
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convergent growth approach to electroactive ferrocene rich carbosilane- and siloxane-based dendrons, dendrimers, and dendronized polymers†‡
Sonia
Bruña
 *ab,
Josefina
Perles
c and
Isabel
Cuadrado
ab
*ab,
Josefina
Perles
c and
Isabel
Cuadrado
ab
aDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Ciudad Universitaria de, Cantoblanco, Calle Francisco Tomás y Valiente, 7, 28049, Madrid, Spain. E-mail: sonia.brunna@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Madrid 28049, Spain
cLaboratorio de Difracción de Rayos X de Monocristal, Servicio Interdepartamental de Investigación (SIdI), Universidad Autónoma de Madrid, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
First published on 6th April 2023
Abstract
The construction of the first members of a novel family of structurally well-defined, ferrocenyl rich, dendritic macromolecules based on carbosilane skeletons and siloxane linkages has been achieved via a convergent growth approach. Starting from triferrocenylvinylsilane Fc3SiCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (1) (Fc = Fe(η5-C5H4)(η5-C5H5) as the key monomer, the sequential utilization of platinum-catalyzed hydrosilylation and alkenylation steps with Grignard reagents (allylmagnesium bromide) can be applied to prepare three different branched structures: multiferrocenyl-terminated dendrons 2 and 3, dendrimers 4 and 5, and dendronized polymers 7n–9n. All of the dendritic metallomacromolecules have been thoroughly characterized using a combination of elemental analysis, multinuclear (1H, 13C, 29Si) NMR spectroscopy, FT-IR and MALDI-TOF mass spectrometry, to establish their chemical structures and properties. The molecular structures of G1-dendron 3 and dendrimer 4, containing six and nine ferrocenyl units, respectively, have been successfully determined by single-crystal X-ray analysis, compound 4 being the branched multiferrocenyl-containing siloxane with the highest number of Fc substituents whose structure has been reported so far. Electrochemical studies (using cyclic voltammetry (CV) and square wave voltammetry (SWV) performed in dichloromethane solution with [PF6]− and [B(C6F5)]4− as supporting electrolyte anions of different coordinating abilities) reveal that all the macromolecular compounds obtained exhibit a three-wave redox pattern, suggesting appreciable electronic interactions between the silicon-bridged triferrocenyl moieties as they are successively oxidized. In addition, dendrimer 5 and dendronized polymers 7n–9n, with 12 and 4 < n < 14 ferrocenyl units, respectively, linked in threes around the periphery, undergo remarkable oxidative precipitation in CH2Cl2/[n-Bu4N][PF6] and are able to form chemically modified electrodes with stable electroactive films.
CH2 (1) (Fc = Fe(η5-C5H4)(η5-C5H5) as the key monomer, the sequential utilization of platinum-catalyzed hydrosilylation and alkenylation steps with Grignard reagents (allylmagnesium bromide) can be applied to prepare three different branched structures: multiferrocenyl-terminated dendrons 2 and 3, dendrimers 4 and 5, and dendronized polymers 7n–9n. All of the dendritic metallomacromolecules have been thoroughly characterized using a combination of elemental analysis, multinuclear (1H, 13C, 29Si) NMR spectroscopy, FT-IR and MALDI-TOF mass spectrometry, to establish their chemical structures and properties. The molecular structures of G1-dendron 3 and dendrimer 4, containing six and nine ferrocenyl units, respectively, have been successfully determined by single-crystal X-ray analysis, compound 4 being the branched multiferrocenyl-containing siloxane with the highest number of Fc substituents whose structure has been reported so far. Electrochemical studies (using cyclic voltammetry (CV) and square wave voltammetry (SWV) performed in dichloromethane solution with [PF6]− and [B(C6F5)]4− as supporting electrolyte anions of different coordinating abilities) reveal that all the macromolecular compounds obtained exhibit a three-wave redox pattern, suggesting appreciable electronic interactions between the silicon-bridged triferrocenyl moieties as they are successively oxidized. In addition, dendrimer 5 and dendronized polymers 7n–9n, with 12 and 4 < n < 14 ferrocenyl units, respectively, linked in threes around the periphery, undergo remarkable oxidative precipitation in CH2Cl2/[n-Bu4N][PF6] and are able to form chemically modified electrodes with stable electroactive films.
Introduction
Dendrons, dendrimers, and dendronized polymers are among the most widespread, well-defined, branched macromolecular structures.1 Specifically, dendrimers constitute a unique class of macromolecular architectures that differ from all other synthetic macromolecules in their perfectly branched topology, which is constructed, in an exceptionally challenging step-wise manner, by a repetitive reaction sequence from a central core and expands to the periphery that becomes denser with increasing generation number. Many synthetic approaches have been developed for dendrimers since the seminal examples reported during the 80s and 90s,2 the divergent2a–c and convergent2g–i growth being the two most widely used strategies.In the divergent approach, dendrimers are grown from the inside to the outside, starting from a multifunctional core to which several layers of branching units are attached progressively. As a different strategy, in the convergent approach the construction of dendrimers starts at what will ultimately become the outer surface of the dendrimer and progresses inwardly. This approach first requires the synthesis of functionalized, progressively larger, dendritic wedges or dendrons, having a single reactive focal point, which is then covalently attached to a polyfunctional core to yield the desired dendrimer. Furthermore, dendrimer approaches imply other accelerated synthetic strategies that can minimize the number of reaction steps in dendrimer synthesis.3 Remarkably, the click reaction concept has proved to be a powerful synthetic tool for the design of highly sophisticated dendritic architectures, for instance to prepare dendrons and dendrimers via thiol–ene chemistry,4 to assemble dendrons in order to construct siloxane dendrimers5 or to prepare liquid-crystalline Janus dendrimers, merging two types of dendromesogens.6
Among the fascinating architectural features of dendrimers, their high overall symmetries and their well-defined nanometer sizes and internal cavities have allowed the rapid development of their new and improved properties. In fact, they are materials with valuable and promising applications in a wide range of fields, such as catalysis, cancer therapy, biosensors for diagnostics, light harvesting scaffolds, controlled drug delivery, medical diagnostics or nanoelectronics, to name just a few examples.7
One of the most active research areas in dendrimer chemistry involves the integration of transition metals into dendritic structures to create metallodendrimers.8 Since the first transition-metal containing dendrimers were reported in the early 1990s, advances in the synthesis and chemistry of these molecules have not ceased to blossom. For instance, the incorporation of electroactive organometallic units into dendrimer structures is an especially attractive target area because such highly branched macromolecules are good multielectron-transfer mediators in electrocatalytic processes of biological and industrial importance, and this remains a topical challenge. In particular, covalent incorporation of ferrocene units into dendrimers,9 polymers,10 and macrocycles11 can greatly modify their physical and chemical properties. Ferrocene is an ideal donor moiety and the ease of functionalization, the chemical stability, and the diamagnetism in the neutral state (allowing NMR characterization) make this metallocene one of the mostly used building blocks to construct metallomacromolecular architectures.12 In addition, this organometallic sandwich molecule exhibits attractive electrochemical behavior, a fast electron-transfer rate, low oxidation potential, and two stable redox states (neutral ferrocene and oxidized ferrocenium). In fact, the achievements in the synthesis of ferrocene-containing organosilicon polymers and oligomers,13 and well-defined branched molecules of different nature, including ferrocene dendrimers, have attracted significant attention over the years due to their beauty, synthetic complexity, and valuable applications.
Likewise, in order to expand the dendrimer chemistry, silicon chemistry has played a dominant role in the field of dendritic polymers, enabling the synthesis of a robust and diverse family of silicon-based dendritic macromolecules.14,15 As a result, silicon-containing dendritic skeletons based on carbosilane (Si–C), siloxane (Si–O–Si) or carbosiloxane (Si–O–C) linkages are among the most widely used ones for the synthesis of transition metal-containing dendrimers, because of their kinetic and thermodynamic stability, chemical inertness, and accessibility. Regarding this point, our research group has also contributed to the design, construction and electrochemical studies of dendritic macromolecules based on silicon, mainly carbosilanes and siloxanes, functionalized with electroactive organometallic units of a very diverse nature.16
On the other hand, dendronized polymers have become an important class of macromolecules, which merges the concepts of dendrimers and linear polymers.17,18 They are composed of a linear polymer backbone to which dendrons of increasing size (i.e., generations G1, G2, G3…) are appended. At high degrees of dendronization, dendronized polymers gain stiffness and adopt a cylindrical shape. This exclusive architecture makes dendronized polymers unique macromolecules and interesting candidates for a variety of nanoscale applications, such as molecular wires, self-adaptive materials, and functional scaffolds for catalysis. The vast majority of dendron-branched polymers present an organic nature. Few examples of branched siloxanes have been published so far.19 Furthermore, in spite of the fact that numerous metallodendrimers have been reported, only very few studies have addressed the synthesis of dendronized polymers containing metallic moieties.20
Herein, we merge the convergent approach, the silicon chemistry and the unique redox properties and ease of functionalization of the ferrocene unit and present full details of the synthesis, characterization (including the solid-state structures of some macromolecules), thermal properties, and electrochemical behavior of first- and second generation, ferrocene-rich and siloxane-based dendrons, dendrimers, and dendronized polymers. Remarkably, the presence of triferrocenyl end moieties, bridged by a silicon atom, has led us to explore the electrochemical behavior and electronic interactions between the redox-active metal sites. Furthermore, we have investigated in detail the chemically modified electrodes formed with some ferrocenyl-dendritic molecules, highlighting their ability to form robustly adsorbed films onto metallic electrode surfaces.
Experimental
General procedures
CH2 (1) was synthesized from monolithioferrocene (generated in situ from the reaction between ferrocene and t-BuLi, at −10 °C) according to the procedure already described.21 The platinum-divinyltetramethyldisiloxane complex in xylene (3–3.5% Pt concentration) (Karstedt's catalyst), chlorophenylsilane, allylmagnesium bromide (1 M in diethyl ether) and poly(methylhydrosiloxane) ((Me3SiO)(MeSiHO)n(SiMe3), Mn 1700–3200) (available from Merck-Sigma-Aldrich), and tetrakis(dimethylsiloxy)silane (ABCR) were used as received. Silica gel (70–230 mesh) (Merck-Sigma-Aldrich) was used for column chromatography purifications. Infrared spectra were recorded on a PerkinElmer 100 FT-IR spectrometer. Elemental analyses were performed on a LECO CHNS-932 elemental analyzer, equipped with a MX5 Mettler Toledo microbalance. All NMR spectra were recorded on Bruker Avance III-HD NANOBAY 300 MHz and Bruker DRX 500 MHz spectrometers. Chemical shifts were reported in parts per million (δ) with reference to CDCl3 residual solvent resonances for 1H (δ 7.26 ppm) and 13C (δ 77.2 ppm). 29Si NMR resonances were recorded with inverse-gated proton decoupling to minimize nuclear Overhauser effects and were referenced externally to tetramethylsilane. For the types of silicon atoms, depending on the number of bonded oxygen atoms, the terminologies M, D, T and Q represent the structures R3SiO0.5, R2SiO, RSiO1.5 and SiO2, respectively (see Scheme S1 in the ESI‡). MALDI-TOF mass spectra were recorded using a Bruker-Ultraflex III TOF/TOF mass spectrometer equipped with a nitrogen laser emitting at 337 nm. Dichloromethane solutions of the matrix (dithranol, 10 mg mL−1) and CH2Cl2 solutions of the corresponding compound (1 mg mL−1) were mixed in a ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. Then, 0.5–1 μL of the mixture was deposited on the target plate using the dried droplet method. The positive ion and the reflectron mode were used for these analyses. Thermogravimetric analyses were performed using a TGA Q-500 instrument coupled with an EGA oven. The samples (5–10 mg) were loaded in platinum pans. The measurements were carried out under N2 (90 mL min−1) with a heating rate of 10 °C min−1. The morphology of the ceramic residues was investigated with SEM using a Philips XL30 instrument coupled with an EDAX DX4i analyzer.
