
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalen, salan and salalen zinc(II) complexes in the interaction with HS−: time-resolved fluorescence applications†
Maria
Strianese
 *a,
Gerard Joseph
D'Auria
a,
Marina
Lamberti
a,
Alessandro
Landi
a,
Andrea
Peluso
a,
Antonio
Varriale
bc,
Sabato
D'Auria
d and
Claudio
Pellecchia
a
*a,
Gerard Joseph
D'Auria
a,
Marina
Lamberti
a,
Alessandro
Landi
a,
Andrea
Peluso
a,
Antonio
Varriale
bc,
Sabato
D'Auria
d and
Claudio
Pellecchia
a
aDipartimento di Chimica e Biologia “Adolfo Zambelli”, Università degli Studi di Salerno, Via Giovanni Paolo II, 132, 84084, Fisciano (SA), Italy. E-mail: mstriane@unisa.it
bInstitute of Food Science, CNR Italy, 83100 Avellino, Italy
cURT-ISA, CNR at Department of Biology, University of Naples Federico II, 80126 Napoli, Italy
dDepartment of Biology, Agriculture, and Food Sciences, National Research Council of Italy (CNR-DISBA), Piazzale Aldo Moro 7, 00185 Rome, Italy
First published on 3rd January 2023
Abstract
In the current work we investigate the route of interaction of a newly synthesized family of zinc complexes with HS− by a plethora of different spectroscopic techniques. A computational analysis on the time dependent density functional theory (TD-DFT) level explored the overall fluorescence properties of the title complexes and their different fluorescence responses to HS−. Time-resolved fluorescence experiments were also performed and highlight the great potential of the current systems to be implemented as HS− fluorescent sensors.
Introduction
In the last decades, along with its traditional toxic role, hydrogen sulfide (H2S) emerged for its beneficial functions for humans, depending on the concentration.1–3 This was a consequence of the discovery that it occurs naturally in the human body4,5 with diverse roles in cardioprotection,6 neuroprotection,7 wound healing,8,9 and mitigating oxidative stress and associated damage.2,10,11Various diseases such as Huntington's disease, asthma, and Crohn's disease seem to be associated with decreased H2S levels,12–16 and establishing whether the downregulation of H2S production is one of the contributing causes or one of the symptoms of these diseases is still a challenging question. And overall the mechanisms by which H2S exerts its biological reactivity are not completely clear yet.17 One molecular mechanism that has been proposed for H2S as a beneficial bio-molecule is the posttranslational modification of protein cysteine residues forming persulfide (RSSH):18–20 this process has been called “sulfhydration” or persulfhydration. This reaction generates S0 that seems involved in several aspects of H2S chemistry, and that has the high potential of transducing signals owing to its unique property of transsulfuration.
From a different point of view, the mechanisms of action of H2S can be envisioned to involve interactions with transition metals and, in this context, the study of its coordination chemistry appears highly interesting. Motivated by our interest in shedding light on the biochemical reactions H2S is involved in, our studies on the coordination of H2S to transition metals started some time ago and focused both on properly tailored low molecular weight compounds and on natural metalloproteins.21–30 More recently we focused on zinc salen-based complexes and targeted these systems as efficient scaffolds for isolating and characterizing hydrosulfido species and as performing HS− sensing constructs via a ‘coordinative-based’ mechanism.31–36 We and others also reported that the fluorescence of ZnSalen complexes can be fine-tuned by an ad-hoc design of the ligands’ electronic states37 and that is both dependent on the electronic states of the diamine moiety bridging the two nitrogens which chelate the zinc center and on the substituents on the salycilaldehyde units.35,38 In addition, we showed that the electronic state of the diamine moiety between the zinc-chelating nitrogens strongly modulates the photophysical properties of the salen complexes so as their fluorescence response to hydrogen sulfide.31,35 Starting from these achievements, in the current work, we decided to explore whether the imine functionalities of the salen complexes play a determining role in the interaction of the sensor molecule and HS−. In other words since, as well acknowledged in the literature, salen stands for N,N′-bis(salicylidene)ethylenediamine and refers to a tetradentate C2-symmetric ligand with both sp2-hybridized nitrogen atoms synthesized from salycilaldehyde (sal) and ethylenediamine (en); we thought of exploring the tetrahydro derivative of salen, named salan (which displays two phenoxyamine functionalities and a sp3 hybridization of the amine nitrogen atoms) together with the salalen (which is the dihydro derivative of salen and is also considered a hybrid derivative of salen/salan ligands with sp2- and sp3-hybridized nitrogen atoms).39,40 and of comparing the reactivity of the related complexes with HS−. We synthesized a new family of zinc complexes derived from salen, salan and salalen ligands with the same substituents on the salycilaldehyde units (Scheme 1) and investigated the possible effects of these ligands on the zinc hydrosulfido stabilization. We also wanted to answer the question whether the different ligand structures would tune the fluorescence properties of the related complexes as HS− sensors: the different hybridization of the nitrogen atoms in these classes of tetradentate ligands may differentiate the behaviour of the corresponding complexes in a substantial way.
