
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and computational investigation of heteroatom substitution in nucleolytic Cu(II) cyclen complexes for balancing stability and redox activity†
Jan
Hormann
a,
Olga
Verbitsky
b,
Xiaoyu
Zhou
c,
Beatrice
Battistella
ad,
Margarete
van der Meer
a,
Biprajit
Sarkar
 ae,
Cunyuan
Zhao
*c and
Nora
Kulak
*ab
ae,
Cunyuan
Zhao
*c and
Nora
Kulak
*ab
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany
bInstitut für Chemie, Otto-von-Guericke-Universität, Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: nora.kulak@ovgu.de
cMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, XinGang Rd. W., Guangzhou 510275, China. E-mail: ceszhcy@mail.sysu.edu.cn
dDepartment of Chemistry, Humboldt Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
eInstitut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
First published on 16th January 2023
Abstract
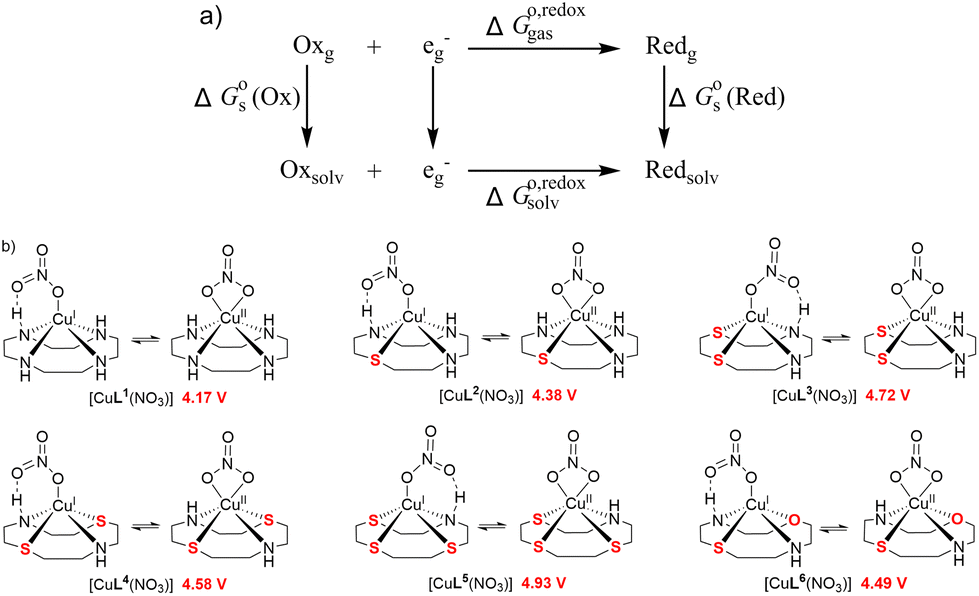
Cu(II) complexes of cyclen-based ligands CuL1–CuL6 were synthesized and characterized. The corresponding ligands L1–L6 comprise different donor sets including S and O atoms. Whereas cyclen (L1) is commercially available, L2–L6 were synthesized according to protocols available in the literature. Cleavage activity of the complexes towards plasmid DNA was tested in the presence and absence of ascorbate as a reducing agent (oxidative vs. hydrolytic cleavage). As previously shown, the substitution of N donor atoms with hard donor O atoms leads to efficient oxidative nucleases, but dissociation of the complex upon reduction. We thus opted for S substitution (soft donors) to stabilize the reduced Cu(I) species. Increasing the S content, however, leads to species that are difficult to reoxidize in order to ensure efficient oxidative DNA cleavage. We are showing by experimental (cyclic voltammetry) and computational means (DFT) that the rational combination of O and S atoms next to two nitrogen donors within the macrocycle (oxathiacyclen complex CuL6) leads to the stabilization of both redox states. The complex thus exhibits the highest oxidative DNA cleavage activity within this family of cyclen-based Cu(II) complexes – without leaching of the metal ion during reduction.
Introduction
Metal complexes of cyclen (1,4,7,10-tetraazacyclododecane) and of the smaller analog 1,4,7-triazacyclononane have been successfully applied in the hydrolytic cleavage of DNA and RNA, with the aim of developing agents for biotechnological purposes.1–5 The oxidative cleavage of DNA by such macrocyclic copper complexes has been investigated less extensively,6–8 despite their potential use as anticancer agents. In cell studies various nucleolytic copper complexes have shown high activity against cancer cells.9–11There are several approaches to increase the cleavage activity of cyclen metal complexes towards nucleic acids: multinuclear complexes,12–14 attachment of nucleobase-affine moieties15 or positively charged groups for better interaction with the phosphate backbone.16
Whereas the 4N donor ligand cyclen has been frequently applied in such studies, heteroatom-substituted analogs are less common. This might be due to difficult synthetic accessibility to the derivatives, whereas cyclen itself is commercially available. By using analogous precursor molecules, however, their syntheses are equally straightforward.
Older and recent examples of macrocyclic ligands with different heteroatoms have focused on the larger analog cyclam (1,4,8,11-tetraazatetradecane), e.g. the Cu(I) and Cu(II) complexes of the trans-dithiacyclam derivative have been suggested as analogs of blue copper proteins and copper monooxygenases,17–19 and its Cu(II) complex with two molecules of diclofenac, a nonsteroidal anti-inflammatory drug (NSAID) at the axial positions, has been used to generate reactive oxygen species (ROS) for killing breast cancer cells and highly resistant breast cancer stem cells.20
In the last couple of years, we have systematically investigated the coordination chemistry and electrochemical properties of Cu(II) cyclen derivatives substituted with one sulfur or one and more oxygen donors.6,7 To find out if exchanging nitrogen donors in Cu(II) cyclen [Cu([12]aneN4)] (CuL1) with several sulfur atoms also had an effect on nuclease activity, CuL2–CuL5 were prepared and their DNA cleavage activity was compared to that of CuL1. Additionally, CuL6 having an O donor as well as an S donor merged in the macrocyclic ligand L6, was synthesized (Fig. 1). Based on our previous findings, an heteroatom exchange N → O/S was expected to have a significant impact on the redox potential and DNA cleavage activity of the respective complexes.6,7
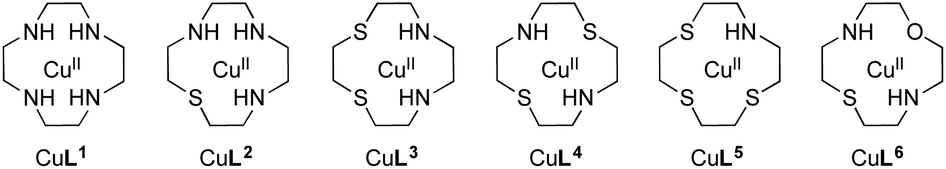 | ||
| Fig. 1 Cu(II) complexes containing the macrocyclic ligands L1–L6 studied in the present work. | ||
We studied the redox chemistry, oxidative and hydrolytic DNA cleavage activity of the Cu(II) complexes CuL1–CuL6 and rationalized their nucleolytic activity with computational results by DFT.
Results and discussion
Synthesis and characterization of ligands and complexes
Ligand L4, 1,7-dithia-4,10-diazacyclododecane or [12]aneNSNS, was synthesized according to the literature22 in a four-step procedure over a dilactame (ESI, S-1†): In contrast to the synthesis of the regioisomeric compound L3 ([12]aneN2S2), where a dichlorinated amide and a dithiol were used to form the dilactame, cyclization happened in the case of L4 by reaction of thioethers of an acid chloride and a diamine. The amine was used in excess in order to trap the released hydrogen chloride. Reduction of the dilactame lead to the desired compound.
Ligand L5, 1,4,7-trithia-10-azacyclododecane or [12]aneNS3, was synthesized according to the same protocol21 as L3 (ESI, S-1†): Therefore, 2,2′-thiodiethanethiol was cyclized with a chlorinated diamine under high-dilution conditions in the presence of cesium carbonate.
Ligand L6, 1-oxa-7-thia-4,10-diazacyclododecane, shortly [12]aneNONS or oxathiacyclen, was synthesized according to a procedure published by Hambley et al. comprising the cyclization of tosylated precursors23 with slight modifications (ESI, S-1†).
