
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe coordination chemistry of 2,4,6-oxy functionalised 1,3,5-triphosphinines†
Anne Sofie
Abels
a,
Frederik
Eiler
 a,
Grégoire
Le Corre
a,
Pascal
Jurt
a,
Michael
Wörle
a,
René
Verel
a,
Zoltan
Benkő
b and
Hansjörg
Grützmacher
*a
a,
Grégoire
Le Corre
a,
Pascal
Jurt
a,
Michael
Wörle
a,
René
Verel
a,
Zoltan
Benkő
b and
Hansjörg
Grützmacher
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog Weg 1, Hönggerberg, 8093 Zürich, Switzerland. E-mail: hgruetzmacher@ethz.ch; abelsa@inorg.chem.ethz.ch
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, 1111 Budapest, Műegyetem rakpart 3, Hungary. E-mail: benko.zoltan@vbk.bme.hu
First published on 6th February 2023
Abstract
A number of stable group 6 metal complexes bearing 2,4,6-oxy functionalised 1,3,5-triphosphinines, phosphorus containing heterocyclic ligands with a central C3P3 core, were synthesised such that a complete series of [M{P3C3(OX)3}(CO)3] compounds is obtained [M = Cr(0), Mo(0), W(0); X = H, SitBuPh2, B(ipc)2]. In all complexes, the triphosphinine coordinates in a η6-binding mode via the delocalized 6π-system of the ring. The ligand properties can be tuned by changing the substituent on the oxygen centre. The π-electron accepting properties of the ligand increases in the following order: P3C3(OH)3 < P3C3(OSitBuPh2)3 < P3C3(OB(ipc)2)3. This trend is reflected in the structures determined by X-ray crystallography, and the ν(CO) stretching frequencies determined by IR spectroscopy. The collected data raise questions with respect to the frequently made assumption that phosphinines act as stronger π-acceptors with respect to arenes and thereby deplete electron density at the metal centres. With P3C3(OH)3 as an η6-coordinated ligand further molecules can be coordinated in the second coordination sphere via hydrogen bonds, which may be of interest for the construction of coordination polymers.
Introduction
Since Märkl and Ashe successfully prepared the first monophosphinines,1,2 these phosphorus heterocycles and their coordination chemistry have been studied extensively.3–10 Nowadays, these compounds serve as ligands in transition metal complexes that are active homogeneous catalysts, for example in hydroformylation, hydrogenation and oligomerisation reactions.6,9,11 In contrast to monophosphinines, the chemistry of phosphinines containing three phosphorus centres, namely triphosphinines, is much less developed.12,13 Generally, 1,3,5-triphosphinines are obtained by transition metal mediated trimerisation of kinetically stabilised phosphaalkynes.14–17 The range of reported 1,3,5-triphosphinines is rather limited and most carry tertiary butyl, tBu, substituents or related bulky hydrocarbon groups attached to the C atom of the ring. In combination, these studies led to the assumption that phosphorus-containing heterocycles have strong π-accepting properties with respect to arenes, making them excellent ligands for binding low-valent electron rich metal centres in a η6-coordination mode.4,18–26In 2017, Grützmacher and co-workers reported on a new synthetic route to access functionalized triphosphinines by the reaction of sodium phosphaethynolate, [Na(OCP)], with commercially available (−)-B-chlorodi(isopinocampheyl)borane, (ipc)2B-Cl, forming the boryloxy-functionalised phosphaalkyne (ipc)2B–O–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P as slightly thermodynamically preferred product that trimerises in absence of a catalyst to form a 1,3,5-triphosphinine with a central six-membered P3C3 ring (Scheme 1, P3C3–B) on a multi-gram scale.27 This compound is the first triphosphinine that can readily be derivatised and thereby the electronic properties can be tuned by changing the substituent at the oxygen atom. Treatment of triphosphinine P3C3–B with an excess of alcohol yields the deprotected tris(hydroxyl) triphosphinine P3C3(OH)3 (P3C3–H), the phosphorus analogue of cyanuric acid. But unlike cyanuric acid, P3C3(OH)3 is rather labile, therefore could not be isolated and was only stable in solution.
P as slightly thermodynamically preferred product that trimerises in absence of a catalyst to form a 1,3,5-triphosphinine with a central six-membered P3C3 ring (Scheme 1, P3C3–B) on a multi-gram scale.27 This compound is the first triphosphinine that can readily be derivatised and thereby the electronic properties can be tuned by changing the substituent at the oxygen atom. Treatment of triphosphinine P3C3–B with an excess of alcohol yields the deprotected tris(hydroxyl) triphosphinine P3C3(OH)3 (P3C3–H), the phosphorus analogue of cyanuric acid. But unlike cyanuric acid, P3C3(OH)3 is rather labile, therefore could not be isolated and was only stable in solution.