 | ||
| Scheme 1 | ||
Results and discussion
Complexes 1–3 were synthesized by the direct reaction of the ligand precursors and ZnEt2 in dry benzene and were first characterized via high-resolution MALDI Fourier transform ion cyclotron resonance mass spectrometry (HR MALDI-FT-ICR) (Fig. S1–S3†). The major peaks at 555.291 m/z units for complex 1, at 586.346 m/z units for complex 2 and at 570.314 m/z units for complex 3 correspond to the mononuclear species: no evidence for binuclear species in solution were found. In a second instance, complexes were characterized via1H NMR experiments (Fig. S4–S7†). The 1H NMR spectra of complexes 1–3 in DMSO-d6 exhibit sharp signals consistent with the expected coordination of the ligand. For complex 1 the solubility in deuterated dimethylsulfoxide was scarce, therefore the 1H NMR spectrum is not well resolved (Fig. S4†) whereas it resulted completely soluble in deuterated methylene chloride: the protonic spectrum of complex 1 in CD2Cl2 is displayed in Fig. S5.† The spectra of complexes 2 and 3 in DMSO-d6 are reported in Fig. S6 and S7.† The presence of AB patterns for the methylenic protons of the ethylene bridge and/or for the methylenic protons between the coordinating nitrogen and the aromatic moiety, highlights the coordination of the nitrogen atoms to the zinc centers in each complex. Furthermore, comparison of the 1H NMR spectra of the free ligands (Fig. S8–S10†) with those of the related complexes points to similar conclusions: by overlapping the spectrum of each ligand with that of the complex a shift of most of the signals is visible; in addition the spectra of the ligands exhibit signals at circa 11 ppm ascribable to the OH of the phenolates (missing in the spectra of the complexes) together with the equivalence of the methylenic protons of the ethylene bridge (Fig. S8–S10†).After characterization of complexes 1–3 we started a study to establish the mechanism of their interaction with HS−. To assess which is the operating route of interaction, we first examined the reactions via high-resolution MALDI-FT-ICR. Fig. S11–S13† display the outcome of our experiments. As for complex 1 and complex 3, binding of the HS− seems the working reaction route (Fig. S11 and S13†): the major peaks at 587.267 and at 603.296 m/z units are consistent with the mononuclear [complex 1/HS]− and [complex 3/HS]− species. MS experiments clearly indicate the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HS− adducts for complexes 1 and 3.
:1 HS− adducts for complexes 1 and 3.
Differently for complex 2 mass experiments suggest displacement of the zinc from the organic ligand (Fig. S12†): the peak at 523.425 m/z units exhibits the isotopic pattern of the salan ligand and the peak at 545.40 corresponds to the [salan ligand + Na]+ (Fig. S12†). Most likely, the different behavior of complex 2 respect to complexes 1 and 3 maybe due to the fact that the presence of the amine backbone results in a more flexible structure.
Aiming at further insights on the mechanism of the reaction between the title complexes and HS−, we analyzed the protonic NMR spectra of complexes 1–3 in the presence of NaSH. Fig. 1 displays the 1H NMR spectrum of complex 3 in the presence of HS−.
 | ||
| Fig. 1 1H NMR spectrum of complex 3 in DMSO-d6 after the addition of an excess of HS−. [Complex 3] = 5 × 10−3 M; [NaSH] = 0.01 M. * = benzene used for the synthesis. | ||
In the presence of an excess of NaSH, the whole pattern of signals of complex 3 undergoes a shift (Fig. 1 and also Fig. S14†). In addition, in the spectrum of the complex 3/HS adduct (see Fig. 1), the high field signal (at ∼−2.7 ppm) which, as well acknowledged in the literature,27,30–33,35,41–43 indicates binding of SH− to the zinc center, is also present.
The hypothesis of HS− coordination to the zinc center could be drawn also when analyzing the 1H NMR spectrum of complex 1 in the presence of NaSH (Fig. S17†). Differently, in case of complex 2 the protonic spectrum in the presence of NaSH points to leaching of the organic ligand (Fig. S9 vs. S15 and S16†). The resulting spectrum is not well resolved, most likely for the concomitant precipitation of ZnS occurring; furthermore, peaks ascribable to the free ligand are also evident (Fig. S16†). Hence, the NMR experiments corroborate the results of the MS experiments.
Next, we tested the optical properties of complexes 1–3 to assess their potential use in the recognition of HS−. Fig. 2 displays the UV-vis spectra of the title complexes before and after the treatment with an excess of HS−.
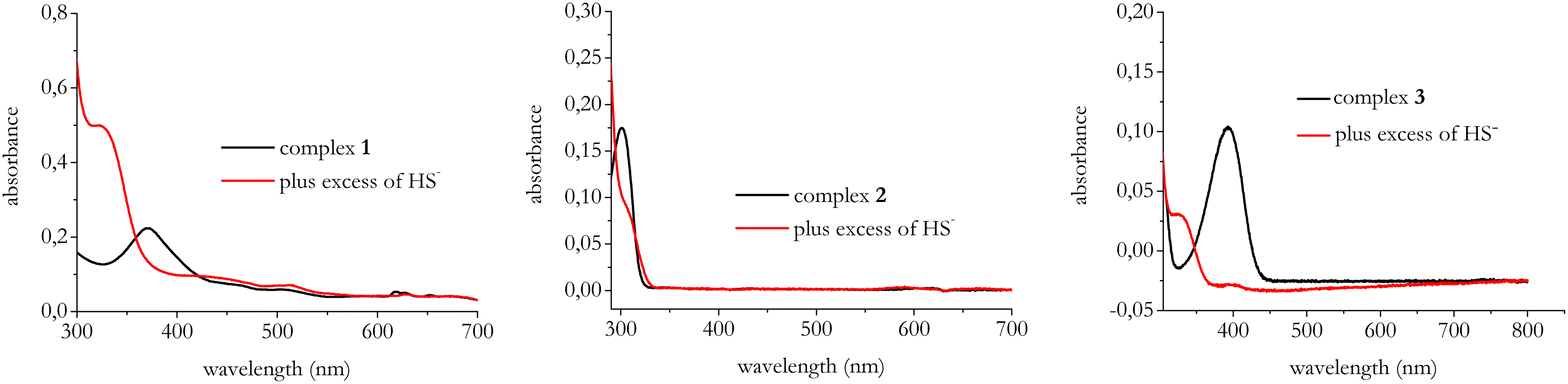 | ||
| Fig. 2 Electronic absorption spectra of complexes 1–3 with and without the addition of 50 μM of NaSH. Spectra were recorded in DMSO at room temperature. [Complexes] = 10 μM. | ||
As evident in Fig. 2, for all the complexes under investigation the interaction with HS− results in a change of the initial UV-visible spectrum which points to the formation of new species.
