
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic and aliphatic hydrocarbon hydroxylation via a formally NiIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O oxidant†
O oxidant†
Philipp
Heim
 a,
Robert
Gericke‡
a,
Giuseppe
Spedalotto
a,
Marta
Lovisari
a,
Erik R.
Farquhar
b and
Aidan R.
McDonald
*a
a,
Robert
Gericke‡
a,
Giuseppe
Spedalotto
a,
Marta
Lovisari
a,
Erik R.
Farquhar
b and
Aidan R.
McDonald
*a
aSchool of Chemistry and CRANN/AMBER Nanoscience Institute, Trinity College Dublin, The University of Dublin, College Green, Dublin 2, Ireland. E-mail: aidan.mcdonald@tcd.ie
bCenter for Synchrotron Biosciences, National Synchrotron Light Source II, Brookhaven, National Laboratory Case Western Reserve University, Upton, NY 11973, USA
First published on 30th January 2023
Abstract
The reaction of (NMe4)2[NiII(LPh)(OAc)] (1[OAc], LPh = 2,2′,2′′-nitrilo-tris-(N-phenylacetamide); OAc = acetate) with 3-chloroperoxybenzoic acid (m-CPBA) resulted in the formation of a self-hydroxylated NiIII-phenolate complex, 2, where one of the phenyl groups of LPh underwent hydroxylation. 2 was characterised by UV-Vis, EPR, and XAS spectroscopies and ESI-MS. 2 decayed to yield a previously characterised NiII-phenolate complex, 3. We postulate that self-hydroxylation was mediated by a formally NiIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxidant, formed from the reaction of 1[OAc] with m-CPBA, which undergoes electrophilic aromatic substitution to yield 2. This is supported by an analysis of the kinetic and thermodynamic properties of the reaction of 1[OAc] with m-CPBA. Addition of exogenous hydrocarbon substrates intercepted the self-hydroxylation process, producing hydroxylated products, providing further support for the formally NiIVO entity. This study demonstrates that the reaction between NiII salts and m-CPBA can lead to potent metal-based oxidants, in contrast to recent studies demonstrating carboxyl radical is a radical free-chain reaction initiator in NiII/m-CPBA hydrocarbon oxidation catalysis.
O oxidant, formed from the reaction of 1[OAc] with m-CPBA, which undergoes electrophilic aromatic substitution to yield 2. This is supported by an analysis of the kinetic and thermodynamic properties of the reaction of 1[OAc] with m-CPBA. Addition of exogenous hydrocarbon substrates intercepted the self-hydroxylation process, producing hydroxylated products, providing further support for the formally NiIVO entity. This study demonstrates that the reaction between NiII salts and m-CPBA can lead to potent metal-based oxidants, in contrast to recent studies demonstrating carboxyl radical is a radical free-chain reaction initiator in NiII/m-CPBA hydrocarbon oxidation catalysis.
Introduction
A direct route to oxidative arene hydroxylation remains a challenge.1 Natural systems provide excellent inspiration, utilising O2 and a metal to insert an oxygen atom into aromatic amino acids. In mammalian cells, pterin dependant amino acid hydroxylases including phenylalanine hydroxylase, tyrosine hydroxylase, and tryptophan hydroxylase, employ a non-heme FeIVO for aromatic oxidation via electrophilic attack.2–5 Synthetic models inspired by these enzymes have emulated this chemistry, although with limited insight into the active oxidant and intermediates in arene hydroxylation.6–14 Mononuclear non-heme Fe systems can undergo self-hydroxylation of a pendant aryl ring.15–17 For Cu systems, intramolecular aryl oxidation occurs mostly via dinuclear oxidants.6,10,13,18,19
Homogeneous NiII complexes when combined with peracids have been shown to be highly effective catalysts, capable of saturated hydrocarbon hydroxylation20–26 and olefin epoxidation.27,28 Mechanistic analysis of these reactions suggests terminal NiIII–oxyl (NiIII–O˙) or NiIVO adducts are the oxidising moiety.22–24,29–32 Such catalytic systems were also found capable of the hydroxylation of benzene.33 Bis-μ-oxo-Ni2III complexes have been implicated in such arene hydroxylation reactivity.34–36 However, mononuclear terminal Ni–O˙ or NiO complexes remain elusive, although a plethora of Ni–OX (OX = OCl, O2CCH3, OCO2H, ONO2) have recently appeared.37–45 The oxo-wall axiom, where the occupation of anti-bonding orbitals in a MO molecular orbital is maximised when the d-electron count >4 (NiIV = d6, NiIII = d7), provides an explanation for the lack of tetragonal NiO complexes.46,47 There remains, thus, considerable lack of clarity as to the identity of Ni–oxygen adducts in hydroxylation catalysis. For example, Hartwig and co-workers recently suggested that NiII/peracid mediated hydroxylation did not involve NiO.48 They postulated, with reasonable experimental support, that an organic free-radical chain mechanism facilitated by carboxyl radical led to essentially ‘Ni-free’ hydroxylation. Herein, we probe the reaction of a NiII complex with 3-chloroperoxybenzoic acid (m-CPBA), showing that a formally NiIVO species forms and is capable of arene and alkane hydroxylation, without the involvement of carboxyl radical.
Results and discussion
1[OAc] (Scheme 1) was prepared according to a previously reported method.49 Addition of m-CPBA (1.0 equiv., CH2Cl2) to 1[OAc] (0.50 mM, CH3CN) at −40 °C led to the formation of new electronic absorption features at λ = 390, 675 and 950 nm, attributed to the formation of a new species (defined as 2, Fig. 1). A colour change from a pale green to a dark green colour was noted and a maximum yield was achieved within 60 s. The new absorption spectrum was markedly different to that obtained when 1[OAc] was reacted with aliphatic peracids,49 indicating a different species had formed. Exactly one equivalent of m-CPBA was required to yield the maximum yield of 2 (Fig. S1†). 2 displayed a half-life (t1/2) of 6000 s at −40 °C (Fig. S2†) and we observed a rapid disappearance of its electronic absorption features upon warming to 25 °C, implicating 2 was a reactive species that could only be stabilised at low temperature. The product of the thermal decay of 2 was the previously characterised square-planar NiII-phenolate complex 3,49 where one of the phenyl groups of the pendant phenylcarboxamidate ligands had undergone oxygen atom insertion at the ortho position (Scheme 1). This led us to postulate that 2 was a precursor to NiII-phenolate 3, and was either a terminal NiO entity or a NiIII-phenolate adduct.
