
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anomalous behaviour of silver catalyst for soot oxidation explained: state of silver when operating and the influence of potassium ions†
Ewa M.
Iwanek (née Wilczkowska)
 *a,
Donald. W.
Kirk
b and
Zbigniew
Kaszkur
c
*a,
Donald. W.
Kirk
b and
Zbigniew
Kaszkur
c
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: ewa.iwanek@pw.edu.pl
bDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College St., M5S3E5, Toronto, ON, Canada
cInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 11th September 2023
Abstract
Two independent steps in which CO2 formed were observed in soot oxidation using silver catalysts supported on cerium-doped zirconia and zirconium-doped ceria. The first was attributed to thermal decomposition of oxidized silver species to metallic silver with simultaneous soot combustion, whereas the second was catalytic soot combustion. The first step was endothermic when the catalyst was heated without soot, whereas it was overall exothermic when the catalyst was heated while in tight contact with soot. Both mechanisms of CO2 formation are described. The influence of potassium ions on the catalytic activity in soot oxidation was found to be substantial for 1 wt% Ag/Zr0.93Ce0.07O2, though negligible for 10 wt% Ag/Zr0.93Ce0.07O2. In contrast, in the case of the zirconium-doped ceria support, the 1.5 wt% K+/Ce0.85Zr0.15O2 sample was equally active in soot combustion with and without silver. Proper interpretation of activity onset of silver catalysts for soot combustion, the state of silver when operating and the influence of potassium ions on the activity was the focus of this study.
1. Introduction
Particulate matter from diesel engines continues to be an environmental issue. Supported silver catalysts for combustion of soot have been often studied for potential application in diesel engine exhaust clean-up.1–8 The harmful nature of particulate matter (PM) in the exhaust of vehicles with diesel engines is well-documented.9–12 In fact, PM has been classified as a carcinogenic substance by the World Health Organization13 and so its removal from the exhaust stream is critical for human health. In the view of our recent findings, in order to design the best catalyst, the interpretation of activity results from laboratory studies must take into account several decisive points. The results of our studies show that a critical assessment of the optimistic diagnosis of activity of silver catalysts for soot oxidation is warranted. Although many issues have been successfully tackled and are commonly agreed on by researchers, some still need to be addressed. One of the points of agreement is that the contact between the soot and catalyst plays an important role.14–17 Another commonly accepted fact is that silver catalysts contain metallic silver, as evidenced by X-ray Diffraction patterns,1–8,18 but also contain some oxidized silver species on the surface. The presence of the latter has been determined by means of Temperature-Programmed Reduction studies, in which low-temperature peaks are assigned to the reduction of oxidized silver species.4,19 Some high-resolution TEM images indicate the presence of silver oxide as a separate phase between metallic silver and CeO2,8 but even when no such phase is observed, the TPR still shows a low-temperature reduction peak attributed to oxidized silver species.2,20 However, the views on which species are more active is divided. Some groups consider the oxidized form to be less reactive, e.g., Aneggi et al.8 They have noted a negligible influence of a pre-reduction treatment on the T50 of the zirconia supported silver catalyst and concluded that in soot combustion the metallic form of silver is more active than oxidized forms of silver. In contrast, Shimizu et al.21 state that “Ag2O completely deactivated after the first catalytic run because of its conversion to Ag metal particle, indicating that Ag2O does not act as a catalyst but as a strong oxidant for soot combustion”.Two other important factors for laboratory-scale tests are the model soot material used in the study and the conditions of the catalyst pre-treatment. Studies of processes such as catalytic gasification of coal and catalytic oxidation of diesel soot are often performed on different carbonaceous materials. Few studies deal with actual coal22,23 or diesel engine soot24 at the laboratory scale. In the case of model soot materials some are easier to obtain than others, some are easier to combust. In order to obtain meaningful and reproducible results using a model material with uniform composition and properties helps to systemize the results and knowledge acquired through catalytic tests. In the case of silver catalysts, the pre-treatment of the catalyst prior to the activity tests is also of critical importance. Some groups, e.g., Aneggi et al.8 have performed soot combustion measurements after maintaining the catalysts in a reducing atmosphere in order to reduce the surface Ag2O prior to activity measurements. Nevertheless, thus far, few papers have properly addressed the issue of the impact of the oxidized forms of silver on the activity of the catalyst, suggested a proper pre-treatment method which is not affected by oxidized forms of silver.
