
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCapsule-based automated synthesis for the efficient assembly of PROTAC like molecules†
Samuele
Bordi
a,
Tuo
Jiang
a,
Anna
Konopka
b,
Guillaume
Coin
ab,
Paula L.
Nichols
ab and
Benedikt M.
Wanner
 *a
*a
aSynple Chem AG, 8310 Kemptthal, Switzerland. E-mail: wanner@synplechem.com
bETH Laboratory for Organic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zürich, Switzerland
First published on 15th May 2023
Abstract
In recent years, the therapeutically beneficial degradation of proteins using PROteolysis Targeting Chimeras (PROTACs) has become an increasingly popular approach in drug discovery. However, the preparation of these larger than average, heavily functionalised molecules can be synthetically challenging and time-consuming, and experience in making and handling the final PROTACs and their precursors is not yet widespread. To overcome these challenges, an existing capsule-based automated synthesis console has been adapted and employed for the automated synthesis of PROTAC-like molecules. Reagent capsules containing a partial PROTAC reagent plus the reagents required for conjugation of the partial PROTAC to the target protein binder, as well as the materials for product isolation, were prepared in order to accelerate the process and simplify PROTAC synthesis. The use of these capsules, in combination with the automated synthesis console, has enabled the safe, automated preparation of a range of different PROTAC-like molecules bearing different linker and E3 ligase functionalities.
Introduction
Advances in genome sequencing have enabled the identification of many of the malfunctioning proteins involved in disease states. Despite this improved knowledge, up to 85% of the human proteome still remains ‘undruggable’. Many proteins lack accessible binding pockets for small molecule therapeutics, rendering them unsuitable targets for traditional drug discovery approaches.1 However, over the last decade, there have been increased efforts to develop novel approaches to “drug the undruggable”.2 One such approach – PROteolysis TArgeting Chimeras (PROTACs) – based on the degradation rather than on the inhibition of the target protein, has become increasingly popular in recent years.3 Due to the promise it offers with respect to undruggable targets, the approach is attracting a great deal of attention in the drug discovery field.4Given the widespread interest in these new chemical modalities across pharma, biotech and academia, numerous different PROTACs have now been reported to effectively degrade the target proteins in pre-clinical studies. In June 2022, an online database was found to list more than 3200 PROTACs, acting on over 100 targets.5 Among those, the degraders ARV-110 (Bavdegalutamide) and ARV-471 from Arvinas, developed as treatments for prostate and breast cancer respectively, have recently moved into phase II trials, while many others are currently following behind in phase I (Fig. 1).6,7
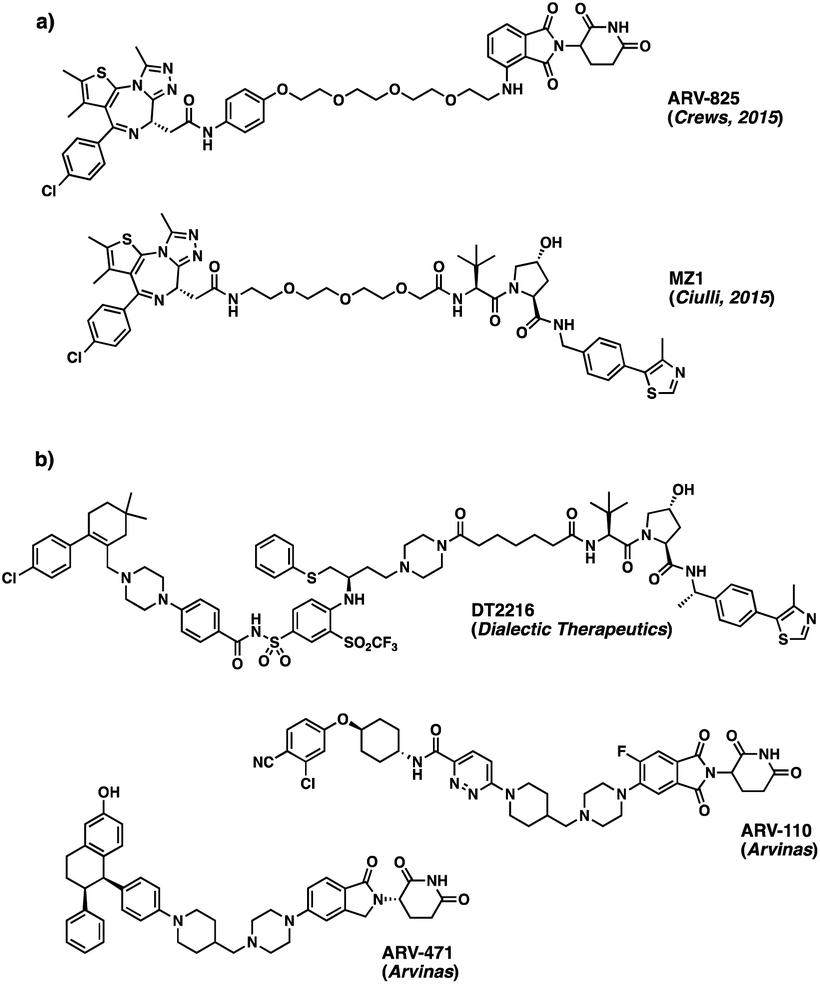 | ||
| Fig. 1 (a) Structures of ARV-825 and MZ1, BRD4 protein degraders based on BET inhibitor JQ1;8,9 (b) disclosed structures of PROTAC molecules currently in phase I (DT2216) and phase II (ARV-110 and ARV-471) clinical trials. | ||
Despite the explosion of interest in this field, the chemical synthesis of these molecules remains challenging, and knowledge of how to handle, purify and analyse the products is not yet widespread. In acknowledgement of the challenges, several CROs have started to offer their expertise in PROTAC synthesis,10 and there are an increasing number of commercial reagents available to support PROTAC synthesis, including Millipore Sigma's range of partial PROTAC building blocks.11
Several groups have also reported on their efforts to accelerate bioactive PROTAC discovery and optimisation via the development of new techniques that simplify the assembly of bifunctional PROTAC molecules.12–15 Other novel synthetic approaches that simplify PROTAC synthesis and handling are also slowly starting to emerge. Jiang16 and Derksen17 recently reported some insights on the synthesis of pomalidomide and lenalidomide-based PROTACs, building small libraries of PROTACs, which, thanks to carefully selected reaction conditions, typically had enhanced yields compared to similar reported structures. Although limited to a single class of E3 ligase ligands, Soural18 and Demizu19 reported interesting solid-phase approaches for the synthesis of PROTACs, which overcome most of the challenges associated with the use of classical solution-phase PROTAC synthesis. Despite these significant developments, all these approaches still need to be effected by highly specialized chemists with a deep knowledge of the field.