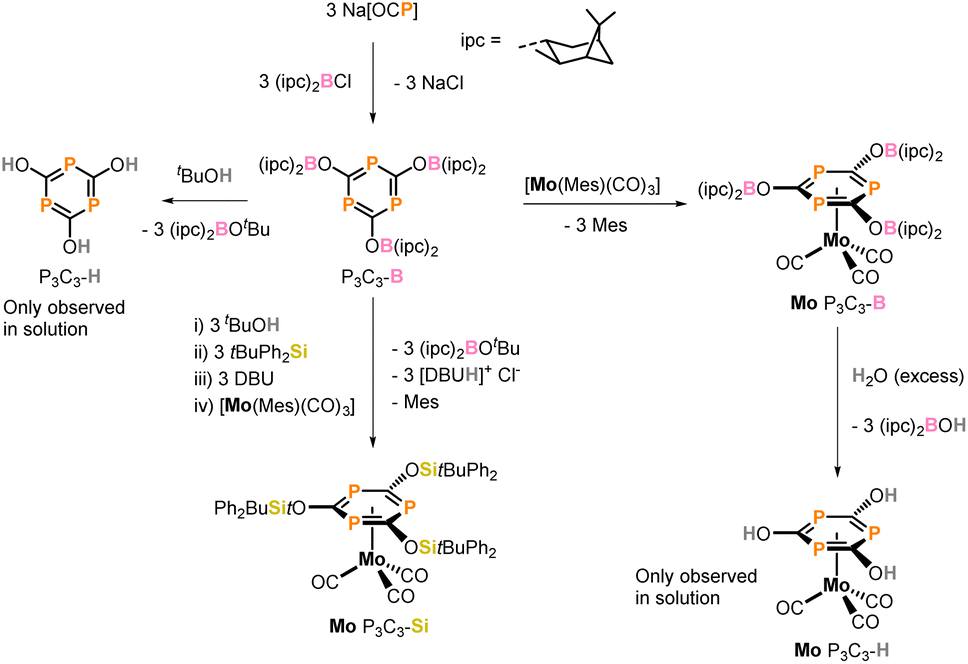 | ||
| Scheme 1 Previous synthesis of borylated compound P3C3–B from the reaction of Na[OCP] with (ipc)2BCl, the formation of the phosphorus analogue of cyanuric acid (P3C3–H) and the synthesis of molybdenum 2,4,6-oxy functionalised 1,3,5-triphosphinine complexes. | ||
Cyanuric acid is an industrially useful chemical and finds use in the production of herbicides,28,29 dyes30 and flame retardants.31–33 Furthermore, its trimeric structure and the ability to form hydrogen bonds makes cyanuric acid an attractive precursor for cross linkers in polymers34,35 and an building block in supramolecular networks.36–41 The formation of a stable P3C3(OH)3 compound may therefore be of interest. Furthermore, these type of P3C3 rings have shown that they are excellent ligands and coordinate in a η6-binding mode through the π-system of the ring to a molybdenum centre (Scheme 1). However, the molybdenum complex with P3C3(OH)3 as ligand could not be isolated, therefore key structural information and spectroscopic data is missing.
In this study we investigated the coordination chemistry of these 2,4,6-oxy functionalised 1,3,5-triphosphinines with group 6 metals in depth and we were able to isolate and analyse the phosphorus analogue of cyanuric acid as ligand in coordination compounds. This series of group 6 metal 2,4,6-tri-oxy-1,3,5-triphosphinine complexes bearing different substituents on the oxygen centre allowed us to study the physical properties of this class of compounds in detail by single-crystal X-ray structure determinations (XRD's), IR-, and NMR- spectroscopic analyses.
Results and discussion
The Mo(0) complexes [Mo{P3C3[OB(ipc)2]3}(CO)3] Mo–P3C3–B and [Mo{P3C3[O(SitBuPh2)]3}(CO)3] Mo–P3C3–Si were previously synthesised from [Mo(Mes)(CO)3] and isolated in crystalline form. The complex [Mo{P3C3(OH)3}(CO)3] Mo–P3C3–H could only be spectroscopically observed in solution.27 In straightforward ligand exchange reactions, the analogous tricarbonyl chromium and tungsten complexes of P3C3–B were synthesised from [M(NCMe)3(CO)3] (M = Cr and W) (Fig. 1). Single crystals of Cr–P3C3–B and W–P3C3–B were grown by slow evaporation of n-hexane solutions and analysed by single crystal diffraction. The silyloxy-substituted compound P3C3[O(SitBuPh2)]3, P3C3–Si, was prepared in solution27 and addition of the group 6 metal precursor to the reaction mixture yielded compounds Cr–P3C3–Si and W–P3C3–Si (Fig. 1). The reaction time varied from 1 hour for Cr–P3C3–Si to 5 days for W–P3C3–Si, the latter was heated to 40 °C to accelerate the reaction. The use of multiple reagents explains the rather low isolated yields (10–11%) in these one-pot syntheses of M–P3C3–Si (M = Cr, W) and it proved to be difficult to separate the desired complexes from a number of side products. Slow evaporation of n-hexane solutions of Cr–P3C3–Si and W–P3C3–Si gave single crystals suitable for X-ray diffraction studies. | ||
| Fig. 1 Synthesis of complexes M–P3C3–B (M = Cr, W), M–P3C3–Si (M = Cr, W) and M–P3C3–H (M = Cr, Mo, W). The crystal structures of W–P3C3–B, W–P3C3–Si and W–P3C3–H are given below the respective chemical structures. Hydrogen atoms and solvent molecules are omitted for clarity. Average interatomic distances are summarised in Table 1. The crystal structures of Cr–P3C3–B, Cr–P3C3–Si, Cr–P3C3–H and Mo–P3C3–H are included in the ESI.† | ||
