
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the mercuration, palladation, transmetalation and direct auration of a C^N^C pincer ligand†‡
Alice Jane
McEllin
 ,
Christopher A.
Goult
,
Adrian C.
Whitwood
,
Jason M.
Lynam
and
Duncan W.
Bruce
*
,
Christopher A.
Goult
,
Adrian C.
Whitwood
,
Jason M.
Lynam
and
Duncan W.
Bruce
*
Department of Chemistry, University of York, Heslington, YORK YO10 5DD, UK. E-mail: duncan.bruce@york.ac.uk
First published on 4th January 2023
Abstract
The C^N^C ligand 2,6-bis(2,3-dialkoxyphenyl)pyridine forms dimercury and orthopalladated complexes, both of which may be transmetallated to gold(III) complexes; the gold complexes may also be formed directly in a Rh(III)-catalysed process, hence it is possible to circumvent the use of organomercury intermediates in the synthesis of this important class of compound.
Since the demonstration in 2005 by Yam and co-workers of room-temperature phosphorescent emission in C^N^C pincer complexes of gold(III),1 there has been significant interest in materials of this type.2,3 As in related complexes of platinum(II),4 the key to unlocking phosphorescence was to employ sufficiently strong field ligands that would raise the energy of the d–d states, which otherwise quench the luminescence excited state by thermal equilibration or energy transfer. In the complexes reported by Yam et al., this was achieved using a phenylacetylide co-ligand (Fig. 1, 2). From these initial reports with quantum yields of <1%, the field has moved on, much higher quantum yields have been realised and the emissive properties have been employed in solvent sensing,5 luminescent gels6 and, of course, as emitters in organic light-emitting diodes.2,7
 | ||
| Fig. 1 Synthesis of luminescent gold(III) complexes (2) described by Yam et al., showing the mercury(II) intermediate (1). Typically, R′ = H and R = 4-substituted phenyl ring. | ||
Our own contribution to this field has looked to functionalise both the pincer ‘backbone’ ligand as well as the phenylacetylide to generate materials that are both triplet emitters and liquid crystals. To that end, we have reported on two series of complexes, one containing simple hydrocarbon chains bound to the phenylacetylide,8 the other employing semiperfluorocarbon chains instead (Fig. 2).9,10 Briefly, the vast majority of the complexes are liquid crystalline, showing almost exclusively columnar phases with the complexes having four chains on the pincer ligand having photoluminescent quantum yields (PLQY) of up to 36%, while those with only two chains had PLQY values of <4%. Encouragingly, OLED devices fabricated from the all-hydrocarbon complexes showed external quantum efficiencies of >7%.
 | ||
| Fig. 2 Emissive gold(III) pincer complexes showing liquid-crystalline properties. | ||
However, one of the great drawbacks in the preparation of all complexes of this type, is that direct auration using, for example, tetrachloroaurate(III) and free ligand does not proceed, and the synthesis (Fig. 1) inevitably goes through an intermediate chloromercury(II) complex (1).11 This is in contrast to the direct auration possible with bidentate 2-phenylpyridine ligands. This was demonstrated initially by Constable and Leese12 who first formed the neutral, N-bound AuCl3 complex of 2-phenylpyridine, which can then convert thermally to the orthometalated product. This general approach persists, for example in preparing gold N^C^C complexes where the final orthometalation is promoted by microwave irradiation.13 Direct reaction using microwaves is also a productive approach,14 and, for example, Hylland et al. have recently published a very extensive study of such preparations starting from Au(OAc)3 and driven by microwave conditions.15 Indeed, these studies were also able to demonstrate direct auration of N^C^C ligands.16 Separate studies have shown that direct auration is also possible if the ligand is pre-functionalised, for example with a diazonium function,17 but this can lead to additional synthetic complexity depending on other substituent requirements on the ligands and also the preparation of bespoke gold precursor complexes.18 Impressively, Martín et al. have recently shown that direct auration of bidentate 2-phenylpyridines can be mediated catalytically by [Cp*RhCl2]2.22
It is therefore evident that to capitalise on the highly promising photophysical properties of the Au complexes, a simple, mercury-free protocol would enable a wider range of modified C^N^C ligands to be employed in this chemistry. This would, in turn, allow for greater flexibility in the informed introduction of new properties into the complexes through modified C^N^C ligands. In order to develop such a route, we have prepared the related ligands 3-n (Fig. 3a), in which there is an alkoxy chain on the phenyl carbon ortho- and meta-to the phenyl-pyridine bond. The compounds are prepared by reaction of 2,6-dibromopyridine with 2,3-dimethoxyphenylboronic acid using a Suzuki–Miyaura protocol (89% yield), with the 2,2′,3,3′-tetraalkyloxy-substituted ligands (butyl and dodecyl) resulting from subsequent demethylation using pyridinium chloride (90% yield) followed by re-alkylation under Williamson ether conditions using 1-bromoalkane (53% yield C4, 65% yield C12). Thus, the dodecyloxy-substituted ligands have been those of greatest interest in relation to generating liquid-crystalline materials,8–10 however these prove challenging to crystallise (as do methoxy derivatives on account of low solubility) and so we have typically deployed butyloxy-substituted ligands to obtain single crystals for study by diffraction. The steric demands of these materials mean that in the free ligand, the phenyl and pyridyl rings cannot be co-planar (Fig. 4b), one consequence of which is that in the preferred anti atropoisomer,§ the meta-pyridyl hydrogens are diastereotopic so that, in the 1H NMR spectrum, the pyridine hydrogens present as two AB spin systems (Fig. 3b). However, when complexed to gold (Fig. 4c and d), the rings are forced to be co-planar and the two hydrogens become equivalent (Fig. 3c). Interestingly, the steric congestion does not affect the stability of the complex adversely and 6-12 melts without decomposition at 129 °C.
