
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-concentration CO2 capture using metal–organic frameworks – current status and future perspectives
Michelle
Åhlén
,
Ocean
Cheung
 * and
Chao
Xu
*
* and
Chao
Xu
*
Division of Nanotechnology and Functional Materials, Department of Materials Science and Engineering, Uppsala University, Ångström Laboratory, SE-751 03 Uppsala, Box 35, Sweden. E-mail: ocean.cheung@angstrom.uu.se; chao.xu@angstrom.uu.se; Tel: +46 18 471 3279 Tel: +46 18 471 3230
First published on 20th January 2023
Abstract
The ever-increasing atmospheric CO2 level is considered to be the major cause of climate change. Although the move away from fossil fuel-based energy generation to sustainable energy sources would significantly reduce the release of CO2 into the atmosphere, it will most probably take time to be fully implemented on a global scale. On the other hand, capturing CO2 from emission sources or directly from the atmosphere are robust approaches that can reduce the atmospheric CO2 concentration in a relatively short time. Here, we provide a perspective on the recent development of metal–organic framework (MOF)-based solid sorbents that have been investigated for application in CO2 capture from low-concentration (<10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) CO2 sources. We summarized the different sorbent engineering approaches adopted by researchers, both from the sorbent development and processing viewpoints. We also discuss the immediate challenges of using MOF-based CO2 sorbents for low-concentration CO2 capture. MOF-based materials, with tuneable pore properties and tailorable surface chemistry, and ease of handling, certainly deserve continued development into low-cost, efficient CO2 sorbents for low-concentration CO2 capture.
000 ppm) CO2 sources. We summarized the different sorbent engineering approaches adopted by researchers, both from the sorbent development and processing viewpoints. We also discuss the immediate challenges of using MOF-based CO2 sorbents for low-concentration CO2 capture. MOF-based materials, with tuneable pore properties and tailorable surface chemistry, and ease of handling, certainly deserve continued development into low-cost, efficient CO2 sorbents for low-concentration CO2 capture.
Introduction
The ever-increasing combustion of fossil fuels, such as energy generation in coal-fired power plants, cement plants, and oil refineries has contributed towards increasing the atmospheric CO2 concentration.1 The atmospheric CO2 level has gone from the pre-industrial value of 280 ppm to a current level of 418 ppm (December 2022).2 The greenhouse effect that is caused by the high atmospheric levels of CO2 is considered to be one of the main reasons for global warming as well as the associated environmental issues. In addition to the irreversible changes to the climate and environment, a high atmospheric CO2 concentration is a big risk to human health, for example, it can trigger respiratory illnesses when the atmospheric CO2 concentration is over 600 ppm.3 Although the move towards non-fossil fuel-based energy generation could be a long-term solution to reduce CO2 emission due to human activities, carbon capture and storage (CCS) is undoubtedly an important current approach to reduce the CO2 emission from point sources of CO2. Alternatively, direct air capture (DAC) of CO2, which implies not only capturing CO2 from emission point sources, but rather from the atmosphere, is also an important complementary approach for reducing the atmospheric CO2 level. DAC is a negative emission technology that has the potential to lower the atmospheric CO2 concentration down to 350 ppm.4 DAC of CO2 would mean capturing CO2 from sources with low-concentrations, or trace amounts of CO2. This type of CO2 capture is important for gas purification, indoor air quality control, and a number of industrial processes. DAC of CO2 has been discussed for indoor settings such as classrooms, hospitals, or offices in order to ensure the well-being of individuals. Air purification devices are needed in ventilators, or in confined spaces such as spacecraft and submarines to keep the CO2 concentration at a safe level. From an industrial point of view, in order to meet the liquefied natural gas (LNG) specifications, CO2 concentration in natural gas has to be reduced down to 50 ppm before its liquefaction.5 These example application areas show that there is currently a great interest and urgent need for the development of efficient technology for low-concentration CO2 capture. For the purpose of this perspective, we operationally define “low-concentration” as below approximately 10000 ppm in CO2 concentration.
Amine scrubbing is probably the most mature and viable CO2 capture technology that has been widely applied in natural gas purification and post-combustion capture of CO2.6 It uses aqueous amine solutions to absorb CO2 from gases via chemical reactions, which offers high separation and purification efficiencies even at ultralow CO2 concentrations (<1000 ppm). However, amine scrubbing suffers from significant drawbacks such as high energy consumption for amine regeneration, risk of amine leakage, and corrosion to the associated equipment. Temperature or pressure swing adsorption (TSA, PSA) technologies have also been developed for CO2 capture. These technologies can be adapted to utilize solid physisorbents. Physisorbents such as porous solids can be engineered to adsorb CO2 selectively over other gases on the internal surfaces of the sorbent. The adsorbed CO2 is then released at elevated temperatures and/or reduced pressures and the sorbent is regenerated for subsequent cycles.7–9 In contrast to chemisorption processes (using chemisorbents), physisorbents adsorb CO2 with relatively low enthalpy of adsorption (ΔHads). The energy cost for regeneration of physisorbents is much lower than for chemisorbents (where chemical bonds form between the sorbent and CO2). Porous physisorbents including activated carbons,10–12 zeolites,13–16 silica,17,18 and porous organic polymers19–21 have been intensively studied for CO2 capture. Some of these sorbents show great potential for post-combustion carbon capture, where CO2 partial pressures are 0.05–0.15 bar. However, it appears to be more challenging to use conventional porous physisorbents for low-concentration CO2 capture, as physisorbents tend to have low CO2 uptake at low-concentrations (i.e. low partial pressures). The advantages of these physisorbents for CO2 capture at low CO2 concentrations fade significantly in terms of uptake capacity, selectivity, and adsorption kinetics. In the ideal case, a suitably engineered CO2 sorbent for DAC would not only have high CO2 uptake capacity at low CO2 concentrations, but should also show high CO2 selectivity under the relevant conditions. This means that the ΔHads of CO2 sorption at zero or low loading must be significantly lower than that typically observed for physisorbents (∼−20 to −40 kJ mol−1). However, very low enthalpies of CO2 sorption (i.e. <∼−60 kJ mol−1), or chemisorption of CO2, may mean that regeneration of the sorbent will be energy-demanding.
Metal–organic frameworks (MOFs) are a type of porous coordination polymers constructed by linking metal ions or clusters with organic linkers via coordination bonds.22,23 They usually have ordered porous channels and high specific surface areas. The rich coordination chemistry and large amount of available organic linkers endow MOFs with synthetic and structural diversity. Consequently, more than 90000+ types of MOFs with defined structures have been synthesized so far.24 The unique advantages of MOFs include tunable pore size and surface chemistry. The possibility to pre-design structures and composition renders MOFs attractive for many applications including low-concentration CO2 capture.25 For example, by judicious selection of the building units, the size, and shape of the pore-aperture of MOFs can be precisely tuned to achieve a high CO2 separation efficiency by the molecular sieving effect.26–28 Formation of ultramicropores,29–31 creation of unsaturated metal centers,32,33 and amine grafting34,35 are effective approaches to introduce strong CO2 adsorption sites on MOFs that can increase the binding affinity between the sorbents and CO2. Such functionalized MOFs usually display high CO2 uptake capacity and high selectivity, even at very low CO2 concentrations. In this perspective, we will give an overview of the recent advances in the development and engineering of MOFs for low-concentration CO2 capture. The relationship between the CO2 capture performances (e.g. uptake capacity, selectivity, enthalpies of adsorption, kinetics, cyclic stability) and the MOF structures, as well as possible approaches to structure and upscale MOF sorbents for applications, will be discussed. We also discuss the prospects and challenges when it comes to the use of MOFs for CO2 capture from low-concentration sources under different circumstances.
Strategies for enhancing CO2 capture performance on MOFs
