
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Heterocyclic carbene-based porous polymer macroligand for the Ni-catalyzed C–H arylation of benzothiophenes†
Partha
Samanta‡
 *a,
Remi
Beucher
a,
Riddhi
Kumari Riddhi
a,
Alisa
Ranscht
a,
Florian M.
Wisser
b,
Elsje Alessandra
Quadrelli
a and
Jerome
Canivet
*a
*a,
Remi
Beucher
a,
Riddhi
Kumari Riddhi
a,
Alisa
Ranscht
a,
Florian M.
Wisser
b,
Elsje Alessandra
Quadrelli
a and
Jerome
Canivet
*a
aUniv. Lyon, Université Claude Bernard Lyon 1, CNRS, IRCELYON – UMR 5256, 2 Av. Albert Einstein, 69626 Villeurbanne Cedex, France. E-mail: partha.samanta@icn2.cat; jerome.canivet@ircelyon.univ-lyon1.fr
bErlangen Center for Interface Research and Catalysis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany
First published on 26th September 2023
Abstract
A porous organic polymer (POP) with imidazolium functionality (IM-POP) has been synthesized through radical polymerization. N-Heterocyclic carbene (NHC) has been generated from an imidazolium moiety via base treatment followed by coordination of nickel salt, giving access to a single-site Ni–NHC catalyst inside the polymeric network. The NiCl2@NHC-POP was found to heterogeneously catalyze the direct selective C2–H arylation of (benzo)thiophenes using various aryl electrophiles with a turnover number (TON) up to 75, reaching cumulative TON of ca. 400 after recycling for six consecutive runs.
Molecular catalysis is a key aspect of organic transformations, and heterogenization of molecular catalytic processes has gained significant interest due to its potential for increasing sustainability through the recycling of the solid catalyst, thus increasing the turnover number, and having easy separation from the solubilized product. Heterogenization can be considered efficient only in the absence of leaching of the active species in the solution and with high accessibility of active sites within solid supports.1
Organic and coordination polymers, such as metal–organic frameworks (MOFs) using molecular ligands as monomers, can be employed as solid porous macroligands for molecular complexes.2 The macroligand strategy allows combining well-defined single-site molecular catalytic mechanisms with site-isolation at the solid surface, avoiding detrimental sintering, bimolecular deactivation processes or undesired binding.3,4
Compared to MOFs made with metal–carboxylate bonds, porous organic polymers (POPs) built around C–C linkages offer advantageous robustness, against hydrolysis for instance, and have been demonstrated to provide a more electron-rich environment when used as macroligands for heterogenized molecular catalysts.5–7 Furthermore, POPs benefit from high synthetic versatility thanks to a wide library of monomers with polymerizable groups, including bipyridine (bpy), phenanthroline and phosphine derivatives available for metal coordination.8,9
Thanks to a better electron-donating effect and a stronger binding ability to late transition metals, N-heterocyclic carbenes (NHC) represent an appealing alternative to bpy and phosphine ligands in molecular catalysts.10–12 NHC-functionalized MOFs and POPs, prepared from imidazolium-based linkers or monomers, were already reported as platform materials for luminescence, sensing, and for NHC- and transition metal-catalysed organic transformations.13 In particular, Pd(NHC) complexes embedded within POPs,14,15 often prepared by Al-mediated Scholl or Friedel–Craft reactions16 between N,N-(dibenzyl)benzimidazolium chloride and benzene followed by reaction with Pd(II) salts, were already reported to catalyse Suzuki–Miyaura cross-coupling reactions.17–19
Among C–C coupling reactions, C–H arylation reactions, whether oxidative or non-oxidative, are considered highly sustainable routes for the synthesis of (hetero)biaryl compounds with broad applications in pharmaceuticals20–22 and optoelectronic materials.23–26 Homogeneous Ni(NHC) complexes are known for C(sp2)–H activation including borylation, alkylation, alkenylation and for arylation of heteroarenes like indoles and azoles.27–29
Considering the high potential of NHC-based porous macroligands, we investigated their implementation in the challenging C–H arylation reaction catalysed by earth abundant nickel complex as an appealing alternative to palladium for C–C coupling reactions.30–32 Molecular Ni catalysts have been already heterogenized within bpy and phenanthroline (phen) based POP macroligands for C–H arylation reactions.33,34 However, Pd residues in bpy-based POPs, originating from the Sonogashira cross-coupling synthetic methodology, have to be proved to be inactive in the targeted reaction.33
Alternatively, phen-based POPs made by radical polymerization circumvented the use of transition metal catalysed synthesis but was performed in dimethylformamide.34 Furthermore in both cases, the use of a reducing agent, typically sodium borohydride, was necessary to reactivate the heterogeneous Ni-based catalyst for reuse.
Here we report the facile synthesis of a POP based on imidazolium vinyl monomer as a precursor of the NHC-functionalized macroligand. After metalation with the Ni(II) precursor, the material efficiently catalyzed the C–H arylation of benzothiophenes without an external activator.
The POP functionalized with pendant imidazolium units (IM-POP) was synthesised via radical polymerization of a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 divinylbenzene and 1-methyl-3-(4-vinylbenzyl)-1H-imidazol-3-ium chloride mixture using azobis(cyclohexanecarbonitrile) (ACHN) as initiator under solvothermal conditions in tetrahydrofuran (THF) and water mixture (Scheme 1). The obtained IM-POP monolith has been washed thoroughly by Soxhlet extraction with THF and, after drying at 80 °C under vacuum, characterized by solid-state 13C NMR spectroscopy, nitrogen and toluene vapor physisorption measurements, FT-IR spectroscopy, thermogravimetry (TGA) and elemental analysis (Fig. S3–S7 and S10 and Table S1†).
:1 divinylbenzene and 1-methyl-3-(4-vinylbenzyl)-1H-imidazol-3-ium chloride mixture using azobis(cyclohexanecarbonitrile) (ACHN) as initiator under solvothermal conditions in tetrahydrofuran (THF) and water mixture (Scheme 1). The obtained IM-POP monolith has been washed thoroughly by Soxhlet extraction with THF and, after drying at 80 °C under vacuum, characterized by solid-state 13C NMR spectroscopy, nitrogen and toluene vapor physisorption measurements, FT-IR spectroscopy, thermogravimetry (TGA) and elemental analysis (Fig. S3–S7 and S10 and Table S1†).
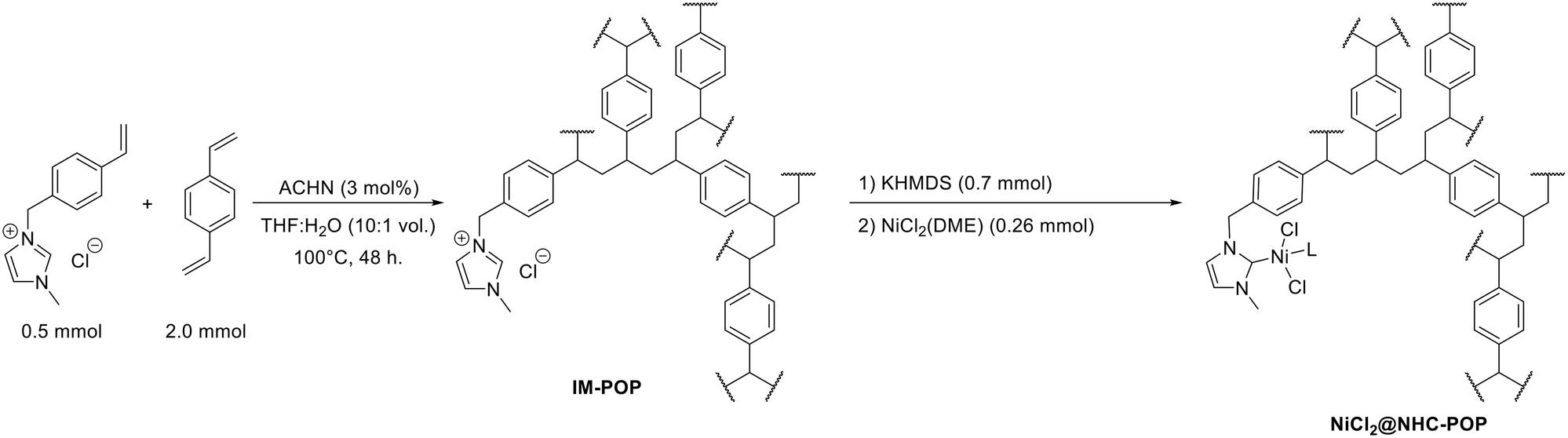 | ||
| Scheme 1 Synthesis of the IM-POP porous macroligand (shown as a single repeating unit) and subsequent nickelation towards NiCl2@NHC-POP catalyst (L = DME or solvent). | ||
In the FT-IR spectrum, the absence of a signal at 1683 cm−1, corresponding to vinylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration, evidenced the successful polymerization.14,17,18,35 Vibrations at 1600–1700 cm−1 were attributed to CN stretching in imidazolium units.36–39
C stretching vibration, evidenced the successful polymerization.14,17,18,35 Vibrations at 1600–1700 cm−1 were attributed to CN stretching in imidazolium units.36–39
The apparent surface area of the IM-POP was estimated from a nitrogen physisorption isotherm at 77 K to be 510 m2 g−1, similar to already reported analogous POPs.17 The IM-POP has a total pore volume of 0.54 cm3 g−1. The N2 physisorption isotherm is characterised by a type H4 hysteresis loop (Fig. S4†), indicative of the presence of micropores. The closure of the loop at around 0.42p/p0 is characteristic for cavitation, i.e. larger mesopores are partially accessible through smaller pores (<5–6 nm).40 The fact that the hysteresis between adsorption and desorption branch does not close below 0.42p/p0 is indicative for swelling of the network.
A toluene vapor physisorption isotherm was recorded to investigate the solvent accessibility. From the toluene vapor isotherm, a pore volume of 1.04 cm3 g−1 was determined. The pore volumes obtained from physisorption measurements highlight the good solvent accessibility of all pores. As for the N2 physisorption isotherm, cavitation occurs also in the toluene physisorption isotherm, albeit at a different partial pressure (at around 0.2p/p0, Fig. S6†). The adsorption profiles showed a H3 hysteresis loop in type IV isotherms which are usually observed for flexible materials having most likely a wide pore size distribution (Fig. S4 and S6†).40
TGA performed under air showed that IM-POP was stable up to 300 °C (Fig. S5†).
The solid state 13C NMR spectrum (Fig. S3†) shows broad peaks of aromatic carbon atoms at 125, 135 and 145 ppm as well as a broad, overlapping signal at approx. 40 ppm which is assigned to carbon atoms from bridging methylene and methine groups. Additional signals at 28 and 13 ppm are attributed to ethylene groups of ethyl-vinyl-benzene, and at 112 ppm to residual dangling vinyl groups. Based on elemental analysis (Table S1†), the chemical composition of IM-POP was confirmed with a loading of 1 mmol of imidazolium per gram of porous polymer.
Prior to metal coordination, the imidazolium moieties of IM-POP have been converted into N-heterocyclic carbenes by base treatment using potassium hexamethyldisilazide (KHMDS). The solid suspension was subsequently exposed to nickel dichloride dimethoxyethane (DME) to afford the NiCl2@NHC-POP catalyst (Scheme 1).
Elemental analyses revealed a nickel loading of 2.8 wt% and a nitrogen content of 2.5 wt% which correspond to a Ni:NHC ratio of 1:2 (Tables S1 and S2†). Furthermore, X-ray photoelectron spectrometry (XPS) analysis showed peaks corresponding to Ni 2p3/2 at 856.2 eV and 2p1/2 at 874.1 eV, along with their shakeup satellite peaks at 862 and 880 eV, respectively (Fig. S10†). In the case of Ni binding energies, the oxidation state cannot be assigned unambiguously.41,42 It has been shown that the binding energy strongly depends on the oxidation state, the nature of the ligand atom or the coordination number.37,41,42 Considering the similarity in electron donating between NHC carbenes and phosphines,43 and the binding energies typically observed in NiCl2(PR3)2 complexes (853.6–857.0 eV),36–38 we tentatively assign the signals observed here to a Ni +II oxidation state, having 2 chloride and one or two NHC ligands.
For molecular complexes, the reaction of NHC and Ni(II) species gave rise to mono-coordinated Ni(II)(NHC) even in the presence of excess NHC, in which the reduction towards bis-coordinated Ni(0)(NHC)2 occurs when using strong reducing agents (such as NaH or BuLi) but not using strong bases (such as NaOtBu).44 Thus, taking into account the site isolation inside the POP network and the reactivity reported above, heterogenized complexes are postulated here to be more likely mono-coordinated Ni(II)(NHC) as shown in Scheme 1, even if bis(NHC) complexes cannot be unambiguously ruled out.
While no change appeared in the solid state 13C-NMR of NiCl2@NHC-POP compared to its parent polymer (Fig. S3†), FT-IR spectra showed the weakening of the peak at 1159 cm−1, attributed to quaternary imidazolium, while a new band at 1233 cm−1 appeared upon nickelation (Fig. S7†), similar to previously reported analogous POP-supported Pd(NHC) catalysts.14
The UV-vis spectrum of NiCl2@NHC-POP, compared to that of pristine IM-POP, showed a new broad band at 350 nm which was assigned to ligand-to-metal charge transfer.45 Additionally, bands at 400 and 670 nm were attributed to the splitting of d orbitals in the Ni complex (Fig. S11†).46 These transitions revealed a more likely tetrahedral geometry for the POP-embedded Ni–NHC complex,47 in line with the geometry previously reported for a Ni(II) mono–NHC complex.44
Upon nickelation, the NiCl2@NHC-POP showed a reduced porosity compared to the parent IM-POP, as found by nitrogen physisorption (Fig. S4†), with an apparent surface area of 390 m2 g−1, in the range of previously reported metalated NHC-based POP catalysts.14 Toluene vapor physisorption measurement showed that NiCl2@NHC-POP has similar solvent uptake compared to pristine POP but with a less pronounced hysteresis during desorption (Fig. S6†).
Based on previously reported Ni-catalysed C–H arylation of benzothiophenes,33,34,48 the NiCl2@NHC-POP was evaluated as a catalyst for the same reaction varying the base, solvent and temperature. As for other reported POP-based catalysts in Ni-catalysed benzothiophene C–H arylation, the unique combination of toluene as the solvent and lithium hexamethyldisilazide (LiHMDS) as the base allowed the reaction between benzothiophene and iodobenzene to proceed with a yield up to 69% for the 2-phenyl-benzothiophene as the sole product at 120 °C after 18 hours. For comparison, previously reported bipyridine-based analogue Ni@BpyMP-1 (ref. 33) and phenanthroline-based analogue POP-Phen@Ni-2 (ref. 34) allowed reaching similar yields with however slightly lower turnover numbers of 50 and 44, respectively, compared to the TON of 75 obtained with NiCl2@NHC-POP under the same conditions (Scheme 2).
 | ||
| Scheme 2 Comparison of POP-based Ni-catalysts for the direct and fully selective C2-phenylation of benzothiophene. | ||
In contrast to the Ni(bpy) analogous system,48 under the same conditions, the in situ combination of NiCl2·glyme and 1-methyl-3-(4-vinylbenzyl)-1H-imidazolium chloride, as a molecular analogue to the IM-POP porous macroligand, in a nickel:imidazolium molar ratio of 1:1 or 1:2, did not lead to arylated products formation despite favourable basic conditions (LiHMDS) allowing the deprotonation of the imidazolium ligand to form the corresponding NHC ligand.
Similarly, a catalyst prepared by physisorption of NiCl2·glyme within the polydivinylbenzene polymer (DVB-POP), that is unequipped of imidazolium and NHC sites, did not allow the reaction to proceed, evidencing that physisorbed Ni species, not coordinated to NHC ligand, could not act as a catalyst here (see the ESI† for details).
The yield of the reaction drastically dropped to 10% in the case of bromobenzene, in line with reactivity reported for other Ni catalysts,34,48 when no product formation was found using chlorobenzene, phenyl p-toluenesulfonate or anisole as the coupling partner (Table S4†).
A hot filtration test performed after four hours of reaction showed no evolution of the 2-phenyl-benzothiophene production from the supernatant solution, evidencing the absence of leached active species from the NiCl2@NHC-POP under catalytic conditions (Fig. S14†).
The stability of the POP-based catalyst was further confirmed with retained textural properties according to FT-IR (Fig. S16†) and solid-state NMR spectroscopy (Fig. S17†) as well as electron microscopy (Fig. S18 and S19†). Furthermore, the XPS spectrum after catalysis is very similar to the XPS spectrum of fresh NiCl2@NHC-POP, with binding energies of 873.7 eV (2p1/2) and 855.9 eV (2p3/2, Fig. S20†). Importantly, no reduced Ni species could be detected with characteristic binding energies around 870 eV (2p1/2) and 854 eV (2p3/2),39 highlighting the high stability of the heterogenized nickel species.
The NiCl2@NHC-POP heterogeneous catalyst was subjected to consecutive reuses after simple filtration and washing with methanol. With a first decrease in TON from 75 to 67 after two runs, the activity remained stable with a TON of ca. 60 for at least five runs, leading to a cumulative TON of 393 (Fig. 1).
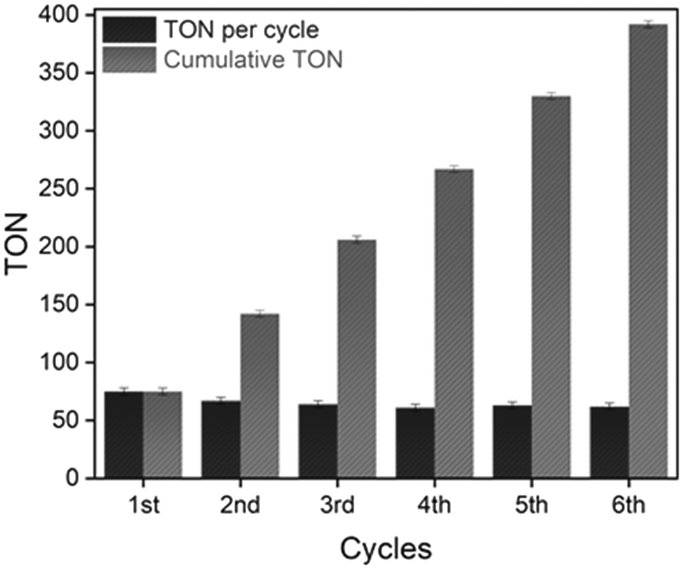 | ||
| Fig. 1 Graphical representation of the TON value per catalytic run (black) and the evolution of the cumulative TON (grey) using the NiCl2@NHC-POP catalyst. Conditions: 10 mg of NiCl2@NHC-POP (4.8 μmol Ni), benzothiophene (0.5 mmol), iodobenzene (0.6 mmol), LiHMDS (1.1 mmol), toluene (4 ml), under argon, 120 °C, 500 rpm, 12 hours. | ||
ICP analysis revealed a slight leaching of Ni from the solid catalyst during the two first catalytic runs; no further loss being observed for the four subsequent runs, with retention of 84% of the initial nickel amount after six consecutive reuses (Table S8†), in line with a previous report on nickel catalyst heterogenization within POPs.33 More importantly, the NiCl2@NHC-POP did not require any additive to maintain its activity upon reuse, in contrast to bipyridine33 and phenanthroline based-systems.34 Thus, thanks to a stronger electron-donating effect and a stronger binding affinity to late transition metals,10–12 the NHC ligand was postulated to provide a more stable environment to the active Ni species than N-donor bipyridine and phenanthroline, also in line with the higher TON reported (Scheme 2).
The applicability of the NiCl2@NHC-POP heterogeneous catalyst was studied on various (benzo)thiophenes derivatives, as well as on aryl iodide derivatives (Scheme 3).
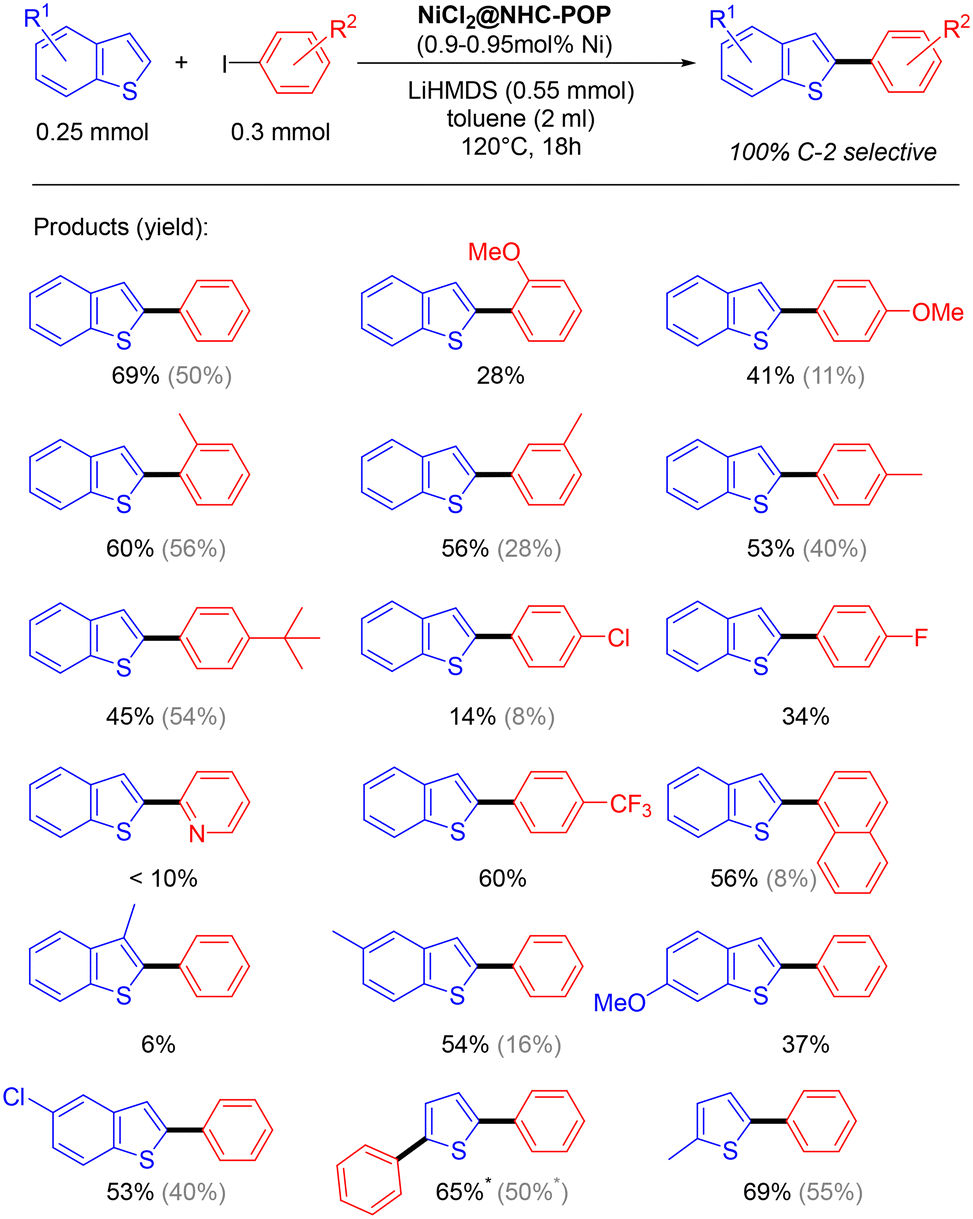 | ||
| Scheme 3 Direct C2–H arylation of benzothiophenes and thiophenes heterogeneously catalyzed by the NiCl2@NHC-POP (* 2.4 equiv. of iodobenzene are used). For comparison, activities of the previously reported Ni@BpyMP-1 catalyst are shown in gray.33 | ||
The C2–H arylation of benzothiophene catalysed by the NiCl2@NHC-POP in the presence of LiHMDS proceeded with moderate to good yields and, in general, with higher yield than the previously reported POP-heterogenized bpy system using the same 1 mol% Ni loading33 (e.g. 2-phenyl-benzothiophene 50% vs. 69%; 2-(4-methoxy)phenyl-benzothiophene 11% vs. 41%; 2-(naphthalen-1-yl)benzothiophene 8% vs. 56%; see Scheme 3).
Phenyl iodo electrophiles with methyl group at the ortho, meta or para-position showed no restriction in the reaction with yields from 53 to 60%.
The benzothiophene arylation also proceeded with sterically demanding para-(tert-butyl)-phenyl and naphthyl electrophiles, giving rise to 45 and 56% yield, respectively, as well as with electron-withdrawing para-(trifluoromethyl)-phenyl coupling partner with 60% yield. In contrast, iodopyridine gave a low yield (<10%), likely due to the possible pyridyl N-coordination to nickel consequently leading to poisoned catalytic sites.
Under the same catalysis conditions, thiophene reacted with 1.2 equivalents of iodobenzene to give together 2-phenylthiophene (30% yield) and 2,5-diphenyl-thiophene (21% yield); when using 2.4 equivalents of iodobenzene, it gave rise to only 2,5-diphenylthiophene with 69% yield.
Also, 3-methylbenzothiophene was not converted (only 6% yield); the methyl on position 3 of benzothiophene likely hindered the reaction as already observed for the homogeneous Ni(bpy) system.48
Finally, with overall similar reactivity in unique combination with LiHMDS, the benzothiophene C2–H arylation catalysed by the NiCl2@NHC-POP can be postulated to proceed following the same catalytic pathway reported for the Ni(bpy) system, with the deprotonation of benzothiophene to form the corresponding lithiated species as the primary step, followed by transmetallation with (aryl)Ni species in a Ni(I)/Ni(III) redox catalytic cycle.33,48
Conclusions
A radical polymerization strategy allowed designing novel NHC-based macroligands for heterogenized molecular catalysis. Starting from an easily accessible imidazolium monomer, the NiCl2@NHC-POP heterogeneous catalyst allowed the C2–H arylation of benzothiophene to proceed with 100% regioselectivity and with yields up to 69% in the presence of LiHMDS as a base. Over simple reuse without any additive, a cumulative TON of 393 was obtained for the production of 2-phenyl-benzothiophene. The catalytic system was applicable to a range of iodoaryl electrophiles as well as to (benzo)thiophene derivatives.The use of NHC ligand within a POP matrix allowed tackling issues raised by bpy and phen analogous systems by increasing the stability of the heterogenized Ni active species. Furthermore, the Ni(NHC) site isolation within the polymer seemed to allow reactivity not reachable by an analogous homogenous system under the same conditions.
With higher intrinsic activity, also maintained during reuse without any additive, the NHC-POP macroligand appears as an optimized platform, compared to bpy and phen analogues, for heterogenized molecular catalysis, and paves the way for further development of heterogenized molecular catalysis for fine chemicals and energy.
Author contributions
P. S.: investigation, formal analysis and writing – review & editing. R. B.: investigation (supporting) and writing – review & editing. R. K. R.: investigation (supporting) and writing – review & editing. A. R.: investigation (supporting) and writing – review & editing. E. A. Q.: supervision and writing – review & editing. F. M. W.: investigation (supporting), formal analysis and writing – review & editing. J. C.: conceptualization, funding acquisition, supervision and writing – original draft, review & editing. The manuscript was written through the contribution of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the C123 project that has received funding from the European Union's Horizon 2020 research, the innovation program under grant agreement no. 814557 and the ANR project FLIPS (ANR-21-CE07-0028). The authors are also grateful to the IRCELYON scientific services.Notes and references
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- F. M. Wisser, Y. Mohr, E. A. Quadrelli and J. Canivet, ChemCatChem, 2020, 12, 1270–1275 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2015, 115, 127–150 CrossRef CAS PubMed.
- M. D. Korzyński and C. Copéret, Trends Chem., 2021, 3, 850–862 CrossRef.
- F. M. Wisser, P. Berruyer, L. Cardenas, Y. Mohr, E. A. Quadrelli, A. Lesage, D. Farrusseng and J. Canivet, ACS Catal., 2018, 8, 1653–1661 CrossRef CAS.
- M. König, M. Rigo, N. Chaoui, T. Tran Ngoc, J. D. Epping, J. Schmidt, P. Pachfule, M.-Y. Ye, M. Trunk, J. F. Teichert, M. Drieß and A. Thomas, Angew. Chem., Int. Ed., 2020, 59, 19830–19834 CrossRef PubMed.
- H. Li, F. Pan, C. Qin, T. Wang and K.-J. Chen, Adv. Energy Mater., 2023, 13, 2301378 CrossRef CAS.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- P. Gäumann, D. Cartagenova and M. Ranocchiari, Eur. J. Org. Chem., 2022, 2022, e202201006 CrossRef.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef CAS PubMed.
- Y. Wang, J.-P. Chang, R. Xu, S. Bai, D. Wang, G.-P. Yang, L.-Y. Sun, P. Li and Y.-F. Han, Chem. Soc. Rev., 2021, 50, 13559–13586 RSC.
- H. Zhao, L. Li, Y. Wang and R. Wang, Sci. Rep., 2014, 4, 5478 CrossRef PubMed.
- S. Let, G. K. Dam, P. Samanta, S. Fajal, S. Dutta and S. K. Ghosh, J. Org. Chem., 2022, 87, 16655–16664 CrossRef CAS PubMed.
- N. Taheri, M. Dinari and M. Asgari, ACS Appl. Polym. Mater., 2022, 4, 6288–6302 CrossRef CAS.
- H. Lin, X. Gao, H. Yao, Q. Luo, B. Xiang, C. Liu, Y. Ouyang, N. Zhou and D. Xiang, Catal. Sci. Technol., 2021, 11, 3676–3680 RSC.
- S. Xu, K. Song, T. Li and B. Tan, J. Mater. Chem. A, 2014, 3, 1272–1278 RSC.
- X. Liu, W. Xu, D. Xiang, Z. Zhang, D. Chen, Y. Hu, Y. Li, Y. Ouyang and H. Lin, New J. Chem., 2019, 43, 12206–12210 RSC.
- S. Dadiboyena, Eur. J. Med. Chem., 2012, 51, 17–34 CrossRef CAS PubMed.
- R. S. Keri, K. Chand, S. Budagumpi, S. Balappa Somappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002–1033 CrossRef CAS PubMed.
- J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- M. R. Reddy, H. Kim, C. Kim and S. Seo, Synth. Met., 2018, 235, 153–159 CrossRef CAS.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed.
- A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed.
- B. C. Lee, C.-F. Liu, L. Q. H. Lin, K. Z. Yap, N. Song, C. H. M. Ko, P. H. Chan and M. J. Koh, Chem. Soc. Rev., 2023, 52, 2946–2991 RSC.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- M. J. West and A. J. B. Watson, Org. Biomol. Chem., 2019, 17, 5055–5059 RSC.
- Y. Mohr, M. Alves-Favaro, R. Rajapaksha, G. Hisler, A. Ranscht, P. Samanta, C. Lorentz, M. Duguet, C. Mellot-Draznieks, E. A. Quadrelli, F. M. Wisser and J. Canivet, ACS Catal., 2021, 11, 3507–3515 CrossRef CAS.
- Y. Tang, Z. Dai, S. Wang, F. Chen, X. Meng and F.-S. Xiao, Chem. – Asian J., 2021, 16, 2469–2474 CrossRef CAS PubMed.
- F. A. Yassin, F. Y. El Kady, H. S. Ahmed, L. K. Mohamed, S. A. Shaban and A. K. Elfadaly, Egypt. J. Pet., 2015, 24, 103–111 CrossRef.
- M. Senō, S. Tsuchiya, M. Hidai and Y. Uchida, Bull. Chem. Soc. Jpn., 1976, 49, 1184–1186 CrossRef.
- G. J. Colpas, M. J. Maroney, C. Bagyinka, M. Kumar, W. S. Willis, S. L. Suib, P. K. Mascharak and N. Baidya, Inorg. Chem., 1991, 30, 920–928 CrossRef CAS.
- J. R. Blackburn, R. Nordberg, F. Stevie, R. G. Albridge and M. M. Jones, Inorg. Chem., 1970, 9, 2374–2376 CrossRef CAS.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, in NIST X-ray Photoelectron Spectroscopy Database, NIST Standard Reference Database Number 20, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2000, DOI:10.18434/T4T88K.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. St, C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- B. Zhao, X.-K. Ke, J.-H. Bao, C.-L. Wang, L. Dong, Y.-W. Chen and H.-L. Chen, J. Phys. Chem. C, 2009, 113, 14440–14447 CrossRef CAS.
- R. Bharti, M. Verma, A. Thakur, R. Sharma, R. Bharti, M. Verma, A. Thakur and R. Sharma, in Carbene, IntechOpen, 2022 Search PubMed.
- K. Matsubara, S. Miyazaki, Y. Koga, Y. Nibu, T. Hashimura and T. Matsumoto, Organometallics, 2008, 27, 6020–6024 CrossRef CAS.
- M. Sharma, A. M. Perkins, A. K. Duckworth, E. J. Rouse, B. Donnadieu, B. Adhikari, S. L. Stokes and J. P. Emerson, Inorganics, 2023, 11, 120 CrossRef CAS.
- Z. Wei, Y. Peng, D. L. Hughes, J. Zhao, L. Huang and X. Liu, Polyhedron, 2014, 69, 181–187 CrossRef CAS.
- D. Lomjanský, C. Rajnák, J. Titiš, J. Moncoľ, L. Smolko and R. Boča, Inorg. Chim. Acta, 2018, 483, 352–358 CrossRef.
- Y. Mohr, G. Hisler, L. Grousset, Y. Roux, E. A. Quadrelli, F. M. Wisser and J. Canivet, Green Chem., 2020, 22, 3155–3161 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Catalyst synthesis, characterization and catalysis details. See DOI: https://doi.org/10.1039/d3cy01263h |
| ‡ Current address: Catalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and The Barcelona Institute of Science and Technology, Campus UAB, Bellaterra, Barcelona 08193, Spain. |
| This journal is © The Royal Society of Chemistry 2023 |