
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of surface conditions on the electrochemical CO2 reduction performance of bimetallic AuPd electrocatalysts†
Daniël
van den Berg
,
Boaz
Izelaar
,
Shilong
Fu
and
Ruud
Kortlever
 *
*
Large-Scale Energy Storage, Process & Energy Department, Faculty of Mechanical, Maritime and Materials Engineering, Delft University of Technology, Leeghwaterstraat 39, 2628 CB Delft, The Netherlands. E-mail: r.kortlever@tudelft.nl
First published on 16th November 2023
Abstract
Most research into electrochemical CO2 conversion focusses on improving electrode materials, but neglects the role of the electrolyte. We show the buffer influence on the selectivity of a bimetallic gold–palladium electrode in an effort to elucidate observed inconsistencies between different studies. While hydrocarbons are produced in the phosphate buffer, they remain absent in the bicarbonate buffer, showing that the electrolyte choice plays a crucial role in the selectivity of the electrode.
In order to shift away from fossil resources as main source for fuels and bulk chemicals, new and sustainable production processes based on renewable energy inputs need to be developed. Interest in the electrochemical reduction of CO2 to base chemicals such as CO, methane and ethylene has therefore significantly increased in the last decades. Copper-based electrode materials have been the main focus of research, ever since the discovery by Hori and co-workers that copper is able to reduce CO2 to hydrocarbons and alcohols.3 However, copper produces a wide range of C1–C3 products, with limited selectivity towards one product.4,5 The selectivity of a monometallic copper electrode can be tuned by controlling the local conditions at the surface and by coating the electrode with organic layers.1,6,7 Alternatively, the design of multimetallic catalysts provides a means to tailor the electronic and structural properties, thereby modifying its catalytic properties. Many recent studies have been devoted to investigating the electrocatalytic properties of copper-based alloys.1,8–11 In most cases, these copper-based alloys were not able to outperform the intrinsic activity of copper towards the production of multi-carbon hydrocarbons. However, some alloys display higher selectivities towards CO or suppress the hydrogen evolution reaction, such as CuAg or CuIn alloys (HER).1,12,13 So far, only two classes of copper-free alloys have been found that are able to produce hydrocarbon products. The first are combinations of nickel and group 13 metals (aluminium, gallium) that produce C1–C3 products,14–16 and the second are gold palladium combinations that have yielded C1–C5 products.17,18 The Ni–Ga combination was chosen following DFT-calculations of oxygen-adsorption energies for several alloys. Here, the oxygen-adsorption energy of a material was found to be determining for its catalytic activity of the thermochemical CO2 reduction towards methanol.19 For AuPd, the combination of metals was chosen to optimally bind COads to the electrode surface. The COads intermediate is important for CO2 reduction towards further reduced products; if the intermediate is bound too weakly, it will desorb before it can be reduced further, which is the case for a gold electrode, while if COads is bound too strongly, which is the case for a palladium electrode, it is not energetically favourable to reduce it further and little to no CO2 reduction products will be observed.20 By bringing both metals together on the same surface, the binding energies of the metals to COads will be “averaged” leading to further reduction of COads to hydrocarbons.21
Interestingly, studies into electrochemical reduction of CO2 on AuPd obtained different findings; Kortlever et al. studied palladium electrodeposited on a polycrystalline gold foil in a 0.1 M KH2PO4 + 0.1 M K2HPO4 buffer (pH = 6.7) and observed a mixture of C1–C5 products from an onset potential of −0.8 V vs. RHE.17 Humphrey et al. investigated the same metal combination as core–shell nanoparticles in a 0.1 M Na2SO4 electrolyte at pH 4 with different shell thicknesses of palladium. In their study, hydrocarbons (C1–C2) and formate were observed with nanoparticles with a Pd shell thickness of 5 nm or higher, while thinner Pd shells only produced CO and H2.18 Contrary to previous studies, Wang et al. did not observe any hydrocarbon formation when studying AuPd nanoparticles with varying Pd surface compositions in 0.1 M KHCO3.22 Neither did Hahn et al. when investigating different compositions of thin film AuPd alloys in a 0.1 M KHCO3 buffer.23 To elucidate the origin of the differences in observed product selectivity between the aforementioned studies, electrodeposited palladium on a gold surface was studied in 0.1 M KHCO3 and 0.1 M KH2PO4 + 0.1 M K2HPO4 electrolytes. As a comparison, electrodeposited palladium on a glassy carbon electrode and a polycrystalline gold electrode were studied in both buffers to investigate the effect on the separate counterparts.