To obtain the tris(hydroxyl)triphosphinine complexes, the group 6 metal tricarbonyl complexes of P3C3–B were dissolved in n-hexane and an excess of methanol was added to give a biphasic system. After shaking the reaction flask, the subsequently formed P3C3(OH)3 complexes could be isolated as orange (M = Cr) or yellow (M = Mo, W) solids from the methanol phase. The n-hexane phase contains the methoxyborane-(ipc)2B(OMe), which could easily be separated. Coordination of the P3C3(OX)3 rings [X = B(ipc)2: δ(31P) = 182.7 ppm, X = SitBuPh2: δ(31P) = 177.5 ppm; X = H: δ(31P) = 133.7 ppm]27 leads to a significant coordination shift Δδ = δ(P3C3[OX]3) − δ([M{P3C3[OX]3(CO)3}]) of the 31P NMR resonances to lower frequencies in the range of 161 to 198 ppm, which is especially pronounced in case of the parent tris(hydroxyl)triphosphinine P3C3(OH)3 (see Table 1, first entry). The Δδ values increase slightly in the order Cr < Mo < W within each series of ligand and SitBuPh2 < B(ipc)2 < H with respect of the group X bound to the oxygen centres of the P3C3(OX)3 ring. Comparable strong coordination shifts (Δδ > 160 ppm to lower frequencies) were also observed when 2,4,6-triphenyl-phosphinine, PC5H2(Ph)3, or 2,4,6-tert-butyltriphosphinine, P3C3(tBu)3, are η6-coordinated to a M(CO)3 fragment.42
| Cr–P3C3–B | Cr–P3C3–Si | Cr–P3C3–H | Mo–P3C3–B | Mo–P3C3–Si | Mo–P3C3–H | W–P3C3–B | W–P3C3–Si | W–P3C3–H | |
|---|---|---|---|---|---|---|---|---|---|
| Δδ | 166 | 161 | 191 | 176 | 167 | 193 | 185 | 177 | 198 |
| M–ct | 1.6407(8) | 1.6499(5) | 1.6817(8) | 1.753(1) | 1.778(6) | 1.8157(11) | 1.7462(11) | 1.7743(19) | 1.802(5) |
| P–C | 1.7578(13) | 1.7577(7) | 1.7597(9) | 1.765(4) | 1.769(2) | 1.7717(18) | 1.7687(18) | 1.767(3) | 1.773(8) |
| M–CO | 1.8947(18) | 1.8714(11) | 1.8667(12) | 2.022(4) | 2.005(2) | 1.989(3) | 2.024(3) | 2.009(5) | 2.001(12) |
| C–O | 1.1370(18) | 1.1453(12) | 1.1487(16) | 1.139(5) | 1.143(2) | 1.156(4) | 1.129(3) | 1.137(5) | 1.130(13) |
| C–P–C | 101.46(7) | 102.25(5) | 102.69(8) | 101.19(2) | 101.93(8) | 102.7(3) | 101.13(11) | 101.37(18) | 102.2(6) |
| P–C–P | 138.53(10) | 137.7(6) | 137.28(8) | 138.7(2) | 137.88(2) | 137.3(18) | 138.8(12) | 138.4(3) | 137.7(6) |
Single crystals of Cr–P3C3–B, W–P3C3–B, Cr–P3C3–Si, and W–P3C3–Si were grown by slow evaporation of n-hexane solutions and analysed by single crystal diffraction (XRD) (Fig. 1). Single crystals of the coordinated P3C3(OH)3 group 6 complexes were obtained by cooling a concentrated n-hexane solution of Cr–P3C3–H, Mo–P3C3–H, or W–P3C3–H in the presence of tBuOH. The structures of the P3C3–H metal complexes determined by XRD show interactions of the OH groups of the ring with the OH groups of tBuOH via hydrogen bonding. Related O–H⋯O hydrogen bonding was previously postulated for Mo–P3C3–H in THF solution.27Table 1 summarises selected spectroscopic data and bond lengths and angles of the solid state structures of the nine different chromium, molybdenum and tungsten triphosphinine complexes, bearing different functionalities at the oxygen centres in 1,3,5 position of the P3C3 ring.
In these piano stool complexes, the P3C3 ring is planar and binds in an η6-coordination mode to the metal centre (Fig. 1). The P–C bond lengths (1.76–1.78 Å) of the coordinated P3C3 rings are slightly elongated compared to the uncoordinated triphosphinine ring (1.736 Å).27 Compared to P3C3[OB(ipc)2]3 (C–P–Cav: 104°; P–C–Pav 136°), the C–P–C angles become slightly more acute (P–C–Pav 101.9°) while the P–C–P angles widen up slightly (P–C–Pav 138.0°) upon coordination to a M(CO)3 fragment. Within the complexes, these values vary rather little [C–P–C 100.6(3)–102.9(3)°; P–C–P 137.0(3)–140.2(4)], but independently of M, the boryloxy substituted complexes show the smallest C–P–C (largest P–C–P) angle, while the hydroxyl substituted show the largest C–P–C (smallest P–C–P) angle.
