and contacted ion-pair species (Scheme 1a, II and III)19,20 are known, with alkali-metal:nickel ratios ranging from 1:1, 2:1 or 3:1.
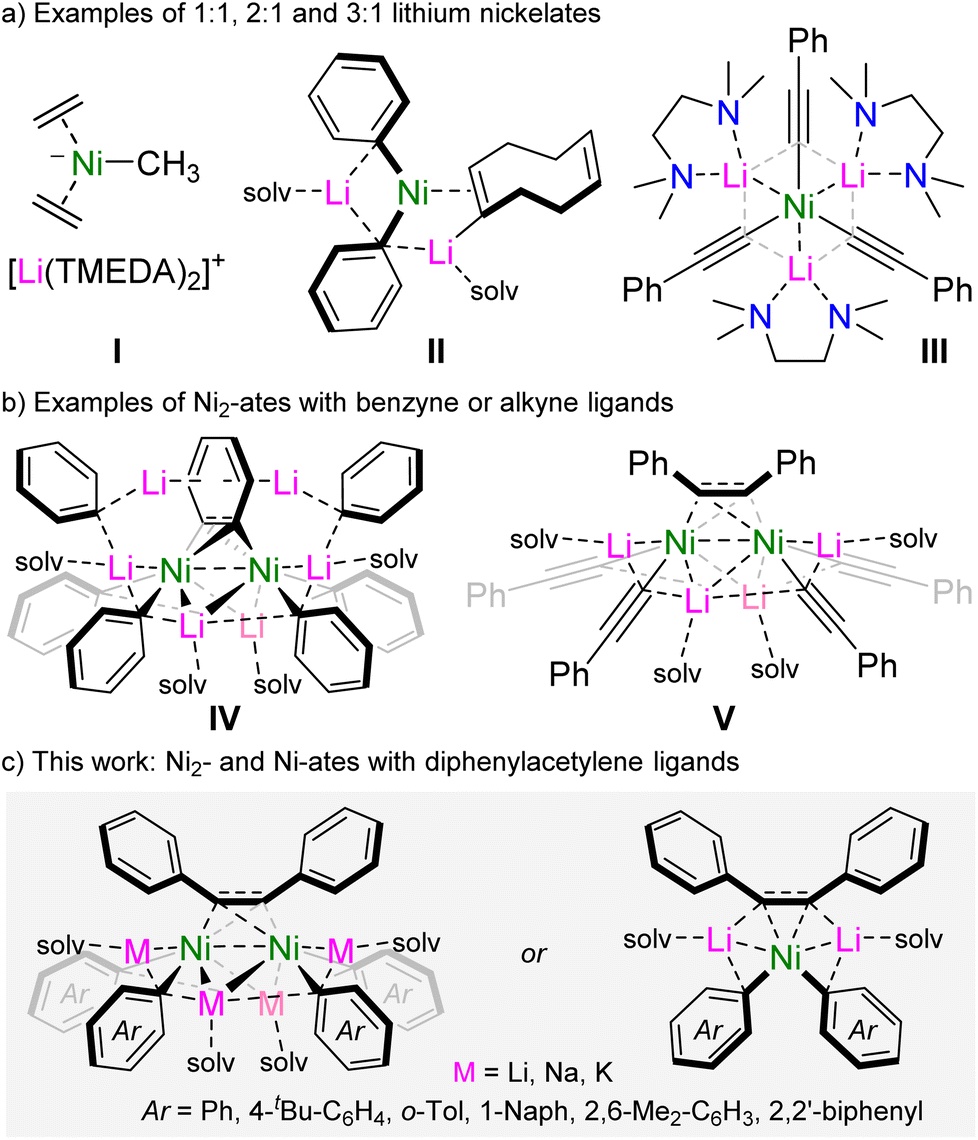 | ||
| Scheme 1 Selected examples of alkali-metal nickelates. | ||
Despite exhibiting a wealth of unique structural features, low-valent anionic nickelates remained dormant in the literature for several decades and were overshadowed by parallel developments on the applications of nickel complexes in catalysis.21 More recently, however, it has been proposed by theoretical studies that low-valent nickelates may be potential intermediates in certain nickel-catalysed cross-coupling reactions involving polar organometallics,22,23 prompting a renewed experimental interest into these overlooked species.
We have previously explored the rich co-complexation chemistry of Ni(COD)2 and PhLi, which gives a series of lithium nickelates including II (Scheme 1), and demonstrated that these are key intermediates in the nickel-catalysed cross-coupling of aryl ethers.19,24 When further assessing these co-complexation reactions, we found that using a larger excess of PhLi led to the formation of a polynuclear dinickel cluster containing a bridging C6H4 dianion as a result of intramolecular C–H activation of a phenyl substituent (Scheme 1, IV).25 The formation of IV suggests that the homoleptic tri-lithium nickelate “Li3NiPh3(solv)3” is too electron-rich to form, leading to the in situ formation of the π-accepting benzyne-type ligand. Only when moving to organolithiums which themselves could serve as π-accepting ligands, namely lithium aryl acetylides, is it possible to isolate homoleptic tri-lithium nickelates III, which were found to be further stabilised by London dispersion interactions.20 Notably, compound III reacts with PhI to give V in which the cross-coupled product (diphenylacetylene) is trapped and coordinated in a bridging motif between two nickel centres. Since V bears similar structural features to IV, we thus considered whether it was possible to use diphenylacetylene as a simple π-accepting ligand to access new families of alkali-metal nickelates. Herein, we detail synthetic and structural insights into dinickel and mononickelate complexes which can be readily accessed by reacting Ni(COD)2 with two molar equivalents of alkali-metal aryls in the presence of diphenylacetylene. Advancing the structure/reactivity correlations, we also assess the catalytic ability of these novel heterobimetallic complexes to promote the cyclotrimerisation of diphenylacetylene, as well as its insertion into biphenylene.