On MOFs, the general approach adopted to increase the CO2 capture performance, especially for low-concentration CO2 capture, is by tuning the sorption affinity for CO2 at low-concentrations.31 The CO2 partial pressures (pCO2) that is of interest range from ∼0.4 to 10 mbar. This partial pressure range can be considered as equivalent to 400 ppm (current atmospheric CO2 concentration) up to 10000 ppm under atmospheric pressure, respectively. The CO2 adsorption affinity at low-concentrations can be achieved through three main routes: (1) via careful control of the pore architecture of the frameworks, (2) through the introduction of high-energy sorption sites (e.g. open-metal sites, anionic groups), and (3) by post-synthetic amine-functionalization. In all cases, the CO2 sorption isotherm of these MOFs should have a very sharp increase in the very low-pressure region (i.e. at 0.4 mbar for atmospheric CO2 concentration). In this section, these three approaches will be discussed. The CO2 capture performance of all the sorbents discussed in this paper are summarized in Table 1 for comparison.
| CO2 partial pressure (mbar) | Temperature (K) | Activation temperature (K) | Uptake (mmol g−1) | Uptake (cm3 g−1) | ΔHads (kJ mol−1) | Ref. | |
|---|---|---|---|---|---|---|---|
| a Data extracted from the original publication, and may be approximate. | |||||||
| NbOFFIVE-1-Ni | 0.4 | 298 | 378 | 1.30 | 29.14 | ∼−50 (1 mmol g−1 CO2 loading) | 44 |
| SIFSIX-3-Ni | 0.4 | 298 | 378 | 0.29 | 6.50 | ∼−49.8 (1 mmol g−1 CO2 loading) | 44 |
| ZU-36-Ni (GeFSIX-3-Ni) | 0.4 | 298 | 373 | 1.07 | 23.98 | ∼−55.5 (near-zero coverage) | 45 |
| ZU-36-Co (GeFSIX-3-Co) | 0.4 | 298 | 373 | 0.30 | 6.72 | ∼−39.1 (near-zero coverage) | 45 |
| SIFSIX-3-Cu | 0.4 | 298 | 323 | 1.24 | 27.79 | −54 (∼0.25 mmol g−1 CO2 loading) | 46 |
| TIFSIX-3-Ni | 0.4 | 298 | 347/433 | 1.15a | 25.78 | ∼−53 (near-zero coverage) | 31 and 47 |
| NbOFFIVE-1-Ni | 0.4 | 298 | 378 | 1.23a | 27.57 | ∼−54.9 (∼0.1 mmol g−1 CO2 loading) | 47 |
| SIFSIX-3-Cu-i | 0.4 | 298 | 293 | 0.684 | 15.33 | −32 (near-zero coverage) | 46 and 48 |
| SIFSIX-3-Zn | 0.4 | 298 | 393 | 0.13 | 2.91 | ∼−45 (near-zero coverage) | 46 and 49 |
| Mg-MOF-74 | 0.4 | 298 | 453 | 0.14a | 3.14 | ∼−41.5 (∼0.1 mmol g−1 CO2 loading) | 47 and 50 |
| Zn(ZnOH)4(bibta)3 | 0.4 | 300 | 373 | 2.20 | 49.31 | ∼−42 (near-zero coverage) | 51 |
| ∼−71 (∼2.0 mmol g−1 CO2 loading) | |||||||
| Pyrazine-functionalized Co-MOF-74 | 0.4 | 298 | 393 | 1.36 | 30.48 | −48.4 (near-zero coverage) | 52 |
| Co-MOF-74 | 0.4 | 298 | 393 | 0.65a | 14.57 | — | 52 |
| ZU-16-Co (TIFSIX-3-Co) | 0.4 | 298 | 373 | 1.05 | 23.53 | — | 53 |
| mmen-Mg2(dobpdc) | 0.4 | 298 | 343 | 2.00a | 44.83 | — | 54 |
| en-Mg2(dobpdc) | 0.4 | 298 | 343 | 2.50a | 56.04 | — | 54 |
| en-Mg2(dobpdc) | 0.39 | 298 | 403 | 2.83 | 63.43 | ∼−22.5 (near-zero coverage) | 55 |
| ∼−49–−51 (∼1.25–2.0 mmol g−1 CO2 loading) | |||||||
| Mg2(dobdc)(N2H4)1.8 | 0.4 | 298 | 403 | 3.89 | 87.19 | −90 (Virial) −118 (Clausius–Clapeyron) | 56 |
| SIFSIX-3-Ni | 1 | 298 | 378 | 0.62 | 13.90 | ∼−49.8 (1 mmol g−1 CO2 loading) | 44 |
| SIFSIX-3-Cu | 1 | 298 | 378 | 1.72 | 38.55 | ∼−53 (∼1.05 mmol g−1 CO2 loading) | 44 |
| ZU-36-Ni (GeFSIX-3-Ni) | 1 | 298 | 373 | 1.55 | 34.74 | ∼−55.5 (near-zero coverage | 45 |
| ZU-36-Co (GeFSIX-3-Co) | 1 | 298 | 373 | 0.75 | 16.81 | ∼−39.1 (near-zero coverage) | 45 |
| SIFSIX-3-Cu | 1 | 298 | 323 | 1.75 | 39.22 | — | 46 |
| NbOFFIVE-1-Ni | 1 | 298 | 378 | 1.68 | 37.66 | — | 44 |
| TIFSIX-3-Ni | 1 | 298 | 347/433 | 1.50a | 33.62 | ∼−53 (near-zero coverage) | 31 and 47 |
| Mg-MOF-74 | 1 | 298 | 453 | 0.33a | 7.40 | ∼−41.5 (∼0.1 mmol g−1 CO2 loading) | 47 |
| Zn(ZnOH)4(bibta)3 | 1 | 300 | 373 | 2.35a | 52.67 | ∼−42 (near-zero coverage) | 51 |
| ∼−71 (∼2.0 mmol g−1 CO2 loading) | |||||||
| Pyrazine-functionalized Co-MOF-74 | 1 | 298 | 393 | 2.10a | 47.07 | −48.4 (near-zero coverage) | 52 |
| Co-MOF-74 | 1 | 298 | 393 | 0.90a | 20.17 | — | 52 |
| ZU-16-Co (TIFSIX-3-Co) | 1 | 298 | 373 | 1.55a | 34.74 | — | 53 |
| mmen-Mg2(dobpdc) | 1 | 298 | 343 | 3.00a | 67.24 | — | 54 |
| en-Mg2(dobpdc) | 1 | 298 | 343 | 3.20a | 71.72 | — | 54 |
| en-Mg2(dobpdc) | 1 | 298 | 403 | 3.10a | 69.48 | ∼−22.5 (near-zero coverage) | 55 |
| ∼−49–−51 (∼0.13–2.0 mmol g−1 CO2 loading) | |||||||
| Mg2(dobdc)(N2H4)1.8 | 1 | 298 | 403 | 4.35a | 97.50 | −90 (Virial) −118 (Clausius–Clapeyron) | 56 |
| SIFSIX-3-Cu | 40 | 298 | 323 | 2.36a | 52.90 | −54 (∼0.25 mmol g−1 CO2 loading) | 46 |
| SIFSIX-3-Zn | 40 | 298 | 323 | 2.19a | 49.09 | ∼−45 (near-zero coverage) | 46 |
| UTSA-16 | 50 | 298 | 363 | 0.95a | 21.29 | −39.7 (near-zero coverage) | 39 and 40 |
| Zn2(Atz)2Ox (MeOH) | 50 | 293 | 373/353 | 1.10a | 24.66 | −40.8 (near-zero coverage) | 41 and 42 |
| Zn2(Atz)2Ox (H2O) | 50 | 283 | 423 | 2.70a | 60.52 | ∼−55 (near-zero coverage) | 43 |
| JLU-MOF56 | 50 | 298 | 303 | 0.25a | 5.60 | ∼30 (near-zero coverage) | 57 |
| JLU-MOF57 | 50 | 298 | 303 | 0.09a | 2.02 | ∼−32.5 (near-zero coverage) | |
| Cu-F-pym | 50 | 298 | 393 | 0.53a | 11.88 | ∼−30 (near-zero coverage) | 58 |
| UTSA-280 | 50 | 298 | 383 | 0.85a | 19.05 | −42.9 (near-zero coverage) | 59 |
| SIFSIX-3-Ni | 50 | 293 | 413 | 2.45a | 54.91 | — | 50 |
| ZU-36-Ni (GeFSIX-3-Ni) | 50 | 298 | 373 | 2.60a | 58.28 | ∼−55.5 (near-zero coverage | 45 |
| ZU-36-Co (GeFSIX-3-Co) | 50 | 298 | 373 | 2.65a | 59.40 | ∼−39.1 (near-zero coverage) | 45 |
| Zn(ZnOH)4(bibta)3 | 50 | 300 | 373 | 3.00a | 67.24 | ∼−42 (near-zero coverage) | 51 |
| ∼−71 (∼2.0 mmol g−1 CO2 loading) | |||||||
| Pyrazine-functionalized Co-MOF-74 | 50 | 298 | 393 | 6.60a | 147.93 | −48.4 (near-zero coverage) | 52 |
| Co-MOF-74 | 50 | 298 | 393 | 5.20a | 116.55 | — | 52 |
| ZU-16-Co (TIFSIX-3-Co) | 50 | 298 | 373 | 2.75a | 61.64 | — | 53 |
| mmen-Mg2(dobpdc) | 50 | 298 | 343 | 3.40a | 76.21 | — | 54 |
| en-Mg2(dobpdc) | 50 | 298 | 343 | 3.60a | 80.69 | — | 54 |
| en-Mg2(dobpdc) | 50 | 298 | 403 | 3.50a | 78.45 | ∼−22.5 (near-zero coverage) | 55 |
| ∼−49–−51 (∼0.13–2.0 mmol g−1 CO2 loading) | |||||||
| Mg2(dobdc)(N2H4)1.8 | 50 | 298 | 403 | 5.10a | 114.31 | −90 (Virial method) | 56 |
| −118 (Clausius–Clapeyron) | |||||||
| SIFSIX-3-Ni | Lab atmosphere, 49% RH | 296.55 | 413 | 0.18a | 4.03 | — | 50 |
| MIL-101(Cr) | 10 vol% CO2 (0.1 atm) | 298 | 473 | 0.48 | 10.78 | — | 60 |
| 100 ppm SO2, 100 ppm NO, 10% RH | |||||||
Engineering pore architecture for CO2 sorption
The typically weak CO2 binding affinity on MOFs is related to physisorption-based processes. The weak interaction between CO2 and the pore surface of MOFs is also indirectly linked to the low CO2 selectivity over other gases, including the gaseous constituents of indoor air, such as O2, N2, and H2O.36 The development of MOF sorbents for low-concentration CO2 capture through pore-size tailoring has been attained with a handful of materials. Ultramicroporous MOFs (i.e. frameworks containing pores with apertures <5–7 Å)37 have remarkable CO2 uptake capacities at low CO2 concentrations due to their topological pore structures. The ultramicroporous MOF UTSA-16 is an example of such a structure with narrow pore apertures of 3.30 × 5.40 Å.38 UTSA-16 was found to be capable of selectively interacting with CO2 over other gases with a reported CO2 uptake capacity of ∼0.95 mmol g−1 at pCO2 = 50 mbar, 298 K.39 The CO2 molecules were found to interact with the terminal water molecules coordinated to K+ ions, through hydrogen bonding, in the interior of the diamond-shaped cages (4.5 Å in diameter) in the framework. This interaction resulted in moderate ΔHads of CO2 adsorption of ∼−39.7 kJ mol−1 (at near zero-coverage).40 The hydrogen bonding interaction between the oxygen atoms in the CO2 molecules and the crystallographically independent oxygen atoms in H2O was also found to account for 74% of the total CO2 uptake capacity.38 Similarly, Vaidhyanathan et al.41,42 and Banerjee et al.43 reported on a series of solvothermally synthesized ultramicroporous zinc-based aminotriazolate/oxalate (Atz/Ox) MOFs (Zn2(Atz)2Ox (solvent)). Specifically, Zn2(Atz)2Ox (MeOH) was observed to possess pore channels with apertures of 3.50 × 4.00 Å along the a-axis, 3.90 × 2.10 Å along the b-axis, and narrow slit-shaped pores (3.00 × 1.60 Å) along the c-axis, respectively. The primary amino-groups on the aminotriazolate ligands were found to protrude into the cube-shaped pore cavities (4.00 × 4.00 × 4.00 Å) along the a-axis. The high CO2 uptake capacity of the framework (∼1.1 mmol g−1 at pCO2 = 50 mbar, 293 K) was found to be due to both pore-size effects as well as from CO2–amine interactions. This was also indicated by the relatively low ΔHads of CO2, ∼−40.8 kJ mol−1 at near zero-coverage.41,42 Similarly, Zn2(Atz)2Ox (H2O) was shown to possess the same oxalate-pillared structure as Zn2(Atz)2Ox (MeOH) with comparable pore channels and apertures − 5.35 × 5.35 Å along the a-axis, 6.40 × 6.40 Å along the b-axis, and 5.80 × 5.25 Å along the [0 1 1] direction, respectively (Fig. 1a).43 However, unlike Zn2(Atz)2Ox (MeOH), the water-solvated Zn2(Atz)2Ox (H2O) framework was observed to undergo a subtle CO2-induced structural rearrangement at pCO2 = 200 mbar, 273 K (Fig. 1b). This gate-opening effect was observed alongside the appearance of new adsorption sites (denoted as site I and site II) in the framework, which could be separated by their ΔHads. The ΔHads pre-gate opening (i.e. corresponding to site I) was shown to increase from −46 kJ mol−1 to −32 kJ mol−1 post-gate opening, where CO2 adsorption occurred on site II. The ΔHads for the two sites mainly corresponded to amine–CO2 (site I) and CO2–CO2 (site II) interactions.43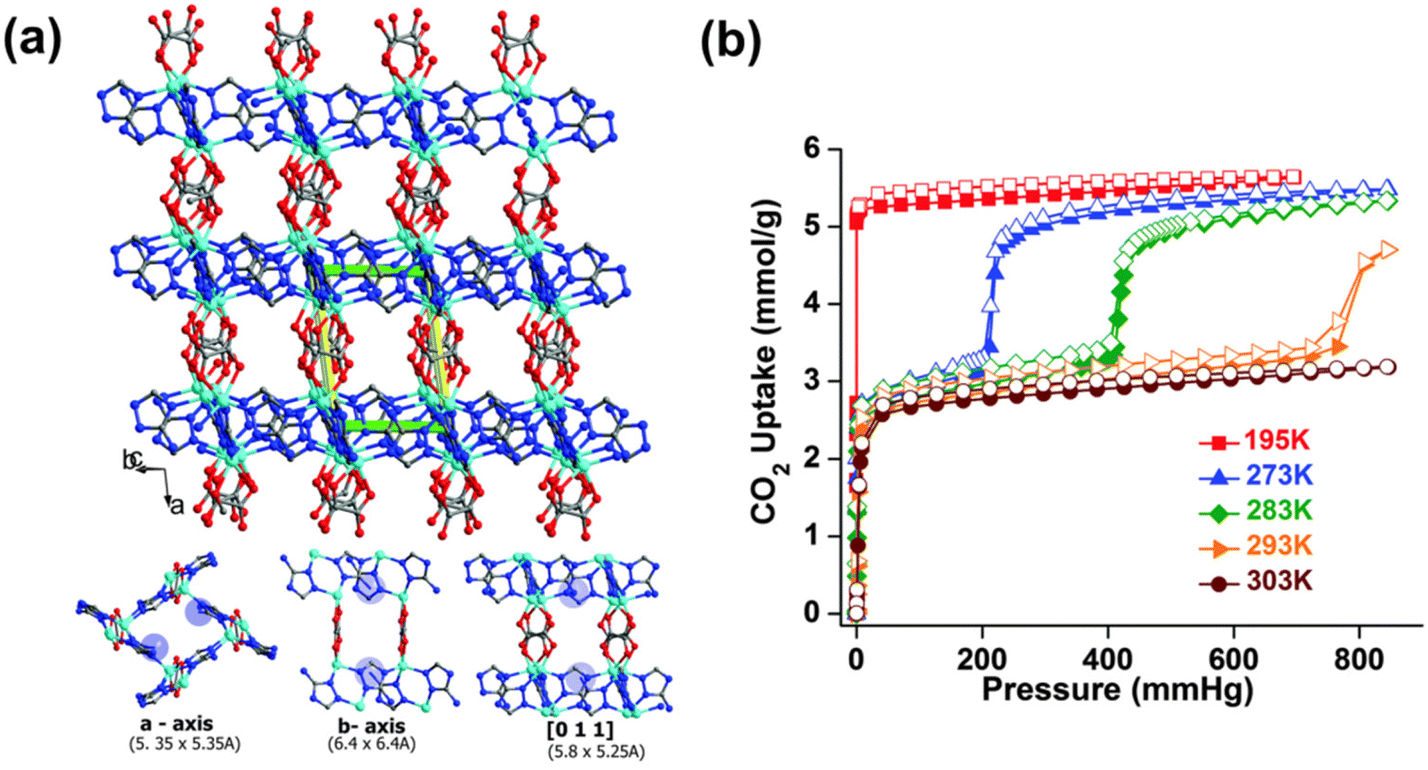 | ||
| Fig. 1 (a) Three-dimensional structure of Zn2(Atz)2Ox showing the ultramicroporous channels along a-, b- and c-axis, and (b) CO2 sorption isotherms of Zn2(Atz)2Ox at different temperatures.43 Reproduced with permission, Copyright 2015 Royal Society of Chemistry. | ||