For every metal, the M–ct distance is the longest with P3C3(OH)3 as ligand (Cr–P3C3–H = 1.682 Å, Mo–P3C3–H = 1.816 Å, W–P3C3–H = 1.802 Å), followed by the siloxylated complexes (Cr–P3C3–Si = 1.650 Å, Mo–P3C3–Si = 1.778 Å, W–P3C3–Si = 1.774 Å) and the boryloxylated P3C3 metal complexes (Cr–P3C3–B = 1.619 Å, Mo–P3C3–B = 1.753 Å, W–P3C3–B = 1.746 Å). Although the differences are small (within 3σ), the Mo-complexes show always slightly longer Mo–ct distances compared to the corresponding W–ct distances despite the increasing covalent radii [rcov(Mo) = 1.54 Å; rcov(W) = 1.62 Å]. Complexes with chromium show expectedly significantly shorter Cr–ct distances [rcov(Cr) = 1.39 Å].43 (The atomic van der Waals radii of Cr, Mo, and W [rvdW(Cr) = 1.89 Å; rvdW(Mo) = 2.09 Å, rvdW(W) = 2.10 Å] reflect better the observed trends in the M–P3C3–X complexes with M in the oxidation state 0, but also suggest that the M–ct bond lengths should steadily increase in the order W > Mo > Cr).44 Indeed this trend is observed for [M[P3C3(tBu)3](CO)3] (M = Cr, Mo, W), whose structures were calculated by DFT methods. Here the increase in M–ct distances corresponds to the van der Waals radii of the metals.23,44
In [M(L)x(CO)y] complexes, a ligand L with π-accepting properties competes with carbonyl co-ligands for M → π*(CO) electron back donation. Consequently the electronic properties of a ligand L in [M(L)x(CO)y] complexes are reflected in the M–C and C–O distances as well as in the CO stretching frequencies ν(CO) of the carbonyl co-ligands.45 A ligand L of high π-electron accepting property will lead to long M–C bonds to the CO ligand, short C–O bonds and ν(CO) at high wave numbers. In case of complexes with Cr(CO)3 or Mo(CO)3 fragments, the ones with P3C3–B rings show the longest M–CO distances while, on the contrary, the M–CO distances are the shortest in complexes with the P3C3–H ligand. As expected, the opposite trends are observed with respect to the C–O bond lengths.
Another frequently used instrument in order to evaluate the electronic properties of L in[M(L)x(CO)y] complexes are the ν(CO) of the carbonyl co-ligands. It is generally accepted that the relative ratio of ligand-to-metal electron donation d (M ← L) to metal-to-ligand back donation b (M → L), denoted here as d/b, decreases in the order CO ≈ alkynes < olefins < carbenes < phosphines < amines (simplified meaning that the π-electron accepting property of the respective ligand decreases in this order).46–48 Consequently a decrease of ν(CO) is expected along this ligand line. In Tables 2–4 a compilation of IR data is given for selected, comparable chromium, molybdenum, and tungsten tricarbonyl complexes with simple monodentate σ-donor/π-acceptor ligands, arene ligands, and phosphinine ligands. In all complexes the M(CO)3 fragment (M = Cr, Mo, W) adopts a facial coordination mode and reference is made only to the symmetrical ν(CO) stretching frequency of A1 symmetry at higher wave numbers (in case of deviation from local C3v symmetry of the M(CO)3 fragment, reference is made to ν(CO) at the highest wave number). As reference compounds the hexacarbonyl complexes [M(CO)6] are chosen, for which ν(CO) is quite similar independently on the metal (M = Cr: 1985 cm−1; Mo = 1985 cm−1; M = W: 1981 cm−1). As expected, the replacement of three CO groups in facial position by ligands with an increasingly stronger σ or π-donating but poorer π-accepting property – that is an increasingly higher d/b ratio – as in the series P(OMe)3 < PMe2Ph < NCMe – will increasingly lower the ν(CO) stretching frequency as a result of increasing electron density on the metal centre (see entries 2–4 in each table). This effect is remarkably similar for each metal and the variation Δν(CO) from [M(CO)6] to [M(NCMe)3(CO)3] is about 70 cm−1 to lower wave numbers. In the benzene complexes [M(C6H6)(CO)3] a slight blue-shift of ν(CO) to higher wave numbers is observed for M = Mo (1987 cm−1) or W (1990 cm−1) (entries 5 in Tables 3 and 4), while in [Cr(C6H6)(CO)3] (1971 cm−1) a red-shift by 14 cm−1 to lower wave numbers occurs (entry 5 in Table 2). σ-Donating substituents like methyl groups in 1,3,5-position of the arene ring increase the d/b ratio and lower the ν(CO) by 6 to 15 cm−1 (entries 6 in Tables 2–4). The IR data for a range of Cr(0) complexes with arene ligands functionalised in 1,3,5-position are available (see entries 7–10 in Table 2). σ-Electron and π-electron donating substituents like tBu or hydroxyl groups shift ν(CO) by about 25 cm−1 to lower wave numbers. Electron accepting groups like carboxy groups, CO2Me, or triflate groups, OSO2CF3, in 1,3,5-position shift ν(CO) to higher wave numbers by 32 cm−1 or 48 cm−1, respectively. Hence, the range by which A1ν(CO) can be changed in these type of complexes is about 70 cm−1.
