
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, spectroscopic and structural properties of Sn(II) and Pb(II) triflate complexes with soft phosphine and arsine coordination†
Kelsey R.
Cairns
 ,
Rhys P.
King
,
Robert D.
Bannister
,
William
Levason
and
Gillian
Reid
*
,
Rhys P.
King
,
Robert D.
Bannister
,
William
Levason
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 23rd January 2023
Abstract
Reaction of the divalent M(OTf)2 (M = Sn, Pb; OTf = CF3SO3) with soft phosphine and arsine ligands, L, where L = o-C6H4(ER2)2 (E = P, R = Me or Ph; E = As, R = Me), MeC(CH2ER2)3 (E = P, R = Ph; E = As, R = Me), PhP(CH2CH2PPh2)2 or P(CH2CH2PPh2)3, affords complexes of stoichiometry M(L)(OTf)2 as white powders, which have been characterised via elemental analysis, 1H, 19F{1H}, 31P{1H} and 119Sn NMR spectroscopy, with the expected 31P–119Sn and 31P–207Pb couplings clearly evident. The crystal structures of nine of these pnictine complexes are reported, in each case revealing retention of one or both OTf anions, which gives rise to a diverse range of coordination environments including monomers, as well as varying degrees of oligomerisation to form weakly associated (OTf-bridged) dimers, trimers and polymers. 19F{1H} NMR spectra indicate that the OTf is essentially anionic (dissociated) in solution. Anion metathesis of [M(OTf)2{MeC(CH2PPh2)3}] with Na[BArF] (BArF = B{3,5-(CF3)2C6H3}4) yields the corresponding [M{MeC(CH2PPh2)3}][BArF]2 salts, the crystal structures of all three (M = Ge, Sn, Pb) reveal pyramidal dications with discrete [BArF]− anions providing charge balance. Density functional theory (DFT) calculations on these [M{MeC(CH2PPh2)3}]2+ (M = Ge, Sn, Pb) dications using the B3LYP-D3 functional show the presence of a directional lone pair, which is a mixture of valence s and pz character, with the valence p-orbital character decreasing down group 14. Natural bond orbital (NBO) analysis also shows that the natural charge at the metal centre increases and the charge on the P centre decreases upon going down group 14.
Introduction
The metallic elements of group 14, germanium, tin and lead have extensive coordination chemistries, for germanium and tin in both the M(II) and M(IV) oxidation states; in contrast very few lead(IV) complexes are known.1–3 Germanium(IV) halides form mostly six-coordinate complexes with neutral N- and O-donor ligands1,2 but whilst GeF4 complexes with phosphine and thioether ligands are well established,2,4 complexes with arsenic ligands have not been obtained, and GeCl4 and phosphines give redox products [R3PCl][GeCl3].4 There appear to be no crystallographically confirmed complexes of GeI4.4 In contrast, tin(IV) halides form many complexes with soft P, As, S and Se ligands; redox chemistry is rarely observed and even SnI4 forms a significant range of complexes.1,3,5 Recent work has focussed on attempts to isolate Sn(IV) cations using halide abstraction reagents such as Na[B{3,5-(CF3)2C6H3}4] (Na[BArF]) or Me3SiO3SCF3 (TMSOTf).5 Coordination complexes of Ge(II) were little known for many years, but have received intensive study in the last twenty years.1,2 Complexes mostly contain halide co-ligands and often a central 3- or 4-coordinate core, with longer interactions to anions in neighbouring molecules in many (but not all) cases producing di-, oligo- or poly-meric structures.1–3,6 A rich chemistry of Ge(II) cations containing oxa-, aza- or thia-macrocycles,7 and group 14 tetryliumylidenes has also emerged.8Pnictine chemistry of the M(II) centres is less developed.3 The [GeX2(diphosphine)] (diphosphine = Me2PCH2CH2PMe2, Et2PCH2CH2PEt2; X = Cl, Br, I) are discrete four coordinate monomers with near linear GeX2 units; the [GeX2{o-C6H4(PMe2)2}] contain four-coordinate Ge weakly associated into dimers via X-bridges.9 The diarsine complexes include [GeCl{o-C6H4(AsMe2)2}][GeCl3] and [GeI2{o-C6H4(AsMe2)2}], whilst the structure of [GeX2{o-C6H4(PPh2)2}] reveals a very asymmetrically coordinated diphosphine, possibly best described as κ1-coordinated.9 Halide-free, three-coordinate pyramidal Ge(II) dications, [Ge(PMe3)3][OTf]2, [GeL][OTf]2 (L = MeC(CH2PPh2)3, MeC(CH2AsMe2)3, κ3-P(CH2CH2PPh2)3), have been described very recently and the electronic structures and bonding probed by DFT calculations.10
Much less effort has been devoted to the study of Sn(II) complexes compared to the detailed results available for Sn(IV) compounds.3 Phosphine complexes of SnF2 have not been prepared, however, a series of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with SnCl2 was isolated, including [SnCl2{o-C6H4(PMe2)2}] and [SnCl2{Me2PCH2CH2PMe2}] which have a SnP2Cl2 core and are weakly associated into dimers via chlorine bridges.11 [SnCl2{o-C6H4(PPh2)2}] contained a pyramidal SnPCl2 core with the second phosphino group ∼3.29 Å from the tin, and like the germanium analogue is best described as containing a κ1-diphosphine. The product of reacting SnCl2 and Ph2PCH2CH2PPh2 is [(SnCl2)2{μ-Ph2PCH2CH2PPh2}], again with a pyramidal SnPCl2 core linked into chains via chlorine bridges and with the diphosphine cross-linking the chains.11 The only reported diarsine complex, [SnCl{o-C6H4(AsMe2)2}][SnCl3], like the Ge(II) analogue, is cationic with a polymeric chloride bridged cation.11In situ31P and 119Sn NMR data have been obtained from MeNO2 solutions of Sn[SbF6]2 and various polydentate phosphines including Ph2PCH2CH2PPh2, PhP(CH2CH2PPh2)2, MeC(CH2PPh2)3, {Ph2PCH2CH2P(Ph)CH2}2 and P(CH2CH2PPh2)3.12,13 Although the data mostly indicated three phosphine donors are bound to the tin, none were isolated and no crystallographic data are available.
:1 complexes with SnCl2 was isolated, including [SnCl2{o-C6H4(PMe2)2}] and [SnCl2{Me2PCH2CH2PMe2}] which have a SnP2Cl2 core and are weakly associated into dimers via chlorine bridges.11 [SnCl2{o-C6H4(PPh2)2}] contained a pyramidal SnPCl2 core with the second phosphino group ∼3.29 Å from the tin, and like the germanium analogue is best described as containing a κ1-diphosphine. The product of reacting SnCl2 and Ph2PCH2CH2PPh2 is [(SnCl2)2{μ-Ph2PCH2CH2PPh2}], again with a pyramidal SnPCl2 core linked into chains via chlorine bridges and with the diphosphine cross-linking the chains.11 The only reported diarsine complex, [SnCl{o-C6H4(AsMe2)2}][SnCl3], like the Ge(II) analogue, is cationic with a polymeric chloride bridged cation.11In situ31P and 119Sn NMR data have been obtained from MeNO2 solutions of Sn[SbF6]2 and various polydentate phosphines including Ph2PCH2CH2PPh2, PhP(CH2CH2PPh2)2, MeC(CH2PPh2)3, {Ph2PCH2CH2P(Ph)CH2}2 and P(CH2CH2PPh2)3.12,13 Although the data mostly indicated three phosphine donors are bound to the tin, none were isolated and no crystallographic data are available.