Liu et al. also reported on two isomorphic triazolate-based ultramicroporous MOFs, namely, JLU-MOF56 ([Ni2(μ2-Cl)(BTBA)2·DMF]·Cl·3DMF) and JLU-MOF57 (([Co2(μ2-Cl)(BTBA)2·DMF]·Cl·3DMF, where BTBA4− = 3,5-bis(triazol-1-yl)benzoate, DMF = N,N′-dimethylformamide).57 Both JLU-MOF56 and -57 featured channels with dimensions of 3.50 × 3.40 Å, 8.50 × 2.80 Å, and 3.50 × 3.40 Å along the a-, b-, and c-axis as well as internal cages 14 Å in diameter. Despite the presence of uncoordinated N-atom sites, the CO2 uptake capacities of JLU-MOF56 (0.25 mmol g−1 at pCO2 = 50 mbar, 298 K) and JLU-MOF57 (0.09 mmol g−1 at pCO2 = 50 mbar, 298 K) were relatively low at low CO2 concentrations. This was attributed to the significantly larger dimension of the cages in the framework as compared to the kinetic diameter of CO2 (3.30 Å).57 Navarro et al.61 and Shi et al.58 investigated the CO2 sieving properties of an ultramicroporous MOF possessing appropriately sized channels as well as surface functionalities, namely, Cu-F-pym ([Cu(F-pymo)2(H2O)1.25]n, where F-pymo = 5-fluorpyrimidin-2-olate)). Cu-F-pym exhibited a 3D structure with GIS-related framework topology and possessed helical channels in the ab-plane with an aperture of 2.90 Å along the c-axis.61 Selective adsorption of CO2 (∼0.53 mmol g−1 at pCO2 = 53 mbar, 298 K) was observed on Cu-F-pym at ambient temperatures (i.e. 293 K) despite the narrow pore aperture. This was attributed to a thermal expansion of the Cu-F-pym framework which enabled the diffusion of CO2 through the structure.61 Pore-size tailoring has also been achieved using small non-functionalized linkers. An example of such was presented for the ultramicroporous MOF UTSA-280 ([Ca(C4O4)(H2O)]·xH2O),59,62 which utilizes squaric acid to form a 3D framework structure with cylindrical 1D channels (3.20 × 4.50 Å and 3.80 × 3.80 Å apertures) along the c-axis. The adsorbed CO2 molecules were found, according to grand canonical Monte Carlo (GCMC) simulations, to interact with the organic linker as well as the coordinating water molecules in the pore channels through van der Waals and electrostatic interactions, giving rise to a ΔHads of CO2 of ∼−42.9 kJ mol−1 from the combined host–guest interactions.59
Hybrid ultramicroporous materials (HUMs) have garnered great attention in the last decade due to their unique structural properties. It is important to note that HUMs may not strictly be classified as MOFs, nevertheless, they will be included in this discussion for comparison. The prototypical HUM structure can broadly be described as 2D square sql nets composed of metal–organic units interconnected by pillaring inorganic anions (e.g. [SiF6]2−, [TiF6]2−, [NbOF5]2−, and [GeF6]2−, see Fig. 2).45,47,63,64 The inherent structure of HUMs provides them with narrow and highly ordered pore channels that are decorated with polarizing atoms.47,64 A comparison between the HUM SIFSIX-3-Ni and TEPA-SBA-15 (tetraethylenepentamine-functionalized mesoporous silica SBA-15) as well as Zeolite-13X was made by Kumar et al.31,50 The authors showed that SIFSIX-3-Ni had superior CO2 uptake capacity (0.18 mmol g−1) as compared to Zeolite13X (0.03 mmol g−1) at 1 bar pure CO2 with 49% RH (296.55 K). However, the performance of the HUM was observed to be worse than TEPA-SBA-15 (3.59 mmol g−1) under the same conditions. On TEPA-SBA-15, chemisorption of CO2 accounted for its high CO2 uptake, especially in the presence of water.50 Fine-tuning of the CO2 uptake properties in SIFSIX-3-M was further attained through the incorporation of different metal cations into the metal–organic unit of the structure. Bhatt et al.44 observed a narrowing of the square channels in SIFSIX-3-Cu (dF⋯F = 6.483(1) Å) when compared with SIFSIX-3-Ni (dF⋯F = 6.694(1) Å) and SIFSIX-3-Zn (dF⋯F = 6.784(1) Å), due to a reduced distance between adjacent [SiF6]2− units. The narrowing of the channel resulted in an enhanced adsorbate–adsorbent interaction at low CO2 concentrations. This enhanced interaction was also indicated by a decrease in the ΔHads from −45 kJ mol−1 on SIFSIX-3-Ni to −54 kJ mol−1 on SIFSIX-3-Cu. As further reported by Bhatt et al. the substitution of the pillaring anion from [SiF6]2− to [NbOF5]2− was also found to decrease the distance between the pendant fluorine moieties due to an increase in the bonding distance for Nb–F (dNb–F = 1.899(1) Å) as compared to Si–F (dSi–F = 1.681(1) Å). An increase in volumetric CO2 uptake at pCO2 = 0.4 mbar by 15 to 340% was subsequently observed for NbOFFIVE-1-Ni (1.3 mmol g−1 at 298 K) over SIFSIX-3-Cu (∼1.25 mmol g−1 at 298 K), SIFSIX-3-Ni (∼0.39 mmol g−1 at 298 K), and SIFSIX-3-Zn (∼0.14 mmol g−1 at 298 K). The increase in CO2 uptake was attributed to a further decrease in distance between pendant F⋯F moieties in NbOFFIVE-1-Ni (dF⋯F = 3.210(8) Å) compared with SIFSIX-3-Cu (dF⋯F = 3.483(1) Å) and SIFSIX-3-Ni (dF⋯F = 3.694(1) Å).
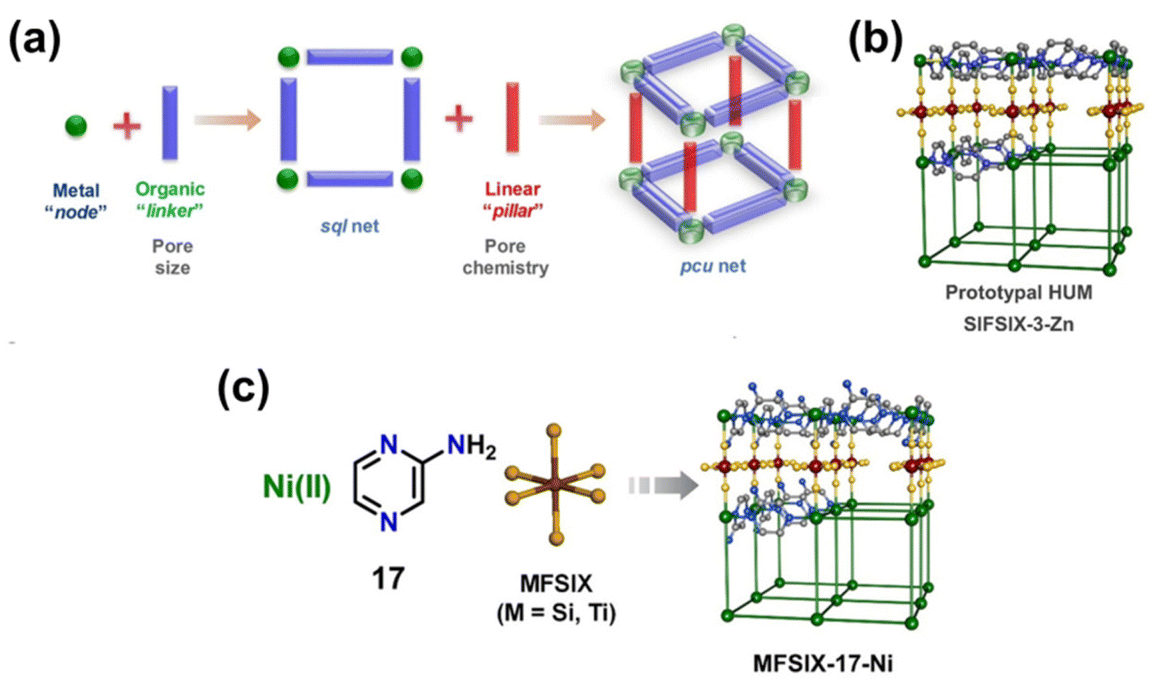 | ||
| Fig. 2 (a) Schematic illustration of the modularity of pillared square grids that form the pcu topology of HUMs, (b) the prototypal pyrazine (pyz) linked HUM [Zn(pyz)2(SiF6)]n, and (c) schematic illustration of the building blocks.64 Reproduced with permission, Copyright 2021 John Wiley & Sons, Inc. | ||
Zhang et al.45 reported the use of [GeF6]2− units to slightly reduce the M–F distance (dGe−F = 1.83 Å) in the inorganic anions in ZU-36-Ni as compared to NbOFFIVE-1-Ni. The formation of an isostructural Co-based HUM, ZU-36-Co, was additionally investigated to assess the pore size and CO2 uptake capacities in these ZU-36 frameworks. A shortening of the bond distance between the metal cation and the pyrazine linker in the square lattice of ZU-36-Co (dNi-pyrazine = 2.12 Å) was achieved in ZU-36-Ni (dNi-pyrazine = 2.08 Å). The decreased metal cation – pyrazine distance led to an enhanced CO2 uptake capacity in the low-pressure range for ZU-36-Ni (1.07 mmol g−1 at pCO2 = 0.4 mbar, 1.55 mmol g−1 at pCO2 = 1 mbar CO2, 298 K) and corresponded to an over 200% increase from ZU-36-Co (0.30 mmol g−1 at pCO2 = 0.4 mbar, 0.75 mmol g−1 at pCO2 = 1 mbar CO2, 298 K). The CO2 uptake capacity of ZU-36-Ni was found to be slightly lower than other HUMs such as SIFSIX-3-Cu (1.24 mmol g−1 at pCO2 = 0.4 mbar, 1.75 mmol g−1 at pCO2 = 1 mbar CO2, 298 K) and NbOFFIVE-1-Ni (1.30 mmol g−1 at pCO2 = 0.4 mbar, 1.68 mmol g−1 at pCO2 = 1 mbar CO2, 298 K). The difference in CO2 uptake may be related to the more optimal F⋯F distances in SIFSIX-3-Cu and NbOFFIVE-1-Ni than in ZU-36.45 Similarly, Kumar et al.47 investigated another HUM structure that was isoreticular with SIFSIX-3-M, namely TIFSIX-3-Ni. The authors utilized [TiF6]2− anionic pillars to further tailor the sorption properties of the SIFSIX-3-M framework. The M–F bond distance in TIFSIX-3-Ni (dTi−F = 1.81 Å) was found to be similar to Zu-36-Ni (dGe−F = 1.83 Å) and NbOFFIVE-1-Ni (dNb−F = 1.899(1) Å). The corresponding CO2 uptake capacity at pCO2 = 0.4 mbar of TIFSIX-3-Ni (∼1.15 mmol g−1 at 298 K) was not found to differ significantly from NbOFFIVE-1-Ni (∼1.23 mmol g−1 at 298 K) or ZU-36-Ni (1.07 mmol g−1 at 298 K).45 The presence of tight CO2 binding sites was also observed in this framework, as indicated by a low ΔHads which was compared to other HUMs – TIFSIX-3-Ni, ∼−49 kJ mol−1 (at 0.1 mmol g−1 CO2 loading), NbOFFIVE-1-Ni, ∼−54.9 kJ mol−1 (0.1 mmol g−1 CO2 loading), ZU-36-Ni, −55.5 kJ mol−1 (at near-zero loading),45 SIFSIX-3-Cu, ∼−53.0 kJ mol−1 (0.1 mmol g−1 CO2 loading).44,50
The impact of increasing the length of the organic molecule in the metal–organic unit in SIFSIX-3-Cu was further investigated by Shekhah et al.46 through the substitution of pyrazine in SIFSIX-3-Cu by dipyridylacetylene. This approach resulted in the formation of the isoreticular HUM SIFSIX-2-Cu-i. The authors reported a decrease in CO2 uptake capacity at pCO2 = 0.4 mbar for SIFSIX-2-Cu-i (0.07 mmol g−1 at 298 K) when compared to SIFSIX-3-Cu (1.24 mmol g−1 at 298 K) and SIFSIX-3-Zn (0.13 mmol g−1 at 298 K). The decrease in CO2 uptake was attributed to an increase in pore size from 3.5 Å in SIFSIX-3-Cu to 5.15 Å in SIFSIX-2-Cu-i. This enlargement of the average distance between the CO2 molecules and the fluorine atoms in the channels of SIFSIX-2-Cu-i further led to a significantly increased ΔHads for CO2 of ∼−32 kJ mol−1 as compared to SIFSIX-3-Cu (−54 kJ mol−1).46 It is therefore clear that the ΔHads in MOF sorbents play a crucial role in their performance to capture CO2. Many sorbents generally exhibit relatively low enthalpies of adsorption (<−50 kJ mol−1) in the absence of ultramicropores or active functional groups. Thus leading to their poor performance at adsorbing CO2 at low-concentrations. Various routes for improving the affinity between CO2 molecules and a framework has been presented in the previous section, however, a compromise is generally required in order for these materials to be utilized in real-world applications. TSA processes were proposed by Lively et al.65 to be more thermodynamically efficient as compared to PSA processes when utilizing sorbents with lower enthalpies of adsorption (i.e. <−50 kJ mol−1) for dilute steams containing ∼100–1000 ppm CO2. The PSA process, on the other hand, was found to have low efficiency even for sorbents with relatively low CO2 affinity (i.e. ΔHads >−35 kJ mol−1). Although sorbents with low enthalpies of CO2 adsorption were found to be more suitable for trace CO2 capture in TSA processes, it is important to note that a too low ΔHads will significantly increase the regeneration costs.
