
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic anisotropy and structural flexibility in the field-induced single ion magnets [Co{(OPPh2)(EPPh2)N}2], E = S, Se, explored by experimental and computational methods†
Eleftherios
Ferentinos
a,
Demeter
Tzeli
 bc,
Silvia
Sottini
d,
Edgar J. J.
Groenen
d,
Mykhaylo
Ozerov
e,
Giordano
Poneti
*f,
Kinga
Kaniewska-Laskowska
g,
J.
Krzystek
*e and
Panayotis
Kyritsis
*a
bc,
Silvia
Sottini
d,
Edgar J. J.
Groenen
d,
Mykhaylo
Ozerov
e,
Giordano
Poneti
*f,
Kinga
Kaniewska-Laskowska
g,
J.
Krzystek
*e and
Panayotis
Kyritsis
*a
aInorganic Chemistry Laboratory, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, GR-15771 Athens, Greece. E-mail: kyritsis@chem.uoa.gr
bPhysical Chemistry Laboratory, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, GR-15771 Athens, Greece
cTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Ave., GR-11635 Athens, Greece
dHuygens-Kamerlingh Onnes Laboratory, Department of Physics, Leiden University, Niels Bohrweg 2, 2333 CA Leiden, The Netherlands
eNational High Magnetic Field Laboratory, Florida State University, Tallahassee, Florida 32310, USA. E-mail: krzystek@magnet.fsu.edu
fInstituto de Química, Universidade Federal do Rio de Janeiro, 21941-909 Rio de Janeiro, Brazil. E-mail: gponeti@iq.ufrj.br
gDepartment of Inorganic Chemistry, Faculty of Chemistry, Gdańsk University of Technology, G. Narutowicza St. 11/12, Gdańsk PL-80-233, Poland
First published on 24th January 2023
Abstract
During the last few years, a large number of mononuclear Co(II) complexes of various coordination geometries have been explored as potential single ion magnets (SIMs). In the work presented herein, the Co(II) S = 3/2 tetrahedral [Co{(OPPh2)(EPPh2)N}2], E = S, Se, complexes (abbreviated as CoO2E2), bearing chalcogenated mixed donor-atom imidodiphosphinato ligands, were studied by both experimental and computational techniques. Specifically, direct current (DC) magnetometry provided estimations of their zero-field splitting (zfs) axial (D) and rhombic (E) parameter values, which were more accurately determined by a combination of far-infrared magnetic spectroscopy and high-frequency and -field EPR spectroscopy studies. The latter combination of techniques was also implemented for the S = 3/2 tetrahedral [Co{(EPiPr2)2N}2], E = S, Se, complexes, confirming the previously determined magnitude of their zfs parameters. For both pairs of complexes (E = S, Se), it is concluded that the identity of the E donor atom does not significantly affect their zfs parameters. High-resolution multifrequency EPR studies of CoO2E2 provided evidence of multiple conformations, which are more clearly observed for CoO2Se2, in agreement with the structural disorder previously established for this complex by X-ray crystallography. The CoO2E2 complexes were shown to be field-induced SIMs, i.e., they exhibit slow relaxation of magnetization in the presence of an external DC magnetic field. Advanced quantum-chemical calculations on CoO2E2 provided additional insight into their electronic and structural properties.
Introduction
Extensive studies are currently being carried out, with an aim of investigating in depth the role of unpaired electrons in assuring specific functions in homogeneous or heterogeneous catalysts,1,2 the active sites of metalloproteins,3 as well as molecular magnetic materials.4,5 A major achievement related to the latter was the discovery, in the early 1990s, of single molecule magnets (SMMs), i.e., multinuclear metal complexes that exhibit magnetic bistability, and hence slow relaxation of magnetization owing to an energy barrier, whose magnitude depends on their total spin S and their magnetic anisotropy.6–8 The latter, conventionally expressed via the zero-field splitting (zfs) axial D component, has been shown to acquire much larger values for mononuclear than for multinuclear metal complexes.9–11 Thus, currently there is an increased interest in exploring slow relaxing mononuclear metal complexes, which are referred to either as mononuclear SMMs or as single ion magnets (SIMs).12–15 Along these lines, intense research efforts have been devoted to mononuclear complexes of lanthanides16–21 and actinides,22 exhibiting large magnetic anisotropy. Potential applications of these materials include information-storage or molecular devices that could be employed for the design of quantum computers.23–25 However, it should be stressed that the slow magnetization relaxation of SMMs and SIMs is limited to temperatures lower than the corresponding blocking temperature (TB),20 setting a target of increasing this limit to technologically manageable values. To that end, major breakthroughs have been reported during the last five years concerning lanthanide complexes, the TB value of which approaches,26–28 or even surpasses,29,30 that of liquid nitrogen (77 K).In parallel to the development of lanthanide/actinide SIMs, there has been an increased interest in exploring mononuclear 3d-metal-based complexes.12,13,15,31–39 For these systems, it is of paramount importance to reliably and accurately determine the sign and magnitude of their zfs D component, as well as their rhombicity E/D (E is the rhombic component of the zfs).40 The experimental methods that are employed for this - rather demanding - task, have been recently reviewed,41 and mainly include DC magnetometry,40 high-frequency and -field EPR spectroscopy (HFEPR),42–44 far-infrared magnetic spectroscopy (FIRMS),45–47 inelastic neutron scattering,48–50 magnetic circular dichroism51–53 and Mössbauer spectroscopy.54–57 Each experimental method has its advantages and limitations.41 Among these methods, direct current (DC) magnetometry has been a conventional method of estimating the magnitude of the zfs D and E components. However, to this end, DC magnetometry cannot compete with the significantly improved accuracy of resonance spectroscopic methods. For that reason, some of the above specific spectroscopic methods, either separate, or in combination, are currently employed for this task41 (vide infra). In that respect, it should be noted that, in combination with other physicochemical methods, FIRMS has been recently employed to probe the magnetic properties of various Co(II) complexes.58–64
The vast majority of 3d-based SIMs involve Co(II) complexes of various coordination spheres and geometries,13,65 including the seminal tetrahedral complex [Co(SPh)4]2−,66,67 and its O- and Se- congeners,67 as well as some outstanding examples of SIMs bearing four-68 or two-69,70 coordinate Co(II) centers. Accordingly, the main target of the present work was the investigation of the tetrahedral Co(II) complexes [Co{(OPPh2)(EPPh2)N}2], E = S,71 Se,72 (abbreviated as CoO2E2), bearing chalcogenated mixed donor-atom imidodiphosphinato ligands.73 The crystal structure of these complexes is shown in Fig. S1 in the ESI.† The zfs parameters originally estimated by DC magnetometry71,72 were not replicated by FIRMS/HFEPR. Therefore, these complexes were re-investigated by DC magnetometry in the present study. Their dynamic magnetic properties were also explored by alternating current (AC) susceptometry. Comparisons are thus made between the DC magnetometry- and FIRMS/HFEPR-derived zfs parameters of CoO2E2, as well as between the dynamic magnetic properties of CoO2E2 and CoE4.
Recently, some of us investigated the S = 3/2 tetrahedral Co(II) complexes [Co{(EPiPr2)2N}2], E = S, Se, bearing two chelating chalcogenated imidodiphosphinato ligands74 (in the following abbreviated as CoE4), by DC and AC magnetic investigations, as well as by HFEPR. These slow-relaxing complexes exhibit D values of −30.5 cm−1 and −30.4 cm−1, as deduced by DC magnetometry, since HFEPR cannot be fully implemented for these complexes, due to the magnitude of D and their negligible rhombicity.74 In the work presented herein, these complexes were studied by FIRMS, providing accurate zfs data, in good agreement with the magnetometry-derived ones.
In the past, some of us carried out high-resolution multifrequency EPR spectroscopy studies on single crystals of the S = 2 tetrahedral Fe(II) complex [Fe{(SPPh2)(SPPh2)N}2] bearing the disulfido analogue of the above ligands.75 This study (at 9, 95 and 275 GHz) showed EPR signals attributed to distinct S = 2 centers and therefore provided evidence for the existence of multiple structural conformations of the compound. The zfs parameters of two such conformations were accurately determined. Since X-ray crystallography has shown that the crystal structure of CoO2Se2 exhibits structural disorder,72 similar studies were performed on the CoO2E2, E = S, Se, complexes, which indeed revealed two conformations of CoO2Se2. Furthermore, the CoO2E2 complexes were investigated by advanced quantum-chemical methods. Overall, our combined experimental and computational work provides a coherent picture of the structural, electronic and magnetic properties of this sub-family of tetrahedral Co(II) complexes, probing both their magnetic anisotropy and structural flexibility.
Results and discussion
DC magnetometry: static magnetic properties
The large discrepancy between the zfs values of the CoO2E2 complexes determined by FIRMS/HFEPR (vide infra) and those reported in the literature based on DC magnetometry (E = S,71 Se,72), prompted us to reinvestigate them by the latter method. The χMT product of CoO2E2, E = S, Se, as a function of temperature, is presented in Fig. 1.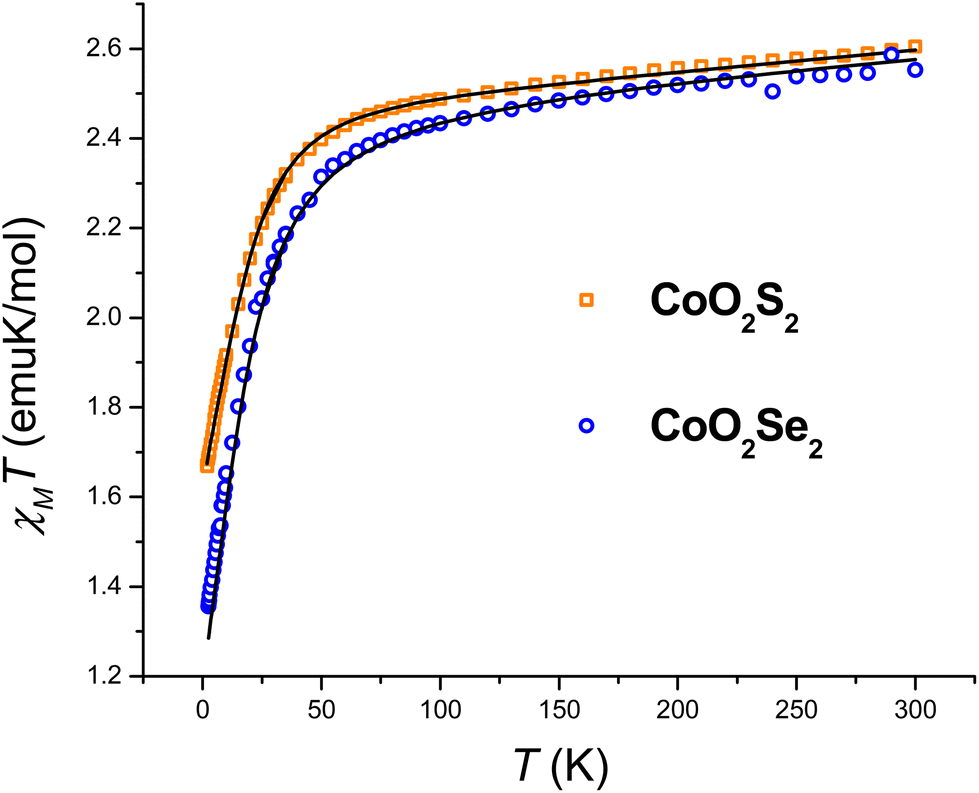 | ||
| Fig. 1 Temperature dependence of the χMT product of CoO2S2 and CoO2Se2, along with the best fitting lines calculated as described in the text. | ||
The investigated systems displayed a room temperature χMT value of 2.60 and 2.55 emu K mol−1, respectively, higher than the one expected for a spin-only S = 3/2 system (1.875 emu K mol−1), suggesting the presence of an unquenched orbital magnetic moment. On cooling down to 100 K, both systems undergo a linear decrease in their χMT product, which is interpreted as arising from a temperature-independent paramagnetism (TIP), previously found for tetrahedral S = 3/2 Co(II) complexes.71,76 Upon further lowering of the temperature, the magnitude of χMT plot decreases more rapidly, to reach 1.67 and 1.35 emu K mol−1 at 2.5 K for CoO2S2 and CoO2Se2, respectively, indicating the presence of magnetic anisotropy and/or antiferromagnetic interactions between neighboring Co(II) sites in their crystal lattice. The shortest distance between these sites in both CoO2S271 and CoO2Se272 is quite large (9.8 Å), and hence antiferromagnetic interactions are expected to be negligible. The zfs parameters D and E and Landé (g) factors were extracted from a fit of the χMT and isothermal magnetization plots shown in Fig. S2 (E = S) and S3 (E = Se) in the ESI,† using the following spin Hamiltonian:
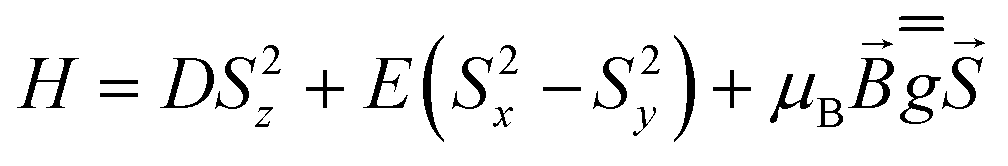
The resulting best fitting parameters were obtained using the PHI software77 and are reported in Table 1, and describe both systems as possessing a negative D parameter exceeding the 20 cm−1 value, in much better agreement with the FIRMS/HFEPR-derived values, (D ∼ −21 cm−1, Table 2, vide infra), than the previously reported |D| values by DC magnetometry (11.9 cm−1,71 and 10.4 cm−1,72 for the E = S and Se, respectively). However, the magnitude of the rhombicity E/D estimated by DC magnetometry (Table 1) differs significantly from that derived by FIRMS/HFEPR, especially for CoO2Se2 (4.2 × 10–4 and 0.08 cm−1, respectively, vide infra), again revealing the limitations of DC magnetometry in the accurate determination of the zfs D and E values.
