
Formation of a cyclooctatetraenylsamarium(III) inverse sandwich that ring-opens tetrahydrofuran†
Ramees
Peedika Paramban
 a,
Zhifang
Guo
a,
Glen B.
Deacon
b and
Peter C.
Junk
*a
a,
Zhifang
Guo
a,
Glen B.
Deacon
b and
Peter C.
Junk
*a
aCollege of Science & Engineering, James Cook University, Townsville, QLD 4811, Australia. E-mail: peter.junk@jcu.edu.au
bSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia
First published on 5th January 2023
Abstract
Reductive trapping of the cyclooctatetraenyl dianion (COT2−) by treatment of [Sm(DippForm)2(thf)2] (DippFormH = N,N′-bis(2,6-diisopropylphenyl)formamidine; thf = tetrahydrofuran) with 1,3,5,7-cyclooctatetraene (C8H8) in toluene yields an inverse sandwich dinuclear complex [Sm2(DippForm)4(COT)] (1), but [Sm(DippForm)(COT)(thf)2] (2) and [Sm(DippForm)2(O-C4H8-DippForm)(thf)] (3) in thf, and 1 yields 2 and 3 on treatment with thf.
The potential of divalent lanthanoid complexes to act as powerful reducing agents and their impact in reduction chemistry has been a hot topic of discussion for decades.1 Bis(pentamethylcyclopentadienyl)samarium(II) [SmCp*2]2 has been a particular favourite among such complexes, and has given rise to a range of notable trivalent samarium complexes from exercising its high reducing power.3,4 However, treatment of cyclooctatetraene with [SmCp*2] in a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mole ratio in toluene in an attempt to make the inverse sandwich [Sm2Cp*4(COT)] instead gave [SmCp*3] (A) and [SmCp*COT] (B) (Scheme 1a)5 and the former was converted by thf into [SmCp*2{O(CH2)4Cp*}(thf)] which has a ring opened thf coordinated.6
:2 mole ratio in toluene in an attempt to make the inverse sandwich [Sm2Cp*4(COT)] instead gave [SmCp*3] (A) and [SmCp*COT] (B) (Scheme 1a)5 and the former was converted by thf into [SmCp*2{O(CH2)4Cp*}(thf)] which has a ring opened thf coordinated.6
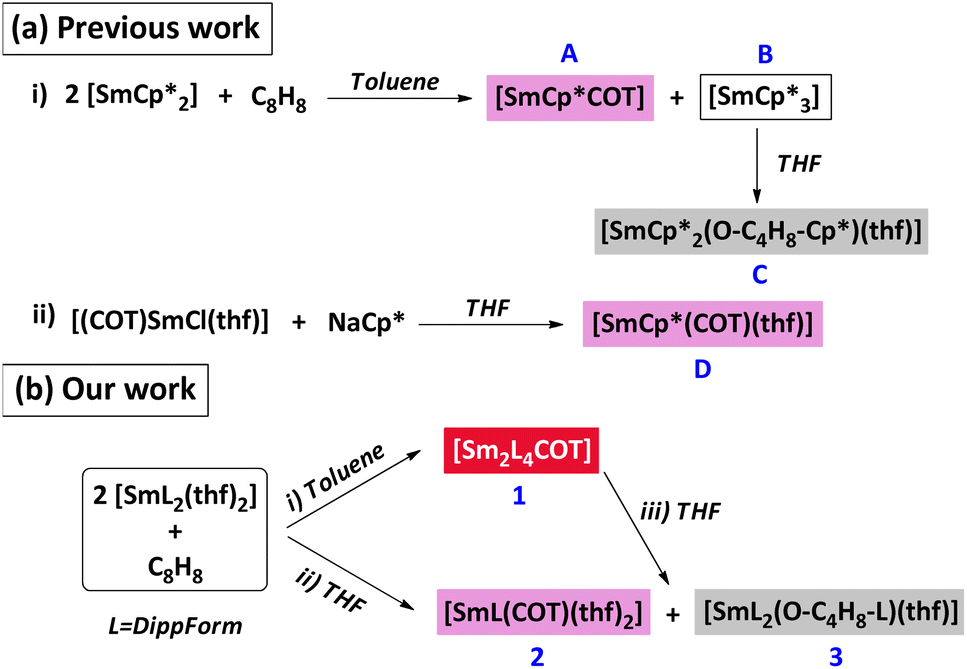 | ||
| Scheme 1 (a) Formation of [SmCp*COT] (i) and its solvated analogue [SmCp*COT(thf)] (ii) (Previous work). (b) Reaction of COT with [Sm(DippForm)2(thf)2] (Our work). | ||