:5. Then, 0.5–1 μL of the mixture was deposited on the target plate using the dried droplet method. The positive ion and the reflectron mode were used for these analyses. Thermogravimetric analyses were performed using a TGA Q-500 instrument coupled with an EGA oven. The samples (5–10 mg) were loaded in platinum pans. The measurements were carried out under N2 (90 mL min−1) with a heating rate of 10 °C min−1. The morphology of the ceramic residues was investigated with SEM using a Philips XL30 instrument coupled with an EDAX DX4i analyzer.
The preparation of electrode surfaces modified with films of dendrimer 5 and polymers 7n–9n was accomplished by electrodeposition of the corresponding species on Pt electrodes. The electroactive films were prepared by cyclically scanning the potential (between +0.0 and +1.3 V vs the SCE) in degassed CH2Cl2 solutions of the corresponding dendritic species at different numbers of times (10 or 20 scans for dendrimer 5 and 10 scans for polymers 7n–9n). The electrochemical behavior of the films was then studied by CV in multiferrocene-free CH2Cl2 or CH3CN solution containing only the supporting electrolyte ([n-Bu4N][PF6] or [n-Bu4N][B(C6F5)4]). From the CVs of the modified electrodes, the surface coverages, Γ (mol cm−2), of the ferrocenyl sites were calculated from the charge, Q, under the voltammetric current peaks, using Γ = Q/nFA.
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH2){(CH2)2SiFc3}2 (3).
To a solution of dendron 1 (1.00 g, 1.64 mmol) in 25 mL of dry and degassed toluene, Karstedt's catalyst (60 μL) was added under argon. The mixture was stirred for 20 min before a toluene solution of chlorophenylsilane (110 μL, 0.82 mmol) was added dropwise. The reaction mixture was heated at reflux temperature until the 1H NMR experiments showed the complete disappearance of the Si–H signal of the starting (C6H5)Si(H)2Cl (18 h). At this moment, the reaction mixture was allowed to cool to room temperature, and the solvent was removed under vacuum. The remaining orange oil (C6H5)SiCl{(CH2)2SiFc3}2 (2) was immediately used in the next reaction step.
CH2){(CH2)2SiFc3}2 (3).
To a solution of dendron 1 (1.00 g, 1.64 mmol) in 25 mL of dry and degassed toluene, Karstedt's catalyst (60 μL) was added under argon. The mixture was stirred for 20 min before a toluene solution of chlorophenylsilane (110 μL, 0.82 mmol) was added dropwise. The reaction mixture was heated at reflux temperature until the 1H NMR experiments showed the complete disappearance of the Si–H signal of the starting (C6H5)Si(H)2Cl (18 h). At this moment, the reaction mixture was allowed to cool to room temperature, and the solvent was removed under vacuum. The remaining orange oil (C6H5)SiCl{(CH2)2SiFc3}2 (2) was immediately used in the next reaction step.
To a three-necked, round-bottomed flask equipped with a gas inlet, a pressure equalizing addition funnel, an Allihn condenser topped with a gas inlet and bubbler, and a Teflon-covered magnetic stir bar, freshly distilled and degassed diethyl ether (30 mL) was added. A solution of recently prepared 2 in 30 mL of diethyl ether was added dropwise, with vigorous stirring. Simultaneously, allylmagnesium bromide (0.9 mL, 0.9 mmol, 1 M in diethyl ether) was added dropwise. The resulting yellow-orange solution was refluxed for 20 h, cooled to 0 °C with an ice bath, and then hydrolyzed with aqueous NH4Cl (10%). The organic layer was separated, washed three times with water, and dried over anhydrous MgSO4. After filtration, the solvent was removed under vacuum yielding an orange-yellow oily product, which was purified by column chromatography on silica gel (3 cm × 10 cm). A first band was observed corresponding to unreacted triferrocenylvinylsilane 1 which was eluted with n-hexane/CH2Cl2 (10:2), and subsequently, a second major orange band was observed with n-hexane/CH2Cl2 (10:5). Solvent removal afforded the desired grown dendron 3, carrying six ferrocenyl units, which was obtained as an analytically pure, air-stable, orange crystalline solid.
Yield: 0.48 g (43%). Anal. Calcd for C73H72Si3Fe6: C 64.06; H 5.30. Found: C 64.10; H 5.21. 1H NMR (CDCl3, 300 MHz, ppm): δ 1.26 (s, 8H, Si–CH2–CH2), 2.11 (m, 2H, –CH2CHCH2), 3.98 (s, 30H, C5H5), 4.24, 4.37 (m, 12H + 12H, C5H4), 5.03 (m, 2H, –CH2CHCH2), 5.98 (m, 1H, –CH2CHCH2), 7.38 (m, 3H, Ph), 7.60 (m, 2H, Ph). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ 5.2, 8.3 (Si–CH2CH2), 19.5 (–CH2CHCH2), 68.5 (C5H5), 69.8 (ipso–Fc), 70.5, 73.8 (C5H4), 114.1 (–CH2CHCH2), 128.0 (m–Ph), 129.3 (p–Ph), 134.3 (o–Ph), 134.7 (–CH2CHCH2), 136.3 (ipso–Ph). 29Si{1H} RMN (CDCl3, 59 MHz, ppm): δ −8.3 (Fc–Si), −0.1 (Si–CH2CHCH2). IR (KBr, cm−1): δ(Si–C) 1258, ν(Si–C) 820. MS (MALDI-TOF): m/z 1368.0 [M+].
:2). Then, upon eluting with n-hexane/CH2Cl2 (10:6), a second major band was observed. Solvent removal afforded dendrimer 4, with nine terminal ferrocenes, as an analytically pure, air-stable, orange, crystalline solid.
Dendrimer 4. Yield: 0.09 g (38%). Anal. Calcd for C104H118Si8Fe9O4: C 57.85; H 5.51. Found: C 58.09; H 5.48. 1H NMR (CDCl3, 500 MHz, ppm): δ 0.28 (s, 24H, CH3), 0.98, 1.22 (m, 6H + 6H, CH2), 4.01 (s, 45H, C5H5), 4.26, 4.37 (m, 36H, C5H4), 4.86 (sep, 1H, Si−H). 13C{1H} NMR (CDCl3, 125 MHz, ppm): δ −0.1 (CH3), 1.2 (CH3SiH), 7.8, 11.2 (CH2), 68.6 (C5H5), 70.2 (ipso-Fc), 70.6, 74.1 (C5H4). {1H−29Si} HMQC (CDCl3, 500, 99 MHz, ppm): δ −5.3 (Si–H). {1H–29Si} HMBC (CDCl3, 500, 99 MHz, ppm): δ −103.0 (SiO4), −8.2 (Fc–Si), −5.3 (Si–H), 10.0 (Si(CH3)2). IR (KBr, cm−1): ν(Si–H) 2137, δ(Si–C) 1258, νas(Si–O–Si) 1033–1105, δ(Si–H) 890, ν(Si–C) 818. MS (MALDI-TOF): m/z 2159.1 [M+].
Dendrimer 5 was obtained by the reaction of 1 (0.3 g, 0.49 mmol) in 6 mL of toluene, with 39 μL of tetrakis(dimethylsiloxy)silane I (0.10 mmol) in the presence of 30 μL of Karstedt's catalyst. The reaction mixture was stirred at 70 °C for 17 h. The oily residue was purified by column chromatography on silica gel (2 cm × 20 cm). After isolating the excess of compound 1 with n-hexane/CH2Cl2 (10:2), a second major band was eluted with n-hexane/CH2Cl2 (1:1), which afforded the desired dendrimer 5 as an analytically pure, air-stable, orange, crystalline solid.
Dendrimer 5. Yield: 0.20 g (74%). Anal. Calcd for C136H148Si9Fe12O4: C 58.98; H 5.39. Found: C 59.22; H 5.29. 1H NMR (CDCl3, 300 MHz, ppm): δ 0.31 (s, 24H, CH3), 0.99, 1.24 (m, 16H, CH2), 4.01 (s, 60H, C5H5), 4.26, 4.37 (m, 24H + 24H, C5H4). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ −0.1 (CH3), 7.8, 11.2 (CH2), 68.6 (C5H5), 70.2 (ipso-Fc), 70.6, 74.1 (C5H4). 29Si{1H} RMN (CDCl3, 59 MHz, ppm): δ−103.1 (SiO4), −8.3 (Fc–Si), 9.8 (Si(CH3)2). IR (KBr, cm−1): δ(Si–C) 1249, νas(Si–O–Si) 1028–1107, ν(Si–C) 820. MS (MALDI-TOF): m/z 2770.1 [M+].
Yield: 0.38 g. 1H NMR (CDCl3, 300 MHz, ppm): δ 0.20–0.26 (m, CH3), 0.88, 1.26 (m, CH2), 3.99 (s, C5H5), 4.24, 4.34 (m, C5H4). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ 0.2, 2.7 (CH3), 7.6, 10.7 (CH2), 68.8 (C5H5), 70.1 (ipso-Fc), 70.7, 74.1 (C5H4). 29Si{1H} RMN (CDCl3, 59 MHz, ppm): δ −21.9 (SiO2) −8.2 (Fc–Si), 7.5 (SiO). IR (KBr, cm−1): δ(Si–C) 1260, νas(Si–O–Si) 1031–1105, ν(Si–C) 815, 799. MS (MALDI-TOF). 7n: m/z 2844 [M+, n = 4], 3514 [M+, n = 5], 4184 [M+, n = 6], 4854 [M+, n = 7], 5524 [M+, n = 8], 6194 [M+, n = 9], 6864 [M+, n = 10], 7534 [M+, n = 11], 8204 [M+, n = 12], 8874 [M+, n = 13], 9544 [M+, n = 14]. 8n: m/z 2681 [M+, n = 4], 3351 [M+, n = 5] y 4021 [M+, n = 6]. 9n: 2904 [M+, n = 4], 3574 [M+, n = 5], 4244 [M+, n = 6], 4914 [M+, n = 7], 5584 [M+, n = 8], 6254 [M+, n = 9], 6924 [M+, n = 10], 7594 [M+, n = 11], 8264 [M+, n = 12], 8934 [M+, n = 13], 9604 [M+, n = 14].
Attempts to synthesize dendrimer 6 and the second generation dendronized polymer from dendron 3 are explained in the ESI.‡
Results and discussion
Convergent growth approach from triferrocenylvinylsilane 1: synthesis of dendrons 2 and 3
The convergent approach1g,2g–i has been selected for the synthesis of a new family of ferrocene rich, silicon-containing, dendrons and dendrimers. We first constructed small ferrocene-based monofunctional dendrons, bearing a single carbon–carbon double bond at the focal point, and subsequently we used them in hydrosilylation chemistry to synthesise either dendrons of higher generations or final dendrimer molecules.Prior to initiating dendrimer synthesis, triferrocenylvinylsilane21,28 Fc3Si–CHCH2 (1) (Fc = Fe(η5-C5H4)(η5-C5H5)) was selected as the key monomer for the synthesis of higher generation dendrons. Triferrocenylvinylsilane 1 fulfils several fundamental requirements and shows features for being used in a convergent methodology: (1) it is experimentally accessible; (2) it bears a single vinylsilyl functional group that can act as the reactive point for Si–H functional backbones through hydrosilylation reactions; (3) from an electronic point of view, the –CHCH2 group of 1, linked to three electron-donating ferrocenyl moieties, is particularly electron rich and, accordingly, is highly reactive toward hydrosilylation;29 (4) it has a very stable chemical nature, which implies high tolerance to the reaction conditions of carbosilane chemistry; (5) it shows good solubility in common organic solvents (such as THF, CH2Cl2 and n-hexane), and finally, (6) 1 is an organometallic molecule with remarkable thermal and air stability, which can be obtained and stored for a long period of time (several years) under an air atmosphere at room temperature without noticeable decomposition (1H NMR spectroscopy).