The coordination chemistry of lead(II) with neutral phosphines is extremely limited.3 Recent examples of neutral diphosphine complexes are the lead(II) thiolates [(2,6-Me2C6H3S)2Pb]2{μ-Ph2P(CH2)2PPh2} and [(2,6-Me2C6H3S)2Pb]3{Me2P(CH2)2PMe2};14 the latter contains a chain of the three lead centres linked by thiolate bridges, with the Me2P(CH2)2PMe2 chelating to the central Pb. The insoluble, intractable lead dihalides have meant that salts with oxo-anions, Pb(ClO4)2, Pb(NO3)2, or fluoroanions have been used.12,13,15 The reaction of Pb(NO3)2 with Me2P(CH2)2PMe2, o-C6H4(PMe2)2 or Et2P(CH2)2PEt2 (L–L) in H2O/MeCN gave white [Pb(L–L)(NO3)2].15 The structures of [Pb{Me2P(CH2)2PMe2}(NO3)2] and [Pb{o-C6H4(PMe2)2}(NO3)2] reveal chelating diphosphines and κ2-NO3 groups occupying one hemisphere about the lead centre, with single oxygen bridges to two further nitrate groups from neighbouring molecules completing a distorted eight-coordinate geometry. [Pb{o-C6H4(PMe2)2}(H2O)(SiF6)]·H2O has a chelating diphosphine, a coordinated water molecule and a coordinated [SiF6]2− group, with further Pb–F interactions to neighbouring molecules producing a chain polymer structure.15 Several polydentate phosphine complexes of Pb[SbF6]2 (expected 1:1 ratio) have also been studied by in situ31P{1H} and 207Pb NMR spectroscopy in MeNO2 solution, although none of these complexes were isolated.12,13
Here we report a systematic study of the synthesis, X-ray crystal structures and multinuclear NMR spectroscopic data on polydentate pnictine complexes of Sn[OTf]2 and Pb[OTf]2, and compare the results with the Ge(II) analogues, and the reported complexes formed with other anions. The phosphine and arsine ligands used in this work are depicted in Scheme 1.
 | ||
| Scheme 1 The pnictine ligands employed in this work. | ||
Experimental
Infrared spectra were recorded as Nujol mulls between CsI plates using a PerkinElmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H, 19F{1H}, 31P{1H} and 119Sn NMR spectra were recorded from CD3CN solutions using a Bruker AV400 spectrometer and referenced to SiMe4via the residual solvent resonance (1H), external CFCl3 (19F), 85% H3PO4 (31P) and SnMe4 (119Sn), respectively. Duplicate microanalyses were undertaken at Medac Ltd, with the majority of measurements within ±0.4% of the theoretical value. However, in a few cases the values are slightly outside this range, reflecting the recognised inherent variability of microanalytical measurements across different facilities.16n-Hexane and benzene were dried by distillation from sodium and CH2Cl2 and MeCN from CaH2. All preparations were carried out under anhydrous conditions via a dry dinitrogen atmosphere and standard Schlenk and glovebox techniques. Tin(II) triflate and lead(II) triflate, MeC(CH2PPh2)3, PhP(CH2CH2PPh2)2, o-C6H4(PPh2)2 and P(CH2CH2PPh2)3 were obtained from Sigma-Aldrich. The other ligands, o-C6H4(PMe2)2,17o-C6H4(AsMe2)2,18 and MeC(CH2AsMe2)318 were prepared by the literature methods. Although formulated as “anhydrous”, the IR spectra of the M(OTf)2 typically showed varying amounts of water, which was removed completely by drying in vacuo for a few hours before using for the synthesis of the complexes.
X-ray crystallography
Crystals suitable for single crystal X-ray analysis were grown either by layering CH2Cl2 solutions with n-hexane ([Sn(OTf)2{o-C6H4(PMe2)2}] (1), [Sn{MeC(CH2PPh2)3}][BArF]2 (4), [Sn(OTf){P(CH2CH2PPh2)3}][OTf] (7), [Sn(OTf)2{o-C6H4(PPh2)2}] (8), [Pb(OTf)2{o-C6H4(AsMe2)2}] (10), [Pb{MeC(CH2PPh2)3}][BArF]2 (12), [Ge{MeC(CH2PPh2)3}][BArF]2 (15)), or by vapour diffusion of diethyl ether into MeCN solutions ([Sn(OTf)2{o-C6H4(AsMe2)2}] (2), [Sn(OTf){PhP(CH2CH2PPh2)2}][OTf] (5), [Pb(OTf)2{o-C6H4(PMe2)2}] (9), [Pb(OTf)2{MeC(CH2PPh2)3}] (11), [Pb(OTf){P(CH2CH2PPh2)3}][OTf] (13)).Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with HF Varimax optics (100 μm focus) with the crystal held at 100 K, or a Rigaku UG2 goniometer equipped with a Rigaku hybrid pixel array detector (Hypix 6000 HE detector) mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) (or copper, λ = 1.5406 Å, for the three [BArF] salts) rotating anode generator with Arc)Sec VHF Varimax confocal mirrors (70 μm focus), with the crystal held at 100 K. Structure solution and refinement were performed using SHELX(S/L)97, SHELX2013, or SHELX-2014/7 via Olex.19 Structure solution and refinement was mostly routine, except for disorder of the OTf and BArF anions in some cases, details of which are provided in the relevant cif files. Details of the crystallographic parameters are given in Table 1.
| M = Ge (15) | M = Sn (4) | M = Pb (12) | |
|---|---|---|---|
| d(M–P)/Å | Ge1–P1 = 2.4239(4) | Sn1–P1 = 2.6438(4) | Pb–P1 = 2.7360(5) |
| Ge1–P2 = 2.4070(4) | Sn1–P2 = 2.6194(4) | Pb1–P2 = 2.7092(6) | |
| Ge1–P3 = 2.4110(5) | Sn1–P3 = 2.6249(4) | Pb1–P3 = 2.7184(6) | |
| <P–M–P/° | P1–Ge1–P2 = 86.609(14) | P1–Sn1–P2 = 82.120(13) | P1–Pb1–P2 = 80.594(17) |
| P1–Ge1–P3 = 85.912(15) | P1–Sn1–P3 = 80.761(14) | P1–Pb1–P3 = 78.676(17) | |
| P2–Ge1–P3 = 85.412(15) | P2–Sn1–P3 = 80.160(14) | P2–Pb1–P3 = 77.868(17) | |
Complex preparations
9 (0.050 g, 0.05 mmol) was suspended in CH2Cl2 (1 mL), Na[BArF] (0.089 g, 0.10 mmol) added, the solution was stirred for ∼10 min, forming a colourless solution with a small amount of precipitate (NaOTf). The supernatant was filtered away from the solid and layered with n-hexane (2 mL). After 24 h colourless crystals formed which were isolated by filtration and dried in vacuo. The crystals were suitable for single crystal X-ray diffraction. Yield: 0.068 mg, 56%. Required for C105H63B2F48P3Ge (2423.59): C, 52.03; H, 2.62. Found: C, 52.26; H, 2.84%. 1H NMR (298 K, CD2Cl2): δ = 7.71–7.75 (m, [16H], Ar–H), 7.55–7.57 (s, [8H], Ar) 7.38–7.43 (m, [6H], Ar–H), 7.20–7.25 (m, [24H], Ar–H), 3.02–3.07 (m, [6H], CH2), 2.12–2.18 (q, [3H], 4JPH = 4.0 Hz, Me). 19F{1H} NMR (298 K, CD2Cl2): δ = –62.8 (s, BArF). 31P{1H} NMR (298 K, CD2Cl2): δ = −4.34 (s).