In ESI† Fig. S1A and B, SEM images of the gold–palladium electrode can be found. The images show a fairly uniform surface except for some bright and dark spots, which is further supported by the XPS data of the electrode (ESI† Section S6). In ESI† Fig. S1A–C, the EDX mapping image shows that these dark spots on the SEM image are not identified as either gold, palladium, or any other element. Therefore, these dark spots are assumed to be small cavities in the electrode. Lighter streaks on the SEM image can be excess islands of gold as they are also observed on the pure gold electrode (see ESI† Fig. S1C and D), taking into account that no other element was detected by the EDX mapping (see Fig. S3†). In contrast, palladium forms small particles when electrodeposited on the glassy carbon rather than forming a uniform monolayer. The difference is clearly visible from the SEM images of the Pd/C (ESI† Fig. S1E and F). The particles were confirmed as palladium using EDX mapping of the surface (ESI† Fig. S2D–F).
Both palladium and gold are identified on the AuPd surface by XPS (see ESI† Fig. S4). The Pd 3d spectra has two predominant metallic Pd peaks at 335 eV and 341.3 eV, which matches the peaks of the palladium foil (ESI† Fig. S5). On the gold–palladium and palladium foil, inevitable formation of Pd-oxide species was observed at 336.1 eV and 337.5 eV. The sharp peaks in the Au 4f7/2 spectra at 83.9 eV do not show a negative shift of 0.6–0.8 eV with respect to the metallic Au state (ESI† Fig. S6). The absence of this shift suggests that a layer of metallic Pd covers the Au foil rather than that the gold and palladium have formed an alloyed phase.2,3
The faradaic efficiencies toward the major products on all three electrodes in the phosphate buffer are shown in Fig. 1A, C and E for gold–palladium, gold, and palladium respectively. The partial current densities towards the major products on the same electrodes are shown in Fig. 2A, C, and E. Using the phosphate buffer, small amounts of hydrocarbons with partial current densities between a few and tens of micro amperes are observed on all electrodes. These results agree fairly well with literature results, as Kortlever et al. observed hydrocarbons within the C1–C5 range with a higher selectivity in the same phosphate buffer.17 The difference in selectivity can be explained by a difference in cell