| CoO2S2 | CoO2Se2 | |
|---|---|---|
| g x | 2.21(3) | 2.20(1) |
| g y | 2.20(1) | 2.52(1) |
| g z | 2.41(1) | 2.13(1) |
| D (cm−1) | −23.9(1) | −20.4(1) |
| E (cm−1) | −0.9(4) | −0.01(18) |
| E/D | 0.04(2) | 4.2(8) × 10–4 |
| TIP (emu mol−1) | 5.5 × 10–4 | 4.2 × 10–4 |
FIRMS and HFEPR studies
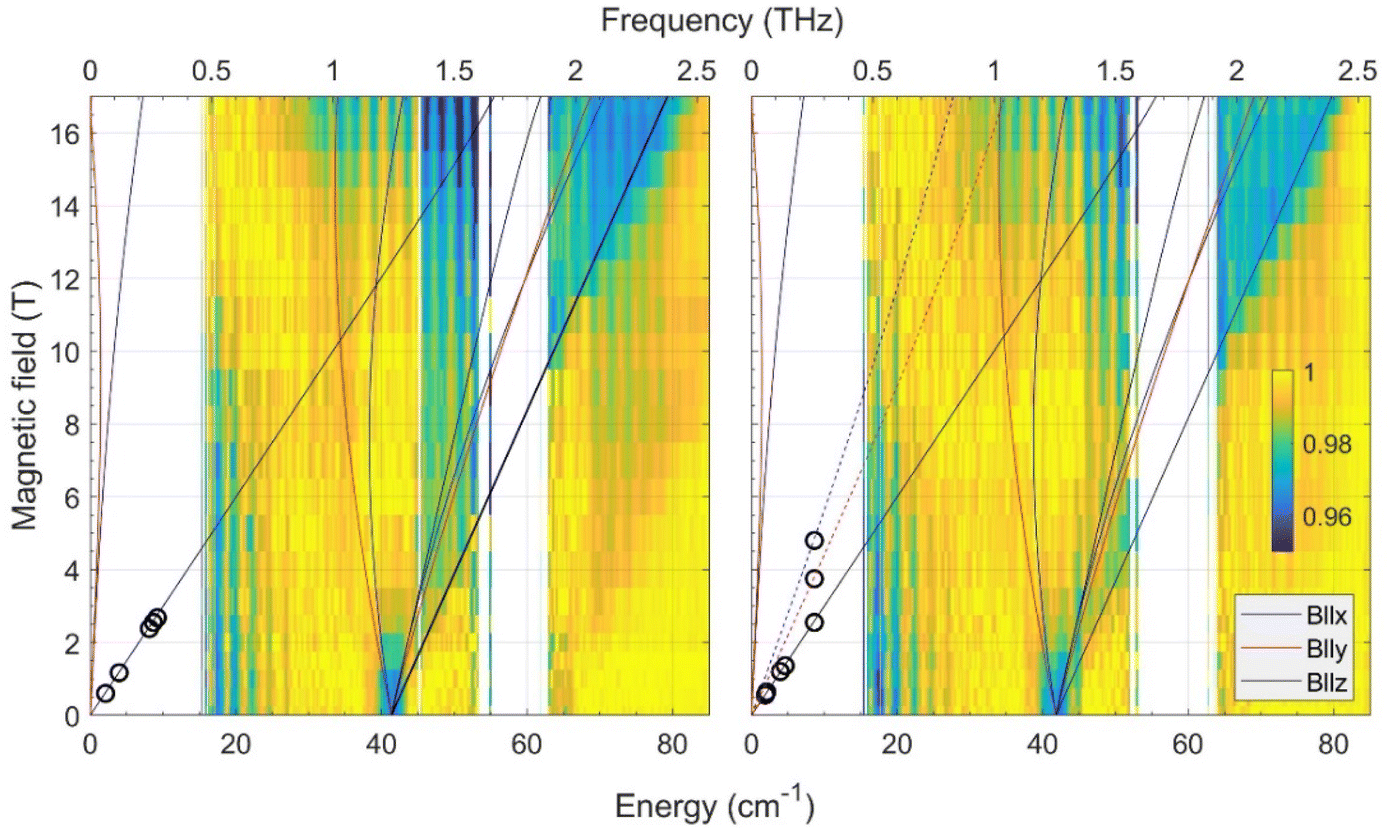 | ||
| Fig. 2 Joint FIRMS and HFEPR plots of magnetic resonances vs. transition energy (frequency) for the CoO2E2 complexes: E = S (left) and E = Se (right.) The color maps represent the magnitude of field-induced changes in the far-IR transmission spectrum (blue: most intense; yellow: least intense). White areas: the data are excluded due to large experimental error. The vertical stripes pattern stems from instrumental artifacts. The circles are observed HFEPR turning points. The lines represent turning points in the powder spectra and were calculated using spin Hamiltonian parameters as in Table 2. Solid lines originating from zero frequency at zero field correspond to the ground (Ms = ±3/2), and dashed lines in the right panel to the excited (Ms = ±1/2) intra-Kramers transitions. | ||
In addition to the FIRMS color maps, resonances detected in the HFEPR spectra of each complex are shown as superimposed circles. In both complexes, the dominant resonance was identified as the B0||z turning point belonging to the intra-Kramers (nominally: ΔMs = ±3) transition within the Ms = ±3/2 doublet. This means that the Ms = ± 3/2 doublet is the ground state, i.e., the sign of D by convention is negative. At elevated temperatures (20–40 K), resonances originating from the excited Ms = ± 1/2 doublet also showed up (Fig. S5† in the ESI†). However, only for CoO2Se2, signals at the perpendicular (B0||x, y) turning points of the ΔMs = ±1 transition within the excited Kramers doublet were strong enough to be used for analysis. These points allowed us to estimate the rhombicity of the zfs tensor for CoO2Se2, which turned out to be impossible for CoO2S2.
By combining the FIRMS and HFEPR data of CoO2Se2, the spin Hamiltonian parameters were determined to be:
| D = −20.9 cm−1, E = −1.7 cm−1, E/D = 0.08, g⊥ = 2.2; g|| = 2.4. |
It should be mentioned that the above values assume an axial symmetry of the g-tensor. Simulations show, however, that if the observed splitting of the perpendicular turning points in the HFEPR originated from rhombic g-values, these values would be unphysical, such as one of the perpendicular g-values equal to about 1.85.
Since the HFEPR response of CoO2S2 was weaker than that of CoO2Se2, we could not reliably identify the perpendicular turning points like we did in the latter system. Hence, we could only determine the gz value as 2.4, which is the same as for CoO2Se2. For the other spin Hamiltonian parameters, we thus had to rely on FIRMS results to deliver them. According to FIRMS/HFEPR, D = –20.9 cm−1 and E/D = 0.08 for CoO2Se2 (Table 2). Previous DC magnetometry/HFEPR74 and current FIRMS studies (vide infra) on the tetrahedral CoE4 complexes showed that the nature of E (either S or Se) has a little bearing on the zfs components of the two complexes (Table 2). Based on this observation, we assume that the rhombicity of the zfs in CoO2S2 is similarly small (∼ 0.1) as for CoO2Se2 (0.08). This assumption was confirmed by the observation of the complete powder pattern belonging to the intra-Kramers transition within the Ms = ±3/2 doublet by high-resolution EPR (Fig. S7,† for text vide infra), owing to the unusual setup of that instrument (combination of an X-band frequency with a superconducting magnet). In particular the resolution of the two perpendicular turning points indicates a rhombicity of the zfs tensor similar to that of CoO2Se2. Based on the above, the D value of CoO2S2 is very close to the corresponding D*, i.e., about 41.6/2 = ∼ 20.8 cm−1, thus the two CoO2E2 complexes have remarkably similar D parameters (Table 2). In addition, the fact that the combination of an X-band frequency and relatively high magnetic field (high-resolution EPR studies, vide infra) at low temperature detects exclusively the intra-Kramers transition within the MS = ±3/2 doublet means that the sign of D is negative.