DFT calculations
The electronic structures of the set of dications, [M{MeC(CH2PPh2)3}]2+ (M = Ge, Sn, Pb; (4), (12), (15)), were investigated using DFT calculations using the Gaussian 16 W software package.20 The density functional used was B3LYP-D3,21 with the basis set 6-311G(d) for H, C, P and Ge atoms22 and the lanl2dz basis set for the Sn and Pb atoms.23 For M = Ge and Sn the initial geometries were taken from their crystal structures, while for M = Pb the initial geometry chosen was from the optimised structure of M = Sn with the tin atom replaced for lead. Calculations for all structures converged with no imaginary frequencies. The calculated structures were found to be in good agreement with the crystallographically-derived metrics (see Table S2†).Results and discussion
The triflate complexes were prepared in good yield by reaction of a suspension of M[OTf]2 (M = Sn, Pb) in an organic solvent with a solution of the di-, tri- or tetra-pnictine ligand in a 1:1 molar ratio (Scheme 2). The BArF salts were prepared from the triflate complexes by metathesis with Na[BArF] in CH2Cl2. The complexes were white powders or colourless crystals with a 1:1 M:pnictine ratio confirmed by microanalysis, with the solids being stable over several weeks in dry air and in daylight. In solution some slow degradation hydrolysis is observed via NMR spectroscopy after 2–3 h. The related literature (Introduction) suggests that many of the complexes are likely to be oligomeric,3,9,11,15 and only very limited data on the solids is provided by spectroscopy. We therefore determined the X-ray crystal structures of six of the tin and five of the lead complexes and discuss these first. The multinuclear NMR spectroscopic behaviour will then be considered to explore the solution speciation.
 | ||
| Scheme 2 Synthesis routes to the pnictine complexes reported in this work. | ||
X-ray crystal structures
The structure of [Sn(OTf)2{o-C6H4(PMe2)2}] (1) shows a distorted four-coordinate tin core, which could be described as tetragonal pyramidal or as a trigonal bipyramid with a vacant equatorial vertex (Fig. 1(a)). The Sn–P distances are not significantly different from those in [SnCl2{o-C6H4(PMe2)2}]11 and the two coordinated triflates have O–Sn–O angles of 144.36(14)°. However, while [SnCl2{o-C6H4(PMe2)2}]11 forms a dimeric unit via chloride bridges, in the triflate complex two triflates from neighbouring molecules also coordinate weakly (Sn⋯O ∼3.0 Å) to form a trimeric assembly (Fig. 1b), well within the sum of the van der Waals radii for Sn + O (3.69 Å).24 Note that we have considered M⋯O distances up to 0.3 Å below the sum of the van der Waals distances to be long, weak interactions. | ||
| Fig. 1 (a) View of the structure of the Sn1-centred [Sn(OTf)2{o-C6H4(PMe2)2}] (1) moiety in the asymmetric unit showing the atom numbering scheme (there is a similar, but crystallographically independent Sn2-centred moiety in the asymmetric unit). H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. There are two slightly different molecules in the cell, only one is shown. Selected bond lengths (Å) and angles (°) for the Sn1-centred unit: Sn1–P1 = 2.6723(14), Sn1–P2 = 2.6682(15), Sn1–O1 = 2.346(4), Sn1–O4 = 2.527(4), Sn1⋯O3′ = 2.967(4), Sn1⋯O8 = 3.002(4), P2–Sn1–P1 = 75.85(5), O1–Sn1–P1 = 77.890(11), O1–Sn1–P2 = 79.14(11), O1–Sn1–O4 = 144.36(14), O4–Sn1–P1 = 72.74(9), O4–Sn1–P2 = 74.50(11); (b) the weakly associated trimeric unit. | ||
In the Ph-substituted diphosphine analogue, [Sn(OTf)2{o-C6H4(PPh2)2}] (8), the two d(Sn–P) are quite similar (2.7179(10), 2.8186(11) Å), which contrasts with the essentially κ1-coordination of the diphosphine present in the reported tin(II) chloride analogue, [SnCl2{o-C6H4(PPh2)2}], where d(Sn–P) = 2.8293(9) and 3.285(1) Å.11 The [Sn(OTf)2{o-C6H4(PPh2)2}] molecules in (8) form weakly associated dimers containing one κ1-coordinated OTf per tin centre (Sn1–O1 = 2.472(3) Å) and two bridging triflates with longer (weaker) Sn⋯OTf contacts (Fig. 2).
 | ||
| Fig. 2 View of the OTf-bridged dimer present in [Sn(OTf)2{o-C6H4(PPh2)2}] (8) showing the atom numbering scheme. H atoms and CH2Cl2 solvent are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles: Sn1–P1 = 2.7179(10), Sn1–P2 = 2.8186(11), Sn1–O1 = 2.472(3), Sn1–O4 = 2.394(3), Sn1⋯O5 = 2.751(3), Sn1⋯O6 = 3.318(4), P1–Sn1–P2 = 69.80(3), O1–Sn1–P1 = 76.66(7), O1–Sn1–P2 = 76.94(8), O4–Sn1–P1 = 91.53(10), O4–Sn1–P2 = 85.14(8), O4–Sn1–O1 = 161.06(11). | ||
The core structure of the diarsine complex, [Sn(OTf)2{o-C6H4(AsMe2)2}] (2) (Fig. 3(a)) is similar to that of its diphosphine analogue, [Sn(OTf)2{o-C6H4(PMe2)2}] (1), although, unlike the phosphorus analogue, the triflates are symmetrically bound (due to crystallographic symmetry). This complex also oligomerises via long Sn⋯OTf contacts (Fig. 3(b)).
 | ||
| Fig. 3 (a) View of core of [Sn(OTf)2{o-C6H4(AsMe2)2}] (2) showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Sn1–As1 = 2.7585(2), Sn1–O1 = 2.4438(12), Sn1⋯ O2 = 3.0094(14), O1–Sn1–O1 = 137.76(6), O1–Sn1–As1 = 73.75(3), As1–Sn1–As1 = 76.755(8); (b) part of the polymeric chain with bridging OTf groups viewed down the b-axis. | ||
The structure of [Sn{MeC(CH2PPh2)3}][BArF]2 (4) (Fig. 4), as expected, shows no cation–anion interaction due to the diffuse nature of the large BArF anion. In this case the tin is in a P3 trigonal pyramidal geometry, with all three Sn–P bond distances in the range 2.6194(4)–2.6438(4) Å, i.e. rather shorter than in the complexes described above with coordinated triflate. This probably reflects the lower coordination number and higher cationic charge.