 | ||
| Fig. 3 (a) Structure of the 2,6-bis(2,2′,3,3′-tetraalkoxy)pyridine ligands; (b) 1H NMR spectrum of the pyridyl hydrogens of 2,6-bis(2,2′,3,3′-tetramethoxy)pyridine (3-1) showing the two AB spin systems; (c) 1H NMR spectrum of the pyridyl hydrogens of the chlorogold(III) complex of 2,6-bis(2,2′,3,3′-tetra-dodecyloxy)pyridine (6-12) showing the AX2 spin system. | ||
 | ||
| Fig. 4 Molecular structure of 2,2′,3,3′-tetramethoxy-substituted 2,6-diphenylpyridine (3-1) (a) viewed from above and (b) viewed from the side; molecular structure of a gold(III) complex (6-4) (c) viewed from above and (d) viewed from the side.¶ | ||
As a control reaction, it was possible to demonstrate that the synthesis of these gold complexes again proceeded through reaction of the ligand with mercury(II) acetate and an aqueous LiCl work up to form intermediate chloromercury(II) complexes. Unexpectedly, however, rather than the low yield (ca. 17%) of analytically impure material obtained in our previous work with related four-chain ligands (which interestingly then leads to a pure gold complex after transmetalation and work up with estimated overall conversion of ca. 8%), we were able to isolate the pure di-mercurated complex 4-12 in 75% yield. The X-ray single crystal structure of the butyloxy derivative, 4-4 (obtained in 55% yield) is shown as Fig. 5.
 | ||
| Fig. 5 Molecular structure of the dimercurated complex 4-4 showing (a) the complex in the asymmetric unit with a short Hg⋯N contact and (b) in a view, butyloxy chains removed, to show the relative disposition of the chloromercury units. | ||
The complex crystallised in the monoclinic space group P2/c (with two complexes present in the asymmetric unit, one of which (shown in Fig. 5) has a short Hg⋯N contact (2.675(9) Å – other Hg⋯N distances are ≥2.75 Å). Each mercury is coordinated in a linear fashion to carbon and a chloride ligand, with the angles at mercury being close to perfectly linear (179.0(3)°, 179.2(4)°, 177.4(4)° and 175.7(3)°). Hg–C bond lengths are found to be 2.016(12) Å, 2.027(14) Å, 2.065(17) Å and 2.062(12) Å, all of which are statistically equivalent and are of a magnitude very similar to those in the literature. However, for the Hg–Cl distances there is a more complex story, which is described in greater detail in the ESI.‡
This dimercury complex can then be transmetalated to give the related gold(III) complex (6-12) in an isolated yield of 24%, which gives an 18% overall yield from the ligand (Scheme 1) In comparison, formation of 6-4 from 5-4 proceeds in 47% yield, which gives an overall conversion of ca. 26%. These represent improvements over yields from the impure mercury intermediates described above,8 but still requires the intermediacy of mercury.