The term “ceria–zirconia” or “ceria–zirconia mixed oxide” is often found in the literature, but is not precise, and the material itself does not contain both oxides as separate phases, but is rather a solid solution of one type of ions incorporated into the matrix of the oxide of the major component.25,26 When the Ce/(Ce + Zr) ratio is large, the structure of pure ceria is kept intact, i.e., the fluorite-type, cubic structure, but the zirconium ions do influence the lattice parameter of the oxide due to ionic radius mismatch. In this study, such oxides are referred to as “zirconium-doped ceria”. In contrast, when the Ce/(Ce + Zr) ratio is small, the structure is that of zirconia with cerium cations inserted into the Zr4+ sites,26 and those will be called “cerium-doped zirconia”. Understanding this distinction is crucial in comprehending why ceria27–31 and zirconium-doped ceria32 are highly reducible oxides with excellent soot oxidation activity, whereas zirconia and cerium-doped zirconia are only used as a support rather than a catalyst for soot oxidation,19 though both are referred to as “ceria–zirconia”. A very important characteristic of zirconia is the existence of three polymorphic types of ZrO2, namely: monoclinic (P21/c), tetragonal (P42/nmc) and cubic (Fm3m).33 The polymorph into which the other two transform at elevated temperature is the monoclinic zirconia, which has the least beneficial properties. In order to keep from turning into the monoclinic polymorph, it is often partially stabilized with magnesia or yttria.34 Stabilization of zirconia with yttrium ions is well known in the application of fuel cells, which have employed yttria stabilized zirconia (YSZ) as the solid electrolyte for decades.35,36 Cubic zirconia is partially stabilized with magnesia to increase strength of the material.36,37 In this study both cerium-doped zirconia and zirconium-doped ceria are investigated and compared.
2. Experimental
2.1 Materials
The zirconia and ceria-doped zirconia supports were synthesized using ammonium ceric nitrate (analytical grade, BDH) and zirconyl nitrate (pure for analysis, Sigma-Aldrich) in order to get the desired compositions. In the case of the zirconium-doped ceria, the concentration of zirconium ions was 15 at% (Zr/(Ce + Zr)), whereas the cerium-doped zirconia was obtained with cerium loadings of 7 at% (Ce/(Ce + Zr)). Such compositions allowed us to have a single-phase support in each case, which makes the interpretation of the results easier than in the case of multiphase supports. The salts were dissolved in water, mixed and precipitated with a 15% NH3 solution (purum p.a. POCh, Gliwice, Poland). The precipitate was washed, dried, and calcined at 550 °C for 4 h. Undoped zirconia and undoped ceria were obtained from the same reagents by the same method as the mixed systems. The supports were impregnated sequentially with a solution of silver nitrate (analytical grade, POCH Gliwice) to give a final loading of 1, 5 and 10 wt% of metallic silver and then with a solution of potassium carbonate (analytical grade, POCH Gliwice) to obtain a loading of potassium ions of either 0.5 or 1.5 wt%. In the case of both steps, the solvent was evaporated, the catalyst was calcined for one hour at 550 °C.2.2 Characterisation studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, transferred into an agate mortar and ground with a pestle for exactly 1 minute directly before the TPO measurements to attain a tight contact of the catalyst with soot. No other pretreatment of samples was implemented. The powder was thoroughly mixed and sample of approx. 50 mg were used for the experiments. Most of the experiments were conducted in premixed synthetic air (N2:O2 = 80:20 ± 1%, Multax). The temperature program consisted of heating the samples to 850 °C at the constant rate of 5 degrees per minute with the flow rate of 100 mL min−1. Mass spectra were collected during the measurement in the scan bar graph mode. An experiment with argon (99.999% purity, Multax) was also performed in accordance with the same temperature program, though with a smaller sample (only 4 mg of soot were used) so that the exothermic effect would not upset the temperature program, with the Multiple Ion Detection MS mode (m/z = 18 and 44 signals were collected using the Channeltron detector).
:200) and LynxEye strip detector, working in Bragg–Brentano geometry. Diffraction patterns were recorded in a continuous mode with rate of 0.02 degree per second.
:1 ratio, transferred into an agate mortar and ground with a pestle for exactly 1 minute directly before the TPO measurements to attain a tight contact of the catalyst with soot. No other pretreatment of samples was implemented. The powder was thoroughly mixed and sample of approx. 50 mg were used for the experiments. Most of the experiments were conducted in premixed synthetic air (N2:O2 = 80:20 ± 1%, Multax). The temperature program consisted of heating the samples to 850 °C at the constant rate of 5 degrees per minute with the flow rate of 100 mL min−1. Mass spectra were collected during the measurement in the scan bar graph mode. An experiment with argon (99.999% purity, Multax) was also performed in accordance with the same temperature program, though with a smaller sample (only 4 mg of soot were used) so that the exothermic effect would not upset the temperature program, with the Multiple Ion Detection MS mode (m/z = 18 and 44 signals were collected using the Channeltron detector).
:200) and LynxEye strip detector, working in Bragg–Brentano geometry. Diffraction patterns were recorded in a continuous mode with rate of 0.02 degree per second.
The in situ PXRD measurements were performed in a lab-build environmental chamber (described in ref. 38) equipped with an automated gas-feeding system, temperature controllers and mass spectrometer (Hiden Analytical). The sample was kept in He (5.0 purity, 20 mL per minute) and heated in 20 °C steps from room temperature to 280 °C. The diffraction patterns were collected for each step in the scattering angle range of 10–140 degrees after the temperature had stabilized.