Thanks to reports, such as that from Steinebach et al., on the hydrolytic susceptibility of Cereblon-based (CRBN) PROTACs,20 awareness of the challenges associated with synthesising and handling PROTAC synthesis is slowly starting to spread. However, it is clear that there is still much to be done before the PROTAC synthesis field can be readily accessed by non-specialized scientists. As in many areas of synthesis, chemists making PROTACs frequently encounter problems or challenges that may have already been solved by others, but due to various factors such as differences in experience, lack of detail in the literature, or quality of reagents, they are forced to troubleshoot these issues again themselves, which is laborious and inefficient.
Innovative synthesis automation that simplifies PROTAC synthesis, and overcomes the difficulties, has the potential to make PROTAC synthesis a simple task for non-experts.21 It is worth noting that although synthesis is just one of the many challenges associated with PROTAC research, the synthetic burden acts as a bottleneck, slowing overall progress in PROTAC discovery. As such, there is a pressing need for new technology that can alleviate or eliminate the synthetic burden for PROTACs.
In terms of addressing this need, our existing platform, which provides scientists with direct access to optimised reaction protocols, would be particularly advantageous for PROTAC synthesis because it would allow researchers to create their compounds without having to tackle the synthesis and handling challenges that are well known to specialised chemists working in the PROTAC field. This would enable them to streamline the synthesis process and ease the synthetic burden, meaning that researchers could focus instead on the value-adding stages of their projects and ultimately accelerate PROTAC discovery.
With this goal in mind, we have adapted and utilized our existing technology for the synthesis of PROTACs (Fig. 2).22
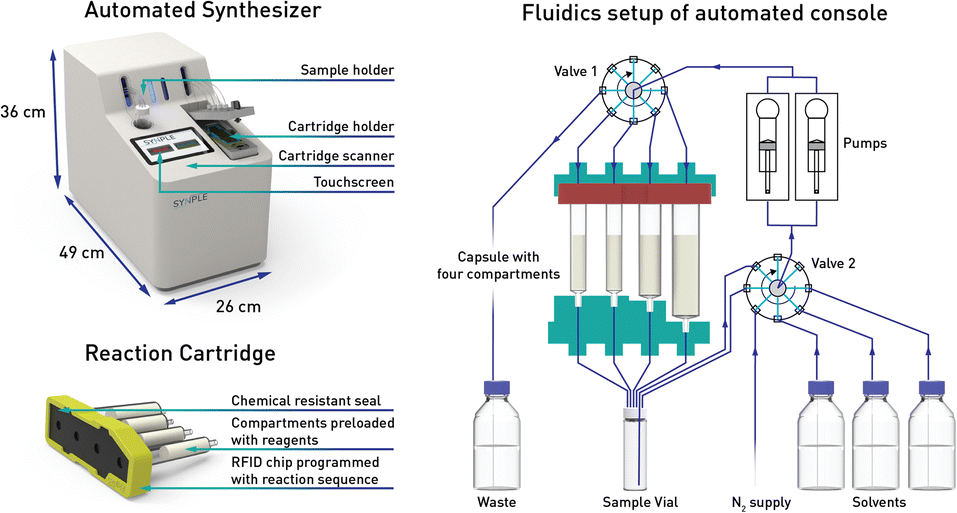 | ||
| Fig. 2 Automated synthesis console, pre-packed reagent capsule and fluidics setup of the console. | ||
In our previous work we described the use of an integrated console for performing fully automated organic reactions (Fig. 3). The practicality of pre-packed capsules, containing all the required reagents and purification materials, appeared well suited for partial PROTAC materials. In addition, the ease with which PROTACs could be prepared using the machine was highly attractive. By simply scanning the capsules on the console, the automation sequence required for that specific reaction and set of reagents is loaded. Following the insertion of the capsule, the whole synthesis and purification process can be effected without further user involvement. Herein, we report the successful development of a new PROTAC synthesis application for the console and on the variety of reagent cartridges available to enable the preparation of a wide range of PROTACs.
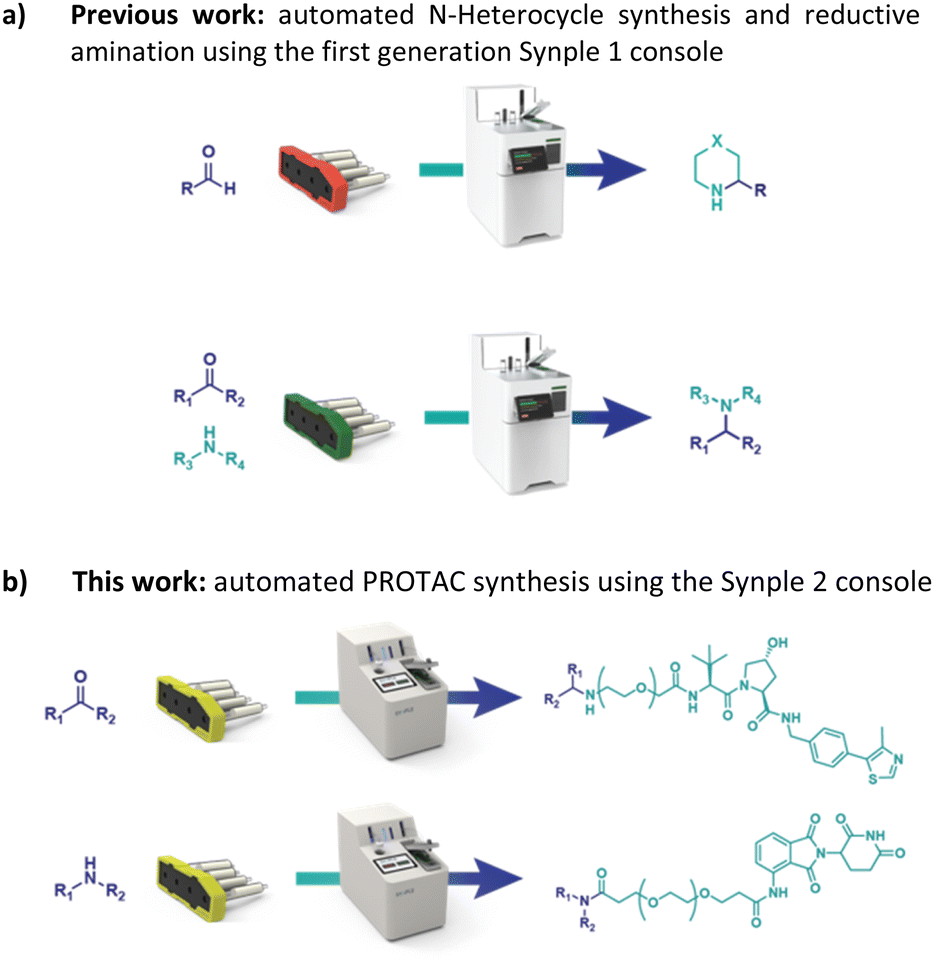 | ||
| Fig. 3 Our previous work on a console for the full automated synthesis of small molecules and the idea of its implementation in PROTAC synthesis. | ||
Results and discussion