Then, we studied the fluorescence responses of the title complexes to HS−. Fig. 3 displays the results of the steady-state fluorescence experiments.
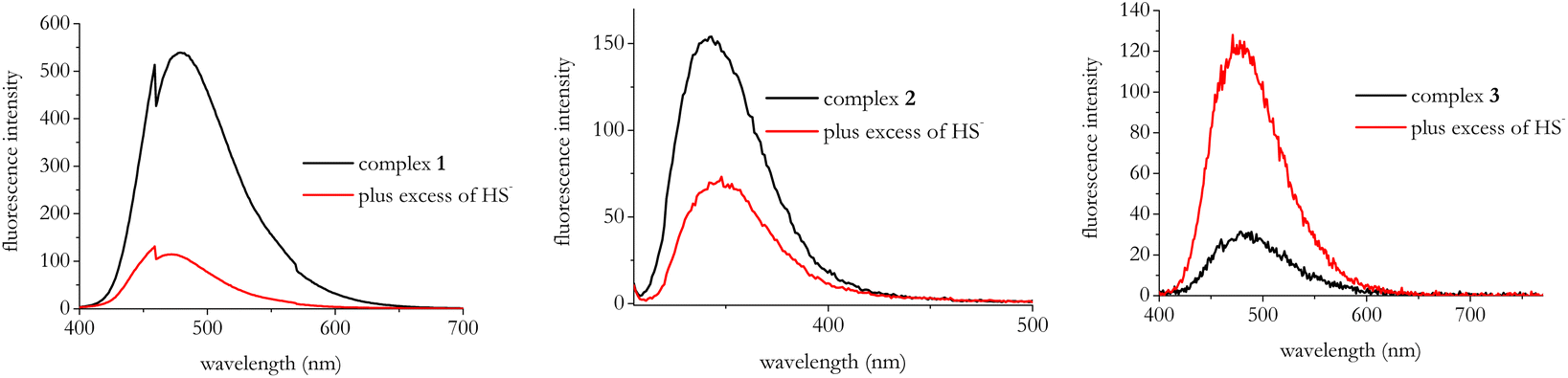 | ||
| Fig. 3 Emission spectra of complexes 1–3 before and after the addition of 5 equiv. of NaSH. [Complexes 1–3] = 1 × 10−5 M; [NaSH] = 5 × 10−5 M. All spectra were measured in DMSO with λexc = 370 nm for complex 1; λexc = 300 nm for complex 2; λexc = 300 nm for complex 3. | ||
In the presence of HS− the fluorescence responses indicate visible quenching effects for complexes 1 and 2 whereas complex 3 undergoes to fluorescence enhancement.
As for complex 2, we also checked the fluorescence of ligand 2 which resulted very weak (see Fig. S17†), most likely as a consequence of its reduced coniugation, thus the fluorescence quenching observed in the presence of HS− further strengthens that displacement of the zinc from the organic ligand takes place upon interaction with HS−.
Willing to rationalize these experimental results and above all to understand why complexes 1 and 3 exhibit a different fluorescence response to HS− while MS and NMR experiments indicate HS− binding to the zinc centers in both cases, we performed a computational analysis at the time dependent density functional theory (TD-DFT) level. Minimum energy geometries of complex 1 and complex 3 and of their HS− adducts (also considering the possibility of multiple adducts) have been computed both for the ground state and for the first excited singlet states. The computed ground state optimum geometries of 1 and of its HS− complex are shown in Fig. 4 (a similar geometry has been also found for 3 and its HS− adduct, Fig. S19†).
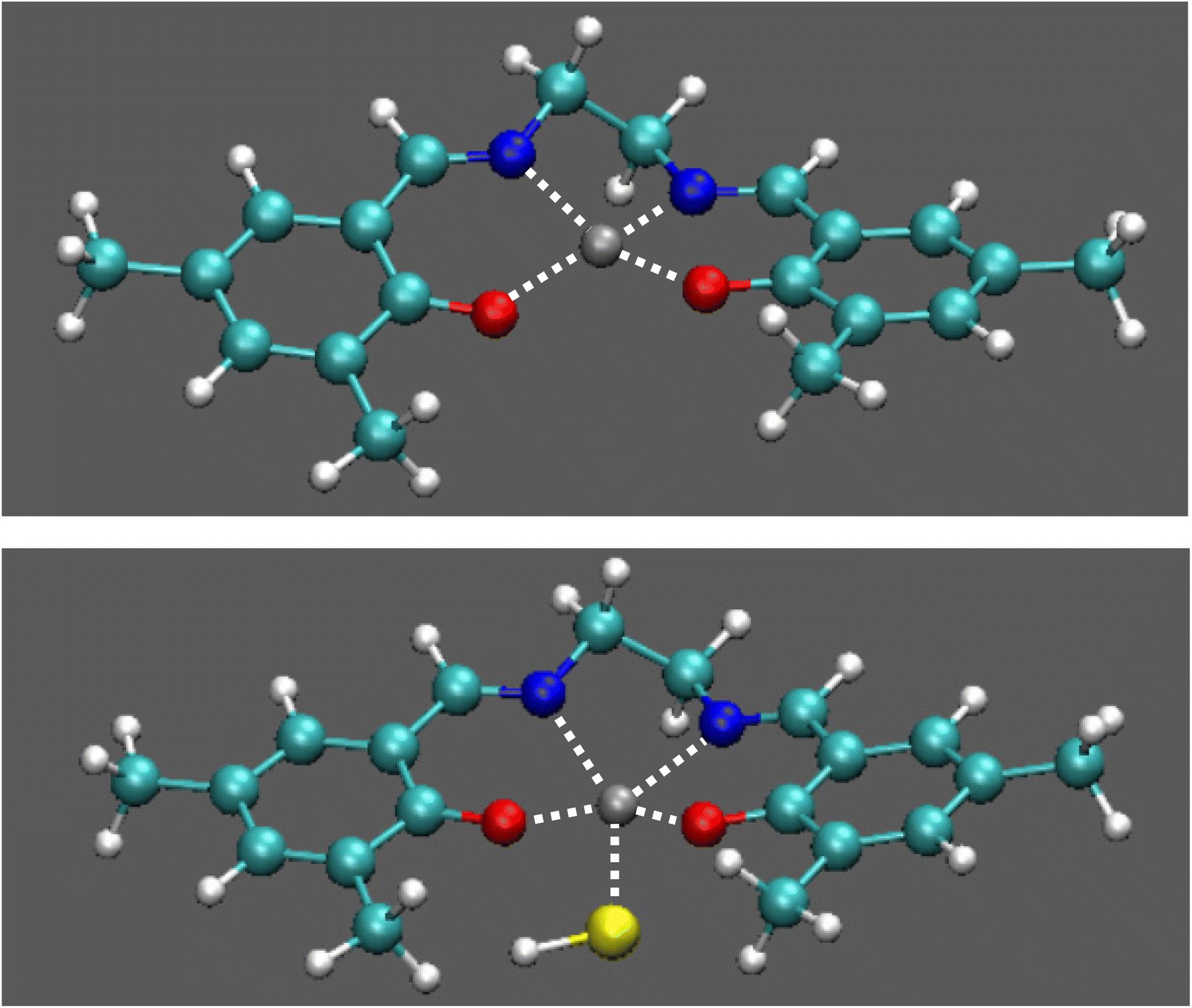 | ||
| Fig. 4 Optimized geometries for complex 1 (top) and its adduct with HS−. | ||
Complex 1 and 3 deviate from the square planar nuclear configuration observed for more rigid analogues,31,35 with the metal atom in the plane of the ligand, see Fig. 4 and Fig. S19.† The formation of the single adduct is predicted to be exoergonic (ΔE = −0.65 eV for complex 1, ΔE = −0.70 eV for complex 3), whereas the double adducts are not predicted to be stable species, as confirmed by DFT computation where the second HS− is moved away from the metal centre during geometry optimization. This finding is also in line with the MS experiments (Fig. S11 and S13†).