 | ||
| Fig. 4 View of the cation in [Sn{MeC(CH2PPh2)3}][BArF]2 (4) showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles: Sn1–P1 = 2.6438(4), Sn1–P2 = 2.6194(4), Sn1–P3 = 2.6249(4), P1–Sn1–P2 = 82.120(13), P1–Sn1–P3 = 80.761(14), P2–Sn1–P3 = 80.160(14). | ||
The complexes [Sn(OTf){PhP(CH2CH2PPh2)2}][OTf] (5) and [Sn(OTf){P(CH2CH2PPh2)3}][OTf] (7) involve coordination to Sn(II) via a P3O donor set, with a further long, weak interaction to the second triflate completing a very distorted five-coordinate geometry (Fig. 5). The Sn–P bond lengths range from 2.6800(12)–2.8412(7) Å, are longer than in the tripodal dication in [Sn{MeC(CH2PPh2)3}][BArF]2 (4), discussed above and the fourth phosphine group (–PPh2) in [Sn(OTf){P(CH2CH2PPh2)3}][OTf] (7) points away from the tin and is not involved in coordination. The Ge(II) analogue adopts a three-coordinate pyramidal structure, with the triflate anions not coordinated.10 In both of these structures, as expected, the P–Sn–P angles involved in the five-membered chelate rings are substantially smaller than the much less constrained P1–Sn–P3 angles.
 | ||
| Fig. 5 (a) View of the structure of [Sn(OTf){PhP(CH2CH2PPh2)2}][OTf] (5) showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Sn1–P1 = 2.6800(12), Sn1–P2 = 2.7655(12), Sn1–P3 = 2.7627(12), Sn–O1 = 2.623(4), Sn⋯O4 = 2.821(4), P1–Sn1–P2 = 74.52(4), P1–Sn1–P3 = 90.23(4), P2–Sn1–P3 = 72.35(4); (b) view of the structure of [Sn(OTf){P(CH2CH2PPh2)3}][OTf] (7) showing the atom numbering scheme. Selected bond lengths (Å) and angles (°): Sn1–P1 = 2.7085(7), Sn1–P2 = 2.7055(7), Sn1–P3 = 2.8412(7), Sn1–O1 = 2.698(2), Sn1⋯O4 = 2.968(3), P1–Sn1–P2 = 74.31(2), P1–Sn1–P3 = 96.68(2), P2–Sn1–P3 = 72.82(2). | ||
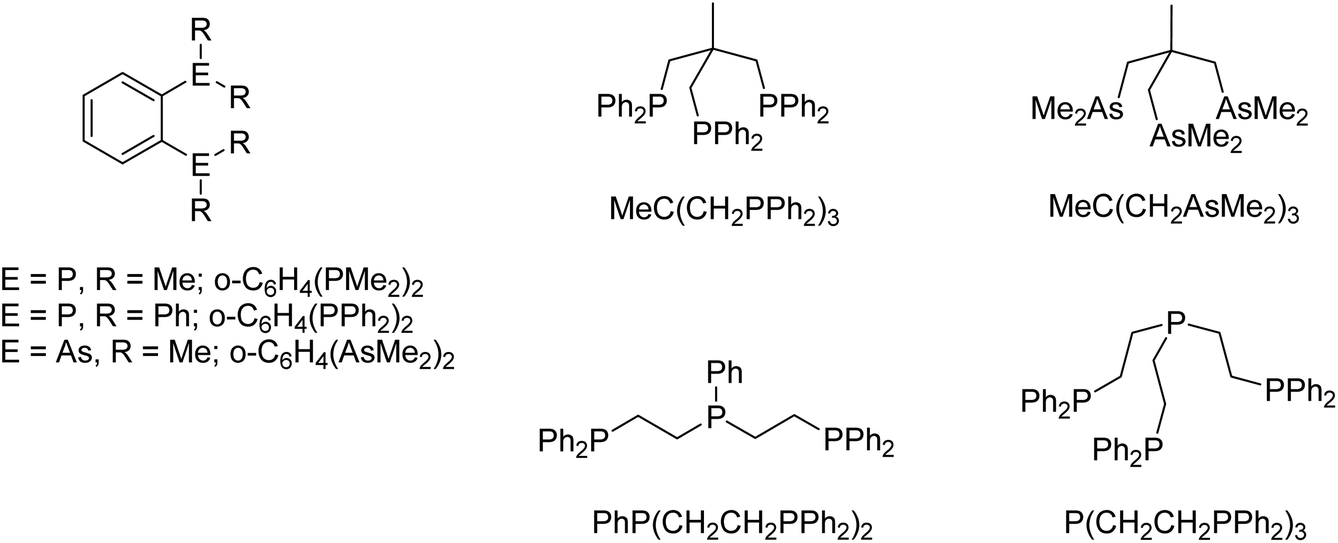
Moving now to the crystal structures determined for the Pb(II) complexes, the four-coordinate core geometry in [Pb(OTf)2{o-C6H4(PMe2)2}] (9) (Fig. 6(a)) is very similar to that in the corresponding tin complex (above), and the coordination through bridging triflate groups (longer Pb⋯OTf contacts) from neighbouring molecules results in a zig-zag polymer chain with (effectively) six-coordination about the lead (Fig. 6(b)) (there is a further OTf group 3.26 Å away from the Pb centre, but this distance is only 0.28 Å within the sum of the van der Waals radii for Pb + O, 3.54 Å,24 and therefore we do not consider this to be a significant interaction).
 | ||
| Fig. 6 (a) View of the of [Pb(OTf)2{o-C6H4(PMe2)2}] (9) core showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.7623(6), Pb1–P2 = 2.7581(6), Pb1–O1 = 2.6740(19), Pb1–O4 = 2.4504(19), Pb1⋯O3 = 2.9394(19), Pb1⋯O5 = 3.0193(19), P2–Pb1–P2 = 72.473(18), O1–Pb1–P1 = 76.99(5), O1–Pb1–P2 = 72.28(5), O4–Pb1–P1 = 79.60(5), O4–Pb1–P2 = 77.71(4), O4–Pb1–O1 = 146.26(7); (b) the OTf-bridged chain structure present in [Pb(OTf)2{o-C6H4(PMe2)2}]. | ||
The lead(II) diarsine complex, [Pb(OTf)2{o-C6H4(AsMe2)2}] (10), has core PbAs2O2 coordination and also forms a chain polymer via weakly bridging OTf groups, with overall six-coordination at Pb(II) and with two very similar Pb–As distances (Fig. 7).