After the first synthesis in 2005,7 [Sm(DippForm)2(thf)2] has been used in reduction chemistry as an N-donor alternative8 to [SmCp*2]9,10 but its possibilities have not yet been fully explored. The value of this reagent is illustrated by its reductive trapping of the bridging ligand [OC(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (C6H5)–C–(Ph)2O]2− upon reaction with benzophenone and the product demonstrated the unexpected, rare head to tail C–C coupling while its reaction with CS2 shows the formation of a C–S bond instead of the more usual C–C coupling and leads to the trapping of the [SCSCS2]2− ligand.9 We now report that [Sm(DippForm)2(thf)2] reacts with 1,3,5,7-cyclooctatraene in toluene to provide an inverse sandwich 1 in contrast to [SmCp*2], but that, in thf ring opening of the solvent occurs owing to reaction of 1 with thf.
(C6H5)–C–(Ph)2O]2− upon reaction with benzophenone and the product demonstrated the unexpected, rare head to tail C–C coupling while its reaction with CS2 shows the formation of a C–S bond instead of the more usual C–C coupling and leads to the trapping of the [SCSCS2]2− ligand.9 We now report that [Sm(DippForm)2(thf)2] reacts with 1,3,5,7-cyclooctatraene in toluene to provide an inverse sandwich 1 in contrast to [SmCp*2], but that, in thf ring opening of the solvent occurs owing to reaction of 1 with thf.
The first target compound was a dinuclear inverse sandwich-type complex, where the COT2− ligand is flanked by two [SmIII(DippForm)2]+ fragments on opposing sides, hence the reaction was carried out in a 2:1 stoichiometric ratio of [Sm(DippForm)2(thf)2] to C8H8 (Scheme 1(b)(i)). The slow addition of C8H8 to [Sm(DippForm)2(thf)2] in toluene resulted in an instant colour change from dark green to red, without heating or stirring. Red crystals of [SmIII2(DippForm)4(COT)] deposited from toluene in good yield after 1 day and red single crystals of [Sm2(DippForm)4(COT)]·4C6D61 (Fig. 1) deposited from C6D6. This outcome contrasts the analogous reaction of [SmCp*2], which yields [SmIIICp*COT] and [SmCp*3] (Scheme 1a).5 However, SmII COT2−/Cp* inverse crowns have been prepared by metathesis reactions.11 The paramagnetic nature of Sm3+ results in a significant broadening of the 1H NMR signals in the low frequency region while the sharp peaks in the high frequency area can be more easily analysed and agree with the composition established by the X-ray crystal structure (Fig. 1). The proton resonances of the COT2− ring