Emission from S1 is predicted to be electric dipole allowed both for 1 and 3 and for their HS− adducts. Computed vertical and adiabatic excitation energies are reported in Table 1, together with the oscillator strengths for the S1 ← S0 transitions.
| Vertical | Adiabatic | Oscillator strength | |
|---|---|---|---|
| 1 | 3.71 | 3.02 | 0.19 |
| 1 + HS− | 3.64 | 2.92 | 0.20 |
| 3 | 3.75 | 3.00 | 0.20 |
| 3 + HS− | 3.62 | 2.93 | 0.23 |
For 1 two electric dipole transition are predicted at around 334 and 321 nm; since they are very close, they are not distinguished in the experimental spectra, which shows only one peak, whereas in the case of 3 the prediction points to one peak at 340 nm, as in the experimental trace (Fig. 2).
However, a meaningful comparison between predicted and observed absorption spectra would require a band shape simulations, with the computations of Franck–Condon integrals44,45 which is far beyond the qualitative purposes of the present computational analysis.
Since for all the species emission from S1 are electric dipole allowed transitions, the different behaviour observed for 1 and 3 and for their HS− complexes has to be related with the possible existence of non-radiative decay paths. We have thus investigated the energy location of the lowest triplet states, which could be responsible of the different fluorescence quantum yields of 1 and 3 and their HS− adducts (see Table S1†).
The energies of the four lowest triplet states are reported in Fig. 5; T5 lies always above in energy than S1 and therefore it should not be involved in non-radiative decay paths. T2 and T1 are significantly lower in energy than S1 for all the species and therefore, based on the energy gap rule, the direct transition S1 → T1 and S1 → T2 should not be an efficient decay path. The triplet states closer in energy to S1 are T3 and T4.
 | ||
| Fig. 5 Computed energies (E, eV) of the ground state (S0) first excited singlet (S1), and four lowest energy triplet states, evaluated at the S1 geometry for complex 1 and 3, and their HS− adducts. | ||
As concerns complex 1, T3 and T4 are almost isoenergetic, both before and after HS− coordination, thus offering a double channel for non-radiative decay from S1. Moreover, these exoergonic S1 → T3 and S1 → T4 transition are more favoured after HS− coordination, as demonstrated by the lower energy difference between the electronic states and by the higher spin–orbit coupling for S1 → T3. Thus, a strong quenching of fluorescence is to be expected in presence of HS−, as indeed observed (Fig. 3).