 | ||
| Scheme 1 Preparation of 2 and 3 from 1[OAc]. | ||
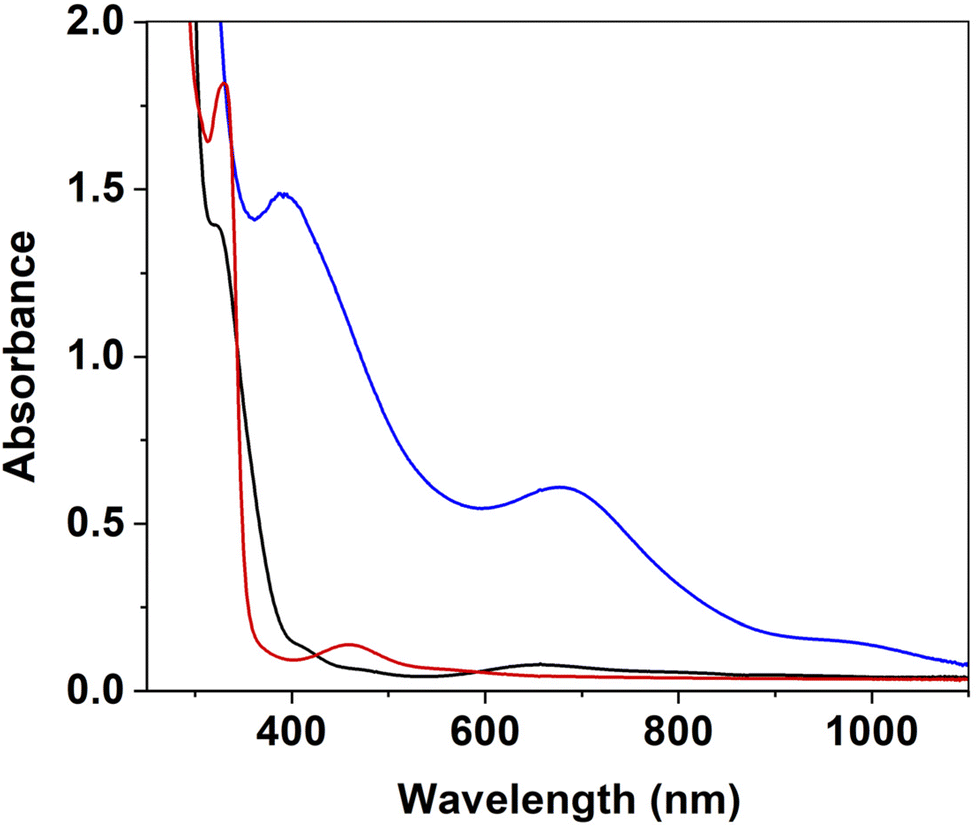 | ||
| Fig. 1 Electronic absorption spectra of 1[OAc] (0.50 mM, black trace), 2 (blue trace) formed from the reaction of 1[OAc] (0.50 mM, CH3CN) with m-CPBA (1.0 equiv.) at −40 °C, and 3 (red trace) from the thermal decay of 2. | ||
ESI-MS of 2 displayed a signal at m/z = 486.0787 with the appropriate isotopic distribution pattern for an ion containing Ni. This mass peak is 15 mass units greater than the parent [(Ni(LPh)]− ion and was assigned to the [(Ni(LPh))–(H) + (O)]− ion (expected m/z = 486.0838, Fig. S3†). This signal shifted by 2 atomic mass units (a.m.u.) when 2 was prepared with the 18O-m-CPBA isotopomer (Fig. S4,† an optimised method for the preparation of 18O-m-CPBA is provided in the ESI†). We concluded that 2 had been oxidised, had incorporated a single oxygen atom, and that the incorporated oxygen atom was derived from m-CPBA. The incorporated O-atom could indicate the formation of a NiO entity or a ligand-hydroxylated product. The loss of an H-atom would suggest that the ligand had been hydroxylated and the resulting alcohol had lost its proton, ruling out a NiO entity. In the reaction between 1[OAc] and aliphatic peracids,49 we observed a product with a mass peak at m/z = 487.10, consistent with that product containing a Ni–oxide core. 2, with a net one hydrogen atom difference, appears to be more likely a ligand-oxidised product, where the loss of a hydrogen atom may indicate ligand oxidation.
The X-band electron paramagnetic resonance (EPR) spectrum of 2 displayed an axial signal (g⊥ = 2.20, g∥ = 2.01, gav = 2.14, Fig. 2) with a three-line hyperfine splitting in g∥. Double integration of the EPR envelope showed a yield of 75 ± 20% compared to a TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl) radical standard analysed under the same conditions. The average g-value (gav = 2.14) was consistent with a d7, S = ½ NiIII species.50,51 The axial signal would suggest that the NiIII ion was located in either a square planar, square pyramidal, or another tetragonally distorted coordination environment. The observed hyperfine coupling value (A = 65 MHz) was consistent with typical values observed for coupling to 14N (I = 1) nuclei. We believe that the observed coupling stems from an axial coordinating N-atom with an unpaired electron in the dz2 Ni-type-orbital.50,51 In contrast, the product of the reaction between 1[OAc] and aliphatic peracids yielded an isotropic EPR signal with no 14N-hyperfine,49 consistent with a NiIII-(hydr)oxide in a highly symmetric (octahedral or trigonal bipyramidal) ligand field. Taken together, these results indicate that the oxidation of 1[OAc] with m-CPBA resulted in the formation of a NiIII entity that had undergone a dramatic symmetry change from a pseudo-trigonal bipyramidal geometry in 1[OAc] into a square planar, square pyramidal, or other tetragonally distorted structure in 2 with 14N-hyperfine in the axial ligand field.