CO2 sorption on high-energy sorption sites
Coordinatively unsaturated or open metal sites in MOFs have additionally been used to tune the CO2 uptake capacity at low CO2 concentrations. Strong interactions between such sites which exhibit electrostatic fields enable them to interact with the π-orbitals of polarizable guest molecules such as CO2. The interaction between CO2 and these high-energy adsorption sites is pivotal for low-concentration CO2 capture.66,67 Notably, MOFs such as M-MOF-74 (e.g. M = Mg2+, Fe2+, Co2+, Zn2+, Ni2+), HKUST-1, and MIL-101 have been proposed for CO2 capture at low CO2 concentrations. Liu et al.60 investigated the CO2 capture performance of MIL-101(Cr), a mesoporous MOF containing Cr(III) with open metal sites. This MOF was observed to have a CO2 uptake of 0.48 mmol g−1 at 298 K and 10% RH from a gas stream containing 100000 ppm (∼pCO2 = 0.1 bar) CO2 as well as 100 ppm SO2, 100 ppm NO.60 Kumar et al.47 investigated the sorption properties of Mg-MOF-74 and HKUST-1 and compared them with TIFSIX-3-Ni and NbOFFIVE-1-Ni. At pCO2 = 0.4 and 1 mbar, the CO2 uptake capacities of Mg-MOF-74 (0.14 mmol g−1 at 0.4 mbar, 0.33 mmol g−1 at 1 mbar, 298 K) and HKUST-1 (no uptake at 0.4 mbar, 0.13 mmol g−1 at 1 mbar, 298 K) were significantly lower as compared to the HUMs. The ΔHads for Mg-MOF-74 (∼−41.5 kJ mol−1 at 0.1 mmol g−1 CO2 loading) and HKUST-1 (∼−23 kJ mol−1 at 0.1 mmol g−1 CO2 loading) further confirmed that the presence of open-metal sites in these frameworks can create strong binding sites for CO2 as compared to conventional physisorption-based interactions. On the other hand, the narrow pore structure of HUMs,44,47,64 such as those found in NbOFFIVE-1-Ni,44 still offers higher energy CO2 sorption sites (ΔHads ∼−54.9 kJ mol−1) than the open-metal sites on some MOFs.
Post-synthetic modification of MOFs for CO2 capture
The introduction of functional groups, commonly through post-synthetic modifications, offers several advantages for increasing the binding interaction with CO2. Typical functional groups include basic groups such as primary and secondary amines, polarizing halogen atoms, and larger hydrocarbon chains. Not only can these functional groups facilitate Lewis acid–base reactions (i.e. chemisorption-based adsorption processes), they can also increase the presence of strong electrostatic interactions, and possibly introduce steric effects to enhance adsorbate–adsorbent van der Waals interactions. As such, numerous studies on functionalized MOFs for trace CO2 capture have been carried out. For example, Bien et al.51 utilized a mild ligand exchange procedure to introduce nucleophilic sites in [Zn(ZnO2CCH3)4(bibta)3] (bibta2− = 5,5′-bibenzotriazolate) to produce [Zn(ZnOH)4(bibta)3], as presented in the schematics shown in Fig. 3.51 The post-synthetic modification resulted in significantly enhanced CO2 uptake. The CO2 uptake capacity of the modified [Zn(ZnOH)4(bibta)3] was 2.20 mmol g−1 (0.4 mbar, 300 K), which was a significant improvement as compared to the negligible uptake on [Zn(ZnO2CCH3)4(bibta)3] under the same conditions. The chemisorption of CO2 in [Zn(ZnOH)4(bibta)3] was further confirmed by the low ΔHads for the MOF (−71 kJ mol−1 at ∼2.0 mmol g−1 CO2 loading) thus indicating that CO2 fixation likely occurred via a reversible Zn-OH/Zn–O(COOH) route (Fig. 3).51 Hu et al.52 also successfully obtained a pyrazine-functionalized Co-MOF-74 by post-synthesis modification. The effective pore size of the Co-MOF-74 was reduced from 11–12 Å down to <7 Å,68 at the same time, Lewis basic sites were introduced from the non-coordination N-atoms on the pyrazine molecules. CO2 uptake for the pyrazine-functionalized Co-MOF reached 1.26 mmol g−1 (0.4 mbar at 298 K), which was significantly higher than its non-functionalized counterpart (∼0.65 mmol g−1 at 0.4 mbar and 298 K). The uptake capacity on the functionalized MOF was however slightly lower as compared to other high-performing MOFs such as TIFSIX-3-Ni (∼1.1 mmol g−1 at 298 K)31 and ZU-16-Co (∼1.05 mmol g−1 at 298 K).53 Correspondingly, the ΔHads of the pyrazine-functionalized Co-MOF-74 was higher (−48.4 kJ mol−1 at zero loading) than other frameworks with highly tailored pore structures (e.g. NbOFFIVE-1-Ni, −54.9 kJ mol−1 at 0.1 mmol g−1 CO2 loading)47 or chemisorption-based sorbents (e.g. [Zn(ZnOH)4(bibta)3], −71 kJ mol−1 at ∼2.0 mmol g−1 CO2 loading).51,52 The moderate ΔHads of the functionalized MOF was however noticeably lower than the ΔHads of typical physisorbents, but not as low as the ΔHads of chemisorbents. This gives pyrazine-functionalized Co-MOF-74 an advantage over other sorbents with respect to energy costs for regeneration.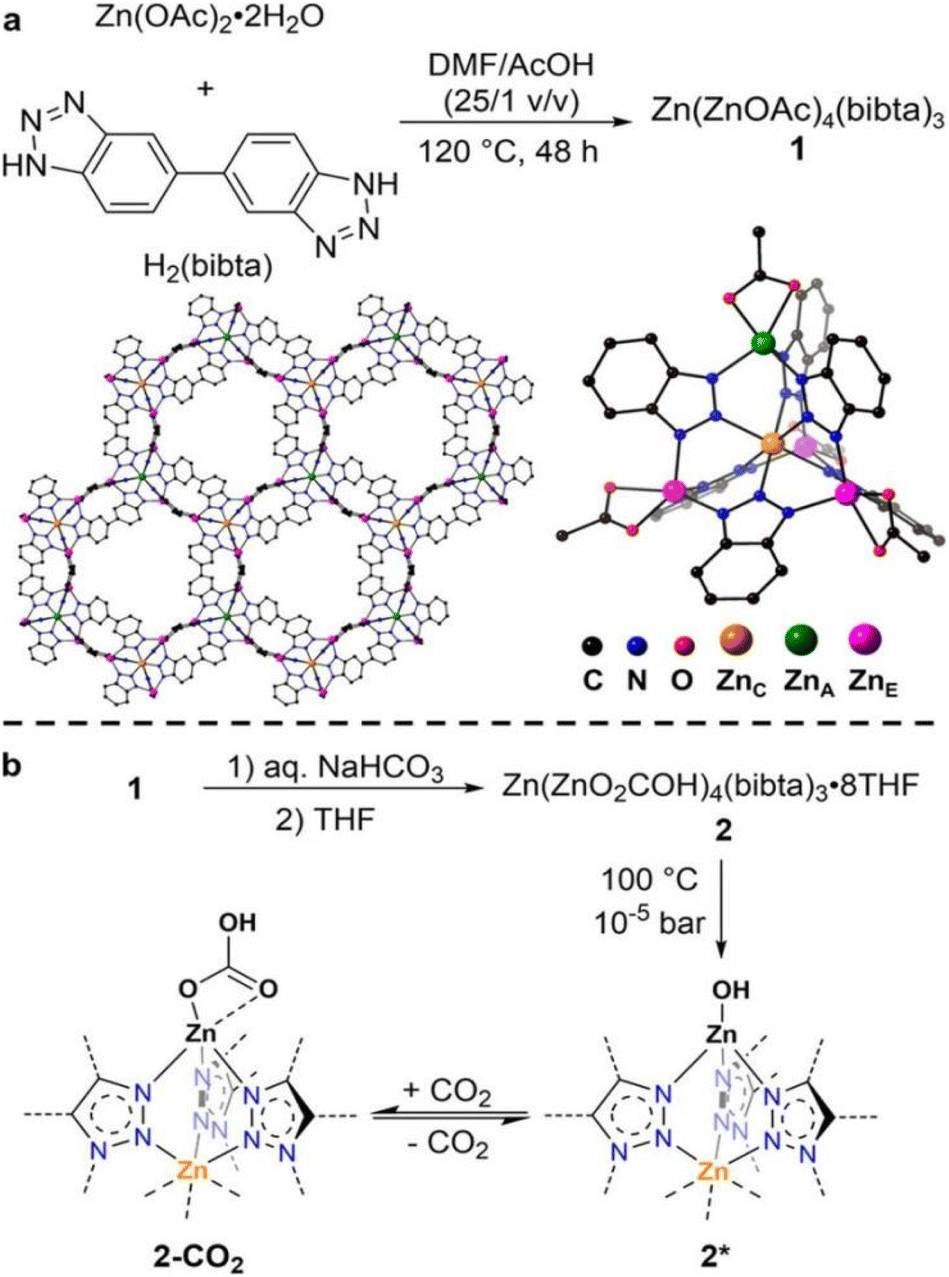 | ||
| Fig. 3 (a) Synthesis structure of the Zn(ZnOAc)4 SBUs. (b) Synthesis of Zn(ZnOH)4(bibta)3 and mechanism of reversible CO2 fixation.51 Reproduced with permission, Copyright 2013 American Chemical Society. | ||
Similarly, MOFs with coordinatively unsaturated sites have commonly been used to post-synthetically introduce amine moieties. Lewis acid–base reactions between the CO2 molecules and the accessible amine groups (e.g. primary or secondary amines) on the pore surfaces generally lead to the formation of carbarmic acid followed by ammonium carbamate in the presence of humidity.35,69 Notable examples include M-MOF-74, Mg2(dobpdc) (dobpdc4− = 4,4′-dioxido-3,3′-biphenyldicarboxylate), and MIL-101(Cr).70 Park et al. grafted various diamines on to the pores of Mg2(dobpdc) to increase the CO2 binding affinity.54 Linear and branched diamines with ethylene and propylene linkages were introduced post-synthetically onto Mg2(dobpdc) (Fig. 4, 5a, and b). The CO2 uptake capacity of N-isopropylethylenediamine-appended Mg2(dobpdc) (mmen-Mg2(dobpdc)) and ethylenediamine-appended MOFs at 400/1000 ppm were approximately 2.30/3.00 mmol g−1 and 2.50/3.20 mmol g−1, respectively, at 298 K. The bulky N-isopropylethylenediamine introduced steric hindrance through the branched isopropyl-substituent, which may have kinetically restricted the CO2 diffusion and the accessibility of the amine sites thus resulting in a slightly lower CO2 uptake.54 Similarly, Lee et al.55 observed that the CO2 uptake of ethylenediamine-appended Mg2(dobpdc) (en-Mg2(dobpdc)) was 2.83 mmol g−1 at 0.39 mbar, 298 K, which was 1.4 times higher than the N-isopropylethylenediamine-functionalized counterpart of the MOF (mmen-Mg2(dobpdc) at 2.00 mmol g−1 (0.39 mbar, 298 K). The higher uptake capacity of en-Mg2(dobpdc) as compared to mmen-Mg2(dobpdc) at low CO2 pressures was hypothesized by Lee et al.55 and McDonald et al.34 to be related to two factors: (1) the higher accessibility of the primary amine moieties in en-Mg2(dobpdc), and (2) due to a large increase in entropy connected with the reorganization of the secondary amines required for chemisorption of CO2. These two factors led to preferential CO2 adsorption onto low-energy sites (i.e. not associated with amine groups) and weak amine sites. The ΔHads between ∼1.25–2.0 mmol g−1 CO2 loading was estimated to range from −49 to −51 kJ mol−1 and corresponded well with the enthalpy of formation (ΔHf) of carbarmic acid (−52.8 kJ mol−1). Additionally, a pressure-induced phase change was also observed in mmen-Mg2(dobpdc), giving rise to a sharp increase in CO2 uptake at ∼0.2 mbar (298 K). This phenomenon was attributed to a cooperative CO2 adsorption process wherein the deprotonation of the metal-bound amine by an adjacent non-coordinating amine moiety triggered a nucleophilic addition of CO2. The resulting formation of a carbamate-ammonium ion pair in turn had a destabilizing effect on the metal-bound amine on the neighboring molecule. This destabilization, in turn, initiated the adsorption of another CO2 molecule through the same process, as described in Fig. 5c.71 Furthermore, adjacent non-coordinating amines were found to interact through hydrogen bonding in the absence of CO272 which may prevent these groups from participating in the CO2 adsorption process at pressures below ∼0.2 mbar.56,71