Our approach involves the use of a capsule composed of four compartments, each containing either a reagent required for the reaction or a material required for product isolation. Initially, we chose to exploit our existing reductive amination strategy for PROTAC synthesis.22 As such, a range of cartridges was developed, containing solid-supported cyanoborohydride (compartment A) as the reducing agent and the partial PROTAC reagent, with a primary or secondary amine (hydrochloride or trifluoroacetate salt) conjugation site (compartment D). Of the remaining two compartments, one contains solid-supported triethylamine (compartment B), used to convert the PROTAC salt starting material into the free amine. The second holds a supported propylsulfonic acid (SCX-2, compartment C) for a catch and release-based purification of the product (Fig. 4).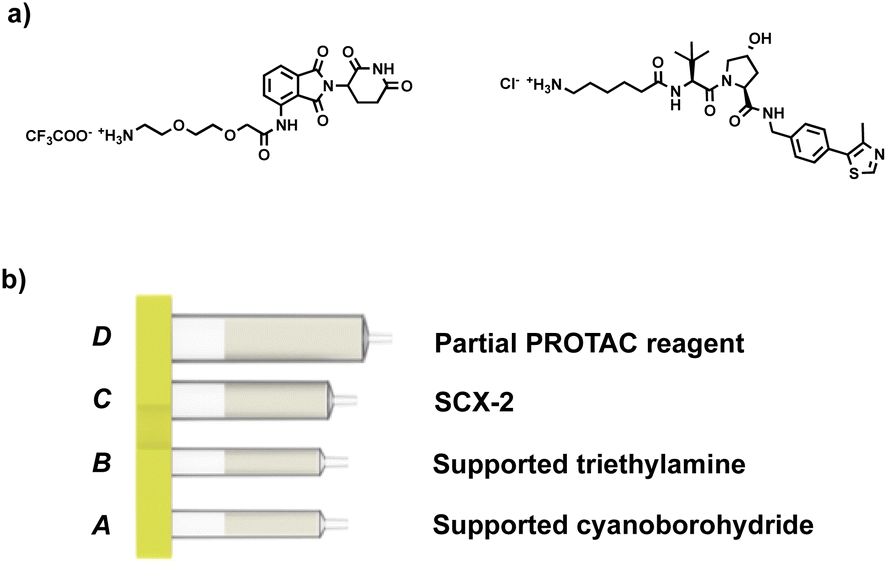 | ||
| Fig. 4 (a) Example of the partial PROTAC reagents used in the experiments; (b) capsule components for the synthesis of PROTACs through reductive amination. | ||
Initially, a range of different partial PROTACs, containing terminal primary or secondary amine conjugation groups were prepared and incorporated into the capsules. These partial PROTACs differed in terms of both the E3 ligase binder and the linker and were prepared by connecting the E3 ligand (1 or 2) to the carboxylic acid bearing linker (3a–g) via amide coupling. Cleavage of Boc group(s) afforded the desired Cereblon (4a–f) and VHL (5a–g) partial PROTAC reagents as trifluoroacetate and hydrochloride amine salts, respectively (Fig. 5).
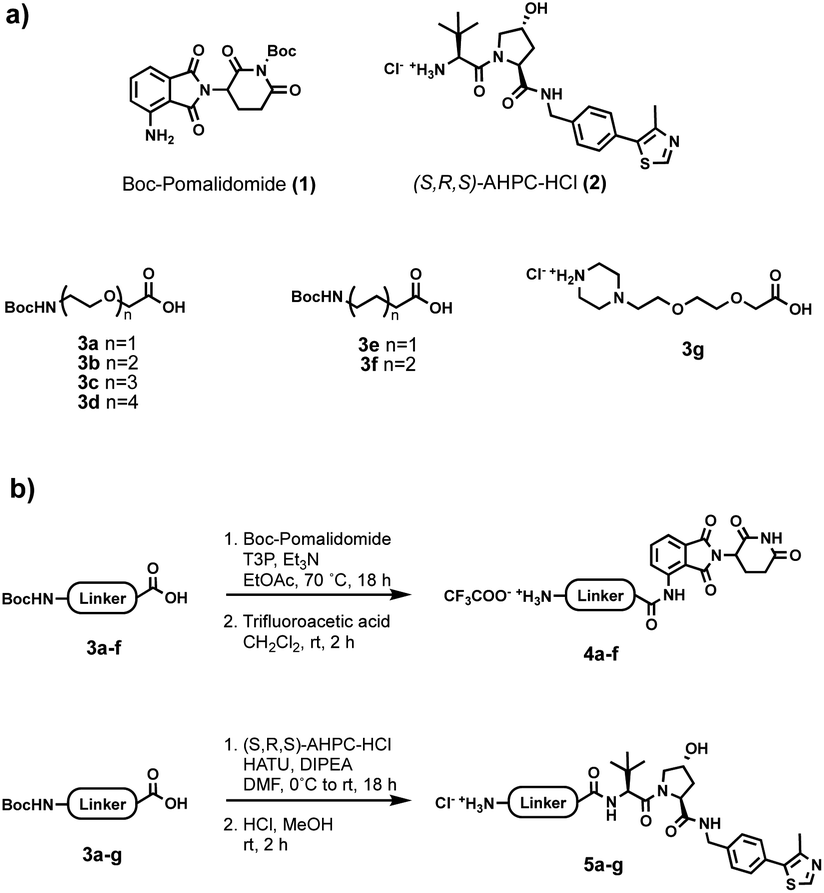 | ||
| Fig. 5 (a) Building blocks used for partial PROTAC reagents synthesis; (b) general synthesis of partial PROTAC reagents. | ||
Using the console and its integrated pre-programmed reaction protocols, along with the partial PROTAC capsules, carbonyl containing target protein binders could be transformed into PROTACs, using very few manual operations. The target protein binder only needed to be weighed into the reaction vial and dissolved in the reaction solvent (3 mL CH2Cl2 + 1 mL 1,1,1,3,3,3-hexafluoroisopropanol). Once the vial was connected to the machine and the reagent capsule scanned and inserted, the reaction could be initiated by simply pressing the start button. In the first step of the automated process, the reaction mixture was repeatedly circulated from the vial through compartment D, containing the partial PROTAC, and back to the vial again. In doing so, the partial PROTAC reagent was dissolved in the reaction solvent and then converted to the free amine via circulation through the supported base in compartment B in the second step. In step three of the process, the solution containing both the partial PROTAC and the target protein binder starting material was circulated through the supported hydride reagent in compartment A, leading to the desired reductive amination product. The purification was then performed using a catch and release strategy, in which the crude product was automatically loaded onto the SCX-2 contained in compartment C to capture the product, followed by washing of the support to remove the non-basic impurities, before finally releasing the product using a 2.5 M diisopropylamine solution in either THF or 2-propanol (Fig. 6).
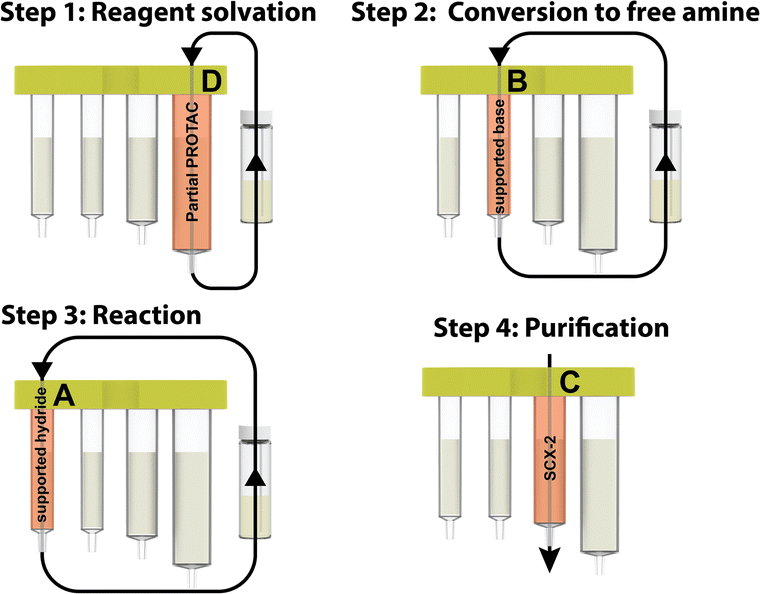 | ||
| Fig. 6 The four steps involved in the capsule-based PROTAC synthesis. | ||
This automated purification is intended to achieve the crude product at approximately 90% purity by removing any known impurities and by-products, thus greatly simplifying any final preparative HPLC or additional flash chromatography, if purities of >95% are required. A range of model carbonyl-containing substrates was tested on a 0.1 mmol scale, using both the CRBN-ligand-based partial PROTAC capsules and the VHL-ligand-based partial PROTAC capsules. The majority of substrates tested afforded the desired PROTAC products in good yield and purity using 3–12 hours of console time (Scheme 1). Pleasingly, and as already observed with our previously reported capsule-based reactions,22 the yields and purities obtained were highly reproducible (see ESI† for details). This was particularly meaningful in this case, due to the small scale on which the reaction was conducted. As such, even for users with only a minimal amount of chemistry training, novel PROTACs can be reliably made using the console and capsules since no prior knowledge of PROTAC synthesis is required and human error is greatly minimised.
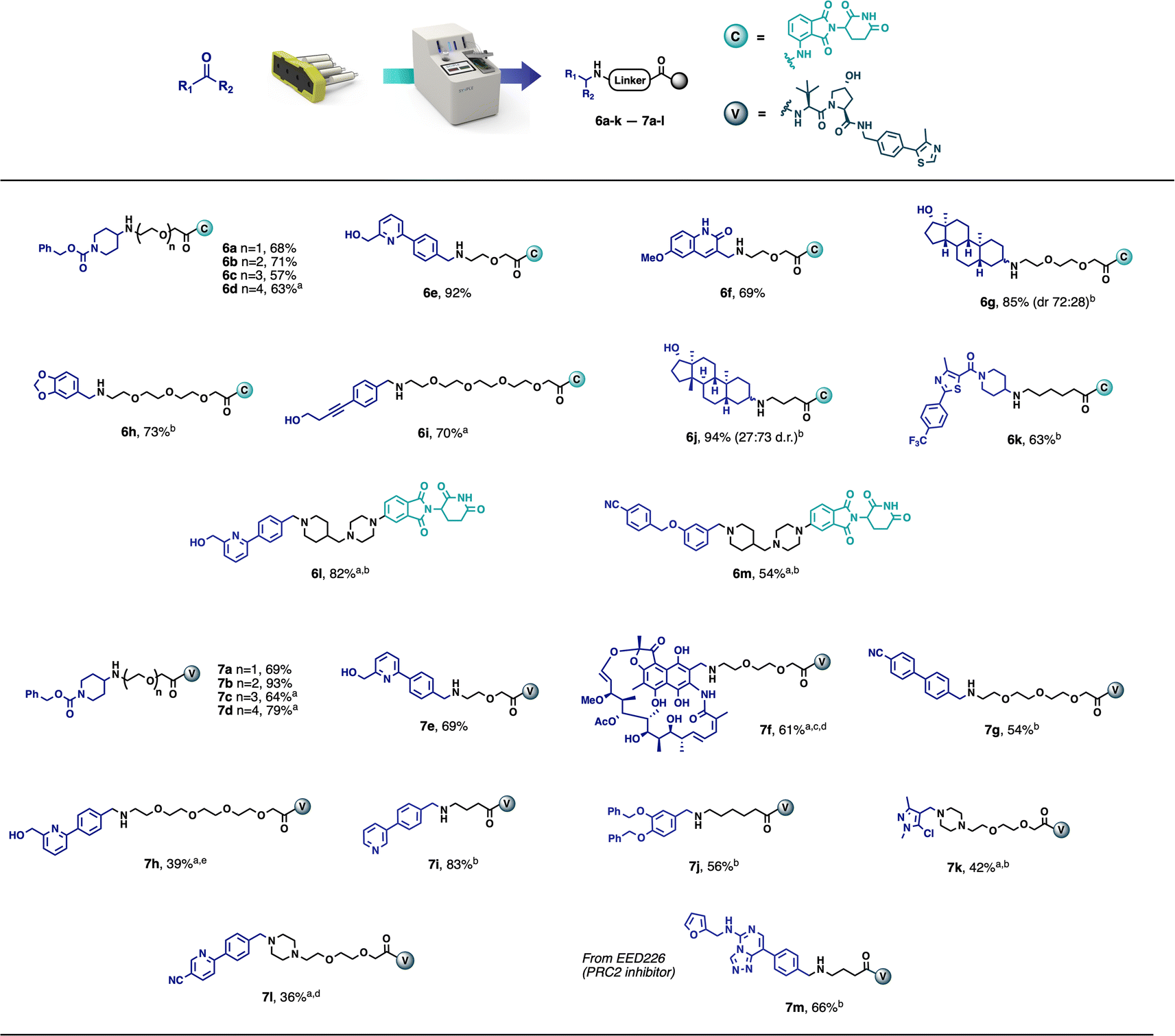 | ||
| Scheme 1 Scope of PROTAC synthesis via reductive amination. Reaction conditions: vial – aldehyde or ketone (0.1 mmol), CH2Cl2 (3 mL), HFIP (1 mL); capsule content – partial PROTAC (0.1 mmol), supported CNBH3 (500 mg, 5.0 equiv.), supported Et3N (300 mg, 3.87 equiv.), SCX-2 (2.0 g); reaction performed at room temperature for 3 h. Isolated yields directly after the automated synthesis are given (purity >90%), unless noted otherwise. (a) reaction time: 12 h. (b) product purified by flash chromatography. (c) SCX-2 purification skipped. (d) NMR yield, product purified by preparative HPLC. (e) isolated yield after preparative HPLC. | ||
Although the model PROTAC compounds prepared during this development are not claimed to be biologically active, the starting materials were selected to demonstrate the applicability of the methodology and technology to a broad range of substrates. Despite the fact that not all final products matched the desired purity (>90%), the preliminary purification performed by the machine made the subsequent flash chromatography or preparative HPLC much easier. Compounds 6h and 7g were purified to a high level of purity after a simple filtration through a silica plug. Compounds 6g and 6j were actually obtained in high purity and the flash chromatography was used only to separate the two diastereoisomers (obtained in a 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28 and 27:73 ratio, respectively).
:28 and 27:73 ratio, respectively).
It was suspected that model PROTAC 7f, synthesised from 3-formyl rifamycin, would be highly sensitive to acid promoted degradation and thus would not be compatible with the acidic SCX-2 resin used in the automated purification step. Therefore, the catch and release step was skipped by selecting this option on the console. Despite this, compound 7f was obtained in acceptable purity (∼85%) and 61% yield as a mixture with a small amount of the aldehyde starting material as an impurity. The product was then further purified by preparative HPLC.
The scope of the methodology also extends to partial PROTACs with non-PEG linkers. Compounds 6j, 6k, 7i and 7j were synthesised in an analogous manner using partial PROTACs with alkyl linkers. Similarly, compounds 6l and 6m were obtained in good yields starting from a partial PROTAC containing a linker composed of a piperazine and a terminal piperidine, leading to structures similar to ARV-110 and ARV-471 (Fig. 1), while compounds 7k and 7l were synthesised, from a partial PROTAC bearing a PEG-based linker with a terminal piperazine group, in 42% and 36% yield respectively, following chromatography. Finally, with the aim of demonstrating the methodology using a known protein binder, compound 7m was synthesised from the polycomb repressive complex 2 (PRC2) inhibitor EED226 in 66% yield after chromatography.23
After proving the effectiveness of our technology using a reductive amination process, the scope was expanded to include a process that linked the target protein binder to the partial PROTAC via an amide coupling reaction. Our initial approach involved the use of an activated ester as a partial PROTAC starting material. As such, a series of CRBN and VHL partial PROTAC derivatives with a 2,3,5,6-tetrafluorophenol ester conjugation site (PROTAC-AE) were synthesised, ready to be coupled with primary or secondary amine-containing target protein binders. Like the partial PROTACs for reductive amination, the PROTAC-AEs were pre-loaded into a capsule, together with silica-supported tetra alkyl ammonium carbonate (supported carbonate), as a scavenger for the tetrafluorophenol byproduct and for any hydrolysed active ester that may be present (Fig. 7). This approach has the advantages of avoiding the use of the common coupling reagents like DCC, EDC and HATU, which are known to be immune sensitisers and sometimes cause severe allergic reactions upon prolonged exposure.24–26
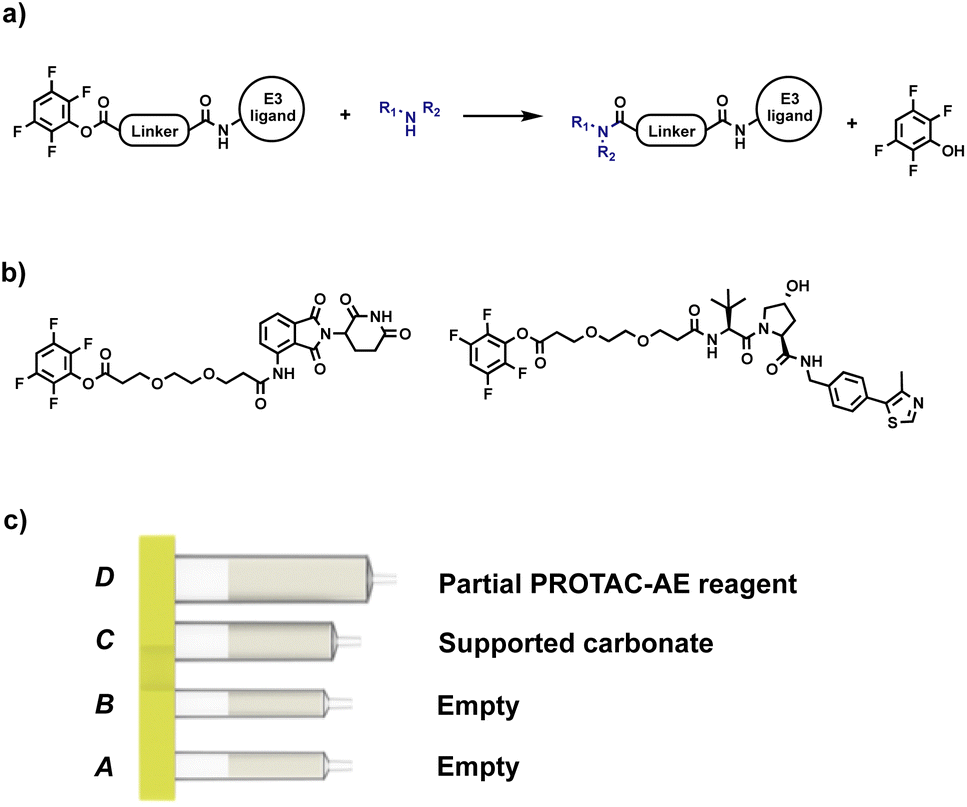 | ||
| Fig. 7 (a) Our initial approach for automated PROTAC synthesis via amide coupling; (b) examples of PROTAC-AEs synthesised for preliminary tests; (c) PROTAC-AE capsule components. | ||
Despite promising early results, with the protocol delivering the desired products in high yield and purity in a short period of time (<4 hours including purification step) in many different solvents such as CH2Cl2, EtOAc, CH3CN and DMF, the PROTAC-AE approach was not without issues. The advantages of high yields and purity of the final PROTAC products and the broad solvent compatibility were unfortunately counterbalanced by some significant drawbacks, such as the tedious, capricious synthesis of the PROTAC-AEs and their low storage stability. In fact, some PROTAC-AEs revealed themselves to be unstable, with a half-life of only a few weeks even when stored cold (−20 °C) or in a desiccator, which made them undesirable for inclusion in a commercial product. In order to ensure that the capsules remained robust and easy to store, we sought other approaches to PROTAC synthesis via amide coupling, which would offer increased reliability while also maintaining most of the advantages that PROTAC-AEs exhibited.