In the current work, we revisited the two CoE4 complexes by employing FIRMS. The results are shown in Fig. 3 as color maps. The transmission spectrum (single beam) and simulation of the magnetic resonance absorption are shown in Fig. S6 in the ESI.† The CoS4 and CoSe4 complexes each show a zero-field transition at 65.0 ± 0.5 and 66.6 ± 0.5 cm−1, respectively. Given the very small rhombicity of the zfs tensor, as established previously,74 these values represent twice the parameter D, which is therefore −32.5 and −33.3 cm−1 for the E = S and Se complexes, respectively. These values are in good agreement (within 8% and 11%, respectively) with the corresponding ones previously derived by DC magnetometry (−30.5 cm−1 and −30.4 cm−1, respectively).74 The previous HFEPR results for CoS4 are plotted in the same Fig. 3 (left panel) as circles. The other spin Hamiltonian parameters, such as E/D and the g-values used to simulate the turning point branches, were taken from the HFEPR investigation of the CoE4 complexes.74
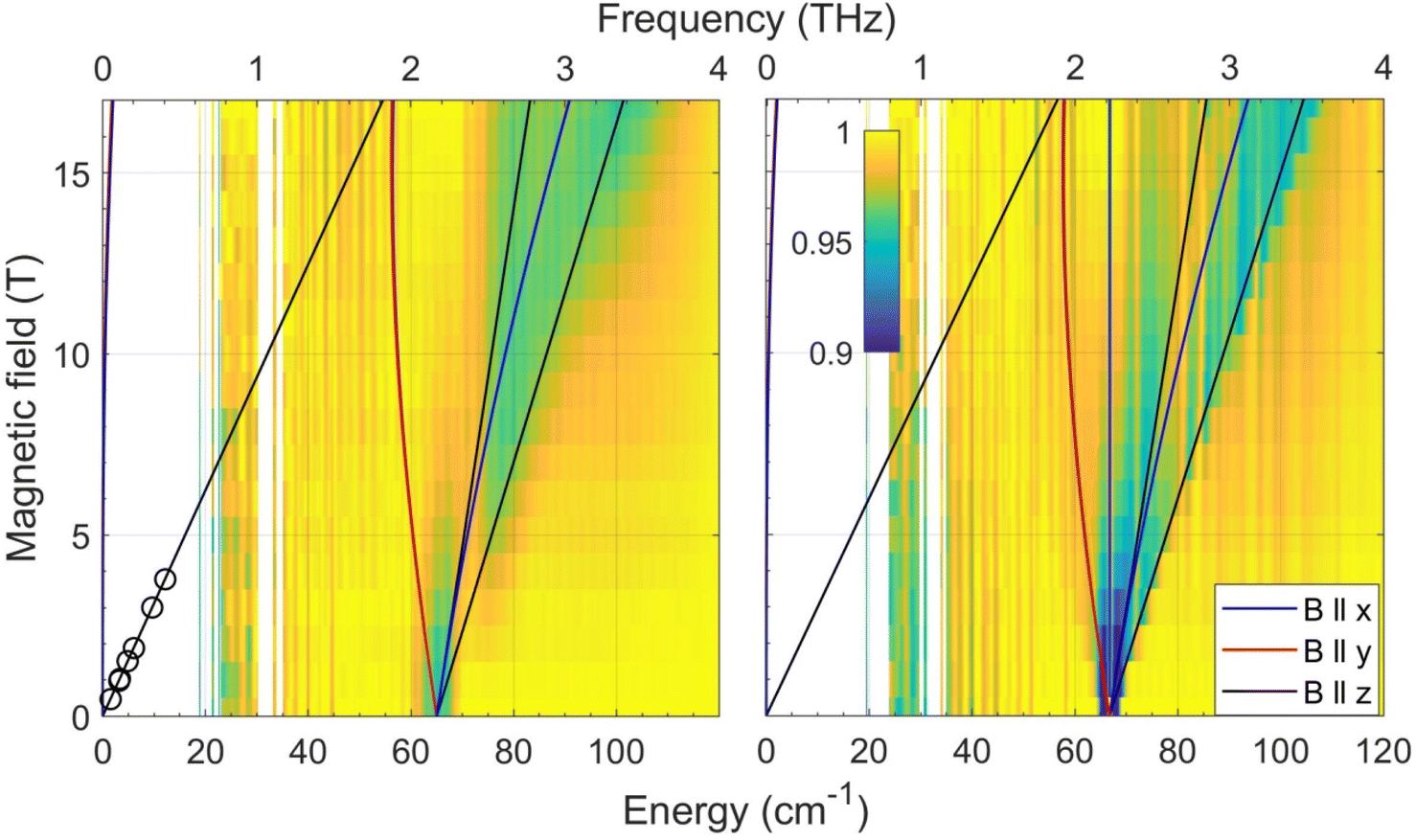 | ||
| Fig. 3 Joint FIRMS and HFEPR plots of magnetic resonances vs. transition energy (frequency) for the CoE4 complexes: E = S (left) and E = Se (right.) The circles in the left panel represent the single turning point branch with B0||z within the MS = ±3/2 Kramers doublet observed by HFEPR in CoS4. No corresponding turning point was detected for CoSe4. The lines represent turning points in the powder spectra and were simulated using the spin Hamiltonian parameters in Table 2. | ||
It should be noted that for both CoE4 and CoO2E2 complexes, the small effect of the nature of the E donor atom (either S or Se) on the magnitude of D in these Co(II) systems is consistent with similar findings for other 3d-metal-based complexes, such as tetrahedral Fe(II) S = 2 FeE4, E = S, Se,55 and [Fe{(EPPh2)2N}2], E = S,75 Se,78 as well as octahedral Ni(II) (S = 1) [Ni{(OPPh2)(EPPh2)N}2(sol)2], E = S, Se; sol = dmf, thf,79 dmso.80
High-resolution EPR spectroscopy at 9, 95, and 275 GHz
The spectrum of a single crystal of CoO2S2 offers higher resolution. The crystal, free to move in the EPR tube, was found to orient itself in a magnetic field of 1.5 T (as previously observed for the [Co{(SPPh2)(SPiPr2)N}2] complex of the same sub-family).81Fig. 4 (top) shows the EPR spectrum at 9 GHz of such an oriented crystal of CoO2S2 at 5 K between 0.02 and 0.40 T. It reveals a narrow line around 0.1 T, which shows a structure that points to the overlap of two EPR transitions of comparable intensity. This structure is maintained upon temperature increase, while the relative intensity of the two transitions changes with temperature.
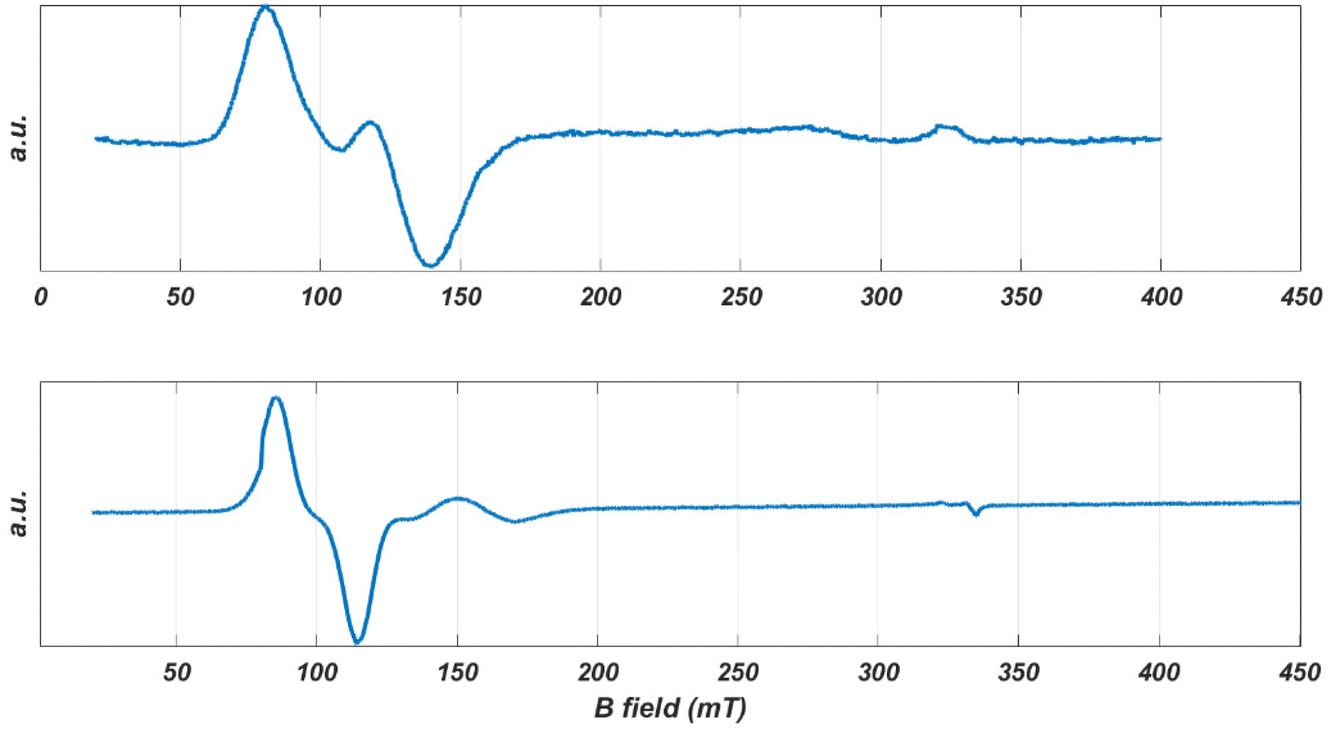 | ||
| Fig. 4 The EPR spectra of oriented single crystals of CoO2S2 (top) and CoO2Se2 (bottom) at 9 GHz and 5 K. | ||
While the EPR spectrum at 9 GHz of the CoO2S2 powder did not resolve the two contributions to the low-field line, the spectra at higher microwave frequencies clearly do, owing to the enhanced g′ resolution. Fig. 5 represents the EPR spectra of the CoO2S2 powder at 9, 95, and 275 GHz on a common g′ axis. The line width at 9 GHz is determined by hyperfine interaction. This interaction is magnetic-field independent, and, at higher microwave frequencies and magnetic fields, transitions characterized by small differences in g′, hidden under the hyperfine-broadened line at low microwave frequency, become resolved.
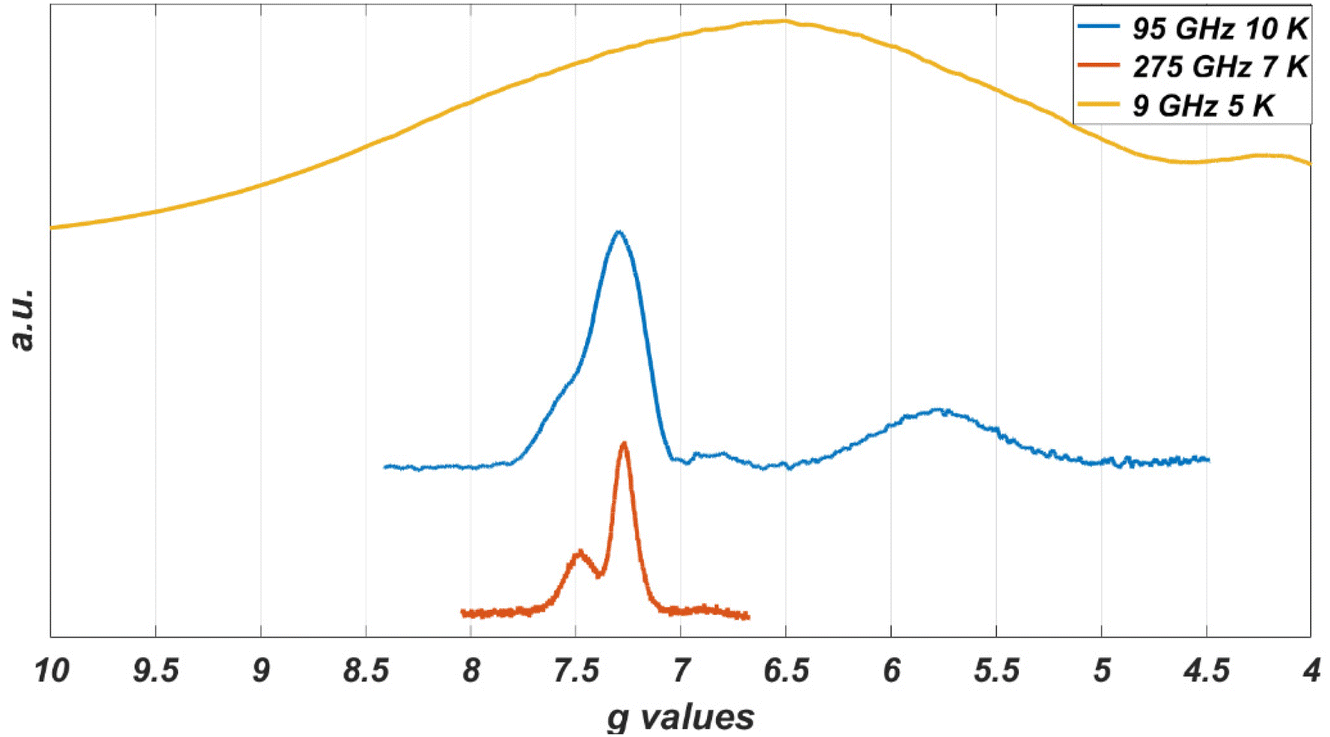 | ||
| Fig. 5 Low-field part of the EPR spectra of a powder sample of CoO2S2 at different microwave frequencies: 9 GHz (upper), 95 GHz (middle), and 275 GHz (lower). The spectrum at 9 GHz covers the magnetic-field range from 10 to 170 mT, the spectrum at 95 GHz from 0.8 to 1.5 T, and the spectrum at 275 GHz from 2.45 to 2.95 T. | ||
A single crystal of CoO2Se2 free to move in the EPR tube orients itself in a high magnetic field, as illustrated in Fig. S10 in the ESI.† The first scan from 0 to 1.5 T shows the spectrum for an arbitrary orientation of the crystal with respect to the magnetic field, the second one for the oriented crystal for which virtually only intensity in the low-field range is left. The latter spectrum is shown in Fig. 4 (bottom). It consists of a narrow line at 0.097 T and a second one of much lower intensity at 0.16 T.
The powder EPR spectra of the CoO2S2 and CoO2Se2 complexes reveal transitions corresponding to g′ values up to 7 and below 1. Such a combination of g′ values points to a negative sign of D, i.e., the Ms = ±3/2 doublet is lowest in energy, and a value of E/D = 0.10 ± 0.05. For CoO2S2, the transitions in the lower doublet show up at g′z ∼ 6.5, g′y ∼ 0.8 and g′x ∼ 0.45, and the transitions in the upper doublet at g′z ∼ 2 and g′ ∼ 4.2. For CoO2Se2, the transitions in the lower doublet show up at g′z ∼ 7.1, g′y ∼ 1 and g′x ∼ 0.5, and the transitions in the upper doublet at g′z ∼ 2 and g′ ∼ 4.9.
Based on the analysis of FIRMS data (vide supra), the value of D for both complexes was determined to be about −21 cm−1 (Table 2). The interpretation of the 9 GHz EPR spectra of both CoO2E2 complexes is compatible with this value of D and a value of about 0.1 for E/D. A more quantitative analysis of the 9 GHz spectra is prohibited by the fact that they depend on too many spin Hamiltonian parameters. The large magnitude of D implies that inter-doublet transitions are not observable at the highest microwave frequency (275 GHz) and magnetic field (13 T) of our EPR experiments.
Subsequently, we consider the EPR data for the single crystals. Both the CoO2S271 and the CoO2Se272 complex crystallize in the P21/c space group, with four molecules in the unit cell. The inversion center connects the molecules two by two, which results in two magnetically inequivalent molecules in the unit cell.
After orientation in the magnetic field, the 9 GHz EPR spectrum of a single crystal of CoO2Se2 becomes easy to interpret. It shows a single narrow line at g′z = 7.09, Fig. 4 (bottom). For this direction of the magnetic field with respect to the crystal, apparently parallel to the principal z axis of the g′ tensor, the signals of the two magnetically inequivalent molecules coincide, which means that the principal g′z axes of the magnetically inequivalent molecules are parallel. In addition, the EPR spectrum contains a small feature at g′ = 3.95, the origin of which will be discussed below.
For the CoO2S2 complex, the single-crystal EPR spectrum of the oriented crystal consists of two lines that strongly overlap corresponding to g′ = 6.81 and g′ = 5.55, Fig. 4 (top). The relative intensity of these lines varies from crystal to crystal, which indicates that the two signals do not refer to the two magnetically inequivalent molecules in the unit cell. Most probably these signals refer to two distinct structures of the CoO2S2 complex with slightly different spin Hamiltonian parameters. In this case, the direction of the magnetic field with respect to the crystal will not be parallel to the principal g′z axis of either of the two structures, and the g′ values of the observed lines will thus be a little smaller than the principal g′z values.
Similarly, we interpret the small signal observed for the oriented CoO2Se2 crystal as to derive from a second conformation of this complex. The g′ value of 3.95 deviates significantly from the g′z value of 7.09 corresponding to the large signal, Fig. 4 (bottom), which indicates that the direction of the magnetic field with respect to the oriented crystal, which is about parallel to the principal g′z axis of the first structure, makes an angle of about 70° with the principal g′z axis of the second structure.
In the EPR spectra at 9 GHz of the powders, the subtleties related to structural variation of the Co(II) coordination are hidden under the broad lines, but they do show up in the powder spectra of the CoO2S2 complex at 95 and 275 GHz (Fig. 5), where the low-field signal splits into two.
The X-ray diffraction study of the CoO2Se2 complex reveals the presence of distinct conformations of this complex. Structural disorder has been observed for one of the two (O, Se) chelates concerning the position of the O and the Se donor atoms (Fig. S1 in the ESI†). Interestingly, this is also the case for the analogous tetrahedral MO2Se2, M = Ni,72 Zn,82 complexes. Although such distinct conformations have not been detected by single-crystal X-ray diffraction for the CoO2S2 complex,71 we speculate that such distinct arrangements of the O and S/Se atoms are responsible for the ‘extra’ signals in the EPR spectra of the single crystals for both complexes. The EPR spectra are sensitive to the electronic structure, while the X-ray diffraction pattern reflects the geometric structure of the complexes. Apparently, the difference in the geometric structure of the two conformations is too small for the CoO2S2 complex to be discernible in the X-ray diffraction pattern. This conclusion is supported by the EPR spectra of the crystals. The directions of the principal g′z axes of the distinct structures are found to (nearly) coincide for the CoO2S2 complex, while these directions differ significantly for the CoO2Se2 complex.
AC susceptometry: dynamic magnetic properties
The dynamics of the magnetic moment of the two complexes CoO2E2, E = S, Se, were probed by AC susceptometry. With no static applied field, both CoO2S2 and CoO2Se2 did not display any out-of-phase susceptibility signal (Fig. S11 in the ESI†). This finding may be ascribed to the presence of a significant tunneling relaxation pathway, which may be triggered by the presence of dipolar magnetic interactions in the solid state, as observed for the structurally related CoE4 complexes, E = S, Se.74 An isothermal field scan of both complexes revealed the onset of slow relaxation upon the application of a static field, and in both cases 120 mT was chosen as the field inducing the slowest magnetization dynamics (Fig. S12†). The temperature dependence of χM′ and χM′′ is shown in Fig. S13† and reports, for both systems, a set of frequency and temperature dependent peaks in the χM′′, highlighting a slow relaxing magnetic moment.The temperature dependence of the magnetic relaxation times τ, represented in Fig. 6, shows a nonlinear behavior: in the low temperature region the relaxation times showed a tiny dependence, while bigger variations appeared upon heating the systems. Fitting of these plots using a mixed relaxation model involving an Orbach/direct mechanism of relaxation12 satisfactorily reproduced the experimental data, with an effective barrier to the reorientation of the magnetic moment (Ueff, Table 3), in line with what is expected on the basis of the static magnetic analysis (Table 1). A mixed Raman/direct relaxation mechanism yielded fits of similar quality, but for parameter values in an unphysical range,83 and were thus discarded. The observed relaxation barriers suggest the presence of additional relaxation pathways lowering the magnetic reorientation barrier with respect to the one observed by FIRMS/HFEPR. When compared with the structurally related CoS4 complex, featuring a barrier of about 54 cm−1 with a D value of −30.5 cm−1 and an E/D value of 0.0023,74 the decrease in the barrier to the Orbach relaxation of CoO2E2, E = S, Se, correlates well with their less negative D and increased E/D values (Table 2). It should be noted that the relaxation of CoSe4 has been shown to take place exclusively through a Raman mechanism, instead of an Orbach one.74
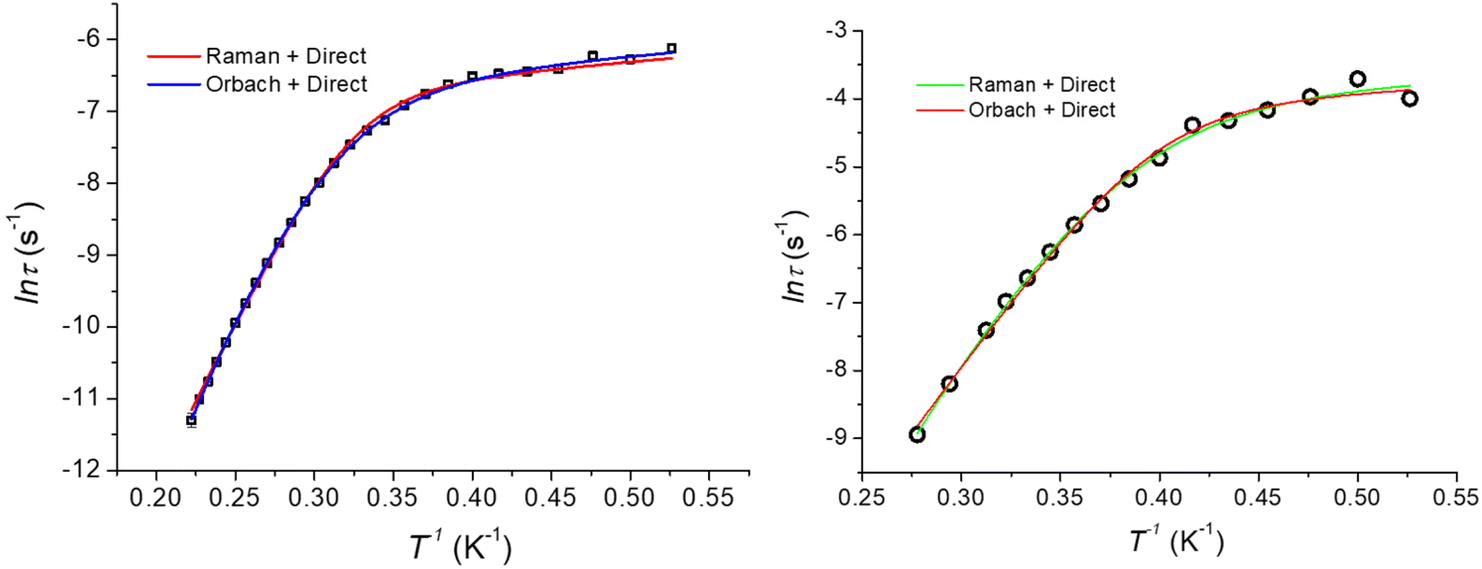 | ||
| Fig. 6 Temperature dependence of the magnetic relaxation times (Arrhenius plot) for CoO2S2 (left) and CoO2Se2 (right), along with the best fitting lines for the two mixed relaxation mechanisms described in the text. | ||
| CoO2S2 | CoO2Se2 | |||
|---|---|---|---|---|
| τ 0 (s) | 8(1) × 10–10 | — | 3(1) × 10–9 | — |
| U eff (cm−1) | 30.5(5) | — | 27(1) | — |
| C (s−1 K−n) | — | 1.9(2) × 10–3 | — | 5(1) |
| n | — | 11.68(6) | — | 12.9(4) |
| B (s−1) | 274(6) | 251(3) | 25(1) | 23(1) |
| R 2 | 0.9998 | 0.9993 | 0.9956 | 0.9969 |
Computational studies
The electronic structure of the crystal structure geometries of CoO2S271 and CoO2Se272 was calculated at the state-average complete active space self-consistent field/N-electron valence perturbation theory 2nd order (SA-CASSCF/NEVPT2) methodology. An active space of seven electrons in five 3d-orbitals, was used, and the orbitals were optimized for 10 quartet and 40 doublet roots. Furthermore, additional calculations were carried out where the orbitals were optimized for 7 quartets and 14 doublets. The spin Hamiltonian properties (zfs parameter D, g-factors) were computed using the effective Hamiltonian approach (HEFF)84 and via the computation of the 2nd order contribution of the hyperfine coupling tensor (HFC) from spin–orbit coupling (SOC), namely SOC-2nd order. For the CoO2Se2 complex, two geometries (a, 88%) and (b, 12%) are experimentally found, see Fig. 1, where the structure (a) in the crystal is more stable than structure (b) by 46.7 kcal mol−1 at NEVPT2. The geometries of the CoO2E2, E = S, Se, complexes were energetically optimized, as described in the Experimental section (vide infra). The lowest in energy structures are shown in Fig. 7. Full geometry optimization of the (a) and (b) structures significantly stabilized both of them and the two structures become almost degenerate, with (b) being lower in energy only by 0.24 kcal mol−1 than (a) (Table S1 in the ESI†). Details about the energetics and the electronic structures of the 50 calculated structures are presented in the ESI.† The main CASSCF electronic configurations of all calculated states are given in Table S3,† the NEVPT2 and NEVPT2-LFT vertical transition energies from the ground state to the excited states are depicted in Fig. S15,† while the NEVPT2-LFT d-orbital splitting and the corresponding field matrix are listed in Table S4.† Finally, the geometries of the CoO2E2, E = S, Se, complexes were energetically optimized, as described in the Computational studies section (vide infra), to investigate the experimental observation of two geometries for CoO2Se2, and, in addition, to examine whether this can also happen in the case of CoO2S2, even though this has not – up to now – been observed experimentally. Moreover, it was checked how the geometry optimization affects the properties of the complexes, including the zfs parameters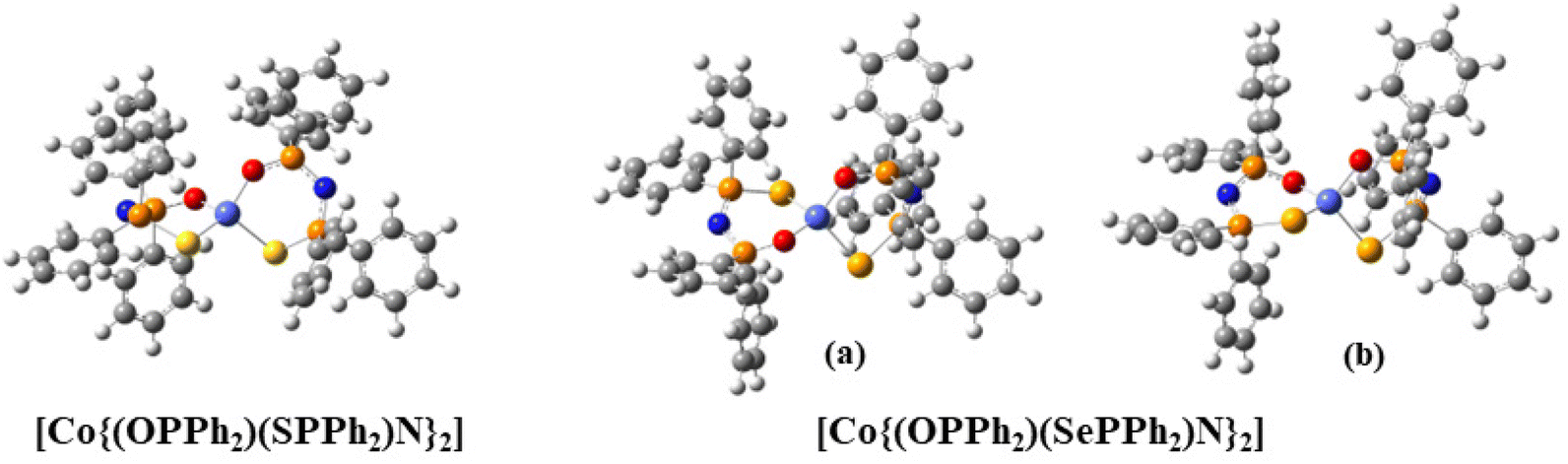 | ||
| Fig. 7 The lowest in energy isomers of the CoO2E2, E = S, Se complexes. Color coding: Co (light blue), O (red), S (yellow), Se (light brown), P (brown), N (blue), C (gray). | ||
Comparing the geometries of the crystal structures with those of the optimized structures, it is found that the former are more distorted than the latter. Specifically, regarding the [Co(OENP2)(O′E′N′P′2)] core, in the lowest-energy optimized structures, the Co–O, Co–S and Co–Se bond distances are similar, i.e., Co–O: ∼1.96 Å, Co–S: 2.37 Å, and Co–Se: ∼2.43 Å. On the contrary, in the crystal structures, only the two Co–S distances are similar at ∼2.33 Å, while the Co–O and Co–Se bond distances are different, i.e., Co–O: 1.95–2.13 Å and Co–Se: 2.38–2.46 Å, see Table S1.† Regarding the phenyl groups, in the optimized structures, the planes of two phenyl groups are approximately parallel to each quasi-plane of the CoOENP2 ring, while in the crystal structures this occurs only for one CoOENP2 ring. Moreover, the PSOP and P′S′O′P′ dihedral angles are similar and about 40° in the optimized structures, while in the crystal geometries they are not equal (33° and 17°, respectively). In addition, the dihedral angle θ between the OCoS and S′CoO′ planes is 88.3°, while in the crystal structure this dihedral angle is 86.3°. Thus, the symmetry is distorted in the experimental crystal structures. The (a) and (b) structures of CoO2Se2 differ in the position of the CoOSe plane with respect to the CoO′Se′plane, see Fig. 7. The OCoEO′ is about −120° for CoO2S2 and CoO2Se2 (b structure), while it is about 120° for CoO2Se2 (a structure), see Table S1.† This distortion is responsible for the energy difference between structure (a) and (b), where (a) is more stable than structure (b) by 46.7 kcal mol−1 at NEVPT2, while in the full geometry-optimized (a) and (b) structures, they become almost energetically degenerate, with (b) being lower in energy only by 0.24 kcal mol−1 than (a) (Table S1 in the ESI†). Finally, it should be noted that concerning the CoO2S2 complex, geometry optimization of the (a) and (b) structures in which Se was replaced by S resulted in the same structure.
The electric dipole moment of the complexes in their crystal structure geometries is 3.74 Debye for CoO2S2, and for the (a) and (b) structures of CoO2Se2 3.57 and 5.46 Debye, respectively (Table S2†). In the energy-optimized geometries, the dipole moments become almost half of the previous values, i.e., the value is 1.90 Debye for CoO2S2, and 1.95 Debye (a) and 2.38 Debye (b) for CoO2Se2, showing that the optimized molecular structures become less polar and more symmetrical than the corresponding crystal structures.71,72
The two complexes have similar g-factors calculated in their crystal geometries and the energy-optimized ones (Table 4). However, regarding the zfs parameters, there are differences in the NEVPT2 values obtained for the crystal and the energy-optimized geometries for the CoO2S2 and CoO2Se2 (a) structures. Finally, comparing the NEVPT2 D values obtained via the HEFF and via the SOC-2nd approach, we observe that the effective Hamiltonian approach results in good agreement with the experimental values derived by FIRMS/HFEPR (Table 2) (vide infra), while the second methodology overestimates the D values by up to 5 cm−1.
| E | Geom. |
D
SOC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
D
SSCc |
D |
E/DSOCb |
E/Dc |
|---|---|---|---|---|---|---|
| a θ: (OCoS)/(S′CoO′). b SOC-HEFF [SOC-2nd order]. c HEFF. d NEVPT2/ZORA-def2-TZVPCo,Se,S,OZORA-def2-SVPP,N,C,H//B3LYP/6-31G(d,p). e NEVPT2/ZORA-def2-TZVPCo,Se,S,OZORA-def2-SVPP,N,C,H//B3LYP/def2-SVP. f NEVPT2/ZORA-def2-TZVPCo,Se,S,OZORA-def2-SVPP,N,C,H//Experimental crystal geometries, E = S,71 Se.72. g NEVPT2/ZORA-def2-TZVPCo,Se,S,OZORA-def2-SVPP,N,C,H//Experimental crystal geometries; roots = 7 (quartets) & 14 (doublets). h NEVPT2/ZORA-def2-TZVPCo,S(-f)OZORA-def2-SVPP,N,C,H//Experimental crystal geometries. | ||||||
| S | −29.7 [−34.2] | −0.18 | −29.85 | 0.07 [0.07] | 0.08 | |
| −30.6 [−35.4] | −0.20 | −30.78 | 0.07 [0.07] | 0.07 | ||
| −19.4 [−21.9]; −19.9g; −19.1h | −0.21 | −19.57 | 0.08 [0.08] | 0.08 | ||
| Se (a) | −23.8 [−26.9] | −0.30 | −24.09 | 0.09 [0.09] | 0.09 | |
| −31.6 [−36.7] | −0.25 | −31.81 | 0.10 [0.09] | 0.10 | ||
| −21.7 [−24.6] | −0.28 | −22.00 | 0.10 [0.11] | 0.10 | ||
| Se (b) | −25.0 [−28.6] | −0.20 | −25.20 | 0.07 [0.07] | 0.08 | |
| −25.8 [−29.8] | −0.33 | −26.10 | 0.22 [0.22] | 0.22 | ||
For CoO2S2, the NEVPT2 D parameter (including only SOC) was computed using the effective Hamiltonian approach, and for the crystal geometry, values between −19.1 and −19.9 cm−1 were obtained depending on the number of the calculated roots (Table 4). These values are in good agreement with those derived by FIRMS (Table 2), as well as with the value of −19.1 cm−1 reported recently by Sarkar et al.85 For the geometry-optimized structure of CoO2S2, the D value (including only SOC) is increased to −29.7 cm−1, most likely due to the significant structural differences compared to the crystal structure (vide supra).
For CoO2Se2, as mentioned above, the (a) geometry, which is the dominant one found in the crystal, is lower in energy by 46.7 kcal mol−1 than the (b) geometry at NEVPT2 (Table S2†). However, full energy optimization results in energetically degenerate (a) and (b) structures. Regarding the (a) structure, the dihedral angle θ between the two planes of OCoSe and Se′CoO′ deviates by about 5° from the orthogonal angle in both calculated and experimental crystal structures. The NEVPT2 calculation yields a D value, including SOC and SSC, of −22.0 cm−1 for the crystal geometry (again in good agreement with the FIRMS/HFEPR-derived value), and −24.1 cm−1 for the energy-optimized structure. Regarding the (b) structure, the NEVPT2 calculations yield values of D for the crystal geometry (−26.1 cm−1) and for the energy-optimized structure (−25.2 cm−1) (Table 4). Our best calculated D values in the crystal structure geometry of CoO2Se2 are −22.0 (a) and −26.1 cm−1 (b) (Table 4), and therefore their average value, considering their corresponding % population, is −22.4 cm−1, in good agreement with the experimental FIRMS/HFEPR-derived value of −20.9 cm−1. Thus, our calculations in the crystal structure geometry confirm the fact that the (a) structure of CoO2Se2 is the favored one.72 Moreover, for CoO2Se2, our calculations support the experimental finding by X-ray crystallography72 - and herein confirmed by EPR spectroscopy (vide supra) - of two different conformations.
It should also be stressed that for both CoO2E2, E = S, Se, the spin–spin coupling (SSC) interactions contribute to the magnitude of D by, approximately, only −0.2 to −0.3 cm−1, which is clearly a minor contribution as compared to that of the corresponding SOC interactions (Table 4).
Regarding the NEVPT2-LFT d-orbital splitting diagram of the calculated complexes, there are differences between the experimental crystal structures and the minimum calculated ones, see Fig. 8, which is expected given that the minimum calculated structures are more symmetrical than the crystallographic ones. Moreover, there are differences between the (a) and (b) structures of CoO2Se2: the lowest doubly occupied orbitals are the dz2 (m![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) = 0) in (a), while in (b) are the dxy and dx2−y2 (m = ± 2). Note also that the CoO2S2 and the (b) structure of CoO2Se2, which have similar relative position of the rings, i.e., similar OCoEE′, OCoEO′, O′CoE′E, and O′CoE′O dihedral angles, have both the dxy and dx2−y2 (m = ± 2) as the lowest occupied orbitals. Finally, the splitting between the doubly occupied d orbitals and single occupied d orbitals, is 1625, 1832 and 1704 cm−1 for CoO2S2, (a) structure of CoO2Se2 and (b) structure of CoO2Se2, respectively, for the energy-minimized structures; while, in the geometry of the crystal structures the splitting is significantly larger up to 400 cm−1, and becomes 2218, 2119 and 2065 cm−1, respectively.
= 0) in (a), while in (b) are the dxy and dx2−y2 (m = ± 2). Note also that the CoO2S2 and the (b) structure of CoO2Se2, which have similar relative position of the rings, i.e., similar OCoEE′, OCoEO′, O′CoE′E, and O′CoE′O dihedral angles, have both the dxy and dx2−y2 (m = ± 2) as the lowest occupied orbitals. Finally, the splitting between the doubly occupied d orbitals and single occupied d orbitals, is 1625, 1832 and 1704 cm−1 for CoO2S2, (a) structure of CoO2Se2 and (b) structure of CoO2Se2, respectively, for the energy-minimized structures; while, in the geometry of the crystal structures the splitting is significantly larger up to 400 cm−1, and becomes 2218, 2119 and 2065 cm−1, respectively.
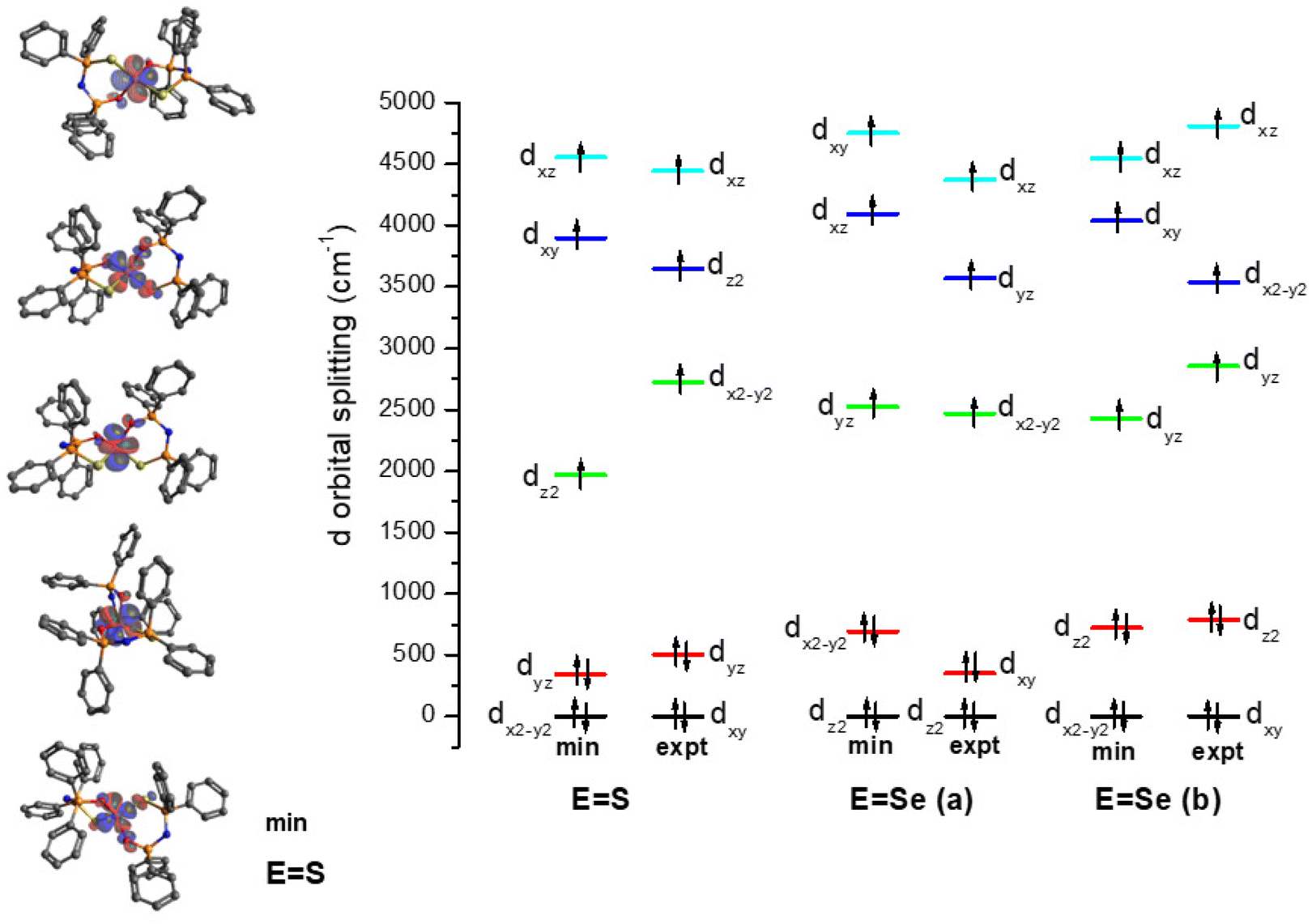 | ||
| Fig. 8 NEVPT2-LFT d-orbital splitting diagram of the calculated structures of CoO2E2, E = S, Se. The structures (a) and (b) correspond to 88 and 12% population, respectively, in the crystal structure of CoO2Se2.72 | ||
Finally, the contribution of the calculated thirty-nine excited states to D and E is presented in Table S5 of ESI.† The three lowest excited states are quartet states, they are lying up to 5 × 103 cm−1 (0.6 eV) above the ground state and have the highest contribution to the D and E parameters, see Table 5 and cf. Table S5.†
| T e |
T
ea |
D | E | T e |
T
ea |
D | E | |
|---|---|---|---|---|---|---|---|---|
| NEVPT2b | NEVPT2c | |||||||
| a NEVPT2-LFT. b NEVPT2/ZORA-def2-TZVPCo,Se,S,OZORA-def2-SVPP,N,C, H//B3LYP/6-31G(d,p). c NEVPT2/ZORA-def2-TZVPCo,Se,S,OZORA-def2-SVPP,N,C, H//Experimental crystal geometries.71,72 | ||||||||
| E = S | ||||||||
| GS | 0 | 0 | 0.00 | 0.00 | 0 | 0 | 0.00 | 0.00 |
| 1 | 2218 | 1815 | 26.30 | 25.84 | 2863 | 2307 | 20.94 | 20.20 |
| 2 | 4726 | 3686 | −23.86 | −0.09 | 4388 | 3412 | −19.85 | −0.88 |
| 3 | 5218 | 4065 | 9.77 | −9.28 | 4993 | 3904 | 6.62 | −8.59 |
| E = Se (a) | ||||||||
| GS | 0 | 0 | 0.00 | 0.00 | 0 | 0 | 0.00 | 0.00 |
| 1 | 2750 | 2154 | 22.20 | 21.51 | 2775 | 2170 | 21.87 | 20.96 |
| 2 | 4380 | 3444 | −12.34 | −2.63 | 4283 | 3291 | −23.51 | −0.13 |
| 3 | 5283 | 4073 | −1.01 | −5.78 | 4847 | 3767 | 9.36 | −8.79 |
| E = Se (b) | ||||||||
| GS | 0 | 0 | 0.000 | 0.00 | 0 | 0 | 0.000 | 0.00 |
| 1 | 2444 | 1952 | 23.61 | 23.13 | 2436 | 1887 | 22.92 | 21.02 |
| 2 | 4775 | 3694 | −19.53 | −0.86 | 4251 | 3202 | −24.04 | −0.15 |
| 3 | 4976 | 3855 | 6.52 | −8.67 | 4678 | 3500 | 8.02 | −5.09 |
To sum up, the computed D and E/D values of both Co2E2 complexes are in good agreement with the ones derived by FIRMS/HFEPR. Additionally, for CoO2Se2, our calculations support the experimental finding by X-ray crystallography72 - and herein confirmed by EPR spectroscopy (vide supra) - of two different conformations.72 Finally, the energetically geometry optimization of the complexes results in more symmetrical structures, with similar g-factors and E/D values.
Conclusions
In this work, the tetrahedral CoO2E2, E = S, Se, complexes were studied by experimental (DC magnetometry and AC susceptometry, as well as FIRMS, high-resolution multifrequency EPR, and HFEPR spectroscopy) and computational methods. In addition, the CoE4, E = S, Se, complexes were studied by FIRMS. One of the main aims of this work was to reliably determine the D and E/D values of all the above S = 3/2 Co(II) tetrahedral complexes. The experimentally determined zfs values, along with the calculated ones better matching the former, are listed in Table 6, and lead to the following conclusions: (i) The combination of FIRMS and HFEPR data on CoO2Se2, exhibiting a non-negligible rhombicity (E/D = 0.08), allowed to reliably determine both zfs components, (ii) A rhombicity E/D ∼ 0.1 is compatible with the high-resolution multifrequency X-band spectra of both CoO2E2 complexes, (iii) Our work has revealed certain limitations of DC magnetometry in providing accurate zfs data, (iv) The computed zfs values of CoO2E2 derived by the NEVPT2 method based on the corresponding crystal structures are in very good agreement with the FIRMS/HFEPR-derived ones. The magnitude of D of both complexes is shaped almost exclusively by SOC interactions, whereas the SSC contribution is negligible.| Complex | D HFEPR/FIRMS | E/DHFEPR/FIRMS | E/DHREPR | D magn | E/Dmagn | D calc | E/Dcalc |
|---|---|---|---|---|---|---|---|
| a Calculated by taking into account the 88 and 12% populations of the (a) and (b) structure of CoO2Se2,72 as described in the text. | |||||||
| CoO2S2 | ∼ −20.8 | ∼ 0.1 | –23.9 | 0.04 | −19.6 | 0.08 | |
| −19.185 | 0.0885 | ||||||
| CoO2Se2 | –20.9 | 0.08 | ∼ 0.1 | –20.4 | 0.0004 | −22.4a | 0.11a |
| CoS4 | –32.5 | –30.574 | 0.02374 | −28.885 | 0.0085 | ||
| CoSe4 | –33.3 | –30.474 | 0.001674 | −25.785 | 0.0285 | ||
Moreover, high-resolution multifrequency EPR on powder and single crystal samples of CoO2E2 revealed the existence of multiple conformations, in accordance with the structural disorder observed previously in the crystal structure of CoO2Se2. Analysis of the EPR data indicate that the structural difference between the conformations is larger for the CoO2Se2 than for the CoO2S2 complex, confirming the crystallographic observations. The existence of two conformations of CoO2Se2 is also supported by advanced quantum chemical calculations.
The second aim of this work was to probe the dynamic magnetic properties of the CoO2E2 complexes, following the previous analogous investigation of the CoE4 complexes. Our AC susceptometry studies reveal a dependence of the barrier to the Orbach relaxation of both CoO2E2, as well as of the previously studied CoS4, on the corresponding D and E/D values.
In more general terms, this work shows that the reliable determination of the zfs D and E components is not an easy task, and points to the necessity of combining as many different experimental methods as possible. In parallel, this work highlights the capacity of a spectroscopic method, namely high-resolution multifrequency EPR spectroscopy, to probe the existence of multiple conformations in a paramagnetic metal complex. In that respect, such high-resolution spectroscopic techniques can be invaluable companions to X-ray crystallography when subtle structural features are investigated.
Experimental
Synthesis of complexes
Complexes CoO2E2, E = S,71 Se72 and CoE4, E = S, Se,74,86 were synthesized following the procedures reported in the literature, in which the corresponding X-ray crystal structures are also reported.DC magnetometry
Samples employed for DC and AC susceptometry analyses consisted of pressed microcrystalline powders of CoO2E2, E = S, Se, wrapped in Teflon (TM) tape. The DC magnetic characterization was performed on Quantum Design MPMS (Magnetic Properties Measurement System) equipment provided with a 5 T magnet. The magnetization (M) dependence on the absolute temperature was investigated between 300 and 35 K using a magnetic field (B) of 1 T, and between 35 and 2 K with a field of 0.1 T to prevent magnetic saturation. Magnetic susceptibility per mole (χM) was then evaluated as χM = MM/B.FIRMS
Far infrared transmission measurements were performed at the National High Magnetic Field Laboratory using a Bruker Vertex 80v FT-IR spectrometer coupled with a 17 T vertical-bore superconducting magnet in a Voigt configuration (light propagation perpendicular to the external magnetic field). The experimental setup employs a mercury arc lamp as a broad-band terahertz radiation source. The radiation transmitted through the sample was detected by a composite silicon bolometer (Infrared Laboratories) mounted at the end of the quasioptical transmission line. Both the sample and bolometer were cooled by a low-pressure helium gas to the temperature of about 5 K. The transmission spectra of the microcrystalline powder sample (∼2 mg) bonded by n-eicosane were measured in the spectral region between 14 and 730 cm−1 (0.42–22 THz) with an instrumental resolution of 0.3 cm−1 (9 GHz). To discern the magnetic absorptions, the transmission spectrum measured at each magnetic field was divided by the reference spectrum, which is the mean of spectra at all magnetic fields, computed after removing the outlier points at each frequency. Such normalized transmittance spectra indicate the small-amplitude changes in the intensity induced by the magnetic field and exclude a strong nonmagnetic contribution due to vibrational absorptions and an instrumental function. All data analysis routine was implemented by an in-house written MATLAB code based on the EPR simulation software package EasySpin.87HFEPR spectroscopy
HFEPR spectra were recorded on polycrystalline samples (20–25 mg), using a homodyne spectrometer at the EMR facility associated with a 15/17-T superconducting magnet and a frequency range from 52 to 426 GHz.88Detection was provided with an InSb hot electron bolometer (QMC Ltd, Cardiff, UK). The magnetic field was modulated at 50 kHz for detection purposes. A Stanford Research Systems SR830 lock-in amplifier converted the modulated signal to dc voltage. The single-frequency spectra as well as their dependencies on frequency were simulated with the SPIN software from A. Ozarowski.
High-resolution EPR spectroscopy at 9, 95, and 275 GHz
These EPR experiments were performed at Leiden University. For the measurements at 9 GHz (X-band) and 95 GHz (W-band) a Bruker Elexsys E680 spectrometer was used. The X-band spectra were obtained using 100 kHz field modulation, with an amplitude of 0.5 to 1.5 mT, depending on the line width. The W-band spectra were obtained at a modulation frequency of 90 kHz, with an amplitude of 1 mT.The X-band spectra of polycrystalline powders were recorded with the sample in the superconducting magnet, belonging to the W-band part of the spectrometer, in combination with the X-band bridge for microwave generation and detection. In this way, X-band spectra have been acquired up to fields of 3 T. Use was made of the dielectric resonator (Flexline, frequency about 9.7 GHz). The probe-head support was modified to position the resonator active volume in the center of the magnet homogeneity zone and to fit into the cryostat of the W-band probe-head.
Temperature calibration was performed as follows: a Lake Shore Carbon-Glass™ sensor (CGR-1 series) was calibrated in a commercial Quantum Design physical properties measurement system (PPMS) and subsequently placed at the sample position in the cryostat. The Oxford temperature unit was then calibrated against this sensor.
The 275 GHz spectrum of polycrystalline powder was measured on a homebuilt spectrometer, using 1.2 kHz field modulation of about 0.2 mT amplitude. In order to avoid self-orientation of the micro-crystals in the high magnetic field (up to 12 T), the powder was pressed into the sample tube using a tiny amount of cotton.
AC susceptometry
Alternating current magnetic susceptibility analyses were performed with a PPMS (Physical Properties Measurement System) platform, also from Quantum Design, with oscillating field frequencies ranging from 10 to 104 Hz, and using static magnetic fields of zero and 0.1 T. The resulting magnetic data were corrected for the diamagnetic contributions of the ligands calculated from Pascal constants,89 together with those measured for the sample container and the wrapping Teflon tape.The isothermal frequency dependence of the out-of-phase magnetic susceptibility (χ′′) data were analyzed within the extended Debye model,90,91 in which a maximum in the out-of-phase component χM′′ of the complex susceptibility is observed when the relaxation time τ equals (2πν)−1:
| χM′′ (ω) = (χT − χS)[(ωτ)1−αcos(απ/2)]/[1 + 2(ωτ)1−αsin(απ/2) + (ωτ)2−2α |
The temperature dependence of the magnetic relaxation times τ has been fitted using the following equation:
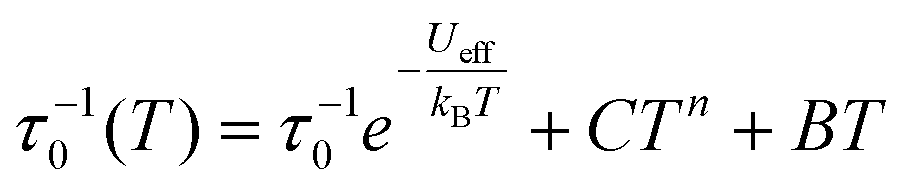
Computational studies
The crystal structure of the CoO2E2, E = S,71 Se,72 complexes was calculated using the CASSCF/NEVPT2 methodologies as implemented in ORCA 4.2.1 suit of codes.92 Additionally, the complexes were optimized energetically optimized using the B3LYP93,94 functional, in conjunction with the 6-31G(d,p)95 and def2-SVP96 basis sets. Additionally, for these optimized structures single point calculations were carried out at the CASSCF/NEVPT2 level of theory. The scalar relativistic Hamiltonian was considered using the ZORA (zeroth-order regular approximation) method.97 The ZORA contracted versions of basis sets ZORA-def2-TZVP were used for Co, Se, S, O and ZORA-def2-SVP(-f) for P, N, C and H.96 For the SA-CASSCF (state-average complete active space self-consistent field) calculations, an active space of seven electrons in five 3d-orbitals, i.e., CAS(7,5) was used; the orbitals were optimized for 10 quartet and 40 doublet roots. Additional calculations were also carried out where the orbitals were optimized for 7 quartets and 14 doublets. Furthermore, the NEVPT2 (N-electron valence perturbation theory 2nd order) methodology was used which incorporates the dynamic correlation additionally to the static of the CASSCF methodology. The SOC was calculated using the spin–orbit mean field SOMF(1×) method,98 while the spin-state mixing was obtained by employing the quasi-degenerate perturbation theory (QDPT) method.99 The spin Hamiltonian properties (zfs parameter D, g-factors) were computed using the effective Hamiltonian approach (HEFF).84 Finally, the contribution of SSC and SOC to the magnitude of D was calculated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Special Account for Research Grants of the National and Kapodistrian University of Athens (P. K.), FAPERJ through grants E-26/202.912/2019, SEI-260003/001167/2020 and E-26/010.000978/2019 (G. P.), and The Netherlands Organization for Scientific Research (NWO) (E. J. J. G) for financial support. Part of this work was performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement No. DMR-1644779 and the State of Florida. E. F. has been awarded a State Scholarship Foundation (IKY) fellowship, by the act “Support for Postdoctoral Researchers”, from the operational program “Human Power Development, Education and Lifelong Learning” (6, 8, 9 priority axes), co-funded by the European Social Fund and the Greek State. K. K.-L. acknowledges the National Science Centre NCN, Poland (ETIUDA 6, no. 2018/28/T/ST5/00120) for financial support during her internship at NHMFL. The Fulbright Foundation in Greece and the NHMFL via its Visiting Scientist Program supported a scientific extended visit of P. K. to the latter. Dr A. Ozarowski (NHMFL) is acknowledged for his EPR fit and simulation software SPIN. We would like to thank the Reviewers of our manuscript for their very constructive comments.References
- A. Brueckner, Chem. Soc. Rev., 2010, 39, 4673–4684 RSC.
- S. Van Doorslaer and D. M. Murphy, EPR Spectroscopy: Applications in Chemistry and Biology, ed. M. Drescher and G. Jeschke, 2012, vol. 321, pp. 1–39 Search PubMed.
- J. Telser, J. Krzystek and A. Ozarowski, J. Biol. Inorg. Chem., 2014, 19, 297–318 CrossRef CAS PubMed.
- K. R. Dunbar, Inorg. Chem., 2012, 51, 12055–12058 CrossRef CAS.
- J. Ferrando-Soria, J. Vallejo, M. Castellano, J. Martinez-Lillo, E. Pardo, J. Cano, I. Castro, F. Lloret, R. Ruiz-Garcia and M. Julve, Coord. Chem. Rev., 2017, 339, 17–103 CrossRef CAS.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- C. J. Milios and R. E. P. Winpenny, Molecular Nanomagnets and Related Phenomena, ed. S. Gao, 2015, vol. 164, pp. 1–109 Search PubMed.
- F. Neese and D. A. Pantazis, Faraday Discuss., 2010, 148, 229–238 RSC.
- S. Gomez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289, 379–392 CrossRef.
- E. Ruiz, J. Cirera, J. Cano, S. Alvarez, C. Loose and J. Kortus, Chem. Commun., 2008, 52–54 RSC.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- M. Feng and M. L. Tong, Chem. – Eur. J., 2018, 24, 7574–7594 CrossRef CAS PubMed.
- Y. S. Meng, S. D. Jiang, B. W. Wang and S. Gao, Acc. Chem. Res., 2016, 49, 2381–2389 CrossRef CAS PubMed.
- A. Zabala-Lekuona, J. M. Seco and E. Colacio, Coord. Chem. Rev., 2021, 441, 213984 CrossRef CAS.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- B. M. Day, F. S. Guo and R. A. Layfield, Acc. Chem. Res., 2018, 51, 1880–1889 CrossRef CAS PubMed.
- Z. H. Zhu, M. Guo, X. L. Li and J. K. Tang, Coord. Chem. Rev., 2019, 378, 350–364 CrossRef CAS.
- K. L. M. Harriman, D. Errulat and M. Murugesu, Trends Chem., 2019, 1, 425–439 CrossRef CAS.
- C. A. P. Goodwin, F. Ortu and D. Reta, Int. J. Quantum Chem., 2020, 120, e26248 CrossRef CAS.
- T. G. Ashebr, H. Li, X. Ying, X. L. Li, C. Zhao, S. T. Liu and J. K. Tang, ACS Mater. Lett., 2022, 4, 307–319 CrossRef CAS.
- K. R. Meihaus and J. R. Long, Dalton Trans., 2015, 44, 2517–2528 RSC.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- P. C. E. Stamp and A. Gaita-Arino, J. Mater. Chem., 2009, 19, 1718–1730 RSC.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamaki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492–8503 RSC.
- F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamaki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E. S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS.
- A. Sarkar, S. Dey and G. Rajaraman, Chem. – Eur. J., 2020, 26, 14036–14058 CrossRef CAS PubMed.
- M. Feng, Z. Y. Ruan, Y. C. Chen and M. L. Tong, Chem. Commun., 2020, 56, 13702–13718 RSC.
- P. S. Perlepe, D. Maniaki, E. Pilichos, E. Katsoulakou and S. P. Perlepes, Inorganics, 2020, 8, 39 CrossRef CAS.
- R. Boča and C. Rajnak, Coord. Chem. Rev., 2021, 430, 213657 CrossRef.
- C. Rajnak and R. Boča, Coord. Chem. Rev., 2021, 436, 213808 CrossRef CAS.
- J. P. Sutter, V. Bereau, V. Jubault, K. Bretosh, C. Pichon and C. Duhayon, Chem. Soc. Rev., 2022, 51, 3280–3313 RSC.
- A. K. Bar, C. Pichon and J. P. Sutter, Coord. Chem. Rev., 2016, 308, 346–380 CrossRef CAS.
- M. Atanasov, D. Aravena, E. Suturina, E. Bill, D. Maganas and F. Neese, Coord. Chem. Rev., 2015, 279, 177–214 CrossRef.
- G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- R. Boča, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef.
- J. Krzystek and J. Telser, Dalton Trans., 2016, 45, 16751–16763 RSC.
- J. Telser, A. Ozarowski and J. Krzystek, Specialist Periodical Reviews of the Royal Chemical Society: Electron Paramagnetic Resonance, ed. B. C. Gilbert, D. M. Murphy and V. Chechik, 2012, 23, 209–263 Search PubMed.
- J. Krzystek, A. Ozarowski and J. Telser, Coord. Chem. Rev., 2006, 250, 2308–2324 CrossRef CAS.
- B. Cahier, M. Perfetti, G. Zakhia, D. Naoufal, F. El-Khatib, R. Guillot, E. Riviere, R. Sessoli, A. L. Barra, N. Guihery and T. Mallah, Chem. – Eur. J., 2017, 23, 3648–3657 CrossRef CAS PubMed.
- J. van Slageren, S. Vongtragool, B. Gorshunov, A. A. Mukhin, N. Karl, J. Krzystek, J. Telser, A. Muller, C. Sangregorio, D. Gatteschi and M. Dressel, Phys. Chem. Chem. Phys., 2003, 5, 3837–3843 RSC.
- P. M. Champion and A. J. Sievers, J. Chem. Phys., 1977, 66, 1819–1825 CrossRef CAS.
- S. D. Jiang, D. Maganas, N. Levesanos, E. Ferentinos, S. Haas, K. Thirunavukkuarasu, J. Krzystek, M. Dressel, L. Bogani, F. Neese and P. Kyritsis, J. Am. Chem. Soc., 2015, 137, 12923–12928 CrossRef CAS PubMed.
- M. A. Dunstan, R. A. Mole and C. Boskovic, Eur. J. Inorg. Chem., 2019, 1090–1105 CrossRef CAS.
- S. E. Stavretis, Y. Q. Cheng, L. L. Daemen, C. M. Brown, D. H. Moseley, E. Bill, M. Atanasov, A. J. Ramirez-Cuesta, F. Neese and Z. L. Xue, Eur. J. Inorg. Chem., 2019, 1119–1127 CrossRef CAS.
- L. Chen, H. H. Cui, S. E. Stavretis, S. C. Hunter, Y. Q. Zhang, X. T. Chen, Y. C. Sun, Z. X. Wang, Y. Song, A. A. Podlesnyak, Z. W. Ouyang and Z. L. Xue, Inorg. Chem., 2016, 55, 12603–12617 CrossRef CAS PubMed.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S. K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y. S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–349 CrossRef CAS PubMed.
- S. Ostrovsky, Z. Tomkowicz, S. Foro, J. Reedijk and W. Haase, Polyhedron, 2020, 187, 114630 CrossRef CAS.
- J. Krzystek, S. Zvyagin, A. Ozarowski, A. T. Fiedler, T. C. Brunold and J. Telser, J. Am. Chem. Soc., 2004, 126, 2148–2155 CrossRef CAS PubMed.
- B. M. Hakey, D. C. Leary, J. Xiong, C. F. Harris, J. M. Darmon, J. L. Petersen, J. F. Berry, Y. S. Guo and C. Milsmann, Inorg. Chem., 2021, 60, 18575–18588 CrossRef CAS PubMed.
- S. A. Stoian, M. Moshari, E. Ferentinos, A. Grigoropoulos, J. Krzystek, J. Telser and P. Kyritsis, Inorg. Chem., 2021, 60, 10990–11005 CrossRef CAS PubMed.
- M. E. Pascualini, N. V. Di Russo, A. E. Thuijs, A. Ozarowski, S. A. Stoian, K. A. Abboud, G. Christou and A. S. Veige, Chem. Sci., 2015, 6, 608–612 RSC.
- V. V. Vrajmasu, E. L. Bominaar, J. Meyer and E. Münck, Inorg. Chem., 2002, 41, 6358–6371 CrossRef CAS PubMed.
- P. Kumar, D. J. SantaLucia, K. Kaniewska-Laskowska, S. V. Lindeman, A. Ozarowski, J. Krzystek, M. Ozerov, J. Telser, J. F. Berry and A. T. Fiedler, Inorg. Chem., 2020, 59, 16178–16193 CrossRef CAS PubMed.
- A. A. Pavlov, D. Y. Aleshin, S. A. Savkina, A. S. Belov, N. N. Efimov, J. Nehrkorn, M. Ozerov, Y. Z. Voloshin, Y. V. Nelyubina and V. V. Novikov, ChemPhysChem., 2019, 20, 1001–1005 CrossRef CAS PubMed.
- A. N. Bone, C. N. Widener, D. H. Moseley, Z. M. Liu, Z. G. Lu, Y. Q. Cheng, L. L. Daemen, M. Ozerov, J. Telser, K. Thirunavukkuarasu, D. Smirnov, S. M. Greer, S. Hill, J. Krzystek, K. Holldack, A. Aliabadi, A. Schnegg, K. R. Dunbar and Z. L. Xue, Chem. – Eur. J., 2021, 27, 11110–11125 CrossRef CAS PubMed.
- J. Vallejo, M. Viciano-Chumillas, F. Lloret, M. Julve, I. Castro, J. Krzystek, M. Ozerov, D. Armentano, G. De Munno and J. Cano, Inorg. Chem., 2019, 58, 15726–15740 CrossRef CAS PubMed.
- E. Y. Misochko, A. V. Akimov, D. V. Korchagin, J. Nehrkorn, M. Ozerov, A. V. Palii, J. M. Clemente-Juan and S. M. Aldoshin, Inorg. Chem., 2019, 58, 16434–16444 CrossRef CAS PubMed.
- A. Landart-Gereka, M. M. Quesada-Moreno, I. F. Díaz-Ortega, H. Nojiri, M. Ozerov, J. Krzystek, M. A. Palacios and E. Colacio, Inorg. Chem. Front., 2022, 9, 2810–2831 RSC.
- D. H. Moseley, S. E. Stavretis, K. Thirunavukkuarasu, M. Ozerov, Y. Q. Cheng, L. L. Daemen, J. Ludwig, Z. G. Lu, D. Smirnov, C. M. Brown, A. Pandey, A. J. Ramirez-Cuesta, A. C. Lamb, M. Atanasov, E. Bill, F. Neese and Z. L. Xue, Nat. Commun., 2018, 9, 2572 CrossRef PubMed.
- S. Tripathi, A. Dey, M. Shanmugam, R. S. Narayanan and V. Chandrasekhar, Organometallic Magnets, ed. V. Chandrasekhar and F. Pointillart, 2019, vol. 64, pp. 35–75 Search PubMed.
- (a) J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS PubMed; (b) E. A. Suturina, J. Nehrkorn, J. M. Zadrozny, J. Liu, M. Atanasov, T. Weyhermuller, D. Maganas, S. Hill, A. Schnegg, E. Bill, J. R. Long and F. Neese, Inorg. Chem., 2017, 56, 3102–3118 CrossRef CAS PubMed.
- J. M. Zadrozny, J. Telser and J. R. Long, Polyhedron, 2013, 64, 209–217 CrossRef CAS.
- Y. Rechkemmer, F. D. Breitgoff, M. van der Meer, M. Atanasov, M. Hakl, M. Orlita, P. Neugebauer, F. Neese, B. Sarkar and J. van Slageren, Nat. Commun., 2016, 7, 10467 CrossRef CAS PubMed.
- X. N. Yao, J. Z. Du, Y. Q. Zhang, X. B. Leng, M. W. Yang, S. D. Jiang, Z. X. Wang, Z. W. Ouyang, L. Deng, B. W. Wang and S. Gao, J. Am. Chem. Soc., 2017, 139, 373–380 CrossRef CAS PubMed.
- P. C. Bunting, M. Atanasov, E. Damgaard-Moller, M. Perfetti, I. Crassee, M. Orlita, J. Overgaard, J. van Slageren, F. Neese and J. R. Long, Science, 2018, 362, eaat7319 CrossRef CAS PubMed.
- M. C. Aragoni, M. Arca, M. B. Carrea, A. Garau, F. A. Devillanova, F. Isaia, V. Lippolis, G. L. Abbati, F. Demartin, C. Silvestru, S. Demeshko and F. Meyer, Eur. J. Inorg. Chem., 2007, 4607–4614 CrossRef CAS.
- E. Ferentinos, D. Maganas, C. P. Raptopoulou, A. Terzis, V. Psycharis, N. Robertson and P. Kyritsis, Dalton Trans., 2011, 40, 169–180 RSC.
- C. Silvestru and J. E. Drake, Coord. Chem. Rev., 2001, 223, 117–216 CrossRef CAS.
- S. Sottini, G. Poneti, S. Ciattini, N. Levesanos, E. Ferentinos, J. Krzystek, L. Sorace and P. Kyritsis, Inorg. Chem., 2016, 55, 9537–9548 CrossRef CAS PubMed.
- G. Mathies, S. D. Chatziefthimiou, D. Maganas, Y. Sanakis, S. Sottini, P. Kyritsis and E. J. J. Groenen, J. Magn. Reson., 2012, 224, 94–100 CrossRef CAS PubMed.
- R. L. Carlin, Magnetochemistry, Springer, Berlin, Heidelberg, 1986 Search PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- E. Ferentinos, S. Chatziefthimiou, A. K. Boudalis, M. Pissas, G. Mathies, P. Gast, E. J. J. Groenen, Y. Sanakis and P. Kyritsis, Eur. J. Inorg.Chem., 2018, 713–721 CrossRef CAS.
- D. Maganas, J. Krzystek, E. Ferentinos, A. M. Whyte, N. Robertson, V. Psycharis, A. Terzis, F. Neese and P. Kyritsis, Inorg. Chem., 2012, 51, 7218–7231 CrossRef CAS PubMed.
- E. Ferentinos, C. P. Raptopoulou, V. Psycharis, A. Terzis, J. Krzystek and P. Kyritsis, Polyhedron, 2018, 151, 177–184 CrossRef CAS.
- D. Maganas, S. Milikisyants, J. M. A. Rijnbeek, S. Sottini, N. Levesanos, P. Kyritsis and E. J. J. Groenen, Inorg. Chem., 2010, 49, 595–605 CrossRef CAS PubMed.
- E. Ferentinos, A. B. Tsoupras, M. Roulia, S. D. Chatziefthimiou, C. A. Demopoulos and P. Kyritsis, Inorg. Chim. Acta, 2011, 378, 102–108 CrossRef CAS.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Oxford University Press, Oxford, U.K., 2012 Search PubMed.
- R. Maurice, R. Bastardis, C. de Graaf, N. Suaud, T. Mallah and N. Guihery, J. Chem. Theory Comput., 2009, 5, 2977–2984 CrossRef CAS PubMed.
- A. Sarkar, S. Tewary, S. Sinkar and G. Rajaraman, Chem. – Asian J., 2019, 14, 4696–4704 CrossRef CAS PubMed.
- L. M. Gilby and B. Piggott, Polyhedron, 1999, 18, 1077–1082 CrossRef CAS.
- J. Nehrkorn, J. Telser, K. Holldack, S. Stoll and A. Schnegg, J. Phys. Chem. B, 2015, 119, 13816–13824 CrossRef CAS PubMed.
- A. K. Hassan, L. A. Pardi, J. Krzystek, A. Sienkiewicz, P. Goy, M. Rohrer and L. C. Brunel, J. Magn. Reson., 2000, 142, 300–312 CrossRef CAS PubMed.
- G. A. Bain and J. F. J. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- C. Dekker, A. F. M. Arts, H. W. Dewijn, A. J. Vanduyneveldt and J. A. Mydosh, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 11243–11251 CrossRef CAS PubMed.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- L. A. Curtiss, M. P. McGrath, J. P. Blaudeau, N. E. Davis, R. C. Binning and L. Radom, J. Chem. Phys., 1995, 103, 6104–6113 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3330 RSC.
- C. V. Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- H. Nakano, J. Chem. Phys., 1993, 99, 7983–7992 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt03335f |
| This journal is © The Royal Society of Chemistry 2023 |