
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural insight into halide-coordinated [Fe4S4XnY4−n]2− clusters (X, Y = Cl, Br, I) by XRD and Mössbauer spectroscopy†
Andreas O.
Schüren
ab,
Benjamin M.
Ridgway
 b,
Florencia
Di Salvo
bc,
Luca M.
Carella
d,
Verena K.
Gramm
a,
Elisa
Metzger
e,
Fabio
Doctorovich
bc,
Eva
Rentschler
d,
Volker
Schünemann
e,
Uwe
Ruschewitz
a and
Axel
Klein
*a
b,
Florencia
Di Salvo
bc,
Luca M.
Carella
d,
Verena K.
Gramm
a,
Elisa
Metzger
e,
Fabio
Doctorovich
bc,
Eva
Rentschler
d,
Volker
Schünemann
e,
Uwe
Ruschewitz
a and
Axel
Klein
*a
aUniversität zu Köln, Mathematisch-Naturwissenschaftliche Fakultät, Department für Chemie, Institut für Anorganische Chemie, Greinstraße 6, D-50939 Köln, Germany. E-mail: axel.klein@uni-koeln.de; Tel: +49-221-470-4006
bINQUIMAE-CONICET-Universidad de Buenos Aires, Intendente Güiraldes 2160, Pabellón 2, Piso 3, C1428EGA, Buenos Aires, Argentina
cUniversidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Química Inorgánica, Analítica y Química Física, Intendente Güiraldes 2160, Pabellón 2, Piso 3, C1428EGA, Buenos Aires, Argentina
dJohannes Gutenberg Universität Mainz, Department Chemie, Duesbergweg 10-14, 55128 Mainz, Germany
eTU Kaiserlautern Department of Physics, 67663 Kaiserlautern, Germany
First published on 14th December 2022
Abstract
Iron sulphur halide clusters [Fe4S4Br4]2− and [Fe4S4X2Y2]2− (X, Y = Cl, Br, I) were obtained in excellent yields (77 to 78%) and purity from [Fe(CO)5], elemental sulphur, I2 and benzyltrimethylammonium (BTMA+) iodide, bromide and chloride. Single crystals of (BTMA)2[Fe4S4Br4] (1), (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Cl2I2] (3), and (BTMA)2[Fe4S4Br2I2] (4) were isostructural to the previously reported (BTMA)2[Fe4S4I4] (5) (monoclinic, Cc). Instead of the chloride cubane cluster [Fe4S4Cl4]2−, we found the prismane-shaped cluster (BTMA)3[Fe6S6Cl6] (6) (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). 57Fe Mössbauer spectroscopy indicates complete delocalisation with Fe2.5+ oxidation states for all iron atoms. Magnetic measurements showed small χMT values at 298 K ranging from 1.12 to 1.54 cm3 K mol−1, indicating the dominant antiferromagnetic exchange interactions. With decreasing temperature, the χMT values decreased to reach a plateau at around 100 K. From about 20 K, the values drop significantly. Fitting the data in the Heisenberg–Dirac–van Vleck (HDvV) as well as the Heisenberg Double Exchange (HDE) formalism confirmed the delocalisation and antiferromagnetic coupling assumed from Mössbauer spectroscopy.
). 57Fe Mössbauer spectroscopy indicates complete delocalisation with Fe2.5+ oxidation states for all iron atoms. Magnetic measurements showed small χMT values at 298 K ranging from 1.12 to 1.54 cm3 K mol−1, indicating the dominant antiferromagnetic exchange interactions. With decreasing temperature, the χMT values decreased to reach a plateau at around 100 K. From about 20 K, the values drop significantly. Fitting the data in the Heisenberg–Dirac–van Vleck (HDvV) as well as the Heisenberg Double Exchange (HDE) formalism confirmed the delocalisation and antiferromagnetic coupling assumed from Mössbauer spectroscopy.
Introduction
Cubane-shaped [Fe4S4] clusters represent a very interesting class of cofactors in biology and are involved in various functions in a cell's life cycle.1–10 Due to their mixed valence character with formally two ferric (Fe3+) and two ferrous (Fe2+) iron atoms, [Fe4S4]2+ clusters show extraordinary electrochemical properties which are crucial for their biological functions in electron transfer and redox catalysis reactions.1–10 During the last 40 years, numerous [Fe4S4]2+ clusters with terminal thiolate ligands have been prepared in order to model the structural, electronic and electrochemical properties as well as the reactivity of the naturally occurring clusters.5–26 Moreover, the fascinating nature of [Fe4S4] clusters has triggered their use as precursors for electrocatalytic materials.27–33 Halide-based [Fe4S4X4]2− with X = Cl, Br, or I are known to be useful precursors because halides can be easily exchanged with thiolate or other ligands by salt metathesis reactions. Homoleptic halide clusters have thus been studied previously and a couple of preparation methods have been published (Scheme 1).12,14,15,32–48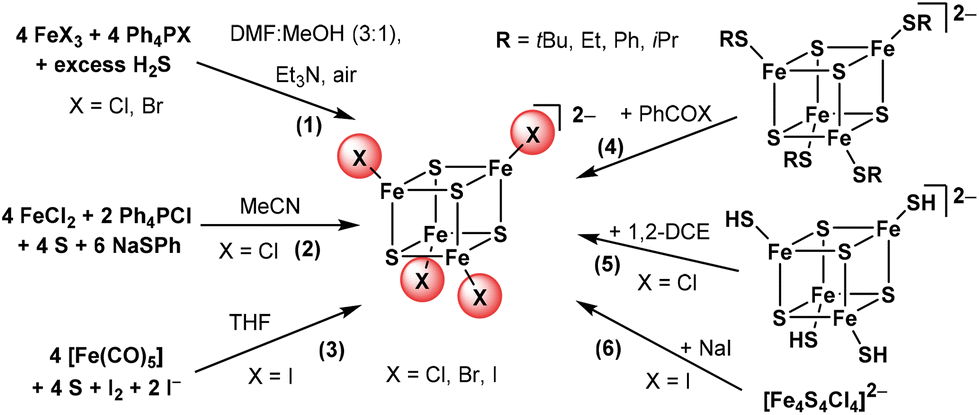 | ||
| Scheme 1 Reported preparation pathways for the [Fe4S4X4]2− clusters. (1) Müller et al.,37 (2) Kanatzidis et al.,32 (3) Pohl et al.,38–40 (4) Holm et al.,36,41 Henderson et al. for X = Cl,43–45 Tuczek et al. for X = Cl or Br,46 (5) Holm et al. for X = Cl,41 and (6) Holm et al. for X = Cl, Br, I,36 Tuczek et al. for X = I.46 | ||
We have recently contributed to this very versatile method (3) starting from [Fe(CO)5] and (BTMA)X (BTMA = benzyltrimethylammonium), (Ph4P)X, and halogens or interhalogens obtaining excellent yields for the compounds (BTMA)2[Fe4S4I4] and (Ph4P)2[Fe4S4Br4] containing the homoleptic [Fe4S4X4]2− clusters.48 By the same method, we obtained the mixed-halide cluster compounds (BTMA)2[Fe4S4BrI3], (BTMA)2[Fe4S4ClI3], and (BTMA)2[Fe4S4Cl2I2]. Using [Fe(CO)5], sulphur, ferrous halides FeX2 and (BTMA)X and adopting method (2), we obtained (BTMA)2[Fe4S4Cl4], (BTMA)2[Fe4S4Br4], (BTMA)2[Fe4S4Cl2I2], (BTMA)2[Fe4S4Br2Cl2] and (BTMA)2[Fe4S4Br2I2]. By replacing the halide FeX2 with tetrahalido ferrates (Ph4P)[FeX4] or (Et4P)[FeX4], we obtained (Et4N)2[Fe4S4Cl4] and (Ph4P)2[Fe4S4Br4]. The compounds were obtained in the pure form as microcrystalline solids, while single crystals were not obtained in this study.48 A careful study of species in solution by UHR-ESI-MS(−) showed that the mixed halide clusters [Fe4S4X2Y2]2− and [Fe4S4XY3]2− scramble their halide ligands in a coordinative disproportionation in THF solution, yielding all possible species [Fe4S4X4−nYn]2− (X/Y = Cl, Br, I). The same was concluded from the UV-vis-NIR absorption spectroscopy results in MeCN solution. Powder X-ray diffraction (PXRD) indicated that the compounds (BTMA)2[Fe4S4Br4], (BTMA)2[Fe4S4Br2Cl2], (BTMA)2[Fe4S4Cl2I2] and (BTMA)2[Fe4S4Br2I2] were isostructural to the previously reported (BTMA)2[Fe4S4I4] (monoclinic, Cc).40
The lack of precise structural information from single crystal XRD was unfortunate since the crystal structures of the [Fe4S4X4]2− clusters are quite interesting due to the frequent observation that the symmetry of [Fe4S4L4]n− clusters strongly depends on the packing in the solid state. Frequently, distortion of the idealised cluster symmetry Td towards D2d was observed (Table 1).22 For the mixed-halide [Fe4S4X2Y2]2− clusters belonging to the vast group of heteroleptic [Fe4S4L2L′2]m− clusters, the Td → C2v symmetry reduction is immanent in the molecular symmetry of the cluster as is the Td → C3v symmetry reduction for [Fe4S4L3L′1]m− clusters. At the same time, the packing in the crystal and thus the size and shape of the counter cation can cause lowering of the cluster symmetry and for all three types even lower symmetry caused by crystal packing was reported (Table 1).37–61 Importantly, with these changes in cluster symmetry, essential properties such as the redox potentials, spin states, magnetic properties, and ligand exchange kinetics vary markedly.1,6,8,13,14,20–23,37–40,47,50,56,62–84
| Cation | Ligand set | Crystal system | Space group | Cluster symmetrya | Ref. |
|---|---|---|---|---|---|
| a Approximate symmetry of the cluster. b Approaches C3v symmetry as one Cl ligand shows strong H bonding to nBu3NH+. c BTMA+ = benzyltrimethylammonium. d Et2Dtc− = N,N-diethyldithiocarbamate. e Selenium clusters [Fe4Se4X4]2−. f Me2Tu = N,N-dimethylthiourea. g tBu2Tu = N,N-di-tert-butyl-thiourea. h R = 2,4,6-(iPr)3C6H2, Et2Tu = N,N-diethylthiourea. | |||||
| Et4N+ | Cl | Monoclinic | P21/c | D 2d | 36 and 42 |
| nPr4N+ | Cl | Monoclinic | P21/n | C 1 | 41 |
| Ph4P+ | Cl | Monoclinic | C2/c | D 2d | 37 |
| nBu3NH+ | Cl | Monoclinic | P21/n | C 3v | 70 |
| [K4[FeCl4](C12H24O6)4]2+ | Cl | Cubic | F23 | T d | 52 |
| [Fe(MeCN)2[(P(OMe)3]4]2+ | Cl | Monoclinic | P21/c | D 2d | 85 |
| Ph4P+ | Br | Monoclinic | C2/c | D 2d | 37 |
| Et4N+ | Bre | Monoclinic | P21/c | S 4 | 87 |
| [(nBu4N)2[Fe(DMF)6]]4+ | Br | Monoclinic | P21/n | T d | 86 |
| Ph4P+ | I | Tetragonal | I41/a | S 4 | 40 |
| BTMA+c | I | Monoclinic | Cc | C 1 | 40 |
| nBu4N+ | Ie | Monoclinic | P21/n | C 3v | 89 |
| [(Et4N)6[Fe2S2I4]]4+ | I | Tetragonal | P42bc | T d | 60 |
| nBu4N+ | SMe | Orthorhombic | Pba2 | T d | 83 |
| HnBu3N+ | SPh | Triclinic |
P |
C 2v | 70 |
| HEt3N+ | SPh | Monoclinic | P21/n | D 2d | 70 |
| BTMA+c | StBu | Monoclinic | P21/c | C 1 | 82 |
| Et4N+ | StBu | Tetragonal |
I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m 2m |
D 2d | 82 |
| Ph4P+ | 2 Br 2 Cl | Monoclinic | C2/c | D 2d | 37 |
| Ph4P+ | 2 Cl 2 SPh | Orthorhombic | Pbcn | D 2d | 88 |
| Ph4P+ | 2 Cl 2 OPh | Orthorhombic | Pbcn | D 2d | 89 |
| Ph4P+ | 2 SPh 2 OpTol | Orthorhombic | P212121 | C 1 | 89 |
| Ph4P+ | 2 Cl 2 S Et2Dtcd | Monoclinic | C2/c | C 2 | 91 |
| Ph4P+ | 2 SPh 2 S Et2Dtcd | Monoclinic | C2/c | C 2 | 91 |
| Ph4P+ | 3 Cl 1 S Et2Dtcd | Monoclinic | P21/c | C s | 91 |
| [FeIL3]+ | 3 I 1 S Me2Tuf | Triclinic |
P |
C 3v | 90 |
| None | 2 I 2 S tBu2Tug | Monoclinic | P21/n | C 2v | 90 |
| None | 2 SR 2 S Et2Tuh | Triclinic | P1 | D 2d | 57 |
Herein, we report the synthesis and detailed study of the materials (BTMA)2[Fe4S4Br4] (1), (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Cl2I2] (3), (BTMA)2[Fe4S4Br2I2] (4), and (BTMA)2[Fe4S4I4] (5) containing cubane type Fe4S4 clusters alongside with (BTMA)3[Fe6S6Cl6] (6) containing the prismane [Fe6S6Cl6]3− structure. After preparation in solution, the compounds were isolated as single crystals and we can provide full structural information from single crystal and PXRD analyses. Additionally, resonance Raman, 57Fe Mössbauer and magnetic data were recorded for the first time on such mixed-halide clusters, with the exception of (Ph4P)2[Fe4S4Br2Cl2] for which the structure and Mössbauer data have previously been reported.37
Results and discussion
Preparations
The materials were prepared as recently described (for details and analyses, see the ESI†). As in our previous study,48 UHR-ESI-MS(−) showed that the mixed halide clusters [Fe4S4X2Y2]2− scramble their halide ligands leading to coordinative disproportionation in solution yielding all possible species [Fe4S4X4−nYn]2− (X/Y = Cl, Br, and I) (see the Experimental data in the ESI†). The elemental analyses of the crystallised products clearly show a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio for X:Y. Of course this does not rule out contributions from the 3:1 and 1:3 cluster species, but then both must contribute with the same amount. The same is true for the 4:0 and 0:4 species. Using methods such as resonance Raman, single crystal XRD, PXRD, magnetic measurements and Mössbauer spectroscopy we not only tried to study the local cluster symmetry but also tried to trace other species than the 2:2 complexes [Fe4S4X2Y2]2−.
:2 ratio for X:Y. Of course this does not rule out contributions from the 3:1 and 1:3 cluster species, but then both must contribute with the same amount. The same is true for the 4:0 and 0:4 species. Using methods such as resonance Raman, single crystal XRD, PXRD, magnetic measurements and Mössbauer spectroscopy we not only tried to study the local cluster symmetry but also tried to trace other species than the 2:2 complexes [Fe4S4X2Y2]2−.
Resonance Raman spectroscopy
The resonance Raman spectra of (BTMA)2[Fe4S4Br4] (1), (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Cl2I2] (3), and (BTMA)2[Fe4S4Br2I2] (4) were recorded at 458 and 514 nm excitation wavelengths (Fig S1, ESI†). Four bands can be distinguished in the NIR range from 180 to 580 cm−1. Unfortunately, these bands are quite invariant on the first view which is expectable, as their main contributions come from the Fe–S bridging modes.46–48,92–94 This is in line with our previous FIR studies and confirms that the absorption bands observed around 500 nm (ε of about 2000 cm−1 M−1) in the UV-vis-NIR absorption spectra in solution have their origin essentially in the Fe4S4 cubane (S → Fe LMCT).48 Unfortunately, direct irradiation of the X → Fe LMCT band at around 700 nm (ε ∼ 1500 cm−1 M−1) was technically not feasible. Nevertheless, two trends were observed. The introduction of the light Cl− led to a slight high-energy shift of all bands and decreased symmetry led to detectable wavelength dependence for all four bands. In homoleptic compound 1, only the bands at about 500 and 400 cm−1 vary with the excitation wavelength, while the bands at 283 and 219 cm−1 are completely invariant. The bands which were most sensitive to variation in the halides and wavelength were ranging from 285 to 276 cm−1 and were assigned to the Tb1 mode in Td symmetry while the resonances from 404 to 398 cm−1 probably fit to Tb2 mode in Td symmetry.46,47,93,94 These modes indicate already a distortion below D2d.46 The intense bands at 220 to 216 cm−1 have clearly their origin in the symmetry reduction lower than D2d for the homoleptic cluster in 1 or lower than C2v for the heteroleptic derivatives 2, 3, and 4.X-ray crystal structures
Single crystals of high quality were obtained for 1 to 4 and (BTMA)3[Fe6S6Cl6] (6) directly from the synthesis procedure or were obtained through the slow diffusion of diethyl ether into saturated MeCN solutions. The crystal structures of 1 to 4 were solved and refined in the monoclinic space group Cc, while that of 5 was found to crystallise in the triclinic space group P (details in Fig. S2 to S6 and Tables S1–S9 in the ESI†). The Fe4S4 compounds are thus isostructural to (BTMA)2[Fe4S4I4] (5),40 regardless if they are pure clusters like 1 or mixed halide species like 2, 3, and 4.
Like in (BTMA)2[Fe4S4I4] (5),40 the iron sulphur clusters in (BTMA)2[Fe4S4Br4] (1) are arranged in planes at the [110] direction packed with the BTMA+ cation in between. The planes are rotated by 90° around the c axis direction (Fig. 1).
 | ||
| Fig. 1 Crystal structure of (BTMA)2[Fe4S4Br4] (1) viewed along the [110] direction (a) and rotated by 90° around the crystallographic c axis to show segregated columns of anions and cations (b). The bottom inset shows atom colouring: H atoms are omitted for clarity. | ||
The intermolecular C–H⋯S and C–H⋯Br hydrogen bonds with the CH3 and CH2 groups of the BTMA+ cations (Fig. 2) exceed 3 Å than the H⋯X distance and are thus weak.95 Br⋯Br interactions between the cluster ions form a chain-like infinite motif along the [110] direction (Fig. 1 and 2) occurring between Br3 and Br4 are 3.814(2) Å long and thus slightly exceed the sum of the van der Waals radii.96,97
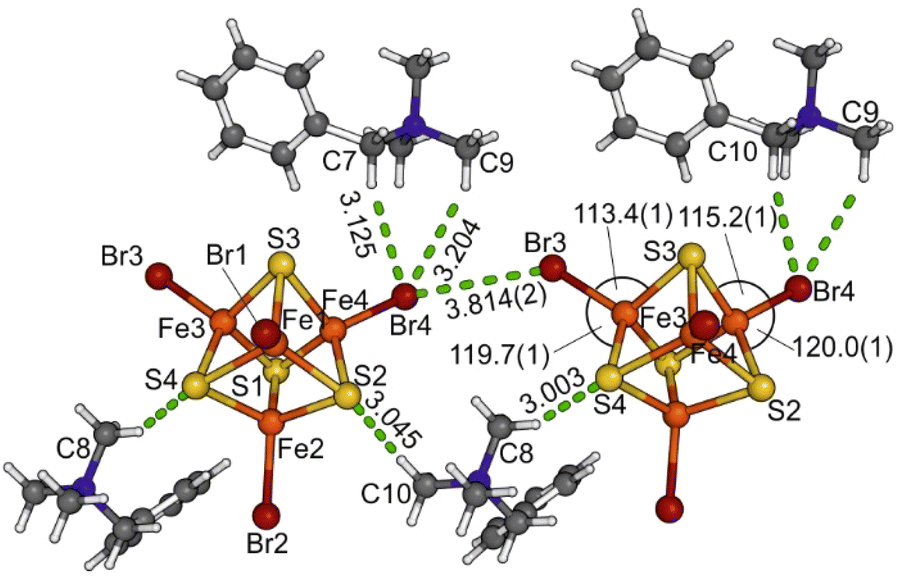 | ||
| Fig. 2 Intermolecular C–H⋯S, C–H⋯Br, and Br⋯Br interactions in (BTMA)2[Fe4S4Br4] (1); distances are in Å and angles are in °. | ||
The idealised Td symmetry of the cubane-shaped [Fe4S4Br4]2− dianion is significantly decreased. The Fe4 cluster core is compressed along the Fe3⋯Fe4 edge with an extraordinarily short distance of 2.717(2) Å (Tables S2, S5 and S6†). This is 0.048 Å shorter than the average of the other Fe⋯Fe distances (2.765 Å). The Fe–S bond lengths vary from 2.249(2) to 2.305(2) Å showing four markedly decreased values, one for each Fe atom (Table S2†). The analysis of the Br–Fe–Fe and S–Fe–Br angles revealing the asymmetrical distortion of the clusters is connected to the intermolecular Br⋯Br interactions between Br3 and Br4 (Fig. 2). The Br3–Fe3–Fe4 (138.53(10)°) and the Br4–Fe4–Fe3 (138.03(10)°) angles are smaller than all other Br–Fe–Fe angles (between 140.06(10)° and 151.03(10)°), whereas the opposite S4–Fe3–Br3 (119.66(12)°) and S2–Fe4–Br4 (120.03(12)°) angles are larger compared with 108.30(11) to 118.87(12)°.
The lengths of the other two terminal Fe–Br bonds are not affected by the compression and the values between 2.332(2) and 2.354(2) Å coincide with those observed for the Ph4P+ structure.37 We thus suspect that the Br⋯Br interactions are rather repulsive in the packing. In contrast to this, the I⋯I interactions in (BTMA)2[Fe4S4I4] (5) seem to be favorable as the I⋯I distance of 3.917 Å is below the sum of the van der Waals radii (4.3 Å).40 In contrast to this, in the PPh4+ derivative, the I⋯I distances are longer which contradicts the idea that the I⋯I interactions might govern the cluster distortion.40
For the mixed-cluster compounds, crystalline material was obtained from (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Cl2I2] (3), and (BTMA)2[Fe4S4Br2I2] (4) and single crystal XRD data were collected at 293 K and 173 K. All three compounds crystallise isostructural (Cc) to the homoleptic 1 and 540 and no phase transition was observed on going from 293 to 173 K. The heteroleptic clusters [Fe4S4X2Y2]2− show mixed occupancies of both halides on each position as reported before for (Ph4P)2[Fe4S4Br2Cl2],37 and we refined the data of the compounds 2, 3, and 4 using the SUMP command (details and data in the ESI†).
In our previous study, we found that in solution and therefore also during synthesis, all possible cubane species [Fe4S4X4−xYx]2− (x = 0 to 4) are present with [Fe4S4X2Y2]2− as the dominating one.48 Since the here reported materials were obtained through slow crystallisation by ether diffusion at −18 °C in the presence of BTMA+ cations, slight deviations of x from the ideal value of 2 in mixed halide clusters [Fe4S4XxYy]2− might be expected.37 But only when we were freely refining the data recorded at 173 K for compound 3 (Cl:I), we obtained a halide ratio of 1.76:2.24 deviating from the ideal 2:2 (Table S10†). Unfortunately, the quality of the data did not allow satisfying refinement. The same problems were reported for the structure solution and refinement of (Ph4P)2[Fe4S4Br2Cl2].37
In line with earlier assumptions, we believe that the cluster composition and thus the cluster symmetry have no effect on the packing and the clusters arrange randomly in the structures.
A comparison of the unit cell volumes of the mixed-ligand compounds (BTMA)2[Fe4S4X2Y2] (2 to 4) with those of the homoleptic derivatives 1 and 5 and the calculated cluster volumes using the so-called Solvent Excluded Volume,98 including the Cl4 cluster derivative (Fig. 3 and Table 2), showed that by exchanging smaller halides with larger ones, the cell volumes and the cluster volumes increase along the series Cl4 < Cl2Br2 < Br4 < Cl2I2 < Br2I2 < I4 in line with the increasing ionic radii99 (Cl−: 1.81 Å, Br−: 1.96 Å, I−: 2.20 Å).
 | ||
| Fig. 3 Comparison of unit cell and the calculated cluster volumes (A) of [Fe4S4X4−xYx]2− (x = 0 or 2). The calculated so-called solvent excluded volumes (VSE) (B).98 Note that the values for 1 to 4 are from data at 173 K, while 5 and “Cl4” are from 298 K data. | ||
| Cell volume 298 Kb | Cell volume 173 Kb | Cluster volume 298 Kc | Cluster volume 173 Kc | |
|---|---|---|---|---|
| a Values in Å3. b From single crystal structures of (BTMA) salts. c Calculated solvent excluded volume (VSE).98 d Data not available for BTMA salts; values for other salts are not comparable. e Derived from the (Et4N)2[Fe4S4Cl4] structure determined at 213 K.36,42 f From ref. 49, at 150 K. g BzEt3N+ salt at 150 K, from ref. 100. h From ref. 40. i BzEt3N+ salt at 173 K, from ref. 101. | ||||
| Cl4 | —d | —d | 221.71e | 221.78f |
| Cl2Br2 | 3269.0(2) | 3201.6(1) | 242.51 | 232.65 |
| Br4 | 3340.8(4) | —d | 244.35 | 244.38g |
| Cl2I2 | 3389.6(1) | 3323.3(2) | 325.62 | 277.02 |
| Br2I2 | 3440.5(2) | 3342.6(1) | 326.00 | 286.03 |
| I4 | 3514.8(2)h | —d | 328.66h | 328.94i |
This underlines that potential contributions from cluster species with 1:3 and 3:1 ratios cannot be detected if they are present in equal amounts or make only minor contributions. So, we cannot exclude mixed-clusters with 3:1 and 1:3 halide ratios to contribute to the mixed-halide cluster compounds, but we have also no evidence for them.
Due to this, the Fe–X bond distances of the mixed-halide clusters do not allow a straightforward comparison with literature data from (BTMA)2[Fe4S4I4] (5),40 (Ph4P)2[Fe4S4I4],40 (Ph4P)2[Fe4S4X4] (X = Br, Cl),37 (Et4N)2[Fe4S4Cl4],36,42 (nPr4N)2[Fe4S4Cl4]41 (Table S7†).
The Fe4S4 cluster cores in the mixed-halide compounds 2 to 4 showed that the characteristic distortion of the [Fe4S4] core is lower than the D2d symmetry (Fig. S5 and S6†). For all three clusters, a significant shortening of the Fe3⋯Fe4 distance (Tables S5 and S6†) and distortion of the X3/4–Fe3/4–Fe4/3 angles and their opposite S–Fe3/4–Br3/4 angles (Table S2†) were found. As for the homoleptic compound 1, the intermolecular halide⋯halide interactions (Table S5†) were observed parallel to the compressions of the cluster core along the Fe3⋯Fe4 axis, while at the same time the Fe1⋯Fe2 axis is slightly elongated (Tables S5 and S6†). For interactions with iodide contribution (i.e. I⋯I, Br⋯I and Cl⋯I), the observed distances are always below the sum of the van der Waal radii pointing to an attractive interaction whereas the Br⋯Br distances are larger as in the homoleptic 1. Br⋯Cl and Cl⋯Cl interactions are not observed.
On the other hand, in [Fe4S4X4]2− (X = Br or I) derivatives containing R4N+ and Ph4P+ cations,9,10,29,30,40,56,69 both Br⋯Br and I⋯I interactions are longer than in the BTMA+ structures by least 0.24 Å (Br) and 0.45 Å (I), respectively (Tables S6 and S7†). The halide⋯halide distances in the homoleptic compounds 1 and 540 are also shorter compared with the published Ph4P+ structures.37,40 This is also in line with the higher density of the BTMA+ structures (Tables S5 and S6†). As a consequence of these shorter halide⋯halide interactions, the BTMA+ structures show markedly higher Fe⋯Fe distortions for both the homoleptic and the mixed-halide clusters. When taking the difference between the largest and the smallest Fe⋯Fe distances, the BTMA+ structures lie at around 0.05 Å (Table S6†), while the Ph4P+ derivatives show values around 0.02 Å (Table S7†). Within the BTMA+ structures, the homoleptic Br4 and I4 show the largest deviations and both these two homoleptic structures and the three mixed-halide structures were found to have the same distortion pattern: two shortened and four extended Fe⋯Fe distances with overall very low cluster symmetry and very similar cluster shapes (Fig. S5 and S6†).
Attempts to crystallise [Fe4S4Cl4]2− with BTMA+ by the same method led to the formation of a prismane structure (BTMA)3[Fe6S6Cl6] (6) which crystallised in the triclinic P space group. The prismane cluster [Fe6S6Cl6]3− is positioned on the inversion centre and is surrounded by three BTMA+ cations (Fig. S2†). Based on this structural feature, the asymmetric unit is represented by two halves of the clusters and three counter ions which can be combined to get two units of 6 per unit cell (Fig. S3†). Pronounced intermolecular H bonding interactions are absent (Fig. S4†). The prismane structure can be described as two alternating layered [Fe3S3] rings in a chair conformation. Its D3d symmetry is only slightly perturbed. The Fe–Cl and Fe⋯Fe distances are found to be very similar (Table S9†) and comparable to bromide and chloride prismanes with Et4N+.87–91 The Cl–Fe–S angles ranging from 108.26(4)° to 115.96(5)° are quite close to an ideal tetrahedral angle. The bulk material of 6 is not completely phase pure but does definitely not contain the (BTMA)2[Fe4S4Cl4] phase isostructural to (BTMA)2[Fe4S4Br4] (1) (see Powder XRD). The [Fe4S4Cl4]2− cluster was previously crystallised with a number of cations, such as Et4N+,36,42nPr4N+,41 or Ph4P+.37 We were unable to detect even traces of (BTMA)2[Fe4S4Cl4] (see powder XRD, Mössbauer, or magnetic measurement). At the moment, we have no explanation other than a superior crystallisation of prismane 6.
Powder XRD studies
In previous investigations,48 we have already shown that the Fe4S4 cluster compounds (1 to 5) are obtained as homogeneous materials with no signs of phase separation. Very weak additional reflections point to some minor impurities of unknown identity. No significant differences between the crystal structures obtained at 170 K and RT are visible excluding the possible phase transitions upon cooling. The diffraction pattern of the Fe6S6 prismane (Fig. S7†) shows that 6 is only a minority phase in the reaction product. It was speculated that the main product could be the still unknown chloride (BTMA)2[Fe4S4Cl4]. The simulated diffraction pattern of (BTMA)2[Fe4S4Br4] (1) is provided for comparison. Assuming that 1 and the unknown chloride crystallise in the isotypic crystal structures, the pattern of the chloride should be shifted to higher 2θ values thus completely excluding this assumption. However, it should be noted that the formation of the chloride (BTMA)2[Fe4S4Cl4] crystallising in another crystal structure cannot completely be ruled out at this point. In addition, another modification of (BTMA)3[Fe6S6Cl6] can possibly make up the majority of the studied material.57Fe Mössbauer spectroscopy
For (BTMA)2[Fe4S4Br4] (1) a symmetric doublet with an isomer shift δ = 0.48 mm s−1 was recorded (Fig. S8A†). Analysis by fitting using a Lorentzian line shape also reveals a quadrupole splitting ΔEQ of 1.77 mm s−1 and a line width at half maximum Γ of 0.42 mm s−1 (Table 3).| n | δ | ΔEQ | Γ | Fe | |
|---|---|---|---|---|---|
|
a Contribution n/%, isomer shift δ/mm s−1, quadrupole splitting ΔEQ/mm s−1, line width at half maximum Γ/mm s−1. Iron oxidation state Fe.
b Two species simulated in a 50:50 ratio.
c Minor species of 11% (see text and Fig. S8†).
|
|||||
| 1 | 100 | 0.48 ± 0.01 | 1.07 ± 0.02 | 0.42 ± 0.01 | 2.5+ |
| 2 | 100 | 0.48 ± 0.01 | 1.07 ± 0.03 | 0.35 ± 0.01 | 2.5+ |
| 3 | A: 50 | 0.48 ± 0.02 | 1.16 ± 0.02 | 0.37 ± 0.02 | 2.5+ |
| B: 50 | 0.46 ± 0.01 | 0.80 ± 0.01 | 0.47 ± 0.02 | 2.5+ | |
| 4 | 100 | 0.48 ± 0.01 | 1.05 ± 0.02 | 0.35 ± 0.01 | 2.5+ |
| 5 | 89 | 0.48 ± 0.02 | 1.02 ± 0.02 | 0.35 ± 0.01 | 2.5+ |
| 6 | A: 96 | 0.48 ± 0.02 | 0.88 ± 0.02 | 0.35 ± 0.01 | 2.5+ |
An isomer shift of 0.48 mm s−1 is in keeping with the previously reported [Fe4S4Br4]2− clusters with different counter ions.86,87 It is also in the range observed for [4Fe–4S]2+ centres occurring in 4Fe–4S proteins. These clusters exhibit complete delocalisation of one electron in the Fe2+–Fe3+ spin pairs. The ferromagnetic double exchange between the iron spins formally leads to Fe2.5+–Fe2.5+ pairs with the pair spin Sp = 9/2.6 In turn, antiparallel (antiferromagnetic) coupling of the pair spins leads to the clusters’ diamagnetic ground state.6,71,75,102,103 The Mössbauer spectrum of 5 was fitted alongside a singlet signal (11%) which can be assigned to monomeric (NEt4)[FeI4]89 (Fig. S8B†). Besides this, the behaviour is the same as for 1. Correspondingly, all Fe atoms in the homoleptic 1 and 5 (Fig. S8†) have an oxidation state of 2.5+.