and NCHN (DippForm) are at δ = 10.71 and 8.34 ppm respectively with an integration ratio of 2:1. In addition, the 13C resonance of the COT ligand is at δ = 67.90 ppm. Comparison of the IR spectrum of 1 with those of [Sm(DippForm)2(thf)2], [Sm(DippForm)3]12 and [Ce2(COT)3] and [Ce(COT)2]13 shows expected features for COT and DippForm ligands A satisfactory microanalysis was obtained. Complex 1 crystallizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with four lattice deuterated benzene molecules in the asymmetric unit, and there is disorder in one of the isopropyl groups of a DippForm ligand. This bimetallic inverse sandwich complex contains a COT2− ligand at the centre which is η8-coordinated to samarium atoms on either side. In addition, each samarium atom is ligated by two chelating DippForm moieties. The distance between Sm and COT2− ring centroid is in the range of 2.217 Å–2.219 Å, which is longer compared to their non-bridging counterparts in complex 2 (1.957 Å). This difference is expected between terminal and bridging COT2− ligands.14
with four lattice deuterated benzene molecules in the asymmetric unit, and there is disorder in one of the isopropyl groups of a DippForm ligand. This bimetallic inverse sandwich complex contains a COT2− ligand at the centre which is η8-coordinated to samarium atoms on either side. In addition, each samarium atom is ligated by two chelating DippForm moieties. The distance between Sm and COT2− ring centroid is in the range of 2.217 Å–2.219 Å, which is longer compared to their non-bridging counterparts in complex 2 (1.957 Å). This difference is expected between terminal and bridging COT2− ligands.14
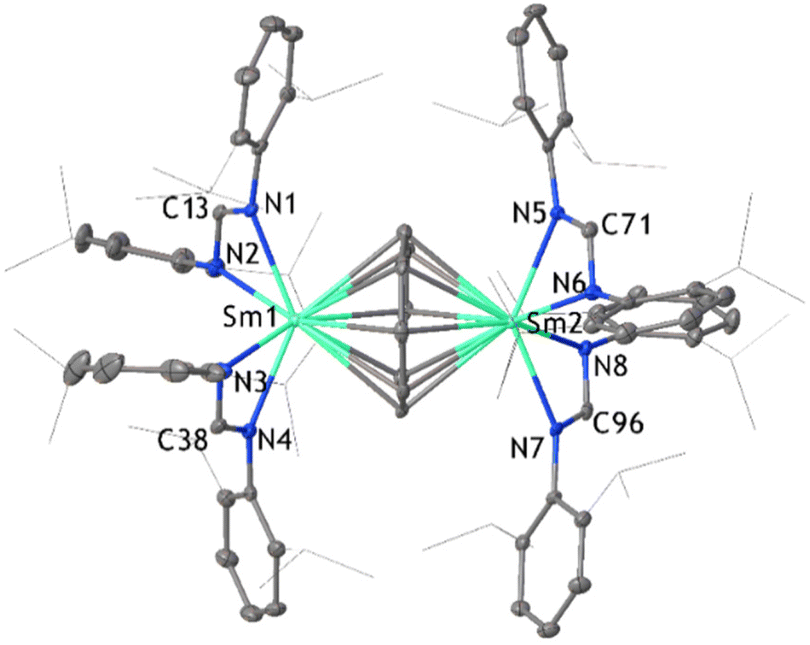 | ||
| Fig. 1 ORTEP diagram of complex 1 in which relevant atoms are labelled. Thermal ellipsoids are drawn at 50% probability level. Isopropyl groups of DippForm are shown as lines and hydrogen atoms along with four lattice deuterobenzene are omitted for clarity. | ||