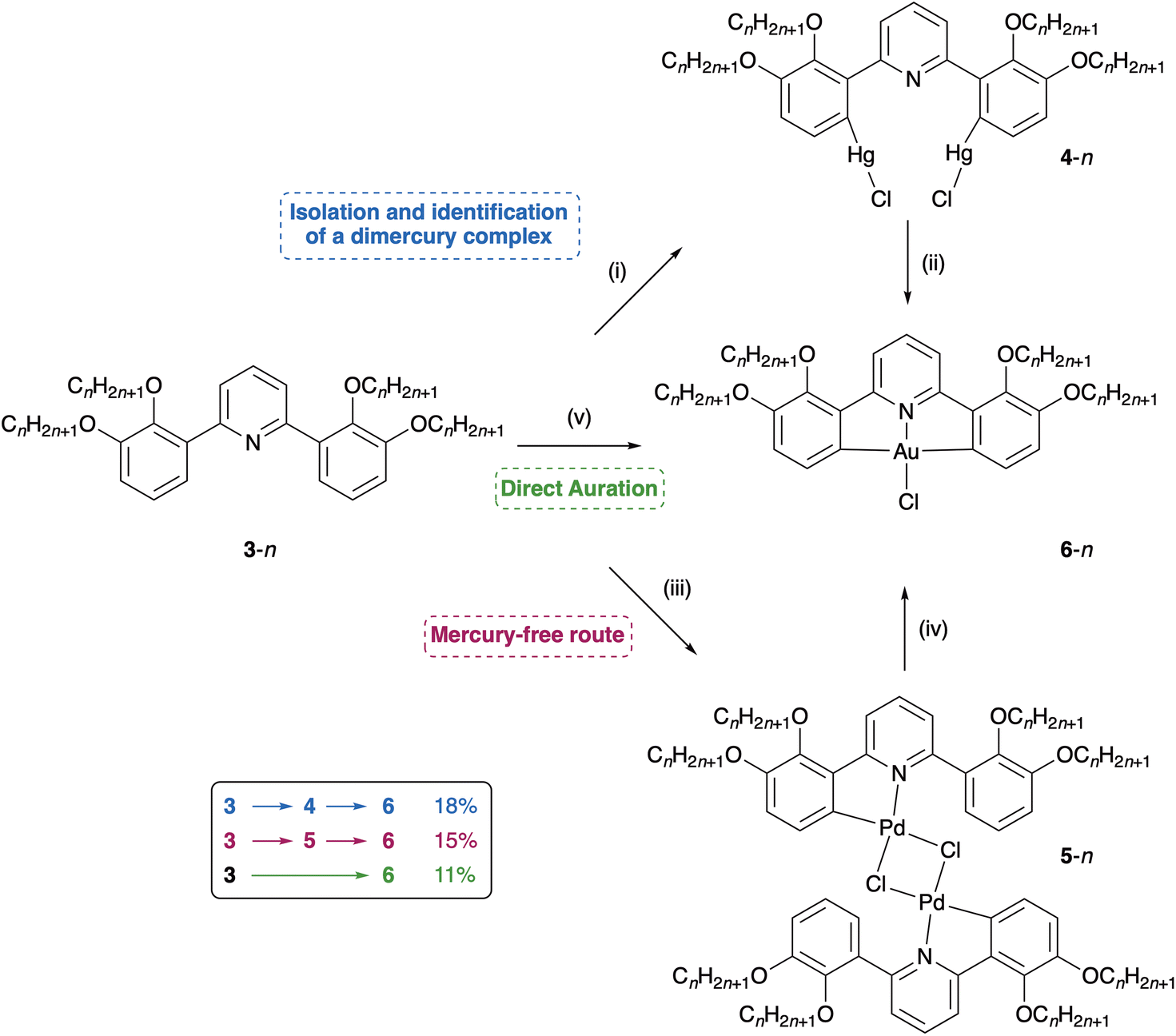 | ||
| Scheme 1 Summary of the reactions and interconversions described in this account. Conditions and reagents: (i) Hg(OAc)2/EtOH/Δ/LiCl(aq); (ii) CHCl3/MeCN/Na[AuCl4]/Δ; (iii) K2[PdCl4]/EtOH/Δ; (iv) CHCl3/MeCN/Na[AuCl4]/Δ; (v) Na[AuCl4]/EtOAc/[Cp*RhCl2]2 (cat.)/Δ. | ||
In then considering alternative approaches, one of the best-known metals for both C–H activation and also ortho-metalation is palladium. This thinking arose in part following the detailed study by Gorunova et al. of a series of palladium-to-mercury transmetalation reactions,19 prompted by the possibility that the simple mercury drop test used to establish the presence of colloidal palladium in catalytic systems could be corrupted by such a reaction. Thus, reaction of ligand 3-12 with K2[PdCl4] in EtOH under reflux leads to isolation of the corresponding di-μ-chloro dimer 5-12, in a yield of 31%, the dimeric nature being confirmed by ESI mass spectrometry (Scheme 1). Dimer 5-12 was then reacted with K[AuCl4] in a mixture of CHCl3/MeCN to give the target gold complex 6-12 in 48% isolated yield as a pure yellow powder, with an overall yield for the two steps of ca. 15% (Scheme 1). This is comparable with that reported using mercury (18%) with the advantage that a toxic metal is not used and thus, while palladium is not cheap, this represents a viable alternative to the use of mercury in the preparation of gold C^N^C pincer complexes of this type. This transmetallation step is accompanied by the appearance of a pale purple colour and the formation of very small amounts of elemental gold (the latter also being observed, again in tiny quantities, in the direct auration reported below). Previously, we have observed that reaction of [PtCl4]2− with [AuCl4]− leads to almost quantitative formation of [PtCl6]2− and either [AuCl2]− or Au0 depending on the stoichiometry,20 while AuI is formed exclusively starting from [PdCl4]2−.21 That very small quantities of gold are recovered from two transformations suggests that an analogous redox reaction is not a major side reaction in these transformations.