hydrodynamics between both studies since both studies used different cell setups. Also, while only hydrocarbons in the C1–C2 range could be quantified in this study, propane/propene was observed qualitatively. However, since the gas chromatograph was not calibrated for propane or propene, these products are not included in the figures. Higher hydrocarbons (C4–C5) are not observed, meaning that if they were produced their concentrations remained under the limit of detection. On gold small amounts of hydrocarbons were observed in the phosphate buffer with a faradaic efficiency of around 0.2% at 0.8 V vs. RHE, which agrees with the findings of Noda and co-workers. Using a phosphate electrolyte of pH 6.8, they observed small amounts of hydrocarbons with a selectivity of 0.6% at −0.8 V vs. RHE on a polycrystalline gold electrode.24 The difference in selectivity could again be attributed to differences in cell hydrodynamics.
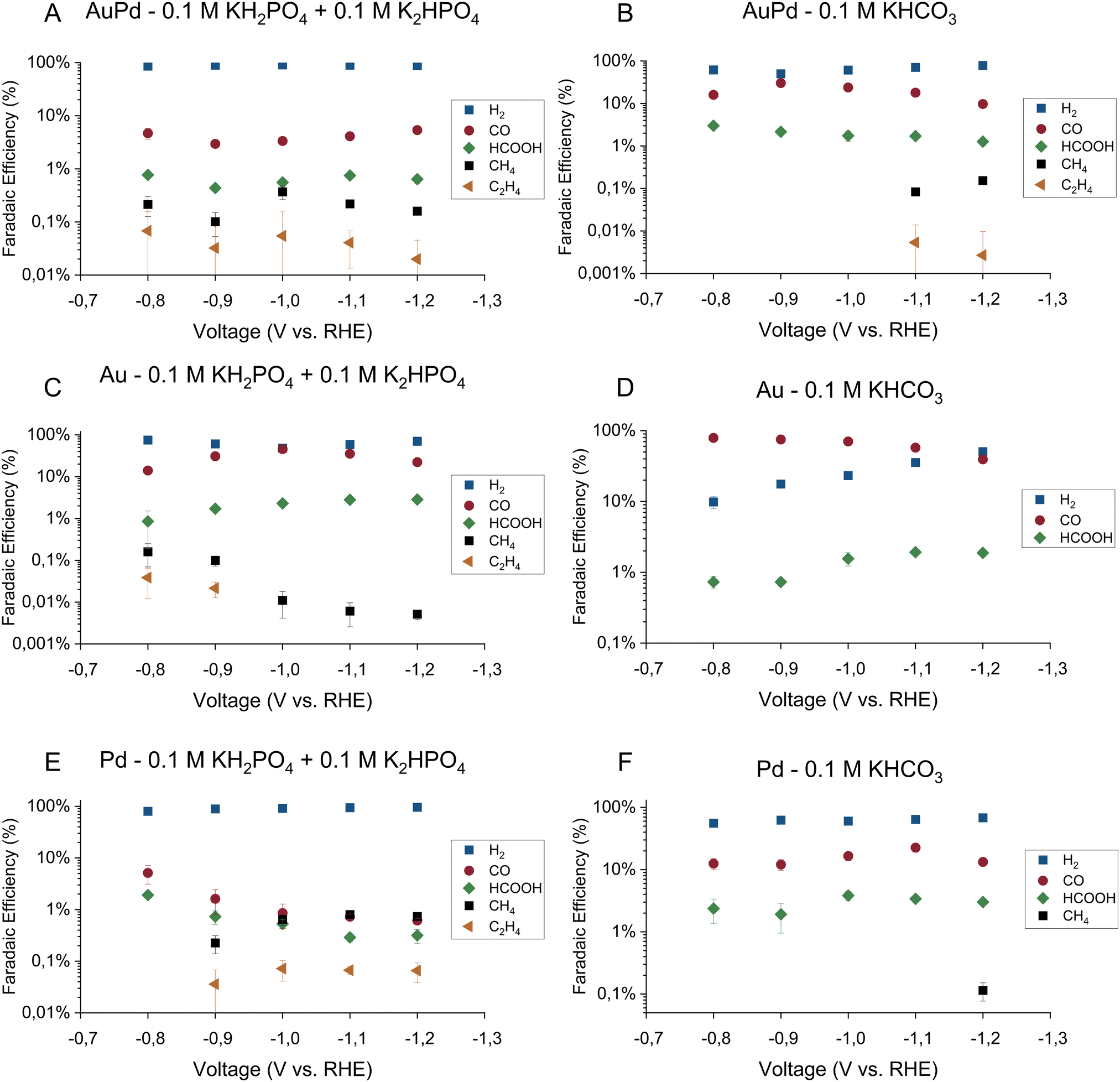 | ||
Fig. 1 Faradaic efficiencies for CO2 reduction for gold–palladium, gold, and palladium electrode in both phosphate (0.1 M KH2PO4/0.1 M K2HPO4) and bicarbonate (0.1 M KHCO3) electrolyte. The major products observed on the electrodes are hydrogen ( ), carbon monoxide ( ), carbon monoxide ( ), formate ( ), formate ( ), methane (■), and ethene ( ), methane (■), and ethene ( ). The error bars show the standard deviation over the triplicate results. ). The error bars show the standard deviation over the triplicate results. | ||
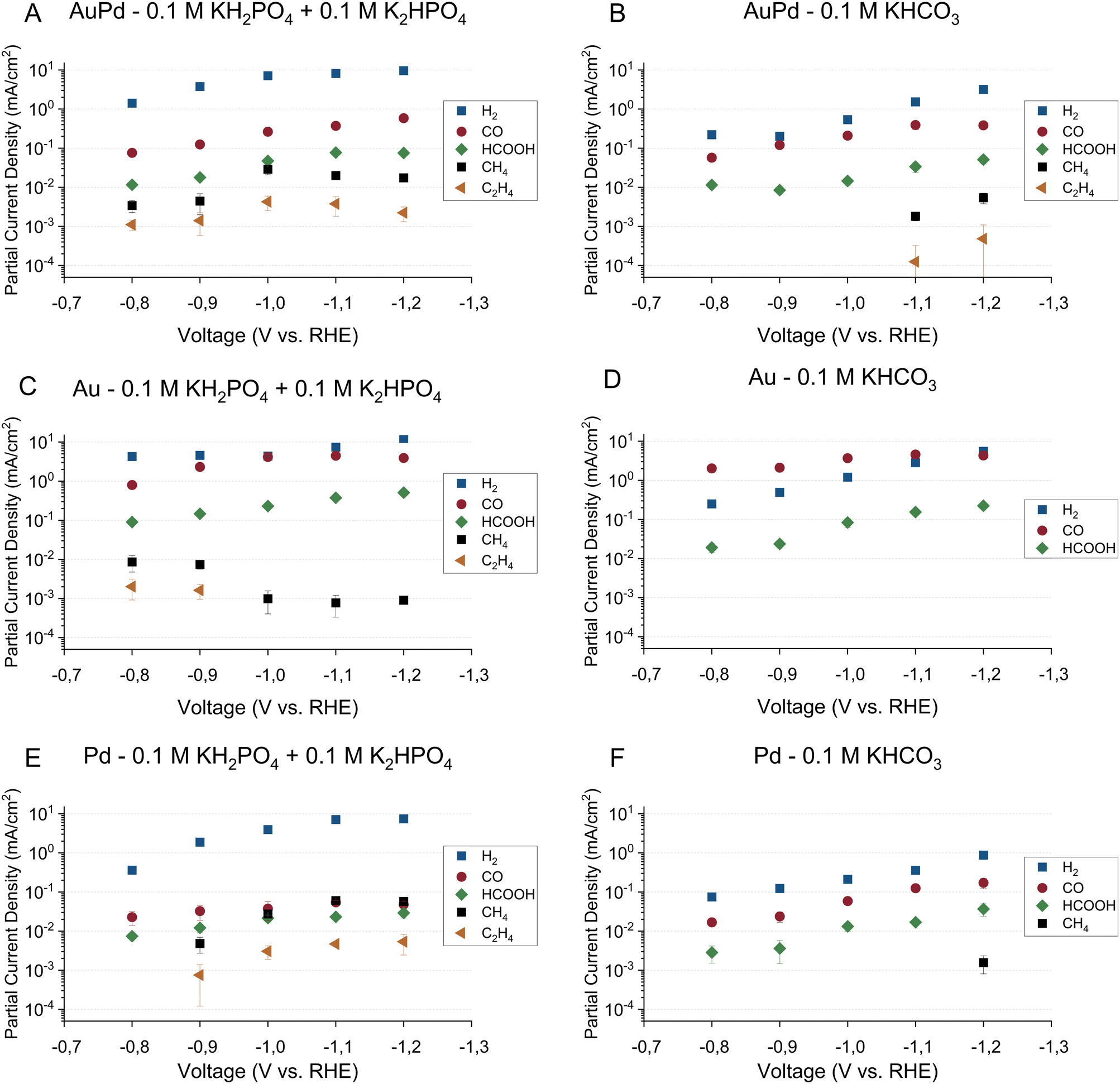 | ||
Fig. 2 Partial current densities for CO2 reduction for gold–palladium, gold, and palladium electrode in both phosphate (0.1 M KH2PO4/0.1 M K2HPO4) and bicarbonate (0.1 M KHCO3) electrolyte. The major products observed on the electrodes are hydrogen ( ), carbon monoxide ( ), carbon monoxide ( ), formate ( ), formate ( ), methane (■), and ethene ( ), methane (■), and ethene ( ). The error bars show the standard deviation over the triplicate results. ). The error bars show the standard deviation over the triplicate results. | ||