| ν CO (cm−1) | ||
|---|---|---|
| 1 | [Cr(CO)6]49 | 1985 |
| 2 | [Cr{P(OMe)3}(CO)3]50 | 1962, 1888, 1874 |
| 3 | [Cr{PMe2Ph}(CO)3]51 | 1920, 1820 |
| 4 | [Cr(NCMe)3(CO)3]52 | 1910, 1782 |
| 5 | [Cr(C6H6)(CO)3]53 | 1971, 1860 |
| 6 | [Cr(Mes)(CO)3]54 | 1965, 1852 |
| 7 | [Cr{C6H3(tBu)3}(CO)3]55 | 1947, 1871 |
| 8 | [Cr{C6H3(OH)3}(CO)3]56 | 1945, 1850 |
| 9 | [Cr{C6H3(CO2Me)3}(CO)3]57 | 2003, 1947 |
| 10 | [Cr{C6H3(OSO2CF3)3}(CO)3]56 | 2019, 1947 |
| 11 | [Cr{PC5H2(Ph)3}(CO)3]58 | 1976, 1915, 1907 |
| 13 | [Cr{P3C3(tBu)3}(CO)3]23 | 1979, 1926 |
| 14 | [Cr{P3C3(OH)3}(CO)3] Cr–P3C3–H | 1980, 1921 |
| 15 | [Cr{P3C3[O(SitBuPh2)]3}(CO)3] Cr–P3C3–Si | 1983, 1927 |
| 16 | [Cr{P3C3[OB(ipc)2]3}(CO)3] Cr–P3C3–B | 2012, 1967 |
| ν CO (cm−1) | ||
|---|---|---|
| 1 | [Mo(CO)6]49 | 1985 |
| 2 | [Mo{P(OMe)3}(CO)3]50 | 1974, 1898, 1884 |
| 3 | [Mo{PMe2Ph}(CO)3]51 | 1939, 1832, 1824 |
| 4 | [Mo(NCMe)3(CO)3]52 | 1915, 1783 |
| 5 | [Mo(C6H6)(CO)3]49 | 1987, 1916 |
| 6 | [Mo(Mes)(CO)3]49 | 1973, 1901 |
| 7 | [Mo{PC5H2(Ph)3}(CO)3]42 | 1996, 1942, 1933 |
| 8 | [Mo{P3C3(OH)3}(CO)3] Mo–P3C3–H | 1988, 1925 |
| 9 | [Mo{P3C3[O(SitBuPh2)]3}(CO)3] Mo–P3C3–Si | 1993, 1941 |
| 10 | [Mo{P3C3[OB(ipc)2]3}(CO)3] Mo–P3C3–B | 2022, 1972 |
| ν CO (cm−1) | ||
|---|---|---|
| 1 | [W(CO)6]49 | 1981 |
| 2 | [W{P(OMe)3}(CO)3]50 | 1970, 1893, 1878 |
| 3 | [W(PMe2Ph)3(CO)3]51 | 1932, 1926 |
| 4 | [W(NCMe)3(CO)3]52 | 1915, 1793 |
| 5 | [W(C6H6)(CO)3]59 | 1990, 1916 |
| 6 | [W(Mes)(CO)3]59 | 1975, 1902 |
| 7 | [W{P3C3(OH)3}(CO)3] W–P3C3–H | 1987, 1921 |
| 8 | [W{P3C3[O(SitBuPh2)]3}(CO)3] W–P3C3–Si | 1989, 1921 |
| 9 | [W{P3C3[OB(ipc)2]3}(CO)3] W–P3C3–B | 2019, 1963 |
What effect will be evoked when one CH unit is replaced by a phosphorus centre? In the monophosphinine complexes [Cr{PC5H2(2,4,6-Ph)3}(CO)3] (1976 cm−1, entry 11 in Table 2) and [Mo{PC5H2(2,4,6-Ph)3}(CO)3] (1996 cm−1, entry 7 in Table 3) ν(CO) shifts slightly (5, 9 cm−1) with respect to the benzene complexes, or modestly (11, 23 cm−1) with respect to the mesitylene complexes to higher wave numbers indicating indeed some depletion of the electron density at the metal centre with respect to an arene ligand. The further substitution of two additional CH units by phosphorus leading to triphosphinine ligands raises the A1ν(CO) to even higher frequencies. A significant blue-shift of 32 cm−1 is observed comparing [Cr{C6H3(tBu)3}(CO)3] (1947 cm−1; entry 7 in Table 2) and [Cr{P3C3(tBu)3}(CO)3] (1979 cm−1; entry 13 in Table 2). As in arene complexes, σ-donating tBu substituents in 1,3,5-position of a triphosphinine ligand have a very similar effect than π-donating hydroxyl groups as a comparison between [Cr{P3C3(OH)3}(CO)3] (Cr–P3C3–H) (1980 cm−1, entry 14 Table 2) and [Cr{P3C3(tBu)3}(CO)3] (1979 cm−1, entry 13 Table 2) shows.
How is ν(CO) influenced by electronically different substituents in 2,4,6-position of a triphosphinine ligand? This question can be partly answered by inspecting the last three entries 14–16 in Table 2. While replacing hydroxyl groups in Cr–P3C3–H by siloxy groups has a marginal influence, the replacement by boryloxy groups has a significant impact. The complex [Cr{P3C3[OB(ipc)2]3}(CO)3] (Cr–P3C3–B) has the highest ν(CO) = 2012 cm−1 (entry 16 Table 2) of all triphosphinine complexes indicating that a B(ipc)2 group efficiently abstracts electron density from the π-electron donating oxygen centres connected to the P3C3 ring thereby depleting the overall electron density at the chromium centre. But the variation between the complex with an “electron rich” P3C3(tBu)3 [or P3C3(OH)3] ligand and the one with an “electron poor” P3C3[OB(ipc)2]3 ligand is smaller [Δν(CO) = 32 cm−1] than the one seen between “electron rich” and “electron poor” arene ligands (70 cm−1; vide supra). This span of Δν(CO) is seen for all [M{P3C3(OX)3}(CO)3] complexes independently on the metal centre M = Cr, Mo, W (see the last three entries in every Tables 2–4).
The nature of the metal centre has also an influence on the ν(CO) stretching frequencies but this is rather small and for all type of ligands the largest step is seen between the Cr(0) and Mo(0) complexes (shift of about 10 cm−1 to higher wave numbers) while there is almost no change between Mo(0) and W(0) complexes.