For both heteroleptic compounds (BTMA)2[Fe4S4Br2Cl2] (2) and (BTMA)2[Fe4S4Br2I2] (4), symmetric Lorentzian-type doublets with δ values of 0.48 mm s−1 were recorded (Fig. S9†). Thus, the average oxidation state of iron in both mixed-halide-clusters is 2.5+ with complete delocalisation.
In contrast, (BTMA)2[Fe4S4Cl2I2] (3) exhibited a slightly asymmetric Mössbauer doublet whose analysis reveals the equal contributions of the two species (Fig. 4).
 | ||
| Fig. 4 57Fe Mössbauer spectrum of (BTMA)2[Fe4S4Cl2I2] (3) at 77 K (dots: collected data, parameters for the simulated species A and B in Table 3). | ||
Species A causes a doublet with δ1 = 0.48 mm s−1 and ΔEQ1 = 1.16 mm s−1. Species B causes an isomer shift with δ2 = 0.46 mm s−1 which is within the experimental error identical to that of species B. Thus, 3 also has all irons in the 2.5+ oxidation state (Table 3). The different values of ΔEQ may be a result of the large differences in the EN of Cl and I.
It should be noted that in contrast to the analysis with two “nested” doublets presented above, the Mössbauer spectrum of 3 can alternatively be fitted with two “crossed” doublets (Fig. S11†). This leads to δA = 0.54 and δB = 0.37 mm s−1 which may indicate electron localisation. However, high spin Fe3+ and Fe2+ sites have significantly different ΔEQ values, and the analysis with two “crossed” doublets shows almost identical ΔEQ values of 1.0 mm s−1 (Fig. S11†) which contradicts electron localisation. Therefore, we conclude that indeed 3 has all irons in the 2.5+ state.
Such a situation has also been observed for [Fe4S4X2Y2]2− clusters with monodentate ligands X, Y = Cl, OPh and SPh show δ values between 0.46 and 0.50 mm s−1 for both coordination sites.56
For (Ph4P)2[Fe4S4Br4], δ = 0.488 mm s−1 and ΔEQ = 0.662 mm s−1 were reported at 77 K.37 A crude measurement of the mixed-halide compound (Ph4P)2[Fe4S4Br2Cl2] was reported with a shift of about 0.00 mm s−1 and significant line-broadening37 similar to that observed for 3.
The high similarity of all clusters in the BMTA+ containing materials in their Mössbauer spectra is in line with the very similar distortion of the clusters from Td to very low symmetry (see the XRD section).
For (BTMA)3[Fe6S6Cl6] (6), an isomer shift of 0.48 mm s−1 was recorded and the oxidation state of each iron inside the prismane core can be assigned to 2.5+. Like the previous prismane cluster, the core of 6 consists of three Fe2+–Fe3+ spin pairs coupling ferromagnetically and resulting in Fe+2.5 on average.86 The observed ΔEQ of 0.88 mm s−1 is smaller than those reported for other prismanes,12,38,85–90,102–108 but overall, the isomer shift and the quadrupole splitting for 6 are very similar to those of the (Et4N)3[Fe6S6Cl6] prismane with δ = 0.495 mm s−1 and ΔEQ = 1.085 mm s−1 at 125 K.86
For fitting the collected data, a second species with a contribution of 4% was necessary. This second doublet was fitted with δ = 1.40, ΔEQ = 1.01, and Γ = 0.40 mm s−1 and is due to the FeCl2 impurities from the starting material (Fig. S10†). For (Ph4P)2[Fe4S4Cl4], a δ value of 0.49 mm s−1 was reported at 77 K with a ΔEQ of 0.67 mm s−1.56,86 Although PXRD showed that the crystal structure of (BTMA)3[Fe6S6Cl6] is only the minor phase in the isolated material, the Mössbauer results provide good evidence that the major component of the material contains the [Fe6S6Cl6]3− prismanes. At the same time, the Mössbauer results do not completely rule out the occurrence of the compound (BTMA)4[Fe4S4Cl4] as the reported data for (Ph4P)2[Fe4S4Cl4] is not very different from our data for 6.
Magnetic measurements
The molar magnetic susceptibilities of all four compounds are shown in Fig. 5 as χMT vs. T plots. All compounds show quite similar magnetic behaviour (Fig. 4 and Fig. S12–S14, ESI†). | ||
| Fig. 5 Temperature dependence of the molar magnetic susceptibility depicted as χMT vs. T of the homoleptic cluster (BTMA)2[Fe4S4Br4] (1, black) and the heteroleptic clusters (BTMA)2[Fe4S4Br2Cl2] (2, green), (BTMA)2[Fe4S4Cl2I2] (3, red), and (BTMA)2[Fe4S4Br2I2] (4, blue). | ||
At 298 K, the observed χMT values are 1.17 cm3 K mol−1, 1.41 cm3 K mol−1, 1.54 cm3 K mol−1 and 1.12 cm3 K mol−1 for 1 to 4, respectively, far below the expected spin-only value of 14.75 cm3 K mol−1 for four uncoupled spins with S1 = S2 = 4/2 (ferrous) and S3 = S4 = 5/2 (ferric).
Upon lowering the temperature, a steady decrease in the χMT-products is observed for all compounds, indicating strong dominant antiferromagnetic interactions between the iron ions. Further cooling below 100 K initially leads to no significant change in the values. However, upon cooling only below 20 K, the χMT values decrease significantly.
For cluster systems with four antiferromagnetically coupled spin centres, an HDvV (Heisenberg–Dirac–van Vleck) spin Hamiltonian,  , with one single exchange interaction parameter J leads to a highly degenerated spin ground state. However, this assumption is incompatible with the results of the Mössbauer investigations and especially with the observed temperature-dependent magnetic moments for the four compounds (Fig. 5).
, with one single exchange interaction parameter J leads to a highly degenerated spin ground state. However, this assumption is incompatible with the results of the Mössbauer investigations and especially with the observed temperature-dependent magnetic moments for the four compounds (Fig. 5).
The application of a full HDdV with two exchange interactions, which includes a stronger antiferromagnetic exchange interaction J1, within the two Fe2+–Fe3+ dimers, and an exchange interaction, J2, accounting for the inter-dimer interaction, allows quite satisfactory simulations for the curves for all four compounds (Fig. 5 and Table 4).
| HDvV | HDE | |||||
|---|---|---|---|---|---|---|
| J 1/cm−1 | J 2/cm−1 | g | J/cm−1 | B/cm−1 | g | |
| a HDvV = Heisenberg–Dirac–van Vleck; HDE = Heisenberg double exchange. | ||||||
| 1 | −129 (3) | −2.6 (3) | 1.775 (16) | −257 (10) | 462.2 (5) | 1.66 (5) |
| 2 | −135 (7) | −3.2 (3) | 2.09 (5) | −256 (10) | 462.5 (5) | 1.91 (6) |
| 3 | −115 (2) | −2.0 (2) | 1.853 (11) | −274 (13) | 554.6 (9) | 1.74 (8) |
| 4 | −132 (1) | −2.8 (2) | 1.759 (6) | −291 (13) | 552.1 (7) | 1.65 (6) |
However, in three of the four fits, a g value considerably below 2.0 was obtained. For the Fe4S4 cluster, this phenomenon is well-known6 and assigned to the presence of spin-dependent delocalisation which is related to the double exchange theory devolved by Anderson and Hasewaga.109 They have shown that resonance delocalisation in a mixed-valence dimer leads to additional splitting ±B(S + 1/2) of the energy-split spin ladder JS(S + 1) for the exchange coupled spin system.
To estimate the double exchange parameter B in the present Fe4S4 tetramers, we followed the approach recently reported by Henthorn, Cutsail et al.110 and applied the solved Bleaney–Bowers equation for dimers of high spin d5–d6 mixed-valence centres (Table 4, eqn (S1), (S2), and Fig. S14†). Note that the data at low temperatures were not taken into account for fitting to E = −JS(S + 1) ± B(S + 1/2). This is in accordance with the literature and the interested reader is referred to the publication and the references therein for further information.110 The values obtained for B are consistent with the reported values6,111–113 in all cases and clearly show the presence of electron delocalisation, which is also evident from the Mössbauer data, showing only a single doublet assigned to a mixed valence state. Most importantly, the ratio |B/J| is relatively small, so the spin ground state of S = 1/2 can be taken as given for the dimers. This justifies the application of the HDdV formalism, which in analogy leads to an S = 1/2 ground state for the antiferromagnetically coupled Fe2+–Fe3+ dimers. Furthermore, with the HDvV-formalism, we are now able to take the inter-dimer interaction into account. Although the values for J2 are almost two orders of magnitude smaller than J1, the resulting final ground states for the Fe4S42+-clusters are diamagnetic with S = 0, which matches perfectly with the decrease in the χMT values at low temperature.
In the temperature-dependent magnetisation measurements for (Et4N)2[Fe4S4Cl4], the magnetic moment μ increased from 0.45 to 2.48 μB upon increasing the temperature from 50 to 338 K,36 which is in good agreement with the behaviour in our compounds.
Conclusions
The iron sulphur halide clusters [Fe4S4Br4]2− and [Fe4S4X2Y2]2− (X, Y = Cl, Br, I) were synthesised in excellent yields and purity on a gram scale using a recently refined method starting from [Fe(CO)5], elemental sulphur, I2 and benzyltrimethylammonium (BTMA+) iodide, bromide and chloride. Through crystallisation, we obtained materials showing a clear 1:1 ratio of X:Y for the mixed-halide cluster compounds. Single crystals of (BTMA)2[Fe4S4Br4] (1), (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Cl2I2] (3) and (BTMA)2[Fe4S4Br2I2] (4) were isostructural to the previously reported (BTMA)2[Fe4S4I4] (5) (monoclinic, Cc). In the same way, we tried to synthesise the previously unknown chloride cubane cluster compound (BTMA)2[Fe4S4Cl4]. But we obtained the prismane-shaped cluster (BTMA)3[Fe6S6Cl6] (6) (P) instead.
No phase transitions were observed for the mixed-halide cluster containing materials from powder XRD at 298 or 170 K or single crystal XRD at 293 and 173 K. Although in solution four different clusters [Fe4S4X4−xYx]2− (x = 0 to 4) might occur for the mixed-halide compounds, the structure refinements gave no indication for the X:Y ratio deviating from 2:2. Also, a closer look on the cluster volumes (solvent excluded volumes, the unit cell volumes and the cluster distortions) gave evidence for other species than X2Y2. On the other hand, our XRD data do not rule out X1Y3 species if they were present in the same amount as X3Y1 species and all cluster species arrange randomly in the structures.
However, Mössbauer spectroscopy rules out larger amounts of such X3Y1 species. For the homoleptic 1 and 5 and the mixed-halide clusters 2, 3, and 4, Lorentzian-type doublets were observed in 57Fe Mössbauer spectroscopy in line with a complete delocalisation with Fe2.5+ oxidation states for all iron atoms.
Magnetic measurements for 1 to 4 showed small χMT values at 298 K ranging from 1.12 to 1.54 cm3 K mol−1. From about 20 K, the values drop significantly. Fitting the data in the Heisenberg–Dirac–van Vleck (HDvV) formalism gave g values markedly below 2 and confirmed the complete delocalisation and antiferromagnetic coupling assumed from Mössbauer spectroscopy.
Thus, the herein described synthesis is reliable and robust and allows the production of mixed-halide clusters [Fe4S4X2Y2]2− (X, Y = Cl, Br, I) on a gram scale and with high purity. In particular, the [Fe4S4Cl2I2]2− derivative might be interesting for further studies as the very different halide ligands might allow the selective substitution of two halide ligands to form new mixed ligand [Fe4S4] clusters.
Experimental section
Materials
The compounds were prepared and crystallised following already published procedures48 by diffusion techniques from layered MeCN/diethyl ether mixtures at −18 °C over a period of 3 weeks. After filtration, washing with diethyl ether and drying under anaerobic and dry atmosphere (argon) using Schlenck techniques the opaque black rod shape crystals were stored and prepared for analyses in an argon-filled glove box at room temperature. The details and analytics are available in the ESI.†Instrumentation
The resonance Raman spectra were recorded either with 457 nm or 514 nm excitation (13 mW, Coherent Innova 70c) in backscattering geometry using a confocal microscope coupled to a single stage spectrograph (Jobin Yvon XY 800) equipped with a CCD detector.For (BTMA)2[Fe4S4Br4] (1), (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Br2I2] (4) and (BTMA)3[Fe6S6Cl6] (6), single crystal data collection at 293(2) K was performed using STOE-IPDS (I, II or IIT) diffractometers using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The samples have been measured in capillaries sealed under an argon atmosphere (0.2 mm diameter). The structures were solved by direct methods using the WinGX114 package and/or Olex2.115 For some of them, first structure proposals were obtained with Sir2014.116 Model refinement was carried out with SHELXL2018/1117,118 by employing full-matrix least-squares methods on F2. All non-hydrogen atoms were treated anisotropic and the hydrogen atoms were included by using appropriate riding models. Numeric absorption correction was performed using X-Red119 and X-Shape120 packages. CCDC Deposition Numbers are 2048374 for (BTMA)2[Fe4S4Br4] (1), 2048953 for (BTMA)2[Fe4S4Br2Cl2] (2), 2055695 for (BTMA)2[Fe4S4Br2I2] (4), and 2048377 for (BTMA)3[Fe6S6Cl6] (6), respectively.†
For (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Cl2I2] (3) and (BTMA)2[Fe4S4Br2I2] (4), LT single crystal data were collected at 173(2) K and for (BTMA)2[Fe4S4Cl2I2] (3) at 293(2) K using an Oxford Diffraction Xcalibur Gemini Eos diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The crystals were transferred under argon to Paratone oil and mounted on a nylon loop. For cooling, cold dry air was provided by an Oxford Cryosystems Desktop Cooler. CrysAlis Pro,121 from Oxford Diffraction, was used to collect the initial frames for determination of the unit cell. Subsequently, the program was used to plan a data collection. In most cases, a full sphere of data were collected. After collection, data reduction was carried out with the Crysalis Pro suite and multiscan absorption correction was carried out (SCALE3 ABSPACK scaling algorithm).122 The space group selection was made using the inbuilt program, GRAL, which selected the monoclinic space group Cc for all structures. The output files were used for initial structure solution using SHELXle123 or Olex2.115 Subsequently, the structures were refined using SHELXL 2018/1.117,118 The mixed-halide clusters showed signs of disorder and this was modelled at first by allowing the occupancies for the halide ligands to refine freely. In the final refinement, the occupancies of the disordered pairs of halides, i.e., bromide and chloride, iodide and chloride, or bromide and iodide, were restrained using the SUMP command (SHELXL).117,118 In all cases, this strategy resulted to be more adequate to treat the data and obtain better quality refinement indicators and realistic Fe–X and X⋯Y distances. The CCDC Deposition Numbers are 2048376, 2048378, 2048375 (173 K) and 2048954 (293 K) for (BTMA)2[Fe4S4Br2Cl2] (2), (BTMA)2[Fe4S4Cl2I2] (3), and (BTMA)2[Fe4S4Br2I2] (4), respectively.†
The selected bond lengths and angles of the compounds were calculated using the PLATON software.124 The graphical representations were done by Mercury125 and PyMOL,126 and the editing of CIF files was performed using the PublCif127 software.
Synchrotron X-ray powder diffraction data collection was performed at DESY (Hamburg, beamline P02.1, storage ring Petra III with λ = 0.207203 Å and a PerkinElmer XRD 1621 flat panel detector). The samples were measured in capillaries sealed under an argon atmosphere (0.5 mm diameter). The collected data was transformed with Fit2d128 and analysed with WinXPow.129
The 57Fe Mössbauer data were recorded in the transmission geometry using a constant acceleration spectrometer operated in conjunction with a 512-channel analyser in the time-scale mode (WissEl GmbH) using Wissoft 2003. The detector consisted of a proportional counter and the source contained 57Co diffused in Rh with an activity of 1.4 GBq. The spectrometer was calibrated against α-iron at room temperature. Sample cooling to 77 K was achieved by placing the samples in a continuous flow cryostat from Oxford Instruments. The spectral data were transferred from the multi-channel analyser to a PC for further analysis employing the public domain program Vinda130 running on the Microsoft Excel 2003® platform. The spectra were analysed by least-squares fits using Lorentzian line shapes.
The magnetic measurements were performed with a Quantum Design MPMS-XL-7 SQUID magnetometer using powdered microcrystalline samples. The variable temperature susceptibility data were collected in the temperature range of 2 to 300 K under an applied field of 0.1 Tesla. The experimental susceptibility data were corrected for the temperature-dependent magnetic contribution of the holder and the underlying diamagnetism using Pascal's constants.
Author contributions
AOS: investigation, data curation, visualisation, writing – original draft, and funding acquisition. BMR: investigation, data curation, and visualisation. FDS: data curation, visualisation, and supervision. LC: investigation, data curation, validation, and visualisation. VKG: investigation, data curation, and visualisation. FD: methodology, validation, project administration, supervision, and funding acquisition. ERE: methodology, validation, and supervision. VS: methodology, validation, and supervision. UR: methodology, validation, project administration, supervision, funding acquisition, and writing – review and editing. AK: methodology, validation, project administration, supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Peter Kliesen, Malgorzata Smolarek and Silke Kremer (Department of Chemistry, University of Cologne) for single crystal X-ray diffraction, IR spectroscopy and microanalysis data collection. We are indebted to HASYLAB at DESY (Hamburg, Germany) for the kind support with synchrotron radiation. AOS acknowledges the German Academic Exchange Programme (DAAD) for traveling grants (EuroBIC 10 Thessaloniki, Greece & EuroBIC 11 Granada, Spain) and CONICET for financial support (Código de Beca: 105 201301 00912 CO).References
- W. Kaim, B. Schwederski and A. Klein, Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life, Wiley Chichester, 2nd edn, 2013 Search PubMed.
- R. Lill and S.-A. Freibert, Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis, Annu. Rev. Biochem., 2020, 89, 471–499 CrossRef.
- C. C. Lee, M. T. Stiebritz and Y. Hu, Reactivity of [Fe4S4] Clusters toward C1 Substrates: Mechanism, Implications, and Potential Applications, Acc. Chem. Res., 2019, 52, 1168–1176 CrossRef CAS.
- M. K. Johnson and A. D. Smith, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, Hoboken NJ, USA, 2011, pp. 2589–2619 Search PubMed.
- C. Greco, M. Bruschi, P. Fantucci, U. Ryde and L. De Gioia, Mechanistic and Physiological Implications of the Interplay among Iron–Sulfur Clusters in [FeFe]-Hydrogenases. A QM/MM Perspective, J. Am. Chem. Soc., 2011, 133, 18742–18749 CrossRef CAS.
- H. Beinert, R. H. Holm and E. Münck, Iron-Sulfur Clusters: Nature's Modular, Multipurpose Structures, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Structure, Function, and Formation of Biological Iron-Sulfur Clusters, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS PubMed.
- H. Beinert, Iron-sulfur proteins: ancient structures, still full of surprises, J. Biol. Inorg. Chem., 2000, 5, 2–15 CrossRef CAS PubMed.
- B. Srour, S. Gervason, B. Monfort and B. D'Autréaux, Mechanism of Iron–Sulfur Cluster Assembly: In the Intimacy of Iron and Sulfur Encounter, Inorganics, 2020, 8, 55 CrossRef CAS.
- S. J. Mayr, R.-R. Mendel and G. Schwarz, Molybdenum cofactor biology, evolution and deficiency, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868, 118883 CrossRef CAS.
- Y. Bai, T. Chen, T. Happe, Y. Lu and A. Sawyer, Iron–sulphur cluster biogenesis via the SUF pathway, Metallomics, 2018, 10, 1038–1052 CrossRef.
- A. McSkimming and D. L. M. Suess, Selective Synthesis of Site-Differentiated Fe4S4 and Fe6S6 Clusters, Inorg. Chem., 2018, 57, 14904–14912 CrossRef CAS.
- S. Ohta and Y. Ohki, Impact of Ligands and Media on the Structure and Properties of Biological and Biomimetic Iron-Sulfur Clusters, Coord. Chem. Rev., 2017, 338, 207–225 CrossRef CAS.
- R. H. Holm and W. Lo, Structural Conversions of Synthetic and Protein-Bound Iron−Sulfur Clusters, Chem. Rev., 2016, 116, 13685–13713 CrossRef CAS PubMed.