 | ||
| Fig. 2 Black trace: X-band EPR spectrum of 2 (10.0 mM) in a frozen CH3CN solution, collected at 77 K, 2.01 mW microwave power, and 0.3 mT modulation amplitude. Blue trace: simulation of experimental data for 2. | ||
Ni-K-edge XANES (X-ray absorption near edge structure) analysis was performed on 1[OAc] and 2 (Fig. 3, S5 and Table S1†). Analysis of a sample of 1[OAc] showed a K-edge energy of 8343.4 eV,49 with a distinct pre-edge feature at 8332.7 eV corresponding to an electronic-dipole forbidden 1s-to-3d transition, which gains intensity in non-centrosymmetric geometries due to p–d mixing.52,53 The Ni-K-edge for 2 exhibited an edge energy of 8344.4 eV, with a +1.0 eV blue shift compared to 1[OAc] (Table 1). Since the edge energy provides a measure of the relative electron density and effective charge on the Ni atom, its variation can be indirectly correlated to a change in the oxidation state and is therefore in line with the oxidation of NiII to NiIII proposed for 2.52 The pre-edge region showed a 1s-to-3d transition at 8333.1 eV (+0.4 eV compared to 1[OAc]) with a peak area of 5.2 × 10−2 eV. The shift and the decrease of area observed reflected a clear change in the ligand field (and possibly oxidation state) between 1[OAc] and 2. The observations suggest a geometry change from a distorted octahedral coordination (in which the forbidden 1s-to-3d transition gains intensity form p–d orbital mixing) in 1[OAc] to a square pyramidal environment in 2 (in which, due to the higher centrosymmetric character, the intensity of the forbidden 1s-to-3d transition is highly reduced). For 2, an additional peak, assigned to a 1s-to-4pz transition with shakedown contributions, was observed at 8338.3 eV, suggesting the presence of a square-pyramidal geometry. These observations are diagnostic of a square pyramidal NiIII complex and are consistent with the proposed structure for 2 and our EPR results.
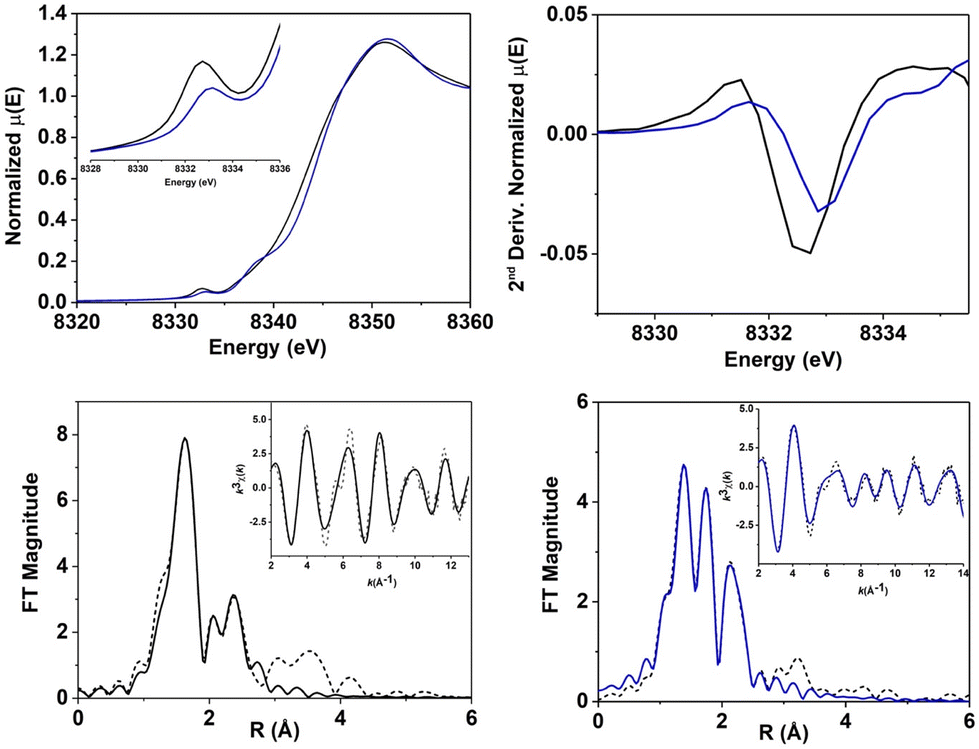 | ||
| Fig. 3 Top left: Ni K-edge XANES spectrum of 1[OAc] (black trace) and 2 (blue trace). Inset: detailed pre-edge region of XANES spectrum. Top right: second derivative of the pre-edge region. Bottom: best fit to k3-weighted EXAFS of 1[OAc] (left) and 2 (right), reported in R-space and k-space (inset). Experimental data are shown as dashed lines, best fits are shown as solid lines. | ||
| μ eff (B.M.) | λ max (nm), ε (mol L−1 cm−1) | g (gav.) | Pre-edge energy (eV) | Edge energy (eV) | Ni–O (Å) | |
|---|---|---|---|---|---|---|
| a Determined by XRD. b Determined by EXAFS. | ||||||
| 1[OAc] | 3.09 | 320 (2800), 405 (170), 657 (100) | — | 8332.7 | 8343.4 | 2.091(3)a 2.324(2)a |
| 2 | — | 390 (3000), 675 (1300) | 2.20, 2.01 (2.14) | 8333.1 | 8344.4 | 1.89b |
[NiIII(O–H⋯OAc)(LPh)]2−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49 49 |
1.47 | 450 (8000), 650 (2300) | 2.13 | 8333.3 | 8344.2 | 1.94b |
| [NiIII(m-CBA)(LPh)]− | — | 455 (4600), 675 (1200) | 2.22, 2.13, 2.07 (2.14) | — | — | — |
Evaluation and analysis of the extended X-ray absorption fine structure (EXAFS) for 1[OAc]49 and 2 was performed (Fig. 3, Tables S2–S5†). EXAFS data for 1[OAc] were previously best fit with a first shell composed of 5 or 6 N/O scatters at 2.05 Å (5 N/O: deviation <0.01 Å; 6 N/O: deviation <0.05 Å). Fitting of EXAFS data for 2 showed a first coordination shell composed of 5 N/O scatterers, divided in two sub-shells: 3 N/O scatterers at 1.90 Å and 2 N/O scatterers at 2.06 Å. A fit including 4 short scatterers and 1 longer scatterer, or indeed any other combination of 5/6 N/O scatterers provided poorer fits. This is consistent with the coordination number of 5 at the Ni center in a square pyramidal geometry, as indicated by XANES and EPR. We postulated that 2 was likely a NiIII-phenolate adduct, based on our ESI-MS results and the observation that the NiII-phenolate complex 3, formed from 2. The first sub-shell was consistent with the proposed structure, matching the Ni–O(Ph) distance (1.89 Å) observed in the crystallographic data obtained for 3 (vide infra).49 The fit was completed by a second shell composed of 2 C at 2.56 Å, 3 C at 2.71 Å and 3 C at 2.84 Å. All Ni/ligand bond lengths were consistent with single bonds and there was no indication of a terminal NiO entity, which would be expected to display a shorter Ni–O distance (∼1.60–1.70 Å). Overall, XAS analysis suggested that 2 was a 5-coordinate complex, likely in a square-pyramidal environment, consistent with its assignment as a NiIII-phenolate adduct (Fig. 4).