Beforehand, perhaps the only structural challenge that monofunctional 1 could offer for a convergent dendrimer synthesis is the sterically hindered environment of the reactive Si–CHCH2 group, surrounded by three bulky ferrocenyl moieties that might well affect the effectiveness of hydrosilylation reactions. However, triferrocenylvinylsilane 1 has already been employed in successful hydrosilylation reactions,29 which led to the preparation of cyclic and cubic macromolecules peripherally decorated with as many interacting ferrocenyl units as chemically possible (24 ferrocenyl moieties). In addition, the presence of three ferrocenes, linked through the same silicon atom, enables an electronic interaction between metal centers and provides strong motivation to investigate their electrochemical processes.
Therefore, by using triferrocenylvinylsilane 1(G0) as the dendritic precursor, the first generation (G1) dendrons 2 and 3 were obtained, by means of a two-step convergent growth methodology (Scheme 1). The reaction sequence began with the hydrosilylation of 1 and (C6H5)Si(H)2Cl. Platinum-catalyzed hydrosilylation was chosen since it has been shown to be a highly efficient process, and Karstedt's catalyst was selected because it is widely used in hydrosilylation chemistry.30
 | ||
| Scheme 1 Dendritic growth from Fc3Si–CHCH2 (1): synthesis of dendritic wedges with Si–Cl (2) and Si–CH2–CHCH2 (3) reactive focal points. | ||
Dendron 2 was obtained as an orange dense oil and was immediately used in the subsequent reaction, avoiding its isolation to prevent any possible decomposition by hydrolysis. In the second step, 2 was successfully alkenylated with allylmagnesium bromide in diethyl ether. A subsequent hydrolytic workup and careful purification by column chromatography afforded an air-stable, orange and crystalline solid (in 43% isolated yield), which was characterized as the desired grown dendron 3 (Scheme 1), carrying six ferrocenyl units linked in three ways through a silicon atom. The coordination sphere of the external silicon atom was completed by a phenyl ring, and an allyl reactive group to allow further dendritic growth.
On the basis of the 1H NMR spectra of both dendrons 2 and 3, it was determined that only the β-isomers formed under the described conditions, and consequently hydrosilylation was anti-Markovnikov. The structural identity of novel dendron 3 was confirmed by IR, multinuclear 1H, 13C and 29Si NMR spectroscopy, MALDI-TOF mass spectrometry and elemental analysis. As can be seen in the 1H NMR spectra of precursor 1 and grown dendron 3 (Fig. 1), the mono-substituted ferrocenyl areas, around δ 4 ppm, are almost identical for both compounds. The allylic group of 3 appears as three resonances: a multiplet at δ 5.98 ppm for the CH unit, another multiplet at δ 5.03 ppm for the terminal –CH2 group, and a doublet at δ 2.11 ppm assigned to the internal –CH2– protons. The hydrogen signals of the two –CH2–CH2– bridges are observed at δ 1.26 ppm under a broad singlet, and those of C6H5 as two multiplets are centered at δ 7.60 and 7.38 ppm.
 | ||
| Fig. 1 1H NMR spectra of dendrons 1 (G0) and 3 (G1) (recorded in CDCl3, 300 MHz). | ||
The same occurs with the 13C NMR experiments. The vinyl resonances at about δ 135 ppm of 1 are replaced with the allyl signals at δ 19.5, 114.1 and 134.7 ppm in 3 (Fig. S3‡). In the 13C NMR spectrum of 3 the corresponding resonances of the phenyl unit (at δ 128.0, 129.3, 134.3 and 136.3 ppm) and of the asymmetric –CH2–CH2– bridges (at δ 5.2 and 8.3 ppm) can also be observed. The 29Si NMR spectrum of the resulting dendron 3 shows two peaks centered at δ −0.1 ppm (for the –Si–CH2–CHCH2 unit) and at δ −8.3 (for the (Fc)3Si–CH2CH2 group). The latter is considerably shifted downfield in comparison with the same signal of precursor 1 (δ −17 ppm), reflecting the change in the electronic environment of the Si units due to the transformation of the adjacent CC bond into a –Si–CH2–CH2– bridge. The MALDI-TOF mass experiment shows a peak at m/z 1368.0 for 3 corresponding to the molecular ion M+, showing an excellent agreement between the experimental and the calculated isotopic patterns (Fig. S7‡). Finally, the solid-state structure of hexaferrocenyl dendron 3 was proved by single-crystal X-ray diffraction, as it will be commented below.
Incorporation of dendrons 1 and 3 into Si–H polyfunctional siloxane backbones: synthesis of dendrimers 4, 5 and 6 and dendronized polymers 7n–9n
The final step of a dendritic molecular construction, following the convergent approach, consists of integrating the previously synthesized monofunctional dendritic wedges into a multifunctional core.1g,2g–i In this case, the availability of free olefinic substituents, at the focal point of dendrons 1 and 3, facilitates the incorporation of interacting organometallic redox-active centers in dendritic structures through hydrosilylation chemistry with Si–H polyfunctional cores. Particularly, two different hydrosiloxanes were carefully selected as multifunctional cores (Fig. 2): star shaped tetrakis(dimethylsiloxy)silane, Si[OSi(CH3)2H]4 (I), which will act as a four-functional core allowing the formation of dendrimers; Si–H-rich linear poly(methylhydrosiloxane), [(Me3SiO)(MeSiHO)n(SiMe3), Mn 1700–3200, II], which will allow the formation of dendronized polymers. Based on our previous synthesis experience, the choice of core units, in which the reactive Si–H sites are located at the ends of extended flexible siloxane spacer chains, is important to reduce steric congestion in the desired final macromolecules. | ||
| Fig. 2 Selected Si–H containing siloxane backbones for the growth of ferrocenyl-rich dendrimers and dendronized polymers. | ||
For the synthesis of the dendrimeric molecules, hydrosilylation assembly reactions of 1 and 3 (in excess) with tetrakis(dimethylsiloxy)silane I were performed at 70 °C, in toluene solutions and in the presence of Karstedt's catalyst (Schemes 2 and 3). Completion of the anchoring reactions was easily monitored by 1H NMR spectroscopy.
 | ||
| Scheme 2 Hydrosilylation reactions of 1 with tetrakis(dimethylsiloxy)silane I leading to the first-generation dendrimers 4 and 5. | ||
 | ||
| Scheme 3 Hydrosilylation between the first generation dendron 3 and the tetra-directional core I. | ||
From the first hydrosilylation reaction between 1 (in stoichiometric excess) and Si[OSi(CH3)2H]4 (I) instead of the target dendrimer 5, only the nonmetallic dendritic molecule 4 was formed and isolated (38% yield, Scheme 2), with three of the four Si–H bonds being functionalized with –(CH2)2SiFc3 units. This result is reproducible under the same reaction conditions (6 hours at 70 °C) and no other functionalization degrees, such as dendrimers with one, two or four –(CH2)2SiFc3 units, were detected by MALDI-TOF experiments of the crude reaction mixtures. It is interesting to note here that the presence of the free single reactive –Si–H functionality in molecule 4 enables further elaboration through, for example, thiol–ene reactions. Therefore, 4 could serve as an excellent candidate to construct two-faced “Janus dendrimers”, which are molecules composed of two dendrimeric wedges (dendrons) and terminated by two different but complementary functionalities.31
We first reasoned that this “structural dendritic defect” was probably caused by a low hydrosilylation time (6 hours of reaction). To test this hypothesis, and to ensure the full substitution of the four Si–H bonds, this reaction was repeated using a larger stoichiometric excess of triferrocenylsilane 1 and it was allowed to react for 17 hours. After appropriate purification, the target dendrimer 5 was isolated as an air-stable, crystalline, and orange solid (74% yield). The 1H NMR and MALDI-TOF experiments confirm its identity. Particularly, there are no other peaks in the mass spectrum, allowing us to exclude the formation of, for instance, other dendrimers with structural defects. In addition, the 1H NMR spectrum of the hydrosilylated dendrimer product proved that only the β-isomer was formed. This ensured a regular dendritic growth and the generation of molecules of maximum symmetry.
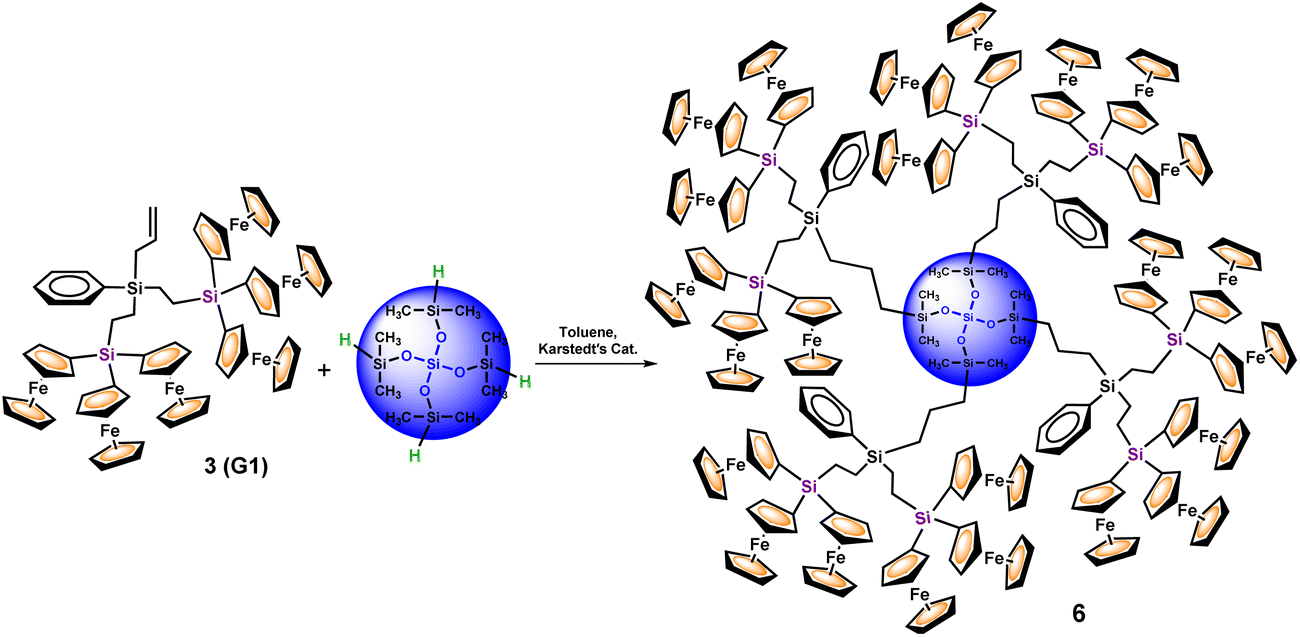
The next task was then focused on the hydrosilylation of G1-dendron 3 with the siloxane core Si[OSi(CH3)2H]4 (I), in order to prepare larger generation dendrimers. The reaction was carried out for 24 hours (Scheme 3), and the orange oil obtained was dissolved in a small amount of CH2Cl2 and precipitated into n-hexane. Both the solution and the oily residue obtained were studied by MALDI−TOF mass spectrometry, showing similar results (see Fig. 3). The most intense peak (at m/z 1369.1) matches with unreacted dendron 3, but additional peaks that correspond to partially functionalized dendrimers can also be observed. Therefore, the peak at m/z 4450.0 matches with the dendrimer containing three functionalized arms and the fourth one ends in a Si–OH group. The one at m/z 3080.2 corresponds to the bifunctionalized dendrimer and two unfunctionalized branches (one Si–H and one Si–OH). This peak is followed by another one at m/z 3096.2 of the bifunctionalized dendrimer with two terminal Si–OH groups. Finally, the peak at m/z 1726.1 correlates with the monofunctionalized dendrimer, two Si–OH and one Si–H groups (the peak of the monofunctionalized dendrimer with three Si–OH groups appears at m/z 1742.1).