The reaction of [Sm(DippForm)2(thf)2] and C8H8 was also carried out in thf with reaction conditions similar to the reaction in toluene. Here again, the slow addition of C8H8 to [Sm(DippForm)2(thf)2] in thf caused a rapid colour change from dark green to red, without heating or stirring. After several hours, the colour became purple. Leaving the reaction mixture unstirred for two days ensured the completion of the reaction. After layering with hexane, purple crystals of the complex [SmIII(DippForm)(COT)(thf)2] 2 (Fig. 2) were isolated (Scheme 1(b)(ii)). It is related to the product ([SmCp*COT]) obtained from Evans’ reaction in toluene,5 whilst the thf complexed analogue [SmCp*COT(thf)] (D),15a,b was obtained by a metathesis reaction of [SmCOT(Cl)thf] (Scheme 1(a)(ii)). 1H and 13C NMR spectra agreed with the composition found in the X-ray crystal structure. The η8-C8H8 ligand protons appear as a singlet at δ = 10.93 ppm while NCHN (DippForm) signal is at δ = 8.31 ppm, and their integrations are in an 8:1 ratio as expected. The CH2 signals of coordinated thf are observed at δ = 3.79 and 1.56 ppm, and the corresponding 13C signals are at δ = 71.82 and 25.95 ppm respectively. These values suggest that the thf is dissociated in solution, as some paramagnetic shifting to the thf resonances is observed in the 1H NMR spectrum of the Cp* analogue.15b The 13C NMR signal for COT2− is visible at δ = 82.58 ppm. After crystallization and collection of 2, the concentrated pale brown filtrate deposited a complex with a ring opened thf ligand, namely [Sm(DippForm)2(O-C4H8-DippForm)(thf)]·thf 3 (Fig. 3). An analogous complex with a ring opened thf product, [SmCp*2(O-C4H8-Cp*)(thf)], was obtained by treatment of [SmCp*3], one of the products of the SmCp*2/C8H8 reaction in toluene,5 with thf (Scheme 1(a), reaction of B)11a This infers that if the SmCp*2/C8H8 reaction was carried out in thf, the products would be similar to those currently observed. Even though the broadening of signals due to paramagnetic effects of trivalent samarium makes it difficult to integrate certain regions of the 1H NMR spectrum of 3, the data is interpretable to an extent and is consistent with the X-ray crystal structure (Fig. 3). The existence of two separate thf and DippForm moieties is evident from the 1H NMR spectrum. The NCHN proton of the DippForm ligand is attributable to the signal at δ = 9.67 ppm with an integration around two while that of DippForm in the ring-opened component is at δ = 7.36 ppm with integration value of one. The four CH2 signals of ring-opened thf are at δ = 7.25 (N–CH2), 5.36, 4.42 and 