Results and discussion
Synthesis of alkali-metal nickelates
The reported dinickel complex [(COD)Ni]2{μ2-η2:η2-Ph–CC–Ph} was selected as a suitable precursor to target alkali-metal dinickelates.26,27 Addition of PhLi (2 equivalents per Ni) to an Et2O solution of in situ prepared [(COD)Ni]2{μ2-η2:η2-Ph–CC–Ph} gave the corresponding hexanuclear lithium nickelate, Li4(Et2O)4Ph4Ni2{μ2-η2:η2-Ph–CC–Ph} (1a), as a dark red crystalline solid in 41% yield (Scheme 2). The formation of dinickelate complex 1a contrasts with the mononickel complex Li2(THF)4Ph2Ni(COD) (II, Scheme 1a) which is prepared from Ni(COD)2 and 2 equivalents of PhLi,19 reflecting the differing coordination ability of alkyne ligands in comparison to traditionally employed olefin ligands. This procedure could be extended to PhNa to give the analogous sodium nickelate, Na4(THF)6Ph4Ni2{μ2-η2:η2-Ph–CC–Ph} (1b), but attempts to prepare the potassium nickelate directly from PhK failed however, likely due to competing side reactions of PhK with the ethereal solvent or 1,5-cyclooctadiene.25 Alkali-metal exchange through the treatment of 1a with 4 equivalents of KOtBu proved successful nevertheless, to give the corresponding potassium nickelate, K4(THF)4Ph4Ni2{μ2-η2:η2-Ph–CC–Ph} (1c) in 79% yield. This represents the first homologous series of alkali-metal nickelates bearing different alkali-metals. Substituted aryl-lithiums such as 4-tBu-C6H4–Li were also compatible to give Li4(Et2O)4(4-tBu-C6H4)4Ni2{μ2-η2:η2-Ph–CC–Ph} (1d). When using o-Tol-Li or 1-naphthyl-Li, the corresponding hexanuclear lithium nickelates Li4(Et2O)4(o-Tol)4Ni2{μ2-η2:η2-Ph–CC–Ph} (1e) and Li4(THF)6(1-Naph)4Ni2{μ2-η2:η2-Ph–CC–Ph} (1f) could be structurally characterised by single-crystal X-ray diffraction, but could not be isolated in pure form (vide infra). Whilst the treatment of [(COD)Ni]2{μ2-η2:η2-Ph–CC–Ph} with aryl-lithiums appeared to be selective and quantitative by NMR spectroscopy (see Fig. S1†), isolated crystalline yields of the alkali-metal nickelates were typically low (33–45%) due to their high solubility in ethereal solvents.
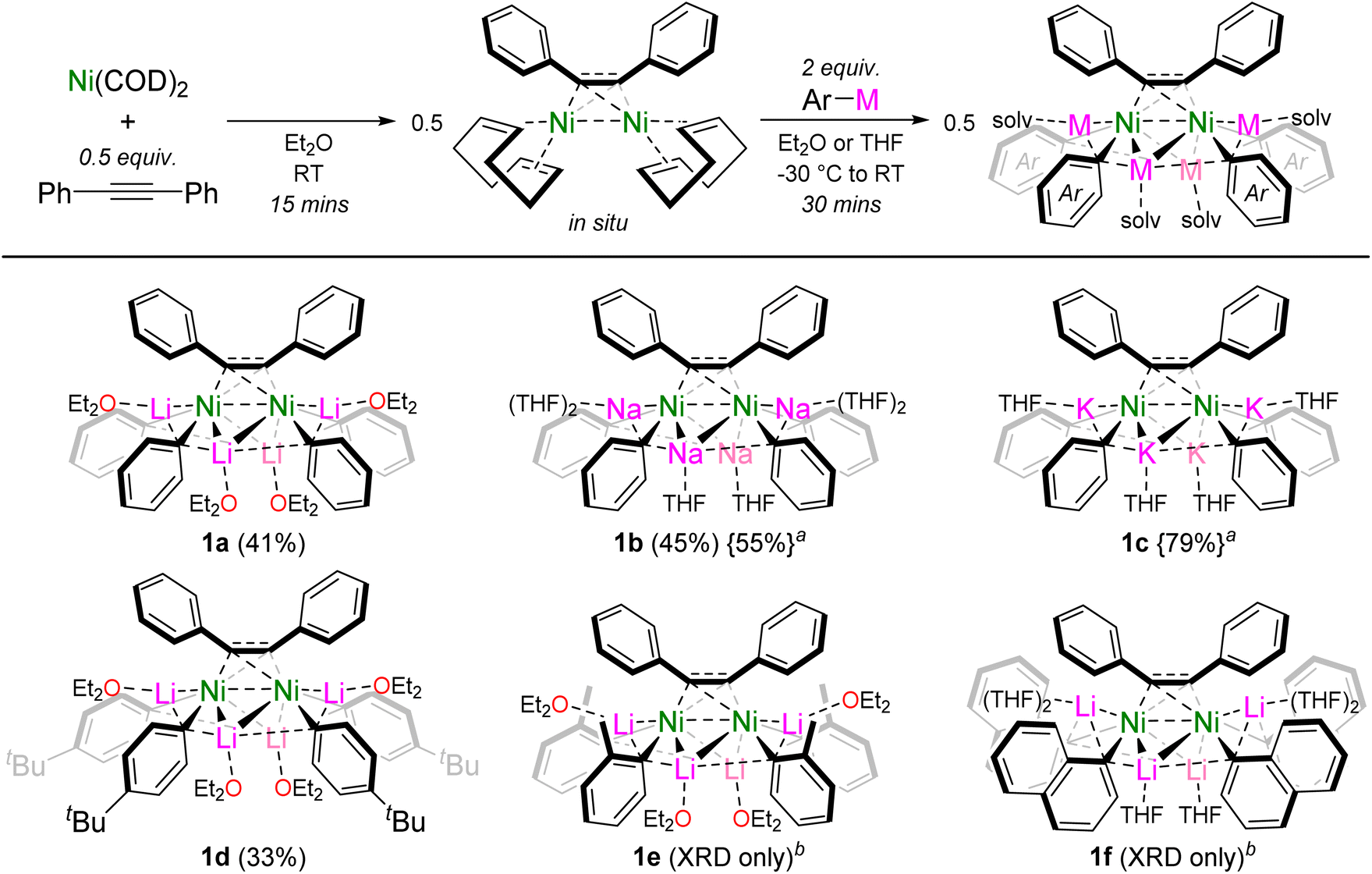 | ||
| Scheme 2 Synthesis of dinickelate complexes 1a–f. Isolated crystalline yields given in parentheses. aPrepared by treating 1a with NaOtBu or KOtBu, respectively. bCould not be isolated in pure form. | ||
Interestingly, we also found that by systematically studying these co-complexation reactions when using ortho-substituted aryl-lithiums, o-Tol-Li and 1-naphthyl-Li, the formation of mononickelate complexes of the formula Li2(solv)n(Ar)2Ni{η2-Ph–CC–Ph} was instead favoured, with the choice of donor solvent playing an important role in dictating the crystallised product. With o-Tol-Li, small quantities of the Et2O solvate (1e) could be crystallised, but samples were always plagued with variable amounts Ni(COD)2 or [(COD)Ni]2{μ2-η2:η2-Ph–CC–Ph}, preventing its isolation in pure form. Contrastingly, performing the reaction with one equivalent of diphenylacetylene in the presence of THF, allowed Li2(THF)2(o-Tol)2Ni{η2-Ph–CC–Ph} (2a) to be reliably prepared and isolated in pure form (Scheme 3). Similarly with 1-naphthyl-Li, small quantities of the THF solvate (1f) could be crystallised, but not isolated in pure form. Performing the reaction with one equivalent of diphenylacetylene in Et2O however, allowed for the reliable isolation of Li2(Et2O)2(1-Naph)2Ni{η2-Ph–CC–Ph} (2b). Further exploring how general the formation of mononickelate alkyne complexes was, the bulky aryl-lithium 2,6-Me2-C6H3-Li also gave Li2(Et2O)2(2,6-Me2-C6H3)2Ni{η2-Ph–CC–Ph} (2c), whilst the structurally constrained aryl-lithium, 2,2′-dilithiobiphenyl, reacted smoothly to give Li2(THF)4(2,2′-biphenyl)Ni{η2-Ph–CC–Ph} (2d). Compound 2d is closely related to the dilithionickelole Li2(THF)4(2,2′-biphenyl)Ni{η2,η2-COD} which was previously reported by Xi and co-workers.28