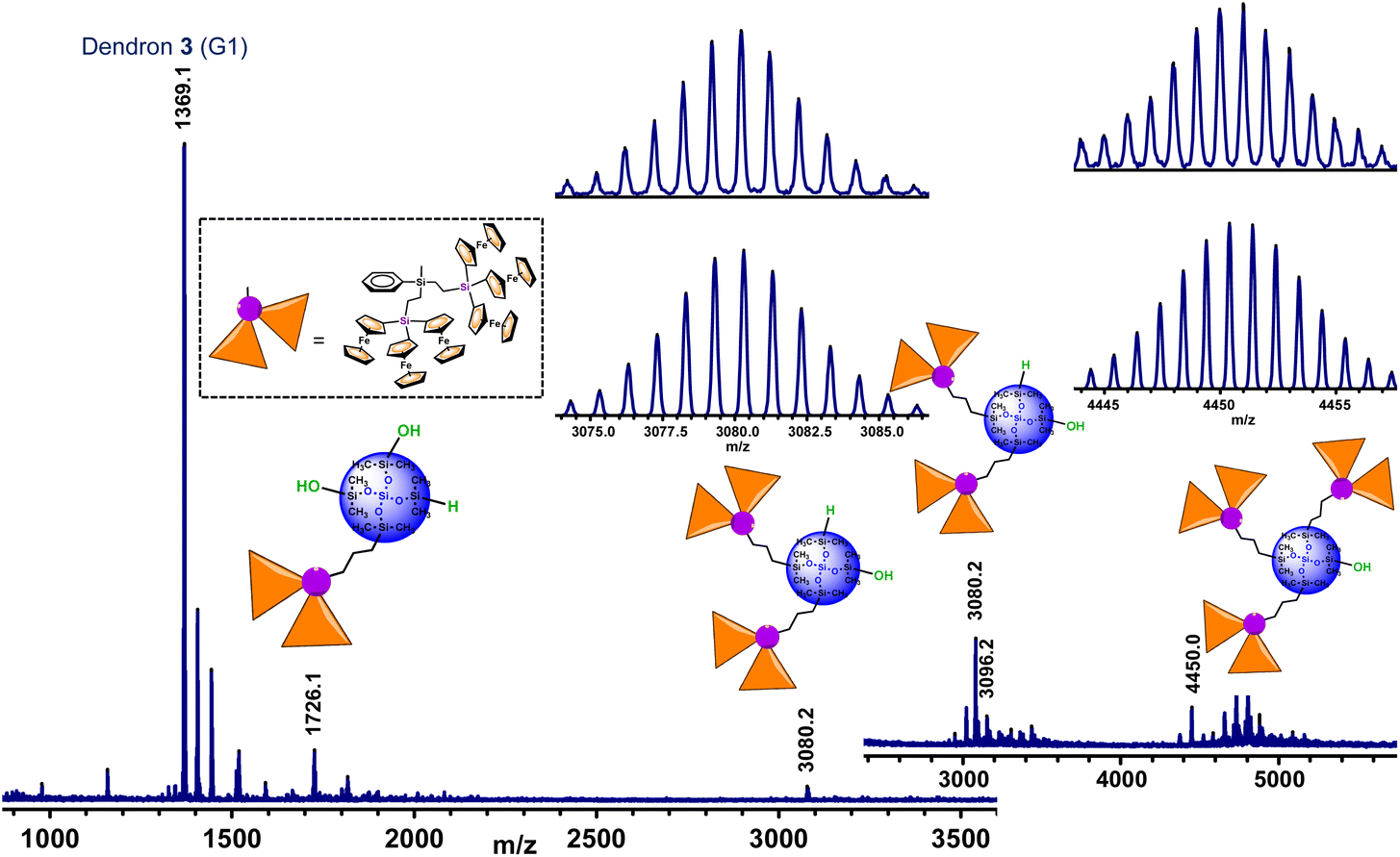 | ||
| Fig. 3 MALDI-TOF mass spectrum of the reaction product resulting from the hydrosilylation of 3 and Si[OSi(CH3)2H]4 (I). The insets show the experimental and calculated isotopic patterns of the peaks at m/z 3080.2 and 4450.0. | ||
As can be observed in Fig. 3, the peak of the target dendrimer 6 was not detected by the MALDI–TOF technique. On the basis of the good results obtained with the reactions of dendron 1 (G0), this last hydrosilylation was repeated with a larger excess of 3 and the increased reaction time (72 hours) and temperature (100 °C), but it was not possible to fully react the four available Si–H groups. It is evident that there is a noteworthy difference in the reactivity of the first generation dendron 3 in comparison with its corresponding precursor 1. Steric congestion appears to be responsible for the inability of the four Si–H groups of the tetradirectional core Si[OSi(CH3)2H]4 (I) to react with G1-dendron 3, which is formed by six ferrocenyl units. Even though the fourth branched core of I is selected for its extended silicon-containing arms, and dendron 3 is functionalized with an allyl group (to generate a long –CH2–CH2–CH2–chain), steric hindrance imposed by the sterically demanding organometallic units seems to be the key factor that dominates this reaction, preventing full functionalization and leading to dendrimers with structural defects. However, it is important to emphasize that the MALDI–TOF technique tends to show considerable molecular weight discrimination for polydisperse samples.32 This means that, although peaks at higher m/z values are not observed in this spectrum (Fig. 3), the formation of higher molecular weight species cannot be ruled out.
Our attention was then turned to the synthesis of dendronized polymers by polyhydrosilylation reactions of 1 and 3 with poly(methylhydrosiloxane) II. The reaction with dendron 1 (G0) as the starting material was accomplished using the methodology mentioned before (see the top of Fig. 4). After 45 hours at 100 °C, analysis of the reaction mixture by 1H NMR experiments confirmed the complete consumption of the Si–H protons of (Me3SiO)(MeSiHO)n(SiMe3) (II) at δ 4.72 ppm. New resonances at δ 0.8–1.3 ppm, corresponding to the new hydrosilylated product with CH2–CH2 linkages, were also observed indicating the quantitative transformation of the Si–H reactive groups.
 | ||
| Fig. 4 Structures of the dendritic products 7n, 8n and 9n formed by platinum-catalyzed hydrosilylation of dendron 1 (G0) onto the multifunctional silicone backbone (Me3SiO)(MeSiHO)n(SiMe3) (II) (top) and MALDI-TOF mass spectrum (bottom). | ||
The oily reaction mixture was dissolved in CH2Cl2 and precipitated twice into methanol. The obtained precipitate was then analyzed by MALDI-TOF mass spectrometry (Fig. 4), showing three different groups of peaks. First of all, the peaks of the expected linear oligomeric species 7n (n = 4–12) appear in the range of m/z 2844–8204 with an interval between the peaks at m/z 670 (this value corresponds to the mass of the repeated fragment –MeSi[–CH2CH2–Si–Fc3]O–). Then, there are peaks corresponding to cyclic species 8n (n = 4–6) without terminal groups, at m/z 2681, 3351 and 4021, also separated by m/z 670. Finally, a group of more intense peaks appears between m/z 2903 and 8264 (equally spaced at m/z 670). These signals that differ from the values of 7n at m/z 60 (mass of one unreacted –H3C–Si(H)O– fragment) (see Fig. 4) form a new family of dendronized oligomers and polymers, 9n (n = 4–12). The isotopic masses of all these peaks (see Fig. S27 in the ESI‡) agree well with the calculated ones, confirming their precisely defined linear or cyclic structures.
In an attempt to obtain a fraction of ferrocenyl-bearing polymers with higher molecular weights, the first precipitate was dissolved again in a small amount of CH2Cl2 and precipitated into methanol. The MALDI-TOF spectrum (Fig. S28‡) proved to follow an identical pattern, when compared to the previous one. However, when the matrix deflection high voltage is set at m/z 2000, species 7n and 9n with n values up to 14 (m/z 9604) can be observed. As shown before, although peaks at higher m/z values are not observed, the formation of higher molecular weight species cannot be ruled out, since MALDI-TOF tends to show a considerable molecular weight discrimination for polydisperse samples.32
The last step in our proposed dendritic growth approach consisted of incorporating the first generation dendron 3 into the polymeric siloxane backbone II. After 72 hours of the hydrosilylation reaction at 100 °C, the oily product was treated, as before, and the precipitates were analyzed by MALDI-TOF. In all the recorded spectra, a major peak was observed at m/z 1369.1, and corresponds to the starting dendron 3, but we were not able to identify any other species. As observed with dendrimer 6, this result can be due to the steric demand of G1-dendron 3, which is responsible for the failure of the Si–CH2–CHCH2 groups to react. Nonetheless, given the proved efficiency of the hydrosilylation processes with G0-dendron 1, we are convinced that both dendrimers and dendronized polymers can be chemically prepared with G1-dendrons of type 3. Obviously, reducing the steric congestion proves to be critical, and this can be achieved by adding spacer units to the focal point in the G1-dendron 3 and/or in the –Si–H arms of the siloxane cores. Indeed, in closely related Si–H rich polysiloxane backbones, it was suggested that steric constraints were responsible for the inability of all Si–H groups to react with the dendrons.19a
Multinuclear NMR (1H, 13C, and 29Si) experiments of dendrimers 4 and 5 and dendronized polymers 7n–9n support their assigned structures. On the basis of their 1H and 13C NMR studies it was determined that the addition of Si–H functionality to the vinyl group of 1 proceeds selectively according to the anti-Markovnikov rule. While the 1H NMR spectrum of dendrimer 4 (Fig. S8‡) shows an informative septuplet at δ 4.86 ppm, confirming the presence of one unreacted Si–H functionality, the 1H NMR spectrum of dendrimer 5 (Fig. S14‡) proves its fully functionalized structure, as no signal appears in this region. The 1H NMR spectrum of the mixture of dendronized polymers 7n–9n (Fig. S21‡) shows broader signals, due to the complex mixture of the species and their large size, but with quite similar chemical shift values as the ones of the already reported dendrimers. The 13C NMR spectrum of dendrimer 4 (Fig. S9‡) shows a representative signal at δ 1.2 ppm for the H–Si–(CH3)2 group.
Regarding the 29Si NMR experiments, the spectra of dendrimers 4 and 5 and polymers 7n–9n display a peak at around δ −8.3 ppm, corresponding to the (Fc)3Si–CH2CH2 group, which supports the incorporation of the triferrocenylsilyl moieties around the dendritic siloxane frameworks via hydrosilylation reactions. Moreover, these spectra show resonances at δ −103.0 (for 4) and −103.1 ppm (for 5) which are consistent with the Q-type silicon atoms (SiQO4), at δ −21.9 ppm (for 7n–9n) of the D-type silicon atoms (SiDO2), and at δ 9.8 (for 5), 10.1 (for 4) and 7.5 ppm (for 7n–9n) of the M-type silicon atoms (SiMO). In the case of 4 an additional resonance can be observed at δ −5.3 ppm corresponding to the unreacted Si–H bond. This last signal was corroborated by a {1H–29Si} HMQC experiment (see Fig. S10‡).
MALDI-TOF mass spectrometry provided further evidence for the formation of the new dendritic macromolecules. The mass spectra of 4 and 5 show the most intense peaks at m/z 2159.1 and 2770.1, respectively, which correspond to the M+ ions. Their isotopic masses agree well with those of the calculated ones, confirming their precisely defined star-shaped dendritic structures and the presence of 9 and 12 pendant ferrocenyl moieties, grouped in three dimensions at the end of each arm. In addition, the IR spectra of dendrimers 4 and 5 and the dendronized polymers 7n–9n are very similar to those of a predominant band corresponding to the ν(Si–O–Si) of the siloxane backbone. The main difference can be observed in the dendrimer 4 spectrum, in which two bands appear at 2137 cm−1 (ν(Si–H)) and at 890 cm−1 (δ(Si–H)) (see Fig. S12‡).