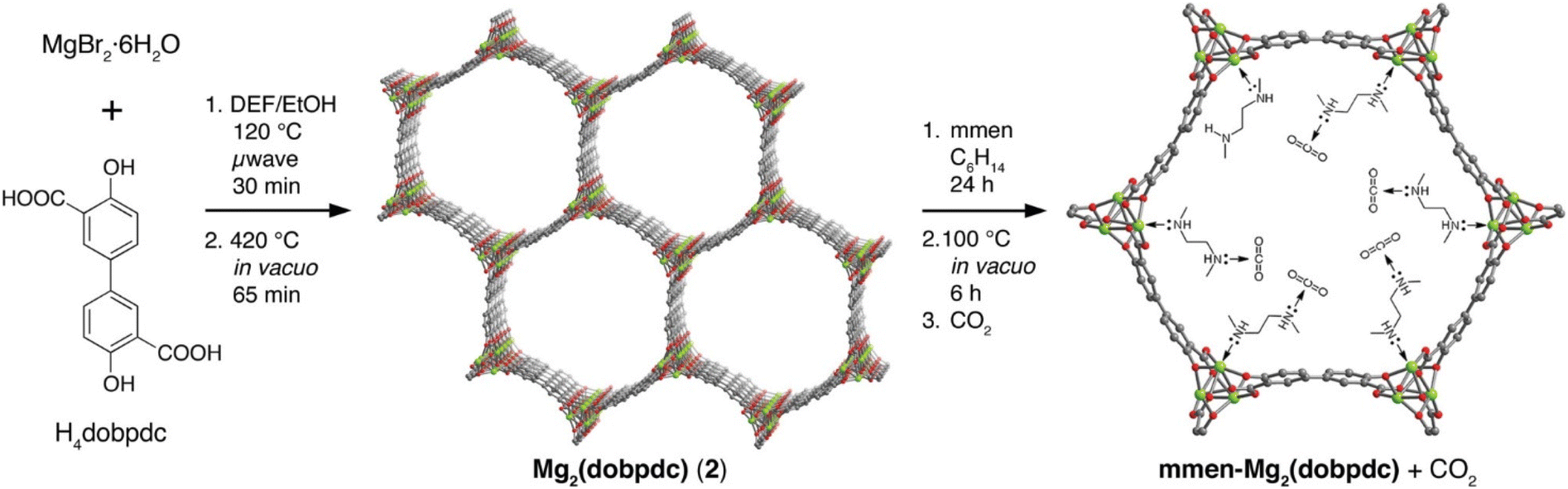 | ||
| Fig. 4 (Left) Schematic of synthesis of Mg2(dobpdc) and (middle) the amine-functionalization process to produce mmen-Mg2(dobpdc), (right) interaction between pre-treated (degassed) mmen-Mg2(dobpdc) and CO2 molecules. Green, red, and gray spheres represent Mg, O, and C atoms respectively; H atoms are omitted for clarity.34 Reproduced with permission, Copyright 2012 American Chemical Society. | ||
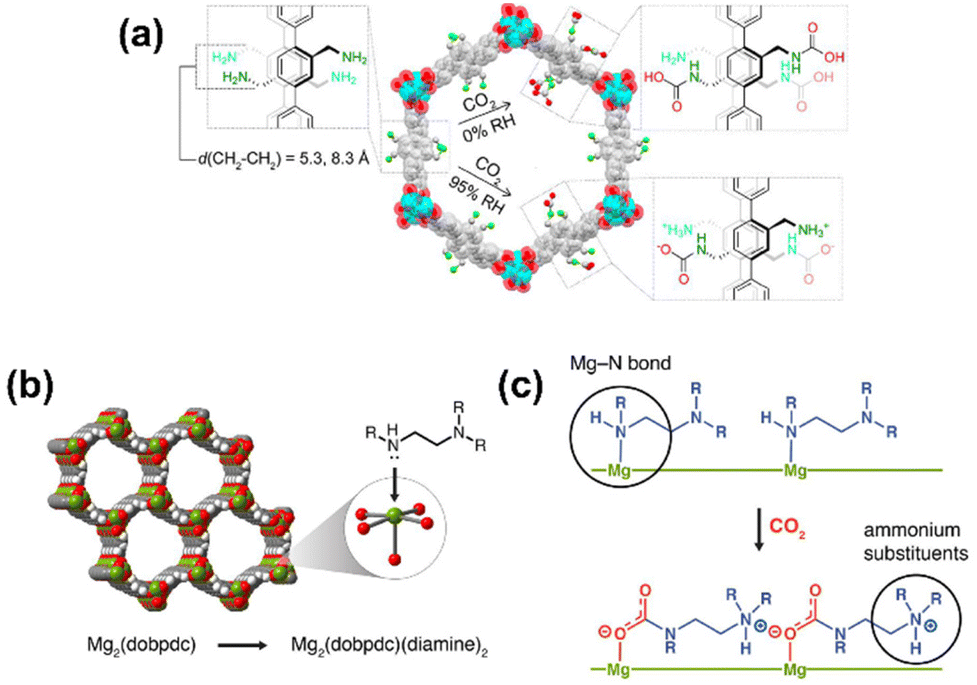 | ||
| Fig. 5 (a) Chemisorption species post-CO2 adsorption in IRMOF-74-III-(CH2NH2)2.35 (b) Representative structure of the metal–organic framework Mg2(dobpdc). Green, red, gray, and white spheres represent Mg, O, C, and H atoms, respectively. (c) Depiction of cooperative CO2 insertion into a row of Mg2+–diamine sites to form ammonium carbamate chains along the pore axis.88 Reproduced with permission, Copyright 2017 American Chemical Society. | ||
Liao et al.56 proposed that the incorporation of smaller diamine-species could increase the intermolecular distance between the uncoordinated amines and thus reduce the energetic favorability of hydrogen bonding between the moieties. An enhanced CO2 uptake capacity in the hydrazine (N2H4)-functionalized Mg2(2,5-dihydroxy-1,4-benzenedicarboxylate – dobdc), [Mg2(dobdc)(N2H4)1.8] of 3.89 mmol g−1 at ∼0.4 mbar (298 K) was observed. The CO2 uptake of the [Mg2(dobdc)(N2H4)1.8] was higher than mmen-Mg2(dobpdc) (2.0 mmol g−1 ∼0.4 mbar at 298 K)55 as well as en-Mg2(dobpdc) (2.85 mmol g−1 ∼0.4 mbar at 298 K).55 The low ΔHads (−118 kJ mol−1 at zero-coverage), in conjunction with observations from in-situ infrared spectroscopy, supported the formation of carbamate, which was found to be possible due to the long intramolecular distances between neighboring amines. An absence of proton-transfer during the adsorption process was also observed from the spectroscopic measurements.56
The three different approaches used to engineer MOFs for low-concentration CO2 capture discussed here mainly aim to increase the interaction between CO2 and the adsorption site. All three approaches have been shown to work effectively. The balance between having sufficiently low ΔHads of CO2 to selectivity capture CO2 at low-concentration, with low enough activation energy for sorption to take place at ambient temperature, and allowing desorption to take place with minimal energy input is perhaps the biggest challenge that deserves continued attention.
CO2 selectivity, adsorption kinetics, and sorbent stability
As discussed, an ideal sorbent for CO2 capture at low CO2 concentrations should have strong binding interactions with CO2, high CO2 uptake capacities at low-concentrations, and relatively low ΔHads of CO2. The utilization of MOFs as CO2 sorbents in real-life applications is strongly dependent on (1) the selective adsorption of CO2 over other gaseous constituents in air (particularly H2O), (2) the framework stability in the presence of guest species, as well as (3) the ΔHads which plays a pivotal role in sorbent regeneration.Various factors affect the gas selectivity of a sorbent, which include the topological structure and surface chemistry of the materials, and the physicochemical properties of the gas in question. The low CO2 partial pressure at low CO2 concentrations (i.e. <1 mbar), in conjunction with the significant presence of other gas molecules (e.g. H2O, N2, O2 – physical properties of these gases are shown in Table 2) present a remarkably challenging problem for the selective capture of CO2 in these conditions. Kinetic (also known as partial molecular sieve action) and thermodynamic effects likely play a crucial role in the overall observed CO2 selectivity of a sorbent.77 Shekhah et al. observed significantly stronger and more rapid adsorption of CO2 in SIFSIX-3-Cu than O2, CH4, and H2. The strong and fast adsorption of CO2 was attributed to a combined kinetic and thermodynamic effect. Partial sieving was achieved in the HUM due to the small size of the CO2 molecules in conjunction with the large quadrupole moment and polarizability of CO2. These factors combined enhanced the favorable interaction between CO2 and the adsorption sites on the HUM.46
| Gas molecule | Kinetic diameter (Å) | Polarizability (10−25 cm3) | Dipole moment (1018 esu−1 cm−1) | Quadrupole moment (1026 esu−1 cm−2) |
|---|---|---|---|---|
| CO2 | 3.30 | 29.1 | 0 | 4.30 |
| H2O | 2.65 | 14.5 | 1.85 | — |
| N2 | 3.68 | 17.4 | 0 | 1.52 |
| O2 | 3.46 | 15.8 | 0 | 0.39 |
On the other hand, amine-modified MOFs rely heavily on the accessibility of the amine-sites on the pore surface as well as the formation of necessary intermediate species that govern the chemisorption process. The formation of carbamate/ammonium ion pairs in MOFs, such as that seen on mme-Mg2(dobpdc) have been found to proceed through the formation of an intermediate species and subsequently followed by a transition state (Fig. 5 and in ref. 71). Although energy is required for the chemisorption process to take place, high temperatures will also simultaneously drive the reaction back towards the starting species, as the formation of the intermediate species is entropically unfavored. Thus, the formation rate of the product is limited by the intermediate species and the rate-limiting step in the reaction is attributed to the chemisorption process.78 Stability of the frameworks in the presence of moisture presents an obstacle from a structural integrity point of view and can severely limit the applicability of MOFs in CO2 capture, especially from humid gas mixtures with low CO2 concentrations. The overall chemical and thermal stability of MOFs could be improved by utilizing high-valence metals (e.g. Zr4+ and Al3+), which tend to form strong coordination bonds with carboxylate-based organic ligands, or similarly, lower-valence metals (i.e. soft metals such as Zn2+ and Cu2+) which form strong coordination bonds with soft basic ligands. These approaches allow the formation of robust frameworks with enhanced stability that can cope with issues related to e.g. ligand displacement by water molecules.79 Furthermore, the incorporation of ligands with hydrophobic functional groups (e.g. fluorine-containing,80 alkyl-,81 or ethyl ester-groups82) or groups that introduce steric effects81,82 may impede the diffusion of water molecules through the framework and prevent hydrolysis. Therefore, careful consideration of the chemical compositions of the frameworks is necessary for the utilization of the MOFs in many real-world applications.