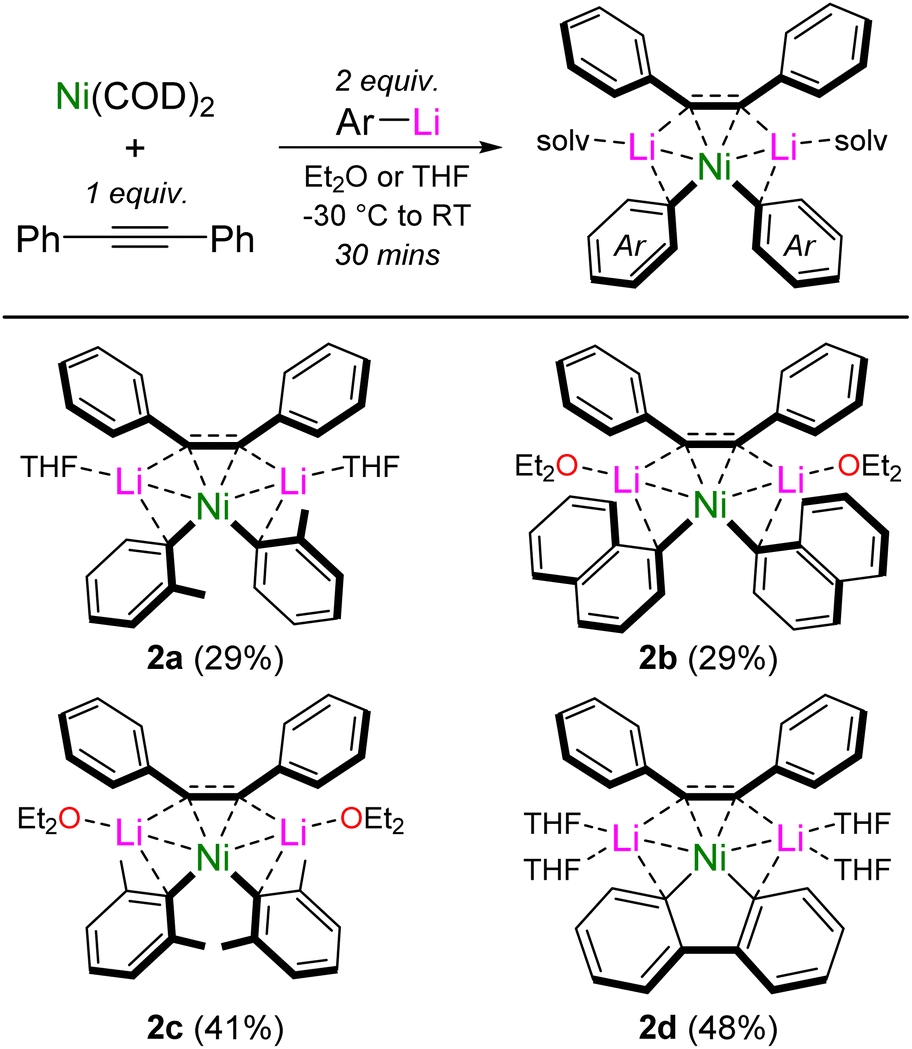 | ||
| Scheme 3 Synthesis of mononickelate complexes 2a–d. Isolated crystalline yields given in parentheses. | ||
Spectroscopic features
The alkali-metal nickelates 1a–d and 2a–d typically show sharp and well resolved 1H, 7Li and 13C NMR spectra, consistent with d10 diamagnetic Ni(0) species. Contrastingly, compound 1d displayed broad signals in the room temperature 1H NMR spectrum for the coordinated 4-tBu-C6H4-substituents (Fig. 1, yellow trace), alongside two broad 7Li NMR resonances at δ 0.26 and −0.59 (ESI, Fig. S3†). Cooling to −40 °C (Fig. 1, purple trace) leads to splitting of the broad ortho and meta-proton signals into two distinct and well-resolved resonances for each, consistent with frozen rotation about the Caryl–Ni bonds. Heating to +60 °C (Fig. 1, red trace) on the other hand, leads to coalescence of the signals in both the 1H and 7Li NMR spectra, indicative of fast rotation about the Caryl–Ni bonds on the NMR timescale. These spectroscopic observations suggest that the electron-rich 4-tBu-C6H4-substituent coordinates stronger to nickel in comparison to other aryl-substituents.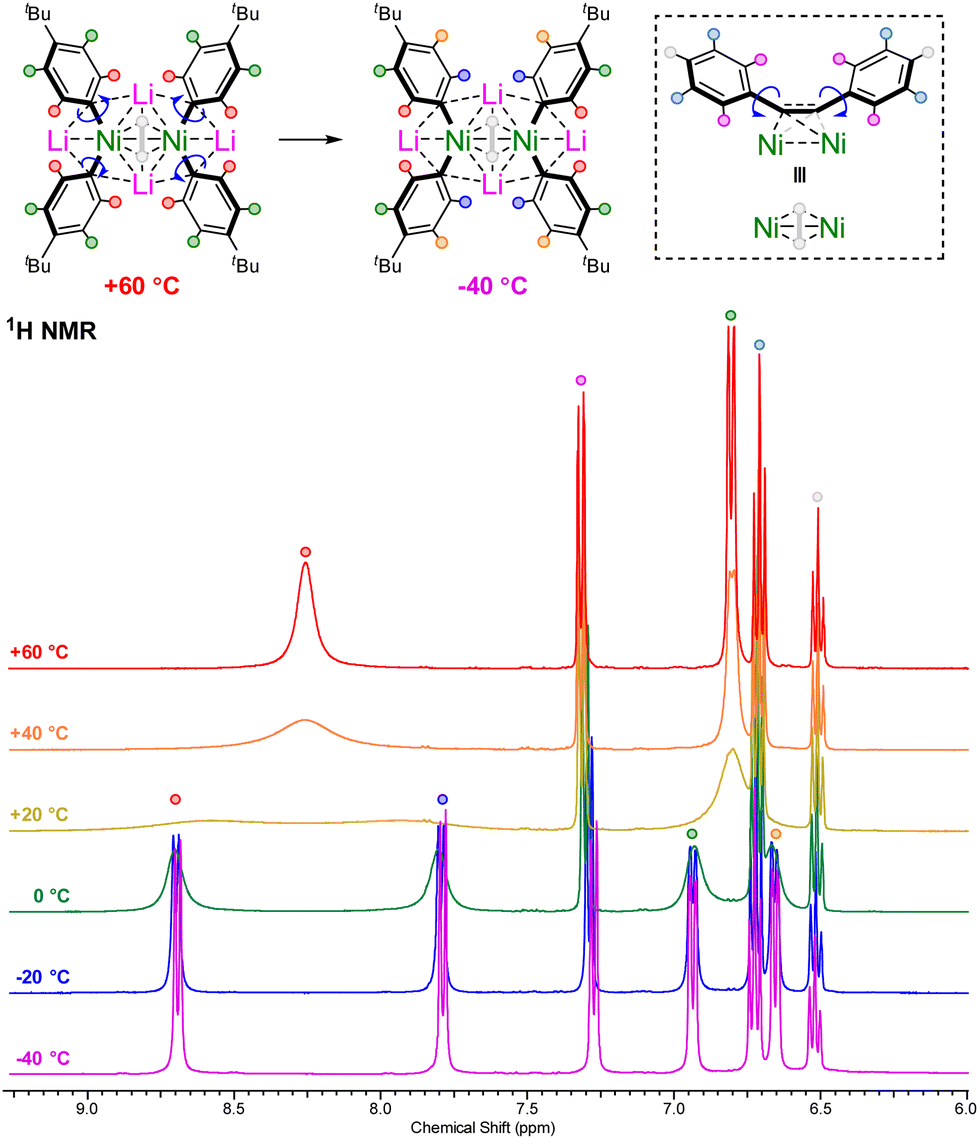 | ||
| Fig. 1 Variable temperature 1H NMR spectra of 1d in THF-d8 illustrating fast or frozen rotation about the Caryl–Ni bonds. | ||
The di- and mono-nickelate complexes can be further distinguished by assessing the 13C{1H} NMR signal of the acetylene carbon. For compounds 1a–d, this signal is observed in the range of δ 68.6–76.6, which is considerably shielded compared to [(COD)Ni]2{μ2-η2:η2-Ph–CC–Ph} (δ 106.9), and consistent with significant back-donation from the electron-rich Ni centres. Contrastingly, the acetylene carbon signal for compounds 2a–d is observed in the range δ 144.9–150.8, which is considerably deshielded compared to free diphenylacetylene (δ 90.8),29 and slightly shielded when compared to other L2Ni{η2-Ph–CC–Ph} complexes [L = N-heterocyclic carbene (δ 139.3)30 or diphosphine (δ 141.3)].31 The Caryl–Ni ipso-carbon signal for compounds 1a, 1d and 2a–d is observed in the range δ 177.2–191.1, which is similar to other phenyl-nickelate complexes such as II and IV (Scheme 1),19,25 and comparable to the free aryl-lithiums (see ESI† for full spectroscopic details). Notably, the ipso-carbon signal for the phenyl-derivatives becomes more deshielded on moving from Li (1a; δ 182.0) to Na (1b; δ 195.8) to K (1c; δ 203.8), consistent with increased charge density at the carbanionic centre.32
Solid-state structures
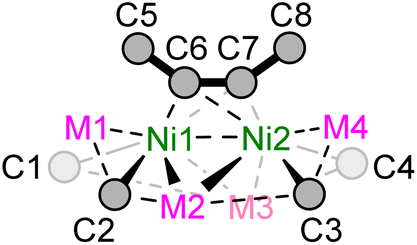
Single crystals suitable for X-ray diffraction studies were obtained for dinickelate complexes 1a–b and 1d–f, and selected crystallographic parameters are shown in Table 1 (see ESI† for further details and full structures). Whilst single crystals of potassium nickelate 1c could be grown, they were found to immediately desolvate and lose crystallinity when removed from the mother liquor, a feature we have previously observed for other alkali-metal nickelates.33 The Ni–Caryl distances are comparable for compounds 1a–b and 1d–f and range from 1.952(2)–2.042(8) Å, which is similar to previously reported nickelates derived from PhLi or PhNa.7,8,16–19,25 The Ni⋯M (M = Li, Na) distances are close to or longer than the sum of covalent radii,34 with increasing distances as the size of the alkali-metal cation increases down group 1. Previous complementary bonding analysis on III (Scheme 1) revealed that there is no direct Ni⋯M bonding,20 and therefore there is likely no bonding interaction between nickel and the alkali-metal cations in compounds 1a–f. Similarly, the Ni⋯Ni distances [range = 2.633(3)–2.8873(7) Å] are longer than observed for reported Ni–Ni bonds (ca. 2.3–2.5 Å),35 and comparable to IV in which no Ni–Ni bond was found from natural bond order (NBO) analysis.25 The CC (C6–C7) distance of the coordinated diphenylacetylene ranges from 1.38(2)–1.394(2) Å; this is significantly longer than free diphenylacetylene [1.198(2) Å]36 and consistent with considerable back-donation from the electron-rich Ni centres. In addition, the coordinated diphenylacetylene shows considerable bending away from linearity [C5–C6–C7 or C8–C7–C6; 125.2(9)–132.6(1)°].
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Distance/Å | Angle/° | Torsion/° | ||||||||
| Ni1–C1 | Ni–C2 | Ni2–C3 | Ni2–C4 | Ni⋯M range | Ni1⋯Ni2 | Ni–C6/7 range | C6–C7 | C5–C6–C7 | C8–C7–C6 | C5–C6–C7–C8 | |
|
a The Ph–CC–Ph is disordered across two positions.
|
|||||||||||
| 1a | 1.986(2) | 1.965(1) | 1.986(2) | 1.965(1) | 2.469(3)–2.663(3) | 2.633(3) | 1.947(1)–1.968(1) | 1.394(2) | 127.6(1) | 127.6(1) | 5.0(2) |
| 1.980(1) | 1.977(2) | 1.980(1) | 1.977(2) | 2.441(1)–2.654(3) | 2.654(3) | 1.936(1)–1.979(1) | 1.388(2) | 127.6(1) | 127.6(1) | 5.8(2) | |
| 1b | 2.042(8) | 1.957(3) | 2.042(8) | 1.957(3) | 2.825(1)–3.099(1) | 2.8873(7) | —a | 1.38(2) | 125.2(9) | 125.2(9) | —a |
| 1d | 1.980(2) | 1.983(2) | 1.973(2) | 1.968(2) | 2.395(5)–2.628(4) | 2.7079(8) | 1.948(2)–1.964(2) | 1.392(3) | 127.7(2) | 127.5(2) | 3.0(4) |
| 1e | 1.958(2) | 1.952(2) | 1.987(2) | 1.965(2) | 2.448(3)–2.758(3) | 2.7906(7) | 1.921(1)–2.013(2) | 1.386(2) | 132.6(1) | 129.8(1) | 20.1(3) |
| 1f | 1.977(3) | 1.959(3) | 1.977(3) | 1.959(3) | 2.674(5)–2.770(5) | 2.8218(8) | 1.931(3)–2.027(3) | 1.381(3) | 127.2(2) | 127.2(2) | 14.8(4) |
| 1.953(3) | 1.975(4) | 1.953(3) | 1.975(4) | 2.587(6)–2.795(6) | 2.7606(6) | 1.925(3)–2.034(2) | 1.392(4) | 126.7(2) | 126.7(2) | 21.8(4) | |
A close inspection into the structures of compounds 1e and 1f, which are derived from o-Tol-Li and 1-naphthyl-Li respectively, reveal considerable structural distortion when compared to lithium nickelates 1a and 1d. In the solid-state structure of 1e, all ortho-CH3 substituents are orientated in the same direction towards the coordinated diphenylacetylene unit with Ni–Caryl–Cortho–CH3 torsion angles ranging from 1.5(3)–6.4(3)° (Fig. 2 & Fig. S10†). Most notably, one of the four Et2O molecules coordinates to Li2 via anagostic interactions (i.e. largely electrostatic) from the CH3 group [C–H⋯Li2 = 2.817–3.189 Å], in contrast to the expected oxygen coordination mode observed for Li1, Li3 and Li4. This is likely a consequence of the shorter Li2⋯C2 and Li2⋯C3 distances [2.257(4) Å and 2.197(4) Å, respectively] when compared to the Li3⋯C1 and Li3⋯C4 distances [2.446(4) Å and 2.563(3) Å, respectively]. Anagostic interactions between CH3 groups and Li have been observed in several crystal structures,37,38 including in lithium nickelates,39 but this is limited to intramolecular examples. In addition, there is considerable torsion in the diphenylacetylene unit [C5–C6–C7–C8 = 20.1(3)°, see Table 1 for general label numbering] to enable additional Carene⋯Li2 interactions [2.727(4)–2.750(4) Å].