Investigations of the properties of the obtained ferrocenyl-rich dendritic molecules were complemented by the evaluation of their thermal stability by thermogravimetric analysis (TGA). We have found that the mixture of dendronized polymers 7n–9n shows a reasonably high thermal stability (Fig. 5). This TGA experiment was performed under nitrogen and was compared with the TGA results previously obtained by our research group29 for the related octasilsesquioxanes (OS) molecules 10 and 11, as also shown in Fig. 5. Ferrocenyl-functionalized cubic macromolecules 10 and 11, peripherally decorated with 24 ferrocenyl units, have also shown good thermal stability.29
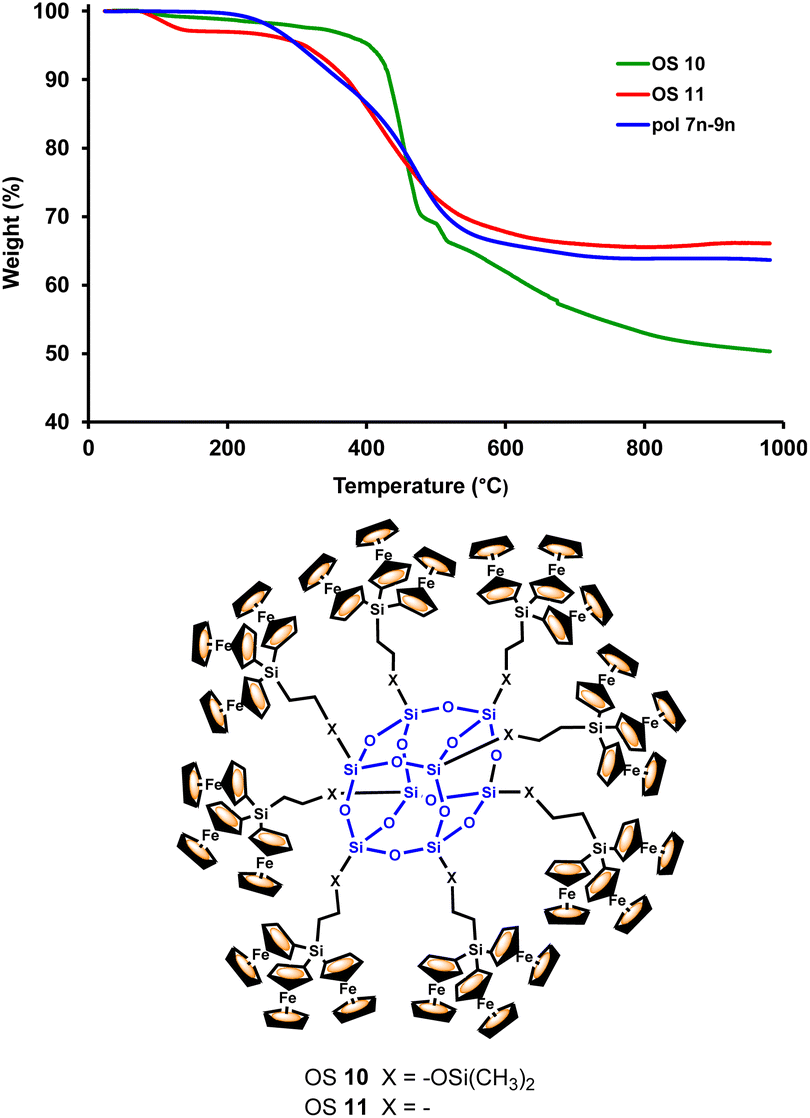 | ||
| Fig. 5 Comparison of the TGA thermogram of dendronized polymers 7n–9n, under N2 at a heating rate of 10 °C min−1, and the TGA thermograms of ferrocenyl-functionalized octasilsesquioxanes 10 and 11 under the same experimental conditions. | ||
As can be seen in Fig. 5, the three multiferrocenyl macromolecules (7n–9n, 10 and 11) show similar weight losses when heated between 0 and 1000 °C. In particular, the mixture of dendronized polymers 7n–9n displays two weight losses. The first loss, beginning near 200 °C, can be attributed to the cleavage of the organic components of the polymeric chains, whereas the second and major weight loss, between 370 and 650 °C, can be ascribed to the breakdown of the inorganic structure. Above this temperature, the TGA curve is almost leveled off and the ceramization yield of 7n–9n, at 1000 °C, is 64 wt% (50 wt% for 10 and 66 wt% for 11). This comparison clearly highlights the great thermal stability of the three ferrocenyl-containing siloxane backbones, regardless of their precisely siloxane nature.
We further examined the residues obtained via pyrolysis of 7n–9n under a nitrogen stream at 1000 °C, by SEM and energy-dispersive X-ray analysis (Fig. S30‡). The pyrolyzed material obtained from 7n–9n in the ceramization process contains iron particles (Fig. S31–S33‡) and can be attracted to a bar magnet, revealing that polymers 7n–9n could be promising precursors for ceramic materials.
Solid-state structures of G1-dendron 3 and dendrimer 4
A key challenge in the synthesis of highly branched molecules is to obtain good single crystals, suitable for SCXRD analysis. Since the dendrons and dendrimers reported here are large and complex molecules, they were very hard to crystallize, due to the difficulty in organizing the molecules in the crystalline network. Fortunately, we were able to obtain suitable crystals for hexametallic dendron 3 (G1) and nonametallic dendrimer 4 by crystallization in CH2Cl2/n-hexane (1:2) at −4 °C. The resulting structures are shown in Fig. 6 and complete structural information is collected in the ESI.‡ Both compounds crystallize in the monoclinic space group P21/n with one molecule per asymmetric unit. The cyclopentadienyl rings in 3 and 4 are essentially parallel and slightly staggered, and the molecular packing in both cases is achieved by C–H⋯π interactions involving the cyclopentadienyl rings.
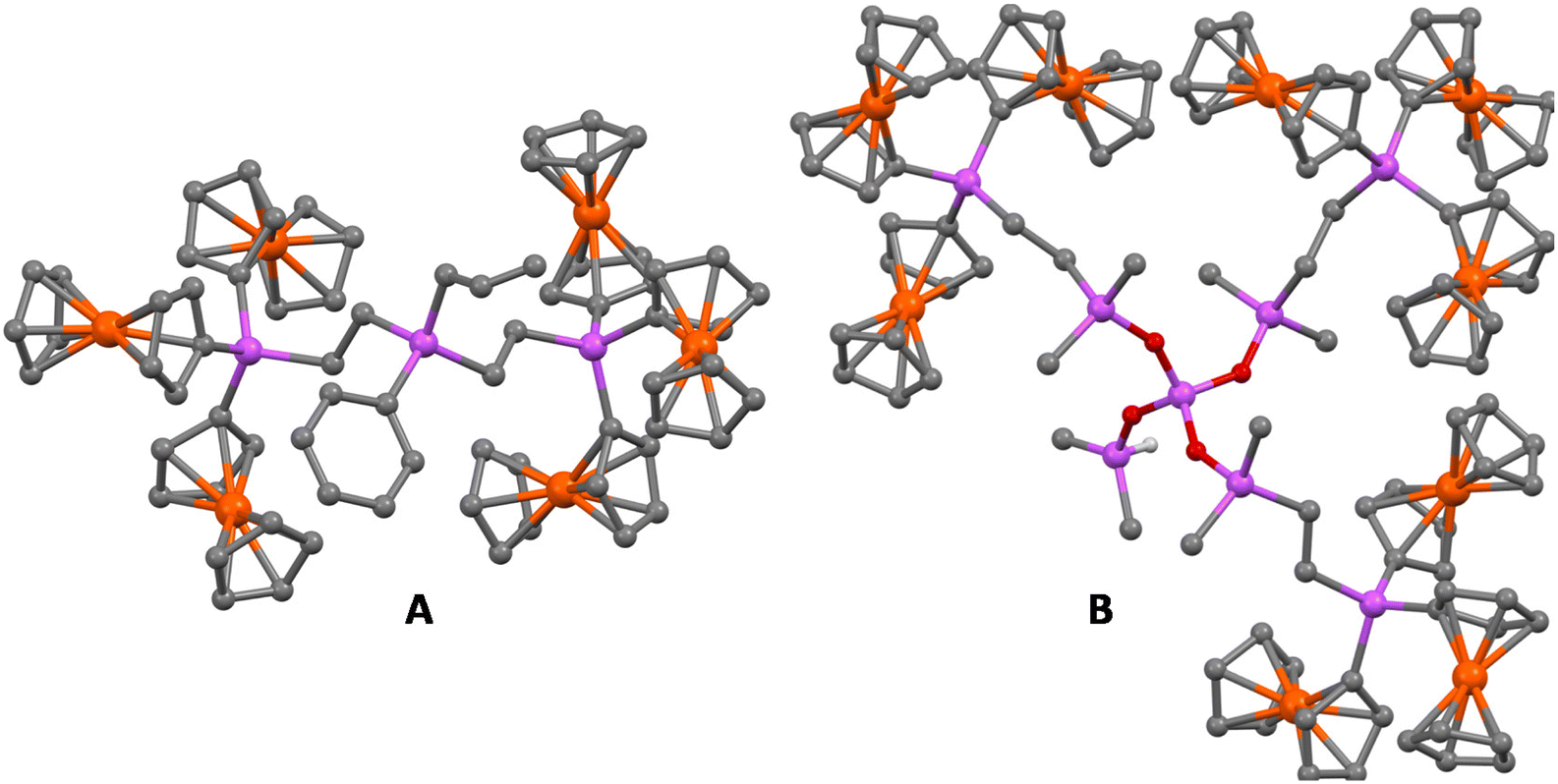 | ||
| Fig. 6 Crystal structures of G1-dendron 3 (A) and dendrimer 4 (B). Hydrogen atoms are omitted for clarity. | ||
The structure of hexaferrocenyl dendron 3 (G1) shows that the two largest (CH2)2SiFc3 substituents of the central silicon atom are located as separated as possible (Fig. 6A). The three silicon atoms in the molecule show a nearly tetrahedral environment, with the C–Si–C bond angles close to 109°, similar to the ones reported for 1.28 The metal atoms of the ferrocenyl substituents attached to the same silicon center are separated by distances ranging from 5.401(1) to 6.164(1) Å, while the Fe centers of the ferrocenyl units attached to different silicon atoms are separated by longer distances (from 6.967(1) to 12.730(1) Å), as can be seen in Table S5.‡
Dendrimer 4 provides a unique opportunity to examine the solid-state structure of a branched siloxane with nine ferrocenyl units. We consider it important to highlight here that a bibliographic search in the CSD has revealed dendrimer 4 as the branched multiferrocenyl-containing siloxane with the highest number of ferrocenes fully characterized by single-crystal X-ray diffraction. The central tetrahedral SiO4 unit displays three large (CH3)2Si–(CH2)2–SiFc3 substituents and a smaller –Si(CH3)2H one. This shorter moiety shows a statistical disorder that was modelled with two sets of positions for the –OSi(CH3)2H fragment, with 60%–40% occupations. The presence of a remaining Si–H reactive group in dendrimer 4 is not only a simple structural defect, but makes it a genuine functional core, being capable to build new types of asymmetric dendrimers by hydrosilylation reactions.
The eight silicon atoms in the molecule display the expected tetrahedral environment with all the C–Si–C, O–Si–O and O–Si–C bond angles close to 109°. The metal atoms of the ferrocenyl substituents attached to the same silicon center are located at distances ranging from 5.806(2) to 6.160(2) Å, and the iron centers of the Fc units attached to different silicon atoms are separated by distances ranging from 6.224(2) to 18.487(2) Å (see Table S5‡). It is remarkable that the large number of ferrocenyl units in this molecule packs them closer to each other, and consequently, the difference between the largest distance for the iron atoms from Fc attached to the same Si atom and the shortest Fe–Fe distance from the metals in adjacent SiFc3 groups is only 0.064 Å.