 | ||
| Fig. 4 Optimized structure with the axis system of the g-tensor orientation (left, hydrogen atoms omitted for clarity) and ChemDraw structure (right) of the mono-anions of 2. | ||
Quantum chemical calculations on 1[OAc]49 and 2 were performed at the density functional theory (DFT; PBE0(B3DJ) – ZORA-def2-TZVPP) level of theory (see ESI for details, Fig. S6–S8 and Tables S6–S8†). The geometry optimised structure of 1[OAc] in the S = 1 state is in good agreement with the molecular structure determined by single-crystal X-ray diffraction analysis (deviation <0.03 Å) and EXAFS (deviation <0.11 Å). This demonstrates that the applied method would be effective for a prediction of the geometry of 2 (S = ½). Our calculations demonstrate that a square pyramidal NiIII-phenolate complex represents an excellent fit with our spectroscopic analyses for 2 (Fig. 4). According to our calculations, the NiIII centre in 2 exhibits a square pyramidal coordination sphere with an average NiN/O distance of 1.87 Å in the basal plane and a Ni–N distance of 2.02 Å in the apical axis. The EXAFS fit of 2 was best described with a 3 + 2 N/O fit. The EXAFS bond lengths are in good agreement with the computational model (deviation <0.06 Å). The fit of the second subshell composed of 2 C at 2.56 Å (deviation −0.09 Å), 3 C at 2.71 Å (deviation 0.01 Å) and 3 C at 2.84 Å (deviation 0.08 Å) is consistent with a highly unsymmetrically coordinating carboxamidate ligand. In order to assess the possibility of a 6-coordinate NiIII core in 2, a CH3CN molecule was introduced in the nickel coordination sphere of the optimised structure for 2. However, geometry optimization of this solvent adduct led to loss of the CH3CN molecule and reformation of 2. The proposed structure is also consistent with the observed axial EPR signal for 2, with 14N-hyperfine observed in gz consistent with an axial carboxamidate donor (Fig. 4). Calculation of the g-tensor at the DFT level of theory supports the axial symmetry around NiIII (calculated: g⊥ = 2.16, g∥ = 2.02; gav = 2.11; experimental: g⊥ = 2.20, g∥ = 2.01, gav = 2.14, Fig. 2) and the calculated 14N hyperfine coupling constant of A = 66.8 MHz (experimental: A = 65 MHz) in the apical direction is in accordance with the experimental EPR findings. The DFT results (structure and g-tensor) suggest that the NiIII ion in 2 remains 5-coordinate in solution and is in agreement with the EPR and XAS results.
In order to assess further the preparation of 2 and its properties, we reacted 1[OAc] with two alternative oxidants. The reaction between 1[OAc] and bis(3-chlorobenzoyl)peroxide (2.0 equiv.) at 25 °C yielded a new complex with an electronic absorption feature at λ = 455 nm within 2000 seconds, which was different to that obtained for 2 (Fig. S9–S11†). The reaction was performed at 25 °C, because the same reaction at −40 °C was too slow to produce meaningful results. ESI-MS of this mixture displayed a peak at m/z = 626.0947, assigned to a NiIII–3-chlorobenzoate (m-CBA) adduct [Ni(m-CBA)(LPh)]− (expected m/z = 626.0867). The signal corresponding to [(Ni(LPh))–(H) + (O)]− observed for 2 was not observed. In contrast to m-CPBA, bis(3-chlorobenzoyl)peroxide is known to undergo homolytic cleavage of the O–O bond,48 generating a benzyloxyl radical that can react with 1[OAc] and presumably lead to the formation of [NiIII(m-CBA)(LPh)]−. Additionally, an X-Band EPR spectrum of this species (Fig. S10†) displayed a more isotropic character, with no N-hyperfine, when compared to 2. The overlay of these spectra revealed a different speciation. In fact, the EPR spectrum of [NiIII(m-CBA)(LPh)]− displayed similarities with that obtained for the NiIII-(hydr)oxide complex (isotropic signal, no 14N hyperfine),49 consistent with a highly symmetrical NiIII environment. Taken together, the two contrasting electronic and spectral properties of [NiIII(m-CBA)(LPh)]− and 2 suggests that the reaction of 1[OAc] with m-CPBA does not result in the generation of a NiIII benzoate adduct.
Importantly, we were also able to generate 2 using peroxybenzoic acid (PBA) as an alternative to m-CPBA. The obtained UV-Vis and EPR features assigned to 2 prepared with PBA matched those of 2 that had been generated using m-CPBA (Fig. S12 and S13†). The identification of the same product suggests a similar oxidation route suggesting that m-CPBA and PBA act to transfer an oxygen atom to 1[OAc] resulting in the formation of 2.
Peroxybenzoic acids such as m-CPBA or PBA can undergo homolytic or heterolytic O–O bond scission upon reaction with metals (Scheme S1†).54 For the reaction of 1[OAc] with m-CPBA, an O–O bond heterolysis mechanism would result in a formally two-electron oxidised NiIVO species and the corresponding carboxylic acid. Alternatively, O–O bond homolysis would yield a formally NiIIIO species and an arylcarboxyl radical (ArCOO˙). This radical would undergo further decarboxylation (loss of CO2) to form a chlorobenzene radical that may abstract H˙, Cl˙, or HO˙ radicals from the solvent or other species in solution to yield chlorobenzene, dichlorobenzene, or 3-chlorophenol respectively. Accordingly, we investigated the organic decay products from the reaction of m-CPBA with 1[OAc] by gas chromatographic flame ionization detection (GC-FID, Fig. S14†). This showed no indication of chlorobenzene, 1,3-dichlorobenzene, or 3-chlorophenol. However, we were able to identify 3-chlorobenzoic acid in the reaction mixture. 1H NMR revealed that 3-chlorobenzoic acid was formed in ∼90% yield (with respect to starting [1[OAc]] (Fig. S15†)). This result was in line with an O–O bond heterolysis mechanism resulting in the formation of the formally NiIVO entity, that we surmise is a precursor to the NiIII-phenolate complex 2. No evidence for O–O bond homolysis was obtained.