On the contrary, the S1 → T4 transition is exoergonic for 3, but it becomes slightly endoergonic (−50 meV) when the HS− is coordinated (Fig. 5). Together with the lowering of the spin orbit coupling element (Table 2), this suggests that quenching of fluorescence via S1 → T4 is possible in the isolated complex, whereas fluorescence is recovered in presence of HS− in solution, in line with experimental observations (Fig. 3).
| SOC T3-S1 | SOC T4-S1 | |
|---|---|---|
| 1 | 1.16473 | 28.86538 |
| 1 + HS− | 18.17608 | 12.30915 |
| 3 | 2.69732 | 6.60415 |
| 3 + HS− | 1.27732 | 1.33585 |
Time-resolved fluorescence experiments
To further explore the sensing abilities of complexes 3 and 1 (the complexes of this family working via the coordinative-based approach) we studied their responses to HS− by means of time-resolved fluorescence spectroscopy.46 The results for complex 3 show an increase of the average lifetime in the presence of NaSH in the sample solution from 2.51 to 4.08 ns (see also Table S2 and Fig. S20 in the ESI†).As proposed in previous work this finding is in line with the enhancement of the fluorescence intensity observed in the steady-state measurements: when probe-analyst interaction results in fluorescence enhancement the increases of the corresponding lifetime is generally found in most cases.47,48
Indeed, when on the contrary there is a fluorescence quenching upon coordination of HS− to the complex, such as in complex 1, we observe a reduction of the average lifetime (from 5.08 to 3.36 ns) upon addition of the NaSH in the sample solution (see Table S3 and Fig. S21 in the ESI†).
Time-resolved fluorescence spectroscopy has been extensively used by some of us for applications in biological contexts thus ensuring a broad applicability of this technology in the field of (bio)sensing.46,49,50
H2S sensing applications
In the following experiments we focused on complex 3 which was the only one of the series undergoing a fluorescence enhancement in the presence of HS− and thus useful for practical sensing measurements (e.g. “turn on” systems are inherently more sensitive than “turn-off” sensors).First, we checked whether we could construct a calibration of the system by monitoring the change of its initial fluorescence intensity after the addition of increasing concentrations of NaSH. Fig. 6A shows the response of the fluorescence switching of complex 3 for a series of subsequent measurements with increased concentrations of NaSH. Fig. 6B displays the fluorescence intensity values at 480 nm fit against HS− concentration.
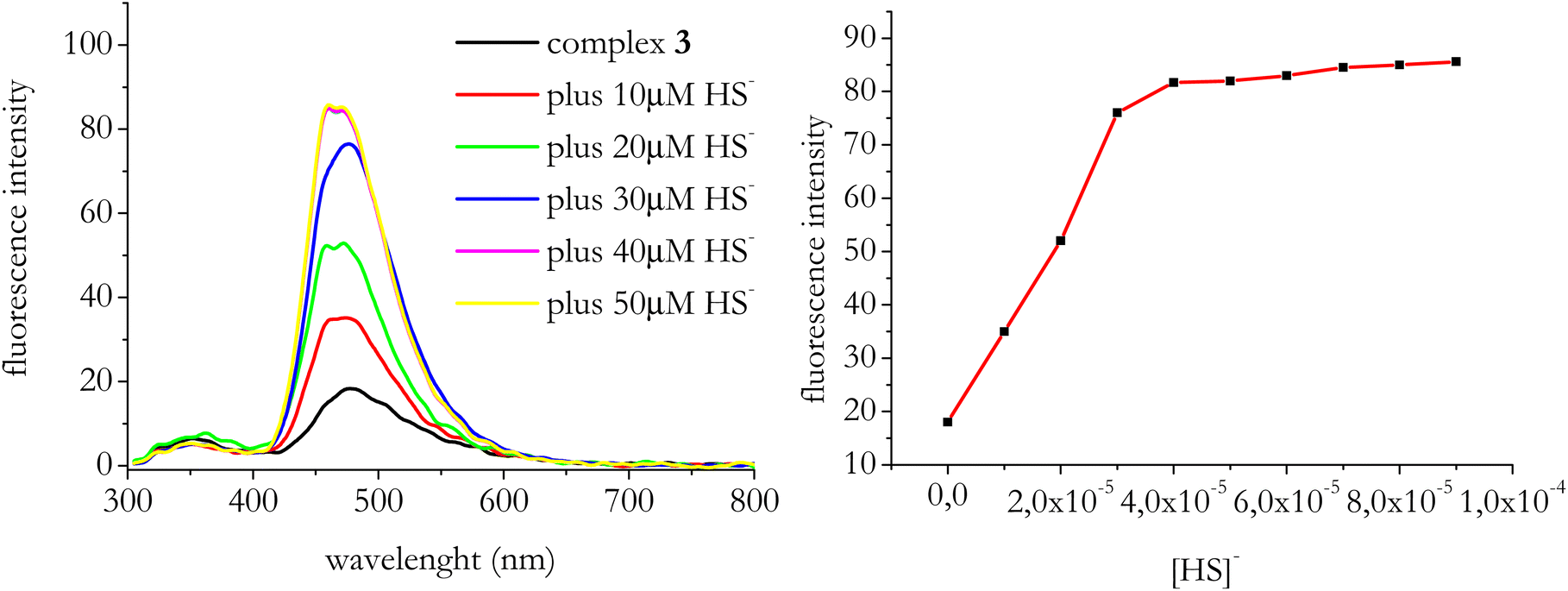 | ||
| Fig. 6 Left: emission spectra of complex 3 (λexc = 300 nm) when titrated with NaSH. [Complex 3] = 1 × 10−5 M; end concentration of NaSH varied in the range (1–5) × 10−5 M. Right: fit of the fluorescence intensity values of complex 3 at 480 nm against HS− concentration. | ||
From Fig. 6 it results that the detection limit of the system is in the micromolar range of H2S.
We also explored the selectivity of complex 3 in the recognition of HS− against biologically relevant and potentially competing thiols (e.g. glutathione (GSH) and L-cysteine (L-cys)). In the presence of both GSH and L-cys we observed a quenching of the initial fluorescence emission of complex 3 (Fig. 7) thus pointing to selectivity of the system towards HS− recognition.