Electrochemical studies of 1 (G0) and 3 (G1), dendrimers 4 and 5 and dendronized polymers 7n–9n
Due to the ferrocenyl-rich nature of the silicon-based dendritic molecules obtained here, a significant property to consider is their electroactivity. The solution electrochemical behavior of isolated ferrocenyl-rich dendritic macromolecules 3–5 and the mixture of dendronized polymers 7n–9n was examined by cyclic (CV) and square wave (SWV) voltammetry and was compared to the redox behavior of the precursor molecule, triferrocenylvinylsilane Fc3SiCHCH2 (1) (G0). The electrochemical responses are shown in Fig. 8 and 9, and the electrochemical data are summarized in Table 1.
| Compounda | 1 E 1/2 | 2 E 1/2 | 3 E 1/2 | ΔE (2E1/2 − 1E1/2)b | ΔE (3E1/2 − 2E1/2)b | ΔE (3E1/2 − 1E1/2)b |
|---|---|---|---|---|---|---|
|
a
E
1/2 for V vs. the SCE, determined by square wave voltammetry in CH2Cl2 solution with [n-Bu4N][B(C6F5)4], or [n-Bu4N][PF6] with CH2Cl2/CH3CN (3:0.5) for values indicated in parentheses.
b Peak potential separation values, ΔE, are given in mV.
|
||||||
| 1 | 0.436 (0.400) | 0.736 (0.568) | 1.048 (0.704) | 300 (168) | 312 (136) | 612 (304) |
| 3 | 0.400 (0.440) | 0.720 (0.604) | 1.068 (0.736) | 320 (164) | 348 (132) | 668 (296) |
| 4 | 0.392 (0.444) | 0.684 (0.620) | 0.896 (0.756) | 292 (176) | 212 (136) | 504 (312) |
| 5 | 0.456 (0.452) | 0.696 (0.624) | 0.819 (0.753) | 240 (172) | 123 (129) | 363 (301) |
| 7n–9n | (0.452) | (0.645) | (0.736) | (193) | (91) | (284) |
It is worth noting that the solution redox behavior of this multiferrocenyl dendritic family is sensitive to the dendrimer generation and to the number of ferrocenyl moieties. In order to check the evolution of the dendritic effect on the voltammetric response, cyclic voltammograms (CVs) were recorded using a Pt-disk working electrode, in dichloromethane solution with 0.1 M of tetra-n-butylammonium hexafluorophosphate ([n-Bu4N][PF6]) as the supporting electrolyte, and containing 10−4 M of the redox-active species, in all cases.
Fig. 7 shows the representative CV responses of dendron 3 and dendrimer 5, as examples, together with the CV of triferrocenyl precursor 1, for comparative purposes. Qualitatively similar responses were observed for dendrimer 4 and dendronized polymers [7n–9n] (see Fig. S38 and S43,‡ respectively). As can be seen, the solubility of all the studied multimetallic dendritic systems is drastically affected by the oxidation and reduction steps. Therefore, the voltammetric response deviates from the typical CV wave shape, characteristic of freely diffusing soluble species undergoing reversible transfer processes.33
 | ||
| Fig. 7 Cyclic voltammograms of multiferrocenyl dendritic molecules 1 (A), 3 (B), and 5 (C) measured in CH2Cl2/[n-Bu4N][PF6] at 100 mV s−1. | ||
Specifically, it is evident from Fig. 7 that for dendritic precursor 3 (bearing 6 ferrocenyl (Fc) units) and dendrimer 5 (12 Fc), the anodic and cathodic peak currents are dissimilar. The anodic waves of macromolecules 3–5 and 7n–9n occur by three poorly resolved redox processes, when compared to those of precursor 1. In addition, the cathodic scan gives rise to extensive precipitation of the oxidized, highly charged species [36+][PF6−]6, [49+][PF6−]9, [512+][PF6−]12 and [7n–9nn+][PF6−]n on the electrode surface, which results in distorted, very sharp peaks. The sharpness of these peaks is strongly related to the stripping nature of the electrochemical processes. As the dendritic material is deposited onto the electrode surface, it is redissolved back in the solution, as a result of the cathodic electron-transfer reaction. Nevertheless, not all the electrodeposited dendritic molecules are stripped from the surface in a single scan. In fact, there are some strong precipitated and/or adsorbed dendrimer molecules remaining on the electrode surface upon reduction.
Likewise, as the number of ferrocenyl moieties per dendritic molecule increases (that is, as the generation starts), the cathodic stripping wave increases in magnitude (see Fig. 7), meaning that the electrogenerated polycationic macromolecules [36+][PF6−]6 and [59+][PF6−]9 become less soluble in this solvent/electrolyte medium.
Taking dendrimer 5 as a representative example, Fig. 8 shows how by enriching the solvent mixture with acetonitrile, the cathodic stripping peaks progressively tend to disappear. Thus, the CVs of dendron 3, dendrimers 4 and 5, and the CV of the mixture 7n–9n, show three well-resolved anodic waves, which are indicative of appreciable electronic interactions between the silicon-bridged triferrocenyl moieties as they are successively oxidized. Under these conditions, the E1/2 values of the three redox processes can be measured for all the compounds and are summarized in Table 1.
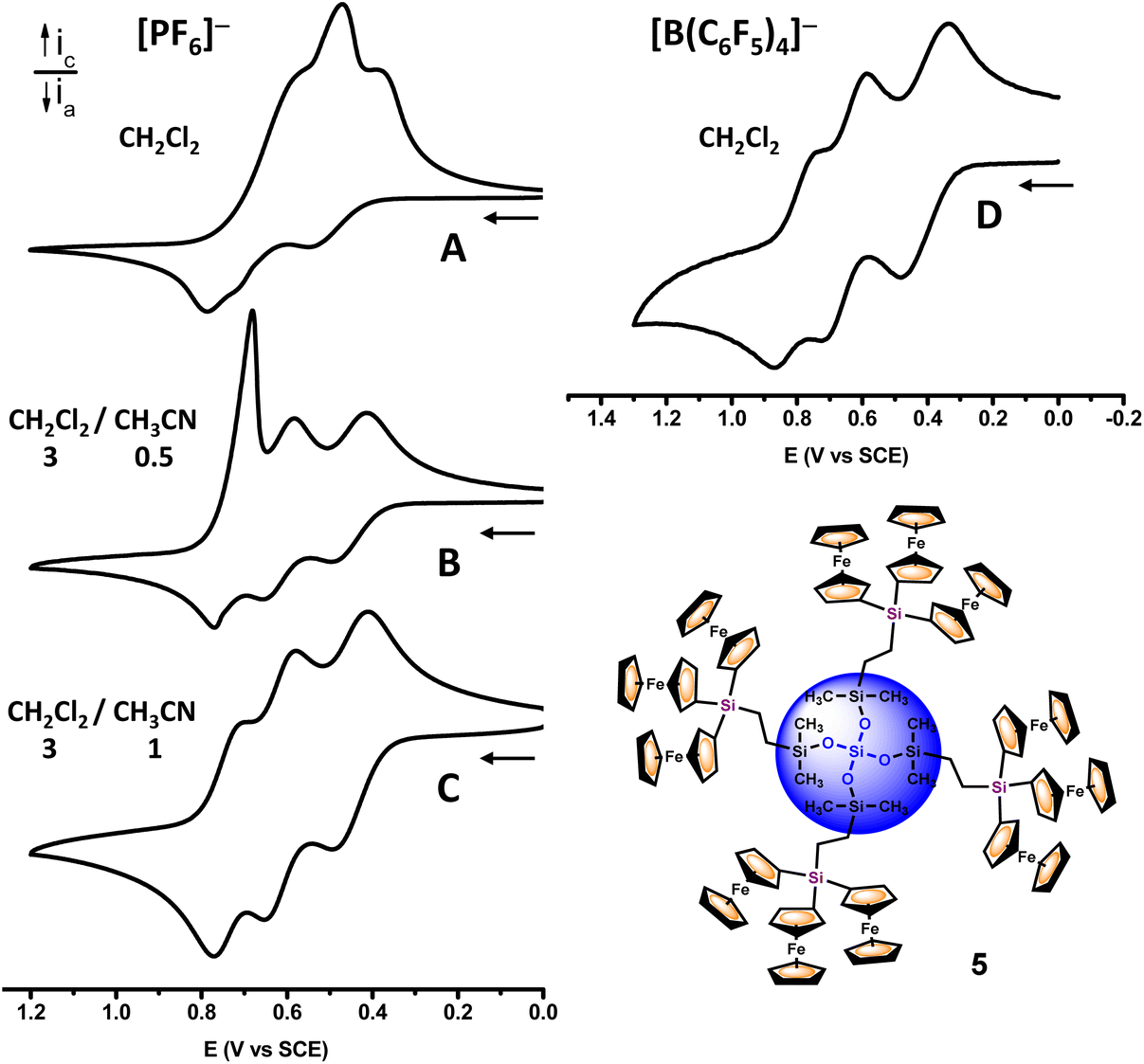 | ||
| Fig. 8 Cyclic voltammograms of dendrimer 5, measured under the conditions indicated therein, at 100 mV s−1. | ||
Fig. 8 also shows the remarkable improvement for the CV response of dendrimer 5 when CH2Cl2 and tetrakis(pentafluorophenyl)borate ([n-Bu4N][B(C6F5)4]) are employed as the solvent/supporting electrolyte. A similar qualitative behavior is observed with dendritic species 3 and 4 and dendronized polymers 7n–9n (see Fig. S37, S39 and S41‡). The weak coordinating counter-anion [B(C6F5)4]− is known to stabilize the highly charged species in solution. Furthermore, the low ion-pairing capabilities lead to a poor shielding of the electrostatic interactions among the redox active sites, resulting in an increase of the observed redox splitting.34 Consequently, in the presence of the less coordinating electrolyte anion[B(C6F5)4]−, a better resolution, improved electrochemical reversibility, and very little electrode adsorption are observed, which is indicative of a better solubility of the multiferrocenium electrogenerated dendritic species 36+, 49+, 512+ and 7n–9nn+ in this solvent/electrolyte medium. Clearly, in agreement with our previous observations,21,29 and with the seminal results reported by Geiger and co-workers,34 the combination of CH2Cl2 and [n-Bu4N][B(C6F5)4] as the solvent/electrolyte medium provides more favorable conditions for the electrochemical studies of multiferrocenyl compounds, minimizing the ion-pairing interactions between the fluoroarylborate anion and the polycationic species generated in the oxidation processes.
In addition to the electrochemical studies of the dendrimers in a homogeneous solution, we have explored the redox properties of the ferrocenyl-containing dendritic molecules confined to electrode surfaces, where the films of the dendritic molecules serve as electrode modifiers. It is well known that the deliberated and controlled modification of electrode surfaces with films of electroactive molecules can produce electrodes with new and interesting properties that may form the basis of new electrochemical applications and novel devices. In addition, such modified electrodes can provide a better insight into the nature of charge transfer and charge transport processes in the films. Therefore, chemically modified electrodes are important due to their potential applications in the areas of photo- and electro-catalysis, ion recognition, biosensing and information storage.33a,35
Remarkably, a valuable feature of the new dendritic macromolecules with the highest number of ferrocenes, dendrimer 5 and dendronized polymers 7n–9n, is their noteworthy tendency to modify electrodes, resulting in dendrimer-modified electrode surfaces that remain persistently attached to the electrodes. Electrodes modified with the films of these multiferrocenyl-containing dendritic molecules were prepared using a 10−4 M solution of the corresponding macromolecule in a 10−1 M CH2Cl2/[n-Bu4N][PF6] mixture (see the Experimental section). The electrochemical deposition could be observed onto the Pt electrodes by continuous increases in both the anodic and cathodic peak currents during consecutive cyclic voltammetric scans. We have evaluated the stability of the films of macromolecules 5 and 7n–9n, by transferring the modified Pt electrodes into fresh CH2Cl2 or CH3CN/electrolyte solutions. As a representative example, Fig. 9 shows the CV responses of an electrodeposited film of dendronized polymers 7n–9n, in both solvents. When CH2Cl2 is used for the fresh solution, three successive well-resolved, reversible oxidation−reduction waves are observed, with formal potential values of 1E1/2 = 0.459, 2E1/2 = 0.600 and 3E1/2 = 0.705 V, vs. the SCE. These values are similar to the ones observed for the dendronized polymers 7n–9n in solution (see Table 1). The surface-confined nature of the electroactive 7n–9n films is proved by the linear dependences of the anodic and cathodic peak currents, of the three waves, on the potential sweep rate v (see the top of Fig. 9A).35,36
 | ||
| Fig. 9 CVs of a Pt-disk electrode modified with a film of 7n–9n (after 10 successive scans) measured in: (A) CH2Cl2/[n-Bu4N][PF6] (Γ = 1.20 × 10−9 mol Fe cm−2) and (B) CH3CN/[n-Bu4N][PF6] (1.10 × 10−9 mol Fe cm−2). Inset (top): plot of the peak currents versus the sweep rate for the CV shown in (A). | ||
As expected, the extent of multiferrocenyl-dendritic molecule adsorption, Γ, is strongly dependent on the number of scans performed (see Fig. S42 and S44‡). The surface coverages, Γ (mol cm−2), of the ferrocenyl sites can be estimated from the charge, Q, under the ferrocene voltammetric current peaks, in the ferrocenyl dendritic molecule-free electrolyte solution, using the following relationship: Γ = Q/nFA, where Q is the integrated charge of the redox peak, n is the number of electrons involved in the redox reaction, F is the Faraday constant, and A is the real surface area of the electrode. In particular, the surface coverage of the electroactive ferrocenyl sites in the film for the studied example of dendronized polymers 7n–9n shown in Fig. 9A is 1.20 × 10−9 mol Fe per cm2, and corresponds to a modified electrode prepared by cyclically scanning the potential (between +0.0 and +1.3 V vs. SCE), in degassed CH2Cl2 solutions of the corresponding dendritic molecule, during 10 scans.