000 and 20000 times. EDX data were acquired with the following parameters: 15 kV, spot size 7. Images were also acquired using a circular backscatter detector.
3. Results and discussion
The zirconium-doped ceria and cerium-doped zirconia supports chosen for further testing were selected based on the results of our previous studies, which have shown that the Ce0.85Zr0.15O2 and Zr0.93Ce0.07O2 supports contain only one phase, the fluorite-type cubic ceria structure and tetragonal zirconia structure, respectively, and have the most beneficial properties.25,26 As in the case of all of the silver catalysts for soot combustion in the literature cited in the Introduction section, the XRD studies have shown that the silver deposited onto Zr0.93Ce0.07O2 is present in the metallic form (Fig. 1), even in the sample which contains only 1 wt% Ag (Fig. 1 enlargement). All of the other peaks belong to the tetragonal zirconia phase with the P42/nmc space group. | ||
| Fig. 1 XRD patterns of the support (Zr0.93Ce0.07O2) with different silver loadings with back-scattered electron (BSE) images (left) of the undoped support and of 10 wt% Ag; enlarged 35–40 deg scattering angle region of the 1 wt% Ag and 5 wt% Ag patterns (right) to show presence of metallic silver peaks. | ||
The values of the specific surface area of the cerium-doped zirconia (ZC) and zirconium-doped ceria supports are 63 and 77 m2 g−1, respectively (Fig. 2), whereas those of undoped zirconia and undoped ceria were only 48 and 35 m2 g−1. Hence it can be seen that the incorporation of zirconium ions into the ceria lattice and vice versa increases the surface area. It can be seen that the deposition of potassium onto the ZC support does not lead to changes in the surface area, whereas the silver loading leads to a substantial decrease of the specific surface area of the catalyst. Already the addition of 1 wt% leads to a decrease of approx. 14% of the original surface. The deposition of 5 and 10 wt% of silver on this support leads to further decrease of the surface area, namely by 41 and 48% respectively. In the case of the zirconium-doped ceria support, the deposition of a large dose (1.5 wt%) of potassium causes a decrease of the surface area, but the drop is not as substantial as that observed for the catalyst with 10 wt% silver. The deposition of potassium carbonate on the catalyst with 10 wt% silver, followed by calcination, causes only a slight further decrease of the surface area.
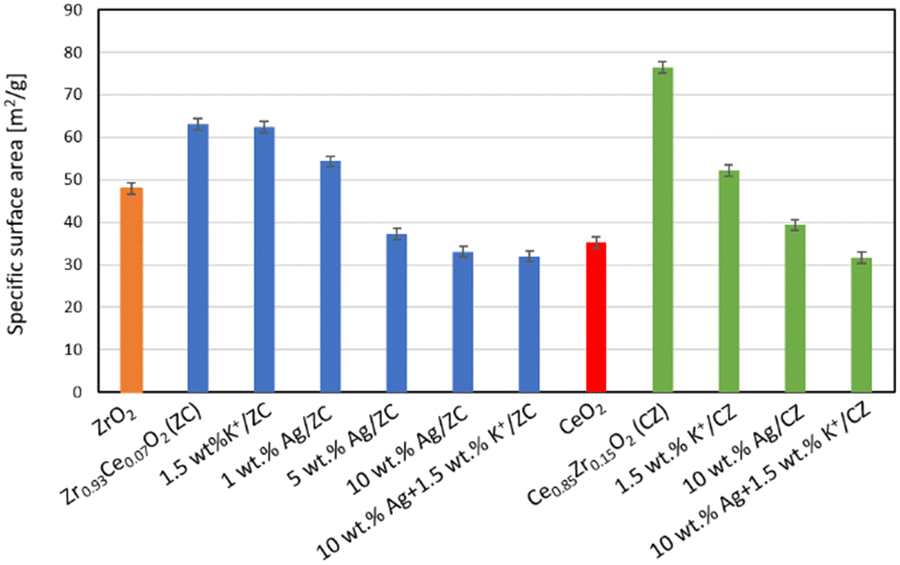 | ||
| Fig. 2 Influence of potassium ions and silver loading on the specific surface area of catalysts. | ||
Fig. 3 shows the compilation of the results of Temperature Programmed Oxidation experiments with both supports, namely: ZC (Fig. 3A) and CZ (Fig. 3B), as well as that of 10 wt% Ag/ZC (Fig. 3C). The weight loss curves for these samples are orange. The back dashed line corresponds to the TGA curve of the pure soot and it has been rescaled and overlayed with the other TGA curves for comparison. The weight loss, DSC signal and CO2 evolution curves of the pure soot are in the ESI† data (Fig. S1) and are typical for the combustion of carbonaceous material-the mass loss is associated with a large exothermic peak and CO2 formation. The blue curves in Fig. 3 show the CO2 evolution from catalysed combustion. The first two samples have qualitatively similar TGA curves with one major mass loss step, which can be attributed to the combustion of soot. The soot combustion on zirconium-doped ceria (Fig. 3B) occurs at a lower temperature than in the presence of cerium-doped zirconia. It can be seen that the temperatures of the beginning and end of CO2 evolution are lower than those noted for the ZC support.