- S. C. Lee, W. Lo and R. H. Holm, Developments in the Biomimetic Chemistry of Cubane-Type and Higher Nuclearity Iron–Sulfur Clusters, Chem. Rev., 2014, 114, 3579–3600 CrossRef CAS PubMed.
- K. Tanifuji, N. Yamada, T. Tajima, T. Sasamori, N. Tokitoh, T. Matsuo, K. Tamao, Y. Ohki and K. Tatsumi, A Convenient Route to Synthetic Analogues of the Oxidized Form of High-Potential Iron–Sulfur Proteins, Inorg. Chem., 2014, 53, 4000–4009 CrossRef CAS PubMed.
- A. Alwaaly, I. Dance and R. A. Henderson, Unexpected explanation for the enigmatic acid-catalysed reactivity of [Fe4S4X4]2− clusters, Chem. Commun., 2014, 50, 4799–4802 RSC.
- I. Dance and R. A. Henderson, Large structural changes upon protonation of Fe4S4 clusters: the consequences for reactivity, Dalton Trans., 2014, 43, 16213–16226 RSC.
- I. Span, K. Wang, W. Wang, J. Jauch, W. Eisenreich, A. Bacher, E. Oldfield and M. Groll, Structures of Fluoro, Amino, and Thiol Inhibitors Bound to the [Fe4S4] Protein IspH, Angew. Chem., Int. Ed., 2013, 52, 2118–2121 CrossRef CAS PubMed.
- D. L. Gerlach, D. Coucouvanis and N. Lehnert, Connecting [4Fe–4S] Clusters and Hemes – Towards Modeling the Active Site of Sulfite Reductase, Eur. J. Inorg. Chem., 2013, 2013, 3883–3890 CrossRef CAS.
- D. L. Gerlach, D. Coucouvanis, J. Kampf and N. Lehnert, Isolation and Characterization of Single and Sulfide-Bridged Double [4Fe–4S] Cubane Clusters with 4-Pyridinethiolato Ligands, Eur. J. Inorg. Chem., 2013, 2013, 5253–5264 CrossRef CAS.
- P. V. Rao and R. H. Holm, Synthetic Analogues of the Active Sites of Iron-Sulfur Proteins, Chem. Rev., 2004, 104, 527–559 CrossRef CAS.
- R. H. Holm, in Comprehensive Coordination Chemistry II, Pergamon, Oxford, 2003, 61–90 Search PubMed.
- R. H. Holm, [1:3] Site-differentiated and sulfide-bridged cubane clusters in chemistry and biology, Pure Appl. Chem., 1998, 70, 931–938 CrossRef CAS.
- T. D. P. Stack, J. A. Weigel and R. H. Holm, The Cavitand Concept in the Synthesis of Subsite-Differentiated Analogues of Biological [4Fe-4S/Se]2+ Clusters: Cluster Capture Reactions, Ligand Conformational Analysis, and the Structure of a Trigonal [4Fe-4Se]2+ Analogue, Inorg. Chem., 1990, 29, 3745–3760 CrossRef CAS.
- R. H. Holm, Synthetic approaches to the active sites of iron-sulfur proteins, Acc. Chem. Res., 1977, 10, 427–434 CrossRef CAS.
- Y. Shim, R. M. Young, A. P. Douvalis, S. M. Dyar, B. D. Yuhas, T. Bakas, M. R. Wasielewski and M. G. Kanatzidis, Enhanced Photochemical Hydrogen Evolution from Fe4S4-Based Biomimetic Chalcogels Containing M2+ (M = Pt, Zn, Co, Ni, Sn) Centers, J. Am. Chem. Soc., 2014, 136, 13371–13380 CrossRef CAS PubMed.
- Y. Shim, B. D. Yuhas, S. M. Dyar, A. L. Smeigh, A. P. Douvalis, M. R. Wasielewski and M. G. Kanatzidis, Tunable Biomimetic Chalcogels with Fe4S4 Cores and [SnnS2n+2]4− (n = 1, 2, 4) Building Blocks for Solar Fuel Catalysis, J. Am. Chem. Soc., 2013, 135, 2330–2337 CrossRef PubMed.
- J. B. Varley, H. A. Hansen, N. L. Ammitzøll, L. C. Grabow, J. Rossmeisl and J. K. Nørskov, Ni−Fe−S Cubanes in CO2 Reduction Electrocatalysis: A DFT Study, ACS Catal., 2013, 3, 2640–2643 CrossRef.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Nitrogenase-mimic iron-containing chalcogels for photochemical reduction of dinitrogen to ammonia, Proc. Natl. Acad. Sci. U. S. A., 2016, 133, 5530–5535 CrossRef.
- V. C.-C. Wang, M. Can, E. Pierce, S. W. Ragsdale and F. A. Armstrong, A Unified Electrocatalytic Description of the Action of Inhibitors of Nickel Carbon Monoxide Dehydrogenase, J. Am. Chem. Soc., 2013, 135, 2198–2206 CrossRef PubMed.
- B. D. Yuhas, A. L. Smeigh, A. P. Samuel, Y. Shim, S. Bag, A. P. Douvalis, M. R. Wasielewski and M. G. Kanatzidis, Biomimetic Multifunctional Porous Chalcogels as Solar Fuel Catalysts, J. Am. Chem. Soc., 2011, 133, 7252–7255 CrossRef PubMed.
- C. J. Pickett, S. K. Ibrahim and D. L. Hughes, Polyferredoxin-based electrode materials, Faraday Discuss., 2000, 116, 235–244 RSC.
- M. Akhtar, J. Akhter, M. A. Malik, P. O'Brien, F. Tuna, J. Raftery and M. Helliwell, Deposition of iron sulfide nanocrystals from single source precursors, J. Mater. Chem., 2011, 21, 9737–9745 RSC.
- P. V. Vanitha and P. O'Brien, Phase Control in the Synthesis of Magnetic Iron Sulfide Nanocrystals From a Cubane-Type Fe−S Cluster, J. Am. Chem. Soc., 2008, 130, 17256–17257 CrossRef CAS.
- G. B. Wong, M. A. Bobrik and R. H. Holm, Inorganic Derivatives of Iron Sulfide Thiolate Dimers and Tetramers: Synthesis and Properties of the Halide Series [Fe2S2X4]2− and [Fe4S4X4]2− (X = Cl, Br, I), Inorg. Chem., 1978, 17, 578–584 CrossRef CAS.
- A. Müller, N. H. Schladerbeck, E. Krickemeyer, H. Bögge, K. Schmitz, E. Bill and A. X. Trautwein, Darstellung von Metall-Schwefel-Clustern durch einfache Reaktion von Metallsalzen mit H2S: R2(NH4)[Cu3S12], R2[Cu4S12].CH3CN, R2[Cu4S12.8], R2[Fe2S2(S5)2], R2[Fe4S4Br4], R2[Fe4S4Br2Cl2], R2[Fe4S4Cl4], R2[Fe2S2Cl4] (R = PPh4) und [Fe(DMF)6][Fe2S2Cl4], Z. Anorg. Allg. Chem., 1989, 570, 7–36 CrossRef.
- W. Saak and S. Pohl, Iodine Substituted Iron-Sulfur-Clusters: Novel Syntheses, Formation and Stability of Fe2S2I42−, Fe4S4I42− and Fe6S6I62−. The Crystal Structure of (Et4N)6(Fe4S4I4)2Fe2S2I4, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40b, 1105–1112 CrossRef CAS.
- W. Saak and S. Pohl, Fe4S4I2(SPPh3)2: a Neutral, Mixed Terminal Ligand Iron-Sulfur Cluster, Z. Naturforsch., B: J. Chem. Sci., 1988, 43b, 813–817 CrossRef.
- S. Pohl and W. Saak, Structural Distortion of Fe4S4I42− Clusters through Iodine-Iodine Contacts: Crystal Structures of (Ph4P)2Fe4S4I4 and (Me3NCH2Ph)2Fe4S4I4, Z. Naturforsch., B: J. Chem. Sci., 1988, 43b, 457–462 CrossRef.
- B. M. Segal, H. R. Hoveyda and R. H. Holm, Terminal Ligand Assignments Based on Trends in Metal-Ligand Bond Lengths of Cubane-Type [Fe4S4]2+,+ Clusters, Inorg. Chem., 1998, 37, 3440–3443 CrossRef CAS.
- M. A. Bobrik, K. O. Hodgson and R. H. Holm, Inorganic Derivatives of Iron-Sulfide-Thiolate Dimers and Tetramers. Structures of Tetrachloro-p-disulfido-diferrate(III) and Tetrakis(chloro-μ3-sulfido-iron) Dianions, Inorg. Chem., 1977, 16, 1851–1858 CrossRef CAS.
- K. Bates, L. Johnson and R. A. Henderson, Mechanism of the Reactions of Synthetic Fe-S-Based Clusters with PhCOCl: Parallel Pathways Involving Free and Coordinated Thiolate as Nucleophiles, Inorg. Chem., 2006, 45, 9423–9433 CrossRef CAS.
- K. Bates, M. Wouldhave and R. A. Henderson, Involvement of thiolate ligands in binding substrates to Fe–S clusters, Dalton Trans., 2008, 2008, 6527–6529 RSC.
- K. Bates and R. A. Henderson, Binding Nucleophiles to [Fe4Y4Cl4]2− (Y = S or Se) can Increase or Suppress the Rate of Proton Transfer to the Cluster, Inorg. Chem., 2008, 47, 5850–5858 CrossRef CAS PubMed.
- A. Kern, C. Näther and F. Tuczek, Application of a Universal Force Field to Mixed Fe/Mo-S/Se Cubane and Heterocubane Clusters. 1. Substitution of Sulfur by Selenium in the Series [Fe4X4(YCH3)4]2−; X = S/Se and Y = S/Se, Inorg. Chem., 2004, 43, 5003–5010 CrossRef CAS PubMed.
- A. Kern, C. Näther and F. Tuczek, Application of a Universal Force Field to Mixed Fe/Mo-S/Se Cubane and Heterocubane Clusters. 2. Substitution of Iron by Molybdenum in Fe4(S/Se)4 Clusters with Terminal Halide and Thiolate Ligands, Inorg. Chem., 2004, 43, 5011–5020 CrossRef CAS.
- A. O. Schüren, V. K. Gramm, M. Dürr, A. Foi, I. Ivanović-Burmazović, F. Doctorovich, U. Ruschewitz and A. Klein, Halide coordinated homoleptic [Fe4S4X4]2− and heteroleptic [Fe4S4X2Y2]2− clusters (X, Y = Cl, Br, I)—alternative preparations, structural analogies and spectroscopic properties in solution and solid state, Dalton Trans., 2016, 45, 361–375 RSC.
- T. M. M. Al-Rammahi, P. G. Waddell and R. A. Henderson, X-ray crystal structures of [NHR3]2[Fe4S4X4] (X = PhS, R = Et or nBu; X = Cl, R = nBu): implications for sites of protonation in Fe–S clusters, Transition Met. Chem., 2016, 41, 1–7 CrossRef.
- P. K. Mascharak, K. S. Hagen, J. T. Spence and R. H. Holm, Structural Distortions of the [Fe4S4]2+ Core of [Fe4S4(S-t-C4H9)4]2− in Different Crystalline Environments and Detection and Instability of Oxidized ([Fe4S4]3+) Clusters, Inorg. Chim. Acta, 1983, 80, 157–170 CrossRef CAS.
- K. S. Hagen, J. G. Reynolds and R. H. Holm, Definition of Reaction Sequences Resulting in Self-Assembly of [Fe4S4(SR)4]2− Clusters from Simple Reactants, J. Am. Chem. Soc., 1981, 103, 4054–4063 CrossRef CAS.
- J. B. Willems and M. Köckerling, Synthesis and structure of an unprecedented high-symmetry [Fe4S4Cl4] cubane-type unit in supramolecular [K4(FeCl4)(C12H24O6)4][Fe4S4Cl4], Chem. Commun., 2001, 2001, 1380–1381 RSC.
- M. Kawano, C. Hoshino, K. Sakai and K. Matsumoto, Crystal Structure of cis-Bis(acetonitrile)tetrakis(trimethylphosphite)iron(II) Tetrachlorotetrakis(μ3-sulfido)tetra-ferrate (2II, 2III), Anal. Sci., 1991, 7, 829–830 CrossRef CAS.
- A. C. M. Young, M. A. Walters and J. C. Dewan, Structure of [N(C4H9)4]2[Fe(C3H7NO)6][Fe4Br4S4]2, Acta Crystallogr., Sect. C: Struct. Chem., 1989, 45, 1733–1736 CrossRef.
- D. Coucouvanis, M. G. Kanatzidis, E. Simhon and N. C. Baenziger, Synthesis, Molecular Structure, and Reactions of Bis(tetraphenylphosphonium) Hexakis(μ-thiophenolato)-tetrachlorotetraferrate(II), (Ph4P)2[Fe4(SPh)6Cl4]. Its Reactions with Dibenzyl Trisulfide and the Synthesis of the [Fe4S4Cl4]2− and [Fe4S4(Cl)2(SC6H5)2]2− “Cubane”-Type Clusters, J. Am. Chem. Soc., 1982, 104, 1874–1882 CrossRef CAS.
- M. G. Kanatzidis, N. C. Baenziger, D. Coucouvanis, A. Simopoulos and A. Kostikas, Synthesis, Structural -Characterization, and Electronic Structures of the “Mixed” Terminal Ligand Cubanes [Fe4S4Cl2(XC6H5)2]2− (X = S, O) and [Fe4S4(SC6H5)2(OC6H4-p-CH3)2]2−. The First Examples of [Fe4S4]2+ Cores with a Noncompressed D2d Idealized Geometry, J. Am. Chem. Soc., 1984, 106, 4500–4511 CrossRef.
- M. G. Kanatzidis, D. Coucouvanis, A. Simopoulos, A. Kostikas and V. Papaefthymiou, Synthesis, Structural Characterization, and Electronic Properties of the Ph4P+ Salts of the Mixed Terminal Ligand Cubanes Fe4S4(Et2Dtc)n(X)4−n2− (X = Cl−, PhS−) (n = 1, 2). Two Different Modes of Ligation on the [Fe4S4]2+ Core, J. Am. Chem. Soc., 1985, 107, 4925–4935 CrossRef.
- M. Kanatzidis, M. Ryan, D. Coucouvanis, A. Simopoulos and A. Kostikas, Synthesis and Structural Characterization of Bis(tetraphenylphosphonium) Bis(diethyldithiocarbamato)bis(thiophenolato)tetrakis(μ3-sulfido)tetraferrate(2II,2III), (Ph4P)2[Fe4S4(SPh)2(Et2dtc)2]. A “Cubane” Type Cluster with Mixed Terminal Ligands and Two Different Modes of Ligation on the Fe4S4 Core, Inorg. Chem., 1983, 22, 179–181 CrossRef.
- S. Rutchik, S. Kim and M. A. Walters, Facile One-Step Synthesis of Fe4Q42+ (Q = S, Se) Cubane-like Centers, Inorg. Chem., 1988, 27, 1513–1516 CrossRef.
- S. Pohl and U. Bierbach, [Fe4S4]2+-Clusters with Thiourea Ligands: [FeIL3][Fe4S4I3L] (L = (Me2N)2CS) and [Fe4S4I2L’2] (L′ = (C4H9NH)2CS), Z. Naturforsch., B: J. Chem. Sci., 1991, 46b, 68–74 CrossRef.
- U. Bierbach, W. Saak, D. Haase and S. Pohl, Neutral and Cationic Iron-Sulfur Complexes and Clusters - Syntheses and Crystal Structures of [Fe(SR)2L2]·PhMe and [Fe4S4(SR)2L2], and on the Formation of [Fe(SR)L3]+, [FeL4]2+, [Fe4S4(SR)L3]+ and [Fe4S4L4]2+ (R = 2,4,6-i-Pr3C6H2; L = SC(NMe2)2), Z. Naturforsch., B: J. Chem. Sci., 1991, 46b, 1629–1634 CrossRef.
- P. Zanello, The competition between chemistry and biology in assembling iron–sulfur derivatives. Molecular structures and electrochemistry. Part V. {[Fe4S4](SγCys)4} proteins, Coord. Chem. Rev., 2017, 335, 172–227 CrossRef.
- T. Terada, K. Hirabayashi, D. Liu, T. Nakamura, T. Wakimoto, T. Matsumoto and K. Tatsumi, [3:1] Site-Differentiated [4Fe−4S] Clusters Having One Carboxylate and Three Thiolates, Inorg. Chem., 2013, 52, 11997–12004 CrossRef.
- L. Deng, A. Majumdar, W. Lo and R. H. Holm, Stabilization of 3:1 Site-Differentiated Cubane-Type Clusters in the [Fe4S4]1+ Core Oxidation State by Tertiary Phosphine Ligation: Synthesis, Core Structural Diversity, and S = 1/2 Ground States, Inorg. Chem., 2010, 49, 11118–11126 CrossRef.
- E. P. L. van der Geer, Q. Li, G. van Koten, R. J. M. Klein Gebbink and B. Hessen, N-Substituted indole-3-thiolate [4Fe–4S] clusters with a unique and tunable combination of spectral and redox properties, Inorg. Chim. Acta, 2008, 361, 1811–1818 CrossRef.
- C. Walsdorff, W. Saak and S. Pohl, A new preorganized tridentate ligand bearing three indolethiolategroups. Preparation of 3:1 subsite-differentiated Fe4S4 clusters, J. Chem. Soc., Dalton Trans., 1997, 1857–1861 RSC.
- C. Zhou and R. H. Holm, Comparative Isotropic Shifts, Redox Potentials, and Ligand Binding Propensities of [1:3] Site-Differentiated Cubane-Type [Fe4Q4]2+ Clusters (Q=S, Se), Inorg. Chem., 1997, 36, 4066–4077 CrossRef.
- J. Zhou, Z. Hu, E. Münck and R. H. Holm, The Cuboidal Fe3S4 Cluster: Synthesis, Stability, and Geometric and Electronic Structures in a Non-Protein Environment, J. Am. Chem. Soc., 1996, 118, 1966–1980 CrossRef.
- F. Osterloh, W. Saak and S. Pohl, Unidentate and Bidentate Binding of Nickel(II) Complexes to an Fe4S4 Cluster Via Bridging Thiolates: Synthesis, Crystal Structures, and Electrochemical Properties of Model Compounds for the Active Sites of Nickel Containing CO Dehydrogenase/Acetyl-CoA Synthase, J. Am. Chem. Soc., 1997, 119, 5648–5656 CrossRef.
- C. Goh, J. A. Weigel and R. H. Holm, The [2:2] Site-Differentiated Clusters [Fe4S4L2(RNC)6] Containing Two Low-Spin Iron(II) Sites, Inorg. Chem., 1994, 33, 4861–4868 CrossRef.
- J. A. Weigel, K. K. P. Srivastava, E. P. Day, E. Münck and R. H. Holm, Isonitrile Binding to a Site-Differentiated Synthetic Analogue of Biological [4Fe-4S] Clusters: Equilibria, Magnetic Interactions and the Spin-Isolated [3Fe-4S] Cluster Fragment, and the Structure of a Low-Spin Iron(II) Subsite, J. Am. Chem. Soc., 1990, 112, 8015–8023 CrossRef.
- J.-M. Mouesca and B. Lamotte, Iron–sulfur clusters and their electronic and magnetic properties, Coord. Chem. Rev., 1998, 178–180, 1573–1614 CrossRef.
- R. E. Johnson, G. C. Papaefthymiou, R. B. Frankel and R. H. Holm, Effects of Secondary Bonding Interactions on the [Fe4S4]2+ Core of Ferredoxin Site Analogues: [Fe4S4(SC6H4-o-OH)4]2−, a Distorted Cubane-Type Cluster with One Five-Coordinate Iron Atom, J. Am. Chem. Soc., 1983, 105, 7280–7287 CrossRef.
- W. E. Cleland, D. A. Holtman, M. Sabat, J. A. Ibers, G. C. DeFotis and B. A. Averill, Effects of Phenoxide Ligation on Iron-Sulfur Clusters. Preparation and Properties of [Fe4S4(OAr)4]2− Ions and the Structure of [(C2H5)4N]2[Fe4S4(OC6H5)4], J. Am. Chem. Soc., 1983, 105, 6021–6031 CrossRef.
- S. J. Yoo, Z. Hu, C. Goh, E. L. Bominaar, R. H. Holm and E. Münck, Determination of the Exchange-Coupling Constant of an Fe3+-Fe3+ Pair in a Cubane-Type Iron-Sulfur Cluster, J. Am. Chem. Soc., 1997, 119, 8732–8733 CrossRef.
- E. L. Bominaar, S. A. Borshch and J.-J. Girerd, Double-Exchange and Vibronic Coupling in Mixed-Valence Systems. Electronic Structure of [Fe4S4]3+ Clusters in High-Potential Iron Protein and Related Models, J. Am. Chem. Soc., 1994, 116, 5362–5312 CrossRef.
- G. C. Papaefthymiou, E. J. Laskowski, S. Frota-Pessoa, R. B. Frankel and R. H. Holm, Antiferromagnetic Exchange Interactions in [Fe4S4(SR)4]2 Clusters, Inorg. Chem., 1982, 21, 1723–1728 CrossRef.
- L. M. Lawson Daku, J. Pecaut, A. Lenormand-Foucaut, B. Vieux-Melchior, P. Iveson and J. Jordanov, Investigation of the Reduced High-Potential Iron−Sulfur Protein from Chromatium vinosum and Relevant Model Compounds: A Unified Picture of the Electronic Structure of [Fe4S4]2+ Systems through Magnetic and Optical Studies, Inorg. Chem., 2003, 42, 6824–6850 CrossRef.