 | ||
| Fig. 7 (a) View of the of [Pb(OTf)2{o-C6H4(AsMe2)2}] (10) core showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Pb1–As1 = 2.8675(6), Pb1–As2 = 2.8752(6), Pb1–O1 = 2.539(4), Pb1–O4 = 2.712(5), Pb1⋯O4 = 2.712(5), Pb1⋯O5 = 2.972(5), As1–Pb1–As2 = 73.297(16), O4–Pb1–As1 = 69.56(9), O4–Pb1–As2 = 69.84(10), O1–Pb1–O4 = 133.97(13), O1–Pb1–As1 = 74.98(10), O1–Pb1–As2 = 72.57(9); (b) view of the polymeric chain in [Pb(OTf)2{o-C6H4(AsMe2)2}]. | ||
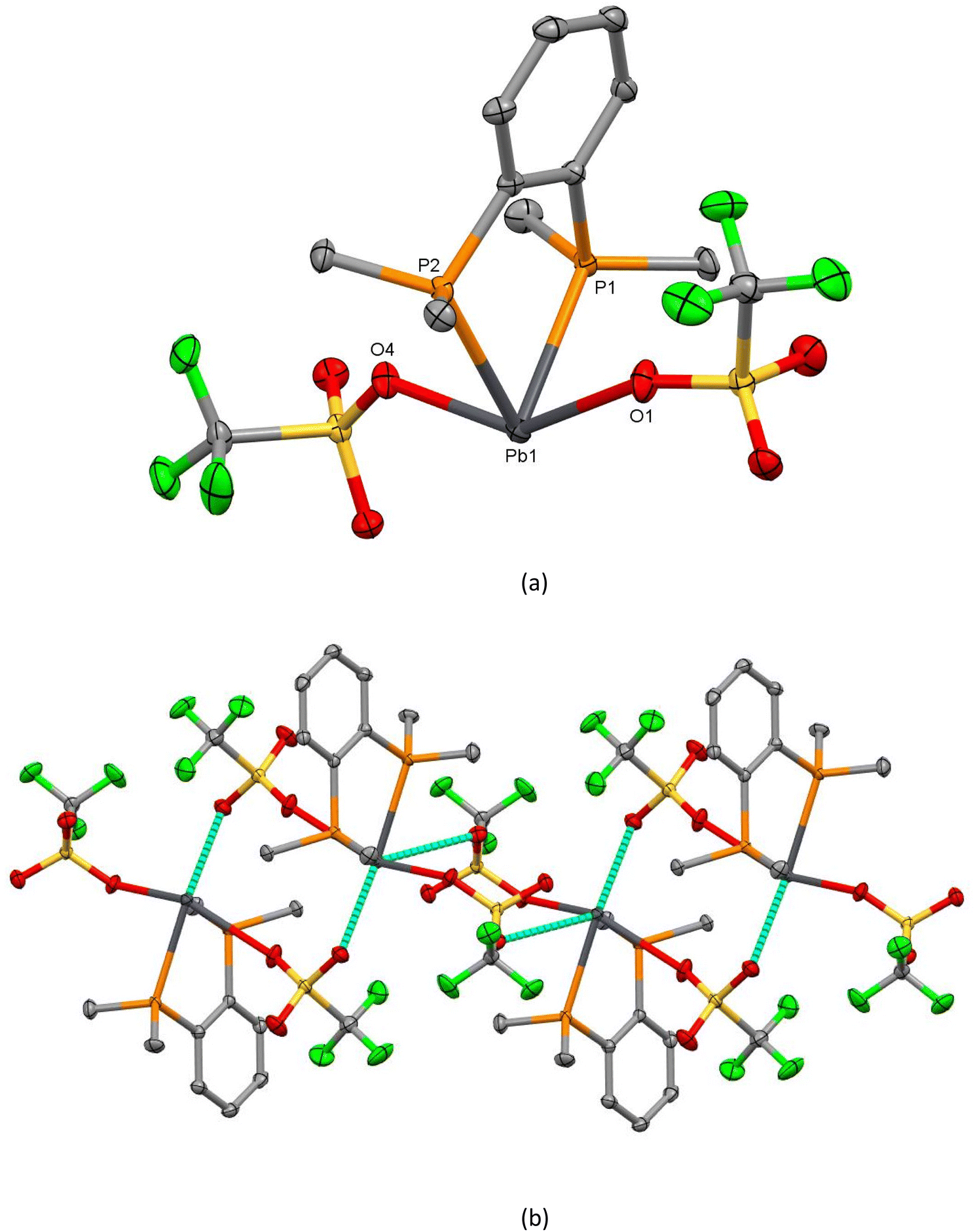
The crystal structure of [Pb(OTf){P(CH2CH2PPh2)3}][OTf] (13) is isomorphous with the tin analogue above, showing P3O2 coordination, with the third pendant –PPh2 group remaining uncoordinated, and with the P1–Pb–P3 angle rather more open than those involving the constrained five-membered chelate rings (Fig. 8).
 | ||
| Fig. 8 View of the structure of [Pb(OTf){P(CH2CH2PPh2)3}][OTf] (13) showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.7771(8), Pb1–P2 = 2.8021(7), Pb1–P3 = 2.9185(7), Pb1–O1 = 2.7514(19), Pb⋯O4 = 2.951(2), P1–Pb1–P2 = 72.95(2), P1–Pb1–P3 = 96.20(2), P2–Pb1–P3 = 70.72(2), O1–Pb1–P1 = 71.18(5), O1–Pb1–P2 = 75.99(4), O1–Pb1–P3 = 146.61(4). | ||
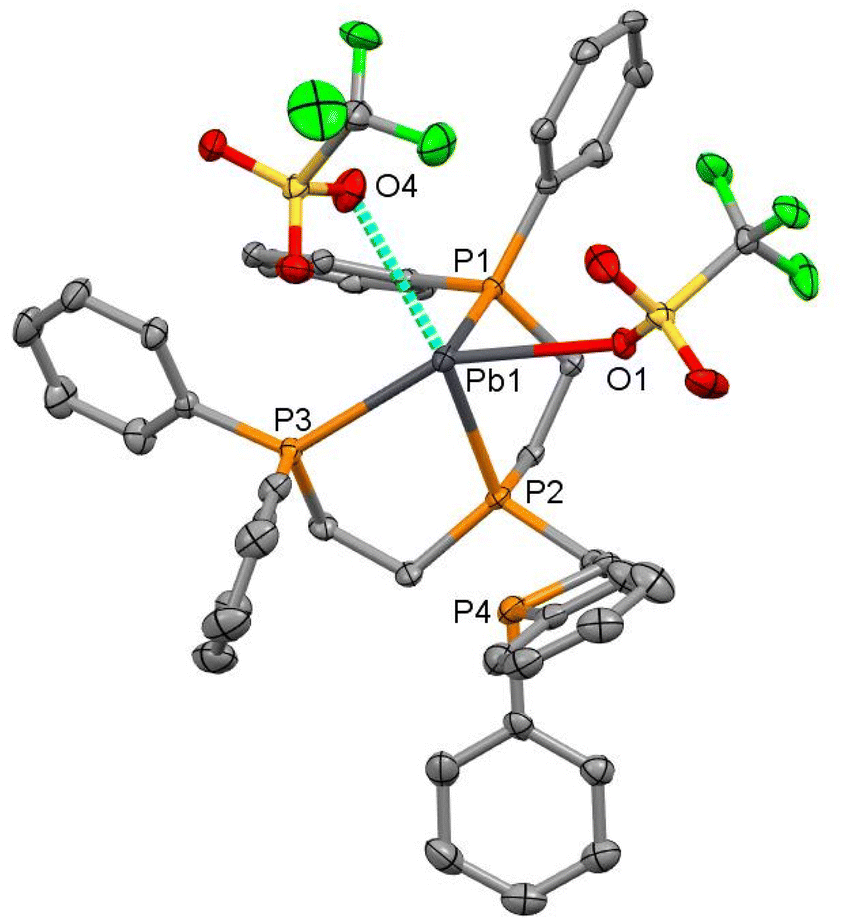
The structure of the lead triflate complex with the tripodal triphosphine, [Pb(OTf)2{MeC(CH2PPh2)3}] (11), reveals a dimer with three bridging (and one ionic) triflates (Fig. 9), and therefore is better formulated as [{Pb{MeC(CH2PPh2)3}}2(μ-OTf)3][OTf]. In contrast, the corresponding BArF salt, [Pb{MeC(CH2PPh2)3}][BArF]2 (12), shows a discrete three-coordinate cation (Fig. 10(a)). Comparison of the [BArF] and OTf structures shows longer Pb–P bonds in the latter, attributable to the higher coordination number. The P–Pb–P bond angles of the triflate bridged species ranged from 71.678(13)–77.348(13), significantly more acute than those seen in the BArF salt (77.868(17)–80.594(17)°).
 | ||
| Fig. 9 View of the structure of the cation in [{Pb{MeC(CH2PPh2)3}2(μ-OTf)3}][OTf] (11) showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Pb1–P1 = 2.8277(4), Pb1–P2 = 2.8844(4), Pb1–P3 = 2.9261(5), Pb1–O1 = 2.6542(15), Pb1–O5 = 2.7722(15), Pb1⋯O8 = 2.8398(15), Pb2–P4 = 2.9075(5), Pb2–P5 = 2.8441(5), Pb2–P6 = 2.9158(5), Pb2–O2 = 2.7889(14), Pb2–O4 = 2.6707(14), Pb2⋯O7 = 2.7375(17), P1–Pb1–P3 = 72.800(13), P2–Pb1–P3 = 77.348(13), P1–Pb1–P2 = 71.678(13). | ||
 | ||
| Fig. 10 (a) View of the cation in [Pb{MeC(CH2PPh2)3}][BArF]2 (12) showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Pb–P1 = 2.7360(5), Pb1–P2 = 2.7092(6), Pb1–P3 = 2.7184(6), P1–Pb1–P2 = 80.594(17), P1–Pb1–P3 = 78.676(17), P2–Pb1–P3 = 77.868(17); (b) view of the cation in [Ge{MeC(CH2PPh2)3}][BArF]2 (15) showing the atom numbering scheme. H atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles: Ge1–P1 = 2.4239(4), Ge1–P2 = 2.4070(4), Ge1–P3 = 2.4110(4), P1–Ge1–P2 = 86.609(14), P1–Ge1–P3 = 85.912(15), P2–Ge1–P3 = 85.412(15). | ||
We also prepared the lighter group 14 congener (M = Ge), [Ge{MeC(CH2PPh2)3}][BArF]2 (15), and determined its structure (Fig. 10(b)). The three salts, [M{MeC(CH2PPh2)3}][BArF]2 (M = Ge, Sn, Pb), are isomorphous (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). As indicated in the Introduction, it is very unusual to find three isomorphous structures for pnictine complexes of these three elements, and as can be seen from Table 1, the M–P bond distances increase in the order Ge < Sn < Pb, and the P–M–P angles decrease in the same order. The increase in d(M–P) down group 14 is consistent with the increase in the covalent radii.25
). As indicated in the Introduction, it is very unusual to find three isomorphous structures for pnictine complexes of these three elements, and as can be seen from Table 1, the M–P bond distances increase in the order Ge < Sn < Pb, and the P–M–P angles decrease in the same order. The increase in d(M–P) down group 14 is consistent with the increase in the covalent radii.25
Spectroscopic data
To probe the solution speciation, multinuclear NMR spectra (1H, 19F{1H}, 31P{1H} and 119Sn) were recorded, usually from CD3CN or CD3NO2, or, if solubility permitted, from CD2Cl2 solutions. The 1H spectra (see Experimental and ESI†) were consistent with the coordinated pnictine, but were otherwise rather uninformative. The 19F{1H} data of the triflate complexes each show a sharp singlet at ca. −79 ppm, assigned to ionic triflate, indicating that the [OTf]− groups are at best weakly associated or exchanging in solution. The 31P{1H} and 119Sn spectra are much more informative and key data are summarised in Table 2, with representative examples shown in Fig. 11 and 12, (full data are in the Experimental section and the ESI†). Table 2 also summarises data on related [M(phosphine)][SbF6]2 taken from the in situ studies by Dean and co-workers.12,13 In some cases three examples with different anions are known for a specific phosphine, for example [Sn{MeC(CH2PPh2)3}]Y2 with Y− = [BArF]−, [SbF6]− and [OTf]−. The 119Sn chemical shifts for these are very similar, indicating the phosphine plays the dominant role. The 31P{1H} NMR chemical shifts and the coupling constants are much more variable, possibly reflecting the different solvents in some cases, and probably some interaction of the anions or phosphine exchange. While coordination by [SbF6]− is viewed as rare,26 examples are known in the solid state, and coordinated triflate is well known. | ||
| Fig. 11 (a) 119Sn NMR spectrum of [Sn(OTf){PhP(CH2CH2PPh2)2}][OTf] (5) showing the 1J119SnP couplings to the two distinct P atoms; (b) 31P{1H} NMR spectrum [Sn(OTf){PhP(CH2CH2PPh2)2}][OTf] (5) showing the 1J117/119SnP and 1J117/119SnP′ couplings. | ||
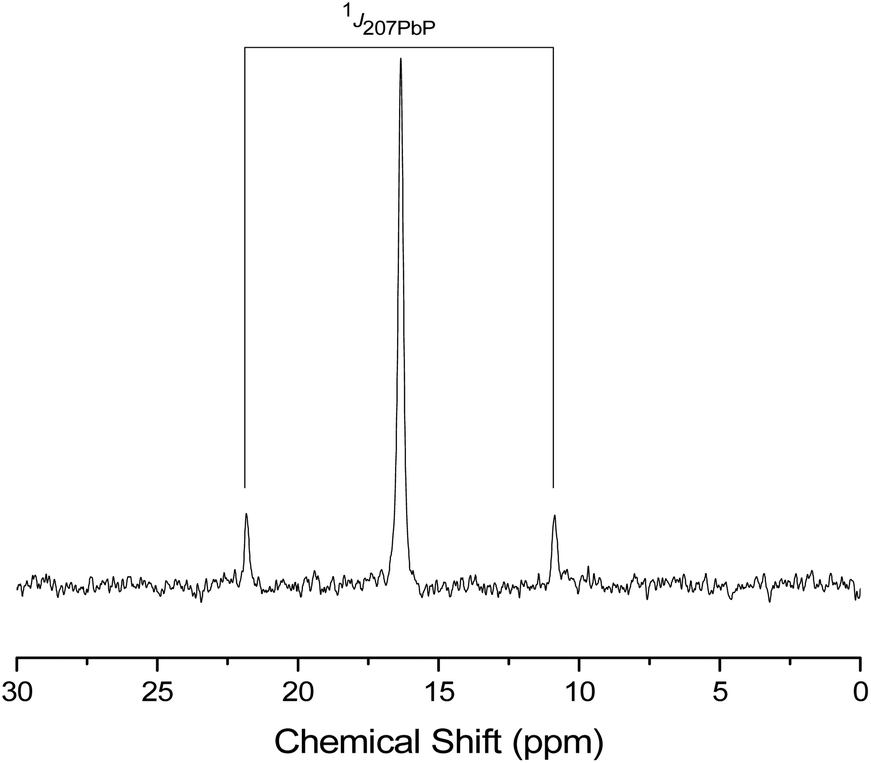 | ||
| Fig. 12 The 31P{1H} NMR spectrum of [Pb{MeC(CH2PPh2)3})][BArF]2 (12) showing the 1J207PbP coupling. | ||
| Complex | δ(31P)/ppm | δ(119Sn)b/ppm | 1 J 119SnP/Hz | 1 J 207PbP/Hz | Ref. |
|---|---|---|---|---|---|
| a Spectra recorded at 298 K in MeCN unless otherwise stated. b Reference SnMe4 (δ = 0). c Spectrum recorded in MeNO2. | |||||
| [Sn(OTf)2{o-C6H4(PMe2)2}] (1) | 14.5 | −690 | 1882 | — | This work |
| [Sn(OTf)2{o-C6H4(AsMe2)2}] (2) | — | Not observed | — | — | This work |
| −886.5 (258 K) | |||||
| [Sn(OTf)2{o-C6H4(PPh2)2}] (8) | 19.9 | −809 | Not observed | — | This work |
| 22.7 (258 K) | −1150 (258 K) | 1550 (258 K) | |||
| [Sn(OTf)2{MeC(CH2PPh2)3}] (3) | −9.4 | −834.0 | 1248 | — | This work |
| [Sn{MeC(CH2PPh2)3}][BArF]2 (4) | −8.9 | −824.3 | 1246 | — | This work |
| −6.0 (258 K) | −843.7 (258 K) | 1252 (258 K) | |||
| [Sn{MeC(CH2PPh2)3}][SbF6]2c |
−11.3 | −792 | 1279 | — | 12 and 13 |
| [Sn(OTf){PhP(CH2CH2PPh2)2}][OTf] (5) | 36.4, 18.5 | −834 | 1549, 1377 | — | This work |
| [Sn{PhP(CH2CH2PPh2)2}][SbF6]2c |
47.7, 24.9 | −686 | 1593, 1381 | — | 12 and 13 |
| [Sn(OTf)2{MeC(CH2AsMe2)3}] (6) | — | Not observed | — | — | This work |
| −920 (258 K) | |||||
| [Sn(OTf){P(CH2CH2PPh2)3}][OTf] (7) | 37.8, 5.5 | −778.6 | 720, 1485 | — | This work |
| [Sn{P(CH2CH2PPh2)3}][SbF6]2c |
36.8, 2.5 | −756 | 702, 1487 | — | 12 and 13 |
| [Pb(OTf)2{o-C6H4(PMe2)2}] (9) | 74.9 | — | — | 1777 | This work |
| [Pb{MeC(CH2PPh2)3}2][(μ-OTf)3][OTf] (11) | 11.6 | — | — | 1150 | This work |
| [Pb{MeC(CH2PPh2)3}][SbF6]2c |
13.8 | — | — | 1786 | 12 and 13 |
| [Pb{MeC(CH2PPh2)3}][BArF]2 (12)c | 15.8 (258 K) | — | — | 1777 | This work |
| [Pb(OTf)2{P(CH2CH2PPh2)3}] (13) | 77.5, 26.1 | — | — | 437, 1870 | This work |
| [Pb{P(CH2CH2PPh2)3}][SbF6]2c |
80.1, 27.5 | — | — | 476, 2136 | 12 and 13 |
| [Pb{PhP(CH2CH2PPh2)2}][SbF6]2c |
90.2, 66.0 | — | — | 1836, 1863 | 12 and 13 |
Also notable are the NMR data on the [Sn{P(CH2CH2PPh2)3}]2+ cation incorporating the tripodal tetraphosphine, whose 31P and 119Sn NMR spectra are consistent with all three PPh2 groups appearing to interact with the tin, suggesting tetradentate coordination on average in solution. This is in contrast to the κ3-phosphine coordination found in the solid state structure of [Sn(OTf){P(CH2CH2PPh2)3}][OTf] (7) described above, in which the one pendant arm remains free. Upon cooling the solution of this complex to 258 K (MeCN), the lines broaden, but do not split, probably indicating fast exchange between coordinated and free pendant –PPh2 groups, which is not frozen out at the lower temperature.
The tin-arsine complexes did not exhibit 119Sn NMR resonances at room temperature, but broad resonances appear upon cooling the solutions to 258 K, with chemical shifts somewhat more negative than the analogous phosphine complexes.
The lead phosphine species incorporating [SbF6]− anions previously reported by Dean et al. were generated in situ, but never isolated.12,13 These solutions did exhibit 207Pb NMR resonances, however, although the [OTf]− and [BArF]− complexes isolated in the present study show clear 207Pb lead satellites in their 31P{1H} NMR spectra, which sharpen at low temperature, we were unable to observe 207Pb NMR resonances in a similar chemical shift range to those in the reported work, either at room temperature or upon cooling in MeCN (258 K). For the triflate complexes it seems most likely that this is a result of rapid reversible coordination of the triflate ions (consistent with coordination of OTf groups in the crystal structures of several examples described above). The 31P{1H} NMR data above also revealed fast phosphine exchange in solutions of some of the Sn(II) and Pb(II) complexes, and this may explain the absence of a 207Pb NMR resonance in the [Pb{MeC(CH2PPh2)3}][BArF]2 (12) complex, where the low temperature limiting spectrum may not have been reached at 258 K, which is the low temperature limit for MeNO2.
DFT calculations
The electronic structures of the set of dications, [M{MeC(CH2PPh2)3}]2+ (M = Ge, Sn, Pb), were investigated using DFT calculations as described in the Experimental section.For the minimum energy structures located, in all cases the HOMO orbital is associated with a valence s–p hybrid orbital on the metal centre, along with ligand-centred lobes. HOMO−1 and HOMO−2 are associated with bonding interactions between the valence p orbitals on the ligand with px/py type orbitals on the group 14 centre and are approximately degenerate in energy (together with ligand-centred lobes); the HOMO orbitals are shown in Fig. 13 below. The degenerate LUMO and LUMO+1 orbitals are px/py orbitals on the metal centre (Fig. 14). The metal HOMOs on [M{MeC(CH2PPh2)3}]2+ are all directional with a mixture of valence s and pz character, with germanium having the highest valence p-character (18.35%), followed by tin (13.75%) and lead (8.12%). This is consistent with the trend expected going down the group (Table 3).
 | ||
| Fig. 13 Representations of the HOMO, HOMO−1 and HOMO−2 orbitals for [Ge{MeC(CH2PPh2)3}]2+ with the orbital energies for each complex shown in brackets (Ge/Sn/Pb). | ||
 | ||
| Fig. 14 Representations of the LUMO and LUMO+1 orbitals for [Ge{MeC(CH2PPh2)3}]2+ with the orbital energies of each complex shown in brackets (Ge/Sn/Pb). | ||
| Complex | HOMO–LUMO gap/eV | %pz character of HOMO on M | Charge at M | Charge at P | Average metal LP to P–Cσ* interaction energy/kJ mol−1 |
|---|---|---|---|---|---|
| [Ge{MeC(CH2PPh2)3}]2+ | 5.17 | 18.35 | 0.26 | 1.11 | 11.36 |
| [Sn{MeC(CH2PPh2)3}]2+ | 4.81 | 13.75 | 0.76 | 0.97 | 7.26 |
| [Pb{MeC(CH2PPh2)3}]2+ | 4.74 | 8.12 | 0.84 | 0.95 | 5.10 |
The lone pair on the metal centre is anti to one of the P–C bonds on each arm of the tripodal phosphine ligand, this leads to a LP → P–C σ* interaction (Fig. 15). Second order perturbation theory was used to quantify the extent of this interaction (Table 3), showing that the interaction gets weaker as the group is descended, with the interaction being about half as strong in the Pb complex when compared to the Ge complex.
 | ||
| Fig. 15 Interaction between the tetral-based lone pair on the metal and the σ* orbital of the P–C bond. | ||
NBO calculations also show that the natural charge at the metal centre increases down group 14, from +0.26 for Ge to +0.76 for Sn and +0.84 for Pb. In contrast, the natural charge on the phosphorus atom decreases as the series is descended, +1.11 (M = Ge) to +0.97 (M = Sn) to +0.95 (M = Pb).
Conclusions
The preparation and characterisation of a series of Sn(II) and Pb(II) triflate complexes with soft, neutral di, tri- and tetra-phosphine and di- and tri-arsine ligands has been described. X-ray structural data on 12 of the complexes confirm that the structures are highly dependent upon the pnictine atom type (P vs. As) and denticity, and show that in the majority of cases, as well as coordinating to two or three P or As donor atoms, one or both of the OTf anions are also retained within the metal coordination sphere, giving rise to a diverse range of structural motifs. These include dicationic and monocationic monomers, weakly associated (OTf bridged) dimers, cyclic trimers or chain polymers, with the degree of association dependent upon the divalent group 14 ion. For example, [M(OTf)2{o-C6H4(PMe2)2}], M = Ge,27 Sn, Pb, shows that upon changing from M = Ge to Sn to Pb, the extended structures go from dimeric to trimeric to polymeric. The triflate-bridged dimer, [Pb{MeC(CH2PPh2)3}2(μ-OTf)3][OTf], undergoes anion metathesis with Na[BArF], affording the pyramidal Pb(II) triphosphine dication, [Pb{MeC(CH2PPh2)3}]2+ as its BArF salt.In solution the 19F{1H} NMR spectra suggest that the OTf groups are dissociated, however, the 31P{1H} NMR spectra show the expected satellite couplings to 117/119Sn and 207Pb, consistent with retention of the pnictine coordination in solution, although typically the solutions require to be cooled to reach the low temperature limiting spectra (with the exception of the tetraphosphine complexes, which are still undergoing fast exchange at 258 K). Tin-119 NMR spectra show the expected multiplet couplings to the phosphine donor groups, and the 119Sn NMR shifts for the arsine complexes occur to low frequency (ca. 200 ppm more negative) than the corresponding phosphine species.
DFT calculations on the [M{MeC(CH2PPh2)3}]2+ homologues show the presence of directional HOMO in each dication, which is a mixture of valence s and pz character, with the valence p-orbital character decreasing on going down group 14. NBO analysis also shows that the natural charge at the metal centre increases and the charge on the P centre decreases on going down group 14.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for support through the ADEPT Programme grant (EP/N035437/1) and through EP/R513325/1. We also thank the EPSRC National Crystallography Service for access to the X-ray diffraction facilities and Professor J. M. Dyke for helpful discussions regarding the DFT calculations.References
- J. Parr, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 3, p. 545 Search PubMed; E. S. Claudio, H. A. Godwin and J. S. Magyar, Prog. Inorg. Chem., 2003, 51, 1 Search PubMed; P. G. Harrison, Comprehensive Coordination Chemistry, Pergamon, Oxford, 1988, vol. 3, p. 183 CrossRef CAS; J. R. Fulton, Comprehensive Coordination Chemistry III, 2021, 3.10, p. 281 CrossRef CAS; The Chemistry of Tin, ed. P. J. Smith, Chapman & Hall, London, 1998 CrossRef CAS; R. L. Davidovich, V. Stavila and K. H. Whitmire, Coord. Chem. Rev., 2010, 254, 2193 CrossRef CAS; P. G. Harrison, The Chemistry of Tin, Blackie London, 1989 Search PubMed.
- W. Levason, G. Reid and W. Zhang, Coord. Chem. Rev., 2011, 255, 1319 CrossRef CAS.
- J. Burt, W. Levason and G. Reid, Coord Chem. Rev., 2014, 260, 65 CrossRef CAS.
- F. Cheng, M. F. Davis, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Eur. J. Inorg. Chem., 2007, 4897 CrossRef CAS; M. F. Davis, W. Levason, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2008, 533 RSC; M. F. Davis, W. Levason, G. Reid and M. Webster, Dalton Trans., 2008, 2261 RSC; R. P. King, W. Levason and G. Reid, Dalton Trans., 2021, 50, 17751 RSC.
- R. P. King, M. S. Woodward, J. Grigg, G. McRobbie, W. Levason and G. Reid, Dalton Trans., 2021, 50, 14400 RSC; V. K. Greenacre, R. P. King, W. Levason and G. Reid, Dalton Trans., 2019, 48, 17097 RSC.
- F. Cheng, J. M. Dyke, F. Ferrante, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2010, 39, 847 RSC.
- P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138 CrossRef CAS PubMed; P. A. Rupar, V. N. Staroverov and K. M. Baines, Science, 2008, 322, 1360 CrossRef PubMed; F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Angew. Chem., Int. Ed., 2009, 48, 5752 CrossRef PubMed; P. A. Rupar, R. Bandyopadhyay, B. F. T. Cooper, M. R. Stinchcombe, P. J. Ragogna, C. L. B. Macdonald and K. M. Baines, Angew. Chem., Int. Ed., 2009, 48, 5755 CrossRef PubMed; F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Chem. Commun., 2008, 5508 RSC.
- For examples, see: F. S. Tschernuth, F. Hanusch, T. Szilvási and S. Inoue, Organometallics, 2020, 39, 4265 CrossRef CAS; S. Hino, M. Brynda, A. D. Phillips and P. P. Power, Angew. Chem., Int. Ed., 2004, 43, 2655 CrossRef PubMed; A. P. Singh, H. W. Roesky, E. Carl, D. Stalke, J.-P. Demers and A. Lange, J. Am. Chem. Soc., 2012, 134, 4998 CrossRef PubMed; M. Bouška, L. Dostál, A. Růžička and R. Jambot, Organometallics, 2013, 32, 1995 CrossRef.
- F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2010, 49, 752 CrossRef CAS PubMed.
- R. P. King, V. K. Greenacre, W. Levason, J. M. Dyke and G. Reid, Inorg. Chem., 2021, 60, 12100 CrossRef CAS PubMed.
- C. Gurnani, A. L. Hector, E. Jager, W. Levason, D. Pugh and G. Reid, Dalton Trans., 2013, 42, 8364 RSC.
- P. A. W. Dean, D. D. Phillips and L. Polensek, Can. J. Chem., 1981, 59, 50 CrossRef.
- P. A. W. Dean, Can. J. Chem., 1983, 61, 1795 CrossRef CAS.
- A. J. Rossini, A. W. Macgregor, A. S. Smith, G. Schatte, R. W. Schurko and G. G. Briand, Dalton Trans., 2013, 42, 9533 RSC.
- J. Burt, W. Grantham, W. Levason and G. Reid, Dalton Trans., 2015, 44, 11533 RSC.
- R. E. H. Kuveke, L. Barwise, Y. van Ingen, K. Vashisth, N. Roberts, S. S. Chitnis, J. L. Dutton, C. D. Martin and R. L. Melen, ACS Cent. Sci., 2022, 8, 855 CrossRef CAS PubMed.
- E. P. Kyba, S. T. Liu and R. L. Harris, Organometallics, 1983, 2, 1877 CrossRef CAS.
- R. D. Feltham, A. Kasenally and R. S. Nyholm, J. Organomet. Chem., 1967, 7, 285 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. L. How, J. A. L. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed; S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- J. Emsley, The Elements, Oxford, 1989 Search PubMed.
- S. H. Strauss, Chem. Rev., 1993, 93, 927 CrossRef CAS.
- R. P. King, W. Levason and G. Reid, Dalton Trans., 2021, 50, 17751 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic parameters for the structures reported (Table S1), a comparison of the bond lengths and angles determined experimentally (X-ray) with those computed by DFT (Table S2), the multinuclear NMR and IR spectra associated with each of the new compounds described (Fig. S1–S18), and the x, y and z coordinates determined from the DFT calculations. CCDC 2216189–2216200. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03687h |
| This journal is © The Royal Society of Chemistry 2023 |