2.73 ppm (O–CH2), all with integrations around 2. One of the CH2 signals of coordinated thf can be observed at δ = 2.89 ppm, but the other signal is expected to be in the broad upfield region of the spectrum where it seems indistinguishable from methyl resonances. IR spectroscopy and elemental analysis also confirmed the same composition. Thus, it is evident that there is some paramagnetic shifting of the thf resonances of 3, in contrast to 2, though to a much smaller extent than observed for [SmCp3(thf)],16 hence some Sm–O(thf) interaction persists in solution.
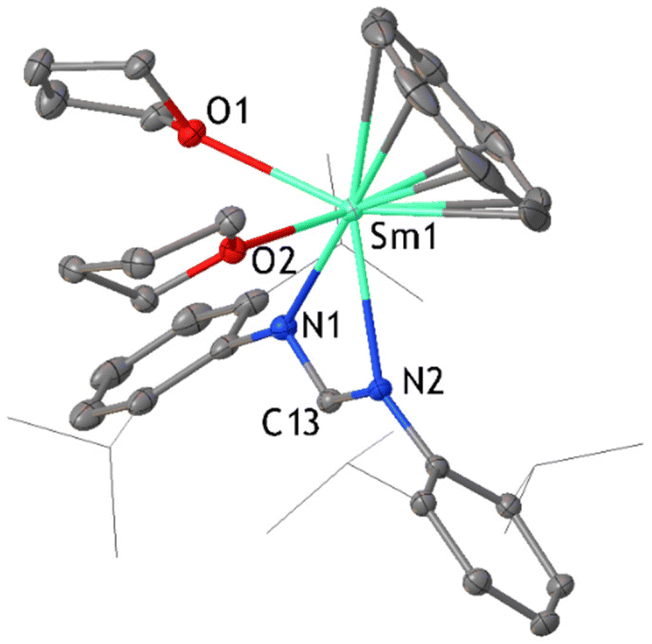 | ||
| Fig. 2 ORTEP diagram of complex 2 in which relevant atoms are labelled. Thermal ellipsoids are drawn at 50% probability level. Isopropyl groups of DippForm are shown as lines and hydrogen atoms are omitted for clarity. | ||
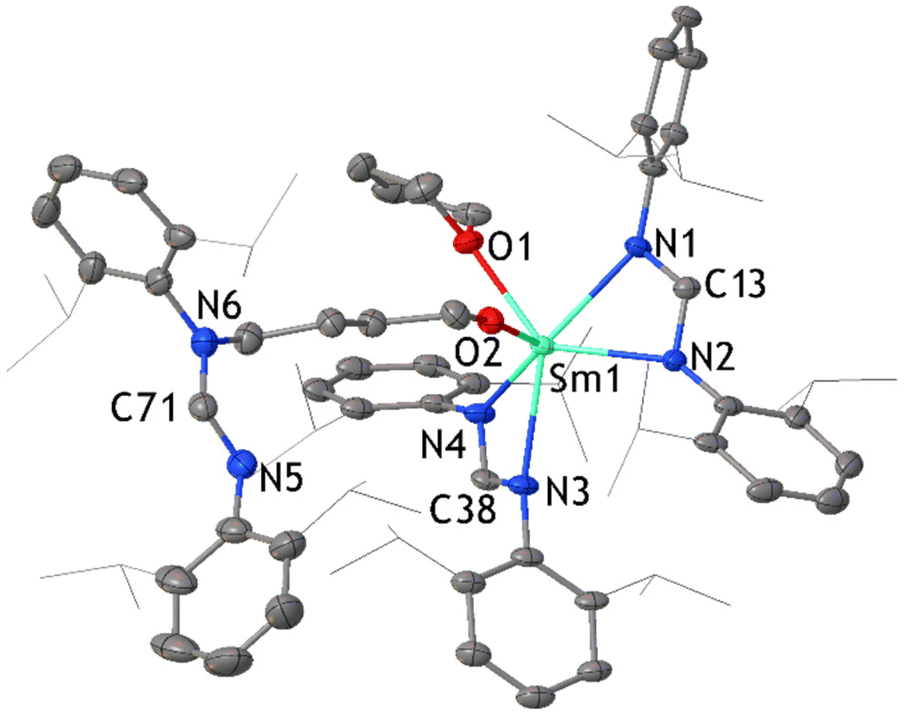 | ||
| Fig. 3 ORTEP diagram of complex 3 in which relevant atoms are labelled. Thermal ellipsoids are drawn at 50% probability level. Isopropyl groups of DippForm are shown as lines and hydrogen atoms along with two lattice thf are omitted for clarity. | ||
Although the reaction of [SmCp*3] with thf provides an explanation of the formation of [SmCp*2(O(CH2)4Cp*)(thf)],5 the current reaction giving 3 (Scheme 1(b)(ii)) cannot be explained in a similar manner, since it has been observed that treatment of the inverse crown 1 with thf yields 2 and the thf opened 3 (Scheme 1(b)(iii)). Thus ring opening of thf in the [Sm(DippForm)2(thf)2]/C8H8 reaction has an origin different from that in the SmCp*2/C8H8 system. It is notable that, in the reaction between [Sm(DippForm)2(thf)2] and C8H8 in thf, the reaction mixture becomes red, the colour of 1, before assuming the purple colour of 2.
Complex 2 crystallizes in orthorhombic space group P212121. The planar COT2− ligand binds in an η8-coordination fashion while one DippForm ligand is chelated to the samarium atom, along with two coordinating thf molecules. The samarium atom to COT2− ring centroid distance is 1.957 Å while the angle between the samarium atom and COT2− plane is 88.83°. The Sm–COT2− (centroid) distance is slightly longer compared to that (1.838 Å) of a similar complex [Sm(COT)Cp*],14 owing to the higher formal coordination number (9) of 2.