In the reaction between 1[OAc] and m-CPBA, we found that the addition of aliphatic hydrocarbon substrates resulted in a decrease in yield of 2 (thus interception of the putative NiIVO oxidant). The obtained yield of 2 appeared to be dependent on the magnitude of the C–H bond dissociation energy of the substrate (Fig. S16†). For example, addition of toluene to 1[OAc], prior to the addition of m-CPBA, resulted in a decrease in the yield of 2 to ∼85% of the original yield of 2 when no hydrocarbon substrate was present. GC-FID analysis revealed the formation of benzaldehyde in the reaction mixture (yield = 10% w.r.t [1[OAc]], Fig. S17†). Similarly, a decreased yield of 2 was observed when tetrahydrofuran (∼70% of original yield of 2), cyclohexene (∼50%), cumene (∼30%) and 1,4-cyclohexadiene (CHD, <25%) were added to the reaction mixture. Interestingly, analysis of the cyclohexene post-reaction mixture products revealed the presence of 1,2-epoxycyclohexane (20% yield) as the major product, with minor products cyclohexene-1-ol (8.0%) and cyclohexene-1-one (5%) (Fig. S18†). The preferential epoxidation of alkene over C–H abstraction of the α-carbon, is a typical outcome for a terminal MO entity,27,55,56 demonstrating that the active oxidant is likely a terminal NiO species. These interception studies and the identification of epoxide products, alongside the observation of arene hydroxylation, support the formation of a formally NiIVO oxidant from the reaction of 1[OAc] and m-CPBA prior to the formation of 2.
The formation of a NiIII-phenolate (2) from a formally NiIVO entity should involve a radical-type aromatic substitution reaction (Scheme 2). An initial attack of the formally NiIVO into the π-system of the pendant arene ligand would result in the formation of a C–O bond and an arene π-radical. This intermediate NiIII-phenoxyl species is postulated to rapidly lose a hydrogen atom regenerating aromaticity in the final product 2. In order to probe this mechanism, we attempted to identify the faith of the hydrogen atom, using the isotopically labelled 1[OAc]-D15.49 A 2H NMR of the post reaction mixture of 1[OAc]-D15 with m-CPBA revealed the presence of a peak at δ = 5.51 ppm that we assigned as CHDCl2 (Fig. S19†). CH2Cl2 is present in the reaction mixture to solubilise m-CPBA. We speculate that a deuterium atom exchange of free D (released in the aromatisation of the phenolate) with CH2Cl2 occurred. Nonetheless, the observation of new 2H resonances would suggest the formation of H/D atom radical species, providing support for the mechanism postulated in Scheme 2, indicating the involvement of a NiIVO adduct in electrophilic aromatic substitution and hydrocarbon oxidation.
 | ||
| Scheme 2 Postulated mechanism of conversion of 1[OAc] to 2. | ||
Varying the concentrations of 1[OAc], while keeping the concentration of m-CPBA and temperature constant, showed little to no difference in the rate of formation of 2 (Fig. S20†). The conversion of NiIVO into 2 should be a unimolecular reaction and therefore the rate of formation of 2 should remain unaffected by changing the concentration of 1[OAc] in the reaction between 1[OAc] and m-CPBA. This is thus indicative of intramolecular ligand oxidation.
We also explored the rate formation of 2 for 1[OAc]-D15. For the reaction of 1[OAc]-D15 with m-CPBA, we observed the same chromophore as observed in the reaction of 1[OAc] and m-CPBA (defined as 2-D14, Fig. S21†). In the 1H NMR of the warmed post reaction mixture we observed the expected methylene peaks that displayed as six inequivalent resonances in the δ = 3.0–5.0 ppm region (Fig. S22†), as well as observing a NH signal at δ = 10.36 ppm, that was previously observed in 3.49 In contrast, none of the aryl CH resonances identified for 3 were present in the post-reaction mixture, consistent with the perdeuteration of the arene. The reaction outcome was thus the same and the rate of formation of the NiIII-phenolate adduct was unchanged whether the proto- or deutero-ligand was employed. The rate of formation of 2-D14 (kobs = 0.059 s−1) was very close to that measured for 2 (0.071 s−1), suggesting no involvement of the aryl H/D-atoms in the rate limiting step for formation of 2. We surmise, supported by the collected evidence, that electrophilic aromatic substitution is rate-limiting. That is, attack of the electrophilic oxo ligand in a formally NiIVO species on the arene ring of one of the pendant arms of the ligand. This is consistent with results obtained elsewhere for analogous Fe and Cu systems.57–62
We probed the activation parameters for the reaction of 1[OAc] with m-CPBA (Fig. S24–S26, Table S9†). The reaction activation enthalpy (ΔH‡) was determined to be 14.5 kcal mol−1. The reaction activation entropy (ΔS‡) was near-zero. This is in agreement with the unimolecular nature of the reaction. Furthermore, analysis of the Eyring plot for 1[OAc]-D15 to 2-D14 showed an almost indistinguishable slope compared to its proto-analogue. By comparing the slopes of the Eyring plot, a KIE (1.02) could be determined, unambiguously demonstrating that arene hydroxylation does not involve rate-limiting H+ or hydrogen atom transfer. The observed activation parameters are in good agreement with literature precedent of arene hydroxylation's involving MO and bis-μ-(M–O–M) complexes (Table 2).60–62 The values are inconsistent with intermolecular (bimolecular) aromatic hydroxylation reactions as observed for FeIV/VO complexes.57–59