The synthesis of the Cu(II) complex of the commercially available cyclic thioether 1,4,7,10-tetrathiacyclododecane was tried by mixing the methanolic solutions of ligand and copper(II) nitrate. However, elemental analysis of the obtained light-blue product revealed the formation of a Cu(I) species of unknown composition. As shown in Table S-4 (ESI†), the stability constant (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K) of the Cu(II) complex is 3.4, whereas the stability constant of the corresponding Cu(I) species is 12.9.24 Since all other complexes were used with copper in oxidation state +II, no attempt was made to further characterize this Cu(I) species. Indeed, it has been reported in the literature that N2S2 donor sets in macrocyclic ligands (here, cis-/trans-dithiacyclen) can accommodate the cupric as well as cuprous state, whereas the former is preferred for a N4 donor set, and the latter for an S4 donor set.17
K) of the Cu(II) complex is 3.4, whereas the stability constant of the corresponding Cu(I) species is 12.9.24 Since all other complexes were used with copper in oxidation state +II, no attempt was made to further characterize this Cu(I) species. Indeed, it has been reported in the literature that N2S2 donor sets in macrocyclic ligands (here, cis-/trans-dithiacyclen) can accommodate the cupric as well as cuprous state, whereas the former is preferred for a N4 donor set, and the latter for an S4 donor set.17
Complexes CuL1–CuL6 were characterized by elemental analysis, UV/VIS, IR and EPR spectroscopy as well as ESI mass spectrometry (ESI, S-1 and S-2†).
In Fig. 2, the molecular structure of complex CuL3 in the solid state is shown. The crystallographic data of the complex can be found in the ESI (S-3),† whereas selected bond lengths and angles can be found in Table 1.
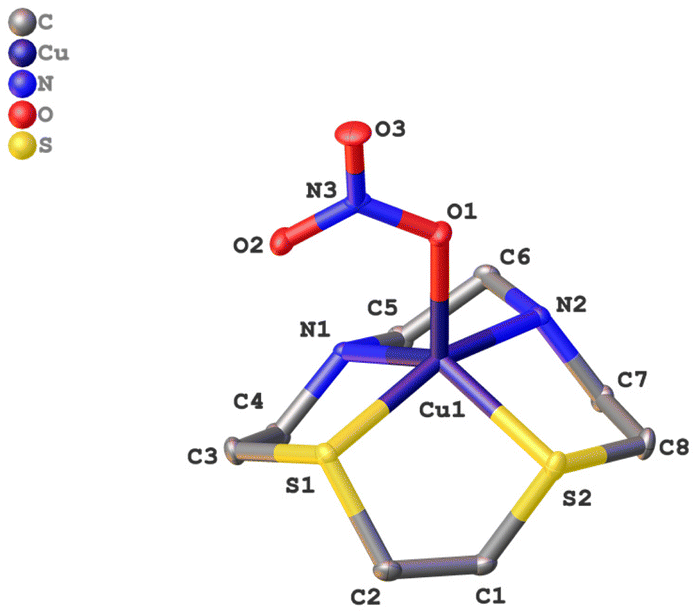 | ||
| Fig. 2 Molecular structure of [CuL3(NO3)]NO3 in the solid state. For clarity reasons the hydrogen atoms and the nitrate counterion have been omitted. | ||
| Bond length [Å] | Bond angle [°] | ||
|---|---|---|---|
| Cu(1)–O(1) | 2.105 | S(1)–Cu(1)–O(1) | 101.396 |
| Cu(1)–S(2) | 2.353 | S(2)–Cu(1)–O(1) | 108.179 |
| Cu(1)–S(1) | 2.349 | N(1)–Cu(1)–O(1) | 113.141 |
| Cu(1)–N(1) | 2.025 | N(2)–Cu(1)–(O1) | 97.379 |
| Cu(1)–N(2) | 2.007 | S(1)–Cu(1)–S(2) | 85.740 |
| C(1)–C(2) | 1.525 | S(2)–Cu(1)–N(1) | 138.655 |
| O(1)–N(3) | 1.285 | S(1)–Cu(1)–N(2) | 161.194 |
| N(1)–Cu(1)–N(2) | 86.051 |
Complex CuL3, like CuL1 and CuL2,6,28 consists of a [CuLNO3]+ cation and a non-coordinating nitrate anion. Due to the small ring size of the cyclen derivatives, a square planar coordination is not possible – in contrast to the cyclam analogs,17,18 but complexes CuL1–CuL3 rather exhibit a distorted square pyramidal coordination. The base of the pyramid is provided by the macrocyclic heteroatoms and the edge of the pyramid by an oxygen atom of the nitrate ligand. The Cu–N bond length is approximately 2 Å (as in CuL1 and CuL2), whereas the Cu–S bond lengths are elongated (ca. 2.3 Å) due to the larger covalent radius of sulfur (this corresponds to the value observed for CuL2).6 The nitrate ligand coordinates in a η1 fashion as it does in complexes CuL1 and CuL2, and as suggested by IR spectroscopy (vide supra). In CuL3, the bond length between the Cu2+ ion and O(1), the top of the pyramid, is 2.11 Å, and it is little larger for CuL2 (2.16) and CuL1 (2.18). Thus, the structures of the cyclen complex CuL1 and its analogs with one and two sulfur donors, respectively, are very similar. Any effects on the reactivity based on the solid state structures can thus be excluded for these three complexes.
DNA cleavage
 | ||
| Fig. 3 Cleavage activities of complexes CuL1–CuL6 and Cu(NO3)2 (0.4 mM) on pBR322 plasmid DNA (0.025 μg μL−1) in Tris-HCl buffer (50 mM, pH 7.4) at 37 °C for 24 h. Illustrated is the average of three experiments, the standard deviations are shown as error bars. | ||
The hydrolysis of the phosphate ester bonds in the small DNA model molecule BNPP (bis(4-nitrophenyl)phosphate) was investigated for comparison, here exemplarily with CuL2 and CuL6 (Fig. 4). The release of the hydrolysis product p-nitrophenolate was detected by UV/VIS spectroscopy for the latter compound but not for the former, even after 8 d incubation. Even CuL6 showed poor reactivity, since the cleavage product was only observed in the spectrum after 48 h (ESI, Fig. S-6.1†), and doubling the concentration (2 mM instead of 1 mM) did not show a significant effect (ESI, Fig. S-6.2†).
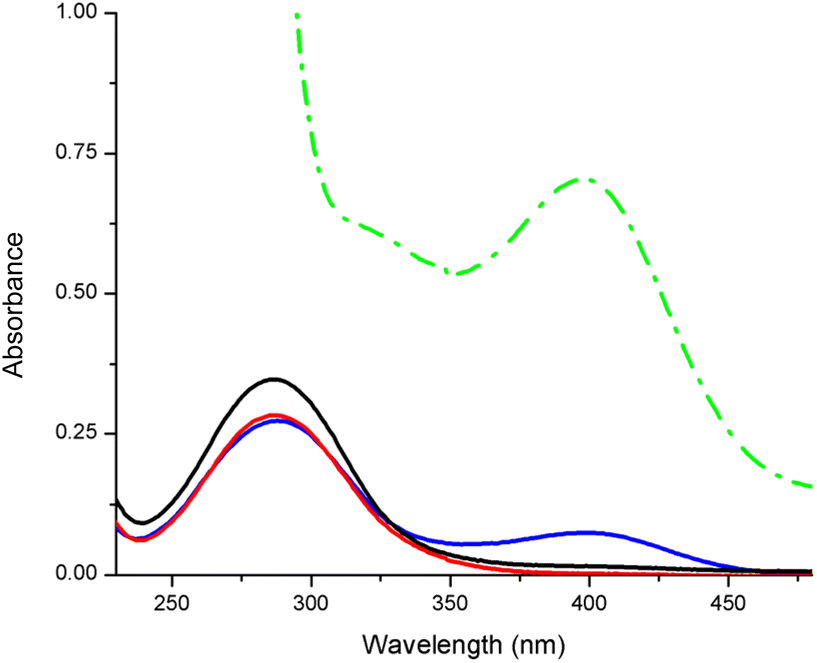 | ||
| Fig. 4 UV/Vis spectra of BNPP (0.08 mM) in the absence (black line) and presence of complexes CuL2 (1 mM, red line), CuL6 (1 mM, blue line) and phosphodiesterase (0.05 U, green line), respectively, in 50 mM Tris-HCl buffer (pH 7.5) after an incubation of 8 d (only 2 d for phosphodiesterase) at 37 °C for monitoring the formation of the BNPP cleavage product p-nitrophenol (λmax = 400 nm). The absorbance of the complexes was subtracted. | ||
 | ||
| Fig. 5 Cleavage activities of complexes CuL1–CuL5 (0.04 mM) on pBR322 plasmid DNA (0.025 μg μL−1) in Tris-HCl buffer (50 mM, pH 7.4) and ascorbic acid (0.32 mM) at 37 °C for 2 h. Illustrated is the average of three experiments with standard deviations shown as error bars (top) and a representative agarose gel (bottom). | ||
A comparison of CuL1, CuL4 and CuL6, i.e. the complexes of the parent compound cyclen, the trans-dithiacyclen and trans-oxathiacyclen, with CuL7 (L7 = trans-dioxacyclen7), is shown in Fig. 6. The order in the diagram is chosen such that the oxygen content in the macrocycle is decreasing from left to the right (dioxacyclen > oxathiacyclen > dithiacyclen), also corresponding to a decrease in DNA cleavage activity. The dioxacyclen complex CuL7 generated up to 80% form III DNA, whereas dithiacyclen CuL4, the most active species among the sulfur-containing cyclen analogs (Fig. 5), only generated 10% form III DNA.§ CuL6, representing a hybrid of the oxacyclen7 and thiacyclen series, concordantly exhibits cleavage activity between that of the dioxa and the dithia analog.
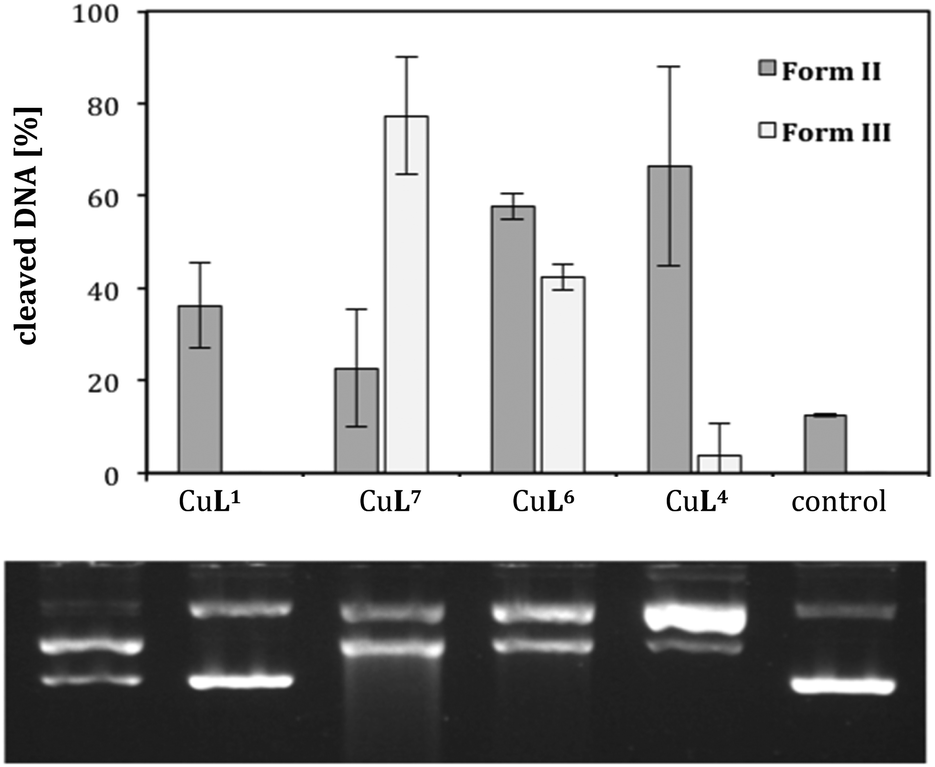 | ||
| Fig. 6 Cleavage activities of complexes CuL1, CuL4, CuL6 and CuL7 (L7 = 1,7-dioxacyclen)7 (0.04 mM) on pBR322 plasmid DNA (0.025 μg μL−1) in Tris-HCl buffer (50 mM, pH 7.4) and ascorbic acid (0.32 mM) at 37 °C for 2 h. Illustrated is the average of three experiments with standard deviations shown as error bars (top) and a representative agarose gel (bottom). | ||
Furthermore, it should be mentioned that, despite the employed Tris buffer is a potential competitive ligand for Cu(II), the stability constants of the complexes under study are higher than the one for the Cu(II)-tris system29 (5.3–6.3 vs. >8, cf. ESI, S-4†). Nevertheless, the large excess of Tris (approx. 100-fold and 1000-fold in the hydrolytic and oxidative cleavage reactions, respectively) in the experiments suggests that the macrocyclic ligands and Tris could compete for Cu(II) and/or form ternary complexes. Experiments with a non-interfering buffer, MOPS (ESI, Fig. S-5.1 and S-5.2,† direct comparison TRIS/MOPS) gave comparable results, and thus indicate that DNA cleavage is not initiated by other Cu(II) species than CuL1–CuL6. As expected from the Pearson's principle, the stability of the Cu(II) complexes decreases with increasing sulfur content of the macrocycle, whereas the stability of the respective Cu(I) complexes increases (ESI, Table S-4†). Since an efficient switching of the oxidation state is decisive for oxidative DNA cleavage, it is required for both the Cu(II) and the Cu(I) species to be sufficiently stable. This is indeed the case for CuL3 and CuL4.
Investigation of redox chemistry
As shown exemplarily for complex CuL5 in Fig. 7, pyruvate (Pyr.) and DMSO exert a significant inhibition effect on DNA cleavage. This is the case also for most of the other complexes (ESI, Table S-5 and Fig. S-5.3–S-5.8†), suggesting the presence of peroxo species and hydroxyl radicals during DNA cleavage (except for CuL1, with very low cleavage activity anyway). An oxidative cleavage mechanism can thus be assumed, where the detected ROS attack the DNA base or ribose moieties and cause degradation of this biomolecule.34
 | ||
| Fig. 7 Cleavage activities of complex CuL5 (0.04 mM) on pBR322 plasmid DNA (0.025 μg μL−1) in Tris-HCl buffer (50 mM, pH 7.4), PBS (1.25×) and ascorbic acid (0.32 mM) at 37 °C for 2 h in the presence of the indicated ROS scavengers. Illustrated is the average of three experiments, the standard deviations are shown as error bars (top) and a representative agarose gel (bottom). | ||
In order to exclude, that potential interactions between Cu(II) and the quencher molecules have an impact on the results, ROS were additionally detected via a fluorescence assay. Herein, fluorogenic compounds, terepthtalate (TPA) and pentafluorobenzenesulfonyl fluorescein (PBSF), get activated in the presence of hydroxyl radicals and hydrogen peroxide,35,36 respectively (ESI, Fig. S-5.9 and S-5.10,† exemplarily for CuL5). Both ROS were detected, corroborating the results of the quenching studies using agarose gel electrophoresis.
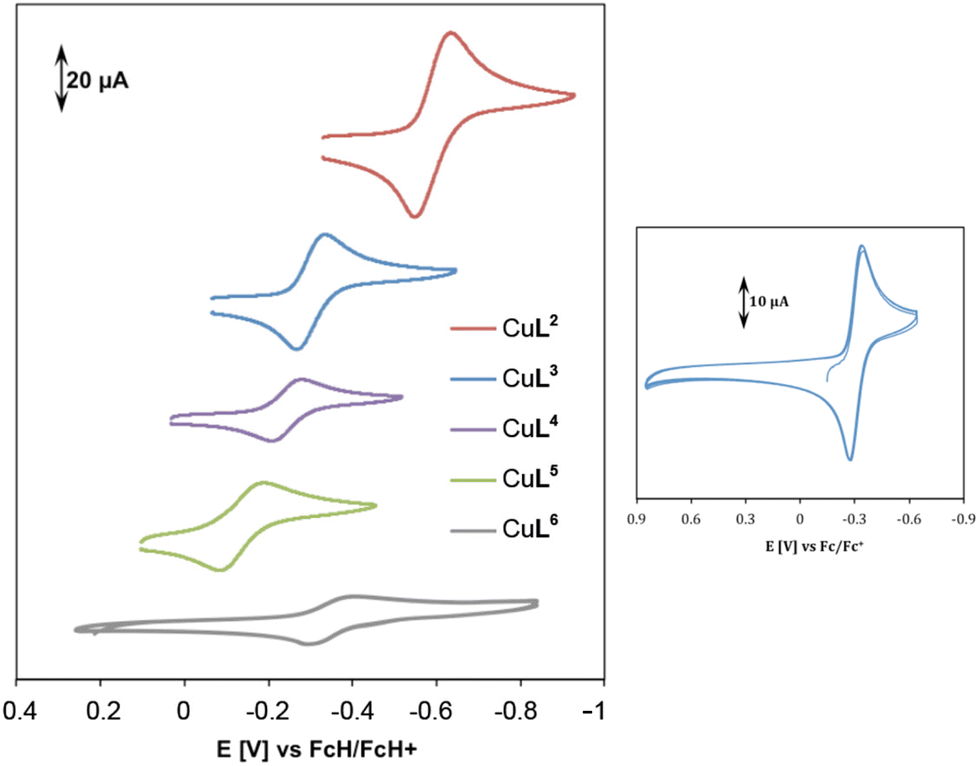 | ||
| Fig. 8 Cyclic voltammetry of complexes CuL2–CuL6 in 0.1 M KCl solution at 100 mV s−1 and room temperature (left). Multiple scanning for complex CuL3 at the same conditions (right). | ||
| E 1/2 [V] | |
|---|---|
| CuL2 | −0.60 |
| CuL3 | −0.31 |
| CuL4 | −0.23 |
| CuL5 | −0.14 |
| CuL6 | −0.35 |
The irreversibility of reduction might be due to the low stability of Cu(I) in cyclen and oxacyclen complexes, whereas a high stability of CuL2–CuL5 in their +I and +II oxidation states is assumed (cf. ESI, Table S-4†).
The cis-dithiacyclen complex CuL3 showing more negative E1/2 indicates higher stability for oxidation state +II in comparison to trans-dithiacyclen complex CuL4, which is reflected in the higher oxidative DNA cleavage activity of the latter compound (Fig. 5), i.e. easier formation of ROS due to Cu(I) generation, but also in the stability constants for the Cu(II) species (ESI, Table S-4,† logK(CuL3) ≈ 14 vs. logK(CuL4) ≈ 12).17
E 1/2 is only −0.14 V vs. FcH/FcH+ for CuL5, thus it should be more easily reduced and a more efficient oxidative nuclease than CuL2–CuL4. Nevertheless, CuL5 was shown to be as active as CuL3. It can be assumed that the Cu(I) complex of L5 is more stable than the respective Cu(II) species (ESI, Table S-4†). The re-oxidation to a Cu(II) species might therefore be thermodynamically less favorable, rendering ROS generation less probable in the case of complex CuL5 in comparison to e.g. CuL4, where Cu(I) and Cu(II) species are equally stable (ESI, Table S-4†).
CuL6 exhibits a completely reversible reduction with a half-wave potential of −0.35 V (Table 2 and Fig. 8), an intermediate value with regard to ease of reduction. Concerning DNA cleavage activity, this complex is, however, much more active than CuL2–CuL5, which might be due to its low dimerization tendency in the cuprous state (vide infra, computational studies). Cu(II) oxathiacyclen CuL6 as a hybrid between Cu(II) trans-dithiacyclen CuL4 and Cu(II) trans-dioxacyclen CuL7, showed DNA cleavage activity which is in between the other two complexes (Fig. 6). Whereas the S atom promotes the Cu(I) stabilization, the O atom favors the Cu(II) oxidation state. The presence of both, S and O donor provides the right balance for Cu(II) reduction and Cu(I) reoxidation, facilitating the switch between the redox states Cu(II) and Cu(I), and thus ensuring efficient ROS generation.38 Consequently, CuL7, which shows the highest cleavage activity, comes with the disadvantage of not being able to stabilize Cu(I), so that copper ions can leach from the ligand and might be responsible for the DNA cleavage instead of the assumed complex. For the oxygen-containing macrocycles, as reported earlier,7 an increase in the number of oxygen atoms promotes metal reduction, but at the same time the affinity of the ligand to Cu(I) decreases. This problem was circumvented within this work by the use of the here presented thiacyclen and oxathiacyclen Cu(II) complexes.
Computational studies and comparison with experimental results
There are some theoretical studies concerning the cleavage of DNA or RNA in the literature by metal complexes,39–42 however, not touching on the here presented compounds.In order to better understand the chemical reactivity of CuL1 –CuL6 regarding hydrolytic and oxidative DNA cleavage, density functional theory (DFT) was used as a complementary method next to the experimental ones. The complexes were studied regarding their dimerization tendency and redox potential. The pKa values of metal-coordinated water molecules (M–OH2) were predicted as well were the reaction mechanisms of the formation of ROS and the BNPP hydrolysis theoretically investigated.
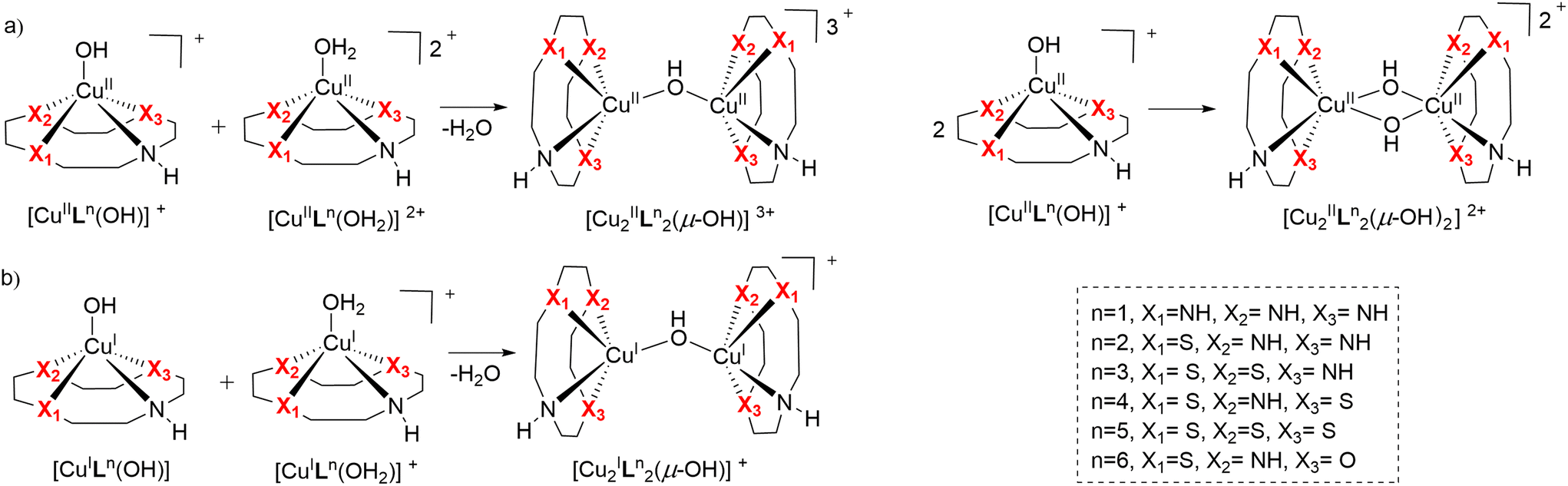 | ||
| Fig. 9 Dimerization schemes of (a) Cu(II)Ln complexes and (b) Cu(I)Ln complexes. | ||
It can be derived from Table 3 that the double-bridged μ-hydroxido complexes [CuII2Ln2(μ-OH)2]2+ (n = 1–6) are in general more stable than their mono-bridged counterparts (ΔGf< 0). The dimerization process is the easiest in case of Cu(II)L1 reflected in the most negative ΔGf. This indicates that the low reactivity of Cu(II)L1 in plasmid DNA degradation might be due to dimer formation.6 The most reactive complex, CuIIL6, concordantly exhibits the least negative value, indicating low stability of its dinuclear complex, and explaining its activity in the BNPP hydrolysis reaction (Fig. 4).
| Structure | [CuII2L12(μ-OH)]3+ | [CuII2L22(μ-OH)]3+ | [CuII2L32(μ-OH)]3+ | [CuII2L42(μ-OH)]3+ | [CuII2L52(μ-OH)]3+ | [CuII2L62(μ-OH)]3+ |
|---|---|---|---|---|---|---|
| ΔGf | −74.7 | −12.2 | 19.0 | 57.7 | −11.3 | −6.1 |
| Structure | [CuII2L12(μ-OH)2]2+ | [CuII2L22(μ-OH)2]2+ | [CuII2L32(μ-OH)2]2+ | [CuII2L42(μ-OH)2]2+ | [CuII2L52(μ-OH)2]2+ | [CuII2L62(μ-OH)2]2+ |
|---|---|---|---|---|---|---|
| ΔGf | −66.7 | −13.1 | −27.7 | −14.9 | −36.9 | −14.7 |
| Structure | [CuI2L12(μ-OH)]+ | [CuI2L22(μ-OH)]+ | [CuI2L32(μ-OH)]+ | [CuI2L42(μ-OH)]+ | [CuI2L52(μ-OH)]+ | [CuI2L62(μ-OH)]+ |
|---|---|---|---|---|---|---|
| ΔGf | −11.1 | −13.1 | −11.7 | −11.3 | −12.2 | −3.5 |
For CuILn (n = 1–5), the [CuI2Ln2(μ-OH)]+ complexes all have ΔGf values in the range of −11 to −13 kcal mol−1. These values are more negative than the ΔGf value of the [CuI2L62(μ-OH)]+ species, again suggesting the least stable dimer for the CuIL6 analog. This result is consistent with CuIIL6 being the most active oxidative cleaving agent within the series, since formation of CuIL6 and ROS is decisive for this process, which could be hindered by any dimerization side reaction.
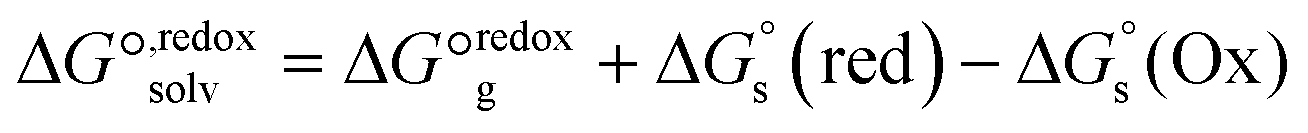 | (1) |
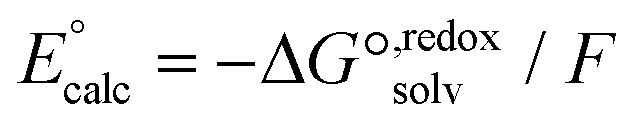 | (2) |
 | ||
| Fig. 10 (a) Thermodynamic Born–Haber cycle of the redox processes, (b) structural variations during the redox processes and corresponding redox potentials for CuI/IILn (in red). | ||
As demonstrated in Fig. 10b, the reduction potentials of the six complexes follow the order CuL5 > CuL3 > CuL4 > CuL6 > CuL2 > CuL1. The higher reduction potential of CuLn (n = 3–5) indicates easier reduction from the Cu(II) complexes to the Cu(I) analogs. This correlates well with the experimental electrochemical results, where CuL5 > CuL4 > CuL3 > CuL6 > CuL2 was found for the half-wave potentials, i.e. only CuL4 and CuL3 are switched, when the two rows are compared. As indicated for the cyclic voltammetric studies, the trends are reflected in the oxidative cleavage activity of the complexes with CuL6 representing an exception (highest cleavage activity despite of intermediate values for E1/2, however, low dimerization tendency, vide supra).
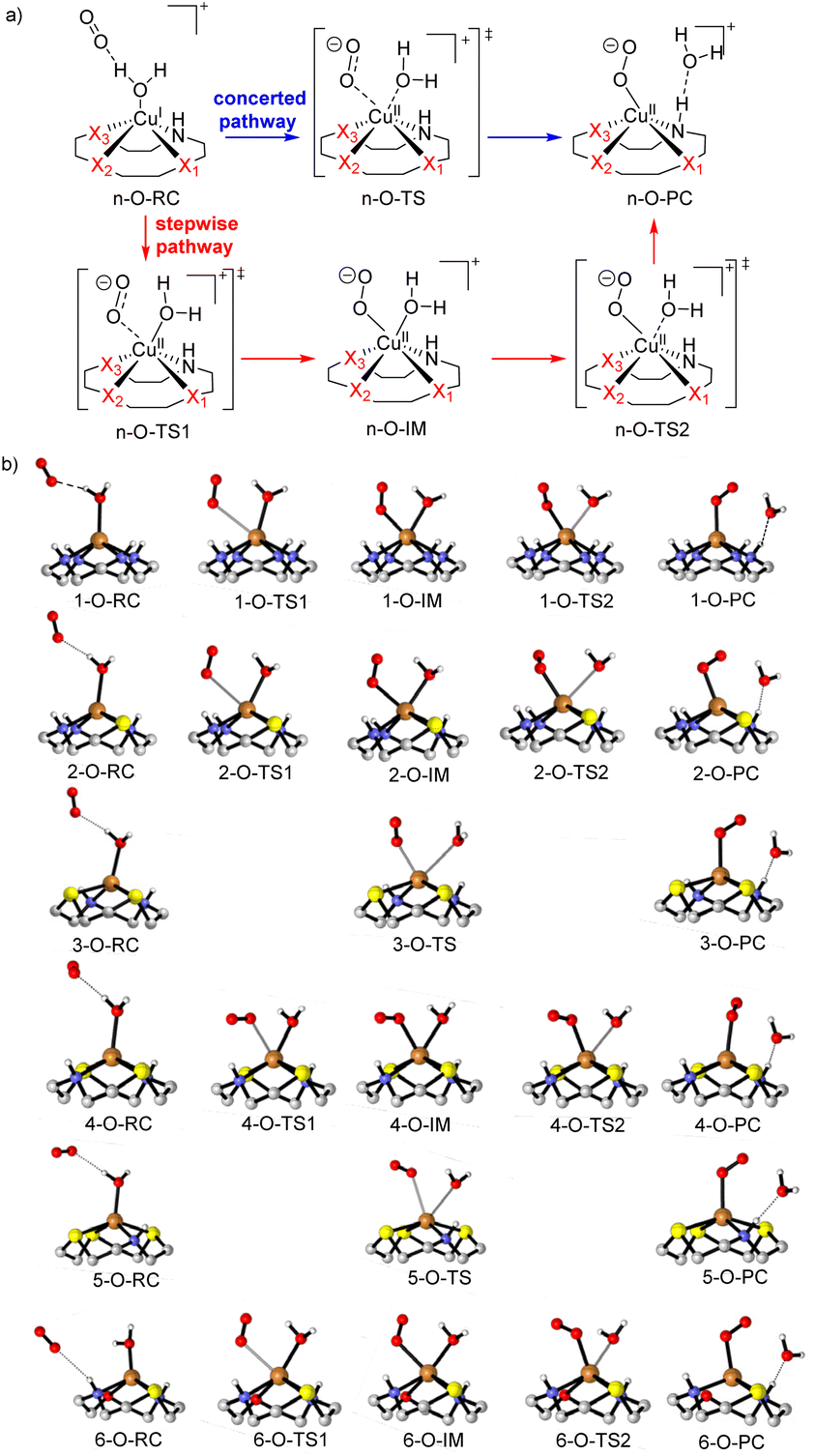 | ||
| Fig. 11 (a) Proposed reaction mechanism of the generation of ROS, (b) optimized complex structures. | ||
Because of the different structures resulting from heteroatom substitution in the cyclen moiety, different mechanisms of the generation of ROS are proposed. For CuLn (n = 1, 2, 4 and 6), complexes with two N atoms in trans position coordinated to the copper center, the mechanism is believed to be a stepwise pathway as depicted in Fig. 11a in red. In the transition state n-O-TS1, it is noted that the copper center is attached to the nucleophilic oxygen molecule. The distance of the Cu–Ooxygen decrease to 2.93, 2.76, 2.47 and 2.51 Å for n-O-TS1 (n = 1, 2, 4 and 6), respectively. The Cu–Ooxygen distances become even smaller in the intermediate n-O-IM. In n-O-IM, the coordination sphere of the copper center consists of two strong O donors (the metal-coordinated oxygen molecule and the water molecule), one strong N-donor and three Xn donors from the cyclen. The bond length of the Cu–Ooxygen is 2.14, 2.16, 2.19 and 2.27 Å for n-O-IM (n = 1, 2, 4 and 6), respectively. The Cu–O coordinated combination of the Cu(II) center with the water molecule is further weakened in the n-O-TS2, and the Cu–Ooxygen bond is more strengthened. Subsequently, the water molecule is dissociated from the Cu(II) center in n-O-PC. However, for the CuL3 and CuL5 complexes, only one transition state was discovered in the generation of ROS, which suggests a concerted pathway (see Fig. 11a in blue). In the transition state n-O-TS (n = 3 and 5, for intrinsic reactions coordinates cf. ESI, Fig. S-7†), it could be seen that Owater and one of the O atoms of the oxygen molecule coordinate to the copper center. The Cu–Owater distance is 2.73 and 2.98 Å, and the Cu–Ooxygen distance is 2.27 and 2.78 Å for 3-O-TS and 5-O-TS, respectively. A tendency for the cleavage of the Cu–Owater bond and simultaneous formation of a Cu–Ooxygen bond is observed upon going from n-O-RC to n-O-TS (n = 3 and 5). At last, a superoxide radical anion (O2˙−) ROS is formed in a metal-bonded manner in the n-O-PC state. All optimized structural parameters are reported in the ESI (ESI, Table S-7.1†).
The calculated relative free energy profiles for the generation of ROS by the CuLn complexes are shown in Fig. 12. It can be established that the nucleophilic attack of the oxygen molecule to the copper center should be the rate-determining step for the CuLn (n = 1, 2, 4 and 6) complexes. A comparison between CuL1, CuL2 and CuL6 reveals that the relative free energy of the CuL2 complex is the highest (1.7 kcal mol−1 in liquid phase), which can be ascribed to the lower electronegativity of the sulfur donor and thus weaker Lewis acidity of the copper center. Moreover, the energy barriers for the CuLn (n = 3, 4 and 5) complexes are higher compared to the CuLn complexes (n = 1, 2 and 6) due to an increase of the sulfur content and an even more distinct reduction of the Lewis acidity of the copper center.
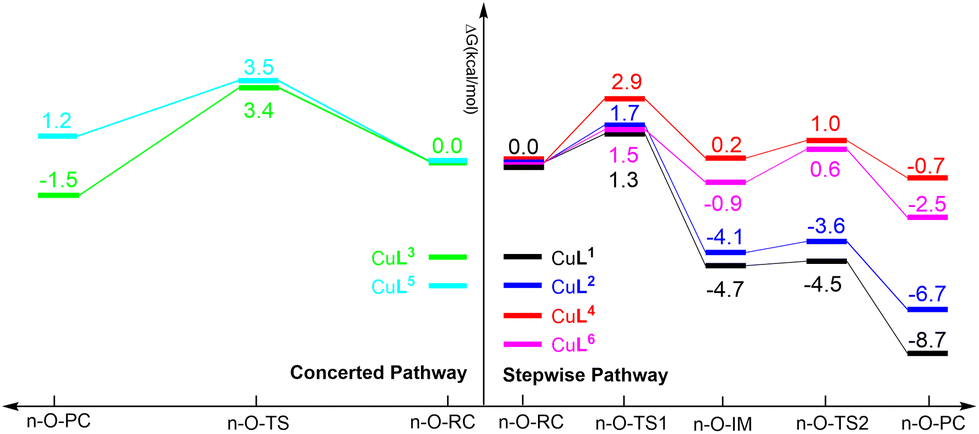 | ||
| Fig. 12 Relative free energy profiles for the generation of ROS by CuLn complexes in liquid phase: CuLn (n = 1, 2, 4 and 6) complexes through a stepwise mechanism and CuLn (n = 3 and 5) complexes through a concerted mechanism. | ||
According to the above presented data for dimerization tendencies and the reduction potential, the higher reduction potential of CuIL6 complex compared to CuIL1 and CuIL2 is considered to be a more important factor in the oxidative cleavage of DNA than the dimerization tendency since the free energy barriers for ROS generation are similar for all above-mentioned complexes.
Also, the lower free energy barrier of CuIL4 compared to CuIL3 and CuIL5 is more favorable in the oxidative cleavage of DNA. Therefore, the CuIL6 complex is expected to be the most efficient DNA oxidative cleaving agent, followed by CuIL4. This is confirmed by the experimental data.
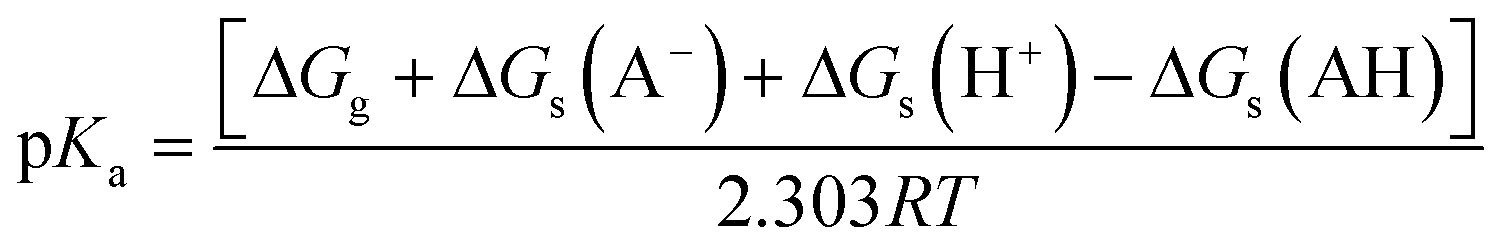 | (3) |
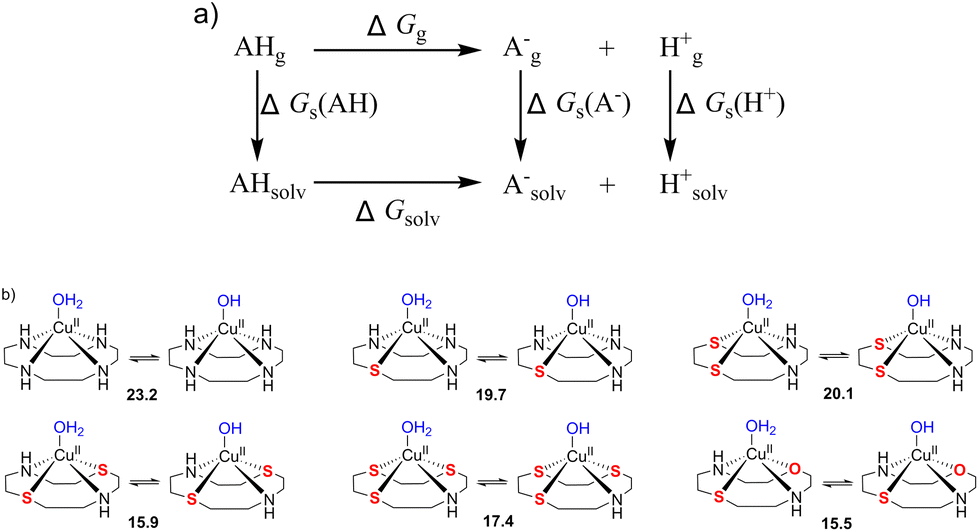 | ||
| Fig. 13 (a) Thermodynamic Born–Haber cycle of the deprotonation processes, (b) the structural variations during deprotonation processes and the calculated pKa values for complexes CuL1–CuL6. | ||
The pKa values of the metal-coordinated water molecules of all the structures of interest are summarized in Fig. 13b. The calculated pKa values of [CuIILn(OH2)]2+ (n = 1–3) systems were close to each other, while a lower M–OH2 pKa was observed for the other three complexes (n = 4–6), which is favorable for the formation of the M–OH nucleophile. This trends can be corroborated with the BNPP cleavage experiment, where CuL6 represents a much better catalyst for phosphoester cleavage than CuL2 (Fig. 4).
Conclusions
Six different Cu(II) complexes of heteroatom-substituted cyclen ligands were synthesized and characterized, among them four new compounds. Their cleavage activity towards plasmid DNA was tested twofold. Under hydrolytic conditions, CuL1–CuL6 were not able to cut DNA within 24 h. We have shown here that relatively low pKa values for their aqua complexes as well as low dimerization tendencies can explain this observation. Under oxidative conditions (reduction of Cu(II) by ascorbate leading to ROS formation) CuL6 was most efficient followed by CuL4, CuL5 and CuL3 with regioisomeric diathiacyclen ligands (CuL3 and CuL4) forming equally stable Cu(II) and Cu(I) complexes and a trithiacyclen ligand (CuL5). The latter one might hinder a reoxidation to Cu(II) due to strong stabilization of the Cu(I) species.Compared to the previously reported oxygen-rich analogs,7 DNA cleavage efficiency is slightly decreased. However, the redox processes of the sulfur-rich and the mixed sulfur/oxygen analogs triggers CuII/CuI redox process reversibility due to higher stability of the corresponding Cu(I) species, which is of fundamental importance for the ROS generation.
Therefore, CuL6 represents the most interesting candidate within this series. The presence of an oxygen donor (stabilizing Cu(II)) as well as an sulfur donor (stabilizing Cu(I)) next to two nitrogen donors within the ligand scaffold, can strike a balance between the two effects, which the pure thia- or oxa-derivatives suffer from. On the one hand, if the Cu(I) state is too stable, as in sulfur-rich systems, the reoxidation, and thus the ROS formation is inhibited. On the other hand, an oxygen-rich system leads to the release of the copper ion once it is reduced to Cu(I) due to its low affinity to O donors.6,7 CuL6 thus represents a compromise between a set of hard (O) and soft donor atoms (S) next to intermediate donor atoms (N) for stabilizing the hard and soft Lewis acids, respectively, Cu(II) and Cu(I). This concept is also used in nature by redox-active blue copper proteins, for instance azurin, however, only with N and S donors.46
Our approach of combining experimental and theoretical data for the evaluation of Cu(II) metallonucleases, and the observed correlation between these data, shows that rational design for this class of biologically active molecules is possible. Decisive parameters like dimerization tendencies, reduction potentials (for oxidative DNA cleavage) and pKa values (for hydrolytic DNA cleavage) can be estimated before elaborate experiments are carried out. We hope this work inspires other groups in the field to use a combinatory approach of theoretical and experimental studies to identify metal complexes being worthwhile to synthesize.
Experimental section
Materials and methods
All chemical reagents were purchased from Sigma–Aldrich if not stated otherwise. Only HPLC-grade solvents were used.UV/VIS absorption spectra were recorded with a Varian Cary 100 spectrophotometer by using precision cells made of quartz (1 cm) at 25 °C.
Fluorescence experiments were carried out at room temperature in Tris and in MOPS buffer (50 mM, pH 7.4). Hydroxyl radicals and hydrogen peroxide were detected by means of the fluorogenic sensors TPA (50 μM) and PBSF (25 μM), respectively. The solutions were composed of Tris (or MOPS) buffer (50 mM, pH 7.4), sensor, L-ascorbic acid (0.25 mM), scavenger compounds (DMSO (400 mM) when using TPA or pyruvate (2 mM) when using PBSF) and Cu(II) complex or Cu(II) salt (40 μM). Samples were incubated at room temperature for 2.5 h and fluorescence spectra were recorded in the range of 350–550 nm (λex = 320 nm) for TPA-containing samples and 490–600 nm (λex = 485 nm) for PBSF-containing samples (slit width 5 nm). Fluorescence spectra were measured in 1000 μL (for TPA) and 500 μL (for PBSF) quartz fluorescence cuvettes with an Agilent Cary Eclipse fluorescence spectrometer the photomultiplier voltage adjusted to 780 V and 720 V using TPA and PBSF, respectively.
Electrospray mass spectra were obtained in positive-ion mode by using an Agilent 6210 ESI-TOF mass spectrometer.
Elemental analyses were performed by using a Vario EL elemental analyzer.
IR spectra were measured with a Nicolet™ iS™ 5 FT-IR spectrometer (Thermo Fisher). The measurement was performed directly from the solid sample without prior preparation.
X-ray diffraction data were collected with a Bruker-AXS SMART CCD system. The structures were solved by direct methods and refined by full-matrix least-squares methods (SHELX-97). CCDC 1569176 (CuL3) contains the supplementary crystallographic data for this paper.†
Cyclic voltammetry experiments were carried out in an aqueous 0.1 M KCl solution. Millipore water was used to prepare the solutions. The water was degassed before by bubbling dinitrogen into it. A three-electrode configuration (glassy carbon working electrode, Pt counter electrode, Ag wire as pseudo-reference) and a PAR VersaSTAT 4 potentiostat were used. The ferrocene/ferrocenium (FcH/FcH+) couple served as internal reference. Due to the poor solubility of ferrocene in water, ferrocene was dissolved in acetonitrile (6.7 mM) and added to the solution after each measurement. The measurement was then repeated to allow for referencing to FcH/FcH+ (see also ref. 47).
EPR measurements were carried out by the use of a Bruker EMXplus (X band) Spectrometer at an average frequency of 9.35 GHz. The samples have been measured as frozen solutions at an average temperature of 12 K by the use of a liquid Helium-recirculating cryostat (ColdEdge-ER4112HV-CF10-H). Samples have been measured at a power of 2 mW, with an attenuation of 20 dB and modulation amplitude of ca. 5 G. EPR simulations were carried out by the use of EasySpin software (version 6.0) supported by Matlab.59
Synthesis
All syntheses are described in detail different batches of plasmid D in the ESI (S-1).†DNA cleavage studies
DNA cleavage activity of complexes CuL1–CuL6 towards pBR322 plasmid DNA (Carl Roth) was monitored by agarose gel electrophoresis. All experiments were carried out in triplicate. In a typical experiment, plasmid DNA (0.025 μg mL−1) in Tris-HCl buffer (50 mM, pH 7.4, Fisher Scientific) was mixed with different concentrations of complexes CuL1–CuL6 in the presence and absence of ascorbic acid (0.32 mM, Acros). Deionized water (Millipore system) was added up to a total reaction volume of 8 μL before the samples were incubated for a given time. After incubation, samples were analyzed directly or stored at −196 °C (liquid nitrogen) before usage in gel electrophoresis. For analysis, 1.5 μL of loading buffer (containing 3.7 mM bromophenol blue, 1.2 M saccharose in deionized water) was added to the incubation solution and loaded onto an agarose (SeaKem LE, Lonza) gel (1% in 0.5× Tris-borate-EDTA (TBE) buffer, Fisher Scientific) containing ethidium bromide as a staining agent (0.2 μg mL−1, Fisher Scientific). Electrophoresis was carried out at 40 V for 2 h with an electrophoresis unit (Carl Roth; power supply: consort EV243) in 0.5× TBE buffer. Bands were visualized by UV light and photographed by using a gel documentation system (GelDoc, Bio-Rad). The quantity of different DNA forms was estimated by using Image Lab™ software (Bio-Rad). To do so, a value of 1 was assigned to the supercoiled control DNA incubated under the same conditions as in the other experiments but without the addition of metal complexes (labeled "control" or "DNA" in the agarose gels). All other values were expressed relatively to that. Taking into account that the supercoiled form I of plasmid DNA has a smaller affinity towards ethidium bromide, its intensity was afterwards multiplied with a correction factor of 1.22.48 Small DNA fragments were added together within form III DNA.To make sure that DNA bands were correctly assigned to forms I, II and III, a DNA ladder was established: 250 μL of pBR322 plasmid DNA (0.025 μg mL−1) were linearized by using EcoRI nuclease, purified by agarose gel electrophoresis and then extracted by using a GenElute™ extraction kit (Sigma–Aldrich). The extract was diluted to 250 μL and mixed with another solution of 250 μL of pBR322 plasmid DNA (0.025 μg mL−1). 2 μL of this solution were loaded into the first pocket of every agarose gel.
Experiments in the presence of ROS scavengers were conducted as described above by using either 200 mM DMSO as hydroxyl radical scavenger, 10 mM NaN3 as singlet oxygen scavenger, 2 mM pyruvic acid as hydrogen peroxide scavenger or 313 units mL−1 superoxide dismutase (SOD, bovine liver, 2000–6000 units mL−1, Sigma–Aldrich) as superoxide radical anion scavenger. Addition of PBS to all samples was necessary, because SOD was preincubated at 37 °C in 10× PBS for 30 min resulting in a 1.25× PBS concentration in the incubation mixture.
Computational studies
Density functional theory (DFT) using the method of B3LYP function49,50 was performed in this work, and all the geometry calculations were accomplished with the Gaussian 09 program package.51 The lanl2dz relativistic effective core potential (ECP) basis52 was used to describe the metal atom (Cu) and the 6-31+G* basis set for nonmetal atoms (C, H, O, N, P and S) was used in the geometry optimization. The calculations of vibrational frequency and the intrinsic reaction coordinates (IRC)53,54 were prepared at the same level as above. The vibrational frequency calculation was given to examine the stable point on the potential energy surface. There was no imaginary frequency for the reactants (RCs) and products (PCs) but only one imaginary frequency for the transition states (TSs) at 298.15 K and 1 atm. The IRC was employed to verify the optimal reaction path from RC to PC. Furthermore, to explore accurate energies, single-point energies for all the optimized structures were also studied at the B3LYP/Stuttgart/Dresden (SDD)55 basis sets with ECP for Cu atoms and 6-311++G** for C, H, O, N, P and S atoms. In addition, solvent effects were investigated by using the SMD56 continuum model with empirical dispersion correction of GD357 (the D3 version of Grimmes dispersion) in water solution (dielectric constant ε = 78.36). All free energies shown were single-point energies in liquid phase.X-ray crystallographic data for CuL2 (CCDC 911568), CuL3 (CCDC 1569176)† and CuL6 (this work, vide supra) were used as initial structures, and the structures of CuL1, CuL4 and CuL5 were constructed based on the former data.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Studienstiftung des deutschen Volkes is acknowledged for a pre-doctoral fellowship and the Dahlem Research School for a postdoctoral fellowship for J. H. We thank K. Licha for a donation of cyclen and S. Hinojosa and P. Liebing for help with X-ray crystallography. Z. C. Y. gratefully acknowledges the National Natural Science Foundation of China (Grant No. 21573292 and 21773312).References
- R. Reichenbach-Klinke and B. König, J. Chem. Soc., Dalton Trans., 2002, 121–130 RSC.
- C. Liu, M. Wang, T. Zhang and H. Sun, Coord. Chem. Rev., 2004, 248, 147–168 CrossRef CAS.
- J. R. Morrow and O. Iranzo, Curr. Opin. Chem. Biol., 2004, 8, 192–200 CrossRef CAS PubMed.
- K. Michaelis and M. Kalesse, Angew. Chem., Int. Ed., 1999, 38, 2243–2245 CrossRef CAS PubMed.
- T. Joshi, B. Graham and L. Spiccia, Acc. Chem. Res., 2015, 48, 2366–2379 CrossRef CAS PubMed.
- J. Hormann, C. Perera, N. Deibel, D. Lentz, B. Sarkar and N. Kulak, Dalton Trans., 2013, 42, 4357–4360 RSC.
- J. Hormann, M. van der Meer, B. Sarkar and N. Kulak, Eur. J. Inorg. Chem., 2015, 4722–4730 CrossRef CAS.
- C. Wende, C. Lüdtke and N. Kulak, Eur. J. Inorg. Chem., 2014, 2597–2612 CrossRef CAS.
- S. Tardito and L. Marchiò, Curr. Med. Chem., 2009, 16, 1325–1348 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- I. Iakovidis, I. Delimaris and S. M. Piperakis, Mol. Biol. Int., 2011, 594529 Search PubMed.
- B. Gruber, E. Kataev, J. Aschenbrenner, S. Stadlbauer and B. König, J. Am. Chem. Soc., 2011, 133, 20704–20707 CrossRef CAS PubMed.
- A. Bencini, E. Berni, A. Bianchi, C. Giorgi, B. Valtancoli, D. K. Chand and H.-J. Schneider, Dalton Trans., 2003, 793–800 RSC.
- Y.-G. Fang, J. Zhang, S.-Y. Chen, N. Jiang, H.-H. Lin, Y. Zhang and X.-Q. Yu, Bioorg. Med. Chem., 2007, 15, 696–701 CrossRef CAS PubMed.
- Y. Zhang, Y. Huang, J. Zhang, D. Zhang, J. Liu, Q. Liu, H. Lin and X. Yu, Sci. China. Chem., 2011, 54, 129–136 CrossRef CAS.
- Q.-L. Li, J. Huang, Q. Wang, N. Jiang, C.-Q. Xia, H.-H. Lin, J. Wu and X.-Q. Yu, Bioorg. Med. Chem., 2006, 14, 4151–4157 CrossRef CAS PubMed.
- K. P. Balakrishnan, T. A. Kaden, L. Siegfried and A. D. Zuberbühler, Helv. Chim. Acta, 1984, 67, 1060–1069 CrossRef CAS.
- T. L. Walker, S. Mula, W. Malasi, J. T. Engle, C. J. Ziegler, A. van der Est, J. Modarelli and M. J. Taschner, Dalton Trans., 2015, 44, 20200–20206 RSC.
- T. Rotärmel, J. Becker and S. Schindler, Faraday Discuss., 2022, 234, 70–85 RSC.
- A. Johnson, L. Iffland-Mühlhaus, J. Northcote-Smith, K. Singh, F. Ortu, U.-P. Apfel and K. Suntharalingam, Dalton Trans., 2022, 51, 5904–5912 RSC.
- M. W. Glenny, L. G. A. van de Water, J. M. Vere, A. J. Blake, C. Wilson, W. L. Driessen, J. Reedijk and M. Schröder, Polyhedron, 2006, 25, 599–612 CrossRef CAS.
- A. H. Alberts, J.-M. Lehn and D. Parker, J. Chem. Soc., Dalton Trans., 1985, 2311–2317 RSC.
- S. Afshar, S. T. Marcus, L. R. Gahan and T. W. Hambley, Aust. J. Chem., 1999, 52, 1–6 CrossRef CAS.
- M. M. Bernardo, R. R. Schroeder and D. B. Rorabacher, Inorg. Chem., 1991, 30, 1241–1247 CrossRef CAS.
- K. B. Yatsimirskii and V. V. Pavlishchuk, J. Coord. Chem., 1996, 37, 341–348 CrossRef CAS.
- M. C. Styka, R. C. Smierciak, E. L. Blinn, R. E. DeSimone and J. V. Passariello, Inorg. Chem., 1978, 17, 82–86 CrossRef CAS.
- B. M. Gatehouse, S. E. Livingstone and R. S. Nyholm, J. Chem. Soc., 1957, 4222–4225 RSC.
- R. Clay, P. Murray-Rust and J. Murray-Rust, Acta Cryst., 1979, B35, 1894–1895 CrossRef CAS.
- J. Nagaj, K. Stokowa-Sołtys, E. Kurowska, T. Frączyk, M. Jeżowska-Bojczuk and W. Bal, Inorg. Chem., 2013, 52, 13927–13933 CrossRef CAS PubMed.
- L. Guilloreau, S. Combalbert, A. Sournia-Saquet, H. Mazarguil and P. Faller, ChemBioChem, 2007, 8, 1317–1325 CrossRef CAS PubMed.
- E. L. Hegg and J. N. Burstyn, Inorg. Chem., 1996, 35, 7474–7481 CrossRef CAS.
- A. Sreedhara, J. D. Freed and J. A. Cowan, J. Am. Chem. Soc., 2000, 122, 8814–8824 CrossRef CAS.
- Y. W. Lee, M. S. Ha and Y. K. Kim, Neurochem. Res., 2001, 26, 1187–1193 CrossRef CAS PubMed.
- J. Hormann, S. Streller and N. Kulak, J. Chem. Educ., 2018, 95, 1848–1855 CrossRef CAS.
- B. Tang, L. Zhang and Y. Geng, Talanta, 2005, 65, 769–775 CrossRef CAS PubMed.
- H. Maeda, Y. Fukuyasu, S. Yoshida, M. Fukuda, K. Saeki, H. Matsuno, Y. Yamauchi, K. Yoshida, K. Hirata and K. Miyamoto, Angew. Chem. Int. Ed., 2004, 43, 2389–2391 CrossRef CAS PubMed.
- K. P. Balakrishnan, T. A. Kaden and A. D. Zuberbühler, Inorg. Chim. Acta, 1983, 79, 202–203 CrossRef.
- J. C. Joyner, J. Reichfield and J. A. Cowan, J. Am. Chem. Soc., 2011, 133, 15613–15626 CrossRef CAS PubMed.
- P. de Hoog, M. J. Louwerse, P. Gamez, M. Pitié, E. J. Baerends, B. Meunier and J. Reedijk, Eur. J. Inorg. Chem., 2008, 612–619 CrossRef CAS.
- H. Gao, Z. Ke, N. J. DeYonker, J. Wang, H. Xu, Z.-W. Mao, D. L. Phillips and C. Zhao, J. Am. Chem. Soc., 2011, 133, 2904–2915 CrossRef CAS PubMed.
- X. Zhang, X. Liu, D. L. Phillips and C. Zhao, ACS Catal., 2016, 6, 248–257 CrossRef CAS.
- T. Miao, Q. Deng, H. Gao, X. Fu and S. Li, J. Chem. Inf. Model., 2018, 58, 859–866 CrossRef CAS PubMed.
- L. E. Roy, E. Jakubikova, M. G. Guthrie and E. R. Batista, J. Phys. Chem. A, 2009, 113, 6745–6750 CrossRef CAS PubMed.
- M. D. Liptak and G. C. Shields, J. Am. Chem. Soc., 2001, 123, 7314–7319 CrossRef CAS PubMed.
- M. D. Liptak, K. C. Gross, P. G. Seybold, S. Feldgus and G. C. Shields, J. Am. Chem. Soc., 2002, 124, 6421–6427 CrossRef CAS PubMed.
- K. Paraskevopoulos, M. Sundararajan, R. Surendran, M. A. Hough, R. R. Eady, I. H. Hillier and S. S. Hasnain, Dalton Trans., 2006, 3067–3076 RSC.
- L. M. P. Lima, D. Esteban-Gómez, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2012, 51, 6916–6927 CrossRef CAS PubMed.
- R. P. Hertzberg and P. B. Dervan, Biochemistry, 1984, 23, 3934–3945 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian,Inc., Wallingford CT, 2009 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 4538–4543 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Schnödt, M. Sieger, B. Sarkar, J. Fiedler, J. S. Manzur, C.-Y. Su and W. Kaim, Z. Anorg. Allg. Chem., 2011, 637, 930–934 CrossRef.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of ligands and complexes, UV/VIS and EPR spectroscopy, X-ray crystallography, summary of stability constants, quench and BNPP assay, intrinsic reaction coordinates, structural parameters and cartesian coordinates of optimized structures. Crystallographic data for [CuL3(NO3)]NO3 (CIF). CCDC 1569176. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03284h |
| ‡ Cleavage activity of the metal salt alone, Cu(NO3)2 is shown in Fig. S-5.1.† Its activity is comparable to the weaker DNA cleaving agents in this study like CuL2. |
| § It should be mentioned that a comparison of Fig. 5 and 6 reveals different cleavage activities for the complexes CuL1 and CuL4. It has to be considered, that a direct comparison between different gel sets is not possible, but only within one single gel set. This can also be derived from the observation that the amount of form II in different batches of plasmid DNA (cf. controls) and the lengths of error bars can be distinct from each other in different gel sets (here, Fig. 5 and 6). This is why we avoid comparison of cleavage activities obtained in different gel experiments in our discussion. |
| ¶ Any effects should be discussed against the background of the fact, that the reduction of oxygen by Cu(I) is an inner-sphere process, whereas the electrochemical reduction represents an outer-sphere process,58 thus DNA cleavage activity does not necessarily correlate with reduction potentials. |
| This journal is © The Royal Society of Chemistry 2023 |