However, having established this alternative reaction route, we then turned our attention to the recent report by Martín et al. of the direct synthesis of bidentate gold C^N complexes via the use of catalytic quantities of [Cp*RhCl2]2.22 First reported by Kang et al. in 1969,23 [Cp*RhCl2]2 is well known for its ability to form C^N chelates and to activate sp2 C–H bonds, and it has also been shown to be capable of transmetalation with gold.24 Thus, Martín et al. showed22 that a series of substituted 2-phenylpyridines and related substrates could be aurated directly in ethyl acetate under reflux using [Cp*RhCl2]2 as the catalyst. They were able to demonstrate that the reaction proceeds via a base-mediated ortho-rhodation to give an intermediate RhIII complex [Cp*Rh{κ2-C,N-(2-phenyl)pyridine}(Cl)], which then transmetalates with Na[AuCl4] to give [Au{κ2-C,N-(2-phenyl)pyridine}Cl2]. Isolated yields ranged from 16% to an optimised value of 69% with 2.5 mol% of catalyst in EtOAc using sodium benzoate as base.
Inspired by this report, we therefore reacted ligand 3-12 with Na[AuCl4] in EtOAc with NaO2CPh using 2.5 mol% [Cp*RhCl2]2 and obtained 6-12 in a yield of 11% (Scheme 1). This is lower than the typical yields reported by Martín et al.22 but is nonetheless remarkable as while the recent reports that use an Au(OAc)3 starting material demonstrate direct metalation in C^C^N chelates,16 this is not reported for the photophysically interesting C^N^C chelate.
These data demonstrate that the successful synthesis of gold complexes containing C^N^C chelates requires an initially well-controlled C–H bond activation step. This may be through both stoichiometric (Pd and Hg) or catalytic (Rh) conditions. Although each of reagents explored probably operates in a mechanistically distinct fashion to achieve selective C–H bond activation,25,26 it is clear that for this ligand-class simple gold salts are not able to perform this transformation. Furthermore, the nature of the ligand has some bearing for, as noted above, 4-12 can be obtained clean in 75% isolated yield whereas using the 3,3′,4,4′-tetrasubstituted ligands (as illustrated complexed to gold in Fig. 2) gives low yields (<25%) of impure, inseparable material on mercuration. Further, we have on one occasion attempted direct auration with the same ligand without success. Therefore, and as is expected, there is a good deal of work to follow relating both to optimisation and to understanding mechanism, for example delineating the possible influences of steric and electronic factors. It is evident, therefore, that there is considerable potential in exploiting the advances in transition metal-catalysed C–H bond functionalisation chemistry that have been applied to the synthesis of complex organic molecules27 to the preparation of metal complexes with novel and important materials properties.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the University of York for funding (AJM & CAG). JML is supported by a Royal Society Industry Fellowship.References
- V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107–3110 CrossRef CAS PubMed.
- M.-C. Tang, M.-Y. Chan and V. W.-W. Yam, Chem. Rev., 2021, 121, 7249–7279 CrossRef CAS PubMed.
- D. L. Zhou, W. P. To, Y. Kwak, Y. Cho, G. Cheng, G. S. M. Tong and C. M. Che, Adv. Sci., 2019, 6, 1802297 CrossRef CAS PubMed.
- See e.g.: J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS.