That for every [M{P3C3(OH)3}(CO)3] (entry 14, entry 8, entry 7 in Tables 2, 3 and 4, respectively) or [M{P3C3[O(SitBuPh2)]3}(CO)3] complex (M = Cr, Mo, W; entry 15, entry 9, entry 8 in Tables 2, 3 and 4, respectively) the A1ν(CO) is quite similar to the one observed in the corresponding parent benzene complex [M(C6H6)(CO)3] (entry 5 in Tables 2, 3 and 4, respectively) may be due to counterbalancing effect of the π-electron donating hydroxyl groups and π-electron withdrawing P3C3 core.
183W satellites (natural abundance 14.3%)60 were observed in the 31P{1H} NMR spectra of the W(0) triphosphinine complexes prepared in this work. The JWP couplings decrease very slightly from W–P3C3–B (21.4 Hz) to W–P3C3–Si (19.1 Hz) and W–P3C3–H (16.4 Hz). These data demonstrate that, as expected, a shorter W–ct distance correlates with a larger 31P–183W coupling constant as a result of a stronger π-accepting ligand (see Fig. 1 and 2, in the ESI† for a correlation of W–ct, JWP, ν(CO), and δ183W). No JWP coupling has been reported for [W(P3C3tBu3)(CO)3] and a η6-coordinated monophosphinine tungsten complex has not been prepared yet. Only the JWP coupling constants of η1-phosphinine tungsten complexes are reported in the literature, as for example in the hexakis-(η1-phosphinine)tungsten complex [W(η1-PC5H5)6], which has a much larger direct P–W coupling of 304 Hz.61
 | ||
| Fig. 2 M(CO)3 complex of bis(imidazolium)-1,3-diphosphete-diide (IPD) (M = Cr, Mo). | ||
Lastly, 183W resonances were measured using a triple resonance broad-band (TBI) probe with 1H/2D/31P/BB channels. A 31P–183W heteronuclear multiple-quantum coherence (HMQC) experiment was performed, enhancing the sensitivity of the low γ183W nucleus. The resonances in the 183W NMR spectrum shift very slightly to higher frequencies in the order W–P3C3–B (δ183W = −3146 ppm) < W–P3C3–Si (δ183W = −3140 ppm) < W–P3C3–H (δ183W = −3110 ppm). The number of reported 183W NMR chemical shifts for tungsten(0) complexes is rather limited and span from about −3500 to −2500 ppm. The most shielded 183W NMR shift is observed for [W(CO)6], which shows a 183W NMR resonance at δ183W = −3446 ppm and is chosen as reference compound here.62 Replacement of three CO groups by a phosphite ligand like P(OMe)3 (a strong σ-donor and medium π-acceptor) leads to a high frequency shift of about 150 ppm to δ183W = −3297 ppm in [W{P(OMe)3}3(CO)3].63 This deshielding effect becomes even more pronounced (630 ppm) in [W(PMe2Ph)3(CO)3] (δ183W = −2818 ppm) where three strong σ-electron donor but relatively weak π-acceptor ligands were incorporated in the ligand sphere.64 To the best of our knowledge, no δ183W NMR shifts are reported for any arene tungsten carbonyl complex. The shifts of the 183W resonances of [W{P3C3(OX)3}(CO)3] to higher frequencies by about 300–336 ppm places them in between [W(CO)6] and [W(PMe2Ph)3(CO)3] indicating that to some extent the electronic effect on the 183W resonance caused by the replacement of three CO groups in facial position by one P3C3(OX)3 ligand may be comparable to three P(OMe)3 ligands.
Conclusions
The successful preparation, and isolation of a 3 × 3 matrix of complexes [M{P3C3(OX)3}(CO)3] using three different metals M = Cr, Mo, W and three different substituents X = B(ipc)2, SitBu Ph2, H allowed to systematically study and evaluate the effects provoked by these variations on a variety of spectroscopic data. The η6-binding mode entails a large coordination shift of >160 ppm to lower frequencies regardless the nature of the phosphinine. As has been observed in the coordination chemistry of 2,4,6-tBu-1,3,5-triphosphinine, the C–P–C angles are narrowed while the P–C–P angles are widened upon binding to a M(CO)3 fragment (note that the bulky and σ-electron donating tBu groups lead to slightly larger C–P–C and more acute P–C–P angles in both the uncoordinated molecule as well as in the complexes).23 As expected, the distances between the Cr centre and the centroid of the P3C3 rings in all complexes are significantly smaller (by about 8%) than the Mo–Ct and W–ct distances, which are almost identical. This is attributed to the contracted d-orbital shell of Cr versus those of Mo and W.65,66 This contraction may also be seen as reason for the fact that in all Cr(0) complexes listed in Table 2 independently of the type of ligand the ν(CO) is somewhat smaller than in analogous Mo and W complexes because efficient M → L electron back donation (which raises ν(CO)) requires an efficient orbital overlap.As mentioned in the introduction, phosphinines are considered as especially good π-electron density accepting ligands. We recalculated the HOMO–LUMO gap of C6H6 (6.60 eV), PC5H4 (5.23 eV), 1,3-P2C4H4 (4.88 eV), and 1,3,5-P3C3H3 (4.63 eV) with density functional theory (DFT) methods (B3LYP/6-311+G**) (see ref. 27 and the ESI† for details). While indeed there is a significant closing of the gap by ΔE = 1.37 eV when converting C6H6 to PC5H5, additional substitution leads to much smaller changes (<0.4 eV). That P3C3(OH)3 has an especially small HOMO–LUMO gap of 4.27 eV is mainly due to the fact that the three hydroxyl groups in 1,3,5-position lift the HOMO in energy. Given these data, it is not surprising that the effect of replacing a monophosphinine by a triphosphinine has only a modest effect on the ν(CO) stretching frequencies and thereby the electron density at the metal centre. The variation of the substituents in 1,3,5-position of an arene ligand or 2,4,6-position of a 1,3,5-triphosphinine allows to modify ν(CO) over an range of about 70 cm−1 or 30 cm−1, respectively, fully in accord with expectations (that is an electron donating ligand lowers ν(CO); an electron accepting ligand raises ν(CO)). However, it remains surprising that in comparison to benzene complexes phosphinines provoke only modest (<10 cm−1 when the parent molecules without strong electronic substituent effects are taken into account) blue-shifts of ν(CO). Under the assumption that these heterocycles are really “strong π-electron acceptor” ligands more pronounced deviations might have been expected. For example, we observed that the coordination of bis(imidazolium)-1,3-diphosphete-diide (IPD) – an unsual molecule with a central 6π-aromatic C2P2 ring of biradicaloid character – to form M(CO)3 complexes A (Fig. 2) in which ν(CO) is shifted by about 100 cm−1 to lower frequencies indicating strong electron donation to the metal centre by these cycles, indeed.67 There is no question that phosphinines act as strongly binding ligands to metals (in their synthesis frequently an arene ligand in a precursor complex is displaced by a phosphinine). But we feel that the characterization of phosphinines as electron accepting ligands is an oversimplification and the electronic structure of phosphinine complexes is not fully understood yet. High-level quantum chemical methods should be applied to gain a better insight into electrostatic interactions and the electron donating properties of phosphinines as well.