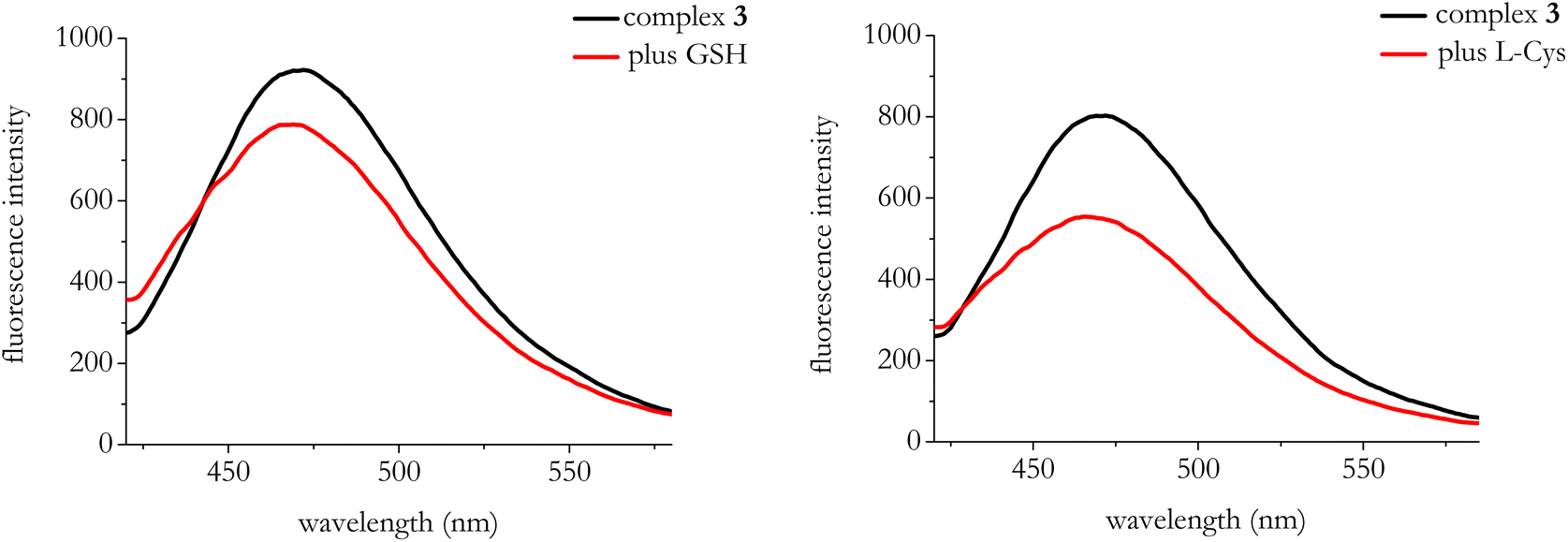 | ||
| Fig. 7 Emission spectra of complex 3 (λexc = 300 nm) in the presence of GSH and L-Cys. GSH = 50 mM; L-Cys = 50 mM. | ||
As for the time-resolved fluorescence experiments, we observed very modest responses in the average lifetimes from 2.1 to 2.75 ns in the presence of GSH and from 1.98 to 2.19 ns in case of L-Cys, again pointing to selectivity of complex 3 in HS− recognition (see Fig. S22, S23, Tables S4 and S5†).
In a second instance we studied the time response of complex 3 by monitoring its fluorescence intensity as a function of time upon HS− addition. Fig. 8 displays a typical time trace of a solution containing 10 μM of complex 3 when excited at 300 nm.
 | ||
| Fig. 8 Fluorescence intensity time trace observed at 480 nm (exc 300 nm) of complex 3, upon addition of HS−. | ||
Conclusions
In conclusion, in the framework of our ongoing studies aiming at shedding light on the coordination chemistry of HS− to bioinorganic targets, and in particular to salen-type zinc complexes, we synthesized and characterized a new family of salen-derived zinc complexes and investigated their reactivity with HS−. By means of different spectroscopic techniques, we assessed coordination of HS− to the zinc center for complex 1 and complex 3 whereas for complex 2 our experiments point to precipitation of ZnS and leaching of the organic ligand. This difference may be due to the different ‘hardness/softness’ of imine and amine nitrogens which chelate the zinc centers: the ‘soft’ imine nitrogens in complex 1 may coordinate the metal center more strongly than the harder amine nitrogens in complex 2.Steady-state fluorescence experiments assessed that the fluorescence intensity of the complexes is modulated by the presence of HS− and the different responses have been rationalized by means of a computational analysis. The different fluorescence trends observed for 1 and 3 in the presence of HS− has been related with the possible existence of non-radiative decay paths.
Preliminary time-resolved fluorescence experiments provided evidences that the current systems experience consistent differences in the average lifetimes in response to HS−, more precisely in the presence of NaSH complex 3 shows an increase of the average lifetime from 2.51 to 4.08 ns conversely complex 1 exhibits a reduction from 5.08 to 3.36 ns. These results are in line with the steady-state fluorescence measurements.
To the best of our knowledge, this is one of the first studies in which zinc compounds are used for sensing HS− by means of time-resolved fluorescence spectroscopy and we do believe that this study could open the doors to new kind of applications for practical measurements in the fast growing field of H2S monitoring.
Experimental section
Materials
Chemicals used for the synthetic work were obtained from Sigma-Aldrich or Strem Chemicals and were of reagent grade. They were used without further purification. NaSH (Alfa Aesar) in aqueous solution was used as HS− source to the end concentration specified in the figure captions. Syntheses of ligands were performed by following literature procedures.40,51–53 Syntheses of complexes 1 and 2 were achieved by modifications of literature procedures.54,55General
HR MALDI and ESI mass spectra were recorded using a Bruker solariX XR Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 7 T refrigerated actively-shielded superconducting magnet (Bruker Biospin, Wissembourg, France). The samples were ionized in positive ion mode using either the MALDI ion or the ESI ion source. The mass range was set to m/z 150–2000. The laser power was 15% and 15 laser shots were used for each scan. Mass spectra were calibrated externally using a mix of peptide clusters in MALDI ionization positive ion mode. As for the ESI mass spectra, they were calibrated externally using a NaTFA solution in negative ion mode. A linear calibration was applied.NMR spectra were recorded on a Bruker AVANCE 400 NMR instrument (1H NMR, 400.13 MHz; 13C NMR, 100.62 MHz) or on a 600 MHz spectrometer [600 (1H NMR) and 150 MHz (13C NMR)] using 5 mm o.d. NMR tubes. The chemical shifts were reported in δ (ppm) referenced to SiMe4. Typically, 5 mg of the complex in 0.5 mL of the solvent were used for each experiment. MilliQ water and is the water filtered with a Millipore filter apparatus.
Fluorescence quantum yield (ΦF) values were measured in optically diluted solutions using as standards the commercial dye Cy3 NHS (ΦF = 0.15 in MilliQ water), according to the equation:46
| ΦFs = ΦFr(Is/Ir)(Ar/As)(ηs/ηr)2 |
The intensity decays were analyzed with Vinci software using the multi-exponential model:
| I(t) = ∑αiexp(−t/τi) |
The average decay lifetime is defined as:
| 〈τ〉i = ∑αiτi/∑αi |
The acceptability of the fits was estimated by χ2 and the mean (average) fluorescence lifetimes were calculated.
Conflicts of interest
There are no conflicts to declare.References
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066 CrossRef CAS PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS PubMed.
- G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587 CrossRef CAS PubMed.
- W. Xuan, C. Sheng, Y. Cao, W. He and W. Wang, Angew. Chem., Int. Ed., 2012, 51, 2282–2284 CrossRef CAS PubMed.
- C. Q. Chen, H. Xin and Y. Z. Zhu, Acta Pharmacol. Sin., 2007, 28, 1709–1716 CrossRef CAS PubMed.
- M. Lavu, S. Bhushan and D. Lefer, Clin. Sci., 2010, 120, 219–229 CrossRef PubMed.
- W. L. Chen, Y. Y. Niu, W. Z. Jiang, H. L. Tang, C. Zhang, Q. M. Xia and X. Q. Tang, Rev. Neurosci., 2015, 26, 129–142 CAS.
- M. Xu, Y. Hua, Y. Qi, G. Meng and S. Yang, Exp. Dermatol., 2019, 28, 776–785 CrossRef CAS PubMed.
- H. Zhao, S. Lu, J. Chai, Y. Zhang, X. Ma, J. Chen, Q. Guan, M. Wan and Y. Liu, J. Diabetes Complicat., 2017, 31, 1363–1369 CrossRef PubMed.
- S. Carballal, M. Trujillo, E. Cuevasanta, S. Bartesaghi, M. a. N. Möller, L. K. Folkes, M. A. Garcìa-Bereguiaìn, C. Gutiérrez-Merino, P. Wardman, A. Denicola, R. Radi and B. Alvarez, Free Radical Biol. Med., 2011, 50, 196–205 CrossRef CAS PubMed.
- C. M. Levinn and M. D. Pluth, Sens. Actuators, B, 2021, 329, 129235 CrossRef CAS PubMed.
- M. Whiteman, S. Le-áTrionnaire, M. Chopra, B. Fox and J. Whatmore, Clin. Sci., 2011, 121, 459–488 CrossRef CAS PubMed.
- S. B. Singh and H. C. Lin, Microrganisms, 2015, 3(4), 866–889 CrossRef CAS PubMed.
- P. Wang, G. Zhang, T. Wondimu, B. Ross and R. Wang, Exp. Physiol., 2011, 96, 847–852 CrossRef CAS PubMed.
- K. F. Chung, Expert Rev. Respir. Med., 2014, 8, 5–13 CrossRef CAS PubMed.
- K. G. Fosnacht, M. D. Hammers, M. S. Earp, A. K. Gilbert and M. D. Pluth, Chem. – Asian J., 2022, 17(16) DOI:10.1002/asia.2022200426.
- Q. Li and J. R. Lancaster, Nitric Oxide, 2013, 35, 21–34 CrossRef CAS PubMed.
- A. K. Mustafa, M. M. Gadalla, N. Sen, S. Kim, W. Mu, S. K. Gazi, R. K. Barrow, G. Yang, R. Wang and S. H. Snyder, Sci. Signaling, 2009, 2, ra72, DOI:10.1126/scisignal.2000464.
- B. D. Paul and S. H. Snyder, Nat. Rev. Mol. Cell Biol., 2012, 13, 499–507 CrossRef CAS PubMed.
- N. Sen and S. H. Snyder, Trends Neurosci., 2010, 2010/09/16, 493–502 CrossRef PubMed.
- S. Mirra, S. Milione, M. Strianese and C. Pellecchia, Eur. J. Inorg. Chem., 2015, 2272–2276 CrossRef CAS.
- M. Strianese, A. Varriale, M. Staiano, C. Pellecchia and S. D’Auria, Nanoscale, 2011, 3(1), 298–302 RSC.
- M. Strianese, S. Milione, V. Bertolasi, C. Pellecchia and A. Grassi, Inorg. Chem., 2011, 50, 900–910 CrossRef CAS PubMed.
- M. Strianese, F. De Martino, C. Pellecchia, G. Ruggiero and S. D’Auria, Protein Pept. Lett., 2011, 18, 282–286 CrossRef CAS PubMed.
- M. Strianese, G. J. Palm, S. Milione, O. Kuhl, W. Hinrichs and C. Pellecchia, Inorg. Chem., 2012, 51, 11220–11222 CrossRef CAS PubMed.
- M. Strianese, S. Mirra, V. Bertolasi, S. Milione and C. Pellecchia, New J. Chem., 2015, 39, 4093–4099 RSC.
- M. Strianese, M. Lamberti and C. Pellecchia, Dalton Trans., 2017, 46, 1872–1877 RSC.
- M. Strianese, S. Mirra, M. Lamberti and C. Pellecchia, Inorg. Chim. Acta, 2017, 466, 426–431 CrossRef CAS.
- M. Strianese, G. J. Palm, D. Kohlhause, L. A. Ndamba, L. C. Tabares and C. Pellecchia, Eur. J. Inorg. Chem., 2019, 2019, 885–891 CrossRef CAS.
- M. Strianese, S. Brenna, G. Ardizzoia, D. Guarnieri, M. Lamberti, I. D'Auria and C. Pellecchia, Dalton Trans., 2021, 50, 17075–17085 RSC.
- M. Strianese, V. Vykhovanets, N. Blal, D. Guarnieri, A. Landi, M. Lamberti, A. Peluso and C. Pellecchia, Sensors, 2022, 22, 3173 CrossRef CAS PubMed.
- M. Strianese, M. Lamberti and C. Pellecchia, Dalton Trans., 2018, 47, 17392–17400 RSC.
- M. Strianese, M. Lamberti, A. Persico and C. Pellecchia, Inorg. Chim. Acta, 2019, 119235 Search PubMed.
- M. Strianese and C. Pellecchia, Coord. Chem. Rev., 2016, 318, 16–28 CrossRef CAS.
- M. Strianese, D. Guarnieri, M. Lamberti, A. Landi, A. Peluso and C. Pellecchia, Inorg. Chem., 2020, 59, 15977–15986 CrossRef CAS PubMed.
- M. Strianese, D. Pappalardo, M. Mazzeo, M. Lamberti and C. Pellecchia, Dalton Trans., 2020, 49, 16533–16550 RSC.
- G. Consiglio, I. P. Oliveri, S. Failla and S. Di Bella, Molecules, 2019, 24, 2514 CrossRef CAS PubMed.
- D. Xie, J. Jing, Y. B. Cai, J. Tang, J. J. Chen and J. L. Zhang, Chem. Sci., 2014, 5, 2318–2327 RSC.
- S. p. Bellemin-Laponnaz and S. Dagorne, Coordination chemistry and applications of salen, salan and salalen metal complexes, in Chemistry of Metal Phenolates, John Wiley Ltd, Chichester, UK, 2014, pp. 263–309 Search PubMed.
- A. Yeori, S. Gendler, S. Groysman, I. Goldberg and M. Kol, Inorg. Chem. Commun., 2004, 7, 280–282 CrossRef CAS.
- M. Rombach and H. Vahrenkamp, Inorg. Chem., 2001, 40, 6144–6150 CrossRef CAS PubMed.
- M. D. Pluth and Z. J. Tonzetich, Chem. Soc. Rev., 2020, 49, 4070–4134 RSC.
- E. Galardon, A. Tomas, M. Selkti, P. Roussel and I. Artaud, Inorg. Chem., 2009, 48, 5921–5927 CrossRef CAS PubMed.
- A. Capobianco, R. Borrelli, A. Landi, A. Velardo and A. Peluso, J. Phys. Chem. A, 2016, 120, 5581–5589 CrossRef CAS PubMed.
- A. Landi, R. Borrelli, A. Capobianco, A. Velardo and A. Peluso, J. Chem. Theory Comput., 2018, 14, 1594–1601 CrossRef CAS PubMed.
- R. Joseph. Lakowicz Principles of Fluorescence Spectroscopy, 2006 Search PubMed.
- B. Das, S. Dey, G. P. Maiti, A. Bhattacharjee, A. Dhara and A. Jana, New J. Chem., 2018, 42, 9424–9435 RSC.
- S. Das, M. Das, S. Laha, K. Rajak, I. Choudhuri, N. Bhattacharyya, B. C. Samanta and T. Maity, J. Mol. Struct., 2022, 1263, 133214 CrossRef CAS.
- S. D'Auria, E. Apicella, M. Staiano, S. Di Giovanni, G. Ruggiero, M. Rossi, P. Sarkar, R. Luchowski, I. Gryczynski and Z. Gryczynski, Anal. Biochem., 2012, 425, 13–17 CrossRef PubMed.
- A. Varriale, V. Marzullo, S. Giovanni, A. Scala, A. Capo, A. Majoli, A. Pennacchio and M. Staiano, Anal. Bioanal. Chem., 2016, 408, 6329–6336 CrossRef CAS PubMed.
- E. Y. Tshuva, N. Gendeziuk and M. Kol, Tetrahedron Lett., 2001, 42, 6405–6407 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed.
- M. Cozzolino, V. Leo, C. Tedesco, M. Mazzeo and M. Lamberti, Dalton Trans., 2018, 47, 13229–13238 RSC.
- Q. Dong, X. Ma, J. Guo, X. Wei, M. Zhou and D. Liu, Inorg. Chem. Commun., 2008, 11, 608–611 CrossRef CAS.
- G. A. Morris, H. Zhou, C. L. Stern and S. T. Nguyen, Inorg. Chem., 2001, 40, 3222–3227 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schelegel. et al. Gaussian 09, revision D.01, Gaussian, Wallingford, 2009. Ref Type: Computer Program Search PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- A. Capobianco, A. Landi and A. Peluso, Phys. Chem. Chem. Phys., 2017, 19, 13571–13578 RSC.
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt03730k |
| This journal is © The Royal Society of Chemistry 2023 |