
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceσ-Hole interactions in organometallic catalysts: the case of methyltrioxorhenium(VII)†
Miriam
Calabrese
 a,
Andrea
Pizzi
a,
Andrea
Daolio
a,
Antonio
Frontera
b and
Giuseppe
Resnati
*a
a,
Andrea
Pizzi
a,
Andrea
Daolio
a,
Antonio
Frontera
b and
Giuseppe
Resnati
*a
aNFMLab, Department of Chemistry, Materials, and Chemical Engineering “Giulio Natta”, Politecnico di Milano, via L. Mancinelli 7, I-20131 Milano, Italy. E-mail: giuseppe.resnati@polimi.it
bDepartment of Chemistry, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain
First published on 26th December 2022
Abstract
Methyltrioxorhenium(VII) (MTO) is a widely employed catalyst for metathesis, olefination, and most importantly, oxidation reactions. It is often preferred to other oxometal complexes due to its stability in air and higher efficiency. The seminal papers of K. B. Sharpless showed that when pyridine derivatives are used as co-catalysts, MTO-catalyzed olefin epoxidation with H2O2 as oxidant, a particularly useful reaction, is accelerated, with pyridine speeding up catalytic turnover and increasing the lifetime of MTO under the reaction conditions. In this paper, combined experimental and theoretical results show that the occurrence of σ-hole interactions in catalytic systems extends to MTO. Four crystalline adducts between MTO and aliphatic and heteroaromatic bases are obtained, and their X-ray analyses display short Re⋯N/O contacts opposite to both O–Re and C–Re covalent bonds with geometries consistent with σ-hole interactions. Computational analyses support the attractive nature of these close contacts and confirm that their features are typical of σ-hole interactions. The understanding of the nature of Re⋯N/O interactions may help to optimize the ligand-acceleration effect of pyridine in the epoxidation of olefins under MTO catalysis.
Introduction
Catalysis plays a primary role in virtually all aspects of life and in the majority of manufacturing processes that produce materials supporting the development of society.1 A transition metal is frequently present at the catalytic active site, and the interactional landscape around the metal substantially impacts the efficiency, selectivity, and other aspects of the catalytic process. For a given metal, this landscape varies as a function of the oxidation state and the nature and number of ligated groups. A detailed understanding of the features of interactions formed by the metal is instrumental in identifying optimal reaction conditions and designing highly efficient catalysts.σ-Hole interactions are weak bonds between an atom donating electron density (e.g., a neutral or anionic atom functioning as the nucleophile) and a region with deficient electron density and positive electrostatic potential (the σ-hole) present on another atom (functioning as the electrophile) opposite to a σ covalent bond it is involved in. The role and relevance of these weak bonding interactions in catalysis have been recently the subject of intense research. For instance, many catalysts accelerating a variety of reactions are based on σ-hole interactions, wherein the electrophile is an element of 17,2 16,3 and 154 groups of the periodic table of elements.
It is well-known that a metal centre can act as an electrophilic site in key steps of the catalytic or stoichiometric processes.5 For instance, OsO4 is a benchmark catalyst in the enantioselective dihydroxylation of olefins, and the reaction rate increases in the presence of pyridine derivatives, which act as co-catalysts.6 We recently reported7 that the binding of pyridine derivatives to osmium tetroxide occurs via σ-hole interactions wherein osmium is the electrophile. Tetroxides of group 7 elements give similar interactions. For instance, manganese and rhenium atoms in MnO4− and ReO4− form the so named matere bond (MaB), namely the σ-hole bonding wherein Re and Mn are the electrophiles, and neutral and anionic atoms are the nucleophiles.8 Rhenium shows an electrophilic character and forms MaBs also in perrhenate silyl esters.9
Several oxorhenium derivatives10,11 are widely employed catalysts thanks to their versatility. They are often preferred to oxometal complexes of Mo, V, or W due to their longer shelf-life and higher efficiency. Their use span isomerization reactions, olefin metathesis, epoxidation, and deoxydehydration.12 Much attention was given to methyltrioxorhenium (MTO).13 The compound was first described in 197914 and it became a widely employed catalyst in olefin epoxidation15 after K. B. Sharpless16,17 showed that MTO-catalyzed olefin epoxidations in the presence of pyridine derivatives as co-catalysts are a remarkable example of effective ligand-accelerated catalysis. In the reaction solution, pyridines form adducts with MTO, and the observed acceleration of the epoxidation rate18–20 occurs thanks to the increased catalyst lifetime and catalytic turnover.16,17
Herein, we report the combined experimental and theoretical studies proving that the relevance of σ-hole interactions to catalytic systems extends to MTO. It is shown how the binding of nitrogen and oxygen Lewis bases to MTO occurs via Re⋯N/O matere bonds. Specifically, the adducts, 3a–d, formed by MTO (1) with 1,2-di(pyridin-4-yl)ethane (2a), 1,4-diazabicyclo[2.2.2]octane (2b), 4,4′-bipyridine-1,1′-dioxide (2c), and pyrazine 1-oxide (2d) have been obtained (Fig. 1) and characterized through single crystal X-ray analyses. All these adducts display short Re⋯N/O contacts‡ on the elongation of one of the covalent bonds involving the metal (Fig. 2). Theoretical investigations (i.e., molecular electrostatic potential studies, quantum theory of “atoms-in-molecules” analyses combined with the noncovalent interaction plot analyses) confirm the attractive nature of Re⋯N/O contacts and the electrophilic character of rhenium. Experiments of competitive self-assembly prove the ability of MaBs formed by MTO to identify the tectons preferentially involved in selective co-crystal formation. The understanding of the nature of the Re⋯N/O bonding gives instrumental information to optimize the ligand-acceleration effect of pyridine by rationally designing the subtle balance between the bindings and equilibria15,20,21 occurring in H2O2-based epoxidation of olefins under MTO/pyridine catalysis.
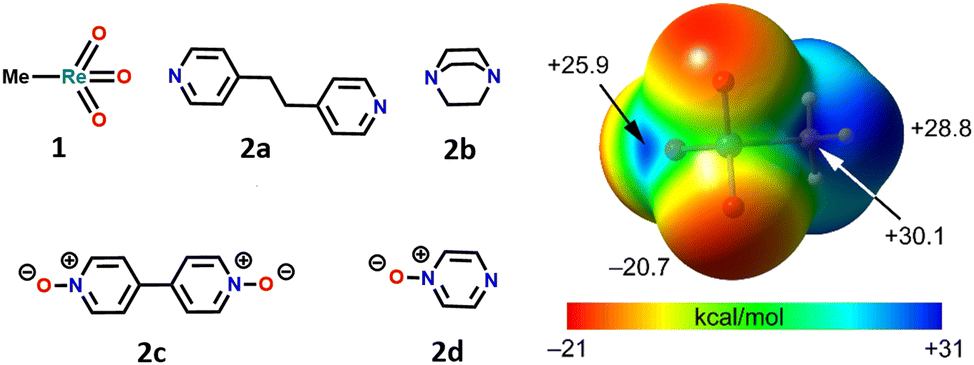 | ||
| Fig. 1 Left: molecular structures of Lewis acids and bases 1 and 2 forming matere bonded adducts studied in this work. Right: MEP surfaces of MTO (1) at the PBE0-D3/def2-TZVP level of theory. Isovalue 0.001 a.u. The energies at selected points of the surface are given in kcal mol−1. | ||
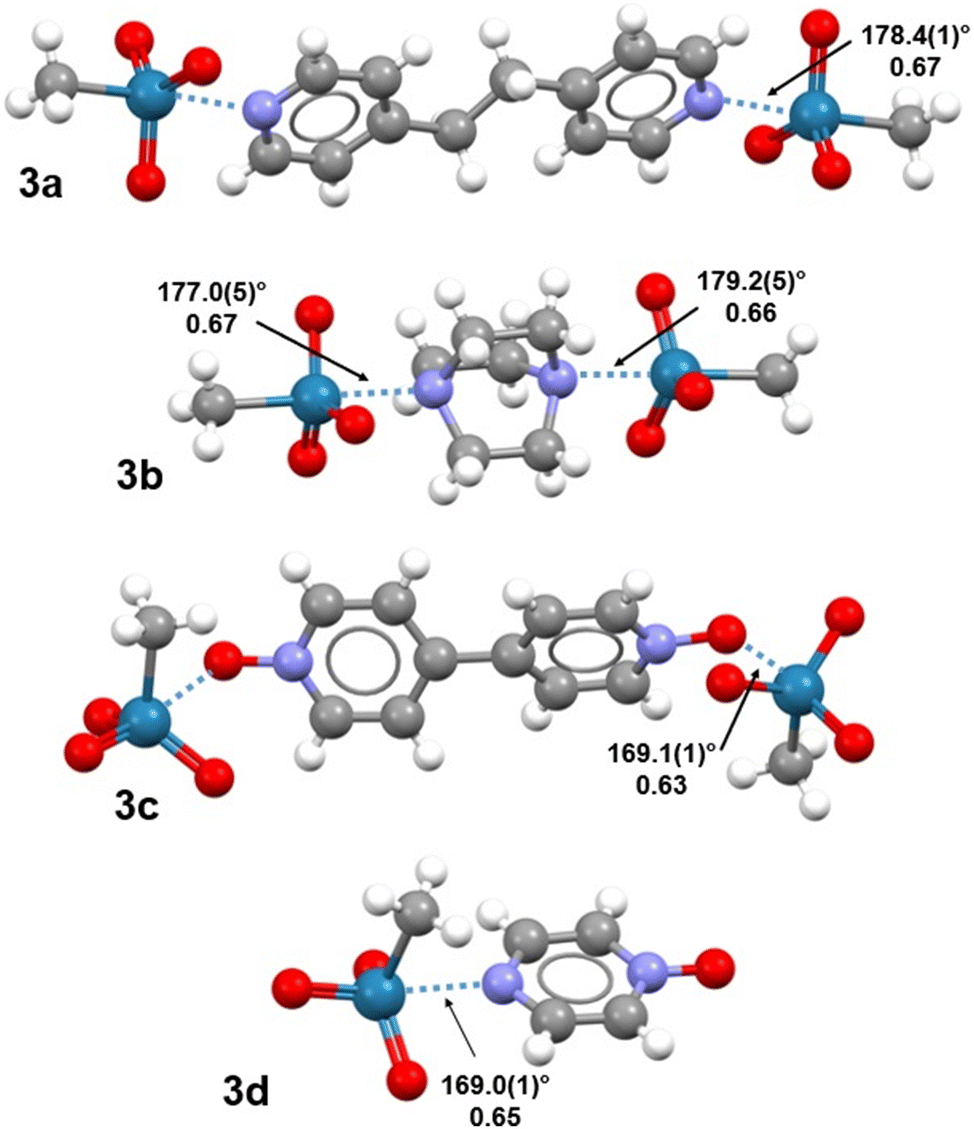 | ||
| Fig. 2 Ball and stick representation (Mercury 4.3.1) of adducts 3a–d. MaBs are blue dotted lines; Nc values and relevant C/O–Re⋯N/O angles are given close to MaBs, a second trimeric adduct is present in the unit cell of 3b, the Nc values and angles are 0.67/178.4(5)° and 0.67/176.9(5)°. Colour code: white, hydrogen; grey, carbon; indigo, nitrogen; red, oxygen; navy, rhenium. | ||
Experimental
Materials and methods
All starting compounds were purchased from commercial suppliers (Merck, TCI, Abcr) and used without further purification. 13C and 19F NMR spectra in solution were recorded at ambient temperature on a Nuclear Magnetic Resonance Spectrometer AVANCE III, Bruker-BioSpin (400 MHz). The Larmor frequency for 13C was 100.61 MHz and for 19F, it was 376.50 MHz. All the chemical shifts are given in ppm, and CDCl3 was used as a solvent. FT-IR spectra were obtained using a Nicolet Nexus FT-IR spectrometer equipped with a UATR unit.Synthesis and characterization
Analogous procedures were used for the synthesis of adducts 3b–d and 1,2-di(pyridine-4-yl)ethane·1,2-diiodotetrafluoro-ethane adducts (see ESI†).
X-ray diffraction analyses
The single crystal X-ray data (SC-XRD) of the adducts were collected at ambient temperature (3a, 3b, 3c) or 100 K (3d) using a Bruker SMART APEX II CCD area detector diffractometer. Data collection, unit cell refinement, and data reduction were performed using Bruker SAINT. Structures were solved by direct methods using SHELXT22 and refined by full-matrix least-squares on F2 with anisotropic displacement parameters for the non-H atoms using SHELXL-2016/6.23 Absorption correction was performed based on multi-scan procedure using SADABS or XABS2.Structural analysis was aided using the program PLATON.24 The hydrogen atoms were calculated in ideal positions with isotropic displacement parameters set to 1.2Ueq of the attached atom.
Results and discussion
X-ray analyses and competitive co-crystallizations
Trimeric or dimeric adducts assembled via short Re⋯N/O contacts are distinctive structural motifs in all four systems 3a–d. In co-crystals 3a,b, these adducts are assembled through Re⋯N MaBs formed opposite to the H3C–Re bond (Fig. 2). These interactions are much shorter than the sum of the van der Waals radii of involved atoms, with normalized contacts (Nc)§ in the range 0.66–0.67. The C–Re⋯N angles are almost linear (>177°), meeting typical requirements of σ-hole interactions. In all previous cases, wherein the electrophilic character of rhenium was reported to result in MaBs formation,8,9 the interaction developed opposite to oxygen, namely a highly electronegative atom. Here, the interaction develops opposite to a carbon, indicating that σ-holes at rhenium can be strong enough to drive the formation of short contacts in the solid also when they are opposite to mildly electronegative elements.Adducts 3c,d display MaBs at the extension of an O–Re bond; these contacts are shorter (Nc 0.63 and 0.65) and expectedly stronger than analogous contacts in 3a,b, consistent with the well-known direct correlation between the electronegativity of an atom and the positive electrostatic potential at the σ-hole opposite to a covalent bond it is involved in (namely the formation of short σ-hole interactions opposite to the atom). In 3c,d, the O–Re⋯N/O angles (169.0(1)° and 169.1(1)°) are less close to linearity than in 3a,b. Of note, the formation of these adducts do not result in a pronounced distortion of the O–Re–O angles involving the oxygen atom opposite to the incoming nucleophile (104.7(1)–105.7(1)°), so that the tetrahedral geometry of pure 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 is substantially maintained.
25 is substantially maintained.
FTIR spectra in the solid state of compound 3a–d (see ESI†) display ν Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands that are red-shifted (e.g., 968 and 926 cm−1 in 3a) compared to pure 1 (994 and 938 cm−1). Similar effects in analogous MTO⋯nucleophile26 and OsO4⋯nucleophile7 adducts have been related to the donation of electron density from the ligand to the metal. Also, the ν H3C–Re band of 1 (570 cm−1 in the pure compound) is red-shifted in 3a,b (546 and 550, respectively), consistent with a charge transfer from the nitrogen lone pair to the C–Re antibonding orbital when MaB is formed. Blue-shifts of signals associated with ν CC of aromatic Lewis bases 2a,c,d (1400–1600 cm−1) and with δ C–H of aliphatic base 2b (1300–1500 cm−1) confirm the n → σ* donation. Analogous hypsochromic shifts have been reported for similar σ-hole bonded adducts wherein the electrophile is a halogen.27
O bands that are red-shifted (e.g., 968 and 926 cm−1 in 3a) compared to pure 1 (994 and 938 cm−1). Similar effects in analogous MTO⋯nucleophile26 and OsO4⋯nucleophile7 adducts have been related to the donation of electron density from the ligand to the metal. Also, the ν H3C–Re band of 1 (570 cm−1 in the pure compound) is red-shifted in 3a,b (546 and 550, respectively), consistent with a charge transfer from the nitrogen lone pair to the C–Re antibonding orbital when MaB is formed. Blue-shifts of signals associated with ν CC of aromatic Lewis bases 2a,c,d (1400–1600 cm−1) and with δ C–H of aliphatic base 2b (1300–1500 cm−1) confirm the n → σ* donation. Analogous hypsochromic shifts have been reported for similar σ-hole bonded adducts wherein the electrophile is a halogen.27
When 13C NMR spectra of pure 1 are registered in different solvents containing N and O atoms, which can function as donors of electron density (i.e., MaB acceptors), the δC(CH3–ReO3) signal moves progressively to lower fields on increasing the electron donor ability of the solvent (Table S1†). Tetrahydrofuran and acetonitrile dissociate 1⋯pyridines adducts28,29 and these δC signal shifts can be mainly attributed to MaB formation rather than to a generic solvent effect. 13C NMR spectra of adducts 3a–d in diluted CDCl3 present a peak, for CH3–ReO3, at lower fields than the peak of pure 1. For instance, the δC chemical shift change shown by 3a and 3b is 4.24 and 4.93 ppm, respectively. These shifts indicate that 3a–d, similar to analogous adducts,18,26,30,31 are present, at least in part, as undissociated species. A single and sharp δC(CH3–ReO3) signal was also observed when pure 1 was added to the adduct solutions, namely when the 1:nucleophile ratio was >1. This proves that, at room temperature, the association equilibrium is rapid at the NMR timescale and supports the rationalization of the observed MaBs as non-covalent interactions.
In order to assess the ability of MaB to prevail over other non-covalent interactions in controlling the self-assembly processes, we performed experiments of competitive co-crystal formation, i.e., slow evaporation of solutions, wherein 2a or 2c was in the presence of equimolar amounts of 1 (MaB donor) and a donor of HB (the benchmark noncovalent interaction) or of HaB (the benchmark σ-hole interaction). The crystallization solvent may affect the relative ability of different interactions to control the co-crystal formation32 and competitive co-crystallization experiments were carried out using two solvents with quite different polarities (Tables S2 and S3†). Dipyridylethane 2a forms halogen bonded adducts with 1,2-diiodotetrafluroethane (see ESI†) and 1,4-diiodotetraflurobenzene.33 From solutions containing 1, 2a, and one of the HaB donors mentioned above, the matere bonded adduct 3a is preferentially formed, independent of the used solvent. This is consistent with the fact that the Nc values of the HaBs in corresponding co-crystals are greater than the Nc value of MaB in 3a (Table S4†). Hydrogen bonded adducts are formed by dipyridylethane 2a with 1,4-dihydroxybenzene33 and 4-cyanophenol34 and by dipyridyl dioxide 2c with pyromellitic35 and tartaric acids.36 The matere bonded adduct 3a is formed from solutions containing 1, 2a, and 1,4-dihydroxybenzene or 4-cyanophenol and the matere bonded adduct 3c from the solution containing 1, 2c, and pyromellitic acid. Differently, the hydrogen bonded co-crystal 2c⋯tartaric acid is preferentially isolated on evaporation of solutions containing 1, 2c, and tartaric acid. While the reliability of Nc values of HBs might be limited, it is interesting to observe that the Nc value of the HB in 2c⋯tartaric acid adduct is smaller than the Nc of the MaB in 3c and it is the smallest of the HBs in co-crystals possibly formed in competitive experiments discussed above (Table S4†).¶
These competitive co-crystallization experiments indicate that 1 has a quite strong tendency to interact with nucleophiles.31 It forms MaBs that prevail over the HaBs and HBs involving various and fairly robust electrophiles, namely, it is particularly effective in directing selective self-assembly processes.
Computational analyses
Calculations were performed to further analyse and characterize the short contacts involving 1. The molecular electrostatic potential (MEP) surface of MTO (Fig. 1) shows the presence of positive σ-holes on the extension of the C–Re and O–Re covalent bonds, consistent with the geometry of the short contacts observed in the co-crystals. Coherent with the electronegativity of the two elements, the MEP at the σ-hole opposite to the methyl group (+25.9 kcal mol−1) is slightly smaller than those opposite to the O-atoms (+30.1 kcal mol−1). These values are similar to those of σ-holes of OsO4 (+36.4 kcal mol−1).7Two adducts have been fully optimized (see ESI† for DFT details) in order to analyse the ability of 1 to establish MaBs with two typical Lewis bases (acetonitrile and pyridine) in the absence of crystal packing effects. Moreover, the quantum theory of “atoms-in-molecules” (QTAIM) combined with the noncovalent interaction plot (NCIPlot) index analyses have been carried out to reveal the NCIs and to identify which molecular regions interact. The results are gathered in Fig. 3, where only the intermolecular interactions are represented. The MaBs are characterized by the corresponding bond critical points (CPs) and bond paths connecting the N atom to the Re atom. Moreover, the MaBs are also revealed by the NCIplot index analysis, showing isosurfaces located between the Re atoms and the Lewis bases. The NCIplot isosurfaces present different colours (light blue or dark blue in line with the interaction energies, which are −4.1 and −11.3 kcal mol−1 for acetonitrile and pyridine, respectively). The interaction energies and Nc values are in good agreement with the relative basicity of acetonitrile and pyridine. For the pyridine complex, a secondary C–H⋯O interaction is also observed, characterized by a bond CP, bond path, and green NCIplot isosurface (weak interaction). We have also estimated the contribution of this ancillary interaction using the energy predictor proposed by Espinosa et al.37 (E = 0.5 × Vr) that is based on the value of the potential energy density (Vr) at the bond CP that connects the hydrogen atom to the donor atom. As a result, the contribution of this H-bond is −2.8 kcal mol−1, thus confirming that the adduct formation is dominated by the matere bond. It can be also observed that in this pyridine complex, the NCIplot index shows that the outer part of the isosurface is yellow, disclosing some N⋯O repulsion between the negative O-atoms and the N-atom of the Lewis base. It is worth highlighting that the theoretical Nc value of the pyridine complex (Nc = 0.68) is similar to those observed experimentally, thus supporting the structure-guiding role of the MaBs in the crystals and eliminates the possibility that the Re⋯N/O contacts simply originate from packing effects. We have analysed orbital charge transfer effects using the NBO analysis and focusing on the second order perturbation energies (E(2) values in Fig. 3). The most important orbital contribution in both complexes is an electron donation from the filled LP orbital at the N atom to the empty σ*(Re–C) orbital (see Fig. 3, bottom). This orbital analysis strongly supports the σ-hole nature of the interaction, similar to the conventional σ-hole interactions involving the p-block elements. The E(2) energies are significant in both acetonitrile and pyridine adducts. In fact, they are larger than the interaction energies. This result discloses the relevance of orbital effects in the matere bonds involving methyltrioxorhenium(VII) as a σ-hole donor molecule. In addition, the large orbital contribution in the pyridine complex suggests some partial covalent character of the MaB in this adduct in line with the small Nc value and dark blue colour of the NCIplot isosurface.
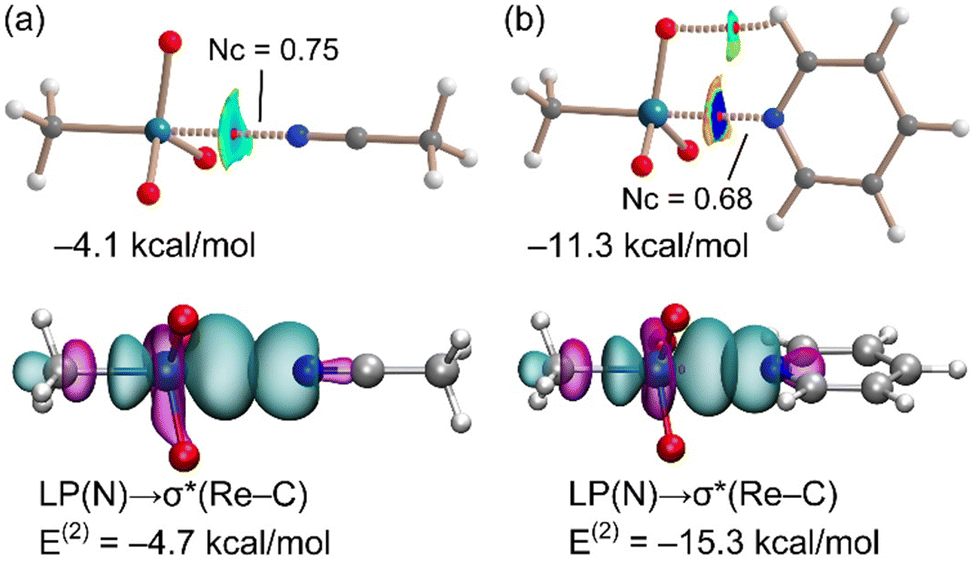 | ||
| Fig. 3 Top: PBE0-D3/def2-TZVP optimized geometries of acetonitrile (a) and pyridine (b) complexes. The QTAIM analysis: bond CPs in red, matere bonds as dashed bonds. NCIPlot: |RGB| isosurface 0.45 a.u.; color range 0.08 a.u. (red) ≤ (signλ2)ρ ≤ −0.08 a.u. (blue). Only the intermolecular interactions are shown. The interaction energies are indicated. Bottom: NBOs corresponding to the LP(N) → σ*(Re–C) donor–acceptor interactions. | ||
A similar QTAIM/NCIPlot study has been performed for the adducts characterized by X-ray analysis in order to investigate the interactions as they stand in the solid state. The results for the MeReO3 adducts are summarized in Fig. 4. In the four adducts, each MaB is characterized by the corresponding bond CP and bond path connecting the N/O atom to the Re atom, thus confirming the existence of the interaction. The analysis also reveals the existence of weak C–H⋯O contacts that are characterized by green NCIplot index isosurfaces. They also reveal that the Re⋯N/O interaction is strong (dark-blue isosurface) in all adducts and discloses the existence of N/O⋯O repulsion characterized by the red-yellow (repulsive) parts of the surfaces. Finally, the interaction energies of the contacts estimated using the QTAIM analysis range from −13.2 kcal mol−1 in 3a to −16.1 kcal mol−1 in 3d. The strong nature of these interactions agrees well with the short distances and dark blue color of the NCIplot isosurfaces, and they are comparable to the OsB energies of similar adducts reported by us.7 It should be noted that the QTAIM energy predictor was initially developed for H-bonds,37 but it has also been found adequate for σ-hole interactions like halogen,38,39 chalcogen,40 and osme bonds.7
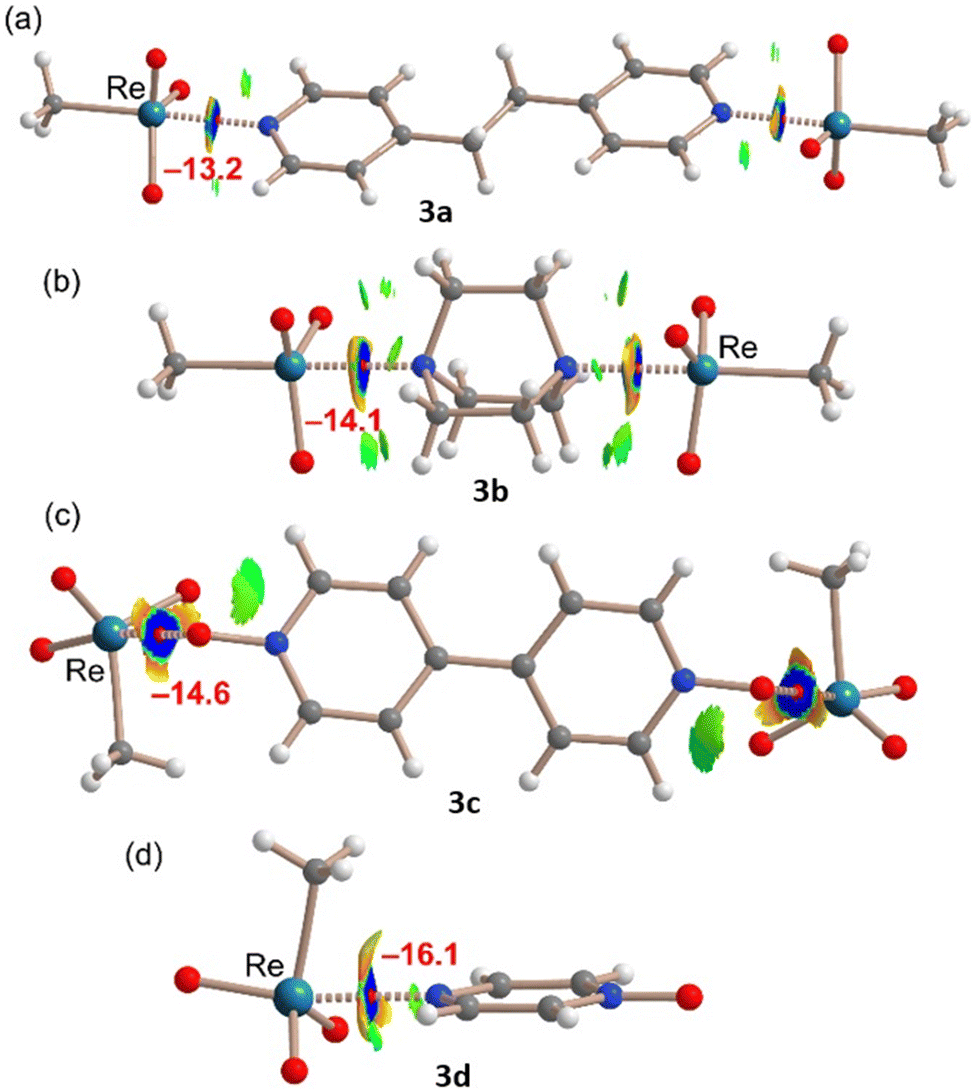 | ||
| Fig. 4 Combined QTAIM/NCIplot analysis for the adducts formed by compounds 3a (a), 3b (b), 3c (c), and 3d (d). The QTAIM analysis: bond CPs are represented as small red spheres. NCIPlot: |RGB| isosurface 0.45 a.u.; color range 0.08 a.u. (red) ≤ (signλ2)ρ ≤ −0.08 a.u. (blue). Only the intermolecular interactions are shown. The estimation of the MaB interaction energy is given in red next to the NCIplot isosurface that characterizes the Re⋯N/O contact. | ||
Conclusions
In conclusion, the combined experimental and theoretical study reported herein demonstrates that the crystal structures of the four MTO⋯Lewis base adducts display short contacts involving rhenium acting as an electron acceptor, geometries being fully consistent with σ-hole interactions. Compounds 3a,b are the first cases wherein MaBs are formed opposite to a C–Re covalent bond. The computations support the attractive nature of these contacts. Moreover, the donor–acceptor orbitals describing the interactions and the positive regions located opposite to the covalent bonds further support the σ-hole nature of the MaBs shown by the adducts in the solid state. The understanding of the nature of Re⋯N/O interactions may help to optimize the epoxidation of olefins under MTO catalysis by rationally optimizing the MTO-pyridine adduct formation and the resulting pyridine-acceleration effect. Ongoing studies prove that other oxorhenium catalysts also display σ-hole interactions.Conflicts of interest
There are no conflicts of interest.Acknowledgements
A. F. acknowledges MICIU/AEI of Spain (PID2020-115637GB-I00, FEDER funds) for financial support and the CTI (UIB) for computational facilities. G. R. and M. C thank PRIN-2020, project NICE, no. 2020Y2CZJ2.References
- G. J. Hutchings, C. R. A. Catlow, C. Hardacre and M. G. Davidson, Philos. Trans. R. Soc., A, 2016, 374, 20150358 CrossRef.
- F. Heinen, D. L. Reinhard, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2021, 60, 5069 CrossRef CAS PubMed.
- T. Kato, B. Lim, Y. Cheng, A.-T. Pham, J. Maynard, D. Moreau, A. I. Poblador-Bahamonde, N. Sakai and S. Matile, JACS Au, 2022, 2, 839 CrossRef CAS PubMed.
- A. Gini, M. Paraja, B. Galmés, C. Besnard, A. I. Poblador-Bahamonde, N. Sakai, A. Frontera and S. Matile, Chem. Sci., 2020, 11, 7086 RSC.
- J. K. Kochi, Organometallic Mechanisms and Catalysis, Elsevier, 1978 Search PubMed.
- D. W. Nelson, A. Gypser, P. T. Ho, H. C. Kolb, T. Kondo, H.-L. Kwong, D. V. McGrath, A. E. Rubin, P.-O. Norrby, K. P. Gable and K. B. Sharpless, J. Am. Chem. Soc., 1997, 119, 1840 CrossRef CAS.
- A. Daolio, A. Pizzi, M. Calabrese, G. Terraneo, S. Bordignon, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 20723 CrossRef CAS PubMed.
- A. Daolio, A. Pizzi, G. Terraneo, A. Frontera and G. Resnati, ChemPhysChem, 2021, 22, 2281 CrossRef CAS.
- R. M. Gomila and A. Frontera, Molecules, 2022, 27, 6597 CrossRef CAS PubMed.
- K. Tadpetch and S. Rychnovsky, Org. Lett., 2008, 10, 4839 CrossRef CAS PubMed.
- W. A. Herrmann, R. W. Fischer, M. U. Rauch and W. Scherer, J. Mol. Catal., 1994, 86, 243 CrossRef CAS.
- C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197 CrossRef.
- J. H. Espenson, Chem. Commun., 1999, 479 RSC.
- I. R. Beattie and P. J. Jones, Inorg. Chem., 1979, 18, 2318 CrossRef CAS.
- F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149 CrossRef.
- J. Rudolph, K. L. Reddy, J. P. Chiang, K. B. Sharpless, N. T. P. Road, L. Jolla and R. V. February, J. Am. Chem. Soc., 1997, 119, 6189 CrossRef CAS.
- C. Copéret, H. Adolfsson and K. B. Sharpless, Chem. Commun., 1997, 1567 Search PubMed.
- W.-D. Wang and J. H. Espenson, J. Am. Chem. Soc., 1998, 120, 11335 CrossRef CAS.
- C. Ehinger, C. P. Gordon and C. Copéret, Chem. Sci., 2019, 10, 1786 RSC.
- S. M. Nabavizadeh, A. Akbari and M. Rashidi, Dalton Trans., 2005, 2423 RSC.
- W. A. Herrmann, H. Ding, R. M. Kratzer, F. E. Kiihn, J. J. Haider and R. W. Fischer, J. Organomet. Chem., 1997, 549, 319 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- C. E. S. Bernardes, M. T. Donato, M. F. M. Piedade, H. P. Diogo, J. N. Canongia Lopes and M. E. Minas da Piedade, J. Chem. Thermodyn., 2019, 133, 60 CrossRef CAS.
- F. E. Kühn, A. M. Santos, P. W. Roesky, E. Herdtweck, W. Scherer, P. Gisdakis, I. V. Yudanov, C. Di Valentin and N. Rösch, Chem. – Eur. J., 1999, 5, 3603 CrossRef.
- A. Priimagi, M. Saccone, G. Cavallo, A. Shishido, T. Pilati, P. Metrangolo and G. Resnati, Adv. Mater., 2012, 24, OP345 CAS.
- S. M. Nabavizadeh, A. Akbari and M. Rashidi, Dalton Trans., 2005, 2423 RSC.
- M. Y. Shatnawi and A. M. Al-Ajlouni, Jordan J. Chem., 2009, 4, 119 CAS.
- S. M. Nabavizadeh and M. Rashidi, J. Am. Chem. Soc., 2006, 128, 351 CrossRef CAS PubMed.
- F. N. Hosseini, K. Kamali and S. M. Nabavizadeh, Polyhedron, 2011, 30, 814 CrossRef CAS.
- C. C. Robertson, J. S. Wright, E. J. Carrington, R. N. Perutz, C. A. Hunter and L. Brammer, Chem. Sci., 2017, 8, 5392 RSC.
- E. Corradi, S. V. Meille, M. T. Messina, P. Metrangolo and G. Resnati, Angew. Chem., Int. Ed., 2000, 39, 1782 CrossRef CAS.
- J. A. Bis, P. Vishweshwar, D. Weyna and M. J. Zaworotko, Mol. Pharmaceutics, 2007, 4, 401 CrossRef CAS PubMed.
- K. K. Arora, M. S. Talwelkar and V. Pedireddi, New J. Chem., 2009, 33, 57 RSC.
- E. C. Escudero-Adan, A. Bauza, C. Lecomte, A. Frontera and P. Ballester, Phys. Chem. Chem. Phys., 2018, 20, 24192 RSC.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170 CrossRef CAS.
- E. V. Bartashevich and V. G. Tsirelson, Russ. Chem. Rev., 2014, 83, 1181–1203 CrossRef CAS.
- M. L. Kuznetsov, Molecules, 2019, 24, 2733 CrossRef CAS.
- A. Bauzá and A. Frontera, ChemPhysChem, 2019, 21, 26 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, spectroscopic, and crystallographic data of examined adducts; details on computational analyses. CCDC 2174961, 2174969–2174971 and 2208886. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03819f |
| ‡ In this paper, contacts are designated short or close when the distance between the involved atoms is smaller than the sum of their van der Waals (vdW) radii of involved atoms. Recommended crystallographic vdW radii (pm) proposed by S. S. Batsanov are used: O, 155; N, 160, Re, 205 (S. S. Batsanov, Inorg. Mater., 2001, 37, 871). There are limitations to the validity of this type of analysis. For instance, the use of vdW radii assumes that atoms in molecules are spherical, while radii along the extension of covalent bonds are typically smaller than perpendicular to the bonds (T. N. G. Row, R. Parthasarathy, J. Am. Chem. Soc., 1981, 103, 477; S. S. Batsanov, Struct. Chem., 2000, 11, 177–183). This type of analysis is nevertheless adopted in this paper, as it is the universally employed approach to identify the hallmarks of interactions. |
| § The “normalized contact” Nc for an interaction involving atoms i and j is the ratio Dij/(rvdW,i + rvdW,j) where Dij is the experimental distance between i and j and rvdW,i and rvdW,j are the van der Waals radii of i and j. Nc allows different interaction lengths to be compared in a more reliable way than by using absolute separation values. Nc values smaller than 1 correspond to attractive interactions, and usually, the smaller the Nc, the stronger the interaction. |
| ¶ The H⋯O separation in 2c⋯tartaric acid adduct is so short that the formation of this adduct might also be rationalized as the result of a proton transfer rather than that of HB formation. |
| This journal is © The Royal Society of Chemistry 2023 |