Conflicts of interest
There are no conflicts to declare.References
- G. Märkl, Angew. Chem., 1966, 78, 907–908 CrossRef.
- A. J. Ashe, J. Am. Chem. Soc., 1971, 93, 3293–3295 CrossRef CAS.
- P. L. Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef.
- P. Le Floch and F. Mathey, Coord. Chem. Rev., 1998, 178–180, 771–791 CrossRef CAS.
- N. Mezailles, F. Mathey and P. Le Floch, Prog. Inorg. Chem., 2001, 49, 455–550 CAS.
- L. Weber, Angew. Chem., Int. Ed., 2002, 41, 563–572 CrossRef CAS.
- L. Kollar and G. Keglevich, Chem. Rev., 2010, 110, 4257–4302 CrossRef CAS PubMed.
- C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 2, 187–202 CrossRef.
- C. Müller and D. Vogt, Dalton Trans., 2007, 47, 5505–5523 RSC.
- C. Müller and D. Vogt, C. R. Chim., 2010, 13, 1127–1143 CrossRef.
- N. T. Coles, A. S. Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 433, 213729–213756 CrossRef CAS.
- H. Heydt, New Asp. Phosphorus Chem. II. 223, 2003, vol. 223, pp. 215–249 Search PubMed.
- R. L. Falconer and C. A. Russell, Coord. Chem. Rev., 2015, 297–298, 146–167 CrossRef CAS.
- A. R. Barron and A. H. Cowley, Angew. Chem., Int. Ed. Engl., 1987, 26, 907–908 CrossRef.
- R. Milczarek, W. Rüsseler, P. Binger, K. Jonas, K. Angermund, C. Krüger and M. Regitz, Angew. Chem., Int. Ed. Engl., 1987, 26, 908–909 CrossRef.
- P. Binger, J. Stannek, B. Gabor, R. Mynott, J. Bruckmann, C. Krüger and S. Leininger, Angew. Chem., Int. Ed. Engl., 1995, 34, 2227–2230 CrossRef CAS.
- F. Tabellion, C. Peters, U. Fischbeck, M. Regitz and F. Preuss, Chem. – Eur. J., 2000, 6, 4558–4566 CrossRef CAS PubMed.
- P. L. Arnold, F. G. N. Cloke, P. B. Hitchcock and J. F. Nixon, J. Am. Chem. Soc., 1996, 118, 7630–7631 CrossRef CAS.
- P. Binger, S. Stutzmann, J. Stannek, B. Gabor and R. Mynott, Eur. J. Inorg. Chem., 1999, 1, 83–86 CrossRef.
- C. W. Tate, P. B. Hitchcock, G. A. Lawless, Z. Benkő, L. Nyulászi and J. F. Nixon, C. R. Chim., 2010, 13, 1063–1072 CrossRef CAS.
- M. D. Francis, C. Holtel, C. Jones and R. P. Rose, Organometallics, 2005, 24, 4216–4225 CrossRef CAS.
- K. Eggers, F. W. Heinemann, M. Hennemann, T. Clark, P. Binger and U. Zenneck, C. R. Chim., 2010, 13, 1203–1212 CrossRef CAS.
- S. B. Clendenning, J. C. Green and J. F. Nixon, J. Chem. Soc., Dalton Trans., 2000, 9, 1507–1512 RSC.
- J. Waluk, H. P. Klein, A. J. Ashe and J. Michl, Organometallics, 1989, 8, 2804–2808 CrossRef CAS.
- C. Elschenbroich, M. Nowotny, B. Metz, W. Massa, J. Graulich, K. Biehler and W. Sauer, Angew. Chem., Int. Ed. Engl., 1991, 30, 547–550 CrossRef.
- R. Gleiter, H. Lange, P. Binger, J. Stannek, C. Krüger, J. Bruckmann, U. Zenneck and S. Kummer, Eur. J. Inorg. Chem., 1998, 1998, 1619–1621 CrossRef.
- R. Suter, Y. Mei, M. Baker, Z. Benkő, Z. Li and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 1356–1360 CrossRef CAS PubMed.