The susceptibility of amine species to thermal and oxidative degradation has also been well-documented in solution, e.g. in the case of monoethanolamine (MEA) which is commonly used in amine scrubbing for CO2 capture and separation.6,83–86 A handful of studies on amine-grafted solid sorbents, such as MCM-41 silica (TRI-PE-MCM-41), has shown primary amine to be more resilient towards oxidative degradation at high temperatures (>100 °C) as compared to secondary amines.87 A cooperative degradation mechanism has also been suggested to take place in diamine-supported MCF silica involving the secondary and terminal primary amines.88 However, the stability of amine-modified MOFs has, on the other hand, been studied to a lesser extent. Siegelman et al.89 observed an improved oxidative stability of 2-ampd-Mg2(dobpdc) (where 2-ampd = 2-(aminomethyl)piperidine) when exposed to a dry gas mixture (∼21% O2 and ∼79% N2 at 1 bar) at 100 °C for 5 h, as compared to many other silica-based materials functionalized with secondary amines. A negligible reduction in CO2 uptake capacity was observed with the modified MOF as compared to the pristine material. Furthermore, no oxidative by-products of 2-ampd were detected by either 1H NMR or IR spectroscopy, in contrast to amine-modified silicas that are prone to excessive oxidative degradation when treated in similar environments. The improved stability of 2-ampd-Mg2(dobpdc) was attributed to the distance between the metal sites in the framework, which separates the amine species by ∼7 Å in the pore channels, thus preventing oxidation reactions from taking place between adjacent amine moieties.89 Furthermore, the incorporation of branched diamines (e.g. 1,1-dimethylethylenediamine and N-ethylethylenediamine) was found to improve the stability of functionalized Mg2(dobpdc) as compared to primary diamines. Signifying that alkyl substituents may play a significant role in improving framework stability in the presence of oxygen and water vapor.90,91
Cost of sorbent production and scalability
Although MOFs show great potential in various application fields, several restrictions such as high synthesis cost, difficulties in scalability, and processability of MOFs need to be addressed before their commercialization and industrial applications can be achieved. In addition to the exploration of MOFs with new structures and applications, great efforts have been devoted to the development of cost-effective, green, and scalable approaches for the synthesis of MOFs. These approaches are especially important for candidate sorbents that are highly feasible for practical applications;92–96 for example, M-MOF-74, UiO-66, HKUST-1, and MIL-series MOFs are relevant for trace CO2 capture.Metal ions and organic linkers are the two components for building a MOF structure. Metal nitrates and chlorides with high solubility and weak interfacial interactions are the most common metal sources for the synthesis of coordination complexes including MOFs.92 However, the by-products containing chloride and nitrate ions are highly corrosive and toxic in general. Recent studies demonstrated that inexpensive metal acetates, hydroxides, and oxides can be used as precursors for low-impact synthesis of MOFs.97 For example, MIL-53(Al)-NH2 layers formed directly on the surface or in the channels of anodized aluminium oxide (AAO) substrates by solvothermal reactions, in which the aluminium oxides served as metal precursors to release aluminium ions for the construction of MOF structures.98–100 The mixed-matrix membranes (MMMs) based on the AAO-MOF nanocomposites exhibited high CO2/N2 selectivities of 34–39 with extremely high CO2 permeance of up to 3000 GPU (1 GPU = 3.35 × 10−10 mol cm−2 s−1 Pa−1) that can potentially be developed for low-concentration CO2 capture. In another example, Majano et al. demonstrated the conversion of Cu(OH)2 into HKUST-1 at room temperature in an aqueous ethanoic solution with high space–time-yields (STY) up to 18127 kg m−3 d−1.101 More importantly, the non-soluble nature of Cu(OH)2 could avoid the release of copper ions into the solution that may ease the purification of the recycled solution. This synthesis approach opens new opportunities for the industrial production of HKUST-1 with low cost, low energy consumption, and minimal environmental impact. In addition, metal acetylacetonates (acac) with low toxicity are considered as green reagents in industrial processes that have been also used for the synthesis of various MOFs. Avci-Camur et al. synthesized several Zr(IV)-carboxylate based MOFs from Zr(acac)4 with relatively high yields in water, including UiO-66-NH2, UiO-66-(OH)2, UiO-66-COOH, and UiO-66-(COOH)2.102
Compared to the low cost and adequate availability of most metal precursors, the organic linkers involved in the synthesis of MOFs are usually costly. For example, polytopic carboxylic acids, the most common linkers for MOF synthesis, are prepared from petrochemical feedstocks through several steps involving the use of massive amounts of organic solvents and the generation of toxic by-products. Therefore, it is greatly desired to develop green MOF synthesis routes from renewable, affordable, and non-toxic linkers. Recently, several studies focused on the synthesis of bio-MOFs using biomolecules and biomass-derived organic compounds.103 Biomolecules such as amino acids, nucleobases, proteins, peptides, cyclodextrins, tannins, and saccharides, which contain coordination sites of carboxylate groups or nitrogen and oxygen atoms with lone pairs of electrons have been successfully used for synthesizing a range of bio-MOFs.104 Fumaric acid, a naturally available organic compound containing two carboxylic acid groups, has been constructed into several fumarate-based MOFs. For example, the aluminum fumarate MOF (Basolite A520), an analogue of MIL-53(Al), has been commercialized by BASF with high STYs of up to 5300 kg m−3 d−1 could be achieved.105,106 Such bio-MOFs with good biocompatibility and low toxicity offer great hope for the development of many biological and medical applications.107 In addition, several recent studies have investigated the potential of bio-MOFs for CO2 capture.108 Some of the bio-MOFs showed relatively high CO2 capacity and selectivity at the conditions that are relevant to post-combustion capture of CO2. For example, Basolite A520 with remarkable water stability and decent CO2 uptake (2.1 mmol g−1 at 1 bar, 303 K) is suitable for CO2 capture under wet conditions.109 Bio-MOF-11, a cobalt-adeninate obtained by the solvothermal reaction between cobalt acetate and adenine (a type of nucleobase), displayed a high CO2 uptake of 6.0 mmol g−1 at 1 bar and 273 K and high calculated CO2/N2 selectivities up to 81 at 273 K.110 However, the performance of bio-MOFs for low-concentration CO2 capture is typically somewhat moderate when compared to the abovementioned top performing MOF-based CO2 sorbents. We anticipate that future studies focusing on amine-modification of bio-MOFs will make these materials suitable for low-concentration CO2 capture.
In addition to the metal sources and organic linkers, the selection of an appropriate solvent is vital for MOF synthesis, especially for the development of green and scalable synthesis approach. The physical properties of the solvent such as polarity, boiling point, viscosity, as well as the cost and environmental impact should be taken into consideration. DMF with a high boiling point and high polarity is widely employed for the synthesis of numerous MOFs under solvothermal conditions. However, the use of DMF not only increases the cost but also generates hazardous amines upon heating, which is not favourable for industrial-scale synthesis of MOFs. Remarkably, recent studies demonstrated that various MOFs including M-MOF-74, NH2-MIL-53(Al), HKUST-1 can be synthesized in green solvents such as ethanol and water, and in some cases even at room temperature and ambient pressure.96,111–115 For example, Huo et al. developed a facile, rapid, and industrially relevant approach for the synthesis of HKUST-1 in water at room temperature with high STY of >2000 kg m−3 d−1. D'Amato et al. performed the reaction of cerium ammonium nitrate and tetrafluoroterephthalic acid in water and afforded a MOF with MIL-140 topology.116 The obtained MOF exhibited an unusual S-shape CO2 isotherm displaying a steep adsorption increase at pressure <0.03 bar at 273 K, which was an indication of the specific interactions of the quadrupolar CO2 molecule (Table 2) with the MOF surface. The high CO2 adsorption capacity at the low partial pressure, the exceptionally high CO2/N2 selectivity of up to 1900, as well as the green synthesis approach, shortlists this MIL-140 type MOF for possible application in low-concentration CO2 capture. It is worth mentioning that MOFs with good CO2 uptake capacity (2.5 mmol g−1 at 1 bar and 273 K) can be synthesized with bio-molecules in water at room temperature, as demonstrated by the synthesis of bismuth-based MOFs [Bi2O(H2O)2(C14H2O8)·nH2O], which was made using ellagic acid as the organic linker.117 Still, solvent-based synthesis will inevitably produce liquid waste containing metal ions and organics that are harmful to the environment. The ideal case would be for the metal precursors and the organic linkers to be entirely converted into desired MOFs via solid-state reactions with no waste/by-products (including solvent waste). Mechanosynthesis has been shown as a green chemistry approach for the synthesis of a variety of functional porous materials such as covalent organic frameworks,118 zeolites,119 as well as MOFs.120 For example, various MOFs have been successfully synthesized with high yields by ball mining or grinding of the starting materials at room temperature without any solvent.121 In some cases, trace-amount of solvent is needed to increase the reactivity of the reagent. The obtained MOFs have high specific surface areas and high crystallinity that are comparable to those prepared by conventional methods. For example, Julien et al. demonstrated the synthesis of highly porous and crystalline Zn-MOF-74 on a gram scale by mechanochemical milling of zinc oxide and 2,5-dihydroxyterephthalic acid without using bulk solvents. Their synthesis approach offers a fast, efficient, cost-effective, and environmentally friendly synthesis method in comparison with conventional solvothermal synthesis.122 The milling reaction was monitored by real-time in-situ X-ray powder diffraction technique and it revealed a stepwise reaction mechanism. The formation of Zn-MOF-74 done by Julien et al. proceeds via a close-packed intermediate. Zhang et al. developed a solvent-free mechanochemical-assisted approach to the synthesis of ZU-36-Ni (GeFSIX-3-Ni).45 Given the ultramicroporous structure, the obtained MOF displayed excellent performance for low-concentration CO2 capture, exhibiting high CO2 uptake of 1.07 mmol g−1 at 0.4 mbar and benchmark CO2/N2 separation selectivity of 4300 at 273 K. More interestingly, Chen et al. showed that mechanochemical methods can be applied in the synthesis of three-component MOFs consisting of mixed organic linkers. The method enables large-scale synthesis of ultramicroporous MOFs of Zn-atz-ipa and Zn-datz-ipa (atz = 3-amino-1,2,4-triazole, datz = 3,5-diamino-1,2,4-triazole, ipa = isophthalic acid) at room temperature with high STY up to 4800 kg m−3 d−1.123 Although not all studies discussed in this section have focused on low-concentration CO2 capture, we believe that the different approaches could be applied to the development of MOFs targeted for low-concentration CO2 capture.
Structuring of sorbents for real-life application
Prior to practical CO2 capture applications, the as-synthesized MOFs sorbents have to be processed into certain shapes (e.g., pellets, granules, films, fibers). However, synthesis of MOFs typically yields solid crystalline powders that can be brittle, insoluble, and infusible. These features of the as-synthesized MOFs provide challenges in processing and shaping these sorbents and have significantly hampered their practical applications. The employment of conventional powder processing methods to shape MOF micro/nanocrystals, such as the use of binders, will block the porous channels and thus compromise the adsorption performances of the MOF sorbents.124 For instance, Chang et al. developed a wet granulation method for shaping a family of MOF nano/microcrystals into mechanically strong granules with the assistance of 5 wt% of mesoporous RHO alumina as the inorganic binder. The shaping process involved wetting and mixing, granulation, and drying procedures, during which the interaction of hydroxyl groups on the MOF and the binder particles assisted in the formation of a monolithic structure. Similarly, Mathe et al. employed sucrose as an organic binder to process zirconium-based MOF powder into pellets via steps of mixing, sieving, and granulation. The MOF pellets can be produced on a kilogram scale in a relatively short operation time of 30 min. However, up to 50% of the porosity was lost during the shaping due to the pore-blocking effect.125 Very recently, Lee et al. reported the scalable synthesis of highly uniform Mg-based MOF (Mg2(dobpdc)) crystals along with the open-porous fiber networks for CO2 capture.126 Specifically, MgO particles were initially loaded into the porous poly(ether imide) (PEI) fiber matrix, and the obtained MgO/PEI precursor fiber was reacted with the organic linker by solvothermal treatment to convert the MgO particles into Mg2(dobpdc) crystals. The Mg2(dobpdc)/PEI composite was further soaked in diamine solution to form the fiber sorbents diamine-Mg2(dobpdc)/PEI. Similarly, Quan et al. developed a straightforward and scalable approach to fabricate hollow fiber sorbents by incorporation of diamine-Mg2(dobpdc) into poly(ethersulfone) (PES) substrate through a conventional “dry-jet, wet-quench” method.127 These porous fiber sorbents showed unprecedented cyclic CO2 capacities in conditions that are relevant to both post-combustion capture of CO2 and low-concentration CO2 capture. In addition, biopolymers such as cellulose, chitosan, have been recently studied as substrates to process MOFs into free-standing films and foams.128–130 The use of such renewable and biodegradable substrates in MOF processing opens new possibilities to develop sustainable CO2 sorbents based on MOF composites.The emerging 3D printing technology, also known as additive manufacturing, could potentially overcome the drawbacks such as loss of surface area and clogging associated with applying conventional granulation and pelletization techniques in MOF processing.131–134 The technique offers an opportunity to process MOFs into desired shapes and geometries in an easy-to-handle form. More importantly, the printed MOF composites could have high MOF loading with preserved MOF structure and properties. To date, various printing techniques including fused filament fabrication (FFF), digital light processing (DLP), selective laser sintering (SLS), and direct ink writing (DIW) have been employed to formulate MOFs for CO2 adsorption and separation studies. Rezaei et al. employed DIW method to fabricate Ni-MOF-74 and UTSA-16(Co) powders into mechanically stable monoliths with the assistance of clay as a binder and poly(vinyl alcohol) (PVA) as a plasticizer.135 The 3D-printed MOF monoliths had high MOF loading of up to 85 wt% and high surface areas of up to 737 m2 g−1. Remarkably, the CO2 adsorption capacities of the MOFs were fully retained during the printing. The MOF monoliths showed relatively high CO2 uptakes of >1.3 mmol g−1 at a low pCO2 = 5 mbar and 298 K. In a similar approach, the same group prepared amine-functionalized MIL-101 monoliths, which showed high CO2 uptakes of up to 1.6 mmol g−1 at pCO2 = 3 mbar at 298 K.136 It is noteworthy that the choices of solvent and binder are extremely important to form stable MOF monoliths via the DIW printing approach. Indeed, there are significant challenges for 3D printing MOFs at the industrial scale with concerns of cost and material stability. It is hoped that these issues will be addressed by future studies and 3D printing will be a powerful approach for processing various sorbents. We expected that the knowledge transfer from 3D printing to MOF processing would overcome the difficulties in shaping and structuring MOFs and promote their practical applications in trace CO2 capture as well as various separation processes.