 | ||
| Fig. 3 TGA (orange) and MS m/e = 44, CO2 evolution, curves (blue) obtained during TPO of a 5:1 mixture of the following three systems with soot: (A) Zr0.93Ce0.07O2, (B) Ce0.85Zr0.15O2, and (C) 10 wt% Ag/ZC; dashed black line: TGA curve of soot alone; inserts: images of catalyst 1 – prior to test, 2 – post test. | ||
In contrast, when looking at the TGA curve of a catalyst containing 10 wt% silver on Zr0.93Ce0.07O2 (Fig. 3C), one can notice a change in its shape, i.e., the onset is more gradual, which is accompanied by a qualitatively different CO2 evolution curve shape. Although CO2 emission begins earlier than in the two other systems, it has two phases of CO2 emission. This could be explained as the catalytic effect of silver, but by definition of a catalyst, it should not be used up during the reaction and should be unchanged at the end of the catalytic cycle. This is not the case because the black/dark grey supported silver catalyst changes colour (Fig. 3C inset 1: colour of catalyst before mixing with soot) during the measurement relative to that of the undoped support (Fig. 3C inset 2). Moreover, the temperature at which CO2 emission ends is not the lowest of the ones for the studied systems, i.e., CO evolution for this system ends at a higher temperature than with CZ, but lower than with ZC. Although the XRD patterns all indicate the presence of metallic silver in the catalysts, as did the TEM measurements (Fig. 4A), the results of techniques which are more sensitive to the surface, such as SEM-EDX (Fig. 4B) and XPS (Fig. 5), indicate that a part of the silver in the studied systems is associated with oxygen, which can be the reason for the observed colour change during activity tests. The application of SEM-EDX allows for precise measurement of specific locations, which is why it was used to investigate the elemental composition of silver agglomerates and their immediate surrounding area. The three points were chosen in the following manner: the first was a part of the support on which no silver agglomerate was visible, whereas the other two were different sized silver particles. The composition of point 1 shows that when very little silver, i.e., 1.7 at%, is present, the Zr:O ratio is close to 2, as expected from the stoichiometry of zirconium oxide. At point 2, in which a larger silver agglomerate is present, the relative zirconium content is only approx. 11 at%, which means that twice the amount of oxygen comes from the support and the rest is associated with the silver agglomerate. In point 3, which contains a larger silver agglomerate and hence the relative at% of silver is even higher and less of it is associated with oxygen. This indicates that silver is mostly metallic, which can be expected after calcination at 550 °C. The larger agglomerates have relatively less oxygen than the smaller ones, which shows that this is a surface phenomenon, and could account for the metallic silver in the XRD results as well as the relative decrease of the oxygen in the elemental maps shown in Fig. 6. The same was noted for the other catalytic systems.
 | ||
| Fig. 4 Investigating silver agglomerates: (A) TEM images of 10 wt% Ag/ZC and (B) SEM-EDX results of 10 wt% Ag supported on undoped zirconia. | ||
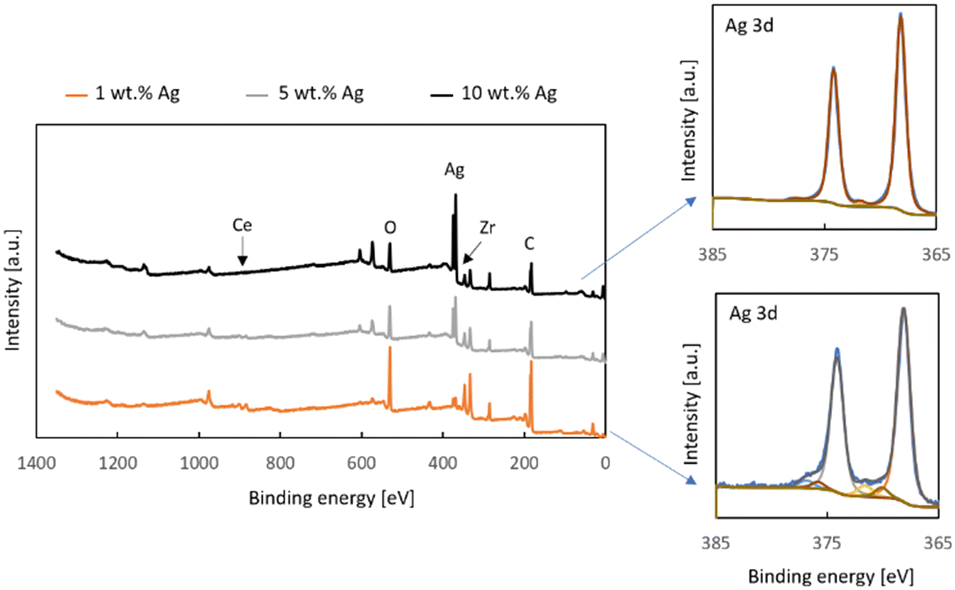 | ||
| Fig. 5 XPS measurement results: survey spectra of x% Ag/Zr0.93Ce0.07O2 (x = 1, 5, and 10) samples, and Ag 3d detailed regions of 10 wt% Ag/ZC and 1 wt% Ag/ZC. | ||
 | ||
| Fig. 6 TPR results: the influence of potassium ions on the reduction curves of different supports and catalysts; (A)–(D) compilations of analogous systems. | ||
The results of the X-ray Photoelectron Spectroscopy measurements of the Zr0.93Ce0.07O2 support with 1, 5 and 10% silver are depicted in Fig. 5. The survey spectra show that the only elements present on the surface of the samples are oxygen, zirconium, cerium, silver and adventitious carbon. It is noteworthy that the support, which contains 7% cerium ions in the tetragonal zirconia lattice shows very little cerium. The effect of surface enrichment in zirconium ions on the surface of the zirconium doped ceria support has been reported by us and other research groups earlier.26 In the case of cerium-doped zirconia, an enrichment of the surface with cerium ions has been reported, though after a much higher thermal treatment than in the present study.39 However, in this case, the low cerium content in the top 10 nm of the surface may be attributed to the presence of silver. It can also be seen that the zirconium signal also decreases along with the increased silver loading and this is in line with the results of the SEM-EDX measurements which indicate a substantial coverage of the surface with silver, as well as the decreased surface area of the samples.
The Ag3d detailed regions of XPS spectra of the samples with 10 wt% Ag and 1 wt% Ag deposited onto Zr0.93Ce0.07O2 can be fitted with two doublets with the Ag 3d5/2 components at 368.2 eV and 372.0 eV (Fig. 5). The first of these is associated with metallic silver and hence both spectra contain a loss feature 4.5 eV away from the main signal, which is typical for metallic silver.40 The second doublet can be attributed to ionic silver. This is the silver associated with oxygen and so it is noteworthy that the smaller silver loading has a greater contribution of this type of species, which is the same conclusion which was reached on the basis of the SEM-EDX results for the different-sized silver particles.
Selected TPR results are shown in Fig. 6. In order to allow a meaningful comparison, the first three images (Fig. 6A–C) have the same y-axis scale, i.e., from 9.8 to 10.8 units, whereas the fourth (Fig. 6D) depicts results from unsupported Ag2O and therefore has a much larger scale despite the smaller overall mass of the sample. The obtained results show that the deposition of silver onto a support completely changes the reduction profile by making the oxygen of the support unavailable to the hydrogen. In the case of the zirconium-doped ceria support (Fig. 6A), the solid solution itself is reduced to a large extent in a mid-temperature region with a smaller reduction at higher temperatures. The presence of 1.5 wt% potassium ions slightly changes the shape of the two peaks and shifts the maxima to slightly lower temperatures, but does not change the overall character of the reduction peaks. In contrast, the addition of 10 wt% silver, with or without potassium ions, onto this support leads to a substantially suppressed reduction of the support (Fig. 6B). Similarly, the suppression of the surface reduction peak of pure ceria upon the deposition of silver has already been reported by Grabchenko et al.4
An interesting finding is that the studied cerium-doped zirconia support (Fig. 6C red curve) shows some reduction (this result has been verified with another measurement due to the unusual shape), although undoped zirconia does not, e.g., ref. 19. This hydrogen consumption peak is also suppressed once loaded with 10 wt% Ag (Fig. 6C blue curve). It can be seen that the catalyst contains oxidized silver species, which are reduced at temperatures even lower than in the case of the CZ-supported catalyst. The fact that the hydrogen uptake signal in the 10 wt% Ag/ZC sample begins at a much lower temperature than that of 10 wt% Ag/CZ indicates a different strength of silver–support interactions. The total hydrogen consumption, however, is lower. Even lower H2 consumption was reported by Montaña et al.19 The TPR curves clearly indicate that there is a different intensity of the metal–support interactions in these two types of catalysts. In order to get the whole picture, a measurement with unsupported Ag2O was performed. The hydrogen uptake signal for Ag2O begins at a higher temperature than in the case of the supported silver catalysts. The presence of potassium ions shifts the beginning of the reduction peak to higher temperatures.