- A. M. Cook and R. Huetter, J. Agric. Food Chem., 1981, 29, 1135–1143 CrossRef CAS.
- H. J. Sanders, Chem. Eng. News, 1981, 59, 20–36 CrossRef CAS.
- Y. Yu, J. C. Yu, C.-Y. Chan, Y.-K. Che, J.-C. Zhao, L. Ding, W.-K. Ge and P.-K. Wong, Appl. Catal., B, 2005, 61, 1–11 CrossRef CAS.
- P. Gijsman, R. Steenbakkers, C. Fürst and J. Kersjes, Polym. Degrad. Stab., 2002, 78, 219–224 CrossRef CAS.
- P. Zhao, M. Zhang, D. Wu and Y. Liu, Korean J. Chem. Eng., 2013, 30, 1687–1690 CrossRef CAS.
- D. Jiang, C. Sun, Y. Zhou, H. Wang, X. Yan, Q. He, J. Guo and Z. Guo, Fibers Polym., 2015, 16, 388–396 CrossRef CAS.
- K. P. Nair, V. Breedveld and M. Weck, Macromolecules, 2008, 41, 3429–3438 CrossRef CAS.
- K. P. Nair, V. Breedveld and M. Weck, Soft Matter, 2011, 7, 553–559 RSC.
- B. Roy, P. Bairi and A. K. Nandi, RSC Adv., 2014, 4, 1708–1734 RSC.
- E. E. Simanek, M. Mammen, D. M. Gordon, D. Chin, J. P. Mathias, C. T. Seto and G. M. Whitesides, Tetrahedron, 1995, 51, 607–619 CrossRef CAS.
- D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229–2260 CrossRef CAS.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154–1196 CrossRef.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS PubMed.
- H. Fenniri, B. L. Deng and A. E. Ribbe, J. Am. Chem. Soc., 2002, 124, 11064–11072 CrossRef CAS PubMed.
- J. Deberitz and H. Nöth, Chem. Ber., 1973, 106, 2222–2226 CrossRef CAS.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, 2019, ch. 4, pp. 98–133 Search PubMed.
- G. Frenking and N. Frohlich, Chem. Rev., 2000, 100, 717–774 CrossRef CAS PubMed.
- G. Frenking, J. Organomet. Chem., 2001, 635, 9–23 CrossRef CAS.
- G. Frenking, Coord. Chem. Rev., 2003, 238–239, 55–82 CrossRef CAS.
- C. J. Breheny, J. M. Kelly, C. Long, S. O'Keeffe, M. T. Pryce, G. Russell and M. M. Walsh, Organometallics, 1998, 17, 3690–3695 CrossRef CAS.
- J. A. S. Howell, P. C. Yates, N. F. Ashford, D. T. Dixon and R. Warren, J. Chem. Soc., Dalton Trans., 1996, 3959–3966 RSC.
- J. M. Jenkins, J. R. Moss and B. L. Shaw, J. Chem. Soc. A, 1969, 2796–2800 RSC.
- B. L. Ross, J. G. Grasselli, W. M. Ritchey and H. D. Kaesz, Inorg. Chem., 1963, 2, 1023–1030 CrossRef CAS.
- R. D. Fischer, Chem. Ber., 1960, 93, 165–175 CrossRef CAS.
- G. Davidson and E. M. Riley, J. Organomet. Chem., 1969, 19, 101–114 CrossRef CAS.
- J. A. S. Howell, C. J. Beddows, P. J. O'Leary, P. C. Yates, P. McArdle, D. Cunningham and H. E. Gottlieb, J. Organomet. Chem., 1997, 527, 21–27 CrossRef CAS.
- Z. Zhao, J. Messinger, U. Schon, R. Wartchow and H. Butenschön, Chem. Commun., 2006, 3007–3009 RSC.
- A. D. Hunter, V. Mozol and S. D. Tsai, Organometallics, 1992, 11, 2251–2262 CrossRef CAS.
- J. Deberitz and H. Nöth, Chem. Ber., 1970, 103, 2541–2547 CrossRef CAS.
- R. B. King and A. Fronzaglia, Inorg. Chem., 1966, 5, 1837–1846 CrossRef CAS.
- P. S. Pregosin, in Encyclopedia of Magnetic Resonance, 2011 Search PubMed.
- C. Elschenbroich, S. Voss, O. Schiemann, A. Lippek and K. Harms, Organometallics, 1998, 17, 4417–4424 CrossRef CAS.
- R. L. Keiter and D. G. Vander Velde, J. Organomet. Chem., 1983, 258, C34–C36 CrossRef CAS.
- G. T. Andrews, I. J. Colquhoun, W. McFarlane and S. O. Grim, J. Chem. Soc., Dalton Trans., 1982, 12, 2353–2358 RSC.
- H. C. E. McFarlene, W. McFarlane and D. S. Rycroft, J. Chem. Soc., Dalton Trans., 1976, 1, 1616–1622 RSC.
- M. Kaupp, J. Comput. Chem., 2007, 28, 320–325 CrossRef CAS PubMed.
- F. Weinhold and C. R. Landis, Valency and bonding: a natural bond orbital donor-acceptor perspective, Cambridge University Press, 2005 Search PubMed.
- Z. Li, X. Chen, L. L. Liu, M. T. Scharnholz and H. Grutzmacher, Angew. Chem., Int. Ed., 2020, 59, 4288–4293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2234729–2234735. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00057e |
| This journal is © The Royal Society of Chemistry 2023 |