O and bis-μ-(M–O–M) mediated aryl hydroxylations
| ΔH‡ (kcal mol−1) | ΔS‡ (cal mol−1 K−1) | Ref. | |
|---|---|---|---|
| xyl-H:N,N‘-(1,3-phenylenebis(methylene))bis(2-(pyridin-2-yl)-N-(2-(pyridin-2-yl)ethyl)ethan-1-amine), H-L-H: 1,3-bis[bis(6-methyl-2-pyridylmethyl)aminomethyl]benzene, N4Py2Ar:1,1-bis(6-(2,6-difluorophenyl)pyridin-2-yl)-N,N-bis(pyridin-2-ylmethyl)methanamine. |
|||
| 1[OAc] | 14.5 ± 0.7 | −1.4 ± 0.1 | — |
| 1[OAc]-D15 | 14.3 ± 0.5 | −2.5 ± 0.1 | — |
| [CuII2(O2)(xyl-H)]2+ | 11.9 ± 0.2 | −8.4 ± 0.5 | 60 |
| [NiIII2(O2)(H-L-H)]2+ | ∼13.9 | ∼ − 15.1 | 61 |
| [FeIV(O)(N4Py2Ar)]2+ | 17.4 ± 0.4 | −12.7 ± 1.4 | 62 |
Having established considerable experimental evidence for a formally NiIVO precursor to 2, formed from O–O heterolysis in the reaction between 1[OAc] and m-CPBA, we performed quantum chemical calculations on the properties of the formally NiIVO unit. Geometry optimisations of [Ni(O)(NTA)]− was performed in three possible spin-states (Fig. 5, S29 and Tables S10–S12†), whereby S = 1 was the lowest in energy (0.00 kcal mol−1) with respect to S = 0 (2.04 kcal mol−1) and S = 2 (6.85 kcal mol−1). Mulliken population analysis of the NiO moiety (S = 1) displayed a charge of 0.67 at Ni and −0.47 at O. Interestingly, the spin was spread over the Ni–O unit (Ni: 0.83, O: 0.95) which points towards a formulation as NiIII–O˙. For comparison, the Mulliken charge (0.61) and spin population (0.79) at NiIII in 2 was slightly reduced even though the same number of ligands are coordinated at the nickel atom. A Wiberg bond order of 1.40 showed that some double bond character between Ni and O was present, whereby the Ni–oxygen adduct was best described as either NiIII–O˙ or NiIVO. We therefore describe the active oxidant as being formally NiIVO, with the understanding that this formalism can also be ascribed to NiIII–O˙.
2 was capable of activating weak C–H bonds at −40 °C, reacting with 1,4-cyclohexadiene (CHD) in CH3CN at −40 °C (Fig. S30–S32†), but could not activate the strong C–H bond of toluene, as the putative NiIVO entity could. Analysis of the CHD post reaction mixture by 1H NMR indicated the formation of benzene. Plotting the change in absorbance at λ = 390 nm versus time and fitting the resulting curve with an exponential decay function, allowed us to determine a pseudo-first order rate constant (kobs). We plotted kobs against a series of substrate concentrations to obtain a linear plot whose slope was used to determine a value for the second order reaction rate constant (k2 = 0.010 M−1 s−1, Fig. S33†). From these reactivity studies, we conclude that the formation of 3 from 2 is likely via the above-mentioned hydrogen atom transfer oxidation by 2 to yield a protonated, 1-electron reduced core (thus 3). 2 displayed k2 values comparable to previous NiIII–OX examples.63–65 The metastable formally NiIVO entity was a superior oxidant, while [NiIII(O–H⋯OAc)(LPh)]49 was also a capable hydrocarbon oxidant.
 | ||
| Fig. 5 Optimized molecular structure of [Ni(O)(NTA)]− (S = 1, left) and electron spin-density plot (right; H atoms omitted for clarity; iso-surface value at 0.005). | ||
In the reactions of NiII catalysts with peroxy acids, the formation of NiIII–OH, NiIII–OOR, NiIII–O˙, and NiIVO entities are all plausible. In the reaction of 1[OAc] with the aryl peracids m-CPBA or PBA, herein, we collated evidence to suggest the formation of a transient formally NiIVO species. In contrast, in the reactions of 1[OAc] with aliphatic peracid (peroxyphenylacetic acid) or NaOCl we have trapped and characterized a masked NiIII=O complex (formally containing a [NiIII(O–H⋯OAc)(LPh)] core).49 Post-reaction mixture analysis showed that the aryl peracids underwent heterolytic O–O bond scission upon reaction with 1[OAc], whereas the aliphatic peracids underwent homolytic O–O bond scission, consistent with the formation of formally NiIVO and NiIIIO products, respectively. This observation was confounding to us, given the similarities in the peracid's properties. There is little difference in the O–O bond dissociation energy when comparing the two sets of peracids.66 The aliphatic peracids have pKa values approximately one pKa unit greater (∼8.2 versus ∼7.5) than those of the aryl peracids. Our tentative hypothesis is that for aryl peracids, O–O bond heterolysis is accelerated by the relatively greater acidity of the H+ catalysing O–O bond scission in the reaction of 1[OAc] with aryl peracids. In contrast, the less acidic protons of the aliphatic peracids appear to play no role, resulting in O–O bond homolysis. Critically, this divergent reactivity shows that while O–O bond homolysis may lead to carboxyl radical formation upon reaction between peracids and NiII, heterolysis does not, but nonetheless yields a potent Ni-based oxidant capable of arene and alkane hydroxylation. Overall, the reactivity of 1[OAc] with peracids demonstrates the nuances associated with NiII/peracid chemistry, showing aryl peracids yield formally NiIVO and carboxylate, while aliphatic peracids yield NiIII–OH and carboxyl radical. Critically, the Ni-based products were more than capable of a variety of oxidative transformations including aliphatic hydrocarbon hydroxylation, arene hydroxylation, and oxygen atom transfer.