Conclusion and future outlook
Significant efforts in developing technologies to combat the high emission of CO2 from point sources have been put in by researchers all over the world in the last few decades. CO2 capture technologies have been implemented with the use of liquid-based amine scrubbing processes. In recent years, there has been notable development on adsorption-based technologies as an alternative to amine scrubbing. At the same time, low-concentration CO2 capture, including direct air capture (DAC) and trace CO2 capture (i.e. from very low partial pressures) through adsorption-based technologies have been put forward as the next step in reducing the atmospheric concentration of CO2. However, for efficient low-concentration CO2 capture, the ideal sorbent must be able to selectivity capture CO2 with high CO2 capacity. This means that the sorbent must have a high affinity for CO2 (i.e. low enthalpy of CO2 sorption), but not so high that the regeneration of the sorbent becomes energy intensive. A summary of the recent advances in the development of metal–organic frameworks (MOFs) and the closely related hybrid ultramicroporous materials (HUMs) was presented here. These potential sorbents have been engineered through different approaches to have many of the desirable properties needed for a good DAC CO2 sorbent. Such approaches include pore size tuning, engineering open metal sites, and the introduction of functional groups with high affinities for CO2 sorption. Significant advances have been made with respect to sorbent properties in recent years. Challenges remain in further improving the CO2 uptake performance of these MOF sorbents, adopting sustainable, and energy-efficient green synthesis routes as well as structuring sorbents via innovative methods such as 3D printing.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors thank the Swedish Research Council (VR, grant no. 2020-04029), the Åforsk foundation (grant no 19-493 and 22-54) and the Swedish Foundation for Strategic Environmental Research (Mistra) (project name: Mistra TerraClean, project number 2015/31) for their financial support.References
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- N. O. O. A. US, Department of Commerce, Global Monitoring Laboratory – Carbon Cycle Greenhouse Gases, GML, https://gml.noaa.gov/ccgg/trends/gl_trend.html, (accessed on December 5, 2022) Search PubMed.
- S. Mukherjee, A. Kumar and M. J. Zaworotko, in Metal-Organic Frameworks (MOFs) for Environmental Applications, ed. S. K. Ghosh, Elsevier, Amsterdam, 2019, ch. 2, pp. 5–61, DOI:10.1016/B978-0-12-814633-0.00003-X.
- R. Custelcean, Chem. Sci., 2021, 12, 12518–12528 RSC.
- T. Maldal and I. M. Tappel, Energy, 2004, 29, 1403–1411 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- R. Ben-Mansour, M. A. Habib, O. E. Bamidele, M. Basha, N. A. A. Qasem, A. Peedikakkal, T. Laoui and M. Ali, Appl. Energy, 2016, 161, 225–255 CrossRef CAS.
- N. Hedin, L. Andersson, L. Bergström and J. Yan, Appl. Energy, 2013, 104, 418–433 CrossRef CAS.
- R. L. Siegelman, E. J. Kim and J. R. Long, Nat. Mater., 2021, 20, 1060–1072 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 RSC.
- G. Singh, K. S. Lakhi, S. Sil, S. V. Bhosale, I. Kim, K. Albahily and A. Vinu, Carbon, 2019, 148, 164–186 CrossRef CAS.
- C. Xu and M. Strømme, Nanomaterials, 2019, 9, 103 CrossRef PubMed.
- O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC.
- S. Kumar, R. Srivastava and J. Koh, J. CO2 Util., 2020, 41, 101251 CrossRef CAS.
- M. Xu, S. Chen, D.-K. Seo and S. Deng, Chem. Eng. J., 2019, 371, 693–705 CrossRef CAS.
- A. A. Dabbawala, I. Ismail, B. V. Vaithilingam, K. Polychronopoulou, G. Singaravel, S. Morin, M. Berthod and Y. Al Wahedi, Microporous Mesoporous Mater., 2020, 303, 110261 CrossRef CAS.
- W. Chaikittisilp, R. Khunsupat, T. T. Chen and C. W. Jones, Ind. Eng. Chem. Res., 2011, 50, 14203–14210 CrossRef CAS.
- C. Chen, S. Zhang, K. H. Row and W.-S. Ahn, J. Energy Chem., 2017, 26, 868–880 CrossRef CAS.
- W. Wang, M. Zhou and D. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H.-C. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
- C. Xu, G. Yu, J. Yuan, M. Strømme and N. Hedin, Mater. Today Adv., 2020, 6, 100052 CrossRef.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit, H. J. Kulik, S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Nat. Commun., 2020, 11, 4068 CrossRef CAS PubMed.
- J. Liu, Y. Wei and Y. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 82–93 CrossRef CAS.
- Q. Wang, J. Bai, Z. Lu, Y. Pan and X. You, Chem. Commun., 2016, 52, 443–452 RSC.
- R.-B. Lin, Z. Zhang and B. Chen, Acc. Chem. Res., 2021, 54, 3362–3376 CrossRef CAS PubMed.
- M. Åhlén, A. Jaworski, M. Strømme and O. Cheung, Chem. Eng. J., 2021, 422, 130117 CrossRef.
- Z. Zhang, S. B. Peh, R. Krishna, C. Kang, K. Chai, Y. Wang, D. Shi and D. Zhao, Angew. Chem., Int. Ed., 2021, 60, 17198–17204 CrossRef CAS PubMed.
- S. Nandi, P. De Luna, T. D. Daff, J. Rother, M. Liu, W. Buchanan, A. I. Hawari, T. K. Woo and R. Vaidhyanathan, Sci. Adv., 2015, 11, e1500421 CrossRef PubMed.
- S. Mukherjee, N. Sikdar, D. O'Nolan, D. M. Franz, V. Gascón, A. Kumar, N. Kumar, H. S. Scott, D. G. Madden, P. E. Kruger, B. Space and M. J. Zaworotko, Sci. Adv., 2019, 5, eaax9171 CrossRef CAS PubMed.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 RSC.
- P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362–7370 RSC.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- R. W. Flaig, T. M. O. Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed.
- D. G. Madden, H. S. Scott, A. Kumar, K.-J. Chen, R. Sanii, A. Bajpai, M. Lusi, T. Curtin, J. J. Perry and M. J. Zaworotko, Philos. Trans. R. Soc., A, 2017, 375, 20160025 CrossRef PubMed.
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- A. Masala, F. Grifasi, C. Atzori, J. G. Vitillo, L. Mino, F. Bonino, M. R. Chierotti and S. Bordiga, J. Phys. Chem. C, 2016, 120, 12068–12074 CrossRef CAS.
- A. Masala, J. G. Vitillo, F. Bonino, M. Manzoli, C. A. Grande and S. Bordiga, Phys. Chem. Chem. Phys., 2016, 18, 220–227 RSC.
- R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- A. Banerjee, S. Nandi, P. Nasa and R. Vaidhyanathan, Chem. Commun., 2016, 52, 1851–1854 RSC.
- P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301–9307 CrossRef CAS PubMed.
- Z. Zhang, Q. Ding, S. B. Peh, D. Zhao, J. Cui, X. Cui and H. Xing, Chem. Commun., 2020, 56, 7726–7729 RSC.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- A. Kumar, C. Hua, D. G. Madden, D. O'Nolan, K.-J. Chen, L.-A. J. Keane, J. J. Perry and M. J. Zaworotko, Chem. Commun., 2017, 53, 5946–5949 RSC.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- K. Uemura, A. Maeda, T. K. Maji, P. Kanoo and H. Kita, Eur. J. Inorg. Chem., 2009, 2329–2337 CrossRef CAS.
- A. Kumar, D. G. Madden, M. Lusi, K.-J. Chen, E. A. Daniels, T. Curtin, J. J. Perry IV and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- P. Hu, H. Liu, H. Wang, J. Zhou, Y. Wang and H. Ji, J. Mater. Chem. A, 2022, 10, 881–890 RSC.
- Z. Zhang, Q. Ding, J. Cui, X. Cui and H. Xing, Sci. China Mater., 2020, 64, 691–697 CrossRef.
- J. Park, J. R. Park, J. H. Choe, S. Kim, M. Kang, D. W. Kang, J. Y. Kim, Y. W. Jeong and C. S. Hong, ACS Appl. Mater. Interfaces, 2020, 12, 50534–50540 CrossRef CAS PubMed.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 RSC.
- P.-Q. Liao, X.-W. Chen, S.-Y. Liu, X.-Y. Li, Y.-T. Xu, M. Tang, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- S. Liu, S. Yao, B. Liu, X. Sun, Y. Yuan, G. Li, L. Zhang and Y. Liu, Dalton Trans., 2019, 48, 1680–1685 RSC.
- Y. Shi, Y. Xie, H. Cui, Z. A. Alothman, O. Alduhaish, R.-B. Lin and B. Chen, Chem. Eng. J., 2022, 446, 137101 CrossRef CAS.
- R.-B. Lin, L. Li, A. Alsalme and B. Chen, Small Struct., 2020, 1, 2000022 CrossRef.
- Q. Liu, L. Ning, S. Zheng, M. Tao, Y. Shi and Y. He, Sci. Rep., 2013, 3, 2916 CrossRef PubMed.
- J. A. R. Navarro, E. Barea, A. Rodríguez-Diéguez, J. M. Salas, C. O. Ania, J. B. Parra, N. Masciocchi, S. Galli and A. Sironi, J. Am. Chem. Soc., 2008, 130, 3978–3984 CrossRef CAS PubMed.
- R.-B. Lin, L. Li, H.-L. Zhou, H. Wu, C. He, S. Li, R. Krishna, J. Li, W. Zhou and B. Chen, Nat. Mater., 2018, 17, 1128–1133 CrossRef CAS PubMed.
- S. Subramanian and M. J. Zaworotko, Angew. Chem., Int. Ed. Engl., 1995, 34, 2127–2129 CrossRef CAS.
- S. Mukherjee, N. Kumar, A. A. Bezrukov, K. Tan, T. Pham, K. A. Forrest, K. A. Oyekan, O. T. Qazvini, D. G. Madden, B. Space and M. J. Zaworotko, Angew. Chem., Int. Ed., 2021, 60, 10902–10909 CrossRef CAS PubMed.
- R. P. Lively and M. J. Realff, AIChE J., 2016, 62, 3699–3705 CrossRef CAS.
- T. Quainoo, S. N. Lavan and Z.-F. Liu, J. Mater. Res., 2021, 37, 334–345 CrossRef.
- Y. Xie, H. Cui, H. Wu, R.-B. Lin, W. Zhou and B. Chen, Angew. Chem., Int. Ed., 2021, 60, 9604–9609 CrossRef CAS PubMed.
- I. Strauss, A. Mundstock, M. Treger, K. Lange, S. Hwang, C. Chmelik, P. Rusch, N. C. Bigall, T. Pichler, H. Shiozawa and J. Caro, ACS Appl. Mater. Interfaces, 2019, 11, 14175–14181 CrossRef CAS PubMed.