As with the reductive amination approach, solid-supported reagents were a good candidate for the coupling step. Commercially available silica-supported carbodiimide (supported DCC), together with Ethyl (hydroxyamino)cyanoacetate (Oxyma Pure), in sub-stoichiometric amount, gave results comparable to the PROTAC-AEs in terms of yield and purity, while still protecting the user from direct contact with the coupling reagent. Oxyma Pure was chosen as an additive, in place of the more commonly used HOBt, due to its known capacity to inhibit racemisation and for being less shock sensitive.27 The reaction utilises partial PROTACs with a carboxylic acid conjugation site (PROTAC-COOH), which are bench stable and easier to synthesise than the PROTAC-AEs. The PROTAC-COOH reagent was again incorporated into the capsule in compartment D, while the coupling reagent (supported DCC) and Oxyma Pure were loaded into compartment A (Fig. 8). As with all the previous reaction processes, the first step involves charging the reaction vial with the desired primary or secondary amine-containing protein binder, and dissolving it in 1:1 CH2Cl2/CH3CN (4 mL total). Following capsule scanning and insertion, the automated sequence starts with the dissolution of the partial PROTAC reagent, followed by repeated circulation through the supported DCC compartment, in which the reaction takes place. After the required reaction time (4–12 hours), the crude mixture was loaded onto compartment C (Silica-supported carbonate), which captured the Oxyma Pure and any residual unreacted PROTAC reagent. In the last purification step, the crude mixture was loaded into compartment B, which contains a buffered SCX resin. In this step, the unreacted starting amine is scavenged from the mixture, resulting in a solution containing only the desired PROTAC product.
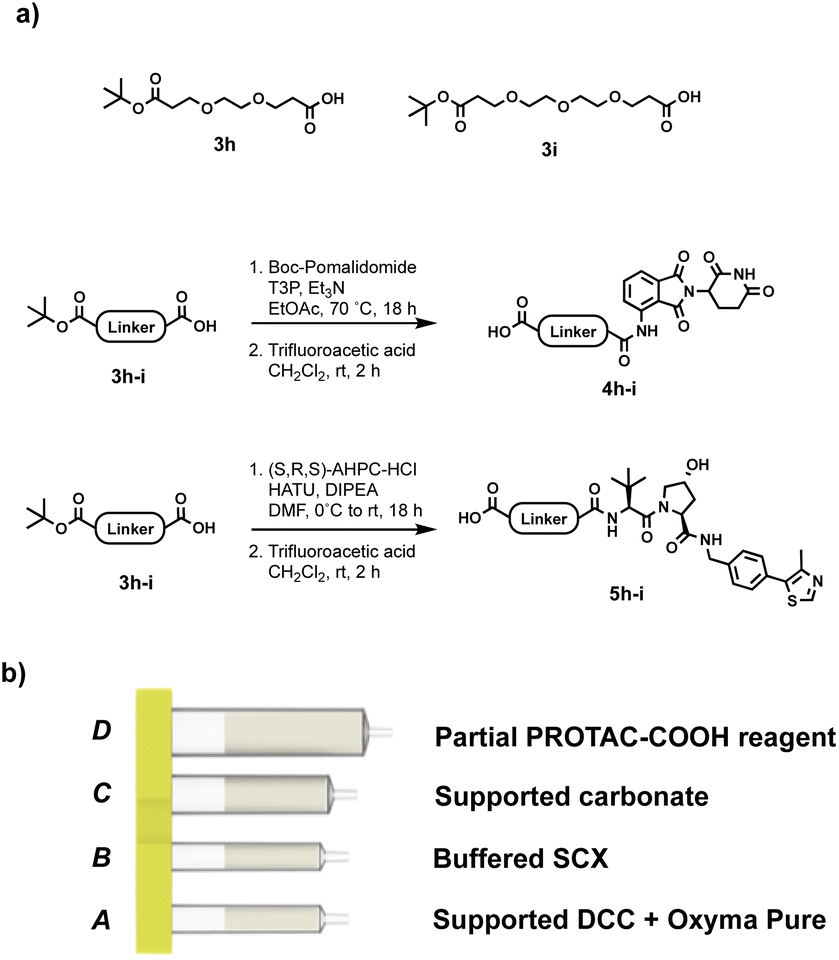 | ||
| Fig. 8 (a) Synthesis of PROTAC-COOH reagents; (b) capsule components for the synthesis of PROTACs through amide coupling from PROTAC-COOH reagents. | ||
This method has been demonstrated to give the desired compounds in 57–92% yield on a 0.1 mmol scale, with broad functional group compatibility (Scheme 2). The products obtained in this way are frequently pure enough (>90%) to be used directly in biological assays. Taking inspiration from existing PROTACs, several compounds were synthesised using known protein binders. Compound 8f was prepared from the anaplastic lymphoma kinase (ALK) inhibitor Ceritinib,28 compound 8g was obtained from Ibrutinib, Bruton's tyrosine kinase (BTK) inhibitor,29 while compounds 9h and 9i were synthesised from a SMARCA2 inhibitor30 and the BET inhibitor JQ1 (ref. 9) respectively. All these compounds were obtained in high yields (81–89%) and high purity without any further purifications. Among the synthesised compounds, only compound 8e needed further purification and was obtained in >95% purity after flash chromatography. The reactions performed most optimally in 1:1 CH2Cl2/CH3CN, but in the case of solubility problems, it was possible to run the reaction in THF instead. However, an extended reaction time of 12 hours was required (8d, 9g).
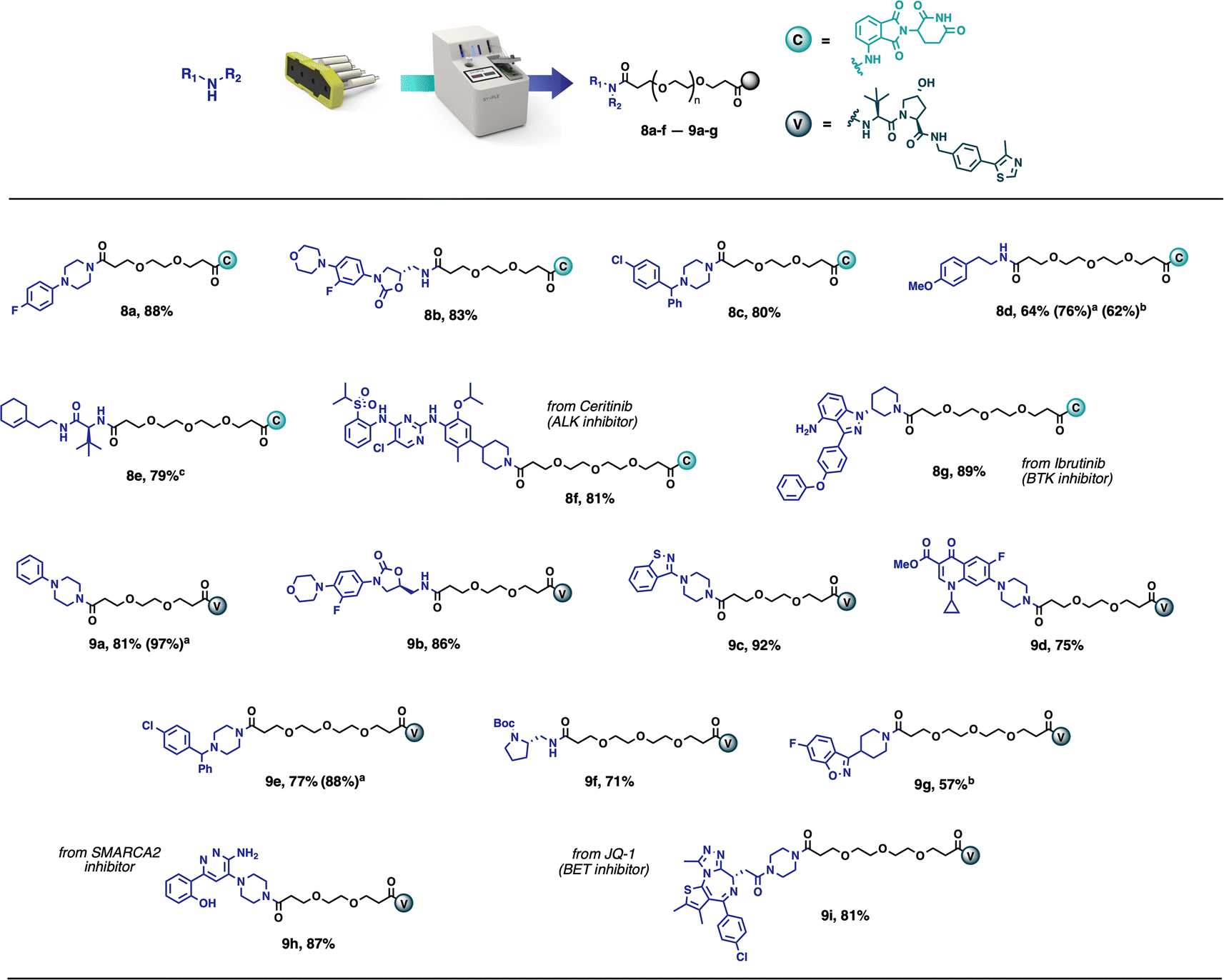 | ||
| Scheme 2 Scope of PROTAC synthesis via amide coupling. Reaction conditions: vial – amine (0.1 mmol), CH2Cl2 (2 mL), CH3CN (2 mL); Capsule content – partial PROTAC (0.1 mmol), supported DCC (280 mg, 2.66 equiv.), Oxyma Pure (5.9 mg, 0.4 equiv.), Buffered SCX (250 mg), supported CO32− (400 mg); reaction performed at room temperature for 4 h. Isolated yields directly after the automated synthesis are given (purity >90%), unless noted otherwise. (a) reaction performed starting from corresponding PROTAC-AE reagent (Fig. 7). (b) THF (4 mL) used as the reaction solvent, reaction time 12 h. (c) product purified by flash chromatography. | ||
As robust commercial products, the capsules need to possess long-term stability. To confirm stability, a reaction performed with a >9 months old capsule, which had been stored at room temperature in a sealed foil bag, still exhibited comparable performance, with no appreciable difference in the purity of the final product (see ESI† for details).
Conclusions
In summary, our capsule-based automated synthesis console was readily adapted to provide a means of preparing PROTACs. By encapsulating the difficult to make and handle partial PROTAC materials inside the capsule, and by providing a pre-optimised automated synthesis method that can be loaded by simply scanning the capsule, this technology enables even the most inexperienced synthetic chemists to enter the PROTAC discovery field, including those with limited practical chemistry experience and also chemists with significant synthesis expertise but with no prior experience of making and handling protein degraders.A series of PROTAC-like compounds were synthesised in this way via both reductive amination, starting from a carbonyl containing substrate, and amide coupling, starting from primary or secondary amine substrates. In most cases, the desired products were obtained in high purity without the need for further purification. Furthermore, the fully automated approach offers a safer alternative for synthesis since the user is protected from direct contact with potentially dangerous reagents. We foresee the opportunity to expand the application further with the development of a chemical biology tool kit consisting of capsules and applications, enabling even non-chemists to carry out more synthesis in the search for new therapeutic treatments.
Data availability
Data for this paper, including detailed synthetic procedures and spectra data is available in the attached electronic ESI† document (PDF).Author contributions
S. B., T. J., A. K., and G. C. did the experimental work. S. B., P. L. N. and B. M. W. wrote the manuscript.Conflicts of interest
The authors declare the following competing financial interest(s): B. M. W. and P. L. N are listed as inventors of a patent (WO Patent WO2017121724A1, 2016) related to this technology and are co-founders of Synple Chem AG.Acknowledgements
Financial support was provided by Innosuisse (Innovation Projects 27406.1 PFLS-LS and 47700.1 PFLS-LS). We thank Prof. Jeffrey W. Bode for his valuable and continuous support. The authors thank the Molecular and Biomolecular Analysis Service (MoBiAS) and the LOC NMR service of ETH Zürich for their analytical support and project students who contributed to this work (Hristo Bonchev and Julien Gaymard).References
- C&EN News, https://cen.acs.org/pharmaceuticals/drug-discovery/quest-drug-undruggable/96/i26, accessed February 2023.
- C. V. Dang, E. P. Reddy, K. M. Shokat and L. Soucek, Nat. Rev. Cancer, 2017, 17, 502–508 CrossRef CAS PubMed.
- M. J. Bond and C. M. Crews, RSC Chem. Biol., 2021, 2, 725–742 RSC.
- H. Gao, X. Sun and Y. Rao, ACS Med. Chem. Lett., 2020, 11, 237–240 CrossRef CAS PubMed.
- G. Weng, C. Shen, D. Cao, J. Gao, X. Dong, Q. He, B. Yang, D. Li, J. Wu and T. Hou, Nucleic Acid Res., 2021, 49, D1381–D1387 CrossRef CAS PubMed.
- Arvinas, https://ir.arvinas.com/news-releases/news-release-details/arvinas-releases-updated-dose-escalation-data-clinical-trial, accessed February 2023.
- A. Mullard, Nat. Rev. Drug Discovery, 2021, 20, 247–250 CrossRef CAS PubMed.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed.
- M. Zengerle, K. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS PubMed.
- (a) Aurigene, https://www.aurigeneservices.com/discovery-chemistry-services/protac-synthesis-and-screening, accessed February 2023; (b) Viva Biotech, https://www.vivabiotech.com/cro/chemicaltechnologyplatforms, accessed February 2023; (c) Syngene, https://www.syngeneintl.com/solutions/protacs, accessed February 2023.
- Merck-Sigma PROTAC building blocks portfolio, https://www.sigmaaldrich.com/CH/en/applications/chemistry-and-synthesis/protein-degradation, accessed February 2023 Search PubMed.
- R. P. Wurz, K. Dellamaggiore, H. Dou, N. Javier, M. Lo, J. D. McCarter, D. Mohl, C. Sastri, J. R. Lipford and V. J. Cee, J. Med. Chem., 2018, 61, 453–461 CrossRef CAS PubMed.
- T. A. Bemis, J. J. La Clair and M. D. Burkart, Chem. Commun., 2021, 57, 1026–1029 RSC.
- L. Guo, Y. Zhou, X. Nie, Z. Zhang, Z. Zhang, C. Li, T. Wang and W. Tang, Eur. J. Med. Chem., 2022, 236, 114317 CrossRef CAS PubMed.
- C. E. Hendrick, J. R. Jorgensen, C. Chaudhry, I. I. Strambeanu, J. F. Brazeau, J. Schiffer, Z. Shi, J. D. Venable and S. E. Wolkenberg, ACS Med. Chem. Lett., 2022, 13, 1182–1190 CrossRef CAS PubMed.
- X. Qiu, N. Sun, Y. Kong, Y. Li, X. Yang and B. Jiang, Org. Lett., 2019, 21, 3838–3841 CrossRef CAS PubMed.
- D. K. Brownsey, B. C. Rowley, E. Gorobets, B. S. Gelfand and D. J. Derksen, Chem. Sci., 2021, 12, 4519–4525 RSC.
- S. Krajcovicova, R. Jorda, D. Hendrychova, V. Krystof and M. Soural, Chem. Commun., 2019, 55, 929 RSC.
- H. Xu, T. Kurohara, R. Takano, H. Yokoo, N. Shibata, N. Ohoka, T. Inoue, M. Naito and Y. Demizu, ChemistryOpen, 2022, 11, e202200131 CrossRef CAS PubMed.
- A. Bricelj, Y. Lam Dora Ng, D. Ferber, R. Kuchta, S. Müller, M. Monschke, K. G. Wagner, J. Krönke, I. Sosič, M. Gütschow and C. Steinebach, ACS Med. Chem. Lett., 2021, 12, 1733–1738 CrossRef CAS PubMed.
- M. Christensen, L. P. E. Yunker, P. Shiri, T. Zepel, P. L. Prieto, S. Grunert, F. Bork and J. E. Hein, Chem. Sci., 2021, 12, 15473–15490 RSC.
- T. Jiang, S. Bordi, A. E. McMillan, K.-Y. Chen, F. Saito, P. L. Nichols, B. M. Wanner and J. W. Bode, Chem. Sci., 2021, 12, 6977–6982 RSC.
- F. Potjewyd, A. M. W. Turner, J. Beri, J. M. Rectenwald, J. L. Norris-Drouin, S. H. Cholensky, D. M. Margolis, K. H. Pearce, L. E. Herring and L. I. James, Cell Chem. Biol., 2020, 27, 47–56 CrossRef CAS PubMed.
- B. B. Hayes, P. C. Gerber, S. S. Griffey and B. J. Meade, Drug Chem. Toxicol., 1998, 21, 195–206 CrossRef CAS PubMed.
- SDS of EDC-hydrochloride, https://www.sigmaaldrich.com/CH/en/sds/sial/e6383, accessed February 2023 Search PubMed.
- K. J. McKnelly, W. Sokol and J. S. Nowick, J. Org. Chem., 2020, 85, 1764–1768 CrossRef CAS PubMed.
- R. Subirós-Fusonas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Chem. - Eur. J., 2009, 15, 9394–9403 CrossRef PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2019, 10, 1102–1103 CrossRef CAS PubMed.
- Y. Sun, X. Zhao, N. Ding, H. Gao, Y. Wu, Y. Yang, M. Zhao, J. Hwang, Y. Song, W. Liu and Y. Rao, Cell Res., 2018, 28, 779–781 CrossRef CAS PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2020, 11, 1797–1798 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures, experimental procedures, analytical data for all new compounds, spectra for all new compounds and NMR spectra of compounds obtained directly from machine. See DOI: https://doi.org/10.1039/d3dd00027c |
| This journal is © The Royal Society of Chemistry 2023 |