- K. Kimura, H. Inokuchi, H. Nanami and T. Yagi, Magnetic Susceptibility of Hydrogenase from Desulfovibrio vulgaris, J. Biochem., 1985, 97, 1831–1833 CrossRef PubMed.
- M. Belinskii, Spin coupling model for tetrameric iron clusters in ferredoxins. II. Hyperfine interactions, magnetism, high-spin systems, Chem. Phys., 1993, 172, 213–238 CrossRef.
- T. D. P. Stack and R. H. Holm, Subsite-Specific Functionalization of the [4Fe-4S]2+ Analogue of Iron-Sulfur Protein Clusters, J. Am. Chem. Soc., 1987, 109, 2546–2547 CrossRef.
- E. Victor and S. J. Lippard, A Tetranitrosyl [4Fe−4S] Cluster Forms En Route to Roussin's Black Anion: Nitric Oxide Reactivity of [Fe4S4(LS3)L′]2−, Inorg. Chem., 2014, 53, 5311–5320 CrossRef PubMed.
- C. Walsdorff, W. Saak, D. Haase and S. Pohl, A preorganised doubly tripodal hexathiol: syntheses and crystal structures of complexes with two 3:1 subsite-differentiated Fe4S4 clusters, Chem. Commun., 1997, 1997, 1931–1932 RSC.
- K. S. Hagen and M. Uddin, A Continuous Symmetry Measure of [4Fe-4S]+ Core Distortions and Analysis of Supramolecular Synthons in Crystal Structures of (Et4N)3[Fe4S4Cl4]·Et4NCl at 100 and 295 K, Inorg. Chem., 2008, 47, 11807–11815 CrossRef.
- M. G. Kanatzidis, A. Salifoglou and D. Coucouvanis, Chemistry of [Fe6S6]3+ Prismatic Cages. Synthesis, Structural Characterization, and Electronic Structures of the [Et4N]3[Fe6S6L6] Clusters (L = p-CH3C6H4O−,Br−), Inorg. Chem., 1986, 25, 2460–2468 CrossRef.
- M. G. Kanatzidis, W. R. Hagen, W. R. Dunham, R. K. Lester and D. Coucouvanis, Metastable Fe/S Clusters. The Synthesis, Electronic Structure, and Transformations of the [Fe6S6(L)6]3− Clusters (L=Cl−, Br−, I−, RS−, RO−) and the Structure of [(C2H5)4N]3[Fe6S6Cl6], J. Am. Chem. Soc., 1985, 107, 953–961 CrossRef.
- D. Coucouvanis, M. G. Kanatzidis, A. Salifoglou and W. R. Dunham, Spectroscopic and Structural Evidence of Temperature Dependent Charge Localization and Structural Differentiation of the Fe Sites within the [Fe6S6X6]2− Clusters (X = Cl, Br), J. Am. Chem. Soc., 1987, 109, 6863–6865 CrossRef.
- M. G. Kanatzidis, W. R. Dunham, W. R. Hagen and D. Coucouvanis, A New Iron-Sulphide Cluster containing the ‘Prismane’ [Fe6(μ-S)6]3+ Core. Synthesis, Structure, and Properties of [Et4N]3[Fe6S6Cl6], J. Chem. Soc., Chem. Commun., 1984, 1984, 356–358 RSC.
- D. J. Evans, G. García, M. D. Santana and M. C. Torralba, About the synthesis of the prismane [NEt4]2[Fe6S6I6], Inorg. Chim. Acta, 1999, 284, 296–299 CrossRef CAS.
- D. Coucouvanis, M. G. Kanatzidis, W. R. Dunham and W. R. Hagen, Oxidative Transformation of the [Fe4S4X4]2− “Cubanes” to the [Fe6S6X6]2− “Prismane” Clusters (X = Cl, Br). The Crystal and Molecular Structure of [(C6H5)4P]2Fe6S6Cl6], J. Am. Chem. Soc., 1984, 106, 7998–7999 CrossRef CAS.
- W. Saak and S. Pohl, Spontaneous Self Assembly of an Octanuclear Iron-Nickel Cluster by Reaction of [Fe6S6I6]2− with [NiI4]2−, Angew. Chem., Int. Ed. Engl., 1991, 30, 881–883 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Part B, John Wiley & Sons, 5. edn, 1997 Search PubMed.
- D. Mitra, S. J. George, Y. Guo, S. Kamali, S. Keable, J. W. Peters, V. Pelmenschikov, D. A. Case and S. P. Cramer, Characterization of [4Fe-4S] Cluster Vibrations and Structure in Nitrogenase Fe Protein at Three Oxidation Levels via Combined NRVS, EXAFS, and DFT Analyses, J. Am. Chem. Soc., 2013, 135, 2530–2543 CrossRef CAS.
- Y. Xiao, M. Koutmos, D. A. Case, D. Coucouvanis, H. Wang and S. P. Cramer, Dynamics of an [Fe4S4(SPh)4]2− cluster explored via IR, Raman, and nuclear resonance vibrational spectroscopy (NRVS)-analysis using 36S substitution, DFT calculations, and empirical force fields, J. Chem. Soc., Dalton Trans., 2006, 2192–2201 RSC.
- T. Steiner, The hydrogen bond in the solid state, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- A. Bondi, van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. S. Rowland and R. Taylor, Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- M. L. Connolly, Computation of molecular volume, J. Am. Chem. Soc., 1985, 107, 1118–1124 CrossRef CAS.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- U. Flörke , CSD Communication 2013 (Private Communication), ID code: ZIXBAL.
- U. Flörke, M. Ayaz and G. Henkel, CSD Communication 2016 (Private Communication). ID code: KABKUW01.
- V. Schünemann, From Small Molecules to Complex Systems: A Survey of Chemical and Biological Applications of the Mössbauer Effect, Top. Appl. Phys., 2021, 137, 173–219 CrossRef.
- A. X. Trautwein, E. Bill, E. L. Bominaar and H. Winkler, Iron-containing proteins and related analogs—complementary Mössbauer, EPR and magnetic susceptibility studies, Struct. Bonding, 1991, 78, 1–95 CrossRef CAS.
- M. Kröckel, A. X. Trautwein, H. Winkler, D. Coucouvanis, A. Kostikas and V. Papaefthymiou, A synthetic iron-containing prismane complex: peculiar electron-delocalization and spin-coupling properties deduced from Mössbauer and magnetization studies, Inorg. Chim. Acta, 1998, 283, 111–117 CrossRef.
- G. P. F. van Strijdonck, J. A. E. H. van Haare, P. J. M. Honen, R. C. G. M. van den Schoor, M. C. Feiters, J. G. M. van der Linden, J. J. Steggerda and R. J. M. Nolte, Cyclotriveratrylene models for [4Fe–4S] proteins: 3:1 subsite differentiation and modulation of the redox potential, J. Chem. Soc., Dalton Trans., 1997, 1997, 449–462 RSC.
- S. Ciurli, M. Carrié, J. A. Weigel, M. J. Carney, T. D. P. Stack, G. C. Papaefthymiou and R. H. Holm, Subsite-Differentiated Analogues of Native [4Fe-4S]2+ Clusters: Preparation of Clusters with Five- and Six-Coordinate Subsites and Modulation of Redox Potentials and Charge Distributions, J. Am. Chem. Soc., 1990, 112, 2654–2664 CrossRef CAS.
- F. Osterloh, B. M. Segal, C. Achim and R. H. Holm, Reduced Mono-, Di-, and Tetracubane-Type Clusters Containing the [MoFe3S4]2+Core Stabilized by Tertiary Phosphine Ligation, Inorg. Chem., 2000, 39, 980–989 CrossRef CAS PubMed.
- D. Coucouvanis, A. Salifoglou, M. G. Kanatzidis, W. R. Dunham, A. Simopoulos and A. Kostikas, Synthesis, Structural Characterization, and Electronic Properties of the [Fe6S6X6(M(CO)3)2]n− Anions (M = Mo, W; n = 3, 4; X = Cl, Br, I). Heteronuclear Clusters of Possible Structural Relevance to the Fe/Mo/S Center in Nitrogenase, Inorg. Chem., 1988, 27, 4066–4077 CrossRef CAS.
- P. W. Anderson and H. Hasegawa, Considerations on double exchange, Phys. Rev., 1955, 100, 675–681 CrossRef CAS.
- J. T. Henthorn, G. E. Cutsail, T. Weyhermüller and S. DeBeer, Stabilization of intermediate spin states in mixed-valent diiron dichalcogenide complexes, Nat. Chem., 2022, 14, 328–333 CrossRef CAS PubMed.
- G. Blondin and J.-J. Girerd, Value of the β transfer integral in Fe-S clusters, JBIC, J. Biol. Inorg. Chem., 1996, 1, 170–172 CrossRef.
- M. Kröckel, M. Grodzicki, V. Papaefthymiou, A. X. Trautwein and A. Kostikas, Tuning of electron delocalization in polynuclear mixed-valence clusters by super-exchange and double exchange, JBIC, J. Biol. Inorg. Chem., 1996, 1, 173–176 CrossRef.
- L. Noodleman, D. A. Case, J.-M. Mouesca and B. Lamotte, Valence electron delocalization in polynuclear iron-sulfur clusters, JBIC, J. Biol. Inorg. Chem., 1996, 1, 177–182 CrossRef.
- L. J. Farrugia, WinGX and ORTEP for Windows: an update, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2 : A Complete Structure Solution, Refinement and Analysis Program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, G. Crystal structure determination and refinement via SIR2014, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef.
- G. M. Sheldrick, SHELXT – Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- STOE X-RED, Data Reduction Program, Version 1.31/Windows, STOE & Cie, Darmstadt, Germany, 2005 Search PubMed.
- STOE X-SHAPE, Crystal Optimisation for Numerical Absorption Correction, Version 1.06/Windows, STOE & Cie, Darmstadt, Germany, 1999.
- CrysAlis: Agilent, CrysAlis PRO, Agilent Technologies Ltd Version 1.171.36.28, Oxfordshire, England, 2013 Search PubMed.
- Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm within CrysAlisPro software, Version 1.171.34.41, Oxford Diffraction Ltd.
- C. B. Hübschle, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Cryst., 2011, 44, 1281–1284 CrossRef PubMed.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, Mercury: visualization and analysis of crystal structures, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC.
- S. P. Westrip, publCIF: software for editing, validating andformatting crystallographic information files, J. Appl. Crystallogr., 2010, 43, 920–925 CrossRef CAS.
- A. P. Hammersley, FIT2D, V10.3, ESRF, Grenoble, France, 1998 Search PubMed.
- STOE WinXPow, version 3.01.03, STOE and Cie GmbH, Darmstadt, Germany, 2010 Search PubMed.
- H. P. Gunnlaugsson, Spreadsheet based analysis of Mössbauer spectra, Hyperfine Interact., 2016, 237, 79 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2048374, 2048953, 2055695, 2048377, 2048376, 2048378, 2048375 and 2048954. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03203a |
| This journal is © The Royal Society of Chemistry 2023 |