Conclusions
The formation of a NiIII-phenolate complex 2 from the reaction of 1[OAc] with m-CPBA was observed. 2 was characterised by a suite of spectroscopic techniques namely UV-Vis, EPR, and XAS spectroscopies and ESI-MS. 2 decayed to yield a previously observed NiII-phenolate complex 3. We postulated that self-hydroxylation was mediated by a formally NiIVO entity and collected a variety of kinetic and reaction product data to support that claim. Critically, addition of exogenous hydrocarbon substrates intercepted the self-hydroxylation process, resulting in hydroxylation of aliphatic substrates and the epoxidation of olefinic hydrocarbons. Overall, the reactivity of 1[OAc] with peracids demonstrates the nuances associated with NiII/peracid chemistry, showing formation of high-valent Ni–oxygen adducts and carboxyl radical that can mediate hydroxylation reactivity.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This publication has emanated from research supported by the European Research Council (ERC-2015-STG-678202). Research in the McDonald lab is supported in part by a research grant from Science Foundation Ireland (SFI/15/RS-URF/3307). Open access funding provided by IReL. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). E.R.F. thanks Dr. Leah Kelly at SSRL for support of remote experiments on beamline 7-3. The authors thank Prof. Robert Barklie for training on and use of an EPR spectrometer.References
- D. A. Alonso, C. Nájera, I. M. Pastor and M. Yus, Chem. – Eur. J., 2010, 16, 5274–5284 CrossRef CAS PubMed.
- T. A. Dix and S. J. Benkovic, Acc. Chem. Res., 1988, 21, 101–107 CrossRef CAS.
- P. F. Fitzpatrick, Annu. Rev. Biochem., 1999, 68, 355–381 CrossRef CAS PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS PubMed.
- K. D. Karlin, J. C. Hayes, Y. Gultneh, R. W. Cruse, J. W. McKown, J. P. Hutchinson and J. Zubieta, J. Am. Chem. Soc., 1984, 106, 2121–2128 CrossRef CAS.
- E. L. Hegg, R. Y. N. Ho and L. Que Jr., J. Am. Chem. Soc., 1999, 121, 1972–1973 CrossRef CAS.
- F. Avenier, L. Dubois and J.-M. Latour, New J. Chem., 2004, 782–784 RSC.
- L. M. Mirica, M. Vance, D. J. Rudd, B. Hedman, K. O. Hodgson, E. I. Solomon and T. D. P. Stack, Science, 2005, 308, 1890–1892 CrossRef CAS PubMed.
- T. Matsumoto, H. Furutachi, M. Kobino, M. Tomii, S. Nagatomo, T. Tosha, T. Osako, S. Fujinami, S. Itoh and T. Kitagawa, J. Am. Chem. Soc., 2006, 128, 3874–3875 CrossRef CAS PubMed.
- W. J. Song, M. S. Seo, S. DeBeer, T. Ohta, R. Song, M.-J. Kang, T. Tosha, T. Kitagawa, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2007, 129, 1268–1277 CrossRef CAS PubMed.
- M. Yamashita, H. Furutachi, T. Tosha, S. Fujinami, W. Saito, Y. Maeda, K. Takahashi, K. Tanaka, T. Kitagawa and M. Suzuki, J. Am. Chem. Soc., 2007, 129, 2–3 CrossRef CAS PubMed.
- I. Garcia-Bosch, X. Ribas and M. Costas, Chem. – Eur. J., 2012, 18, 2113–2122 CrossRef CAS PubMed.
- S. Sahu, M. G. Quesne, C. G. Davies, M. Dürr, I. Ivanović-Burmazović, M. a. Siegler, G. N. L. Jameson, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2014, 8–11 Search PubMed.
- A. Thibon, V. Jollet, C. Ribal, K. Sénéchal-David, L. Billon, A. B. Sorokin and F. Banse, Chem. – Eur. J., 2012, 18, 2715–2724 CrossRef CAS PubMed.
- A. Ansari, A. Kaushik and G. Rajaraman, J. Am. Chem. Soc., 2013, 135, 4235–4249 CrossRef CAS PubMed.
- L. Cheng, H. Wang, H. Cai, J. Zhang, X. Gong and W. Han, Science, 2021, 374, 77–81 CrossRef CAS PubMed.
- V. Mahadevan, M. J. Henson, E. I. Solomon and T. D. P. Stack, J. Am. Chem. Soc., 2000, 122, 10249–10250 CrossRef CAS.
- M. P. Jensen, E. L. Que, X. Shan, E. Rybak-Akimova and L. Que Jr., Dalton Trans., 2006, 3523–3527 RSC.
- W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235–244 CrossRef.
- T. Nagataki, Y. Tachi and S. Itoh, Chem. Commun., 2006, 4016–4018 RSC.
- T. Nagataki, K. Ishii, Y. Tachi and S. Itoh, Dalton Trans., 2007, 21, 1120–1128 RSC.
- T. Nagataki and S. Itoh, Chem. Lett., 2007, 36, 748–749 CrossRef CAS.
- M. Balamurugan, R. Mayilmurugan, E. Suresh and M. Palaniandavar, Dalton Trans., 2011, 40, 9413–9424 RSC.
- S. Hikichi, K. Hanaue, T. Fujimura, H. Okuda, J. Nakazawa, Y. Ohzu, C. Kobayashi and M. Akita, Dalton Trans., 2013, 42, 3346–3356 RSC.
- J. Nakazawa, T. Hori, T. D. P. Stack and S. Hikichi, Chem. – Asian J., 2013, 8, 1191–1199 CrossRef CAS PubMed.
- J. Koola and J. K. Kochi, Inorg. Chem., 1987, 26, 908–916 CrossRef CAS.
- S. Kim, H. Y. Jeong, S. Kim, H. Kim, S. Lee, J. Cho, C. Kim and D. Lee, Chem. – Eur. J., 2021, 14, 4700–4708 CrossRef PubMed.
- J. F. Kinneary, J. S. Albert and C. J. Burrows, J. Am. Chem. Soc., 1988, 110, 6124–6129 CrossRef CAS PubMed.
- M. Sankaralingam, M. Balamurugan, M. Palaniandavar, P. Vadivelu and C. H. Suresh, Chem. – Eur. J., 2014, 20, 11346–11361 CrossRef CAS PubMed.
- I. Terao, S. Horii, J. Nakazawa, M. Okamura and S. Hikichi, Dalton Trans., 2020, 49, 6108–6118 RSC.
- S. Itoh, T. Shinke, M. Itoh, T. Wada, Y. Morimoto, S. Yanagisawa, H. Sugimoto and M. Kubo, Chem. – Eur. J., 2021, 27, 14730–14737 CrossRef PubMed.
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS PubMed.
- S. Itoh, H. Bandoh, S. Nagatomo, T. Kitagawa and S. Fukuzumi, J. Am. Chem. Soc., 1999, 121, 8945–8946 CrossRef CAS.
- K. Shiren, S. Ogo, S. Fujinami, H. Hayashi, M. Suzuki, A. Uehara, Y. Watanabe and Y. Moro-oka, J. Am. Chem. Soc., 2000, 122, 254–262 CrossRef CAS.
- S. Itoh, H. Bandoh, M. Nakagawa, S. Nagatomo, T. Kitagawa, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 11168–11178 CrossRef CAS PubMed.
- F. F. Pfaff, F. Heims, S. Kundu, S. Mebs and K. Ray, Chem. Commun., 2012, 48, 3730–3732 RSC.
- P. Pirovano, E. R. Farquhar, M. Swart, A. J. Fitzpatrick, G. G. Morgan and A. R. McDonald, Chem. – Eur. J., 2015, 21, 3785–3790 CrossRef CAS PubMed.
- T. Corona, F. F. Pfaff, F. Acuña-Parés, A. Draksharapu, C. J. Whiteoak, V. Martin-Diaconescu, J. Lloret-Fillol, W. R. Browne, K. Ray and A. Company, Chem. – Eur. J., 2015, 21, 15029–15038 CrossRef CAS PubMed.
- P. Pirovano, E. R. Farquhar, M. Swart and A. R. McDonald, J. Am. Chem. Soc., 2016, 138, 14362–14370 CrossRef CAS PubMed.
- T. Corona and A. Company, Chem. – Eur. J., 2016, 22, 13422–13429 CrossRef CAS PubMed.
- T. Corona, A. Draksharapu, S. K. Padamati, I. Gamba, V. Martin-Diaconescu, F. Acuña-Parés, W. R. Browne and A. Company, J. Am. Chem. Soc., 2016, 138, 12987–12996 CrossRef CAS PubMed.
- N. Lau, Y. Sano, J. W. Ziller and A. Borovik, Polyhedron, 2017, 125, 179–185 CrossRef CAS PubMed.
- P. Pirovano, A. R. Berry, M. Swart and A. R. McDonald, Dalton Trans., 2018, 47, 246–250 RSC.
- P. Pirovano, B. Twamley and A. R. McDonald, Chem. – Eur. J., 2018, 24, 5238–5245 CrossRef CAS PubMed.
- C. J. Ballhausen and H. B. Gray, Inorg. Chem., 1962, 1, 111–122 CrossRef CAS.
- J. Winkler and H. Gray, in Struct Bond, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer, Berlin Heidelberg, 2012, vol. 142, ch. 55, pp. 17–28 Search PubMed.
- Y. Qiu and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 19239–19248 CrossRef CAS PubMed.
- P. Heim, G. Spedalotto, M. Lovisari, R. Gericke, J. O'Brien, E. R. Farquhar and A. R. McDonald, Chem. – Eur. J., 2023 DOI:10.1002/chem.202203840.
- P. Pirovano, B. Twamley and A. R. McDonald, Chem. – Eur. J., 2018, 24(20), 5238–5245 CrossRef CAS PubMed.
- J. Rajpurohit, P. Shukla, P. Kumar, C. Das, S. Vaidya, M. Sundararajan, M. Shanmugam and M. Shanmugam, Inorg. Chem., 2019, 58, 6257–6267 CrossRef CAS PubMed.
- G. J. Colpas, M. J. Maroney, C. Bagyinka, M. Kumar, W. S. Willis, S. L. Suib, P. K. Mascharak and N. Baidya, Inorg. Chem., 1991, 30, 920–928 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- D. S. Nesterov and O. V. Nesterova, Catalysts, 2021, 11, 1148 CrossRef CAS.
- J. F. Kinneary, J. S. Albert and C. J. Burrows, J. Am. Chem. Soc., 1988, 110, 6124–6129 CrossRef CAS PubMed.
- K. H. Bok, M. M. Lee, G. R. You, H. M. Ahn, K. Y. Ryu, S. J. Kim, Y. Kim and C. Kim, Chem. - Eur. J., 2017, 23, 3117–3125 CrossRef CAS PubMed.
- S. P. de Visser, K. Oh, A.-R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632–4641 CrossRef CAS PubMed.
- O. V. Makhlynets and E. V. Rybak-Akimova, Chem. – Eur. J., 2010, 16, 13995–14006 CrossRef CAS PubMed.
- S. Xu, A. Draksharapu, W. Rasheed and L. Que Jr., J. Am. Chem. Soc., 2019, 141, 16093–16107 CrossRef CAS PubMed.
- K. D. Karlin, M. S. Nasir, B. I. Cohen, R. W. Cruse, S. Kaderli and A. D. Zuberbuehler, J. Am. Chem. Soc., 1994, 116, 1324–1336 CrossRef CAS.
- K. Honda, J. Cho, T. Matsumoto, J. Roh, H. Furutachi, T. Tosha, M. Kubo, S. Fujinami, T. Ogura, T. Kitagawa and M. Suzuki, Angew. Chem., Int. Ed., 2009, 48, 3304–3307 CrossRef CAS PubMed.
- S. Sahu, B. Zhang, C. J. Pollock, M. Dürr, C. G. Davies, A. M. Confer, I. Ivanovic-Burmazovic, M. A. Siegler, G. N. Jameson, C. Krebs and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 12791–12802 CrossRef CAS PubMed.
- F. F. Pfaff, F. Heims, S. Kundu, S. Mebs and K. Ray, Chem. Commun., 2012, 48, 3730–3732 RSC.
- D. Unjaroen, R. Gericke, M. Lovisari, D. Nelis, P. Mondal, P. Pirovano, B. Twamley, E. R. Farquhar and A. R. McDonald, Inorg. Chem., 2019, 58, 16838–16848 CrossRef CAS PubMed.
- G. Spedalotto, M. Lovisari and A. R. McDonald, ACS Omega, 2021, 6, 28162–28170 CrossRef CAS PubMed.
- R. D. Bach and H. B. Schlegel, J. Phys. Chem. A, 2020, 124, 4742–4751 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt03949d |
| ‡ Current address: Helmholtz-Zentrum Dresden-Rossendorf e.V., Institute of Resource Ecology, Bautzner Landstraße 400, 01328 Dresden, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |