
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasonically controlled synthesis of UO2+x colloidal nanoparticles†
Manon
Cot-Auriol
a,
Matthieu
Virot
 *a,
Thomas
Dumas
b,
Olivier
Diat
a,
Xavier
Le Goff
a,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
*a,
Thomas
Dumas
b,
Olivier
Diat
a,
Xavier
Le Goff
a,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
aICSM, Univ Montpellier, CEA, CNRS, ENSCM, Marcoule, France. E-mail: matthieu.virot@cea.fr
bCEA, DES, ISEC, DMRC, Univ Montpellier, Marcoule, France
First published on 6th January 2023
Abstract
Actinide colloids and nanoparticles (NPs) currently constitute a topic of strong interest due to their potential role in advanced nuclear energetics and the environmental migration of radioactivity. A better understanding of the physico-chemical properties of nanoscale actinide oxides requires robust synthesis approaches. In this work, UO2+x NPs were successfully prepared by sonochemistry from U(IV) solutions previously stabilised in a hydrochloric medium (20 kHz, 65 °C, Ar/(10%)CO). Colloidal suspensions were found to be composed of crystalline and spherical NPs showing a UO2-like structure and measuring 18.0 ± 0.1 nm (SAXS, HR-TEM and PXRD techniques). In comparison with the controlled hydrolysis approach used as a reference, sonochemistry appears to be a simple and original synthesis route providing larger, better defined and more crystalline UO2+x NPs with a narrower size distribution. These well-defined NPs offer new opportunities for the preparation of reference actinide materials devoted to fundamental, technological and environmental studies.
1. Introduction
Much effort is underway to optimize or develop technological processes that would enhance the sustainability and safety of nuclear energetics. Increasing attention is especially given to the preparation and characterization of actinide nanomaterials whose nanoscale properties could improve, for instance, the preparation, flowing, burning and reprocessing of nuclear fuel.1,2 At the same time, producing nuclear electricity requires ensuring the safety of the spent fuel under surface or deep underground storage. One of the major concerns deals with the corrosion and subsequent leaching of radionuclides when water comes into contact with nuclear wastes.3,4 Particular attention is paid to the possible formation of actinide colloids which are currently being investigated for their role in the migration of radioactivity in the geosphere.5–7 A better understanding of the speciation and reactivity of nanosized actinide oxides, including UO2 which constitutes the major component of the nuclear fuel worldwide,8 is of paramount interest for fundamental studies (size vs. property relationship), energetics (development of nuclear standards and fuels) and environmental purposes (migration, decontamination, safety storage).Under natural geochemical conditions, uranium principally exists under +IV and +VI oxidation states. In an aerated aqueous medium, U(IV) tends to rapidly oxidize into the uranyl ion UO22+, and the process can be limited under anoxic conditions.3,9,10 Several authors have studied the oxidation kinetics of U(IV) into U(VI) in the presence of O2 or H2O2.11–13 Halpern et al. reported that the oxidation process was dependent on the acidity of the medium (lower rate with a higher concentration of HClO4) and was sensitive to the amount of O2 in solution.11 UO22+ forms hydrolysed soluble complexes at slightly acidic pHs (2–5)14,15 differing from U(IV), which rather yields stable and non-soluble species supposed to be immobilized under aqueous reducing conditions.16,17 The sorption of U(IV) ions onto natural colloids (e.g. clay mineral, silica particles, humic acid, etc.) and their subsequent mobility have been highly reported in the literature.6,18–20 U(IV) also shows a strong Lewis acid behaviour depending on the pH conditions of the aqueous media which results in the hydrolysis properties according to eqn (1).21 Intrinsic U(IV) colloids resulting from the condensation of the as-formed hydrolysed species represent another potential source of mobility for the radioactivity and should be taken into account to correctly estimate their behaviour in an environmental context (speciation, solubility, reactivity, redox, etc.).22
m[U(OH2)n]4+ + hH2O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [Um(OH)h(OH2)n−h](4m−h)+ + hH+ [Um(OH)h(OH2)n−h](4m−h)+ + hH+ | (1) |
Few approaches have been reported for the preparation of UO2 nanoparticles (NPs) including sol–gel methods, electrochemistry or radiation-induced syntheses. The synthesis route can directly influence the physico-chemical properties of the obtained material such as the particle size distribution, shape, crystallinity or reactivity.22–24 Other routes involved the thermal decomposition of uranium(IV) hydroxides, U(VI) reduction or more original processes (e.g. microwaves, combustion).24–28 Studies also emphasized that the conversion of hydrolysed species to polynuclear ones is a highly temperature-sensitive process.29 Recently, the hydrothermal decomposition of U(IV) oxalates under 250 °C allowed the preparation of spherical agglomerates of over-stoichiometric uranium oxide NPs (UO2+x) whose size and crystalline properties were found to be strongly dependent on the experimental conditions (e.g. pH, temperature, pressure or concentration).29–32 Using this approach, Popa et al. avoided the typical platelet morphology arising from oxalate precursors also responsible for technological issues and reported the synthesis of particles ranging from 4.5 ± 0.5 nm to 7.8 ± 0.9 nm.31 Recently, the approach allowed the group to successfully propose homogeneous NPs of mixed oxides (U,Pu)O2, (U,Am)O2 and (U,Pu,Am)O2.1,33 Also, the recent work of Tabata et al. showed the formation of UO2+x nanoparticles under hydrothermal treatment (100–450 °C) from U(IV) or U(VI) nitrates in the presence of organic additives (ethanol, aldehyde, carbonate, etc.).34,35 The experimental conditions have an influence on the size, crystallinity and shape of the NPs, which can be spherical or rectangular.
Gerber et al. prepared UO2 NPs measuring 2–3 nm in diameter in the presence of ammonia under a nitrogen atmosphere (pH 8–11). Using microscopy and spectroscopy techniques, the authors demonstrated that freshly prepared UO2 NPs have the same structural and electronic properties as those of their bulk counterparts which crystallize in a fluorite-type fcc structure.36 However, the large surface-to-volume atomic ratio resulting from the nanometric size of the particles induces a high contribution of surface atoms which increased the reactivity of the NPs. The UO2 NPs were found to oxidize in the air or during synchrotron X-ray exposure but also under an inert or even reducing atmosphere (slow process). The high reactivity of these NPs led to the observation of U4O9 which was accompanied by their significant growth reaching ca. 6 nm.36,37 The integration of oxygen occurs as interstitial defects leading to the formation of hyper-stoichiometric phases exhibiting the general formula UO2+x (with 0 > x > 0.25).27,38 Oxygen incorporation does not affect the cubic crystal structure as long as the O/U ratio is lower than 2.25.36,39,40 The oxidation of UO2+x can continue until UO3 is obtained, passing through the formation of UO2.5 (U2O5) and UO2.67 (U3O8).36,39,40
In the present study, sonochemistry was used for the preparation of uranium(IV) oxide NPs. Sonochemistry deals with the effects of ultrasound on chemical reactions which do not directly result from the interactions between ultrasonic waves and dissolved species. The observed effects rather originate from the acoustic cavitation phenomenon, which consists of the successive nucleation, growth and violent implosion of gas- and vapour-filled micrometric bubbles.41 The final stage of collapse yields localized extreme conditions characterized by the formation of reactive radical species and the generation of physical effects typically observed when heterogeneous solid/liquid systems are considered (surface erosion of solids, fragmentation, depassivation, etc.). Sonochemistry has been reported to provide interesting alternative approaches for the dissolution of refractory actinide materials under soft conditions (e.g. UO2, ThO2, PuO2)42–46 or the preparation of Pu(IV) or U(VI) intrinsic colloids during the low frequency (20 kHz) sonolysis of powdered PuO2 and UO3 in pure water.47–50 In particular, it was observed that the sonication of powdered UO3 led to the polymerization of hydrolysed uranyl species into intrinsic colloids of 3.8 ± 0.3 nm exhibiting a schoepite-like structure.48 The current study focuses on the bottom-up preparation of UO2 NPs in an aqueous solution under a low frequency ultrasound (20 kHz) under an Ar/(10%)CO atmosphere.
2. Materials and methods
2.1. Chemicals
The used reagents were of analytical grade and were provided by Sigma-Aldrich. All aqueous solutions were prepared with Milli-Q water (18.2 mΩ cm at 25 °C). Ar and the Ar/(10%)CO gas mixture used during experiments were supplied by Air Liquide. Uranium metal chips were supplied by CETAMA.2.2. Preparation of U(IV) chloride solution
The uranium metal chips were first rinsed with dichloromethane, acetone and water and washed with 1 M HCl solution to remove possible traces of uranium oxides on the solid surface. The metal pieces were then dissolved in 6 M hydrochloric acid. The obtained solution was centrifuged to eliminate the solid residues and diluted with pure water to obtain a final concentration of HCl equal to 1 M. Such a high hydrochloric acid concentration allowed stabilization of the tetravalent oxidation state of uranium in the solution for several months.51 A typical UV-Vis absorption spectrum of the as-stabilized uranium solution is presented in Fig. S1 (ESI†). The concentration of U in the solution was 0.16 ± 0.01 M according to the characterization studies performed with ICP-OES and UV-Vis absorption spectroscopy (λ = 650 nm, ε = 58 L mol−1 cm−1).13 The final U(IV) solution was stored in an inert glove box until use.2.3. Experiments under stirring or ultrasound irradiation
Sonochemical experiments were carried out under a low frequency ultrasound (20 kHz) with a 1 cm2 titanium alloy horn connected to a 750 W generator (Sonics & Materials, Vibracell VCX 750). 50 mL of the solution poured into a tight homemade glass reactor (Fig. S2, ESI†) was sparged with Ar/(10%)CO at a controlled flow rate (110 mL min−1) for about 20 minutes before sonication and maintained during the whole experiment (2 hours). The use of CO as a mixture with Ar avoids the sonochemical formation of H2O2 in the medium due to the scavenging of OH˙ radicals formed during water splitting inside the cavitation bubble.52,53 The related phenomenon will be presented in more detail in the section “Results and discussion”. The temperature inside the reactor was measured with a thermocouple immersed in the solution and it reached 65 ± 2 °C. The calorimetric method was used to determine the specific acoustic power (Pac, W mL−1) delivered to the solution that reached 0.36 W mL−1 at 20 kHz.54 Experiments were also carried out without ultrasound under magnetic stirring to provide a reference experiment under analogous conditions (700 rpm, 50 mL, Ar, 65 ± 2 °C).For all of the experiments, 1.6 mL of the U(IV) chloride mother solution (0.16 M) was injected into water at 65 °C under sonication (20 kHz) or magnetic stirring providing a 50 mL solution of 5 mM U in 32 mM HCl (Fig. 1). During the experiments, sample aliquots were taken from the treated solutions every 30 minutes for analyses with UV-Vis absorption spectroscopy and SAXS. The experiments were stopped after 120 min of sonication or stirring which was accompanied by the formation of black colloidal suspensions (Fig. 1). Then, in the first study, the stability of the colloidal suspensions was evaluated as a function of time (0–120 min). In the second study, the properties of the colloidal nanoparticles were evaluated by precipitating them by slowly adding 0.1 M NaOH (pH 9–10). The solid phase was centrifuged (9000 rpm) and dried at room temperature under vacuum before characterization with PXRD and HR-TEM, while the corresponding supernatant was sampled for UV-Vis absorption spectroscopy. To stabilize the redox behaviour of the particles towards oxidation, all of the as-synthesized U(IV) solid samples were stored in an inert glove box.
 | ||
| Fig. 1 Scheme of the experimental method applied for the treatment of U(IV) chloride solutions (5 mM, 32 mM HCl) under 20 kHz ultrasound (US) or stirring at 65 °C and after addition of 0.1 M NaOH. | ||
2.4. Analyses and characterization
3. Results and discussion
Sonication of U(IV) chloride solutions (5 mM U, 32 mM HCl) was carried out under an atmosphere of Ar/(10%)CO gas mixture. It is known that CO avoids the formation of hydrogen peroxide in sonicated aqueous solutions by scavenging OH˙ radicals generated inside the cavitation bubbles as shown in eqn (2)–(4).52 These experimental conditions prevented the oxidation of uranium(IV) into U(VI) by HO˙ radicals or H2O2 molecules.9,12,13| H2O →)))→ HO˙ + ˙H | (2) |
| H˙ + ˙H → H2 | (3) |
| HO˙ + CO → CO2 + H˙ | (4) |
The UV-Vis absorption spectra acquired during sonolysis of the 5 mM U(IV) solutions at 65 °C are presented in Fig. 2. At t = 0 min, the characteristic bands of U(IV) are noticeable at λ = 650 nm, 550 nm, 500 nm and 430 nm.13 In comparison with the reference spectrum provided in Fig. S1 (ESI†), some modifications can be attributed to the presence of hydrolysis products of U(IV). In the literature, studies showed that the main forms of U(IV) in aqueous solution at room temperature are U4+, U(OH)3+ and U(OH)22+ for pH < 2.22,55 The spectrum of concentrated U(IV) acquired in the chloride solution (Fig. S1, ESI†) was used as a standard and subtracted from the experimental spectra obtained at t = 0 min (i.e. just after injecting U(IV) into the sonicated water, Fig. S3 ESI†). The resulting spectra exhibited main absorption bands located at λ = 621 nm, 529 nm, 500 nm, 471 nm and 405 nm (Fig. S3, ESI†), which agreed with the spectrum of the U(OH)3+ hydrolysed species reported in the work of Cha et al.55 It is important to mention the absence of oxidation of U(IV) into U(VI) under these conditions. A similar approach was applied in the reference experiment performed under stirring and it revealed similar conclusions (Fig. S3, ESI†). The difference observed in the spectra acquired at t = 0 min illustrated in Fig. 2 can be attributed to different degrees of hydrolysis in both experiments.
 | ||
| Fig. 2 UV-Vis absorption spectra acquired during the treatment of U(IV) chloride solutions (5 mM, 32 mM HCl) under (a) 20 kHz sonolysis (65 °C, Ar/(10%)CO, 0.36 W mL−1, pH = 1.5) and (b) mechanical stirring (65 °C, Ar, pH = 1.5). | ||
Strong absorption in the near UV range can be noticed after 30 min of sonication. This observation is attributed to Mie scattering evidencing the presence of suspended particles in the solution.56 The evolution of the absorption spectra is also accompanied by a strong modification of the colour of the U(IV) solution from turquoise before experiments to deep black after 120 min of treatment (Fig. 3). Such behaviour is opposed to the observations resulting from sonolyses performed at 20 °C (20 kHz, Ar/(10%)CO) where the absorption spectra remained unchanged (spectra not shown). In the literature, different studies illustrated the effect of temperature on the hydrolysis kinetics of U(IV).57,58 Shock et al. demonstrated that an increase in the temperature favoured this reaction.58 The experiments performed under magnetic stirring (65 °C, without ultrasound) led to similar behaviour and absorption spectra (Fig. 2b). In both cases, the final solution presented a black colour at the end of the treatment. The evolution of the pH of the solutions treated in the presence or absence of ultrasound is presented in Table S2 (ESI†). A slight acidification of the medium can be observed in both cases. This phenomenon can be attributed to hydrolysis reactions going with the release of protons in solution.
 | ||
| Fig. 3 Photos of the glass reactor during the sonication of the U(IV) chloride solution (5 mM) under 20 kHz ultrasound (Ar/(10%)CO, 0.36 W mL−1, pH = 1.5). | ||
The normalized SAXS diagrams acquired on the U(IV) solutions during treatments in the presence or absence of ultrasound at 65 °C are given in absolute units in Fig. 4. For both cases, the absence of signal noticed at t = 0 min demonstrates the lack of electronic contrast in the solution before starting the experiment indicating the absence of detectable polynuclear entities in the initial samples. However, both ultrasonic and stirring treatments enable a significant increase in the scattering of SAXS signals. Regardless of the conditions, the slope of the scattering curves is Q0 for low wave vectors and tends towards Q−4 at higher wave vectors. Such a scattering pattern is typical for three-dimensional objects such as compact particles.59 Micheau et al. recently observed a similar SAXS profile for intrinsic plutonium colloids prepared by hydrolysis of Pu(IV).60
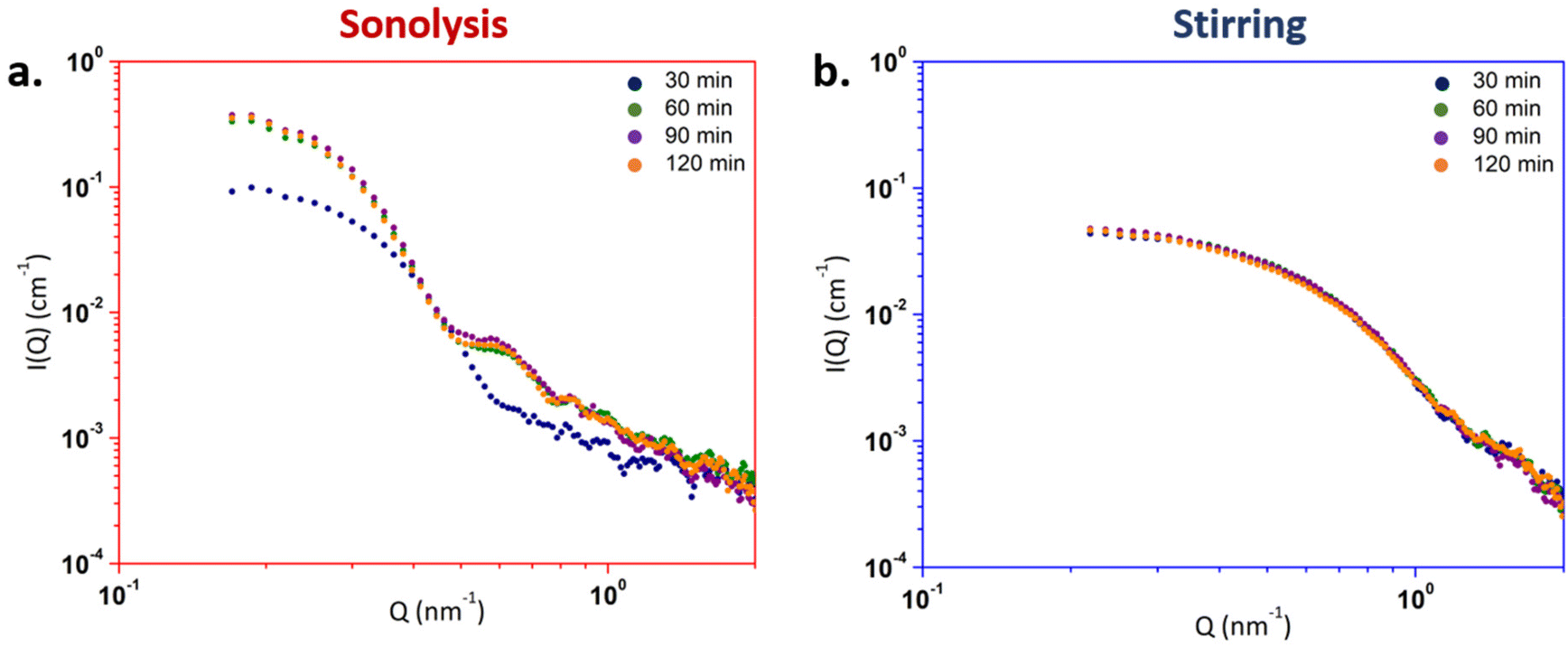 | ||
| Fig. 4 SAXS diagrams acquired as a function of time under (a) 20 kHz sonolysis (65 °C, Ar/(10%)CO, 0.36 W mL−1, pH = 1.5) and (b) mechanical stirring (65 °C, Ar, pH = 1.5). | ||
Significant differences can however be noticed on the SAXS diagrams when comparing both approaches. The scattering curve of the sonicated solutions evolves between 30 and 60 minutes of treatment (Fig. 4a) before remaining stable. This evolution is characterized by an increase in intensity and a shift of the curve inflexion towards lower wave vectors which demonstrate an increase in the concentration and size of the particles, respectively. By contrast, the scattering diagrams obtained under mechanical stirring (hydrolytic colloid) remain stable during the experiment and are shifted to higher wave vectors when compared to ultrasound experiments (Fig. 4b). Such a difference is attributed to a lower concentration and particle size of the particles prepared under stirring. Also, it is important to note the appearance of an oscillation on the SAXS diagram acquired for the sonicated solutions after 30 min. This feature is not observed in the stirred solution and can be attributed to a lower size polydispersity in the colloidal suspensions prepared under ultrasound.61,62 More generally, the characteristics of the normalized SAXS diagrams over time for both treatments confirm the colloidal nature of the samples in agreement with the UV-Vis absorption spectra. The SAXS diagrams obtained for both samples after 120 min of treatment are compared in Fig. 5.
 | ||
| Fig. 5 Normalized SAXS diagrams acquired on the final solutions (red and blue dots) and corresponding sphere fit models (orange and blue lines) after 120 min under 20 kHz sonolysis (65 °C, Ar/(10%)CO, 0.36 W mL−1, pH = 1.5) and mechanical stirring (hydrolytic colloid, 65 °C, Ar, pH = 1.5), respectively. | ||
A sphere form factor model was applied to fit the experimental data (procedure, Table S1 and eqn (S1)–(S3) described in the ESI†). In Fig. 5, the fits of the experimental data (orange and blue curves) match well with the normalized experimental diagrams, which generally confirm the spherical shape of the colloidal particles and their narrow size distribution for the experiment carried out under sonication. The radii R were determined considering spherical and filled NPs (ESI†). The results of the fits are presented in Table 1 and generally agree with the evolution of the normalized SAXS diagram previously discussed. For sonolysis, the particle radius increases between 30 and 60 min of treatment from 7.2 ± 0.1 nm to 9.4 ± 0.1 nm, respectively, before remaining stable. By contrast, the stirred solution only shows a slight increase in the radius value from 3.6 ± 0.1 nm to 3.9 ± 0.1 nm during the 120 min of treatment. Hence, the measured value is twice as small as the one observed under ultrasound. Also, the volume fractions Φp were calculated from I0 values from the sphere model (Table 1). For both treatments, the values appear much lower than the theoretical one (Φth = 1.2 × 10−4) when considering a total U(IV) conversion into the UO2 colloidal species. Such a difference can be attributed to the additional presence of soluble U(IV) oligomers or monomers in the analysed solutions.
| SAXS – sphere model | ||||
|---|---|---|---|---|
| Time (min) | I 0 (10−2 cm−1) | ΦP (10−7) | R (nm) | |
| Sonolysis | 0 | — | — | — |
| 30 | 14 | 2.2 | 7.2 ± 0.1 | |
| 60 | 64 | 4.4 | 9.4 ± 0.1 | |
| 90 | 83 | 5.1 | 9.7 ± 0.1 | |
| 120 | 74 | 4.6 | 9.6 ± 0.1 | |
| Stirring | 0 | — | — | — |
| 30 | 4.7 | 5.9 | 3.6 ± 0.1 | |
| 60 | 5.1 | 5.9 | 3.7 ± 0.1 | |
| 90 | 5.3 | 5.8 | 3.7 ± 0.1 | |
| 120 | 5.0 | 4.8 | 3.9 ± 0.1 | |
The stability of the colloidal suspensions formed after 120 min in the presence or absence of ultrasound was monitored over time by SAXS under room temperature without further treatment or addition of any chemicals (Fig. 1). A similar fitting approach to the one described above was applied (Fig. 5, Table S1, ESI†); the determined particle sizes are provided in Table S3 (ESI†).59 In the case of the sonicated solution (Fig. 6a), the absence of evolution of the diagram intensity or profile was noticed during the 150 min following the end of the experiment. This behaviour was even faster for the colloids obtained under stirring which completely dissolved after 90 min of storage (Fig. 6b). After these periods, SAXS data were not usable anymore for both solutions reflecting the instability of the as-prepared colloids over time. Such behaviour confirmed the dissolution of the colloids into smaller and ionic U(VI) species as revealed by UV-vis absorption spectroscopy (Fig. S4, ESI†). In both cases, the spectra are characteristic of the U(VI) aquo ion which is in agreement with the literature for such conditions (acidic pH < 2).63,64 These results confirmed that the as-prepared uranium colloids are not stable over time.
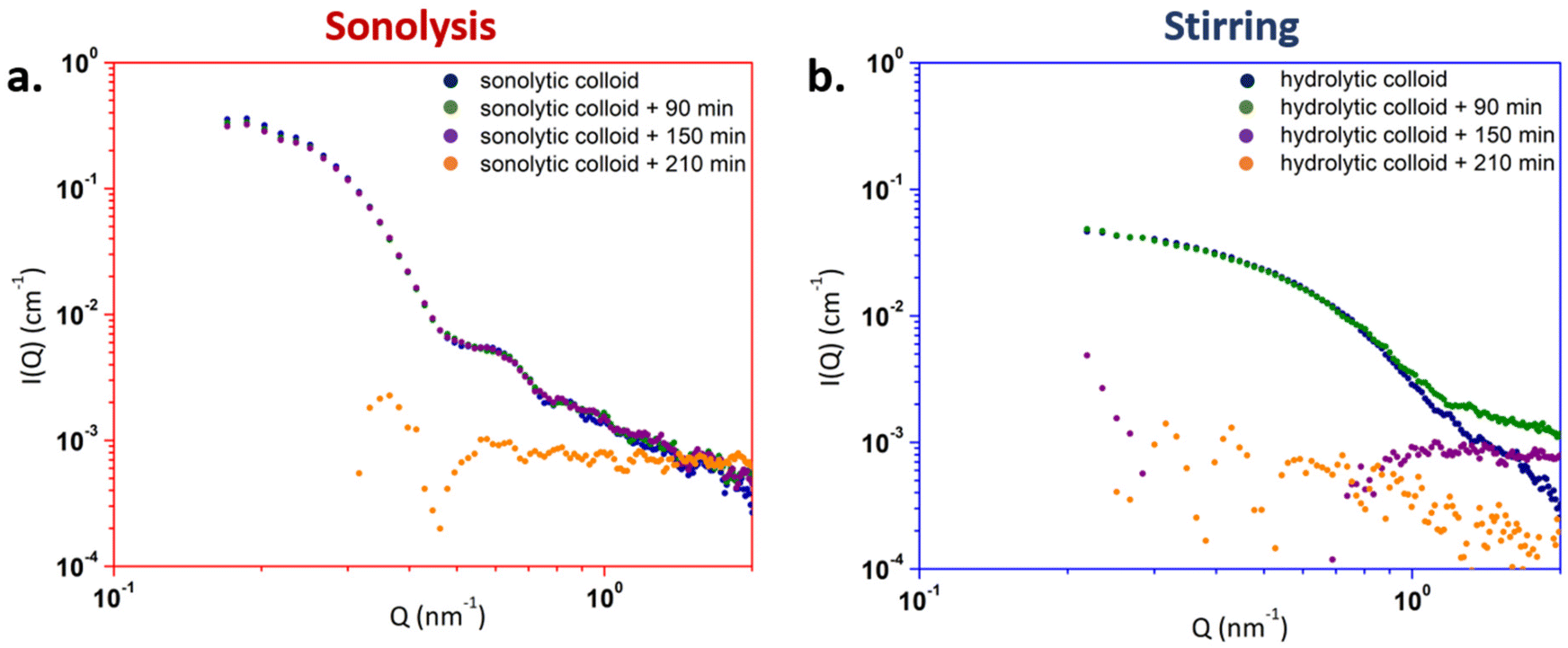 | ||
| Fig. 6 SAXS diagrams acquired as a function of time at the end of (a) 20 kHz sonolysis (65 °C, Ar/(10%)CO, 0.36 W mL−1, pH = 1.5) and (b) mechanical stirring (65 °C, Ar, pH = 1.5). | ||
We prevented the dissolution of the particles by adding a 0.1 M NaOH solution to the colloidal suspension after 120 min of treatment (stirring or sonication). This increased the pH of the solution to a value of approx. 9–10 (Table S2, ESI†) which provoked the precipitation of the black particles (Fig. 1). The analysis of the supernatants obtained after centrifugation by UV-Vis absorption spectroscopy (spectra not shown) indicated the absence of uranium in the solutions evidencing the complete precipitation of U under these conditions. The formation of a precipitate in both cases (after sonolysis or stirring) can be due to a modification of the surface charges of the repelling particles provoking their agglomeration. Some studies reported the precipitation of An(IV) colloids with an increase in the pH to alkaline values.65
The PXRD characterization of these precipitates is presented in Fig. 7. The diffraction pattern of the sonicated system was identified to correspond to shifted XRD diagrams of stoichiometric UO2 which better corresponded to UO2.2 (ICSD 00-047-1879).36,38 Elorrietta et al. illustrated in their study that the oxidation of UO2 into UO2+x leads to a shift of the diffraction peaks agreeing with our observations. The observation of hyperstoichiometric UO2+x is related to the low stability of the particles and their surface oxidation that likely occurred during their exposure to the room atmosphere for several hours during analyses.36 For comparison, the solid obtained after stirring exhibited very weak and noisy diffraction peaks that were attributed to smaller and less crystalline particles.
 | ||
| Fig. 7 Normalized PXRD diagrams (background corrected) acquired on uranium precipitates obtained by the addition of 0.1 M NaOH after 120 min of sonolysis (20 kHz, Ar/(10%)CO, 0.36 W mL−1, pH = 9.5) or mechanical stirring (absence of ultrasound, pH = 9.8). | ||
HR-TEM demonstrated that the precipitate obtained after sonolysis is composed of quasi-spherical and monodisperse well-defined NPs measuring ca. 25.2 ± 0.7 nm in diameter (Fig. 8a). The observed NP size is slightly larger than what is obtained from SAXS (ca. 18 nm in diameter). The high-resolution mode indicated the superimposed presence of small crystalline NPs measuring ca. 5 nm. These particles most probably result from the additional precipitation of the aforementioned soluble hydrolysed U(IV) species (oligomers) upon addition of NaOH (Fig. 8b). This can also explain the slightly bigger size of the particles measured with TEM in comparison with SAXS due to the additional presence of these smaller particles that precipitated on the bigger ones. The analogous sample obtained after stirring was far less defined and showed large and polydispersed agglomerates (Fig. 8d). Related magnifications demonstrated the presence of different populations of ill-defined polycrystalline NPs measuring approx. 7.5 ± 0.8 nm and 4.6 ± 0.5 nm in diameter, close to the sizes determined from SAXS analyses (Fig. 8e). However, the observations contrast with the XRD measurements which suggest the presence of poorly crystalline particles. Such discrepancy can be related to the low concentration of U particles during measurements (diffraction peaks can be distinguished in the noisy baseline), but also to TEM conditions involving vacuum and high voltage which have been reported to compete in the modification of the crystallinity of some materials.66
 | ||
| Fig. 8 HR-TEM analyses of the precipitates obtained by adding 0.1 M NaOH to the U solutions after sonolysis (20 kHz, Ar/(10%)CO, 0.36 W mL−1, pH = 9.5) or mechanical stirring (pH = 9.8). Dashed squares represent the selected areas analyzed by electron diffraction (SAED). (a) and (b) U(IV) precipitate after sonolysis at different magnifications and (c) the corresponding SAED pattern. (d) and (e) U(IV) precipitate after stirring at different magnifications and (f) the corresponding SAED pattern. | ||
The important difference in size between sonolytic and hydrolysed colloids agrees with the SAXS observations. The aggregation of the NPs is most probably related to the neutralization of their surface charge occurring with the addition of NaOH but also to the drying step during HR-TEM sample preparations. The diffraction ring distributions observed by selected area electron diffraction (SAED) are similar whatever the synthesis procedure (Fig. 8c and f) confirming the presence of crystalline NPs. The different distances measured on the patterns are given in Table S4 (ESI†) and are characteristic of the UO2 structure in agreement with the literature. The calculations of the d1/dn values confirmed the cubic crystal structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group) as UO2-like compounds.23 In addition, the d-spacing values directly measured in Fig. 8b and e correspond to the [1 1 1] crystal plane of UO2. These results are in agreement with the one obtained by PXRD.
m space group) as UO2-like compounds.23 In addition, the d-spacing values directly measured in Fig. 8b and e correspond to the [1 1 1] crystal plane of UO2. These results are in agreement with the one obtained by PXRD.
The synthesis and characterization of bulk UO2+x have been highly reported in the literature in contrast to analogous NPs.67,68 Nevertheless, Priyadarshini et al. studied the formation of intrinsic U(IV) colloids as a function of the uranium concentration and the pH value.67 For a 5 mM uranium solution, they observed colloids with an average diameter of 15.46 nm (light scattering measurements) at pH = 2.74. In our study (pH = 1.5, Table S2, ESI†), the size of the sonolytic colloids is twice that of the hydrolytic ones and is more concentrated. This may be due to the effect of ultrasound irradiation which can enhance the reactivity of U(IV) ions and their hydrolysis.43 Cha et al. reported a similar morphology for 20 nm U(IV) NPs synthesized by a hydrothermal route at pH 2.55 They also observed additional smaller crystalline U(IV) particles (2–3 nm), which may have a uraninite-like structure.55 Sonochemistry has been widely used for the preparation of nanomaterials and significant differences in terms of size, morphology, formation kinetics, and yields have been reported when compared to conventional approaches. In particular, hydrolysis of metal ions in aqueous solutions has been reported to be enhanced under ultrasound due to local heating occurring in the vicinity of the cavitation bubbles.69,70 The presence of this transient superheated liquid shell that surrounds the cavitation bubble has been described to yield hydrothermal-like conditions.71,72 By comparison with the work of Cha et al., ultrasonically induced hydrothermal conditions could be invoked to explain the particular morphology and crystallinity of the NPs synthesized in the current study. Cha et al. proposed a two-step mechanism for the formation of crystalline UO2 NPs based on the ability of U(IV) ions to form oxo and hydroxo bonds and the formation of an intermediate entity (noted M) from the U(IV) hydrolysed species (eqn (5)).55,73 It is worth noting that an analogous mechanism has been recently suggested for Pu(IV).74
| U4+ U(OH)3+ → M → UO2(NP) | (5) |
4. Conclusion
This study reports the ultrasonically assisted synthesis (20 kHz, 65 °C, Ar/10%CO) of the poorly described U(IV) intrinsic colloids from a uranium(IV) hydrochloric solution (5 mM U, 32 mM HCl). These species, and their corresponding precipitates obtained by the addition of 0.1 M NaOH, were characterized by SAXS, PXRD and HR-TEM. The colloidal suspensions were found to be composed of spherical and crystalline well-defined NPs measuring 18.0 ± 0.1 nm (diameter). Although both precipitates exhibited a UO2-like structure, in comparison with the hydrolytic synthesis performed under stirring (65 °C, Ar, NP size: 7.8 ± 0.1 nm), the sonochemical approach provides better defined NPs also offering a better crystallinity with a narrower particle size distribution. The application of ultrasound provides an original and facile synthesis route for the preparation of UO2 NPs with controlled properties of nuclear interest or for environmental investigation purposes (e.g. solubility studies, reactivity, redox, etc.).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the French programme NEEDS for its financial support to the project as well as the RTA/RCHIM program (cross-cutting basic research program).References
- J.-F. Vigier, D. Freis, O. Walter, O. Dieste Blanco, D. Bouëxière, E. Zuleger, N. Palina, T. Vitova, R. J. M. Konings and K. Popa, CrystEngComm, 2022, 24, 6338–6348 RSC.
- S. N. Kalmykov and M. A. Denecke, Actinide Nanoparticle Research, Springer Science & Business Media, Berlin, Heidelberg, 1st edn, 2011 Search PubMed.
- A. Yu. Romanchuk, I. E. Vlasova and S. N. Kalmykov, Front. Chem., 2020, 8, 630 CrossRef CAS PubMed.
- G. J.-P. Deblonde, A. B. Kersting and M. Zavarin, Commun. Chem., 2020, 3, 167 CrossRef CAS.
- C. Walther and M. A. Denecke, Chem. Rev., 2013, 113, 995–1015 CrossRef CAS PubMed.
- H. Zänker and C. Hennig, J. Contam. Hydrol., 2014, 157, 87–105 CrossRef PubMed.
- A. B. Kersting, Inorg. Chem., 2013, 52, 3533–3546 CrossRef CAS PubMed.
- R. C. Ewing, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
- K. Maher, J. R. Bargar and G. E. Brown, Inorg. Chem., 2013, 52, 3510–3532 CrossRef CAS PubMed.
- The chemistry of the actinide and transactinide elements, ed. L. R. Morss, N. M. Edelstein, J. Fuger and J. J. Katz, Springer, Dordrecht, 2006 Search PubMed.
- J. Halpern and J. G. Smith, Can. J. Chem., 1956, 34, 1419–1427 CrossRef CAS.
- A. Landgren and H. Ramebäck, Radiochim. Acta, 2001, 89, 75–82 CrossRef CAS.
- J. J. Katz and E. Rabinowitch, Chemistry of uranium: collected papers, United States Atomic Energy Commission, Technical Information Service Extension, 1958 Search PubMed.
- M. Steppert, C. Walther, M. Fuss and S. Büchner, Rapid Commun. Mass Spectrom., 2012, 26, 583–591 CrossRef CAS PubMed.
- F. Quilès, C. Nguyen-Trung, C. Carteret and B. Humbert, Inorg. Chem., 2011, 50, 2811–2823 CrossRef PubMed.
- V. Neck and J. I. Kim, Radiochim. Acta, 2001, 89, 1–16 CrossRef CAS.
- Y. Wang, M. Frutschi, E. Suvorova, V. Phrommavanh, M. Descostes, A. A. A. Osman, G. Geipel and R. Bernier-Latmani, Nat. Commun., 2013, 4, 2942 CrossRef PubMed.
- H. Zänker, K.-U. Ulrich, K. Opel, V. Brendler and F. Dresden-Rossendorf, in IMWA Symposium 2007: Water in Mining Environments, Cagliari, Italy, 2007 Search PubMed.
- P. E. Reiller, N. D. M. Evans and G. Szabó, Radiochim. Acta, 2008, 96, 345–358 CrossRef CAS.
- M. D. Kaminski, N. M. Dimitrijevic, C. J. Mertz and M. M. Goldberg, J. Nucl. Mater., 2005, 347, 77–87 CrossRef CAS.
- I. Grenthe, J. Fuger, R. J. M. Konings, R. J. Lemire, A. B. Muller, C. Nguyen-Trung and H. Wanner, Chemical thermodynamics of uranium, Elsevier, Amsterdam, 1992, vol. 1 Search PubMed.
- K. Opel, S. Weiß, S. Hübener, H. Zänker and G. Bernhard, Radiochim. Acta, 2007, 95, 143–149 CrossRef CAS.
- T. M. Nenoff, B. W. Jacobs, D. B. Robinson, P. P. Provencio, J. Huang, S. Ferreira and D. J. Hanson, Chem. Mater., 2011, 23, 5185–5190 CrossRef CAS.
- M. C. Rath, S. Keny and D. B. Naik, J. Nanosci. Nanotechnol., 2016, 16, 9575–9582 CrossRef CAS.
- R. J. Abril, R. Eloirdi, D. Bouëxière, R. Malmbeck and J. Spino, J. Mater. Sci., 2011, 46, 7247–7252 CrossRef CAS.
- J. Leduc, J. I. Pacold, D. K. Shuh, C. Dong and S. Mathur, Z. Anorg. Allg. Chem., 2018, 644, 12–18 CrossRef CAS.
- J. M. Roach, K. V. Manukyan, A. Majumdar, S. Dede, A. G. Oliver, P. C. Burns and A. Aprahamian, Inorg. Chem., 2021, 60, 18938–18949 CrossRef CAS PubMed.
- H. Wu, Y. Yang and Y. C. Cao, J. Am. Chem. Soc., 2006, 128, 16522–16523 CrossRef CAS PubMed.
- K. Popa and O. Walter, in Comprehensive Nuclear Materials, Elsevier, 2020, pp. 579–592 Search PubMed.
- O. Walter, K. Popa and O. D. Blanco, Open Chem. J., 2016, 14, 170–174 CAS.
- K. Popa, O. Walter, O. D. Blanco, A. Guiot, D. Bouëxière, J.-Y. Colle, L. Martel, M. Naji and D. Manara, CrystEngComm, 2018, 20, 4614–4622 RSC.
- J. Manaud, J. Maynadié, A. Mesbah, M. O. J. Y. Hunault, P. M. Martin, M. Zunino, D. Meyer, N. Dacheux and N. Clavier, Inorg. Chem., 2020, 59, 3260–3273 CrossRef CAS PubMed.
- G. Kauric, O. Walter, A. Beck, B. Schacherl, O. Dieste Blanco, J.-F. Vigier, E. Zuleger, T. Vitova and K. Popa, Mater. Today Adv., 2020, 8, 100105 CrossRef.
- C. Tabata, K. Shirasaki, A. Sunaga, H. Sakai, D. Li, M. Konaka and T. Yamamura, CrystEngComm, 2021, 23, 8660–8672 RSC.
- C. Tabata, K. Shirasaki, H. Sakai, A. Sunaga, D. Li, M. Konaka and T. Yamamura, CrystEngComm, 2022, 24, 3637–3648 RSC.
- E. Gerber, A. Yu. Romanchuk, S. Weiss, S. Bauters, B. Schacherl, T. Vitova, R. Hübner, S. Shams Aldin Azzam, D. Detollenaere, D. Banerjee, S. M. Butorin, S. N. Kalmykov and K. O. Kvashnina, Inorg. Chem. Front., 2021, 8, 1102–1110 RSC.
- G. Leinders, J. Pakarinen, R. Delville, T. Cardinaels, K. Binnemans and M. Verwerft, Inorg. Chem., 2016, 55, 3915–3927 CrossRef CAS PubMed.
- J. M. Elorrieta, L. J. Bonales, N. Rodríguez-Villagra, V. G. Baonza and J. Cobos, Phys. Chem. Chem. Phys., 2016, 18, 28209–28216 RSC.
- G. C. Allen and P. A. Tempest, J. Chem. Soc., Dalton Trans., 1983, 2673–2677 RSC.
- T. Gouder, R. Eloirdi and R. Caciuffo, Sci. Rep., 2018, 8, 8306 CrossRef CAS PubMed.
- K. S. Suslick, Y. Didenko, M. M. Fang, T. Hyeon, K. J. Kolbeck, W. B. McNamara, M. M. Mdleleni and M. Wong, Philos. Trans. R. Soc. London, Ser. A, 1999, 357, 335–353 CrossRef CAS.
- S. I. Nikitenko, L. Venault, R. Pflieger, T. Chave, I. Bisel and P. Moisy, Ultrason. Sonochem., 2010, 17, 1033–1040 CrossRef CAS PubMed.
- S. Lahiri, R. L. Bhardwaj, D. Mandal and P. R. Gogate, Ultrason. Sonochem., 2020, 65, 105066 CrossRef CAS PubMed.
- L. Bonato, M. Virot, X. Le Goff, P. Moisy and S. I. Nikitenko, Ultrason. Sonochem., 2020, 69, 105235 CrossRef CAS PubMed.
- X. Beaudoux, M. Virot, T. Chave, G. Leturcq, G. Jouan, L. Venault, P. Moisy and S. I. Nikitenko, Dalton Trans., 2016, 45, 8802–8815 RSC.
- M. Virot, S. Szenknect, T. Chave, N. Dacheux, P. Moisy and S. I. Nikitenko, J. Nucl. Mater., 2013, 441, 421–430 CrossRef CAS.
- E. Dalodière, M. Virot, V. Morosini, T. Chave, T. Dumas, C. Hennig, T. Wiss, O. Dieste Blanco, D. K. Shuh, T. Tyliszcak, L. Venault, P. Moisy and S. I. Nikitenko, Sci. Rep., 2017, 7, 43514 CrossRef PubMed.
- M. Cot-Auriol, M. Virot, C. Micheau, T. Dumas, X. Le Goff, C. Den Auwer, O. Diat, P. Moisy and S. I. Nikitenko, Dalton Trans., 2021, 50, 11498–11511 RSC.
- M. Virot, T. Dumas, M. Cot-Auriol, P. Moisy and S. I. Nikitenko, Nanoscale Adv., 2022, 4, 4938–4971 RSC.
- S. I. Nikitenko, M. Virot and P. Moisy, Radiochim. Acta, 2022, 110, 453–470 CrossRef CAS.
- N. Dacheux, V. Brandel and M. Genet, New J. Chem., 1995, 19, 1029–1036 CAS.
- S. I. Nikitenko, P. Martinez, T. Chave and I. Billy, Angew. Chem., Int. Ed., 2009, 48, 9529–9532 CrossRef CAS PubMed.
- H. Harada, Ultrason. Sonochem., 1998, 5, 73–77 CrossRef CAS PubMed.
- S. I. Nikitenko, C. Le Naour and P. Moisy, Ultrason. Sonochem., 2007, 14, 330–336 CrossRef CAS PubMed.
- W. Cha, H.-K. Kim, H. Cho, H.-R. Cho, E. C. Jung and S. Y. Lee, RSC Adv., 2020, 10, 36723–36733 RSC.
- Y. Xu and B. A. S. Gustafson, J. Quant. Spectrosc. Radiat. Transfer, 2001, 70, 395–419 CrossRef CAS.
- R. H. Betts, Can. J. Chem., 1955, 33, 1775–1779 CrossRef CAS.
- E. L. Shock, D. C. Sassani and H. Betz, Geochim. Cosmochim. Acta, 1997, 61, 4245–4266 CrossRef CAS.
- L. Boldon, F. Laliberte and L. Liu, Nano Rev., 2015, 6, 25661 CrossRef PubMed.
- C. Micheau, M. Virot, S. Dourdain, T. Dumas, D. Menut, P. L. Solari, L. Venault, O. Diat, P. Moisy and S. I. Nikitenko, Environ. Sci.: Nano, 2020, 7, 2252–2266 RSC.
- J. S. Pedersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 657–665 CrossRef PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed.
- G. Meinrath, J. Radioanal. Nucl. Chem., 1997, 224, 119–126 CrossRef CAS.
- K. Müller, V. Brendler and H. Foerstendorf, Inorg. Chem., 2008, 47, 10127–10134 CrossRef PubMed.
- S. Yariv and H. Cross, in Geochemistry of Colloid Systems, Springer Berlin Heidelberg, Berlin, Heidelberg, 1979, pp. 335–377 Search PubMed.
- M. Schindler, F. C. Hawthorne, M. S. Freund and P. C. Burns, Geochim. Cosmochim. Acta, 2009, 73, 2471–2487 CrossRef CAS.
- N. Priyadarshini, M. Sampath, S. Kumar, U. K. Mudali and R. Natarajan, J. Nucl. Chem., 2014, 2014, 1–10 CrossRef.
- C. Degueldre, P.-Y. Favarger, R. Rossé and S. Wold, Talanta, 2006, 68, 623–628 CrossRef CAS PubMed.
- A. Gedanken, Ultrason. Sonochem., 2004, 11, 47–55 CrossRef CAS PubMed.
- H. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555–2567 RSC.
- T. J. Mason, T. J. Mason and J. P. Lorimer, Applied sonochemistry: the uses of power ultrasound in chemistry and processing, Wiley, Ann Arbor, Michigan, 2002 Search PubMed.
- A.-L. Morel, S. I. Nikitenko, K. Gionnet, A. Wattiaux, J. Lai-Kee-Him, C. Labrugere, B. Chevalier, G. Deleris, C. Petibois, A. Brisson and M. Simonoff, ACS Nano, 2008, 2, 847–856 CrossRef CAS PubMed.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944–994 CrossRef CAS PubMed.
- M. Cot-Auriol, M. Virot, T. Dumas, O. Diat, D. Menut, P. Moisy and S. I. Nikitenko, Chem. Commun., 2022, 58, 13147–13150 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt03721a |
| This journal is © The Royal Society of Chemistry 2023 |