The X-ray crystal structure of complex 3 was solved and refined in the triclinic space group P with two lattice thf molecules (each with 50% occupancy) in the asymmetric unit. Two DippForm ligands are chelated to the samarium atom whereas a third DippForm creates a new N–C bond with a butoxide component, which is a result of the ring-opening of one coordinating thf molecule. There is a significant difference in the Sm–O bond lengths between coordinating thf (2.481(4) Å) and ring-opened thf unit (2.064 (4) Å). The latter is comparable to the usual Sm–O bond distance for samarium-amidinate alkoxides.17 This difference reflects the anionic character of the ring-opened thf component. For all three products, the Sm–N bond lengths are shorter (between 2.542(4) Å and 2.545(4) Å for compound 2, 2.436(4) Å and 2.531(4) Å for compound 3, and 2.437(4) Å and 2.541(4) Å for compound 1) compared with the divalent starting material [Sm(DippForm)2(thf)2] (2.529(4) and 2.617(4) Å), consistent with the trivalent oxidation state for all the samarium atoms of 1–3.
The considerable difference between {Sm(DippForm)2(thf)2} and [SmCp*2] in their behaviour with C8H8 is best seen by the outcomes from reactions in toluene, where the former gives an inverse crown [Sm2(DippForm)4COT] 1 and the latter [SmCp*COT] and [SmCp*3] from the same reaction stoichiometry (Scheme 1(a)(i) and (b)(i)). The difference is unlikely to be steric in origin, as, despite the difference in shape between DippForm and Cp*, their steric coordination numbers18 are much the same, namely 2.54 (ref. 19) and 2.49 (ref. 18) respectively. The general pattern of reactivity of [SmCp*2] and [Sm(DippForm)2(thf)2]2–4,7–9,10,12 suggests that the latter is a weaker reductant and NMR data for [Sm(DippForm or Cp*)COT(thf)n] suggest that the DippForm species is a weaker Lewis acid towards binding thf. These features may lead to isolation of the inverse crown 1 without subsequent rearrangement.
In conclusion, we demonstrated the remarkably different reactivity between the well-known divalent complex [SmCp*2] and its N-donor alternative [Sm(DippForm)2(thf)2] in their reactions with C8H8 in toluene (Scheme 1), enabling the synthesis of the inverse crown complex 1 in the DippForm system, thereby encouraging further exploration of the N-donor system as an alternative reductant. Further, the reaction of 1 with thf yielding 2 and a ring opened butoxidosamarium(III) species 3 provides a route to such species different from the reaction of [SmCp*3] with thf.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
GBD and PCJ gratefully acknowledge the ARC for funding (DP190100798 and DP230100112). Parts of this research were undertaken on the MX1 beamline at the Australian Synchrotron, part of ANSTO.20References
- (a) M. Bochkarev, L. Zakharov and G. Kalimina, Organoderivatives of rare earth elements, Kluwer Academic Publishers, Dordrecht, 1995; (b) Lanthanides: Chemistry and Use in Organic Synthesis, in Topics in Organometallic Chemistry, ed. S. Kobayashi, Springer, Berlin, 1999, vol. 2 Search PubMed; (c) C. Huang, Rare earth coordination chemistry, Wiley-Blackwell, Oxford, 2010 CrossRef; (d) L. R. Morss, Chem. Rev., 1976, 76, 827 CrossRef CAS; (e) F. Ortu, Chem. Rev., 2022, 122, 6040 CrossRef CAS PubMed; (f) K. Gopalaiah and H. B. Kagan, Chem. Rec., 2013, 13, 187 CrossRef CAS PubMed; (g) K. Gopalaiah and H. B. Kagan, New J. Chem., 2008, 32, 607 RSC, and references therein; (h) M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959 CrossRef CAS PubMed.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1981, 103, 6507 CrossRef CAS.
- (a) W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877 CrossRef CAS; (b) W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 9880 CrossRef CAS; (c) W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Chem. Soc., Chem. Commun., 1992, 1138 RSC; (d) W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728 CrossRef CAS.
- (a) S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, H. Schnöckel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740 CrossRef CAS PubMed; (b) C. Schoo, S. Bestgen, A. Egeberg, S. Klementyeva, C. Feldmann, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 5912 CrossRef CAS PubMed; (c) C. Schoo, S. Bestgen, A. Egeberg, J. Seibert, S. N. Konchenko, C. Feldmann and P. W. Roesky, Angew. Chem., Int. Ed., 2019, 58, 4386 CrossRef CAS PubMed; (d) S. N. Konchenko, T. Sanden, N. A. Pushkarevsky, R. Köppe and P. W. Roesky, Chem. – Eur. J., 2010, 16, 14278 CrossRef CAS PubMed; (e) N. Arleth, S. Bestgen, M. Gamer and P. W. Roesky, J. Am. Chem. Soc., 2014, 136, 14023 CrossRef CAS PubMed.
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 7423 CrossRef CAS.
- W. J. Evans, K. J. Forrestal and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 9273 CrossRef CAS.
- M. L. Cole and P. C. Junk, Chem. Commun., 2005, 2695 CAS.
- (a) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657 RSC; (b) F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253 RSC.
- G. B. Deacon, P. C. Junk, J. Wang and D. Werner, Inorg. Chem., 2014, 53, 1255 CrossRef PubMed.
- (a) R. Yadav, M. E. Hossain, R. Peedika Paramban, T. Simler, C. Schoo, J. Wang, G. B. Deacon, P. C. Junk and P. W. Roesky, Dalton Trans., 2020, 49, 7701 RSC; (b) C. Schoo, S. Bestgen, R. Köppe, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2018, 54, 4770 RSC; (c) B. Y. Savkov, T. S. Sukhikh, S. N. Konchenko and N. A. Pushkarevsky, Aust. J. Chem., 2022, 75, 732 CrossRef CAS.
- (a) W. J. Evans, R. D. Clark, M. A. Ansari and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 9555 CrossRef CAS; (b) O. T. Summerscales, S. C. Jones, F. G. N. Cloke and P. B. Hitchcock, Organometallics, 2009, 28, 5896–5908 CrossRef CAS.
- M. L. Cole, G. B. Deacon, C. M. Forsyth, P. C. Junk, D. C. Polo and J. Wang, Dalton Trans., 2010, 39, 6732–6738 RSC.
- M. D. Walter, C. H. Booth, W. W. Lukens and R. A. Andersen, Organometallics, 2009, 28, 698–707 CrossRef CAS.
- W. J. Evans, M. A. Johnston, R. D. Clark and J. W. Ziller, J. Chem. Soc., Dalton Trans., 2000, 1609 RSC.
- (a) A. Recknagel, M. Noltemeyer and F. T. Edelmann, J. Organomet. Chem., 1991, 410, 53 CrossRef CAS; (b) H. Schumann, R. D. Koehn, F. W. Reier, A. Dietrich and J. Pickardt, Organometallics, 1989, 8, 1388 CrossRef CAS.
- H. Schumann and G. Jeske, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40, 1490 CrossRef.
- C. Schoo, S. Bestgen, M. Schmidt, S. N. Konchenko, M. Scheer and P. W. Roesky, Chem. Commun., 2016, 52, 13217 RSC.
- J. Marçalo and A. P. De Matos, Polyhedron, 1989, 8, 2431 CrossRef.
- Z. Guo, V. L. Blair, G. B. Deacon and P. C. Junk, Dalton Trans., 2020, 49, 13588 RSC.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, spectra, and crystallographic information. CCDC 2219717–2219719. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04164b |
| This journal is © The Royal Society of Chemistry 2023 |