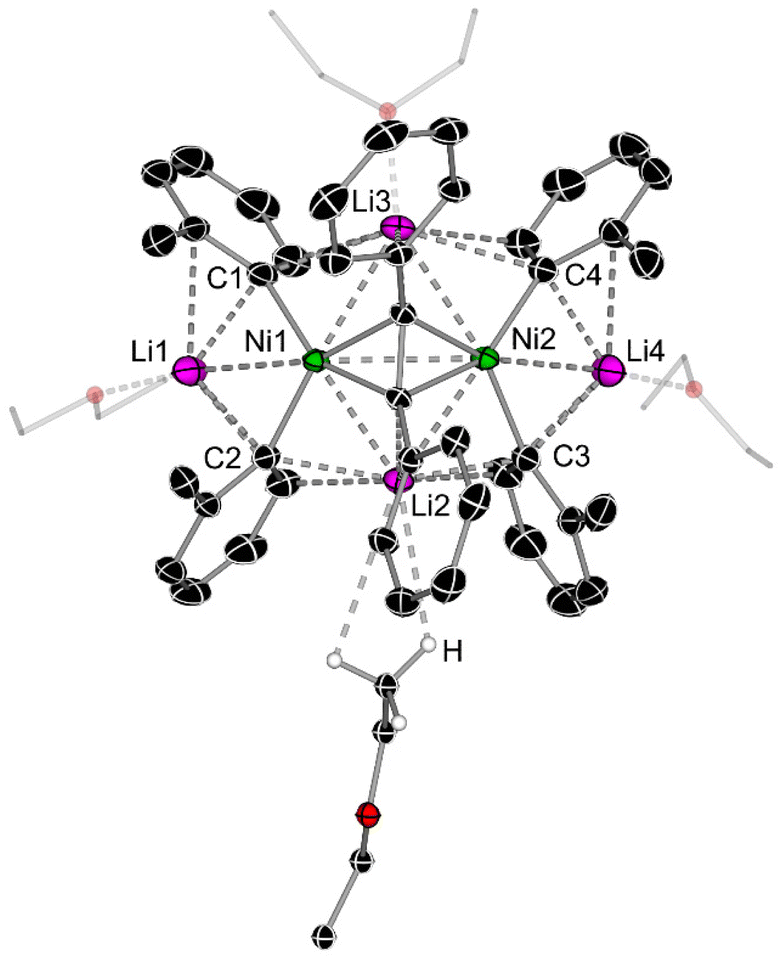 | ||
| Fig. 2 Molecular structure of 1e. Thermal ellipsoids shown at 30% probability and hydrogen atoms (except those on Et2O showing anagostic interactions to Li2) omitted for clarity. See Fig. S10† for alternative view. | ||
In the solid-state structure of 1f (see Fig. S11† for full structure), the 1-naphthyl substituents are similarly all orientated in the same direction towards the coordinated diphenylacetylene unit with Ni–Caryl–Cortho–CH3 torsion angles ranging from 2.9(5)–5.4(5)°. As with 1e, there is also significant torsion in the coordinated diphenylacetylene unit [C5–C6–C7–C8 = 14.8(4)° or 21.8(4)°] to enable additional Carene⋯Li2/3 interactions [2.671(5)–2.998(6) Å]. The central core of 1f shows considerable distortion when compared to 1a (Fig. 3). For example, in 1a, the four ipso-carbons (C1–C4) lie in an approximate plane which sits co-planar below the mean plane of all four lithium atoms (Li1–Li4). In 1f, neither the four ipso-carbons (C1–C4) or the four Li atoms (Li1–Li4) reside in an approximate plane. Whilst Li2 and Li3 are approximately co-planar with Ni1 and Ni2 (∼0.1 Å deviation), Li1 and Li4 sit considerably lower than Li2/Li3 and Ni1/Ni2, as well as the four ipso-carbons (C1–C4). This exposes the Li1/Li4 cations such that two molecules of THF can coordinate to each lithium, in contrast to only one molecule of coordinated Et2O observed in 1a, 1d and 1e. Whilst the identity of the ethereal solvent clearly impacts the isolation and crystallisation of compounds 1e and 1f over their mononickelate analogues 2a and 2b, it is unclear whether the unique solvent coordination in 1e and 1f is the cause or simply a consequence of the observed structural distortions.