The faradaic efficiencies toward the major products on all three electrodes in the bicarbonate buffer are shown in Fig. 1B, D and F for gold–palladium, gold, and palladium respectively. The partial current densities towards the major products on the same electrodes are shown in Fig. 2B, D, and F. Interestingly, these results show that while hydrocarbons are produced in the phosphate electrolyte, they remain mostly absent in the bicarbonate electrolyte or are produced at much smaller rates. For AuPd, hydrocarbons were observed at more negative potentials in the bicarbonate electrolyte in comparison to the phosphate electrolyte, with methane and ethylene production at −1.1 V and −1.2 V vs. RHE. In the literature, AuPd in a bicarbonate electrolyte was only tested up to a potential of −1.0 V vs. RHE in the case of Hahn et al.,23 while Wang and co-workers only tested up to a potential of −0.8 V. Hori et al. report that a small amount of methane (2.9% selectivity) is produced on palladium in 0.1 M KHCO3, with a total faradaic efficiency balance of 60%.20 This discrepancy in the total faradaic efficiency was explained by the absorption of hydrogen into the electrode. Additionally, Azuma and co-workers observed small amounts of hydrocarbons (C1–C6) on a palladium electrode in a 0.05 M KHCO3 electrolyte at −2.16 vs. RHE.25 Their obtained selectivities of the hydrocarbons decrease with increasing hydrocarbon size from 0.3% for methane to about 0.001% for the highest hydrocarbons.
Other major reduction productions also follow a similar trend in the two electrolytes for all electrodes. The HER activity and formate production rate are higher in the phosphate electrolyte compared to the bicarbonate electrolyte. On the other hand, the activity towards CO production remains mostly unaffected by the electrolyte choice. However, we note that on the gold electrode, CO production tapers off around 4.5 mA cm−2 in both buffers. A stagnation in production rate could indicate either a kinetic limitation of the catalyst or a limitation of CO2 transfer to the electrode. To assess this hypothesis, the hydrodynamics of the cell setup were measured using the ferro-/ferricyanide redox couple. The experimental approach and results can be found in the ESI† (Section S3). From the calculations, it is estimated that when the CO2 consumption rate exceeded 27 nmol s−1, the concentration of CO2 at the surface reaches zero and transport rate becomes mass transfer limited. The consumption rate of CO2 on gold in both buffers approached 25 nmol s−1 (see ESI† Table S2). Hence, the CO2 reduction on the gold electrode was mass transfer limited for both buffers over the whole potential range.