For comparison purposes, the surface coverages obtained from the electrochemical responses of the dendrimer 5 film (Γ = 4.19 × 10–11 mol Fe per cm2, see Fig. S42‡) and the octaferrocenyl silsesquioxane 10 film (Γ = 6.05 × 10–12 mol Fe per cm2),29 measured under identical conditions (CH2Cl2/[n-Bu4N][PF6]) and prepared with the same Pt-disk electrode and with identical number of scans (10 scan), are considerably lower. These differences in the values of the surface coverages of the modified electrodes could be related to the attachment mode of the electroactive triferrocenyl groups to the corresponding siloxane backbone. For dendronized polymers 7n–9n the electroactive silicon-bridged triferrocenyl units (–SiFc3) append from the main linear and flexible siloxane polymeric chain (pendant ferrocenyl units). In contrast, in dendrimer 5 and octaferrocenyl silsesquioxane 10 (see Fig. 5), the –SiFc3 units are attached to more rigid spherical and cubic cores. Perhaps, the increased flexibility of the –Si–O–Si– siloxane chain in the polymer backbone affects the redox behavior of dendronized polymers 7n–9n, allowing more effectiveness during the multiferrocenyl-dendritic molecule electrodeposition and/or electroprecipitation process onto the Pt electrode, resulting in modified electrodes with considerably higher values of the surface coverages, Γ (mol cm−2). It therefore appears from these data that dendronized polymers 7n–9n is a better electrode modifier than dendrimer 5 and octasilsesquioxane 10.
On the other hand, the Pt electrode modified with dendronized polymers 7n–9n shows a very different response in a fresh CH3CN/[n-Bu4N][PF6] solution. As can be seen in Fig. 9B, the first anodic peak in this CV is atypically sharp and narrow, something that is not observed in CH2Cl2 solution. This peak can be detected in the first four scans and corresponds to the process mentioned before, known as break-in. This means that the electrodeposited film is activated during the first scans, as it breaks to allow the flow of both the solvent and the [PF6]− anions.34 This behavior is typical of redox-active films into which the diffusion of the solvent molecules and supporting electrolyte ions is slow, as observed for thick redox-active films or films that do not swell easily in the solvent employed for the electrochemical study. The anodic peak current decreases gradually with each successive scan. After the “break-in” period of four scans, the film reaches a steady state, at which stable and reproducible, and well-defined reversible current−potential curves are obtained.
Finally, the electrodeposited films of dendrimer 5 and dendronized polymers 7n–9n are quite robust, persisting after rinsing or soaking the electrode in CH2Cl2 (with or without a dissolved electrolyte). The stability of these electroactive films was further demonstrated by their nearly quantitative persistence after continuous CV scans (for more than 4 h) in ferrocenyl-macromolecule-free CH2Cl2 solutions with [PF6]− as the electrolyte anion.
Conclusions
In summary, we have designed and successfully synthesized, via a convergent approach, three different multiferrocenyl-terminated branched molecules: dendrons, dendrimers, and dendronized polymers, starting from triferrocenylvinylsilane (1) as the key monomer, and using Pt-catalyzed hydrosilylation and alkenylation with Grignard reagents, as consecutive reaction steps. Remarkably, just by varying the reaction conditions between 1 and the tetradirectional siloxane core Si[OSi(CH3)2H]4, the fully functionalized dendrimer 5 (with 12 Fc moieties) or the dendrimer 4 (with 9 Fc units) might be prepared. This last one is a certainly outstanding achievement, because one of its branch-ends is a Si–H group, making it a reactive macromolecule for further convergent growth. Unfortunately, all the attempts to use G1-dendron 3 in this convergent methodology, to prepare larger generation dendrimers and dendronized polymers, were hampered by its high steric hindrance. However, dendrimers with structural defects were detected by MALDI-TOF experiments, suggesting that by adding spacer units to the focal point in the G1-dendron 3 and/or in the –Si–H arms of the siloxane cores, the desired macromolecules may be obtained. Despite the complex nature of these macromolecules, the obtention of good quality crystals has allowed the detailed structure solution of G1-dendron 3 and dendrimer 4 by SCXRD and consequently, a unique insight and a better understanding of the stereochemistry of these remarkable species. In particular, dendrimer 4, peripherally decorated with nine Fc units, has been revealed as the branched multiferrocenyl-containing siloxane with the highest number of ferrocenes fully characterized by SCXRD. Furthermore, solution electrochemical studies have confirmed that the ferrocenyl units in all the obtained dendrons, dendrimers, and dendronized polymers show electronic interaction in threes through a silicon atom. Finally, we have demonstrated the feasibility of modifying Pt-disk electrode surfaces with dendritic molecules such as dendrimer 5, bearing a predetermined number of redox centers, and a mixture of dendronized polymers 7n–9n. A noteworthy feature of platinum electrodes modified with films of such multiferrocenyl dendritic molecules is that they are extremely durable and reproducible.Author contributions
Conceptualization, S. B. and I. C.; methodology, S. B., I. C., and J. P.; X-ray analysis, J. P.; electrochemical studies, S. B. and I. C.; formal analysis, S. B., I. C., and J. P.; investigation and data curation, S. B., I. C., and J. P.; writing, original draft preparation, and editing, S. B., I. C., and J. P.; supervision, S. B.; project administration and funding acquisition, I. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministerio de Ciencia e Innovación (MICINN) of Spain for its financial support through projects PGC2018-094644-B-C21 and PID2021-125207NB-C31.References
- (a) D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719–2728 CrossRef CAS; (b) J. M. J. Fréchet and D. A. Tomalia, Dendrimers and other Dendritic Polymers, Wiley, Chichester, U.K., 2001 CrossRef; (c) G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendrimers and Dendrons: Concepts, Syntheses, Applications, Wiley-VCH, Weinheim, 2001 CrossRef; (d) J.-P. Majoral and A.-M. Caminade, Chem. Rev., 1999, 99, 845–880 CrossRef CAS PubMed; (e) G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendritic Molecules-Concepts, Syntheses, Perspectives, VCH, Weinheim, 1996 CrossRef; (f) G. R. Newkome and C. Shreiner, Chem. Rev., 2010, 110, 6338–6442 CrossRef CAS PubMed; (g) S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819–3868 CrossRef CAS PubMed; (h) D. Messmer, M. Kröger and A. D. Schlüter, Macromolecules, 2018, 51, 5420–5429 CrossRef CAS; (i) M. Malkoch and S. García-Gallego, Introduction to Dendrimers and Other Dendritic Polymers, in Dendrimer Chemistry: Synthetic Approaches Towards Complex Architectures, ed. M. Malkoch and S. García Gallego, Royal Society of Chemistry, 2020, pp. 1–20 RSC; (j) S. E. Seo and C. J. Hawker, Macromolecules, 2020, 53, 3257–3261 CrossRef CAS; (k) A.-M. Caminade, Chem. Soc. Rev., 2016, 45, 5174–5186 RSC.
- (a) D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS; (b) D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Macromolecules, 1986, 19, 2466–2468 CrossRef CAS; (c) G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003–2004 CrossRef CAS; (d) Y. H. Kim and O. W. Webster, Macromolecules, 1992, 25, 5561–5572 CrossRef CAS; (e) L. L. Zhou and J. Roovers, Macromolecules, 1993, 26, 963–968 CrossRef CAS; (f) E. M. M. de Brabander-van den Berg and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1308–1311 CrossRef; (g) C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS; (h) K. L. Wooley, C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4252–4261 CrossRef CAS; (i) C. J. Hawker, R. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4583–4588 CrossRef CAS; (j) N. Launay, A.-M. Caminade, R. Lahana and J. P. Majoral, Angew. Chem., Int. Ed. Engl., 1994, 33, 1589–1592 CrossRef.
- See for example: (a) M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593–4609 RSC; (b) G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC; (c) J. Ruiz, G. Lafuente, S. Marcen, C. Ornelas, S. Lazare, E. Cloutet, J. C. Blais and D. Astruc, J. Am. Chem. Soc., 2003, 125, 7250–7257 CrossRef CAS PubMed; (d) C. Ornelas and M. Weck, Chem. Commun., 2009, 38, 5710–5712 RSC; (e) L. Niu, X. Zhang and S. Ding, Macromolecules, 2022, 55, 3469–3475 CrossRef CAS; (f) Z. Geng, J. J. Shin, Y. Xi and C. J. Hawker, J. Polym. Sci., 2021, 59, 963–1042 CrossRef CAS; (g) S. García-Gallego, O. C. J. Andrén and M. Malkoch, J. Am. Chem. Soc., 2020, 142, 1501–1509 CrossRef PubMed; (h) S. P. Amaral, J. Correa and E. Fernandez-Megia, Green Chem., 2022, 24, 4897–4901 RSC.
- (a) K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS PubMed; (b) C. Rissing and D. Y. Son, Organometallics, 2009, 28, 3167–3172 CrossRef CAS; (c) A. B. Lowe and C. N. Bowman, Thiol-X Chemistries in Polymer and Materials Science, RSC Publishing, Cambridge, UK, 2013 RSC; (d) A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC; (e) M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS; (f) E. Fuentes-Paniagua, C. E. Peña-González, M. Galán, R. Gómez, F. J. de la Mata and J. Sánchez-Nieves, Organometallics, 2013, 32, 1789–1796 CrossRef CAS; (g) M. Galán, E. Fuentes-Paniagua, F. J. de la Mata and R. Gómez, Organometallics, 2014, 33, 3977–3989 CrossRef; (h) E. Fuentes-Paniagua, J. M. Hernandez-Ros, M. Sanchez-Milla, M. A. Camero, M. Maly, J. Perez-Serrano, J. L. Copa-Patino, J. Sanchez-Nieves, J. Soliveri, R. Gómez and F. J. de la Mata, RSC Adv., 2014, 4, 1256–1265 RSC; (i) E. Arnáiz, E. Vacas-Córdoba, M. Galán, M. Pion, R. Gómez, M. A. Muñoz-Fernández and F. J. de la Mata, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1099–1112 CrossRef; (j) M. I. Montañez, L. M. Campos, P. Antoni, Y. Hed, M. V. Walter, B. T. Krull, A. Khan, A. Hult, C. J. Hawker and M. Malkoch, Macromolecules, 2010, 43, 6004–6013 CrossRef; (k) A. B. Cook, R. Barbey, J. A. Burns and S. Perrier, Macromolecules, 2016, 49, 1296–1304 CrossRef CAS; (l) P. Antoni, M. J. Robb, L. Campos, M. Montanez, A. Hult, E. Malmström, M. Malkoch and C. J. Hawker, Macromolecules, 2010, 43, 6625–6631 CrossRef CAS.
- (a) Z. Zhang, S. Feng and J. Zhang, Rapid Commun., 2016, 37, 318–322 CrossRef CAS PubMed; (b) D. A. Lokteva, Y. N. Kononevich, M. N. Temnikov, P. A. Nezhnyy, E. E. Kim, D. A. Khanin, G. G. Nikiforova and A. M. Muzafarov, Polymer, 2022, 256, 25–203 CrossRef.
- S. Guerra, T. L. A. Nguyen, J. Furrer, J.-F. Nierengarten, J. Barberá and R. Deschenaux, Macromolecules, 2016, 49, 3222–3231 CrossRef CAS.
- See for example: (a) A.-M. Caminade, C. O. Turrin, R. Laurent, A. Ouali and B. Delavaux-Nicot, Dendrimers: Towards Catalytic, Material and Biomedical Uses, Wiley, Chichester, UK, 2011 CrossRef; (b) D. Astruc, Nat. Chem., 2012, 4, 255–267 CrossRef CAS PubMed; (c) D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed; (d) A. B. Cook and S. Perrier, Adv. Funct. Mater., 2020, 30, 1901001 CrossRef CAS.
- (a) I. Cuadrado, M. Morán, C. M. Casado, B. Alonso and J. Losada, Coord. Chem. Rev., 1999, 193–195, 395–445 CrossRef CAS; (b) P. A. Chase, R. J. M. Klein and G. van Koten, J. Organomet. Chem., 2004, 689, 4016–4054 CrossRef CAS; (c) D. Astruc and J. Ruiz, J. Inorg. Organomet. Polym., 2015, 25, 2–11 CrossRef CAS; (d) S.-H. Hwang, C. D. Shreiner, C. N. Moorefield and G. R. Newkome, New J. Chem., 2007, 31, 1192–1217 RSC; (e) W. Wang, C. Ornelas, A. K. Diallo, C. Deraedt, Y. Wang, F. Lu, H. Gu and D. Astruc, Polymer, 2022, 246, 124714 CrossRef CAS; (f) N. Sanz del Olmo, R. Carloni, P. Ortega, S. García-Gallego and F. J. de la Mata, Adv. Organomet. Chem., 2020, 74, 1–52 CrossRef CAS; (g) M. Maroto-Díaz, B. T. Elie, P. Gómez-Sal, J. Pérez-Serrano, R. Gómez, M. Contel and F. J. de la Mata, Dalton Trans., 2016, 45, 7049–7066 RSC; (h) B. J. Ravoo, Dalton Trans., 2008, 1533–1537 RSC.
- (a) A. Astruc, New J. Chem., 2011, 35, 764–772 RSC; (b) C.-O. Turrin, J. Chiffre, D. de Montauzon, J.-C. Daran, A.-M. Caminade, E. Manoury, G. Balavoine and J.-P. Majoral, Macromolecules, 2000, 33, 7328–7336 CrossRef CAS; (c) C.-O. Turrin, E. Manoury and A.-M. Caminade, Molecules, 2020, 25, 447–458 CrossRef CAS PubMed.
- (a) X. Liu, A. Rapakousiou, C. Deraedt, R. Ciganda, Y. Wang, J. Ruiz, H. Gu and D. Astruc, Chem. Commun., 2020, 56, 11374–11385 RSC; (b) R. Pietschnig, Chem. Soc. Rev., 2016, 45, 5216–5231 RSC.
- (a) L. Xu, Y. X. Wang, L. J. Chen and H. B. Yang, Chem. Soc. Rev., 2015, 44, 2148–2167 RSC; (b) S. A. Sheppard, T. L. R. Bennett and N. J. Long, Eur. J. Inorg. Chem., 2022, e202200055 CAS.
- (a) P. Štěpnička, Dalton Trans., 2022, 51, 8085–8102 RSC; (b) D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29 CrossRef CAS; (c) P. Štěpnička, Eur. J. Inorg. Chem., 2017, 215–216 CrossRef; (d) K. Heinze and H. Lang, Organometallics, 2013, 32, 5623–5625 CrossRef CAS.
- (a) R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC; (b) M. Kumar, A. J. Metta-Magana and K. H. Pannell, Organometallics, 2008, 27, 6457–6463 CrossRef CAS.
- (a) P. R. Dvornic and M. J. Owen, Silicon-Containing Dendritic Polymers, Vol. 2, Springer, 2009 CrossRef; (b) H. Frey and C. Schlenk, Silicon-Based Dendrimers, in Dendrimers II, ed. F. Vögtle, Topics in Current Chemistry, Springer, Berlin, Heidelberg, 2000, vol. 210, pp. 69–129 Search PubMed; (c) T. Lozano-Cruz, P. Ortega, B. Batanero, J. L. Copa-Patiño, J. Soliveri, F. J. de la Mata and R. Gómez, Dalton Trans., 2015, 44, 19294–19304 RSC.
- See for example: (a) P. Ortega, J. Sánchez-Nieves, J. Cano, R. Gómez and F. J. de la Mata, Poly(carbosilane) Dendrimers and Other Silicon-containing Dendrimers, in Dendrimer Chemistry: Synthetic Approaches Towards Complex Architectures, ed. M. Malkoch and S. García-Gallego, Royal Society of Chemistry, 2020, pp. 114–145 Search PubMed; (b) B. Rasines, J. Sánchez-Nieves, M. Maiolo, M. Maly, L. Chonco, J. L. Jiménez, M. A. Muñoz-Fernández, F. J. de la Mata and R. Gómez, Dalton Trans., 2012, 41, 12733–12748 RSC; (c) E. Fuentes-Paniagua, J. Sánchez-Nieves, J. M. Hernández-Ros, A. Fernández-Ezequiel, J. Soliveri, J. L. Copa-Patiño, R. Gómez and F. J. de la Mata, RSC Adv., 2016, 6, 7022–7033 RSC; (d) N. Sanz del Olmo, J. C. García, R. Gómez, F. J. de la Mata and P. Ortega, RSC Adv., 2022, 12, 10280–10288 RSC; (e) S. A. Milenin, E. V. Selezneva, P. A. Tikhonov, V. G. Vasil'ev, A. I. Buzin, N. K. Balabaev, A. O. Kurbatov, M. V. Petoukhov, E. V. Shtykova, L. A. Feigin, E. A. Tatarinova, E. Y. Kramarenko, S. N. Chvalun and A. M. Muzafarov, Polymers, 2021, 13, 606 CrossRef CAS PubMed; (f) J. C. Furgal, T. Goodson III and R. M. Laine, Dalton Trans., 2016, 45, 1025–1039 RSC; (g) J. Guan, J. J. R. Arias, K. Tomobe, R. Ansari, M. de F. V. Marques, A. Rebane, S. Mahbub, J. C. Furgal, N. Yodsin, S. Jungsuttiwong, D. Hashemi, J. Kieffer and R. M. Laine, ACS Appl. Polym. Mater., 2020, 2, 3894–3907 CrossRef CAS; (h) J. Guan, Z. Zhang and R. M. Laine, Macromolecules, 2022, 55, 5403–5411 CrossRef CAS; (i) R. M. Laine, Chem. Commun., 2022, 58, 10596–10618 RSC; (j) N. Xu, E. J. Stark, P. I. Carver, P. Sharps, J. Hu and C. Hartmann-Thompson, J. Appl. Polym. Sci., 2013, 130, 3849–3861 CAS.
- (a) I. Cuadrado, Organometallic Silicon-Containing Dendrimers and Their Electrochemical Applications, in Silicon-Containing Dendritic Polymers, ed. P. R. Dvornic and M. J. Owen, Springer, Berlin, 2009, pp. 141–196 Search PubMed; (b) S. Bruña, A. M. González-Vadillo, D. Nieto, C. J. Pastor and I. Cuadrado, Macromolecules, 2012, 45, 781–793 CrossRef; (c) I. Cuadrado, C. M. Casado, B. Alonso, M. Morán, J. Losada and V. Belsky, J. Am. Chem. Soc., 1997, 119, 7613–7614 CrossRef CAS; (d) B. Alonso, I. Cuadrado, M. Morán and J. Losada, J. Chem. Soc., Chem. Commun., 1994, 2575–2576 RSC.
- (a) A. D. Schlüter, A Covalent Chemistry Approach to Giant Macromolecules with Cylindrical Shape and an Engineerable Interior and Surface, in Functional Molecular Nanostructures. Topics in Current Chemistry, ed. A. D. Schlüter, Springer, Berlin, 2005, vol. 245, pp. 151–191 CrossRef; (b) R. Freudenberger, W. Claussen, A.-D. Schlüter and H. Wallmeier, Polymer, 1994, 35, 4496–4501 CrossRef CAS; (c) H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325–384 CrossRef CAS.
- (a) X. Liu, W. Lin, D. Astruc and H. Gu, Prog. Polym. Sci., 2019, 96, 43–105 CrossRef CAS; (b) G. I. Dzhardimalievaa and I. E. Uflyand, Dalton Trans., 2017, 46, 10139–10176 RSC.
- (a) J. Morgan, T. Chen, R. Hayes, T. Dickie, T. Urlich and M. A. Brook, Polym. Chem., 2017, 8, 2743–2746 RSC; (b) J. B. Grande, T. Urlich, T. Dickie and M. A. Brook, Polym. Chem., 2014, 5, 6728–6739 RSC; (c) M. Liao, Y. Chen and M. A. Brook, Polymers, 2021, 13, 859 CrossRef CAS PubMed.
- (a) M. Zamora, S. Bruña, B. Alonso and I. Cuadrado, Macromolecules, 2011, 44, 7994–8007 CrossRef CAS; (b) Y. Wang, D. Astruc and A. S. Abd-El-Aziz, Chem. Soc. Rev., 2019, 48, 558–636 RSC.
- S. Bruña, A. M. González-Vadillo, M. Ferrández, J. Perles, M. M. Montero-Campillo, O. Mó and I. Cuadrado, Dalton Trans., 2017, 46, 11584–11597 RSC.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Version 2.03, Bruker AXS Inc., Madison, WI, USA, 2001 Search PubMed.
- G. M. Sheldrick, SAINT+NT, Version 6.04, SAXS Area-Detector Integration Program, Bruker Analytical X-ray Instruments, Madison, WI, USA, 1997 Search PubMed.
- Bruker, AXS SHELXTL, Version 6.10, Structure Determination Package, Bruker Analytical X-ray Instruments, Madison,WI, USA, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- S. Bruña, A. M. González-Vadillo, D. Nieto, C. J. Pastor and I. Cuadrado, Organometallics, 2010, 29, 2796–2807 CrossRef.
- S. Bruña, D. Nieto, A. M. González-Vadillo, J. Perles and I. Cuadrado, Organometallics, 2012, 31, 3248–3258 CrossRef.
- B. Marciniec, Hydrosilylation. A Comprehensive Review on Recent Advances. Advances in Silicon Science, Springer Science, 2009, Vol. 1 Search PubMed.
- A.-M. Caminade, R. Laurent, B. Delavaux-Nicota and J.-P. Majoral, New J. Chem., 2012, 36, 217–226 RSC.
- M. W. F. Nielen, Mass Spectrom. Rev., 1999, 18, 309–344 CrossRef CAS.
- (a) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed; (b) A. J. Bard, J. Chem. Educ., 1983, 60, 302–304 CrossRef CAS; (c) P. Zanello, C. Nervi and F. Fabrizi de Biani, Inorganic Electrochemistry. Theory, Practice and Application, Royal Society of Chemistry, Cambridge, 2013 Search PubMed.
- W. E. Geiger and F. Barrière, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS PubMed.
- R. W. Murray, Techniques of Chemistry: Molecular Design of Electrode Surfaces, Wiley, Chichester, 1992 Search PubMed.
- H. D. Abruña, Coord. Chem. Rev., 1988, 86, 135–189 CrossRef.
Footnotes |
| † Dedicated to Prof. David Tudela, an outstanding inorganic chemist and exceptional colleague, on the occasion of his retirement from Universidad Autónoma de Madrid. |
| ‡ Electronic supplementary information (ESI) available: Supplementary figures referenced in the text; additional synthesis, and spectroscopic and electrochemical data. CCDC 2105207 and 2105208. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03983d |
| This journal is © The Royal Society of Chemistry 2023 |