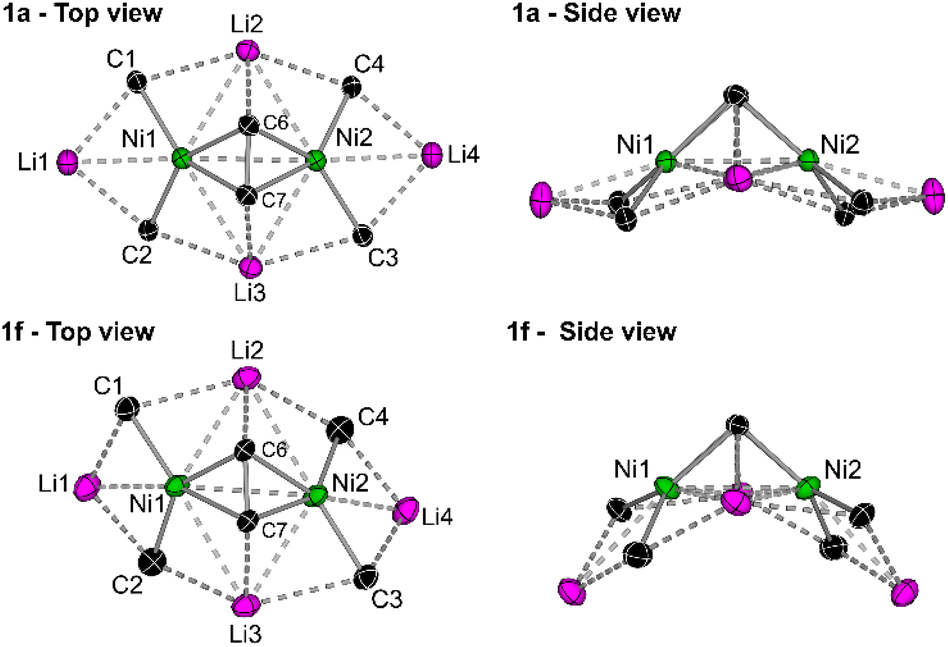 | ||
| Fig. 3 Simplified top and side views of the hexanuclear cores of 1a and 1f illustrating the structural distortion in 1f when compared to 1a. | ||
In the solid-state structure of mononickelate complexes 2c and 2d (Fig. 4), the Ni centre adopts a pseudo trigonal planar geometry in which the diphenylacetylene coordinates in a η2-fashion. The Ni⋯CC distances are shorter [1.892(1)–1.932(1) Å for 2c; 1.877(2)–1.881(2) Å for 2d] when compared to dinickelate complexes 1a–f (see Table 1). In addition, the CC bond lengths [1.318(2) Å for 2c; 1.324(2) for 2d] are shorter, and the Cipso–CC angles are closer to 180° [134.8(1)–135.4(1)° for 2c; 136.1(1)–137.6(1)° for 2d], indicative of weaker overall back-bonding from Ni to the coordinated diphenylacetylene. The Li⋯Ni distances for 2c and 2d range from 2.408(3)–2.515(3) Å, which is comparable to dinickelate complexes 1a and 1d–f, and other structurally characterised lithium nickelates.19,20,25
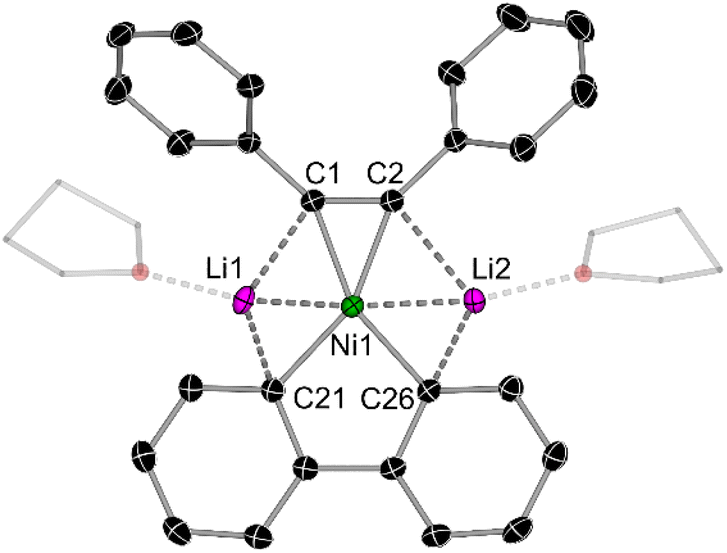 | ||
| Fig. 4 Molecular structure of 2d. Thermal ellipsoids shown at 30% probability. Hydrogen atoms and two molecules of coordinated THF omitted for clarity. | ||
Catalytic cyclotrimerisation of diphenylacetylene
The unique coordination modes of diphenylacetylene observed in alkali-metal nickelates 1a–d and 2a–d, and significant backdonation prompted us to assess their catalytic activity in the cyclotrimerisation of diphenylacetylene. Numerous transition-metal complexes have been reported to catalyse the [2 + 2 + 2] cyclotrimerisation of alkynes to give poly-substituted benzene derivatives.40–42 Following on from the pioneering work of Reppe into the nickel catalysed oligomerisation of acetylene,43–45 several nickel complexes have now been developed for the catalytic oligomerisation or cycloaddition of substituted alkynes and related substrates.46,47 Selected examples include work by Guan which showed that Ni(COD)2/PPh3 is a simple but highly efficient catalyst for the cyclotrimerisation of terminal and internal alkynes.48 Uyeda has employed chelating diamine ligands to assess how metal nuclearity (i.e. mononickel vs. dinickel) impacts the activity and selectivity of alkyne oligomerisation.49The catalytic activity of the neutral dinickel olefin complexes [(COD)Ni]2{μ2-η2:η2-Ph–CC–Ph}27 and Ni2COT2 (COT = cyclooctatetraene)50 was first evaluated (Table 2 and Table S1†). Using 5 mol% of dinickel catalyst, only modest conversions (38% and 41%, respectively) of diphenylacetylene was observed after heating to 80 °C for 4 hours (entries 1 and 2). Moving to the lithium nickelate Li4(Et2O)4Ph4Ni2{μ2-η2:η2-Ph–CC–Ph} (1a, entry 3) led to a considerable increase to 74% conversion. Contrasting with other catalytic studies using alkali-metal magnesiates,51–55 no apparent alkali-metal effect was observed, with comparable conversions observed for the sodium nickelate 1b (73%, entry 4) and potassium nickelate 1c (78%, entry 5), indicating that the alkali-metal does not appear to play an active role in catalysis. Using compound 1d as a catalyst gave a slightly improved conversion (85%, entry 6) suggesting that the more electron-rich 4-tBu-C6H4 substituents enhances the catalytic activity when compared to 1a. Supporting this claim, the acetylide substituted dinickel compound Li4(Et2O)4(Ph–CC)4Ni2{μ2-η2:η2-Ph–CC–Ph} (V, Scheme 1) was completely inactive for the catalytic cyclotrimerisation of diphenylacetylene (entry 7), but could catalyse the cyclotrimerisation of more activated terminal alkynes such as phenylacetylene (see ESI† for further details).
|
|
||
|---|---|---|
| Entry | Catalyst | Conversiona (%) |
| a Spectroscopic conversion of diphenylacetylene monitored using hexamethylbenzene as an internal standard. b Reaction performed in THF-d8. | ||
| 1 | 5% [(COD)Ni]2{PhCCPh} |
38 |
| 2 | 5% Ni2COT2 | 41 |
| 3 | 5% Li4(Et2O)4Ph4Ni2{PhCCPh}, 1a |
74 |
| 4 | 5% Na4(THF)6Ph4Ni2{PhCCPh}, 1b |
73 |
| 5 | 5% K4(THF)4Ph4Ni2{PhCCPh}, 1c |
78b |
| 6 | 5% Li4(Et2O)4(4-tBu-C6H4)4Ni2{PhCCPh}, 1d |
85 |
| 7 | 5% Li4(Et2O)4(CCPh)4Ni2{PhCCPh}, V |
Trace |
| 8 | 10% Li2(THF)2(o-Tol)2Ni{PhCCPh}, 2a |
>95 |
| 9 | 10% Li2(Et2O)2(1-Naph)2Ni{PhCCPh}, 2b |
74 |
| 10 | 10% Li2(Et2O)2(2,6-Me2-C6H3)2Ni{PhCCPh}, 2c |
>95 |
| 11 | 10% Li2(THF)4(2,2′-biphenyl)Ni{PhCCPh}, 2d |
26b |
Full conversion (>95%) of diphenylacetylene was observed when using Li2(THF)2(o-Tol)2Ni{μ2-η2:η2-Ph–CC–Ph} (2a, entry 8) or Li2(Et2O)2(2,6-Me2-C6H3)2Ni{μ2-η2:η2-Ph–CC–Ph} (2c, entry 10) as the catalyst, demonstrating that the mononickelate complexes show superior catalytic activity compared to the dinickelate complexes 1a–d. The electron-deficient derivative Li2(Et2O)2(1-Naph)2Ni{μ2-η2:η2-Ph–CC–Ph} (2b) showed reduced conversions (74%, entry 9), again illustrating how the electronic properties of the aryl-substituents can influence catalytic activity.
Interestingly, when using Li2(THF)4(2,2′-biphenyl)Ni{μ2-η2:η2-Ph–CC–Ph} (2d) as the catalyst, only low conversions of diphenylacetylene (26%, entry 11) were achieved, and characteristic signals consistent with 9,10-diphenylphenanthrene could be observed by 1H NMR spectroscopy. The formation of this product has been previously reported when treating (L)nNi(2,2′-biphenyl) [where L = (Et3P)2 or iPr2PCH2CH2PiPr2] complexes with diphenylacetylene,31,56 and this could be upgraded to catalytic regimes when using biphenylene and diphenylacetylene in the presence of O2.31 Compound 2d was also found to catalyse the insertion of diphenylacetylene into the strained C–C bond of biphenylene (Scheme 4), however this process is slow (10 mol%, 40 hours, 80 °C), particularly when compared to a Ni(NHC)2 catalyst reported by Radius (2 mol%, 30 min, 80 °C).30 Attempts to spectroscopically identify or isolate possible intermediates for the [2 + 2 + 2] cyclotrimerisation of diphenylacetylene, or insertion of diphenylacetylene into biphenylene failed.
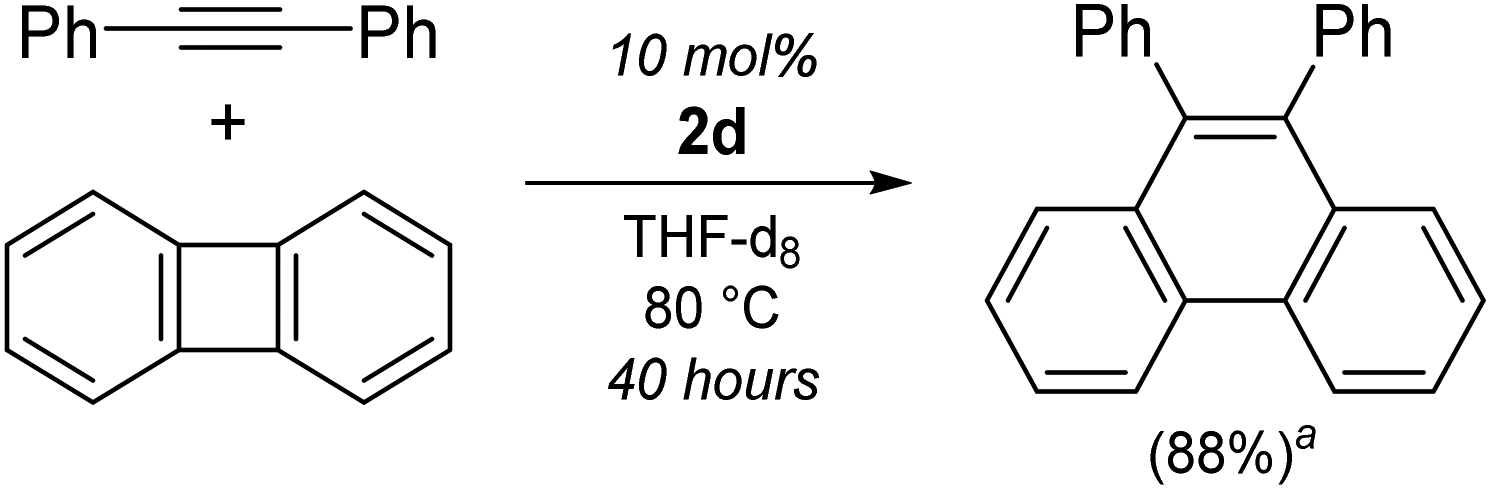 | ||
| Scheme 4 Insertion of diphenylacetylene into biphenylene using catalytic Li2(THF)4(2,2′-biphenyl)Ni{μ2-η2:η2-Ph–CC–Ph} (2d). aSpectroscopic yield of 9,10-diphenylphenanthrene determined against hexamethylbenzene as an internal standard. | ||
Conclusions
In conclusion, we have demonstrated that diphenylacetylene is a versatile π-accepting ligand which enables the facile synthesis of a family of mono- and dinickelate complexes. This methodology is compatible with a range of aryl substituents and alkali-metals, allowing the first homologous alkali-metal (AM = Li, Na, K) nickelate series to be isolated and characterised. These well-defined heterobimetallic complexes provide a rich platform to investigate and compare the unique spectroscopic and structural features of alkali-metal nickelates and help us understand the design principles required to access new low-valent nickelates. In addition, the catalytic ability of the diphenylacetylene stabilised alkali-metal nickelates has also been investigated for the [2 + 2 + 2] cyclotrimerisation of diphenylacetylene, or the insertion of diphenylacetylene into biphenylene. Overall, these studies advance our understanding on how highly reactive mixed alkali-metal/Ni(0) complexes can be stabilised and structurally defined, as well as expanding on their potential to mediate catalytic C–C bond forming processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The X-ray crystal structure determination service unit at Universität Bern is acknowledged for measuring, solving, refining, and summarising all new structures. The Synergy diffractometer used for X-ray single crystal structure determination was partially funded by the Swiss National Science Foundation (SNSF) within the R'Equip programme (project number 206021_177033). We thank Dr Ilche Gjuroski (NMR) and Andrea Bill (CHN) for analytical services, and Universität Bern and the SNSF (project grant 188573 to EH) for generous sponsorship of this research.Notes and references
- K. Fischer, K. Jonas, P. Misbach, R. Stabba and G. Wilke, Angew. Chem., Int. Ed. Engl., 1973, 12, 943–1026 CrossRef.
- V. K. Jonas and C. Krüger, Angew. Chem., Int. Ed. Engl., 1980, 19, 520–537 CrossRef.
- K. Fischer, K. Jonas and G. Wilke, Angew. Chem., Int. Ed. Engl., 1973, 12, 565–566 CrossRef.
- K. Jonas, P. Heimbach and G. Wilke, Angew. Chem., Int. Ed. Engl., 1968, 7, 949–950 CrossRef CAS.
- V. B. Bogdanović, M. Kröner and G. Wilke, Justus Liebigs Ann. Chem., 1966, 669, 1–23 CrossRef PubMed.
- K. Jonas, K. R. Pörschke, C. Krüger and Y. H. Tsay, Angew. Chem., Int. Ed. Engl., 1976, 15, 621–922 CrossRef.
- K. Jonas, Angew. Chem., Int. Ed. Engl., 1976, 15, 47 CrossRef.
- D. J. Brauer, C. Krüger, P. J. Roberts and Y. H. Tsay, Angew. Chem., Int. Ed. Engl., 1976, 15, 48–49 CrossRef.
- K. R. Pörschke, K. Jonas, G. Wilke, R. Benn, R. Mynott and R. Goddard, Chem. Ber., 1985, 118, 275–297 CrossRef.
- K. R. Pörschke, K. Jonas and G. Wilke, Chem. Ber., 1988, 121, 1913–1919 CrossRef.
- W. Kaschube, K. R. Pörschke, K. Angermund, C. Krüger and G. Wilke, Chem. Ber., 1988, 121, 1921–1929 CrossRef CAS.
- K. R. Pörschke, W. Kleimann, Y. H. Tsay, C. Krüger and G. Wilke, Chem. Ber., 1990, 123, 1267–1273 CrossRef.
- K. R. Pörschke and G. Wilke, Chem. Ber., 1985, 118, 313–322 CrossRef.
- K. R. Pörschke and G. Wilke, J. Organomet. Chem., 1988, 349, 257–261 CrossRef.
- K. R. Pörschke and G. Wilke, J. Organomet. Chem., 1988, 358, 519–524 CrossRef.
- C. Krüger and Y. H. Tsay, Angew. Chem., Int. Ed. Engl., 1973, 12, 998–999 CrossRef.
- K. Jonas, Angew. Chem., Int. Ed. Engl., 1973, 12, 997–998 CrossRef.
- K. Jonas, D. J. Brauer, C. Krüger, P. J. Roberts and Y. H. Tsay, J. Am. Chem. Soc., 1976, 98, 74–81 CrossRef CAS.
- A. M. Borys and E. Hevia, Angew. Chem., Int. Ed., 2021, 60, 24659–24667 CrossRef CAS PubMed.
- A. M. Borys, L. A. Malaspina, S. Grabowsky and E. Hevia, Angew. Chem., Int. Ed., 2022, 61, e202209797 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- H. Ogawa, H. Minami, T. Ozaki, S. Komagawa, C. Wang and M. Uchiyama, Chem. – Eur. J., 2015, 21, 13904–13908 CrossRef CAS PubMed.
- K. Kojima, Z. K. Yang, C. Wang and M. Uchiyama, Chem. Pharm. Bull., 2017, 65, 862–868 CrossRef CAS PubMed.
- A. M. Borys and E. Hevia, Synthesis, 2022, 2976–2990 CrossRef CAS.
- R. J. Somerville, A. M. Borys, M. Perez-Jimenez, A. Nova, D. Balcells, L. A. Malaspina, S. Grabowsky, E. Carmona, E. Hevia and J. Campos, Chem. Sci., 2022, 13, 5268–5276 RSC.
- E. L. Muetterties, W. R. Pretzer, M. G. Thomas, B. F. Beier, D. L. Thorn, V. W. Day and A. B. Anderson, J. Am. Chem. Soc., 1978, 100, 2090–2096 CrossRef CAS.
- V. W. Day, S. S. Abdel-Meguid, S. Dabestani, M. G. Thomas, W. R. Pretzer and E. L. Muetterties, J. Am. Chem. Soc., 1976, 98, 8289–8291 CrossRef CAS.
- J. Wei, W. X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2015, 54, 5999–6002 CrossRef CAS PubMed.
- A. Enachi, D. Baabe, M. K. Zaretzke, P. Schweyen, M. Freytag, J. Raeder and M. D. Walter, Chem. Commun., 2018, 54, 13798–13801 RSC.
- T. Schaub and U. Radius, Chem. – Eur. J., 2005, 11, 5024–5030 CrossRef CAS PubMed.
- B. L. Edelbach, R. J. Lachicotte and W. D. Jones, Organometallics, 1999, 18, 4660–4668 CrossRef CAS.
- H. J. Reich, D. P. Green, M. A. Medina, W. S. Goldenberg, B. Ö. Gudmundsson, R. R. Dykstra and N. H. Phillips, J. Am. Chem. Soc., 1998, 120, 7201–7210 CrossRef CAS.
- A. M. Borys and E. Hevia, Organometallics, 2021, 40, 442–447 CrossRef CAS.
- B. Cordero, A. E. Platero-prats, M. Rev, J. Echeverr, E. Cremades and F. Barrag, Dalton Trans., 2008, 2832–2838 RSC.
- C. A. Murillo, Multiple Bonds Between Metal Atoms, Springer-Verlag, New York, 2005, pp. 633–667 Search PubMed.
- M. Bolte, CSD Commun., 2011 DOI:10.5517/ccxmqs0.
- W. Uhl, E. Er and M. Matar, Z. Anorg. Allg. Chem., 2006, 632, 1011–1017 CrossRef CAS.
- D. T. Carey, F. S. Mair, R. G. Pritchard, J. E. Warren and R. J. Woods, Eur. J. Inorg. Chem., 2003, 3464–3471 CrossRef CAS.
- D. Walther, M. Stollenz and H. Görls, Organometallics, 2001, 20, 4221–4229 CrossRef CAS.
- S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2915 CrossRef CAS.
- S. Kotha, E. Brahmachary and K. Lahiri, Eur. J. Org. Chem., 2005, 4741–4767 CrossRef CAS.
- P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS.
- W. Reppe, O. Schlichting, K. Klager and T. Toepel, Justus Liebigs Ann. Chem., 1948, 560, 1–92 CrossRef CAS.
- W. Reppe and W. J. Schweckendiek, Justus Liebigs Ann. Chem., 1948, 560, 104–116 CrossRef CAS.
- B. F. Straub and C. Gollub, Chem. – Eur. J., 2004, 10, 3081–3090 CrossRef CAS PubMed.
- J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890–3908 CrossRef CAS PubMed.
- A. Thakur and J. Louie, Acc. Chem. Res., 2015, 48, 2354–2365 CrossRef CAS PubMed.
- S. K. Rodrigo, I. V. Powell, M. G. Coleman, J. A. Krause and H. Guan, Org. Biomol. Chem., 2013, 11, 7653–7657 RSC.
- S. Pal and C. Uyeda, J. Am. Chem. Soc., 2015, 137, 8042–8045 CrossRef CAS PubMed.
- D. J. Brauer and C. Krüger, J. Organomet. Chem., 1976, 122, 265–273 CrossRef CAS.
- M. Fairley, L. Davin, A. Hernán-Gómez, J. García-Álvarez, C. T. O'Hara and E. Hevia, Chem. Sci., 2019, 10, 5821–5831 RSC.
- L. Davin, A. Hernán-Gómez, C. McLaughlin, A. R. Kennedy, R. McLellan and E. Hevia, Dalton Trans., 2019, 48, 8122–8130 RSC.
- M. De Tullio, A. M. Borys, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Chem Catal., 2021, 1, 1308–1321 CrossRef.
- T. X. Gentner, A. R. Kennedy, E. Hevia and R. E. Mulvey, ChemCatChem, 2021, 13, 2371–2378 CrossRef CAS.
- A. W. J. Platten, A. M. Borys and E. Hevia, ChemCatChem, 2022, 14, 2–7 CrossRef.
- J. J. Eisch, A. M. Piotrowski, K. I. Han, C. Krüger and Y. H. Tsay, Organometallics, 1985, 4, 224–231 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic details, crystallographic information, and NMR spectra. CCDC 2221333–2221339. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00069a |
| This journal is © The Royal Society of Chemistry 2023 |