In summary, the dependency of the product selectivity upon the choice of electrolyte shows that apart from the catalyst material, the local conditions at the electrode play a key role. The cause of this dependency could be due to the different anions that enhance or impede the formation of products on the electrode. Alternatively, since the buffer strength of the phosphate buffer is higher than the bicarbonate buffer, the buffer strength of the electrolyte could influence the formation of hydrocarbons by causing different surface conditions (for instance, differences in local pH and CO2 availability). To determine the cause for the observed differences, the AuPd catalyst activity was measured in a series of electrolytes with increasing bicarbonate concentration at a fixed potential. The purpose of these experiments is two-fold: if the cause for the observed difference is due to buffer strength, increasing the buffer strength will mimic the conditions of the phosphate buffer and an enhancement in the formation of hydrocarbons is expected. However, if the observed differences are due to the different anions, an increased presence of HCO3− or CO32− would not yield any significant differences. The potential chosen for these experiments was −1.1 V vs. RHE, since this is the lowest potential at which hydrocarbons were observed initially in the 0.1 M bicarbonate electrolyte. In Fig. 3, the partial current densities are shown for AuPd at −1.1 V vs. RHE in bicarbonate buffers with increasing KHCO3 concentration. Hydrogen, formate, and hydrocarbon production rates all increase linearly with KHCO3 concentration. Meanwhile, the CO activity remains mostly unaffected by the changing buffer strength. If the presence of HCO3− or CO32− would impede the reactions towards certain products, the production rates towards hydrogen, formate, and hydrocarbons should not have increased with increasing buffer concentration. Therefore, based on these results, we conclude that it is the difference in buffer strength between the phosphate and bicarbonate electrolyte that leads to the earlier observed differences in product activities.
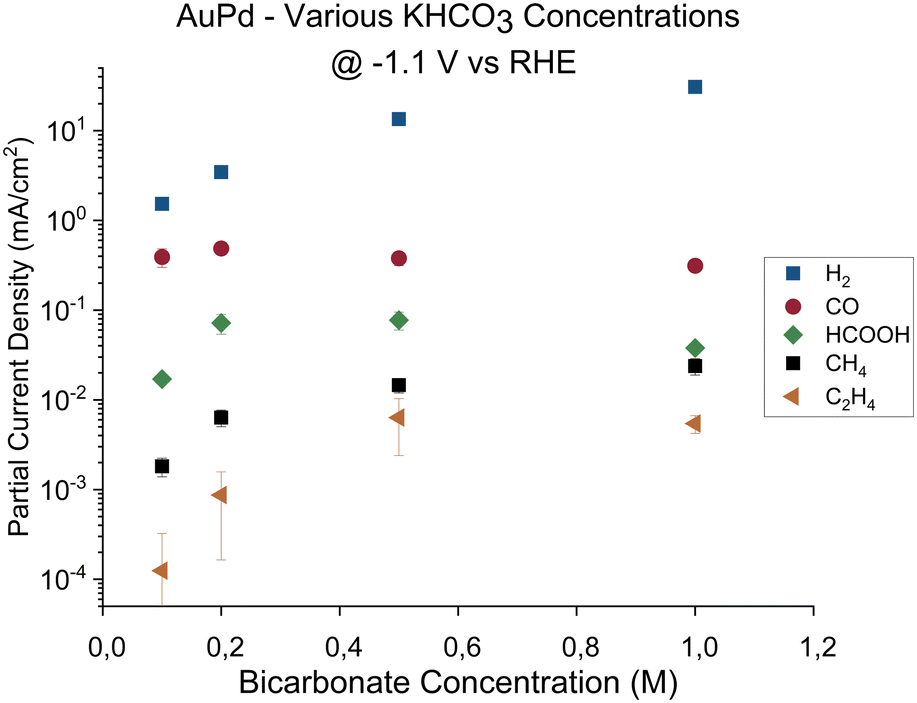 | ||
Fig. 3 Partial current densities for CO2 reduction for gold–palladium, gold, and palladium electrode in increasingly more concentrated bicarbonate (0.1 M KHCO3) electrolyte. The major products observed on the electrodes are hydrogen ( ), carbon monoxide ( ), carbon monoxide ( ), formate ( ), formate ( ), methane (■), and ethene ( ), methane (■), and ethene ( ). The error bars show the standard deviation over the triplicate results. ). The error bars show the standard deviation over the triplicate results. | ||
Since stronger buffers will result in a lower surface pH during reaction, the rate determining step towards hydrocarbon products on these three catalysts can assumed to be pH-dependent. On copper, ethylene is formed via two different reaction mechanisms: CO-dimerization, which is pH-independent, and a pH dependent route that shares a common intermediate with methane formation CHO*.26 On other transition metals, higher hydrocarbon formation has been observed as well, often following a Schulz–Flory distribution. This mechanism was first suggested by Cook et al. based on the correlated production of hydrocarbons on various transition metals27 and subsequently by Kudo and co-workers on iron, cobalt, and nickel.28 The same conclusion was also drawn by Kortlever et al. for the gold palladium electrode discussed in this article.17 Which exact intermediate(s) participate in the C–C bonding step is however unclear. From the observed pH-dependency of the hydrocarbon formation, it is clear that C–C bonding intermediate(s) of this mechanism share a common intermediate the pH-dependent formation of methane (CHO*). The articles above all suggest that the formation of higher hydrocarbons proceeds through chain-coupling of the 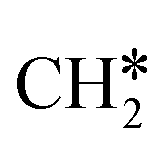 intermediate following a Fischer–Tropsch mechanism, but no experimental proof of the intermediate on these catalysts exists in literature. Based on DFT calculations on a polarised nickel surface, Zhou and co-workers suggest a reaction between COOH* and
intermediate following a Fischer–Tropsch mechanism, but no experimental proof of the intermediate on these catalysts exists in literature. Based on DFT calculations on a polarised nickel surface, Zhou and co-workers suggest a reaction between COOH* and 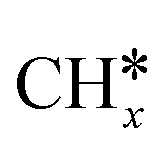 (x = 1, 2) as the favoured C–C coupling step.2 It is clear, however, that on non-copper metals the formation of C2+ products follows a different mechanism than the preferred CO-dimerization on copper. This is a logical conclusion based on the nature of the metals; only on Cu(100), the CO* intermediate has an intermediate binding strength that makes the dimerization step possible,29 while on the other metals, the CO-binding strength is either too strong or weak to make its dimerization reaction kinetically favourable. Therefore, C–C bonding is more likely through other intermediates than CO*, see Fig. 4. Therefore, to enhance the formation of C2+ products on a non-copper alloy, it would be more advantageous to study these materials in a stronger buffer. The stronger buffer will enhance the production of C2+ products making it more likely to pass the limit of detection and easier to identify promising materials. However, this reaction mechanism is an hypothesis and further in situ spectroscopic characterization is needed to confirm it or give further insights.
(x = 1, 2) as the favoured C–C coupling step.2 It is clear, however, that on non-copper metals the formation of C2+ products follows a different mechanism than the preferred CO-dimerization on copper. This is a logical conclusion based on the nature of the metals; only on Cu(100), the CO* intermediate has an intermediate binding strength that makes the dimerization step possible,29 while on the other metals, the CO-binding strength is either too strong or weak to make its dimerization reaction kinetically favourable. Therefore, C–C bonding is more likely through other intermediates than CO*, see Fig. 4. Therefore, to enhance the formation of C2+ products on a non-copper alloy, it would be more advantageous to study these materials in a stronger buffer. The stronger buffer will enhance the production of C2+ products making it more likely to pass the limit of detection and easier to identify promising materials. However, this reaction mechanism is an hypothesis and further in situ spectroscopic characterization is needed to confirm it or give further insights.
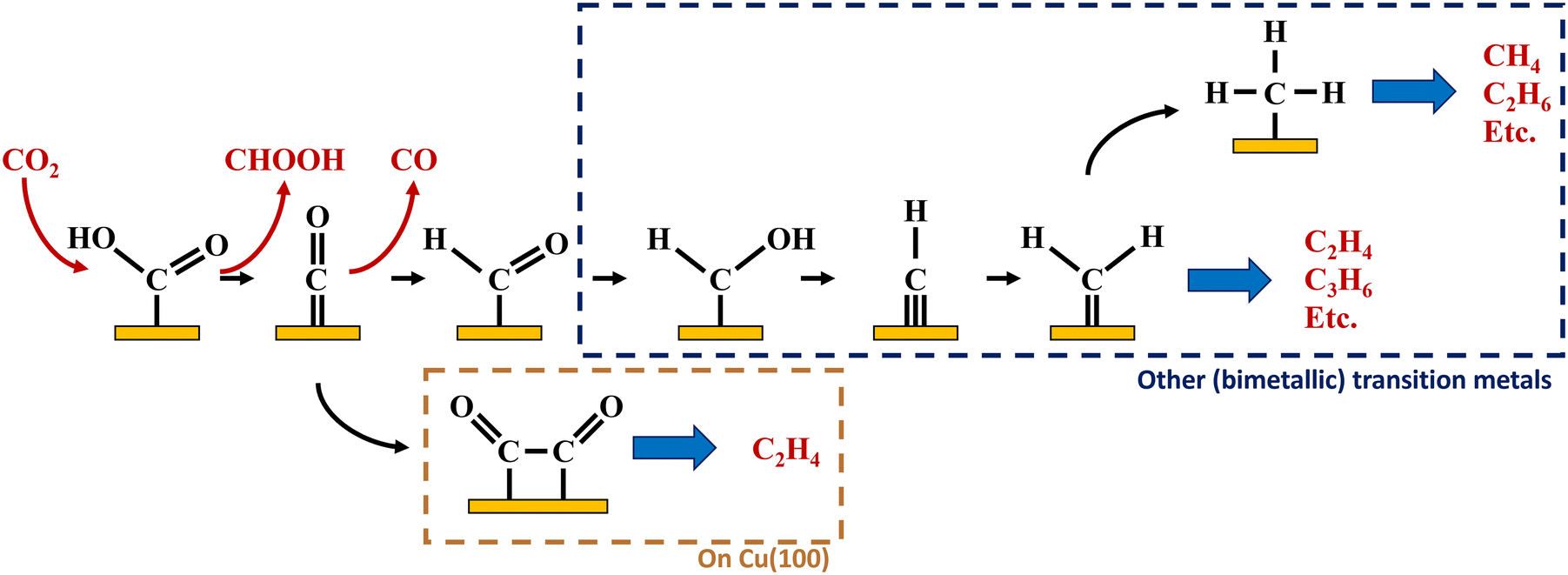 | ||
| Fig. 4 Possible pathways towards multi-carbon hydrocarbons on Cu(100) and other (bimetallic) transition metals based on literature and data in this article.1,2 | ||
Conclusions
In conclusion, a AuPd electrocatalyst has a higher activity towards hydrocarbons in a phosphate buffer compared to a bicarbonate buffer around the same pH. This observation also holds for a polycrystalline gold electrode and electrodeposited palladium on glassy carbon. Through the study of CO2 reduction on AuPd in bicarbonate buffers of varying buffer strengths, we conclude that the observed differences in product activities are caused by the difference in buffer strength, not the presence of certain anions at the electrode interphase. The results presented here show a strong link between the buffer choice and product activity, further demonstrating the strong interaction between the catalyst activity and the local reaction conditions at the surface. Our data suggests that ethylene and higher hydrocarbons are formed via a pH-dependent reaction mechanism that is different from the pH independent CO-dimerization mechanism on a copper surface. These observations highlight the importance of the buffer choice on the catalytic activity. In conclusion, the study gives additional evidence that catalyst design cannot be a one-sided effort solely optimizing the catalyst material. During the catalyst design process, the material should be evaluated under multiple reaction conditions to investigate their electrocatalytic response before conclusions are drawn on their selectivity.Author contributions
Daniël van den Berg: conceptualization, data curation, formal analysis, investigation, writing – original draft. Boaz Izelaar: XPS data curation, XPS formal analysis, writing – XPS material analysis & review. Shilong Fu: SEM data curation, writing – review. Ruud Kortlever: conceptualization, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld and S. Horch, et al., Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119(12), 7610–7672 CrossRef CAS.
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López and J. Pérez-Ramírez, et al., Long-chain hydrocarbons by CO2 electroreduction using polarized nickel catalysts, Nat. Catal., 2022, 5(6), 545–554 CrossRef CAS.
- Y. Hori, K. Kikuchi and S. Suzuki, Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution, Chem. Lett., 1985, 14(11), 1695–1698 CrossRef.
- S. Asperti, R. Hendrikx, Y. Gonzalez-Garcia and R. Kortlever, Benchmarking the Electrochemical CO2 Reduction on Polycrystalline Copper Foils: The Importance of Microstructure Versus Applied Potential, ChemCatChem, 2022, e202200540 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5(5), 7050–7059 RSC.
- M. Moura de Salles Pupo and R. Kortlever, Electrolyte effects on the electrochemical reduction of CO2, ChemPhysChem, 2019, 20(22), 2926–2935 CrossRef CAS.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, CO2 reduction selective for C≥2 products on polycrystalline copper with N-substituted pyridinium additives, ACS Cent. Sci., 2017, 3(8), 853–859 CrossRef CAS.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, Electrocatalytic alloys for CO2 reduction, ChemSusChem, 2018, 11(1), 48–57 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch and J. I. Gold, et al., Electroreduction of carbon dioxide to hydrocarbons using bimetallic Cu–Pd catalysts with different mixing patterns, J. Am. Chem. Soc., 2017, 139(1), 47–50 CrossRef CAS.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, Cu–Sn bimetallic catalyst for selective aqueous electroreduction of CO2 to CO, ACS Catal., 2016, 6(5), 2842–2851 CrossRef CAS.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad and K. Chan, et al., Electrochemical activation of CO2 through atomic ordering transformations of AuCu nanoparticles, J. Am. Chem. Soc., 2017, 139(24), 8329–8336 CrossRef CAS.