- A. C. Forse, P. J. Milner, J.-H. Lee, H. N. Redfearn, J. Oktawiec, R. L. Siegelman, J. D. Martell, B. Dinakar, L. B. Zasada, M. I. Gonzalez, J. B. Neaton, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 18016–18031 CrossRef CAS PubMed.
- A. Sinha, L. A. Darunte, C. W. Jones, M. J. Realff and Y. Kawajiri, Ind. Eng. Chem. Res., 2017, 56, 750–764 CrossRef CAS.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- N. Planas, A. L. Dzubak, R. Poloni, L.-C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed.
- Y. A. Majid, S. K. Garg and D. W. Davidson, Can. J. Chem., 1968, 46, 1683–1690 CrossRef CAS.
- G. Sethia, R. S. Somani and H. C. Bajaj, RSC Adv., 2015, 5, 12773–12781 RSC.
- X. Wang, L. Zhong, Y. Cressault, A. Gleizes and M. Rong, J. Phys. D: Appl. Phys., 2014, 47, 495201 CrossRef.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, in Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks, ed. O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019, ch. 14, pp. 313–314, DOI:10.1002/9783527821099.ch14.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. D. Martell, P. J. Milner, R. L. Siegelman and J. R. Long, Chem. Sci., 2020, 11, 6457–6471 RSC.
- X. Zhang, B. Wang, A. Alsalme, S. Xiang, Z. Zhang and B. Chen, Coord. Chem. Rev., 2020, 423, 213507 CrossRef CAS.
- C. Yang, U. Kaipa, Q. Z. Mather, X. Wang, V. Nesterov, A. F. Venero and M. A. Omary, J. Am. Chem. Soc., 2011, 133, 18094–18097 CrossRef CAS PubMed.
- Y. Chen, B. Wang, X. Wang, L. H. Xie, J. Li, Y. Xie and J. R. Li, ACS Appl. Mater. Interfaces, 2017, 9, 27027–27035 CrossRef CAS PubMed.
- J. M. Taylor, R. Vaidhyanathan, S. S. Iremonger and G. K. Shimizu, J. Am. Chem. Soc., 2012, 134, 14338–14340 CrossRef CAS PubMed.
- S. A. Bedell, C. M. Worley, K. Darst and K. Simmons, Int. J. Greenhouse Gas Control, 2011, 5, 401–404 CrossRef CAS.
- K. Veltman, B. Singh and E. G. Hertwich, Environ. Sci. Technol., 2010, 44, 1496–1502 CrossRef CAS PubMed.
- K.-S. Zoannou, D. J. Sapsford and A. J. Griffiths, Int. J. Greenhouse Gas Control, 2013, 17, 423–430 CrossRef CAS.
- H. Lepaumier, D. Picq and P.-L. Carrette, Ind. Eng. Chem. Res., 2009, 48, 9068–9075 CrossRef CAS.
- A. Heydari-Gorji, Y. Belmabkhout and A. Sayari, Microporous Mesoporous Mater., 2011, 145, 146–149 CrossRef CAS.
- P. Bollini, S. Choi, J. H. Drese and C. W. Jones, Energy Fuels, 2011, 25, 2416–2425 CrossRef CAS.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef CAS PubMed.
- H. Jo, W. R. Lee, N. W. Kim, H. Jung, K. S. Lim, J. E. Kim, D. W. Kang, H. Lee, V. Hiremath, J. G. Seo, H. Jin, D. Moon, S. S. Han and C. S. Hong, ChemSusChem, 2017, 10, 541–550 CrossRef CAS PubMed.
- W. R. Lee, J. E. Kim, S. J. Lee, M. Kang, D. W. Kang, H. Y. Lee, V. Hiremath, J. G. Seo, H. Jin, D. Moon, M. Cho, Y. Jung and C. S. Hong, ChemSusChem, 2018, 11, 1694–1707 CrossRef CAS PubMed.
- S. Kumar, S. Jain, M. Nehra, N. Dilbaghi, G. Marrazza and K.-H. Kim, Coord. Chem. Rev., 2020, 420, 213407 CrossRef CAS.
- A. Garcia Marquez, P. Horcajada, D. Grosso, G. Férey, C. Serre, C. Sanchez and C. Boissiere, Chem. Commun., 2013, 49, 3848–3850 RSC.
- T. He, X. Xu, B. Ni, H. Wang, Y. Long, W. Hu and X. Wang, Nanoscale, 2017, 9, 19209–19215 RSC.
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47, 2071–2073 RSC.
- J. Huo, M. Brightwell, S. El Hankari, A. Garai and D. Bradshaw, J. Mater. Chem. A, 2013, 1, 15220–15223 RSC.
- G. Zhan and H. C. Zeng, Chem. Commun., 2016, 53, 72–81 RSC.
- C. Yim, M. Lee, M. Yun, G.-H. Kim, K. T. Kim and S. Jeon, Sci. Rep., 2015, 5, 10674 CrossRef CAS PubMed.
- K. Xie, Q. Fu, C. Xu, H. Lu, Q. Zhao, R. Curtain, D. Gu, P. A. Webley and G. G. Qiao, Energy Environ. Sci., 2018, 11, 544–550 RSC.
- Y. Yu, X.-J. Wu, M. Zhao, Q. Ma, J. Chen, B. Chen, M. Sindoro, J. Yang, S. Han, Q. Lu and H. Zhang, Angew. Chem., 2017, 129, 593–596 CrossRef.
- G. Majano and J. Pérez-Ramírez, Adv. Mater., 2013, 25, 1052–1057 CrossRef CAS PubMed.
- C. Avci-Camur, J. Perez-Carvajal, I. Imaz and D. Maspoch, ACS Sustainable Chem. Eng., 2018, 6, 14554–14560 CrossRef CAS.
- I. Imaz, M. Rubio-Martínez, J. An, I. Solé-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287–7302 RSC.
- S. L. Anderson and K. C. Stylianou, Coord. Chem. Rev., 2017, 349, 102–128 CrossRef CAS.
- E. Alvarez, N. Guillou, C. Martineau, B. Bueken, B. Van de Voorde, C. Le Guillouzer, P. Fabry, F. Nouar, F. Taulelle, D. de Vos, J.-S. Chang, K. H. Cho, N. Ramsahye, T. Devic, M. Daturi, G. Maurin and C. Serre, Angew. Chem., Int. Ed., 2015, 54, 3664–3668 CrossRef CAS PubMed.
- B. Yilmaz, N. Trukhan and U. Müller, Chin. J. Catal., 2012, 33, 3–10 CrossRef CAS.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- A. Zulys, F. Yulia, N. Muhadzib and N. Nasruddin, Ind. Eng. Chem. Res., 2020, 60, 37–51 CrossRef.
- J. A. Coelho, A. M. Ribeiro, A. F. P. Ferreira, S. M. P. Lucena, A. E. Rodrigues and D. C. S. d. Azevedo, Ind. Eng. Chem. Res., 2016, 55, 2134–2143 CrossRef CAS.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38–39 CrossRef CAS PubMed.
- M. Sánchez-Sánchez, N. Getachew, K. Díaz, M. Díaz-García, Y. Chebude and I. Díaz, Green Chem., 2015, 17, 1500–1509 RSC.
- L. H. Wee, M. R. Lohe, N. Janssens, S. Kaskel and J. A. Martens, J. Mater. Chem., 2012, 22, 13742–13746 RSC.
- J.-L. Zhuang, D. Ceglarek, S. Pethuraj and A. Terfort, Adv. Funct. Mater., 2011, 21, 1442–1447 CrossRef CAS.
- J. Shi, J. Zhang, D. Tan, X. Cheng, X. Tan, B. Zhang, B. Han, L. Liu, F. Zhang, M. Liu and J. Xiang, ChemCatChem, 2019, 11, 2058–2062 CrossRef CAS.
- I. Pakamorė, J. Rousseau, C. Rousseau, E. Monflier and P. Á. Szilágyi, Green Chem., 2018, 20, 5292–5298 RSC.
- R. D'Amato, A. Donnadio, M. Carta, C. Sangregorio, D. Tiana, R. Vivani, M. Taddei and F. Costantino, ACS Sustainable Chem. Eng., 2019, 7, 394–402 CrossRef.
- E. S. Grape, J. G. Flores, T. Hidalgo, E. Martínez-Ahumada, A. Gutiérrez-Alejandre, A. Hautier, D. R. Williams, M. O’Keeffe, L. Öhrström, T. Willhammar, P. Horcajada, I. A. Ibarra and A. K. Inge, J. Am. Chem. Soc., 2020, 142, 16795–16804 CrossRef CAS PubMed.
- G. Das, D. B. Shinde, S. Kandambeth, B. P. Biswal and R. Banerjee, Chem. Commun., 2014, 50, 12615–12618 RSC.
- D. N. Rainer and R. E. Morris, Dalton Trans., 2021, 50, 8995–9009 RSC.
- A. M. Fidelli, B. Karadeniz, A. J. Howarth, I. Huskić, L. S. Germann, I. Halasz, M. Etter, S.-Y. Moon, R. E. Dinnebier, V. Stilinović, O. K. Farha, T. Friščić and K. Užarević, Chem. Commun., 2018, 54, 6999–7002 RSC.
- S. Głowniak, B. Szczęśniak, J. Choma and M. Jaroniec, Mater. Today, 2021, 46, 109–124 CrossRef.
- P. A. Julien, K. Užarević, A. D. Katsenis, S. A. J. Kimber, T. Wang, O. K. Farha, Y. Zhang, J. Casaban, L. S. Germann, M. Etter, R. E. Dinnebier, S. L. James, I. Halasz and T. Friščić, J. Am. Chem. Soc., 2016, 138, 2929–2932 CrossRef CAS PubMed.
- T. Gao, H.-J. Tang, S.-Y. Zhang, J.-W. Cao, Y.-N. Wu, J. Chen, Y. Wang and K.-J. Chen, J. Solid State Chem., 2021, 303, 122547 CrossRef CAS.
- Z. Wang, L. Liu, Z. Li, N. Goyal, T. Du, J. He and G. K. Li, Energy Fuels, 2022, 36, 2927–2944 CrossRef CAS.
- J. Ren, N. M. Musyoka, H. W. Langmi, A. Swartbooi, B. C. North and M. Mathe, Int. J. Hydrogen Energy, 2015, 40, 4617–4622 CrossRef CAS.
- Y. H. Lee, Y. Kwon, C. Kim, Y.-E. Hwang, M. Choi, Y. Park, A. Jamal and D.-Y. Koh, JACS Au, 2021, 1, 1198–1207 CrossRef CAS PubMed.
- W. Quan, H. E. Holmes, F. Zhang, B. L. Hamlett, M. G. Finn, C. W. Abney, M. T. Kapelewski, S. C. Weston, R. P. Lively and W. J. Koros, JACS Au, 2022, 2, 1350–1358 CrossRef CAS PubMed.
- S. Sultan, H. N. Abdelhamid, X. Zou and A. P. Mathew, Adv. Funct. Mater., 2019, 29, 1805372 CrossRef.
- S. Zhou, X. Kong, B. Zheng, F. Huo, M. Strømme and C. Xu, ACS Nano, 2019, 13, 9578–9586 CrossRef CAS PubMed.
- X.-H. Ma, Z. Yang, Z.-K. Yao, Z.-L. Xu and C. Y. Tang, J. Membr. Sci., 2017, 525, 269–276 CrossRef CAS.
- E. R. Kearns, R. Gillespie and D. M. D'Alessandro, J. Mater. Chem. A, 2021, 9, 27252–27270 RSC.
- S. Lawson, X. Li, H. Thakkar, A. A. Rownaghi and F. Rezaei, Chem. Rev., 2021, 121, 6246–6291 CrossRef CAS PubMed.
- C. A. Grande, R. Blom, V. Middelkoop, D. Matras, A. Vamvakeros, S. D. M. Jacques, A. M. Beale, M. Di Michiel, K. A. Andreassen and A. M. Bouzga, Chem. Eng. J., 2020, 402, 126166 CrossRef CAS.
- K. A. Evans, Z. C. Kennedy, B. W. Arey, J. F. Christ, H. T. Schaef, S. K. Nune and R. L. Erikson, ACS Appl. Mater. Interfaces, 2018, 10, 15112–15121 CrossRef CAS PubMed.
- H. Thakkar, S. Eastman, Q. Al-Naddaf, A. A. Rownaghi and F. Rezaei, ACS Appl. Mater. Interfaces, 2017, 9, 35908–35916 CrossRef CAS PubMed.
- S. Lawson, C. Griffin, K. Rapp, A. A. Rownaghi and F. Rezaei, Energy Fuels, 2019, 33, 2399–2407 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |