
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tweaking the bridge in metallocene Zr(IV)/W(IV) bimetallic hydrides†
Selwin
Fernando‡
 a,
Martina
Landrini‡
b,
Alceo
Macchioni
b,
David L.
Hughes
a,
Peter H. M.
Budzelaar
*c and
Luca
Rocchigiani
*ab
a,
Martina
Landrini‡
b,
Alceo
Macchioni
b,
David L.
Hughes
a,
Peter H. M.
Budzelaar
*c and
Luca
Rocchigiani
*ab
aSchool of Chemistry, University of East Anglia, Norwich Research Park, NR47TJ, Norwich, UK
bDepartment of Chemistry, Biology and Biotechnology and CIRCC, University of Perugia, I-06123, Perugia, Italy. E-mail: luca.rocchigiani@unipg.it
cDepartment of Chemistry, University of Naples Federico II, Via Cintia, I-80126, Naples, Italy. E-mail: p.budzelaar@unina.it
First published on 8th December 2022
Abstract
Zirconocene cations react with Cp2WH2 affording the bimetallic [Cp2Zr(μ-H)(μ–η1:η5-C5H4)WHCp]+ bridging hydride 1 (Cp = cyclopentadienyl anion, C5H5−) via σ-bond metathesis. Complex 1 features an atypical out of plane Zr(μ-H)W moiety, where no intermetallic interaction is involved, and a fluxional core. Coordination geometry and bond distances of the bridging hydride interaction can be modulated upon reaction with Lewis bases and unsaturated substrates. PMe3, P(p-tol)3, 3,5-dimethylpyridine and THF bind to 1 and shift the hydride bridge on the coordination plane of Zr. Insertion of olefins and alkynes into the Zr–C bond of 1 leads instead to alkyl and vinyl species where the Zr and W coordination planes are perpendicular to each other. Such alterations of the Zr(μ-H)W arrangement are reflected in the average 1H NMR chemical shift values of the hydride, which correlate linearly with computed Zr–H distances. Reactivity experiments with H2 showed that the bridging hydride interaction prevents bimetallic cooperativity and that σ-bond metathesis between Zr–C and H–H bonds is the preferred pathway for all the investigated complexes.
Introduction
Heterobimetallic complexes have attracted considerable interest over the past decades for their potential in triggering chemical reactivity that would not occur with a single specific transition metal or main group element.1–3 In this respect, the combination of two fragments with complementary Lewis acid/base properties is particularly interesting, as it can lead to polarization and cleavage of small molecules bypassing the classic monometallic pathways and enable important catalytic applications.4,5There are multiple ways to make two metal fragments interact with each other, which go beyond the simple formation of unsupported metal–metal bonds. One strategy is to use bridging ligands to provide additional thermodynamic stabilisation to the intermetallic bonding. Bridging hydrides are among the most widespread examples and constitute a common structural feature of many heterobimetallic complexes, ranging from cluster compounds to metalloenzymes.6–8 Such interactions are generally described as 3-center–2-electron (3c2e) bonds9 and show usually higher stability than their terminal M–H counterparts. Nonetheless, they may retain chemical reactivity arising from the electron deficiency at the metal centres, which makes bimetallic hydrides, for instance, good models for catalytically relevant hydrogenation10 and hydroelementation intermediates.11,12
Bimetallics based on highly Lewis acidic zirconocenes of the type Zr(μ-H)nTM or Zr(μ-H)nE (TM = transition metal, E = main group element) form an important class of bridging hydride complexes and take part in many stoichiometric and catalytic reactions. Notable examples include cationic [L2Zr(μ-H)3(AliBu2)2][X] complexes (a, Fig. 1, L2 = bis-cyclopentadienyl or rac-bridged bis-indenyl), which have been investigated by Bercaw and Brintzinger in the context of olefin polymerisation,13–15 or the neutral Cp2Zr(H)(μ-H)(N = tBu)IrCp* (b, Fig. 1) reported by Bergman as the product of cooperative H2 activation by Zr–Ir bimetallics.16 More recently, Cp2Zr(H)(μ-H)2Zn(diketimine) (c, Fig. 1) complexes have found application in olefin isomerisation catalysis.17,18 A peculiar combination originates when electropositive zirconocenes are combined with nucleophilic metal hydrides based on the middle-late portion of the transition series, as the two metals have complementary electronic characteristics. There are a few examples of bimetallics of such kind beyond Bergman's complex c, like the neutral bridging polyhydrides Cp2ZrCl(μ-H)3MLn (MLn = Os(PMe2Ph)3,19 IrCp*,20d and e, Fig. 1) and Cp2ZrCl{H5W(PMe3)3},21 or the chloro-hydrido bridging Cl2Zr(Cp*PMe2)2(μ-H)Ru(H)(Cl)PPh3 complex (f, Fig. 1).22 Despite their stability, the reactivity of these species towards small molecules remains a largely unexplored topic.
 | ||
| Fig. 1 Contextualisation of the present work. | ||
In terms of bonding, it has been proposed that some of these species may contain intermetallic interactions approaching what can be called a “closed” bridging hydride situation.23 This would occur upon populating one empty orbital on the zirconocene with electron density from the second metal, ideally in the plane perpendicular to the Cpcentroid–Zr–Cpcentroid plane.24 The presence of intermetallic interactions is generally invoked to account for short metal–metal distances obtained by X-Ray diffraction analysis or to explain peculiar trends in the 1H NMR chemical shift values of the hydride in solution. As these examples are quite scattered and involve different metals and ligands, unequivocal trends on the bonding situation in this family of compounds cannot be drawn.
Herein we aimed at exploring more systematically the interaction between zirconocenes and nucleophilic hydrides by using Cp2WH2 as a prototypical electron-rich hydride donor. Cp2WH2 has been successfully used as ligand for many Lewis acidic transition metals and main group elements,25–29 owing to its strong nucleophilicity arising from the formal d2 electron configuration at the metal. This makes such complex a potential hydride and lone pair donor, and it becomes interesting to understand how it interacts with electropositive cationic metallocenes Cp2ZrX+ having, in theory, two empty orbitals available for bonding. In this scenario, several arrangements may be active, ranging from typical in-plane interactions to out-of-plane bridges, passing through metal–metal stabilised structures, with or without bridging hydride (Fig. 1).
To investigate such scenarios, we used zirconocenium alkyl cations to metalate Cp2WH2 and synthesize the novel cationic bimetallic bridging hydride [Cp2Zr(μ-H)(μ–η1:η5-C5H4)WHCp]+1 (Fig. 1). The geometry of 1 is peculiar, as the metalation of one Cp ring of tungstenocene makes the bimetallic complex flexible enough to probe the selectivity for establishing a bridging hydride interaction or the metal–metal bond. Therefore, it makes a very useful platform to evaluate the tendency of zirconocene cations to accept electron density from a metal hydride. Moreover, complex 1 contains three coexisting functionalities (metal hydride, Zr–C bond and potential intermetallic interaction) and it becomes intriguing to probe its reactivity towards small molecules like alkenes, alkynes or H2 and test what is the most reactive site. To investigate these aspects, we use NMR spectroscopy, X-Ray diffraction and DFT calculations, and we rationalize how different Zr(μ-H)W arrangements affect the spectroscopic properties and bonding in this class of complexes.
Results and discussion
σ-Bond metathesis reactions
To explore reactions between zirconocenes and tungstenocenes, [Cp2ZrMe+⋯MeB(C6F5)3−]30 and [Cp2Zr(η2-CH2–NMePh)][B(C6F5)4]31,32 were chosen as starting materials as they can be straightforwardly prepared in situ from Cp2ZrMe2 and B(C6F5)3 or [HNMe2Ph][B(C6F5)4]. These ion pairs were reacted with 1 equivalent of Cp2WH2 in C6D6/orthodifluorobenzene (ODFB, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at 297 K. 1H NMR indicated that a fast and quantitative reaction takes place upon mixing the reagents, leading to the formation of the bimetallic ion pair 1X (X− = MeB(C6F5)3−, B(C6F5)4−), featuring the tungstenocene fragment covalently bound to the Zr centre (Fig. 2). Concurrently, the formation of CH4 or Me2NPh was observed. 13C NMR spectroscopy indicated that the σ-metalated carbon atom is high-frequency shifted at δC = 152.8 for 1B(C6F5)4 and δC = 153.2 ppm for 1MeB(C6F5)3 (δC Cp2WH2ca. 75.0 ppm), while the hydride signal appears as a singlet in the 1H NMR spectrum for both complexes at δH = −11.7 ppm, with 183W satellites (1JWH = 76.5 Hz). Broadening of this resonance was observed upon cooling down a sample of 1B(C6F5)4 in C7D8/ODFB to −70 °C, but no decoalescence was obtained. The methylborate anion in 1MeB(C6F5)3 shows NMR fingerprints which are in between those of an inner and an outer sphere ion pair (δH(B–Me) = 0.82 ppm, Δδ(m-F/p-F) = 3.5 ppm),33 suggesting that the anion is sterically prevented from establishing a strong coordination to the Zr. Analogously, the aniline released upon the formation of 1B(C6F5)4 does not coordinate to the metal centre.
:1 v/v) at 297 K. 1H NMR indicated that a fast and quantitative reaction takes place upon mixing the reagents, leading to the formation of the bimetallic ion pair 1X (X− = MeB(C6F5)3−, B(C6F5)4−), featuring the tungstenocene fragment covalently bound to the Zr centre (Fig. 2). Concurrently, the formation of CH4 or Me2NPh was observed. 13C NMR spectroscopy indicated that the σ-metalated carbon atom is high-frequency shifted at δC = 152.8 for 1B(C6F5)4 and δC = 153.2 ppm for 1MeB(C6F5)3 (δC Cp2WH2ca. 75.0 ppm), while the hydride signal appears as a singlet in the 1H NMR spectrum for both complexes at δH = −11.7 ppm, with 183W satellites (1JWH = 76.5 Hz). Broadening of this resonance was observed upon cooling down a sample of 1B(C6F5)4 in C7D8/ODFB to −70 °C, but no decoalescence was obtained. The methylborate anion in 1MeB(C6F5)3 shows NMR fingerprints which are in between those of an inner and an outer sphere ion pair (δH(B–Me) = 0.82 ppm, Δδ(m-F/p-F) = 3.5 ppm),33 suggesting that the anion is sterically prevented from establishing a strong coordination to the Zr. Analogously, the aniline released upon the formation of 1B(C6F5)4 does not coordinate to the metal centre.
 | ||
| Fig. 2 Left: synthesis of 1X (X−=B(C6F5)4− or MeB(C6F5)3−). Right: two views of the molecular structure of 1B(C6F5)4 (one of the two independent molecules found in the asymmetric unit is shown). Non-hydride hydrogen atoms and anion are omitted for clarity, thermal ellipsoids are drawn at 50%. Selected distances [Å] and bond angles [°]: Zr2–W2 3.1395(6), Zr2–C21 2.268(4), Cpcentroid–Zr–Cpcentroid 129.45, Cpcentroid–W–Cpcentroid 144.85, Zr2–C1–Cpcentroid 146.84. | ||
The formation of 1MeB(C6F5)3 was also probed at low temperature, by adding 1 equivalent of Cp2WH2 to a solution of [Cp2ZrMe+⋯MeB(C6F5)3−] in C6D5Cl at −50 °C. Under these conditions, the formation of the bimetallic ion pair [Cp2ZrMe(H2WCp2)][MeB(C6F5)3] (I) was observed, implying that Cp2WH2 coordinates to the zirconocene before being σ-metalated. According to 1H and 19F NMR, I is an outer sphere ion pair (δH(BMe) = 1.34 ppm, ΔδF(m–F, p–F) = 2.7 ppm), where the hydride 1H chemical shift is close to that of pure Cp2WH2 (δH(I) = −12.7, 1JWH = 77 Hz) and does not show any temperature dependence. Conversion of I into 1MeB(C6F5)3 and CH4 elimination occurred simultaneously over the period of 4 hours at −40 °C, most likely through a σ-bond metathesis pathway.34
Emerald crystals of 1B(C6F5)4 suitable for X Ray diffraction were obtained by layering a C6D6/ODFB solution with light petrol ether. Two independent molecules with very similar structural parameters were found in the asymmetric unit; they showed an average Zr–W distance of 3.126 Å, which is below the sum of the van der Waals or the covalent radii of Zr and W.35 The tungstenocene fragment is tilted towards the zirconocene, with a Zr2–C21–Cpcentroid angle of about 147° and a W2–Zr2–C21 angle of 46° (Fig. 2). The arrangement of the hydride moieties is asymmetric, with one hydrogen atom oriented towards the zirconium at an estimated average Zr⋯H distance of 2.2 Å and describing a Zr–H–W angle of about 104°. The second hydride is oriented far away from the Zr atom, at over 4 Å from the metal centre. The η1:η5 Cp is almost perpendicular to the zirconocene fragment, with the hydride moieties oriented out of the Zr2–W2–C21 plane by 48°. Both Zr–W and Zr–H bond distances are compatible with the presence of an interaction between the two metal fragments that leads to the bending of the Zr–C21–Cpcentroid angle, even though the coordination geometry of the bridging hydride is considerably twisted with respect to the ideal planar arrangement. The nature of this interaction was dissected by DFT calculations (see below).
The Zr(μ–η1:η5–Cp)M motif is well-known and there are many examples of neutral species that have been obtained, for instance, by lithiation/transmetalation of ferrocene36 or hydride expulsion from trimetallic Zr–Ru2 carbonyl complexes.37 On the other hand, reactions involving cationic zirconocenes are less common. Seminal cases are the zirconation of ferrocene by σ-bond metathesis with the Zr–Me bond of [Cp2ZrMe+⋯MeB(C6F5)3−],38 or the metalation of Rh indenyl by transient alkyl metallocenes generated by AlMe3.39 Even though metalation of Cp2WH2 was observed previously in Ir polyhydride systems by Venanzi40 and Moore,41,42 as well as in lanthanide complexes by Tilley and co-workers, the “tilted” bridging hydride coordination mode on 1 seems to be less common.
Unlike 1B(C6F5)4, 1MeB(C6F5)3 is not thermally stable in C6D6/ODFB and transforms into the orange zwitterion 2 upon CH4 elimination over the course of 2 weeks at 297 K (Fig. 3). This is corroborated by the loss of one hydrogen from the C5H5 ligand bound to tungsten and a moderate high-frequency shift of the hydride signal (δH = −9.7 ppm, 1JWH = 76.5 Hz) in the 1H NMR spectrum. 19F NMR shows the appearance of a new set of signals for the pentafluorophenyl rings, characterised by a very broad resonance for the ortho-F at δF = −136.6 ppm suggesting the presence of Zr⋯F interactions.43 The chemical shift of the σ-metalated carbon atom is slightly high-frequency shifted with respect to 1MeB(C6F5)3 (δC = 160.2 vs. 152.8 ppm). The molecular structure (Fig. 3) is compatible with NMR observations and shows that B(C6F5)3 attacked the second cyclopentadienyl ligand bound to tungsten. Consistently, one of the ortho-fluorine atoms in 2 shows a close interaction with the Zr centre (dZr–F32 = 2.562 Å). Despite the presence of this additional interaction at the Zr, the asymmetric hydride arrangement of the metalated tungstenocene remains unaltered with respect to 1, with a Zr–W distance of 3.104 Å. The roughly estimated Zr⋯H distance of 2.38 Å is slightly longer than that in 1 and the Zr–H–W angle is 100°, just 4° lower than that observed in 1.
 | ||
| Fig. 3 Left: conversion of 1MeB(C6F5)3 into 2. Right: molecular structure of 2. Non-hydride hydrogen atoms and solvent molecules are omitted for clarity, thermal ellipsoids are drawn at 50%. Selected distances [Å] and bond angles [°]: Zr–W 3.1044(6), Zr–C10 2.248(18), B–C1 1.662(3), Zr–F32 2.5626(10), Cpcentroid–Zr–Cpcentroid 128.02, Cpcentroid–W–Cpcentroid 147.08, Zr1–C10–Cpcentroid 147.91. | ||
The transformation of 1MeB(C6F5)3 into 2 resembles previous reactivity reported by Braunschweig, who showed that B(C6F5)3 reacts with Cp2WH2 leading to a W(VI) zwitterion of the type [{C5H4B−(C6F5)3}CpWH3+].44 In our case, it can be speculated that transient concentrations of B(C6F5)3 are generated upon anion coordination at the Zr and breakage of the B–Me bond, in analogy to what is often observed for zirconocene methylborate ion pairs.30 The formed borane would then attack the W–Cp unit, form a C–B bond and release a proton, which would eventually lead to CH4 elimination from the Zr–Me group generated upon loss of borane. Alternatively, σ-bond metathesis between Zr–C and Me–B bonds could take place, leading to the formation of a borane-functionalized tungstenocene, which would eliminate CH4 upon rotation/remetalation.
Such reactivity was not observed when the solvent was C6D5Cl. In the latter medium, 1MeB(C6F5)3 showed indeed to be stable over a week at 297 K. Strikingly, freshly prepared samples of 1X in C6D6 or C6D6/ODFB change colour from bright green to orange upon drying and redissolving in C6D5Cl. The same is observed when 1MeB(C6F5)3 is synthesised in situ using C6D5Cl as solvent. The hydride signal is unaffected by the change of medium (δH = −11.8 ppm, 1JWH = 76.5 Hz), while the metalated carbon atom is slightly low frequency shifted at δC = 144.0 ppm with respect to C6D6 solutions (δCca. 153.0 ppm). It is reasonable to assume that C6D5Cl coordinates strongly at the Zr centre,45 pushing the MeB(C6F5)3− anion in the second coordination sphere and preventing its degradation. Most likely, the electronic properties, hence the colour, of the bimetallic complex are affected by the solvent coordination as well. Anyway, as the spectroscopic fingerprints of the bimetallic core are basically unaltered, it can be assumed that C6D5Cl coordination does not disrupt the interaction between the two metal fragments.
Reactions with Lewis bases
The presumed ability of chlorobenzene to coordinate at the Zr centre of complex 1 confirms that the latter is not “saturated” by the Zr⋯H–W interaction and a coordinative vacancy is available for further reactivity. The lack of appreciable coordination of the MeB(C6F5)3− anion (or Me2NPh) mentioned above may be simply due to a mismatch in basicity or sterics. To probe whether using stronger or less encumbered Lewis bases leads to a different outcome, we investigated the reactions between 1MeB(C6F5)3 and prototypical Lewis bases, such as PR3 (R = Me, p-tolyl), 3,5-lutidine (3,5-dimethylpyridine) and THF.46Unlike NMe2Ph, PMe3 reacts instantaneously with 1 and affords a mixture of two isomers in a 85:15 molar ratio: a W-outside complex (3a), where PMe3 is coordinated close to the metalated Cp, and a W-inside complex (3b), where PMe3 binds close to the bridging hydride (Scheme 1). The coordination of PMe3 induces a considerable low-frequency shift of the hydride signal (δH(1) = −11.7 ppm), which appears at δH = −17.9 and δH = −14.3 ppm for 3a and 3b, respectively. Splitting of the hydride signals due to coupling with 31P is observed, with JPH values of 6.4 Hz for 3a and 10.7 Hz for 3b. The 1JWH values for the two isomers are appreciably different: while 1JWH for 3b is very similar to that of 1 (72.0 Hz), the hydride-tungsten coupling is reduced somewhat in 3a (1JWH = 66.0 Hz). Based on these spectroscopic fingerprints, it can be inferred that the bridging hydride interaction is maintained in 3. As in the previous case, the two hydride moieties appear as a singlet in the 1H NMR spectrum, likely due to the fast averaging of chemical shift and coupling constant values over the two hydride environments (see DFT calculations).
 | ||
| Scheme 1 Reactions of 1MeB(C6F5)3 with Lewis bases. | ||
The crystal structure of 3a (Fig. 4) is consistent with the NMR data and shows that the planes described by the two Cp–M–Cp units are almost perpendicular, with a Zr–W distance of around 3.4 Å and a Zr–C1–Cpcentroid angle of 154°. The Zr–P distance of 2.71 Å is unexceptional and lies in the typical range observed for Cp2Zr–PMe3 fragments.47–49 The observation of a longer Zr–W distance in 3a compared to 1 (δdZr–W = 0.3 Å) suggests that metal–metal interactions play a negligible role in its bonding (see DFT section). Consistently, electron density corresponding to a bridging hydride was located in the Zr coordination plane, at an approximate distance of 2.0 Å from Zr and 1.7 Å from W, establishing a Zr–H–W angle of about 130° (average between the two independent molecules in the cell). Clearly, increasing the coordination number at the Zr centre affects the geometry of the bridging hydride, which is now oriented in the same plane as the other two ligands bound to Zr.
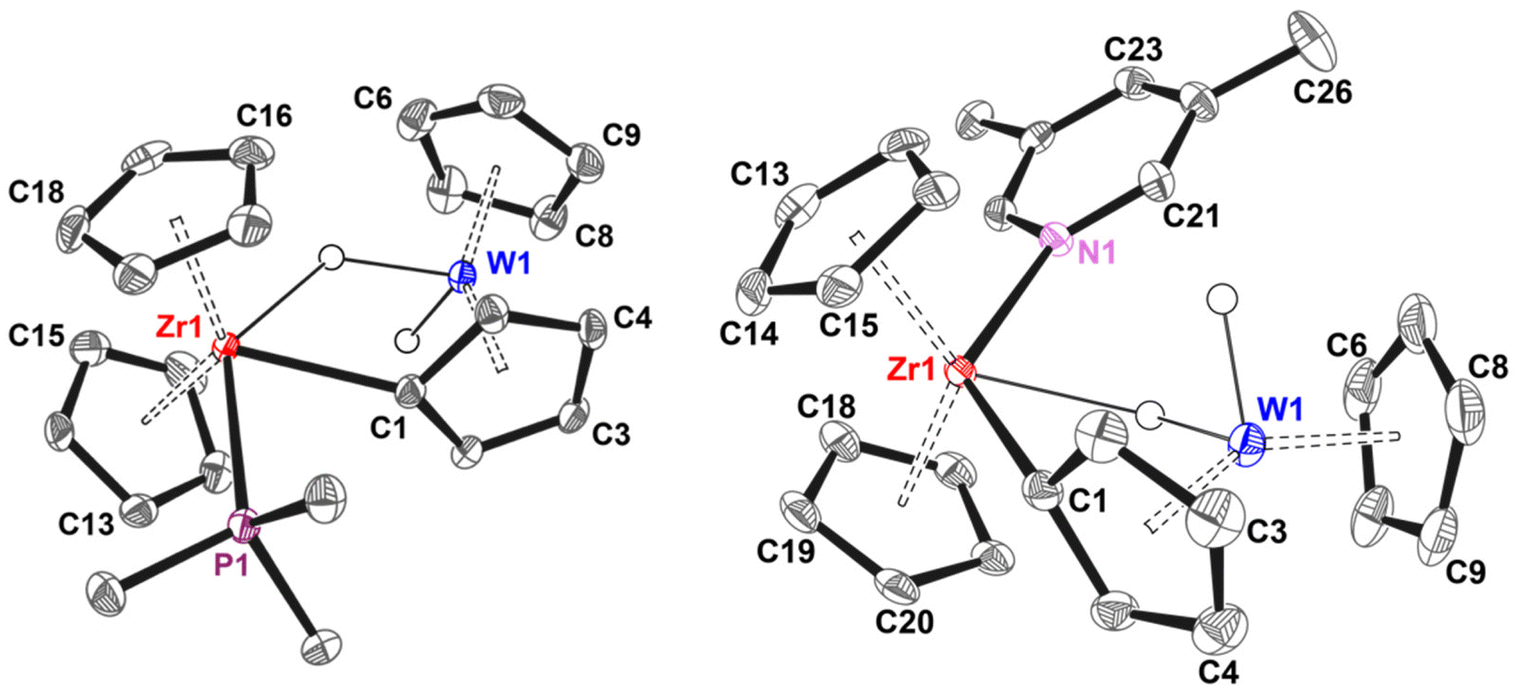 | ||
| Fig. 4 Molecular structures 3a (left, one of the two independent molecules found in the asymmetric unit) and 6b (right). Non-hydride hydrogen atoms and anion are omitted for clarity, thermal ellipsoids are drawn at 50%. Selected distances [Å] and bond angles [°], 3a: Zr1–P1 2.7088(8), Zr1–C1 2.316(4), Zr1–C1–Cpcentroid 154.72, Cpcentroid–W1–Cpcentroid 147.03, Cpcentroid–Zr1–Cpcentroid 129.68; 6b: Zr1–N1 2.441(2), Zr1–C1 2.307(2), Zr1–C1–Cpcentroid 153.87, Cpcentroid–W1–Cpcentroid 145.86, Cpcentroid–Zr1–Cpcentroid 128.49. | ||
The low abundance of 3b makes its complete NMR assignment more difficult. In any case, the presence of a strong phosphorous-hydride coupling (stronger than 3a) led us to formulate 3b as a bridging hydride as well. This hypothesis is also backed by DFT calculations (see below).
The bulkier tris-p-tolyl-phosphine binds 1MeB(C6F5)3 as well, but the regiochemical outcome of the reaction is reversed with respect to PMe3. In fact, almost exclusive formation of the W-inside isomer 4b (4a:4b = 6:94) was observed. The latter shows a dihydride signal at δH = −14.8 ppm (2JPH = 11.3, 1JWH = 69.0 Hz) and a 31P NMR signal located at δP = 18.2 ppm. The stereochemistry of 4b was elucidated by NOE experiments, which revealed the presence of strong dipolar contacts between the aromatic protons of Ptol3 and the hydride moieties. Consistently, no interactions were observed between tolyl rings and the σ-metalated Cp ring.
Remarkably, the 1H and 13C NMR spectra of 4b show the presence of two different sets of signals for the p-tolyl rings in a 2:1 ratio, suggesting that the rotation of the phosphine about the Zr–P bond is slow (or frozen) on the NMR timescale. Most reasonably, two tolyl rings are oriented towards the zirconocene, while the other is pointing towards the tungstenocene, making them magnetically not equivalent (see ESI†). As only 2 sets of ortho and meta C–H groups are observed in total, a fast process interconverting 4b into its mirror image must be active. Most likely, the fast hydride interconversion process averaging the two W–H chemical shifts is also responsible for such an effect.
In contrast with 3a–b, 1H NOE NMR also indicates that 4a and 4b are in chemical exchange. For example, intense exchange cross peaks were observed between the hydrides of 4a and 4b or between the W–Cp rings of the two isomers. Reasonably, 4a and 4b form a thermodynamic mixture equilibrating through phosphine dissociation/migration. In agreement with the latter hypothesis, the addition of a second equivalent of Ptol3 to the mixture enables chemical exchange between coordinated and free phosphine, corroborating the presence of a labile Zr–P bond in 4 (ESI†).
Preference for W-inside coordination was also observed with THF and 3,5-lutidine (selectivity >95%). In the case of the former, the hydride signal of the inside adduct 5b is considerably broad at room temperature (δH = −13.8 ppm), while it sharpens into a well-defined singlet with 183W satellites (1JWH = 73 Hz) when the sample is cooled down to 253 K. By using 1H NOE NMR at this temperature it is possible to detect chemical exchange between 5a and 5b, as observed in the case of Ptol3, confirming that binding of THF to Zr is loose. Moreover, if a small excess of THF (1.33 mol) is used, free and coordinated THF signals are averaged at room temperature and can be separated only at 253 K. This indicates that broadening of the W–H signals is due to THF fluxional coordination, rather than internal hydride dynamicity. The identity of 5b was confirmed by 1H NOESY, which revealed selective interactions between the hydride signals and α- and β-THF protons, while the latter do not interact with the σ-metalated Cp. As in the previous cases, the relative concentration of 5a was too low for a compelling NMR assignment.
3,5-Lutidine reacts with 1MeB(C6F5)3 forming the stable adduct 6b in 97% selectivity. Contrary to the results obtained with THF, the hydride resonance appears as a sharp singlet located at δH = −14.4 ppm, with 1JWH = 73 Hz. 1H NOESY NMR showed dipolar contacts between the pyridine protons and the hydride signal, while there is no interaction with the σ-metalated tungsten-cyclopentadienyl, as observed for 5b. Moreover, chemical exchange between inside and outside isomers was observed, implying labile coordination of nitrogen as in the cases of THF and Ptol3. The solid-state molecular structure of 6b (Fig. 4) is in perfect agreement with the NMR data, showing that the coordination of 3,5 lutidine occurs in proximity of the hydride moieties and opposite to the σ-metalated cyclopentadienyl. The Zr1–N1 distance of 2.441(2) Å is in line with other previously reported zirconocenium pyridine adducts50 and the Zr1–C1–Cpcentroid angle of 154° matches that of complex 3a. Electron density compatible with the presence of a bridging hydride was located at an approximate Zr⋯H distance of 2.1 Å showing a Zr–H–W angle of 123°, which is slightly lower than that observed in 3a.
The picture emerging from the reactivity of 1MeB(C6F5)3 with Lewis bases is that the Zr centre of the bimetallic complex can accommodate ligands with sufficient basicity and different steric demand, originating adducts with a regiochemistry that depends on the nature of the base. Irrespective of the W-inside or W-outside configuration, the geometry of the bimetallic core is considerably affected by the coordination of the base with respect to 1 and the clear formation of in-plane bridging hydride interaction is observed. In the case of the 4, 5 and 6, the coordination of the base is labile and exchange between the two regioisomers occurs. On the other hand, with the PMe3 complex 3 such exchange does not take place (or is too slow to be observed by NMR), most likely owing to its higher Lewis basicity.
Insertion reactions
The presence of an accessible coordinative vacancy in cis position to the σ-metalated Cp opens to the possibility to exploit insertion of double and triple bonds into the Zr–C bond to tweak the interaction between Zr and the W–H bonds.
1MeB(C6F5)3 reacts with internal alkynes affording products whose structure depends on the substituents on the triple bond. Electron rich alkynes, such as 2-butyne, insert exclusively into the Zr–C bond leading to the corresponding cis-vinyl-cyclopentadienyl products 7 (Fig. 5). In the case of asymmetric substrates (R ≠ R′), mixtures of insertion products are observed. Generally, the bulkiest group is preferentially inserted close to the Cp bound to tungsten: with 1-phenyl propyne, a molar ratio of 70:30 was observed while this increased to 80:20 when 4,4′dimethyl-2-pentyne was used. In the latter case, the mixture evolves to 100% of the 2,3 insertion products over the course of 48 h, suggesting that a kinetic mixture was initially obtained. The interconversion between the two isomers is likely to occur through a β-carbon elimination–reinsertion sequence, which is not unprecedented in zirconocene alkyne chemistry.51 In the case of the bulky bis-adamantyl acetylene, no insertion was observed over the period of 7 days at room temperature.
 | ||
| Fig. 5 Left: reactivity between 1 and alkynes/alkenes. Right: molecular structure 7c. Non-hydride hydrogen atoms and MeB(C6F5)3− anion are omitted for clarity, thermal ellipsoids are drawn at 50%. Selected distances [Å] and bond angles [°]: Zr–W 3.0878(2), Zr–C27 2.268(2), C26–C27 1.349(3), Cpcentroid–Zr–Cpcentroid 126.21, Cpcentroid–W–Cpcentroid 144.42, C26–C16–Cpcentroid 164.60. | ||
Alkyne insertion products 7a–c display a high-frequency shift of the hydride resonance of about 2 ppm with respect to 1 and a moderate increase of the 1JWH values to around 80 Hz. This is consistent with a further alteration in the interaction mode between the Zr centre and the WH2 moiety. The molecular structure of 7c (Fig. 5), obtained by X-Ray diffraction, shows that the orientation of the two metallocene fragments is almost perpendicular, with the whole Zr–vinyl–tungsten unit lying in the same plane and the two W–H groups oriented above and below this plane. Despite the different orientation, the W–Zr distance is only marginally shorter than that in 1 (3.087 Å), suggesting the presence of an interaction between the two fragments that is not much stronger than that of the starting complex. However, the coordinative vacancy in these insertion products is less accessible with respect to 1. For example, 7c does not react with excess PMe3 and the 31P NMR signal of free phosphine can be detected in solution for at least 24 h at room temperature. After a few days, slow decomposition processes to unidentified species took place.
Using electron-poor alkynes, such as dimethyl-acetylene-dicarboxylate (DMAD), flips the reactivity and insertion occurs into the W–H bond. The outcome of the reaction is composed of a complex mixture of isomeric bimetallic complexes where no W–H signals are detected by 1H NMR. Such a complex mixture evolves over the course of 1 week, generating two main sets of signals in 85:15 ratio, with a spectroscopic yield of about 60%. The 1H NMR spectrum of the major product showed the presence of two magnetically inequivalent doublets (3JHH = 10.8 Hz) at δH = 1.67 and δH = 3.33 ppm, together with two –OMe singlets (δH = 3.31 and δH = 3.54 ppm), showing long-range correlations with two signals at δC = 7.7 and δH = 8.6 ppm in the 13C NMR spectrum. This pattern is consistent with a double insertion of one DMAD molecule into both the W–H bonds of 1MeB(C6F5)3, which leads to the formation of tungstenocycle 8, in analogy to what previously observed by Herberich and Barlage on Cp2WH2.52 The stereochemistry of 8 was determined by using 1H NOE experiments, which revealed the presence of a dipolar interaction between the C–H group of the metallacyclopropane at δH = 1.67 ppm and a W–Cp singlet at δH = 4.40 ppm. On the other hand, the second C–H group (at δH = 3.33) interacted with the Zr–Cp oriented towards the opposite direction, thus confirming the mutual trans arrangement of the two C–H groups. According to 13C NMR, the η1:η5 Cp ring remains intact, with its quaternary carbon atom resonating at δC = 142.0 ppm, in analogy to 1. The second set of signals was attributed to the cis isomer 8′, again based on 1H NOE interactions. Unfortunately, no single crystals of 8 or 8′ could be obtained and the nature of the interactions between the two metal centres could not be ascertained.
1MeB(C6F5)3 reacts also with 1-hexene, undergoing 1,2 insertion into the Zr–C bond and affording the corresponding bimetallic alkyl-bridged derivative 9 (Fig. 5). Even when a large excess of olefin was used, no trace of polymerisation was observed at room temperature. Complex 9 shows a Zr–CH2 moiety at δC = 53.7 ppm bearing two magnetically inequivalent hydrogen atoms located at δH = 1.44 and δH = −0.44 ppm in the 1H NMR spectrum. Interestingly, the two hydrides were also found to generate two different signals δH = −10.7 and δH = −10.8 ppm, coupled with each other with a 2JHH = 6.3 Hz. Most likely, this splitting is due to the diastereotopic character of the two hydride moieties. These two signals show two different satellite couplings with 183W of 83.2 (H anti to the butyl chain) and 76.4 Hz (H syn to the butyl chain), implying that the degree interaction between the two hydrides and the Zr centre is not symmetric, as proposed for all the other complexes above.
Reactions with H2
We previously reported that zirconaziridinium salts react with H2 undergoing hydrogenolysis of the Zr–CH2 bond leading to an intermediate zirconocene hydride amine complex that splits H2 heterolytically.53 Complexes 1X can be seen as analogues of zirconaziridium ion pairs, in which the σ-metalated Cp and the tungsten atom replace the CH2–NR2 group. In this respect, it is interesting to test whether hydrogenolysis leads to the same outcome or the presence of the tungsten centre enables other reaction pathways. In addition, it is also of interest to probe how the different structures that have been derived from 1 upon reaction with bases or alkynes/alkenes affect H2 activation.1B(C6F5)4 reacts slowly with H2 (298 K, 1 atm), producing initially a 1H NMR spectrum with broad resonances that evolves to two major sets of signals over the course of 2 days. The first was assigned to [Cp2WH3][B(C6F5)4], based on the appearance of the typical pattern of W(VI) trihydrides with a doublet at δH = −7.13 ppm and a triplet located at δH = −6.55 ppm (2JHH = 9.6 Hz).54 The second set of signals was assigned to the trimetallic complex 10, containing a hydride bridged zirconocene dimer bearing a doubly σ-metalated tungstenocene dihydride facing the two Zr centres (Fig. 6). Complex 10 showed two different hydride resonances in the 1H NMR: a broad triplet, without 183W coupling, resonating at δH = −5.50 ppm and a sharp doublet with 183W satellites (1JWH = 70.5 Hz) at δH = −12.7 ppm. Single crystals of complex 10 were obtained from double layering a concentrated C6D5Cl solution with light petrol ether. The corresponding molecular structure revealed that the metalated tungstenocene orients the hydrides towards the coordinative vacancies of both the Zr centres, at tungsten–zirconium distances of 3.413 Å (W1–Zr2) and 3.313 Å (W1–Zr3). The Zr2–Zr3 distance is 3.762 Å and the estimated Zr⋯H distances are comprised between 2.5 and 2.9 Å. While the intrinsic uncertainty in the estimation of H positions does not allow to precisely understand the nature of the Zr⋯H bonding, the presence of a 2.7 Hz coupling between Zr–H and W–H signals implies the presence of a residual interaction between them.
 | ||
| Fig. 6 Left: reaction of 1B(C6F5)4 with H2. Right: molecular structure of 10, non-hydride H atoms and anion are omitted for clarity, thermal ellipsoid are shown at 50% probability. Selected distances [Å] and bond angles [°]: Zr3–C1 2.263(3), Zr2–C6 2.260(4), Cpcentroid–W1–Cpcentroid 141.05. | ||
From a mechanistic standpoint, the formation of 10 requires two extra H atoms, hence splitting of one H2 molecule. Even though an hydridic Zr(μ-H) and a protic W(VI) hydride are formed, it is likely that this reactivity is not due to heterolytic hydrogen splitting at the bimetallic core. More reasonably, an hydrogenolysis route analogous to that of zirconaziridium salts takes place. In this scenario, coordination of H2 to the Zr of 1 would enable σ-bond metathesis between the H–H and the Zr–C bonds, leading to the regeneration of Cp2WH2 and formation of a zirconocenium hydride. The latter could react quickly with residual 1 establishing a bridging interaction and forming an intermediate that would undergo deprotonation by transient Cp2WH2 to give 10 and Cp2WH3+. Complex 10 seems to be a thermodynamic sink, as it does not react further in the presence of excess H2, at least for 2 weeks at RT.
Consistently, saturated complexes are much less reactive with H2 than 1. In the case of complex 6b, traces of 10 formed, together with the formation of Cp2WH3+ and Cp2WH2, suggesting that hydrogenolysis takes place also for this compound. However, only a 15% conversion was observed after 7 days at RT. Complexes 3a–b were also found to be rather unreactive and only minor side reactivity was observed after 2 weeks at RT.
On the other hand, quantitative reactivity with H2 was observed for the 1-hexene insertion product 9. After 9 days at RT, the signals of the starting complex disappeared from the 1H NMR spectrum to afford a mixture of products with rather similar spectroscopic fingerprints to those obtained for the reaction of 1. Several hydride signals were observed at δH = −5.34 to −5.37 ppm (Zr μ-H), δH = −11.79, −11.81, −12.50, −13.12, −13.15 ppm (WIV–H) and δH = −6.04, −6.36, −6.70 (WVI–H), whose presence suggests that a hydrogenolysis–deprotonation sequence may be active also for complex 9. 2D NMR methods allowed the formed W(VI) species to be assigned as the cationic hydride 12, featuring a 1-methylpentyl substituent on one Cp ring. The presence of a stereocenter makes the three hydride moieties in 12 magnetically inequivalent, so they appear as three distinct pseudotriplets with 2JHH values of 8.2 and 8.7 Hz and typical 1JWH of 47.4 and 68.7 Hz. While their complete NMR assignment is hampered by extensive overlapping, the two main Zr-containing products can be formulated as the isomeric trimetallic complexes 11a–b, in which the substituted Cp ring is metalated either in α or β position with respect to the 1-methylpentyl substituent. Complexes 11 show strong dipolar contact between the W–H signals, which are magnetically inequivalent, and the Zr μ-H ones. Moreover, NOE interactions between the Zr–Cp rings and the aliphatic signals of the alkyl chain were observed. Quaternary carbon signals compatible with σ-metalated Cp were identified at δC = 140.0 and δC = 139.8 ppm in the 13C{1H} NMR spectrum (ESI†).
The formation of 1-methylpentyl chains from 9 indicates that σ-bond metathesis between the Zr–CH2 moiety and H2 most likely takes place, forming Zr–H moieties and Cp(1-methylpentyl-Cp)WH2. At this stage, it is possible that the functionalised tungstenocene is remetalated at either the C5H5 or the C5H4R ring producing an intermediate that resembles 1. The latter would undergo the same reactivity with H2 described above, where the W(IV) dihydride acts as a base and is protonated upon a second Cp metalation, affording 11 and 12. In any case, reactivity is less straightforward than that of 1, and the trimetallic complexes are obtained in approximately 30% spectroscopic yield (Scheme 2).
 | ||
| Scheme 2 Hydrogenolysis of complex 9. | ||
The reactivity with H2 of the alkyne insertion product 7c is even more complex. After 3 weeks at RT, the signals of the starting complex disappeared from the 1H NMR spectrum to originate a plethora of species. In the hydridic region of the spectrum, two main tungsten species were observed along with other secondary products showing chemical shift values similar to that of 11a–b (δH = −5.33, −5.55, −11.27, −12.60 and −13.12 ppm). 1D and 2D NMR experiments allowed to assign the two tungsten species to W(VI) cyclopentadienyl-vinyl and alkyl trihydrides, which formed in 37% and 18% respectively (ESI†). The formation of the latter suggests that, as in the case of 1, Zr–C hydrogenolysis takes place. However, the increased steric demand of 7c prevents the efficient trapping of transient Zr–H into bridging species and the formation of trimetallic species occurs only in traces. As previously reported by Jordan and co-workers, cationic zirconocene hydrides react in chlorobenzene under ambient light to give mixtures of dimeric bridging hydrides and chlorides.55 It is likely that the formation of such species triggers further collateral reaction with H2 and tungstenocene species. The formation of 11, where a fully hydrogenated alkyl chain is present, may be related to a reinsertion–hydrogenolysis of 10 into one of the transient Zr–H mentioned above.
DFT calculations
Density functional calculations were performed to shed some light on the formation and potential dynamic behaviour of 1, and to explain the bonding in these bimetallic complexes. First, formation of complex 1via σ-bond metathesis of I or its hydride analogue seems plausible from the energetic point of view, and it is found to occur via the typical 4-membered transition state, whose energy depends on the group that is eliminated from Zr (Me or H), see Fig. 7. | ||
| Fig. 7 Energy profile for formation of 1. | ||
Then, we find that the Zr/W core of 1 is extremely flexible. The preferred arrangement is highly asymmetric, with two clearly inequivalent W-bound hydrides as experimentally observed in the solid state. The Cp2Zr fragment is located outside the WH2 wedge and interacts with only one of the two hydrides (Fig. 8a). However, a more symmetric structure is accessible that has Zr sitting inside the WH2 wedge, still interacting mostly with one of the two hydrides (Fig. 8c). Connecting this structure with its mirror image is the Cs-symmetric inversion transition state (Fig. 8d). The whole asym–sym–asym sequence is calculated to happen within a band of <2 kcal mol−1, explaining the effective equivalence of the two hydrides as observed by NMR.
 | ||
| Fig. 8 (A) Asymmetric calculated “out” structure of 1; (B) TS for in–out rearrangement; (C) “in” local minimum, and (D) Cs-symmetric inversion TS, with Zr–W bond lengths. Distances in Å. Relative free energies in kcal mol−1. Cp H atoms omitted for clarity. | ||
Structures of Lewis base (PMe3, Ptol3, THF, 3,5-lut) adducts were also optimised using DFT, which confirmed the existence of separate outside and inside local minima (see Fig. 9 for 3a/b). In agreement with experimental observations, PMe3 has the largest binding energy (see Table 1) and is also the only base to prefer the outside arrangement. The calculated binding energies for 4–6 are compatible with fluxional behaviour (reversible base dissociation) although the stability of 4b is probably overestimated at the computational level employed.
 | ||
| Fig. 9 Calculated structures of (A) 3a and (B) 3b, with Zr–W bond lengths. Distances in Å. Non-hydride H atoms omitted for clarity. | ||
| Outside | Inside | |
|---|---|---|
| PMe3 (3) | −18.7 | −17.9 |
| Ptol3 (4) | −11.7 | −17.0 |
| THF (5) | −6.3 | −8.5 |
| 3,5-lut (6) | −12.4 | −15.2 |
The nature of the interaction between the two metallocene fragments of 1 is not immediately obvious. In a seminal 1976 paper, Lauher and Hoffmann24 analysed the bonding capabilities of bent-metallocene fragments. They concluded that such fragments have available for bonding three valence orbitals lying in the coordination plane between the two Cp rings. In Cp2WH2, two of them are used to form the W–H bonds. The third contains a metal-centered lone pair (LP) and can be protonated to form the well-known Cp2WH3+ cation. In the same way, Cp2WH2 could react with Lewis acids to form metal–metal dative bonds.56–58 Alternatively, the W–H bonds could act as donors to form one or two 3c2e bonds. Recent computational work suggests that the latter bonding mode is preferred.59,60 (Cp2MoH2 has also been found to form metal complexes,29,60–63 for which similar bonding options are conceivable). We have used IBOview (IBO: Intrinsic Bond Orbital)64,65 to analyse the situation; Fig. 10 shows relevant orbital plots, and Table 2 shows the numbers of electrons from W, H and Zr contributing to the most relevant IBOs. Shown in the figure are the W–H bonding IBO (Fig. 10A and B) as well as the lone pair (10C) of Cp2WH2. It can be seen immediately that the LP has most of its density away from the mouth of the wedge and is not ideal for mixing with the Zr fragment.
 | ||
| Fig. 10 (A–C) IBO for Cp2WH2, W–H bonds and lone pair; (D–F) IBO for the asymmetric (“out”) structure of 1: 3c2e bond, W–H bond and lone pair; (G–I) IBO for symmetric inversion TS of 1, W–H bonds and lone pair. | ||
| IBO: | W LP | W–H | W–H–Zr | |||||
|---|---|---|---|---|---|---|---|---|
| contr. from: | W | Zr | W | H | Zr | W | H | Zr |
| a Contributions larger than 0.02 e. b Zr interacts with both hydrides. | ||||||||
| Cp2WH2 | 1.546 | — | 0.733 | 1.175 | — | 0.733 | 1.175 | |
| Cp2ZrH⋯H⋯WCp2 | 1.588 | — | 0.729 | 1.167 | — | 0.476 | 1.332 | 0.106 |
| 1, out | 1.639 | — | 0.811 | 1.108 | — | 0.464 | 1.341 | 0.107 |
| 1, ioTS | 1.624 | — | 0.746 | 1.157 | — | 0.386 | 1.416 | 0.115 |
| 1, in | 1.613 | — | 0.712 | 1.179 | — | 0.426 | 1.382 | 0.110 |
| 1, iiTSb | 1.635 | — | 0.591 | 1.248 | 0.066 | 0.574 | 1.261 | 0.072 |
| 3a | 1.596 | — | 0.727 | 1.175 | — | 0.422 | 1.368 | 0.104 |
| 3b | 1.600 | — | 0.684 | 1.203 | — | 0.472 | 1.334 | 0.082 |
| 7a | 1.609 | 0.046 | 0.674 | 1.181 | 0.059 | 0.612 | 1.220 | 0.079 |
Fig. 10D–F show the corresponding IBOs for the asymmetric (“out”) structure of 1. The W lone pair (10F) has negligible mixing in of Zr, while one W–H bonding IBO mixes with 0.107 e of Zr, conferring some 3c2e character on this bond. On rearrangement to the Cs symmetric inversion TS, the Zr mixing is spread out over the two W–H bonds (0.066 and 0.072 e), but the W lone pair (Fig. 10I) remains mostly undisturbed.
Alkyne insertion into the Zr–C bond was also probed using DFT (see Fig. 11). Insertion of the parent alkyne ethyne is barrierless and highly exergonic (−31.2 kcal mol−1, see Table 3). In all other cases the reaction proceeds from the separated reactants directly to the insertion TS, without an intermediate π-complex. Addition of 2-butyne has a significant barrier (13.4 kcal mol−1) and is sufficiently exergonic (−20 kcal mol−1) that the reverse reaction is not accessible at or around room temperature; the same holds for phenylpropyne. For the more hindered alkynes 4,4-dimethyl-2-pentyne and adamantylpropyne, initial insertion is not very regioselective, but reaction to the non-thermodynamically-preferred product (bulky group close to Zr) is only ∼6 kcal mol−1 exergonic, making the insertion effectively (slowly) reversible.
 | ||
| Fig. 11 Free energy profile for insertion of 2-butyne and methyl-adamantyl-acetylene. | ||
| R1/To Zr | R2/To W | ΔG‡ | ΔGrxn |
|---|---|---|---|
| a Free energies relative to separated 1 and alkyne/alkene. | |||
[Zr]–C(R1)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R2)–[W] C(R2)–[W] |
|||
| H | H | n/a | −31.5 |
| Me | Me | 13.4 | −20.2 |
| Me | Ph | 14.5 | −19.4 |
| Ph | Me | 15.2 | −19.7 |
| Me | tBu | 16.5 | −14.1 |
| tBu | Me | 18.2 | −6.8 |
| Me | Ad | 16.2 | −11.2 |
| Ad | Me | 15.3 | −6.1 |
| tBu | tBu | 29.1 | +14.3 |
| Ad | Ad | 28.6 | +17.7 |
| [Zr]–(R1)–(R2)–[W] | |||
| CH2 | CH2 | 14.4 | −16.6 |
| CH2 | CHnBu | 17.4 | −14.3 |
| CHnBu | CH2 | 22.0 | −9.4 |
| CH2 | CMe2 | 19.3 | −8.7 |
| CMe2 | CH2 | 28.9 | −0.1 |
| CMe2 | CMe2 | 36.2 | +12.6 |
In terms of bonding, alkyne adduct 7a, perhaps surprisingly, shows a core arrangement similar to the symmetric inversion TS of 1: Zr interacts comparably with both hydrides (0.059/0.079 e, see Table 2 and Fig. 12A and B). Presumably due to geometric constraints, the two hydrides are tilted out of the Zr coordination plane. This is not an ideal geometry for donation to Zr (although “mismatched” Cp2WH2 orientation has also been reported for a Cu(I) complex59), and perhaps due to this we find for complex 7a a small amount of Zr mixing into the W LP (.046 e). Still, the overall pattern of dominant WH donation and relative inertness of the W lone pair seems to hold also for the alkyne adducts.
 | ||
| Fig. 12 IBOs for adduct 7a: (A and B) the W–H bonds; (C) lone pair. | ||
1-Hexene insertion was also checked. Consistent with the results for alkyne insertion, the lowest insertion barrier (17.1 kcal mol−1, only slightly higher than alkyne insertion) is found for the hexene “bulk” (nBu group) approaching the W centre. Hexene insertion is also exergonic enough to make it irreversible. To put this reaction in context, calculated energetics for insertion of ethene, isobutene and 2,3-dimethyl-2-butene are included in Table 3. The trend follows that of alkyne insertion, going from clearly exergonic (ethene) to highly endergonic (2,3-dimethyl-2-butene).
Correlations between structure and spectroscopic parameters
NMR spectroscopic parameters of transition metal hydrides are strongly affected by the electronic structure of the complex and M–H bonding situation.66–69 Even though all hydrides reported in this work seem to follow the same bonding pattern, there is a discrete variability in their δH and 1JWH values (see above). By plotting the experimentally obtained δHversus the computed closest Zr–H distance in the bridging hydrides (Fig. 13a), a linear correlation is obtained (R = 0.89). Similarly, a rough linear correlation is also found between chemical shift values and optimised Zr–W distances (ESI†). The 1JWH values are instead less responsive and the data are much more scattered (linear fit R = 0.67, ESI†).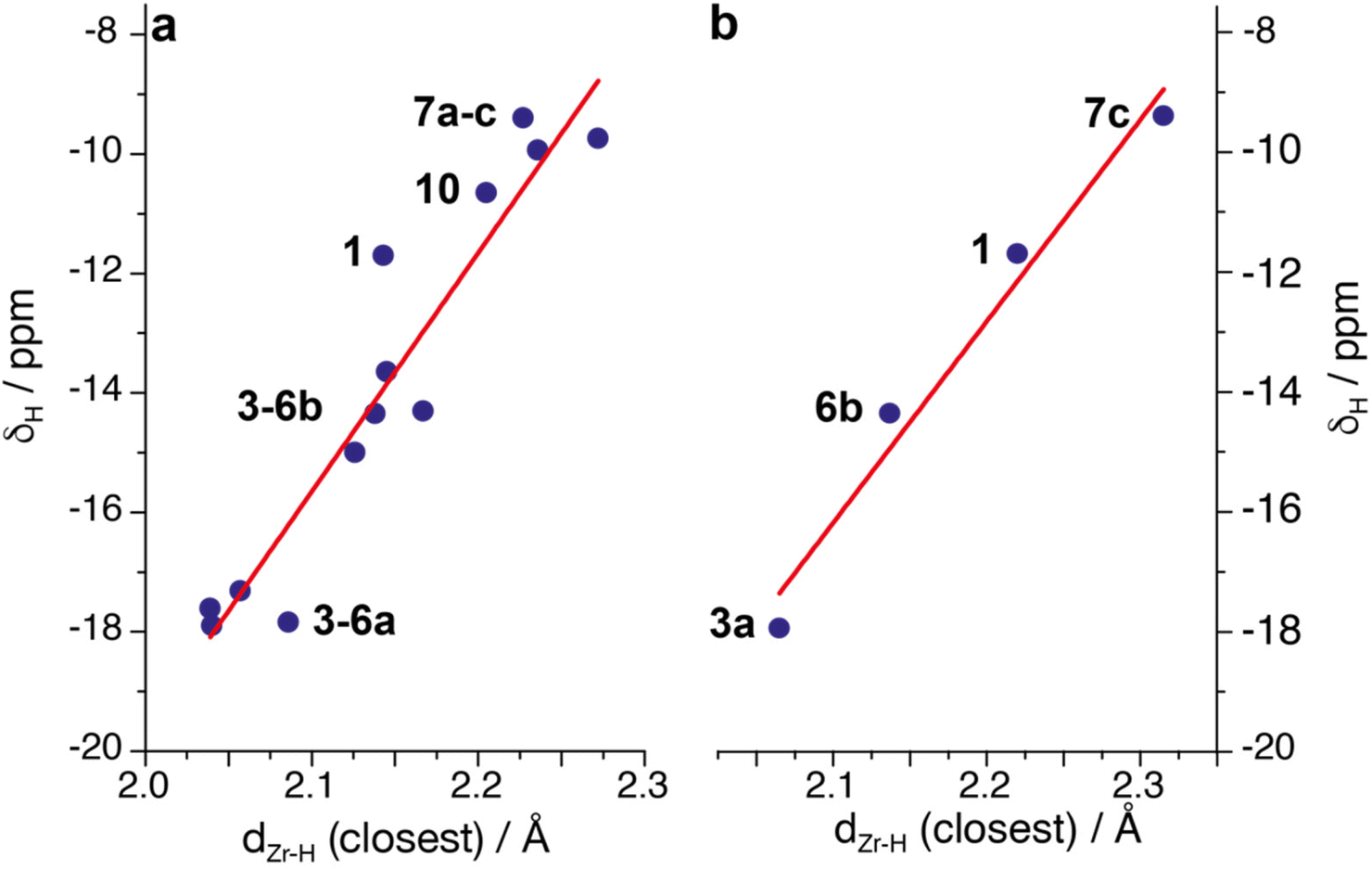 | ||
| Fig. 13 (a) Plot of experimental chemical shift values versus the closest Zr–H distance obtained by DFT optimised structures in complexes 1–7; (b) plot of chemical shift versus the approximate experimental Zr–H distances obtained by X Ray diffraction. | ||
In the approximation that bond length reflects the interaction degree between the Zr centre and the hydride ligand, we could expect that stronger bridging interactions would affect more strongly the NMR observables. Generally, the smaller the Zr–H distance in the bridging hydride, the more negative the δH and the lower the 1JWH. The opposite trend is observed with the Zr–W distance: the higher the intermetallic separation, the lower the hydride δH. The same linear trend was obtained also when δH values were plotted against the experimentally available Zr–H distances (Fig. 13b). Even though the presumed large uncertainty of the experimental M–H determination must be considered, there is good agreement with the theoretical data.
Given that δH values are averaged between bridging and non-bridging hydrides by their very fast interconversion at RT, the validity of this correlation over a range of 8 ppm is remarkable. The lower chemical shift at shorter Zr–H distances seems to fit with the general rule of thumb that bridging group IV metallocene hydrides are more shielded than the corresponding terminal ones. It is likely that the weak overlap between W–H and Zr empty orbitals described above may increase the paramagnetic shielding contribution70 or enhance the effect of spin–orbit coupling, as previously observed by some of us in Cp2WH2/Au(III) bridging hydrides.25
Irrespective of the quantum mechanical origin of such an effect, we can use this correlation to categorise all the complexes synthesised in this work. From the data reported in Fig. 13a, three main clusters of compounds can be identified: (i) the outside Lewis adducts, with Zr–H distances between 2.0 and 2.1 Å and δH around −18 ppm, (ii) the inside Lewis adducts (Zr–H 2.1 to 2.2 Å, δHca. −15 to −14 ppm) and (iii) the alkyne/alkene insertion products (Zr–H over 2.2 Å, δHca. −10 to −9 ppm). Complex 1 seems to fall in between class (ii) and class (iii), even though it fits better the experimental trend than the theoretical one. Such a linear response of δH for the whole series is a clear sign that minor alterations in the geometry of the adducts lead to a fine tuning of the Zr–H–W bridge, which can be assessed by simply using the 1H NMR chemical shift of the hydride atom.
Conclusion
Reactions of zirconocene alkyl cations with Lewis basic Cp2WH2 lead to the formation of the bridging hydride [Cp2Zr(μ-H)(μ–η1:η5-C5H4)WHCp]+ (1) upon σ-bond metathesis of one W–Cp ring. 1 is characterised by an unusual out of plane bridging hydride interaction, which makes the core of the complex very flexible. The thermodynamically preferred asymmetric configuration is in fast interconversion with a symmetric arrangement, with a calculated barrier of <2 kcal mol−1. Despite being floppy, the bridging interaction in 1 is not broken by other ligands. 1 reacts, indeed, with Lewis bases affording adducts where the bridging hydride is either close (W-inside) or opposite to the coordinated base (W-outside). With PMe3, the latter is preferred while with other more labile bases, such as Ptol3, THF or 3,5-lutidine, the inside configuration is obtained almost exclusively. In forming such adducts, the bridging hydride switches to an in-plane configuration and the average Zr–H distances decrease with respect to 1.Despite the proximity between the Lewis acidic Zr and the lone pair of W, reactions of 1 with alkynes, alkenes and H2 proceed without any bimetallic cooperativity. In reacting with unactivated internal alkynes and olefins, insertion proceeds selectively into the Zr–C bond leading to vinyl or alkyl-metallocene complexes. In these species, the hydrides still interact with the Zr centre, but assume a perpendicular orientation with respect to its coordination plane. With H2, a trimetallic complex is obtained upon σ-bond metathesis of the H–H bond.
DFT studies revealed that metal–metal interactions in all these species are negligible, and the lone pair remains localised on tungsten. On the other hand, considerable overlap between W–H and Zr empty orbitals is observed, suggesting that the most reasonable description of these species is an “open” 3c2e bonding interaction. The computed Zr–H bond distances correlate linearly with the experimentally observed 1H chemical shift values of the hydrides, indicating that NMR parameters can be used to assess the proximity of the bridging hydride to the Zr centre.
The results reported above strongly point to the fact that short metal–metal distances in the solid state or hydride 1H NMR trends in solution should not be used as univocal parameters to imply the presence of an M–M bond in heterobimetallic dihydrides. Moreover, very small alterations in the coordination environment of these complexes modulate the degree of interaction between the W–H bond and the Zr centre, with important effects on reactivity, as it is shown in the case of hydrogenolysis. We are currently trying to introduce further modifications in this class of compounds aiming at triggering bimetallic cooperation in small molecule activation and enable novel reactivity in zirconocene chemistry.
Experimental section
Materials and methods
All manipulations of air-sensitive materials were performed in flamed Schlenk glassware on a Schlenk line or in nitrogen-filled Mbraun Unilab gloveboxes with a high-capacity recirculator (<1 ppm O2 and H2O). Deuterated solvents were freeze–pump–thaw degassed, distilled over the appropriate drying agent (CaH2 or Na/K alloy), and stored over activated 4 Å molecular sieves in the glovebox.Cp2ZrMe271 and B(C6F5)372 were synthesized according to literature procedures. [HNMe2Ph][B(C6F5)4] was purchased from Strem and used as received. Cp2WH2 was purchased from Sigma Aldrich and further purified by vacuum sublimation when required. Trimethylphosphine (1 M in THF, Sigma Aldrich) was used as received. Tris-para-tolylphosphine and 3,5-lutidine (Sigma Aldrich) were dried under vacuum and stored in the glovebox. 2-Butyne, 4-4′-dimethyl-2-pentyne (Alfa Aesar), dimethylacetylene dicarboxylate, 1-phenylpropyne, and 1-hexene (Sigma Aldrich) were freeze–pump–thawed degassed and stored over activated 4 Å molecular sieves in the glovebox.
Experiments with H2 were performed in J-Young NMR tubes on a dedicated Schlenk line interfaced with a H2 gas supply (>99.5% purity) at 1 atmosphere. In the typical procedure, NMR solutions were initially freeze–pump–thaw degassed three times to remove the headspace. Successively, H2 was introduced in the NMR tube at −78 °C and the solution was left to equilibrate at the desired temperature.
1H, 19F, 31P{1H}, 13C{1H}, 1H NOESY, 1H,13C HMQC, 1H,13C HSQC, 1H,13C HMBC, 19F,1H HOESY NMR experiments have been recorded on Bruker Avance III HD 400 and Bruker DPX-300 spectrometers equipped with 1H, BB smartprobes and Z-gradients. 1H NMR spectra are referenced to residual protons of the deuterated solvent. 13C NMR spectra are referenced to the D-coupled 13C signals of the solvent. 19F NMR are referenced to an external standard of CFCl3. 31P NMR are referenced to an external standard of H3PO4.
Crystallographic details
CCDC 2212595–2212600† contain the supplementary crystallographic data for this paper.DFT calculations
DFT calculations were performed using Gaussian 1673 coupled to an external optimizer.74 All structures were fully optimised, without constraints or symmetry restrictions, as minima or transition states, using the dispersion-aware functional MN1575 and the cc-pVDZ(PP) basis set.76–79 Analytical frequency calculations were done to check the nature of all stationary points (minima without imaginary frequencies, transition states with exactly one with the correct movement). IRC calculations failed in several cases for the flat energy surfaces associated with cluster rearrangement, so connectivity of each transition state was checked by moving each TS along the reaction coordinate (in both directions, 0.03 to 0.20 Bohr) and re-optimizing. Thermal corrections (enthalpy and entropy) at 298 K were calculated from the vibrational analyses and were scaled by 0.67 to account for reduced freedom in solution.80,81 Improved single-point energies were calculated at the MN15/cc-pVTZ(PP) level including a solvent (benzene) correction using the SMD model,82 and were combined with the thermal corrections to obtain final free energies; all energies mentioned in the text are Gibbs free energies. Optimisations including the solvent model were also attempted, but in many cases these were found to fail spectacularly due to gradient discontinuities apparently caused by discretization issues. In any case, the solvent used here (benzene) is not very polar and is not expected to affect the results much.The “intrinsic bond orbital” formalism introduced by Knizia64,65 was used to analyse bonding in the bimetallic cores of Zr/W bimetallic complexes. IBOview was used to calculate (exponent: 4) IBOs from the MN15/cc-pVDZ(PP) wave functions that were produced by Gaussian and converted to Molden83 format.
Data availability
Experimental details, NMR spectra, crystallographic and DFT details are collected in the Electronic ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
LR acknowledges the University of East Anglia and the Royal Society of Chemistry (R21-1292108967) for financial support. We thank MIUR (AMIS, “Dipartimenti di Eccellenza 2018–2022” program). We also thank the UK National Crystallography Service84 for the acquisition of crystallographic data of 1B(C6F5)4, 2, 7a and 10, while Dr Leonardo Tensi (University of Perugia) is acknowledged for assistance with the X-Ray structures of 3a and 6b.References
- L. M. Venanzi, Coord. Chem. Rev., 1982, 43, 251–274 CrossRef CAS.
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS.
- M. Navarro and J. Campos, in Advances in Organometallic Chemistry, Academic Press Inc., 2021, vol. 75, pp. 95–148 Search PubMed.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev.: Sci. Eng., 2012, 54, 1–40 CrossRef CAS.
- E. Bodio, M. Picquet and P. le Gendre, in Homo- and Heterobimetallic Complexes in Catalysis: Cooperative Catalysis, ed. P. Kalck, Springer International Publishing, Cham, 2015, pp. 139–186 Search PubMed.
- G. G. Hlatky and R. H. Crabtree, Coord. Chem. Rev., 1985, 65, 1–48 CrossRef CAS.
- M. D. Fryzuk, J. B. Love, S. J. Rettig and V. G. Young, Science, 1997, 275, 1445–1447 CrossRef CAS.
- X. Zhao, I. P. Georgakaki, M. L. Miller, R. Mejia-Rodriguez, C. Y. Chiang and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 3917–3928 CrossRef CAS PubMed.
- J. C. Green, M. L. H. Green and G. Parkin, Chem. Commun., 2012, 48, 11481–11503 RSC.
- R. E. Adams, T. A. Grusenmeyer, A. L. Griffith and R. H. Schmehl, Coord. Chem. Rev., 2018, 362, 44–53 CrossRef CAS.
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed.
- R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506–8544 CrossRef CAS PubMed.
- S. M. Baldwin, J. E. Bercaw, L. M. Henling, M. W. Day and H. H. Brintzinger, J. Am. Chem. Soc., 2011, 133, 1805–1813 CrossRef CAS PubMed.
- S. M. Baldwin, J. E. Bercaw and H. H. Brintzinger, J. Am. Chem. Soc., 2010, 132, 13969–13971 CrossRef CAS PubMed.
- S. M. Baldwin, J. E. Bercaw and H. H. Brintzinger, J. Am. Chem. Soc., 2008, 130, 17423–17433 CrossRef CAS PubMed.
- A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 3822–3835 CrossRef CAS.
- M. J. Butler, A. J. P. White and M. R. Crimmin, Organometallics, 2018, 37, 949–956 CrossRef CAS.
- M. J. Butler, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2016, 55, 6951–6953 CrossRef CAS PubMed.
- J. W. Bruno, J. C. Huffman, M. A. Green and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 8310–8312 CrossRef CAS.
- M. Oishi and H. Suzuki, Inorg. Chem., 2009, 48, 2349–2351 CrossRef CAS PubMed.
- A. Berry, M. L. H. Green, J. A. Bandy and K. Prout, J. Chem. Soc., Dalton Trans., 1991, 2185–2206 RSC.
- V. I. Bakhmutov, M. Visseaux, D. Baudry, A. Dormond and P. Richard, Inorg. Chem., 1996, 35, 7316–7324 CrossRef CAS PubMed.
- G. Parkin, Metal-metal bonding in bridging hydride and alkyl compounds, 2010, vol. 136 Search PubMed.
- J. W. Lauher and R. Hoffmann, J. Am. Chem. Soc., 1976, 98, 1729–1742 CrossRef CAS.
- L. Rocchigiani, W. T. Klooster, S. J. Coles, D. L. Hughes, P. Hrobárik and M. Bochmann, Chem. – Eur. J., 2020, 26, 8267–8280 CrossRef CAS PubMed.
- J. W. Bruno, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 444–445 CrossRef CAS.
- H. Brunner and D. Mijolovic, J. Organomet. Chem., 1999, 577, 346–350 CrossRef CAS.
- A. Albinati, R. Naegeli, A. Togni and L. M. Venanzi, Organometallics, 2002, 2, 926–928 CrossRef.
- H. Brunner, M. Muschiol, T. Neuhierl and B. Nuber, Chem. – Eur. J., 1998, 4, 168–171 CrossRef CAS.
- X. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10015–10031 CrossRef CAS.
- L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. Macchioni, Angew. Chem., Int. Ed., 2011, 50, 11752–11755 CrossRef CAS PubMed.
- L. Rocchigiani, G. Bellachioma, G. Ciancaleoni, A. Macchioni, D. Zuccaccia and C. Zuccaccia, Organometallics, 2010, 30, 100–114 CrossRef.
- A. D. Horton and J. de With, Organometallics, 1997, 16, 5424–5436 CrossRef CAS.
- R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, J. Chem. Soc., Dalton Trans., 2008, 2832–2838 RSC.
- A. Bartole-Scott, A. J. Lough and I. Manners, Polyhedron, 2006, 25, 429–436 CrossRef CAS.
- C. P. Casey, R. E. Palermo, R. F. Jordan and A. L. Rheingold, J. Am. Chem. Soc., 1985, 107, 4597–4599 CrossRef CAS.
- A. Ramos, E. Otten and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 15610–15611 CrossRef CAS PubMed.
- D. Takeuchi, J. Kuwabara and K. Osakada, Organometallics, 2003, 22, 2305–2311 CrossRef CAS.
- A. Albinati, A. Togni and L. M. Venanzi, Organometallics, 1986, 5, 1785–1791 CrossRef CAS.
- O. W. Howarth, C. H. McAteer, P. Moore, G. E. Morris and N. W. Alcock, J. Chem. Soc., Dalton Trans., 1982, 541–548 RSC.
- N. S. Radu, P. K. Gantzel and T. D. Tilley, J. Chem. Soc., Chem. Commun., 1994, 1175–1176 RSC.
- F. Hannig, R. Fröhlich, K. Bergander, G. Erker and J. L. Petersen, Organometallics, 2004, 23, 4495–4502 CrossRef CAS.
- H. Braunschweig and C. Kollann, Z. Naturforsch., B: J. Chem. Sci., 1999, 54, 839–842 CrossRef CAS.
- L. Sian, A. Dall'Anese, A. Macchioni, L. Tensi, V. Busico, R. Cipullo, G. P. Goryunov, D. Uborsky, A. Z. Voskoboynikov, C. Ehm, L. Rocchigiani and C. Zuccaccia, Organometallics, 2022, 41, 547–560 CrossRef CAS.
- Reactions with 1B(C6F5)4 gave the same outcome.
- K. Yan, A. Pindwal, A. Ellern and A. D. Sadow, Dalton Trans., 2014, 43, 8644–8653 RSC.
- R. F. Jordan, C. S. Bajgur, W. E. Dasher and A. L. Rheingold, Organometallics, 1987, 6, 1041–1051 CrossRef CAS.
- M. Dahlmann, R. Fröhlich and G. Erker, Eur. J. Inorg. Chem., 2000, 2000, 1789–1793 CrossRef.
- A. D. Miller, S. A. Johnson, K. A. Tupper, J. L. McBee and T. D. Tilley, Organometallics, 2009, 28, 1252–1262 CrossRef CAS.
- V. H. Gessner, J. F. Tannaci, A. D. Miller and T. D. Tilley, Acc. Chem. Res., 2011, 44, 435–446 CrossRef CAS PubMed.
- G. E. Herberich and W. Barlage, Organometallics, 1987, 6, 1924–1930 CrossRef CAS.
- P. H. M. Budzelaar, D. L. Hughes, M. Bochmann, A. Macchioni and L. Rocchigiani, Chem. Commun., 2020, 56, 2542–2545 RSC.
- P. Legzdins, J. T. Martin, F. W. B. Einstein and A. C. Willis, J. Am. Chem. Soc., 2002, 108, 7971–7981 CrossRef.
- F. Wu and R. F. Jordan, Organometallics, 2005, 24, 2688–2697 CrossRef CAS.
- M. P. Johnson and D. F. Shriver, J. Am. Chem. Soc., 1966, 88, 301–304 CrossRef CAS.
- J. W. Bruno, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 444–445 CrossRef CAS.
- M. P. Johnson and D. F. Shriver, J. Am. Chem. Soc., 1966, 88, 301–304 CrossRef CAS.
- A. Hicken, A. J. P. White and M. R. Crimmin, Dalton Trans., 2018, 47, 10595–10600 RSC.
- M. Amati and F. Lelj, Can. J. Chem., 2009, 87, 1406–1414 CrossRef CAS.
- H. Brunner, A. Hollman and M. Zabel, J. Organomet. Chem., 2001, 630, 169–176 CrossRef CAS.
- H. Brunner, M. Muschiol, T. Neuhierl and B. Nuber, Z. Naturforsch., B: J. Chem. Sci., 1999, 54, 337–340 CrossRef CAS.
- H. Brunner, A. Hollman, B. Nuber and M. Zabel, J. Organomet. Chem., 2001, 633, 1–6 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobárik and M. Bochmann, J. Am. Chem. Soc., 2018, 140, 8287–8302 CrossRef CAS PubMed.
- L. Rocchigiani, P. H. M. Budzelaar and M. Bochmann, Chem. Sci., 2019, 10, 2633–2642 RSC.
- Z.-L. Xue, T. M. Cook and A. C. Lamb, J. Organomet. Chem., 2017, 852, 74–93 CrossRef.
- P. Hrobárik, V. Hrobáriková, F. Meier, M. Repiský, S. Komorovský and M. Kaupp, J. Phys. Chem. A, 2011, 115, 5654–5659 CrossRef PubMed.
- Y. Ruiz-Morales, G. Schreckenbach and T. Ziegler, Organometallics, 1996, 15, 3920–3923 CrossRef CAS.
- E. Samuel and M. D. Rausch, J. Am. Chem. Soc., 1973, 95, 6263–6267 CrossRef CAS.
- S. J. Lancaster, ChemSpider 10.1039/SP215.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. v Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. v Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT.
- P. H. M. Budzelaar, J. Comput. Chem., 2007, 28, 2226–2236 CrossRef CAS PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108 CrossRef PubMed.
- R. Raucoules, T. de Bruin, P. Raybaud and C. Adamo, Organometallics, 2009, 28, 5358–5367 CrossRef CAS.
- S. Tobisch and T. Ziegler, J. Am. Chem. Soc., 2004, 126, 9059–9071 CrossRef CAS PubMed.
- J. B. Foresman, T. A. Keith, K. B. Wiberg, J. Snoonian and M. J. Frisch, J. Phys. Chem., 1996, 100, 16098–16104 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2212595–2212600. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03833a |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |