
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of tetrel functionalized heptapnictogen clusters towards heteroallenes†
William D.
Jobbins
 a,
Bono
van IJzendoorn
a,
Inigo J.
Vitorica-Yrezabal
b,
George F. S.
Whitehead
b and
Meera
Mehta
*a
a,
Bono
van IJzendoorn
a,
Inigo J.
Vitorica-Yrezabal
b,
George F. S.
Whitehead
b and
Meera
Mehta
*a
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: meera.mehta@manchester.ac.uk
bX-ray Diffraction Facility, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 31st January 2023
Abstract
Despite being known for decades, the solution-state molecular chemistry of heptapnictogen ([Pn7]3−; Pn = P, As) clusters is not well established. Here we study heavy element derivatives of tetrel functionalized heptapnictogen clusters towards heteroallene capture, specifically isocyanates, an isothiocyanate and CO2 are probed. Clusters (Me3Ge)3P7 (1), (Et3Ge)3P7 (2), (nBu3Sn)3P7 (3), and (Me3Si)3As7 (4) were all found to capture isocyanates between all three of their tetrel–pnictogen bonds. In the case of phenyl isocyanate insertion, tetrel coordination at the isocyanate nitrogen atoms is preferred, while in the case of p-toluenesulfonyl isocyanate insertion, tetrel coordination at oxygen is preferred. Furthermore, the reaction of (Me3Si)3P7 with CO2 gave NMR spectra consistent with the capture of the greenhouse gas. Heteroallene insertion at these clusters was also studied using density functional theory.
Introduction
Zintl clusters, clusters of p-block elements, are interesting because they can be structurally related to heterogenous materials,1,2 and can thus be considered their prototypes. For example, the heptaphosphide cluster ([P7]3−) can be regarded as a fragment of red phosphorus.1,3–5 Red phosphorus is inexpensive and earth-abundant but is also amorphous and polymeric, making it insoluble and thus difficult to study. Meanwhile, [P7] clusters, particularly once functionalized, are soluble in common laboratory solvents allowing for straightforward assessment of in situ reactivity. Of the group 15 clusters, [P7]3− is also the most well-studied because of its synthetic accessibility and the presence of a 31P NMR active handle.1,6–8 The heavy pnictogen analogue [As7]3− can also be easily prepared on a multi-gram scale.9Historically, the molecular chemistry of these clusters has been focused on the preparation of heteroatomic or new cluster morphologies, their coordination chemistry with d- and f-block metals, and salt metathesis with group 14 electrophiles.1,6,7,10,11 The emphasis of these investigations has largely been on understanding the structure, bonding, and physical properties of the material. However, the application of [Pn7] (Pn = P, As) clusters in small molecule activation is in its early stages. (Fig. 1) In 2012 and 2014, Goicoechea and co-workers found that protonated [HPn7]2− clusters hydropnictinated carbodiimides and isocyanates.12–14 The Goicoechea group also found that the reaction of [Pn7]3− with alkynes gave 1,2,3-tripnictolides ([R2C2P3]−),15,16 whereas the reaction with CO gave the phosphaethanyloate anion ([PCO]−).17,18 Recently, we have found that boron-functionalized [P7] cages can be applied as transition-metal free catalysts in hydroboration reactions.19,20
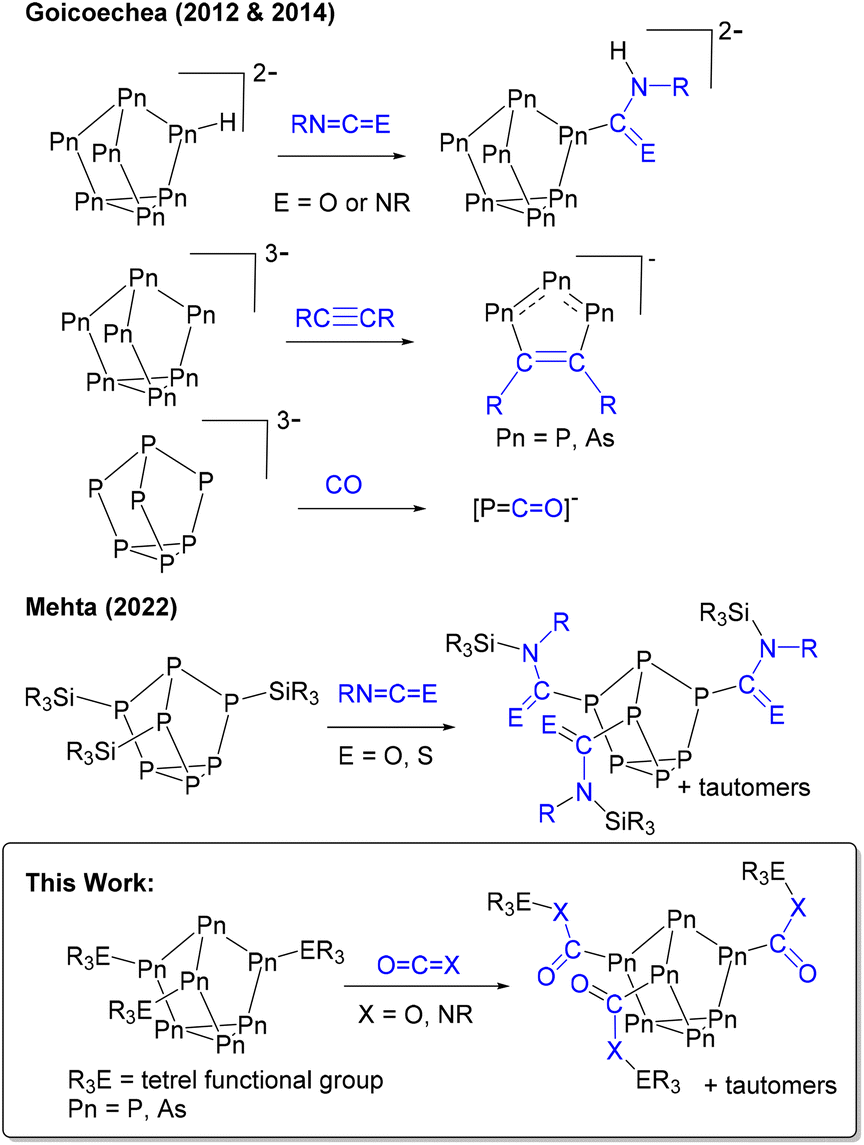 | ||
| Fig. 1 Previously reported reactions of [Pn7] clusters with small molecules and this work. | ||
Heteroallene insertion between labile non-cluster tetrel–pnictogen bonds has previously been reported.21–26 In 2022 we found for the first time this reactivity with Zintl-derived clusters.27 Neutral tris-silyl functionalized heptaphosphorus clusters with the general formula (R3Si)3P7 (R = Ph, Me) were found to capture and exchange isocyanates and an isothiocyanate (Fig. 1). Here, we show the broad application of this insertion, and expand the scope of functionalized heptapnictogen clusters investigated with heteroallenes to include (R3E)3Pn7 (E = Si, Ge, Sn; Pn = P, As) derivatives. Isocyanates (RN=C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) are interesting substrates for capture because of their structural relationship with the greenhouse gas carbon dioxide (OCO). In addition, this family of tetrel functionalized heptapnictogen clusters was reacted with CO2.
O) are interesting substrates for capture because of their structural relationship with the greenhouse gas carbon dioxide (OCO). In addition, this family of tetrel functionalized heptapnictogen clusters was reacted with CO2.
Results and discussion
Synthesis of functionalized clusters and reactivity with isocyanates
First, [P7]3− was prepared by following a known literature method.28 By adapting this method, using grey arsenic and potassium instead of red phosphorus and sodium, K3As7 was prepared. Next, [Pn7]3− clusters were reacted with an appropriate alkyl group 14 chloride to give the known and new neutral clusters (Me3Ge)3P7 (1),29 (Et3Ge)3P7 (2), (nBu3Sn)3P7 (3), and (Me3Si)3As7 (4),30 as shown in Scheme 1.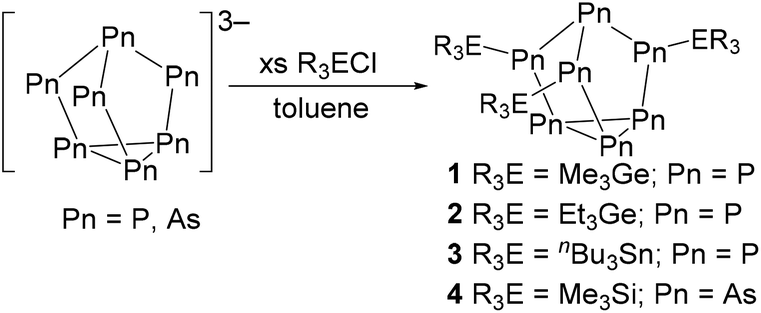 | ||
| Scheme 1 Synthesis of neutral (R3E)3Pn7 clusters. | ||
Efforts were also made to prepare germanium and tin-functionalized arsenic clusters (Et3Ge)3As7 and (nBu3Sn)3As7 by the reaction of the group 14 halide with [As7]3−. When Et3GeCl was reacted with [As7]3−, crystals of the new eleven-atom arsenic cluster (Et3Ge)5As11 (5) were obtained (Fig. 2). Single crystal X-ray diffraction (XRD) studies of 5 revealed an As11 core with 5 exo germanium groups. The arsenic core appears to contain an As7 unit with one basal As–As bond cleaved and further coordination of an [As4] unit to two of the bridging and one of the basal As atoms. This As11 core is distorted compared to previously reported [As11]3− and [P11]3− cores, which appear to contain four fused [Pn5] faces.31–33 An average Ge–As bond length of 2.443(3) Å was observed. Mass spectrometry (MS) studies on the bulk isolated material confirmed the presence of (Et3Ge)5As11 (5) and the targeted product (Et3Ge)3As7. To better understand how compound 5 was formed, the [As7]3− precursor was studied by mass spectrometry, which confirmed the presence of [As11]5−. The extent of this contamination was found to be batch dependent. This [As11]5− contaminant is thought to react with Et3GeCl to form 5. As only resonances from the ethyl signals can be observed by 1H and 13C{1H} NMR spectroscopy, the percentage of 5vs. (Et3Ge)3As7vs. other by-products could not be determined. The reaction of Et3GeCl with a batch of [K3(DME)x][As7] which had no [As11]5− impurity detected by MS still showed minor impurities in the 13C{1H} NMR spectrum. The reaction conditions that lead to contamination of the [K3(DME)x][As7] precursor with [As11]5− are not yet understood. Furthermore, when nBu3SnCl was reacted with [As7]3−, the unreacted tin chloride precursor could be observed by 1H, 119Sn, and 13C{1H} NMR spectroscopy, even after multiple purification efforts including distillation under dynamic vacuum and washing with solvents including toluene, ether, and pentane. Difficulties in clean formation of germanium and tin functionalized arsenic clusters precluded further studies of their reactivity.
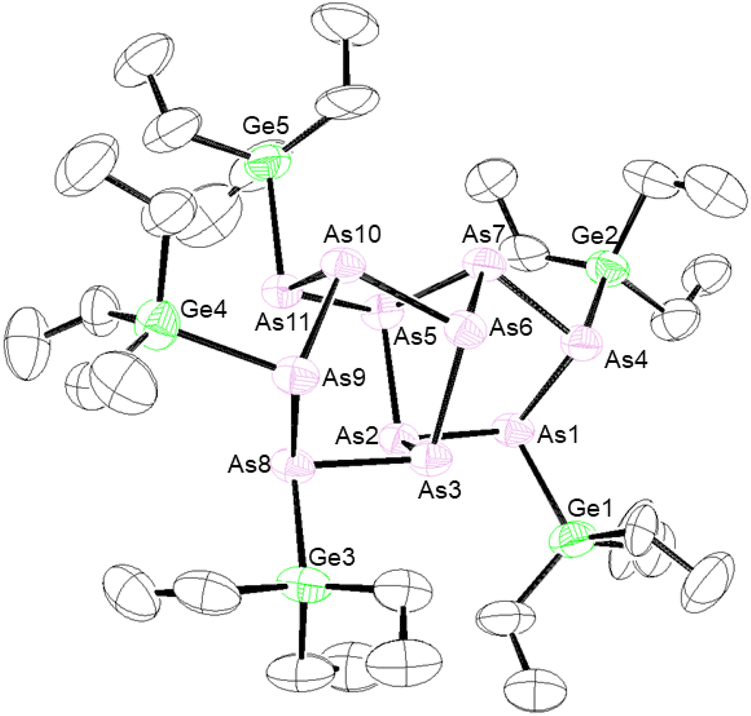 | ||
| Fig. 2 Molecular structure of 5. Anisotropic displacement ellipsoids pictured at 50% probability. Arsenic: plum; germanium: green; and carbon: white. | ||
Clusters 1–4 were then reacted with heteroallenes phenyl isocyanate, p-toluenesulfonyl isocyanate, and phenyl isothiocyanate. In the case of phenyl isothiocyanate, no reactions in any cases were observed, whereas reactions with phenyl isocyanate and p-toluenesulfonyl isocyanate were much more fruitful.
In the case of phenyl isocyanate, reactions of clusters 1 and 2 both gave complete conversion to the heteroallene inserted products 6 and 7, respectively (Scheme 2). Heteroallene insertion between the tetrel–pnictogen bonds of the cluster can lead to the formation of symmetric and/or asymmetric isomers, with the symmetric isomer being thermodynamically favourable (shown in Fig. 3 and discussed in more detail in ESI section 3†).27 In the case of the symmetric isomer, 3 resonances are expected in the 31P NMR spectrum for the basal, apical and bridging P atoms, whereas for the asymmetric isomer all 7 P atoms are magnetically inequivalent. The NMR spectra of both 6 and 7 showed three new resonances in the 31P NMR spectrum consistent with the exclusive formation of the symmetric isomer. In the case of compound 6, the 13C{1H} NMR resonance for the carbon bound to phosphorus could not be observed, while for compound 7 this resonance appears as a doublet at 176.0 ppm with a 1JCP of 49 Hz. Single crystal XRD studies further elucidated the solid-state structures of 6 and 7 (Fig. 4 and 5). In both structures, it was found that the Ge centre coordinates to the isocyanate nitrogen atoms. Average Ge–N bond lengths of 1.921(3) Å and 1.936(7) Å were observed for 6 and 7, while average CO double bond lengths of 1.227(5) Å and 1.231(7) Å were observed for 6 and 7, respectively. This coordination is in contrast to our previously reported reactivity between phenyl isocyanate and (Me3Si)3P7, where upon tris-insertion the Si centres coordinated to two of the isocyanates via the nitrogen atoms and one via the oxygen atom.27 This difference in coordination is presumably due to the lower oxophilicity of Ge than that of Si.34 Unfortunately, combining phenyl isocyanate with clusters 3 and 4 did not lead to reactions.
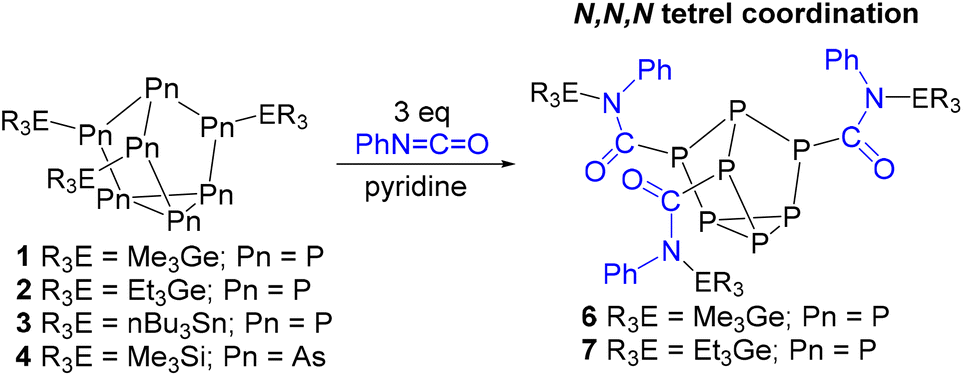 | ||
| Scheme 2 Reaction of neutral (R3E)3Pn7 clusters with phenyl isocyanate. | ||
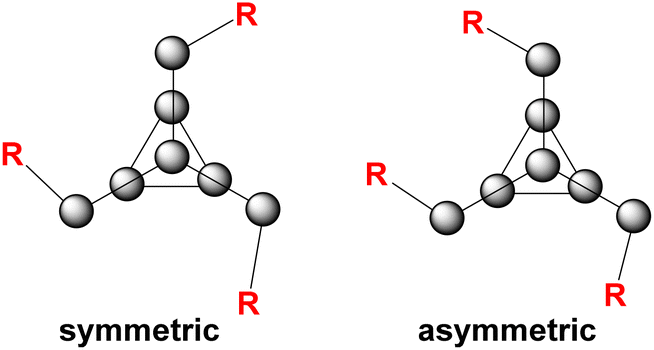 | ||
| Fig. 3 Symmetric vs. asymmetric isomers of functionalized [P7] clusters. | ||
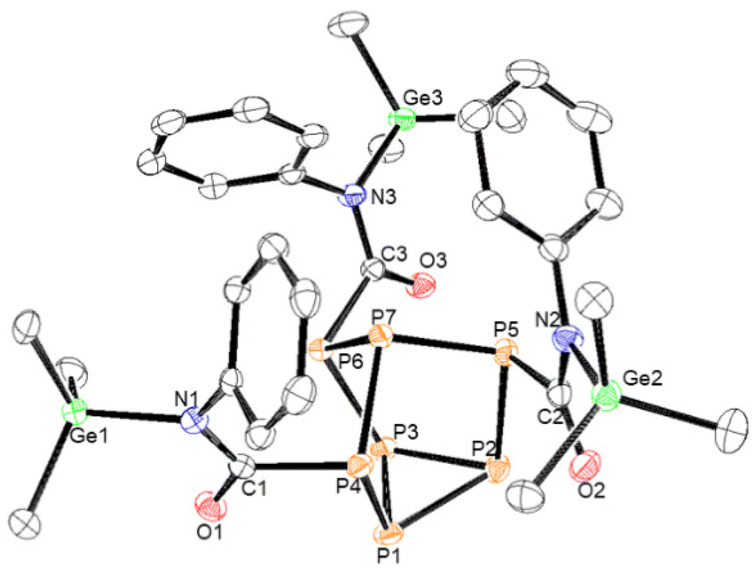 | ||
| Fig. 4 Molecular structure of 6. Anisotropic displacement ellipsoids pictured at 50% probability. Phosphorus: orange; nitrogen: blue; oxygen: red; germanium: green; and carbon: white. | ||
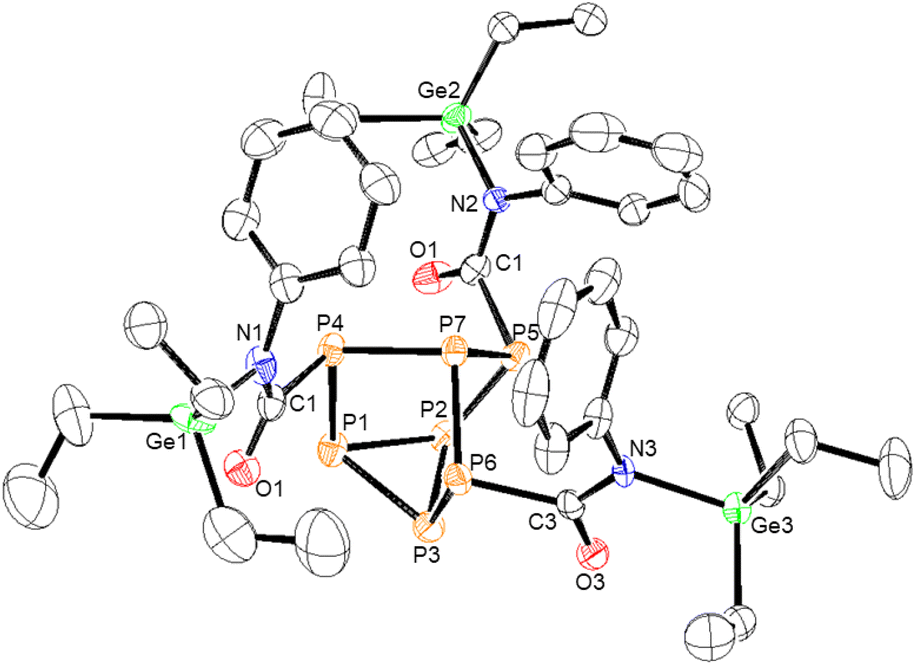 | ||
| Fig. 5 Molecular structure of 7. Anisotropic displacement ellipsoids pictured at 50% probability. Phosphorus: orange; nitrogen: blue; oxygen: red; germanium: green; and carbon: white. | ||
Next, p-toluenesulfonyl isocyanate was allowed to react with clusters 1–4 (Scheme 3). In the case of germanium functionalized phosphorus cluster 1, after 4 days 81% conversion to the inserted product 8 was observed by 31P NMR spectroscopy. The single crystal XRD data of 8 confirmed that the Me3Ge groups were coordinated to the isocyanate oxygen atoms. This coordination is in line with the previously reported reactivity of the (Me3Si)3P7 cluster with p-toluenesulfonyl isocyanate,27 where the electron-withdrawing group of the isocyanate is thought to decrease the basicity of nitrogen and favour oxygen coordination for the tetrel. The XRD data of 8 revealed an average Ge–O bond length of 1.889(2) Å and a CN bond length of 1.294(5) Å (Fig. 6).
 | ||
| Scheme 3 Reaction of neutral (R3E)3Pn7 clusters with p-toluenesulfonyl isocyanate. | ||
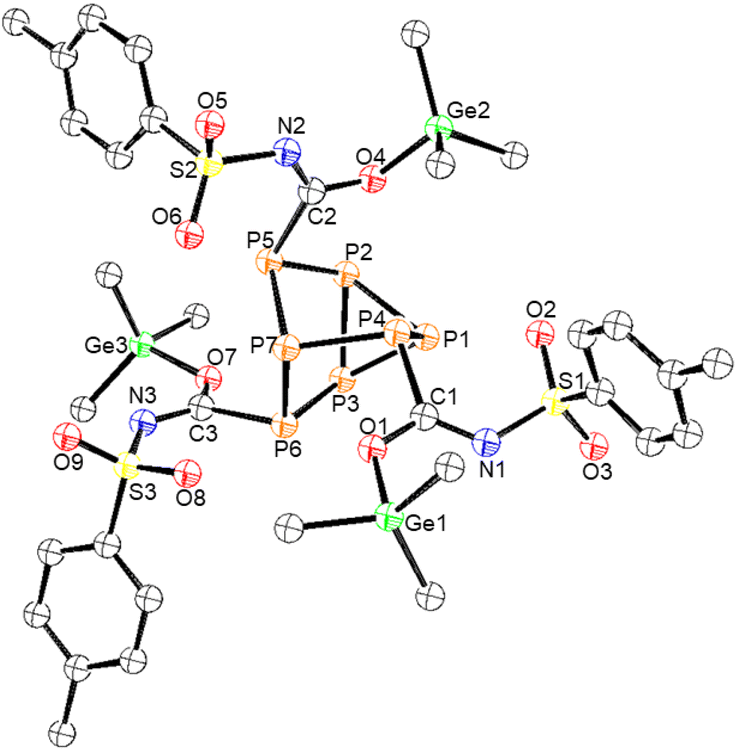 | ||
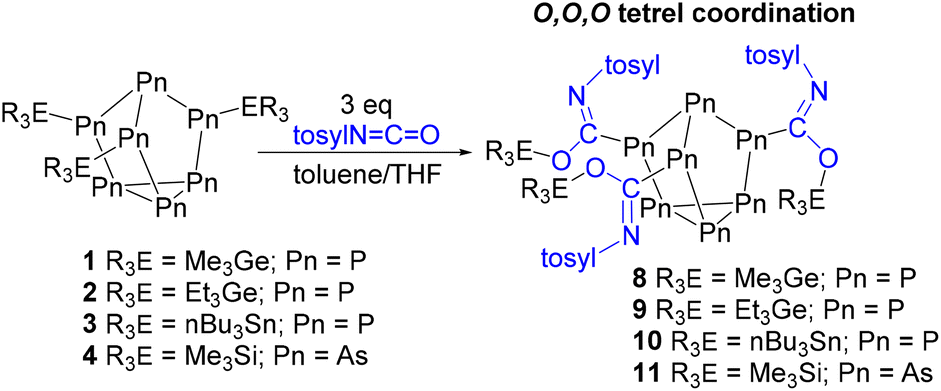
| Fig. 6 Molecular structure of 8. Anisotropic displacement ellipsoids pictured at 50% probability. Phosphorus: orange; nitrogen: blue; oxygen: red; germanium: green; and carbon: white. | ||
In a similar fashion, the reaction of cluster 2 with p-toluenesulfonyl isocyanate gave 55% conversion to 9. Single crystals suitable for XRD studies could not be obtained. Similarly, the reaction of 3 with p-toluenesulfonyl isocyanate gave inserted product 10 in 41% conversion after 1 week. However, when the arsenic cluster 4 was reacted with p-toluenesulfonyl isocyanate complete conversion to 11 was observed after the same amount of time. The structure of 11 was crystallographically verified and again the silicon groups were found to be coordinated to the isocyanate oxygen atoms (Fig. 7). The XRD data of 12 showed an average Si–O bond length of 1.727(3) Å and a CN bond length of 1.283(5) Å.
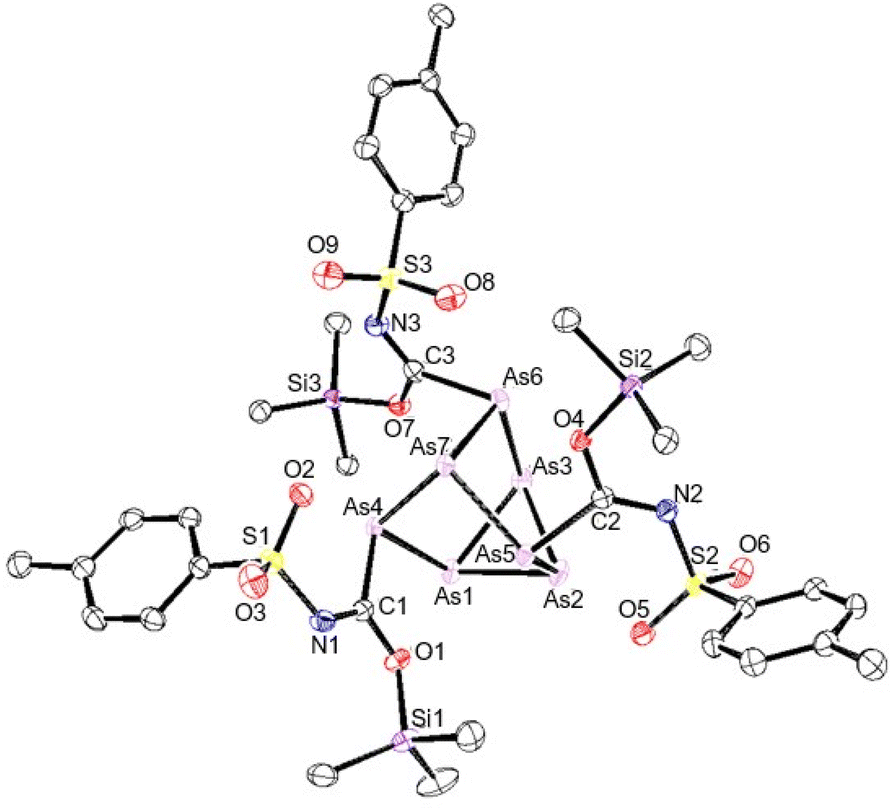 | ||
| Fig. 7 Molecular structure of 11. Anisotropic displacement ellipsoids pictured at 50% probability. Arsenic: plum; nitrogen: blue; oxygen: red; silicon: purple; and carbon: white. | ||
The formation of compounds 8–10 gave 31P NMR spectra consistent with the presence of both symmetric and asymmetric isomers (Fig. 3), with 10 resonances observed in the 31P NMR spectrum. The symmetric and asymmetric isomers were identified using 31P COSY NMR experiments and as expected the symmetric isomer was favoured. In the case of compound 11, because the cluster 31P NMR handle is lost, the symmetric and asymmetric isomers cannot be detected.
For compounds 9 and 10, since crystals suitable for XRD studies could not be obtained, NMR spectroscopy, infrared (IR) spectroscopy, and density functional theory (DFT) were used to better understand their tetrel coordination modes (see below).
For compounds 8–10, the carbon bound to phosphorus could not be observed by 13C{1H} NMR spectroscopy, whereas for compound 11 this resonance appears at 185.9 ppm. We have previously observed with our (R3Si)3P7 isocyanate insertions that27 (1) the quaternary carbon bound to phosphorus is not always observed by 13C{1H} NMR spectroscopy; (2) when it is observable and there is O,O,O-coordination of the silyl groups, the imine carbon resonances appear between 176.2 and 181.3 ppm; and (3) when it is observable and there is N,N,N-coordination of the silyl groups, the carbonyl carbon signals appear between 182.5 and 216.3 ppm. Comparison of these values and that of imine carbon of 11 shows that the resonance for the imine carbon of 11 is downfield from this range, while the resonance for the carbonyl carbon of 7 is upfield from where it would be expected. These observations suggest that the electronic properties of the isocyanate and the electrophile bound to either the N or O significantly affect the 13C{1H} NMR resonance of the carbon on phosphorus. Thus, discerning this carbon as either an imine carbon or carbonyl-like from 13C{1H} NMR spectroscopy alone is difficult. However, it is worth noting that the 31P NMR spectrum of 9 is nearly identical to that of compound 8, where O,O,O tetrel coordination was confirmed by XRD studies.
Compounds 6–11 were also investigated by IR spectroscopy, in an effort to observe either the imidate [E–O–C(NR)] CN and C–O stretches or the amide [E–NR–C(O)] CO and C–N stretches. Additionally, the structures of 6–11 with both N,N,N and O,O,O tetrel coordination modes were computed at the PBE1PBE/6-311G(d,p) (the SV(p) basis set was used for Sn atoms) level of theory to obtain predicted IR stretches and thermodynamic data. Predicted and observed IR data are summarized in Table 1 and ESI section 4.† In the case of compounds 6 and 7 both imidate C–N and CO stretches could be observed, consistent with N,N,N-coordination of the tetrel and their XRD structures. However, for compounds 8–11 the observed and predicted IR data were in agreement with the presence of amide CN and C–O stretches, consistent with O,O,O-coordination of the tetrel. Furthermore, the IR data for compounds 8 and 11 are in line with the XRD structures of both compounds. However, it is important to recognize that IR data are not diagnostic in distinguishing between the presence of imidate vs. amide moieties, because first C–N and C–O stretches appear near the fingerprint region and second the expected regions for CO and CN stretches overlap.35–38
| Cluster | Label | Computeda (cm−1) | Observed (cm−1) |
|---|---|---|---|
| a Computed at the PBE1PBE/6-311G(d,p) level of theory, and the SV(p) basis set was used for Sn atoms (10). | |||
| 6 | CO |
1709 | 1601 |
| C–N | 1366 | 1376 | |
| 7 | CO |
1705 | 1570 |
| C–N | 1228 | 1187 | |
| 8 | CN |
1580 | 1598 |
| C–O | 1343 | 1300 | |
| 9 | CN |
1569 | 1570 |
| C–O | 1438 | 1340 | |
| 10 | CN |
1540 | 1559 |
| C–O | 1343 | 1360 | |
| 11 | CN |
1587 | 1587 |
| C–O | 1293 | 1298 | |
Next, the free energy difference between the tetrel O,O,O- and N,N,N-coordination modes was calculated for compounds 6–11 (Table 2). Because ΔG is calculated by subtracting the free energy of the N,N,N-coordination mode from that of the O,O,O-coordination mode, a positive value means that the N,N,N-coordination is thermodynamically favoured and a negative value means the opposite. In the case of compounds 6 and 7, the tetrel N,N,N-coordination is thermodynamically favoured by 54.2 kJ mol−1 and 31.4 kJ mol−1, respectively. Meanwhile, the free energy difference between the two tetrel coordination modes of compounds 8 and 10 is not significant. And in the case of compounds 9 and 11, the computed energy differences are consistent with O,O,O-coordination being thermodynamically favourable by −18.9 kJ mol−1 and −48.2 kJ mol−1, respectively.
| Cluster | ΔG [O,O,O – N,N,N]a (kJ mol−1) |
|---|---|
| a Computed at the PBE1PBE/6-311G(d,p) level of theory, and the SV(p) basis set was used for Sn atoms. | |
| (Me3Ge-PhNCO)3P7 (6) | 54.2 |
| (Et3Ge-PhNCO)3P7 (7) | 31.4 |
| (Me3Ge-tosylNCO)3P7 (8) | −1.1 |
| (Et3Ge-tosylNCO)3P7 (9) | −18.9 |
| (nBu3Sn-tosylNCO)3P7 (10) | 7.2 |
| (Me3Si-tosylNCO)3As7 (11) | −48.2 |
Thus, for compound 9, based on the 31P NMR spectrum, the close structural relationship between 8 and 9, the IR data, and the computed energy differences, we postulate that the Ge groups coordinate to the three oxygens of p-toluenesulfonyl isocyanate. For compound 10, based on the IR data and that all the other tetrels coordinate to the oxygens upon p-toluenesulfonyl isocyanate insertion, including in our previous work,27 we propose that the Sn groups also coordinate to the isocyanate oxygen atoms.
Additional computational studies
The fluoride ion affinities (FIAs) of the Si, Ge and Sn groups on the phosphorus and arsenic clusters were studied using DFT to probe their relative electrophilicities (Table 3). These values were computed at the BP86/SV(p) level of theory, following a literature method reported by Greb and co-workers.39 Consistent with the literature precedent,40 the electrophilicities decrease in the order of Si > Ge > Sn. These groups are also marginally more electrophilic when on arsenic than on phosphorus. This could be because element–phosphorus bonds are generally stronger than the corresponding element–arsenic bonds.34 Furthermore, Si–F bond formation is highly favourable, and thus hydride ion affinities (HIAs) were computed as a second method to compare the tetrel electrophilicities. The HIA data follow the same trend as the FIA data, except now the difference in electrophilicities of the tetrels when on phosphorus vs. arsenic is even less significant. Compared to the reactivity of isocyanates with (Me3Si)3P7,27 the heavy germanium and tin functionalized phosphorus clusters 1–3 required longer reaction times to give the heteroallene inserted products. Additionally, changing the cluster from phosphorus to arsenic (4) also decreased the reactivity and required longer reaction times to give insertion. However, similar to the chemistry of (Me3Si)3P7, tris-insertion of isocyanates is preferred over mono- or bis-insertion.| Cluster | FIAa (kJ mol−1) | HIAa (kJ mol−1) |
|---|---|---|
| a Computed at the BP86/SV(p) level of theory. b HIA values could not be calculated as [Me3Si]+ was used as the reference. | ||
| (Me3Si)3P7 | 324 | —b |
| (Me3Ge)3P7 (1) | 265 | 331 |
| (Et3Ge)3P7 (2) | 277 | 340 |
| (nBu3Sn)3P7 (3) | 266 | 333 |
| (Me3Si)3As7 (4) | 332 | —b |
| (Et3Ge)3As7 | 281 | 344 |
| (nBu3Sn)3As7 | 269 | 335 |
Encouraged by the insertions observed with isocyanates, the thermodynamic stability of capturing CO2 with clusters (Me3Si)3P7, 1–4, (Et3Ge)3As7 and (nBu3Sn)3As7 was investigated computationally (Table 4). The ΔG for inserting 3 equivalents of CO2 into all three tetrel–pnictogen bonds was computed at the PBE1PBE/6-311G(d,p) level of theory, and the SV(p) basis set was used for Sn atoms. It was found that only the silicon functionalized clusters (Me3Si)3P7 and (Me3Si)3As7 (4) gave CO2 inserted products that are thermodynamically favourable, by −27.1 kJ mol−1 for (Me3Si)3P7 and −32.2 kJ mol−1 for (Me3Si)3As7. However, CO2 insertions with the germanium and tin functionalized phosphorus and arsenic clusters gave products that are significantly unfavourable with ΔG values for the reactions ranging between 91.9 and 136.5 kJ mol−1.