- K. C. Yim, V. K. M. Au, L. L. Hung, K. M. C. Wong and V. W. W. Yam, Chem. – Eur. J., 2016, 22, 16258–16270 CrossRef CAS PubMed.
- H. L.-K. Fu and V. W.-W. Yam, Chem. Lett., 2018, 47, 605–610 CrossRef CAS.
- M.-C. Tang, M.-Y. Chan and V. W.-W. Yam, Chem. Rev., 2021, 121, 7249–7279 CrossRef CAS PubMed; W. P. To, G. Cheng, G. S. M. Tong, D. L. Zhou and C. M. Che, Front. Chem., 2020, 8, 653 CrossRef PubMed.
- R. R. Parker, D. H. Liu, X. K. Yu, A. C. Whitwood, W. G. Zhu, J. A. G. Williams, Y. F. Wang, J. M. Lynam and D. W. Bruce, J. Mater. Chem. C, 2021, 9, 1287–1302 RSC.
- R. R. Parker, A. J. McEllin, X. B. Zeng, J. M. Lynam and D. W. Bruce, Liq. Cryst., 2022, 49, 1162–1173 CrossRef CAS.
- R. R. Parker, R. F. Stracey, A. J. McEllin, X. Chen, Y. Wang, J. A. G. Williams, J. M. Lynam and D. W. Bruce, ACS Omega, 2022, 7, 24903–24917 CrossRef CAS PubMed.
- R. V. Parish, J. P. Wright and R. G. Pritchard, J. Organomet. Chem., 2000, 596, 165–176 CrossRef CAS.
- E. C. Constable and T. A. Leese, J. Organomet. Chem., 1989, 363, 419–424 CrossRef CAS.
- R. Kumar, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 14287–14290 CrossRef CAS PubMed.
- A. P. Shaw, M. Tilset, R. H. Heyn and S. Jakobsen, J. Coord. Chem., 2010, 64, 38–47 CrossRef.
- K. T. Hylland, I. L. Schmidtke, D. S. Wragg, A. Nova and M. Tilset, Dalton Trans., 2022, 51, 5082–5097 RSC.
- M. S. M. Holmsen, A. Nova, K. Hylland, D. S. Wragg, S. Oien-Odegaard, R. H. Heyn and M. Tilset, Chem. Commun., 2018, 54, 11104–11107 RSC.
- D. Eppel, M. Rudolph, F. Rominger and A. S. Hashmi, ChemSusChem, 2020, 13, 1–6 CrossRef PubMed.
- A. Tlahuext-Aca, M. N. Hopkinson, C. G. Daniliuc and F. Glorius, Chem. – Eur. J., 2016, 22, 11587–11592 CrossRef CAS PubMed.
- O. N. Gorunova, I. M. Novitskiy, Y. K. Grishin, I. P. Gloriozov, V. A. Roznyatovsky, V. N. Khrustalev, K. A. Kochetkov and V. V. Dunina, Organometallics, 2018, 37, 2842–2858 CrossRef CAS.
- I. H. Silalahi, N. K. Sethi, M. Z. Shafikov, V. Chechik, A. C. Whitwood and D. W. Bruce, Chem. Commun., 2018, 54, 13682–13685 RSC.
- N. K. Sethi, PhD Thesis, University of York, 2013 (available from https://bit.ly/3SSsTgT).
- J. Martín, E. Gomez-Bengoa, A. Genoux and C. Nevado, Angew. Chem., Int. Ed., 2022, 61, e202116755 CrossRef PubMed.
- J. W. Kang, K. Moseley and P. M. Maitlis, J. Am. Chem. Soc., 1969, 91, 5970–5977 CrossRef CAS.
- Y. Shi and S. A. Blum, Organometallics, 2011, 30, 1776–1779 CrossRef CAS.
- J. Milani, N. E. Pridmore, A. C. Whitwood, I. J. S. Fairlamb and R. N. Perutz, Organometallics, 2015, 34, 4376–4386 CrossRef CAS.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Peter Maitlis. |
| ‡ Electronic supplementary information (ESI) available: Synthetic and analytical details relating to the new complexes; cif and cifcheck files for 1, 3 and 4. CCDC 2195198–2195200. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04114f |
| § Evidence that the preferred atropoisomer has an anti arrangement comes not from the single crystal structure, rather from the observation of the nature of the NMR signal (2× AB signals) as the syn isomer would have a plane of symmetry and so the meta-hydrogens would no longer be related as diastereotopic. |
| ¶ Crystal structures have been deposited at CCDC with deposition numbers 2195198–2195200. |
| This journal is © The Royal Society of Chemistry 2023 |