In order to investigate the impact of the presence of oxygen associated with silver on the onset of soot combustion, TPO-MS experiments were performed with Ag2O mixed with soot in a 5:1 ratio. The DSC and the TGA curves obtained in the measurement in air both clearly show two exothermic mass loss steps (Fig. 7A top panel) and each of them is associated with the evolution of carbon dioxide (Fig. 7A bottom panel). Therefore, the requirement for of oxygen in the atmosphere was probed by running the same experiment in argon. The results (Fig. 7B) show that the first exothermic step takes place even in an inert gas atmosphere, which means it is due to the interaction of the silver oxide sample with the soot. It may be argued that the exothermic peak is only due to the decomposition of the silver compounds from the sample, but such a decomposition reaction would require heat rather than give off heat. Indeed, a measurement with only Ag2O shows a sharp endothermal signal with a mass loss when soot is absent (Fig. 7C). The magnitude of the CO2 evolution is much smaller than that observed in the experiments with soot. The fact that CO2 evolves at all is a result of the fact that surface carbonate can be formed in the presence of CO2 in the air. Such surface species on silver oxide have been reported in other studies.41 Hence, another measurement was performed in the DTA-TGA-MS mode, which allowed for a larger sample of Ag2O to be analysed. The results are depicted in Fig. S4.† The TGA curve shows two weight loss signals, one at approximately 200 °C and another which ends at 400 °C. The DTA curves reveal they are both endothermic. The first is smaller and is typical for solids which desorb chemisorbed species such as water (m/e = 18), and CO2 (m/z = 44). This is confirmed by the MS results shown in Fig. S2.†
 | ||
| Fig. 7 TGA and DSC curves (top panel) and MS m/e = 44 (CO2 evolution) curves obtained during Temperature Programmed Oxidation of a 5:1 mixture of Ag2O with soot performed in air (A) and argon (B), and of Ag2O without soot in air (C). | ||
The next step is associated with the evolution of some CO2, which again evolves over a narrow time range. It is accompanied by the evolution of water, as well as oxygen due to the decomposition of the silver oxide to metallic silver. The evolving oxygen should be capable of oxidizing some soot, which would account for a larger emission of CO2 and the change from endo- to an overall exothermic type of reaction. It is known from literature that silver carbonate decomposes into silver oxide and then metallic silver,42 both of which occur below the temperature at which the majority of soot is combusted. Hence, the presence of such species on the surface of silver catalysts for soot combustion will lead to the same sequence of reactions.
In situ XRD studies were performed to confirm that in the presence of soot the same processes take place as without soot, i.e., the thermal decomposition of silver oxide and silver carbonate. The results obtained in the temperature range from 30 to 280 °C are compiled in Fig. 8A. For convenience, the diffraction patterns recorded at three distinct temperatures are shown in Fig. 8B with the reflexes assigned to each of the three phases. It can be seen that at the beginning of the measurement, the sample contains two phases, namely silver oxide and some silver carbonate, whose peaks can be seen up to 140 °C. Metallic silver can be observed already at 100 °C, though peaks from silver oxide are observed all the way up to 200 °C. At temperatures above 220 °C, so those at which the combustion of soot occurs, the only form of silver which is present is metallic silver. This means that regardless of the amount of the oxidized forms of silver or surface carbonate, the active form is metallic silver. Hence, the metallic core with some surface oxide and carbonate is actually a precursor of the catalyst, which is activated prior to soot combustion. It has been found, however, that the form of the silver is dependent on whether the activation is performed in the presence of soot or not. XRD studies of 10 wt% Ag/Zr0.93Ce0.07O2 have shown that the size of the silver particles is much larger when the activation takes place with soot, i.e., several nanometers vs. >40 nm.
 | ||
| Fig. 8 In situ XRD studies of Ag2O with soot: (A) in the temperature range 30–280 °C and (B) at three selected temperatures, i.e., 30, 160 and 220 °C. | ||
The analysis of the obtained results can be summed up in the following way: two steps of CO2 formation were identified, i.e., (1) a stoichiometric reaction of the oxygen emitted during the thermal decomposition of oxidized silver species and (2) catalytic soot oxidation. These pathways are depicted in Fig. 9. During the thermal decomposition of the silver oxide, oxygen was detected by mass spectrometry. This is not reversible, as evidenced by the studies which have shown a loss of activity of silver oxide in the second cycle.21 However, when only a single cycle of activity measurements is performed, it is easy to confuse this step with a catalytic reaction because at these temperatures, the presence of oxygen and soot alone is not enough to combust any soot. Therefore, there has to be a reason why the oxygen evolving from the silver oxide is more active than that found in the air which flows during the entire measurement. This reason has been provided by Ertl et al., who have shown that there are single oxygen atoms on the surface.43 These are most likely responsible for the soot combustion which has been evidenced by the MS data in Fig. 7 for the step called “Exo 1”. Hence, the soot combustion which occurs in this step starts when the oxidized silver species start to decompose and ends when they are all transformed into metallic silver. The extent of the soot combustion in this step strongly depends in the amount of oxidized silver species in the catalyst:soot mixture. Since our studies used an unusually low, i.e., 5:1 ratio, the results of studies with much higher catalyst:soot ratios can be expected to be more impacted by the presence of oxidized silver species.