Reactivity with CO2
In order to experimentally test the capture of CO2 with these clusters, clusters that previously showed isocyanate capture [(Me3Si)3P7 and 1–4] were pressurized with 1 atm of CO2 in toluene, THF, pyridine, diethyl ether and chloroform solvents. Clusters 1–4 showed no evidence of reactivity with CO2 in any of these solvents, even after several weeks. (Me3Si)3P7 was independently prepared,28 and when reacted with 1 atm of CO2 in toluene, pyridine, and diethyl ether no reaction was observed. However, the same reaction in THF showed a new doublet resonance in the 13C{1H} NMR spectrum at 174.8 ppm with a coupling constant of 40 Hz. The 31P and 29Si NMR spectra were also consistent with the formation of new products that do not feature P–Si bonds, in line with insertion between these bonds. After several days, these resonances disappeared suggesting the decomposition of the product. However, when the reaction was repeated in chloroform, the resonance in the 13C{1H} NMR spectrum now at 176.3 ppm with a coupling constant of 44 Hz was stable (Fig. 8). This chemical shift and coupling constant are consistent with the formation of a product with P–C connected by one bond.41–44 The 31P NMR spectrum revealed a distinctive downfield shift of the resonance for the bridging phosphorus atoms to 110 ppm, characteristic of P–C bond formation, similar to the heteroallene inserted products 6–11. Furthermore, the 29Si NMR spectrum showed resonances indicative of Si–O bond formation, as reported previously for the insertion of isocyanates using (Me3Si)3P7.27 Furthermore, the IR spectrum of the reaction mixture revealed new stretches at 1623 cm−1 and 1122 cm−1, consistent with the CO and C–O stretches of esters and frustrated Lewis pair CO2 sequestered products.38,45,46 These spectroscopic features together are consistent with the capture of CO2 between the Si–P bonds of (Me3Si)3P7, as shown in Scheme 4 with the formation of 12.
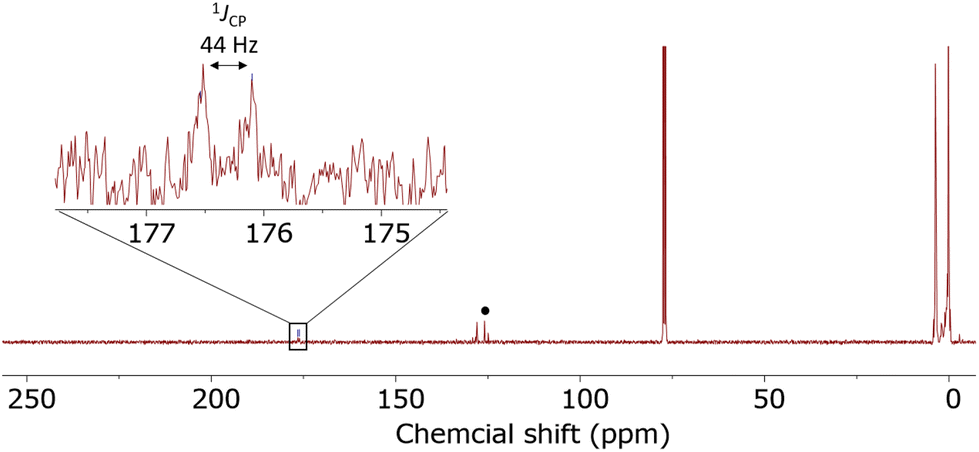 | ||
| Fig. 8 13C{1H} NMR spectra for the reaction of (Me3Si)3P7 with CO2. Dissolved CO2 marked with •. | ||
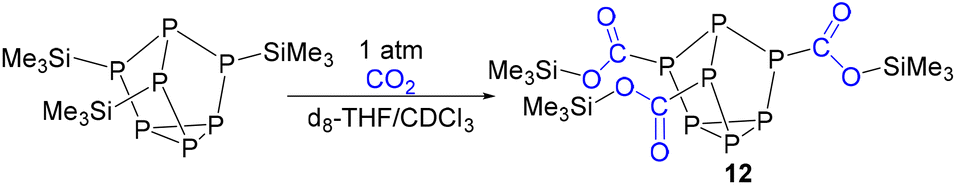 | ||
| Scheme 4 Reaction of (Me3Si)3P7 with CO2. | ||
Clusters 1–3 showed no reactivity towards CO2, which is in line with our computational findings. However, CO2 capture with 4 should yield a thermodynamically favourable product, but experimentally no reaction is observed even after several weeks. This is postulated to be because of (Me3Si)3As7 (4) being less reactive towards heteroallene insertion than (Me3Si)3P7. For example, we observed that (Me3Si)3As7 (4) reacted much slower towards p-toluenesulfonyl isocyanate than (Me3Si)3P7 (7 days vs. 1 day). Additionally, we previously reported that (Me3Si)3P7 reacted with phenyl isocyanate; however, under the same reaction conditions (Me3Si)3As7 showed no reaction even after several weeks.
Conclusions
In conclusion, we prepared tetrel functionalized heptapnictogen cages (Me3Ge)3P7 (1), (Et3Ge)3P7 (2), (nBu3Sn)3P7 (3) and (Me3Si)3As7 (4) and investigated the capture of isocyanates between the three tetrel–pnictogen bonds. We found that phenyl isocyanate reacted with clusters 1 and 2 to give the tris-germanylphosphinated products 6 and 7, but showed no reactivity towards clusters 3 and 4. However, p-toluenesulfonyl isocyanate showed reactivity with all of the tetrel functionalized clusters 1–4 to give the respective tris-inserted products 8–11. Isocyanates are interesting substrates for small molecule capture because they are isostructural with the greenhouse gas CO2. Encouraged by the isocyanate chemistry, CO2 was pressurized with (Me3Si)3P7 and clusters 1–4. In the case of (Me3Si)3P7, evidence for a CO2 captured product 12 could be detected by NMR spectroscopy. This work advances the profile of small molecule activation chemistry with Zintl-derived clusters.Experimental
The general information, experimental procedures, characterization data, and computational details are available in the ESI.†Deposition Numbers 2202018 (for 5), 2202019 (for 6), 2202020 (for 7), 2202022 (for 8), and 2202021 (for 11) contain the supplementary crystallographic data for this paper.†
Author contributions
The experimental and computational work was carried out by W. J. and B. v. I. Crystallographic data were collected and solved by I. J. V.-Y. and G. F. S. W. The manuscript was written by M. M. and edited by W. J. and B. v. I.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for funding (EP/V012061/1) and for supporting a DTA studentship (B. v. I.). The EPSRC is also thanked for supporting W. J. on the iCAT CDT Program (EP/S023755/1). We thank the Royal Society (RGS/R1/211101) for supporting a consumables budget. We also thank Gareth Smith for mass spectrometric analyses, Anne Davies and Martin Jennings for elemental analyses, and Ralph Adams for NMR spectroscopic investigations.References
- B. van IJzendoorn and M. Mehta, Dalton Trans., 2020, 49, 14758–14765 RSC.
- O. P. E. Townrow, C. Chung, S. A. Macgregor, A. S. Weller and J. M. Goicoechea, J. Am. Chem. Soc., 2020, 142, 18330–18335 CrossRef CAS PubMed.
- M. Jo, A. Dragulescu-Andrasi, L. Z. Miller, C. Pak and M. Shatruk, Inorg. Chem., 2020, 59, 5483–5489 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, L. Z. Miller, B. Chen, D. T. McQuade and M. Shatruk, Angew. Chem., Int. Ed., 2016, 55, 3904–3908 CrossRef CAS PubMed.
- M. Ruck, D. Hoppe, B. Wahl, P. Simon, Y. Wang and G. Seifert, Angew. Chem., Int. Ed., 2005, 44, 7616–7619 CrossRef CAS PubMed.
- R. S. P. Turbervill and J. M. Goicoechea, Chem. Rev., 2014, 114, 10807–10828 CrossRef CAS PubMed.
- R. J. Wilson, B. Weinert and S. Dehnen, Dalton Trans., 2018, 47, 14861–14869 RSC.
- R. J. Wilson, N. Lichtenberger, B. Weinert and S. Dehnen, Chem. Rev., 2019, 119, 8506–8554 CrossRef CAS PubMed.
- S. Mandal, A. C. Reber, M. Qian, R. Liu, H. M. Saavedra, S. Sen, P. S. Weiss, S. N. Khanna and A. Sen, Dalton Trans., 2012, 41, 5454–5457 RSC.
- S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed.
- S. C. Sevov and J. M. Goicoechea, Organometallics, 2006, 25, 5678–5692 CrossRef CAS.
- R. S. P. Turbervill and J. M. Goicoechea, Chem. Commun., 2012, 48, 1470–1472 RSC.
- R. S. P. Turbervill and J. M. Goicoechea, Eur. J. Inorg. Chem., 2014, 2014, 1660–1668 CrossRef CAS.
- R. S. P. Turbervill and J. M. Goicoechea, Organometallics, 2012, 31, 2452–2462 CrossRef CAS.
- R. S. P. Turbervill, A. R. Jupp, P. S. B. McCullough, D. Ergöçmen and J. M. Goicoechea, Organometallics, 2013, 32, 2234–2244 CrossRef CAS.
- R. S. P. Turbervill and J. M. Goicoechea, Inorg. Chem., 2013, 52, 5527–5534 CrossRef CAS PubMed.
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed.
- B. van Ijzendoorn, S. F. Albawardi, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, J. E. McGrady and M. Mehta, J. Am. Chem. Soc., 2022, 144, 21213–21223 CrossRef PubMed.
- B. L. L. Réant, B. van Ijzendoorn, G. F. S. Whitehead and M. Mehta, Dalton Trans., 2022, 51, 18329–18336 RSC.
- E. W. Abel, R. A. N. McLean and I. H. Sabherwal, J. Chem. Soc. A, 1968, 2371–2373 RSC.
- K. Issleib, H. Schmidt and H. Meyer, J. Organomet. Chem., 1980, 192, 33–39 CrossRef CAS.
- E. W. Abel and I. H. Sabherwal, J. Chem. Soc. A, 1968, 1105–1108 RSC.
- R. Appel and M. Poppe, Angew. Chem., Int. Ed. Engl., 1989, 28, 53–54 CrossRef.
- G. Becker, J. Härer, G. Uhl and H. J. Wessely, Z. Anorg. Allg. Chem., 1985, 520, 120–138 CrossRef CAS.
- H. Schumann, Angew. Chem., Int. Ed. Engl., 1969, 8, 937–950 CrossRef CAS.
- B. van IJzendoorn, I. Vitorica-Yrezabal, G. Whitehead and M. Mehta, Chem. – Eur. J., 2021, 28, e202103737 Search PubMed.
- M. Cicač-Hudi, J. Bender, S. H. Schlindwein, M. Bispinghoff, M. Nieger, H. Grützmacher and D. Gudat, Eur. J. Inorg. Chem., 2016, 649–658 CrossRef.
- G. Fritz, K. D. Hoppe, W. Hönle, D. Weber, C. Mujica, V. Manriquez and H. G. v. Schnering, J. Organomet. Chem., 1983, 249, 63–80 CrossRef CAS.
- W. Hönle, J. Wolf and H. G. v. Schnering, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 219–223 CrossRef.
- H. G. von Schnering, M. Somer, G. Kliche, W. Hönle, T. Meyer, J. Wolf, L. Ohse and P. B. Kempa, Z. Anorg. Allg. Chem., 1991, 601, 13–30 CrossRef CAS.
- F. Emmerling and C. Röhr, Z. Anorg. Allg. Chem., 2003, 629, 467–472 CrossRef CAS.
- W. Wichelhaus and H. G. v. Schnering, Naturwissenschaften, 1973, 60, 104–104 CrossRef CAS.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, Taylor & Francis, 2007 Search PubMed.
- R. Keese, F. Berdat and P. Macchi, J. Org. Chem., 2013, 78, 1965–1970 CrossRef CAS PubMed.
- H. Bredereck, F. Effenberger and E. Henseleit, Chem. Ber., 1965, 98, 2754–2761 CrossRef CAS.
- W. H. Prichard and W. J. Orville-Thomas, J. Chem. Soc. A, 1967, 1102–1105 RSC.
- P. Larkin, Infrared and Raman Spectroscopy: Principles and Spectral Interpretation, Elsevier Science, 2011 Search PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- M. Wiesemann and B. Hoge, Chem. – Eur. J., 2018, 24, 16457–16471 CrossRef CAS PubMed.
- K. Takeuchi and D. W. Stephan, Chem. Commun., 2012, 48, 11304–11306 RSC.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed.
- N. Szynkiewicz, A. Ordyszewska, J. Chojnacki and R. Grubba, RSC Adv., 2019, 9, 27749–27753 RSC.
- N. Szynkiewicz, Ł. Ponikiewski and R. Grubba, Chem. Commun., 2019, 55, 2928–2931 RSC.
- D. A. Dickie, M. T. Barker, M. A. Land, K. E. Hughes, J. A. C. Clyburne and R. A. Kemp, Inorg. Chem., 2015, 54, 11121–11126 CrossRef CAS PubMed.
- K. Mentoor, L. Twigge, J. W. H. Niemantsverdriet, J. C. Swarts and E. Erasmus, Inorg. Chem., 2021, 60, 55–69 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General information, DFT information, XRD information, IR information, synthetic procedures, analytical data, and NMR spectra. CCDC 2202018–2202022. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt04074c |
| This journal is © The Royal Society of Chemistry 2023 |