- J. He, K. E. Dettelbach, D. A. Salvatore, T. Li and C. P. Berlinguette, High-throughput synthesis of mixed-metal electrocatalysts for CO2 reduction, Angew. Chem., 2017, 129(22), 6164–6168 CrossRef.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, Electrochemical CO2 reduction over compressively strained CuAg surface alloys with enhanced multi-carbon oxygenate selectivity, J. Am. Chem. Soc., 2017, 139(44), 15848–15857 CrossRef CAS.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson and B. S. Brunschwig, et al., Nickel–gallium-catalyzed electrochemical reduction of CO2 to highly reduced products at low overpotentials, ACS Catal., 2016, 6(3), 2100–2104 CrossRef CAS.
- A. R. Paris and A. B. Bocarsly, Ni–Al films on glassy carbon electrodes generate an array of oxygenated organics from CO2, ACS Catal., 2017, 7(10), 6815–6820 CrossRef CAS.
- A. R. Paris and A. B. Bocarsly, Mechanistic insights into C2 and C3 product generation using Ni3Al and Ni3Ga electrocatalysts for CO2 reduction, Faraday Discuss., 2019, 215, 192–204 RSC.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon and G. Mul, et al., Palladium–gold catalyst for the electrochemical reduction of CO2 to C1–C5 hydrocarbons, Chem. Commun., 2016, 52(67), 10229–10232 RSC.
- J. J. L. Humphrey, D. Plana, V. Celorrio, S. Sadasivan, R. P. Tooze and P. Rodríguez, et al., Electrochemical Reduction of Carbon Dioxide at Gold-Palladium Core–Shell Nanoparticles: Product Distribution versus Shell Thickness, ChemCatChem, 2016, 8(5), 952–960 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj and S. Dahl, et al., Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol, Nat. Chem., 2014, 6(4), 320–324 CrossRef CAS PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media, Electrochim. Acta, 1994, 39(11–12), 1833–1839 CrossRef CAS.
- F. Gao and D. W. Goodman, Pd–Au bimetallic catalysts: understanding alloy effects from planar models and (supported) nanoparticles, Chem. Soc. Rev., 2012, 41(24), 8009–8020 RSC.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li and Y. Wang, et al., Ensemble Effect in Bimetallic Electrocatalysts for CO2 Reduction, J. Am. Chem. Soc., 2019, 141(42), 16635–16642 CrossRef CAS.
- C. Hahn, D. N. Abram, H. A. Hansen, T. Hatsukade, A. Jackson and N. C. Johnson, et al., Synthesis of thin film AuPd alloys and their investigation for electrocatalytic CO2 reduction, J. Mater. Chem. A, 2015, 3(40), 20185–20194 RSC.
- H. Noda, S. Ikeda, A. Yamamoto, H. Einaga and K. Ito, Kinetics of electrochemical reduction of carbon dioxide on a gold electrode in phosphate buffer solutions, Bull. Chem. Soc. Jpn., 1995, 68(7), 1889–1895 CrossRef CAS.
- M. Azuma, K. Hashimoto, M. Watanabe and T. Sakata, Electrochemical reduction of carbon dioxide to higher hydrocarbons in a KHCO3 aqueous solution, J. Electroanal. Chem. Interfacial Electrochem., 1990, 294(1–2), 299–303 CrossRef CAS.
- K. J. P. Schouten, E. P. Gallent and M. T. Koper, The influence of pH on the reduction of CO and CO2 to hydrocarbons on copper electrodes, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- R. L. Cook and A. F. Sammels, Electrocatalysis and Novel Electrodes for High Rate CO2 Reduction under Ambient Conditions, in Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, ed. B. P. Sullivan, K. Krist and H. E. Guard, Elsevier, 1992, pp. 217–262 Search PubMed.
- A. Kudo, S. Nakagawa, A. Tsuneto and T. Sakata, Electrochemical reduction of high pressure CO2 on Ni electrodes, J. Electrochem. Soc., 1993, 140(6), 1541–1545 CrossRef CAS.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 reduction: a classification problem, ChemPhysChem, 2017, 18(22), 3266–3273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01411h |
| This journal is © The Royal Society of Chemistry 2024 |