 | ||
| Fig. 9 Mechanisms of CO2 formation in (A) Exo 1 and (B) Exo 2. | ||
The mechanism of the catalytic soot combustion over supported metallic silver can be broken down into four distinct steps (Fig. 9B): step 1: adsorption of oxygen from air, step 2: migration of Oads onto the soot, step 3: formation of a CO2 molecule, and step 4: desorption of the CO2 molecule from the surface. The first step, i.e., adsorption of oxygen from air, can be easily reversed by the desorption of O2 when recombination occurs. Hence, the key is the successful delivery of the adsorbed oxygen onto the soot or to the three-phase boundary of air, silver and soot. It is possible that the successful adsorption would have to occur near or at the three-phase boundary if the migration of oxygen on the silver surface is limited. The adsorbed oxygen then reacts with the soot to form carbon dioxide (step 3), which then desorbs from the surface of the soot restoring the catalyst and leaving the soot more depleted with each cycle until all of it is oxidized.
When potassium carbonate is deposited onto the silver catalysts and calcined, its distribution follows that of silver (Fig. 10), as seen in the elemental maps. Apart from maps of single elements, there are maps which are the sum of the individual signals. In all of the maps the same colours were used: silver is coloured yellow, zirconium is red, cerium is purple, oxygen is blue and potassium is pink. This is why the background of the two maps of the cerium-doped zirconia-supported catalysts have a red background, whereas the zirconium-doped ceria supported catalyst has a purple background. What can be seen in the maps is a substantial difference in the distribution of silver on these two supports. The two cerium-doped zirconia supported catalysts whose images are shown in Fig. 10A and B, differ in the silver loading, which is 1 and 10 wt%, respectively. The silver is distributed as visible particles already at 1 wt% and even larger ones when the loading is higher. This indicates that the interaction of zirconia with silver is not very strong. In the case of a ceria-type support (Fig. 10C), i.e., Ce0.85Zr0.15O2, the silver is distributed differently: some of it is distributed evenly across the support with a few very large particles.
 | ||
| Fig. 10 SEM-EDX results for catalyst containing 1.5 wt% potassium ions deposited onto the following catalysts: (A) 1 wt% Ag/Zr0.93Ce0.07O2 and (B) 10 wt% Ag/Zr0.93Ce0.07O2, and (C) 10 wt% Ag/Ce0.85Zr0.15O2. | ||
The Temperature Programmed Oxidation studies have shown numerous interesting effects and factors which are critical for the proper design of a soot combustion catalyst. The results of these studies are compiled in Fig. 11. For all these studies the catalyst:soot ratio was 5:1. Since the onset temperature of catalytic soot combustion is problematic due to the thermal decomposition of oxidized silver species, the final temperature was used as an indicator of catalytic performance. Fig. 11A contains a comparison of the TGA curves obtained for Ag2O with (1.5 wt%) and without potassium ions. It can be seen that the addition of potassium led to a very pronounced (almost 100 °C) lowering of the end temperature of soot combustion. Therefore, it is clear that in the case of unsupported Ag2O, the potassium ions can act as a promoter or even a catalyst, as in coal gasification, due to their high mobility. It has been observed (Fig. 11B) that the zirconium-doped ceria support doped with 1.5 wt% K+ exhibits similar activity with and without silver. This is not surprising since pure ceria has been studied as a stand-alone catalytic system for soot combustion.21,24,28,31 Nevertheless, such a comparison is a valuable piece of information because it shows that the presence of potassium ions is not the only reason for obtaining such excellent activity in this reaction in the Ag2O + 1.5 wt% K+ system. These two sets of results tell us that the stabilization of oxygen by the potassium species is beneficial for the overall activity of a silver catalyst when no oxygen from the support is available. In fact, the combination of silver and potassium ions is the most effective catalytic system from those studied in this paper. Therefore, it can be seen that the potassium ions are not responsible for the soot combustion, but they help the silver. Otherwise, the results of all systems with 1.5 wt% K+ would be the same.
 | ||
| Fig. 11 TGA curves of mixtures of soot with: (A) potassium carbonate, Ag2O without potassium ions and with 1.5 wt% K+, (B) CZ with 1.5 wt% K+ and 10 wt% Ag + 1.5 wt% K+, (C) 1 wt% Ag/ZC with 0.5 wt% K+ and 1.5 wt% K+, and (D) 10 wt% Ag/ZC with 0.5 wt% K+ and 1.5 wt% K+. | ||
Another interesting result is that with the lowest silver loading, i.e., 1 wt% Ag, supported on the cerium-doped zirconia, the effect of the potassium loading is substantial, as seen in the curves presented in Fig. 11C. There is a 45 deg difference between the temperature of the end of soot combustion upon the increase of the potassium ion loading from 0.5 to 1.5 wt%. No such influence is observed when 10 wt% of silver is deposited onto this support. This is indeed an interesting result, which shows that although the Fig. 11B and C indicate that the beneficial impact of shape of the TGA curve indicates a lower onset temperature, the final temperature is higher for the higher silver loading, which is in line with the proposed mechanisms of CO2 formation, which is enhanced when the silver loading is higher due to a higher content of oxidized silver species. Considering the fact that potassium ions are mobile and associated with oxygen44,45 and taking into account the analysis of results presented in figure potassium ions on the catalytic soot combustion may be attributed to facilitated migration of oxygen from the silver to the soot (step 2 Fig. 9b).
4. Conclusions
Despite the fact that the silver catalysts used in this study, as well as those described in the literature, are calcined at temperatures above 220 °C and hence are predominantly in the form of metallic silver, some oxidized forms of silver are present on the surface. The Temperature Programmed Reduction curves unambiguously show low temperature reduction peaks, which can only be attributed to oxidized forms of silver. The result of our research has clearly shown that catalysts operating at temperatures above this temperature are solely in the metallic form, as the heating to this temperature leads to their thermal decomposition. This means that the active form of silver in soot oxidation is metallic silver and the system which contains oxidized forms of silver is, in fact, a catalyst precursor, which is activated during the first stage of heating. What is more, our studies have shown that when the precursor is heated in the presence of soot, the silver particles are larger than those when the precursor is activated alone. During activation the thermal decomposition of the oxidized forms of silver release oxygen, which can oxidize soot and lead to artificially lower soot combustion onset temperature. The unsupported silver oxide showed to be the best, especially when doped with potassium ions. Instead of pre-activation of the precursor, it was concluded that the end temperature is the least impacted by the presence of oxidized species and is a good indicator of the activity of a given catalyst, provided that all of the tests are conducted with the same heating rate. The impact of the presence of potassium ions was more beneficial in the case of 1 wt% Ag/Zr0.97Ce0.03O2 than for higher silver loadings. For Ce0.85Zr0.15O2 the results obtained for samples with 1.5 wt% K+ either with or without silver are very similar.Author contributions
Conceptualization, D. W. K. and E. M. I.; methodology, Z. K. and E. M. I.; validation, D. W. K. and E. M. I.; formal analysis Z. K. and E. M. I.; investigation, Z. K. and E. M. I.; resources, E. M. I.; data curation, Z. K. and E. M. I.; writing—original draft preparation, D. W. K. and E. M. I.; writing—review and editing, D. W. K. and E. M. I.; visualization, E. M. I.; funding acquisition, E. M. I. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Scientific Discipline Board (RDN) for Chemical Engineering of the Warsaw University of Technology, grant number: 2022 I-CHEM.3-1.3. The authors would like to thank Peter Brodersen from the Ontario Centre for the Characterization of Advanced Materials (OCCAM) for performing the X-ray Photoelectron Spectroscopy measurements.References
- J.-H. Lee, S. H. Lee, J. W. Choung, C. H. Kim and K.-Y. Lee, Appl. Catal., B, 2019, 246, 356 CrossRef CAS.
- A. Serve, A. Boreave, B. Cartoixa, K. Pajot and P. Vernoux, Catal. Today, 2021, 363, 93 CrossRef CAS.
- X. Deng, M. Li, J. Zhang, X. Hu, J. Zheng, N. Zhang and B. H. Chen, Chem. Eng. J., 2017, 313, 544 CrossRef CAS.
- M. V. Grabchenko, G. V. Mamontov, V. I. Zaikovskii, V. La Parola, L. F. Liotta and O. V. Vodyankina, Appl. Catal., B, 2020, 260, 118 CrossRef.
- L. Nossova, G. Caravaggio, M. Couillard and S. Natis, Appl. Catal., B, 2018, 225, 538 CrossRef CAS.
- H. Liang, B. Jin, M. Li, X. Yuan, J. Wan, W. Liu, X. Wu and S. Liu, Appl. Catal., B, 2021, 294, 120271 CrossRef CAS.
- K. Yamazaki, T. Kayama, F. Dong and H. Shinjoh, J. Catal., 2011, 282, 289 CrossRef CAS.
- E. Aneggi, J. Llorca, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Appl. Catal., B, 2009, 91, 489 CrossRef CAS.
- W. W. Hsiao, Z.-Y. Mo, M. Fang, X.-M. Shi and F. Wang, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2000, 471, 45 CrossRef CAS PubMed.
- S. Steiner, C. Bisig, A. Petri-Fink and B. Rothen-Rutishauser, Arch. Toxicol., 2016, 90, 1541 CrossRef CAS PubMed.
- C. Zhang, D. Yu, C. Peng, L. Wang, X. Yu, Y. Wei, J. Liu and Z. Zhao, Appl. Catal., B, 2022, 319, 121946 CrossRef CAS.
- A. Zerboni, T. Rossi, R. Bengalli, T. Catelani, C. Rizzi, M. Priola, S. Casadei and P. Mantecca, Environ. Pollut., 2021, 297, 118767 CrossRef PubMed.
- World Health Organization, Health effects of transport-related air pollution, ed. M. Krzyzanowski, B. Kuna-Dibbert and J. Schneider, WHO Regional Office Europe, 2005 Search PubMed.
- B. A. A. L. van Setten, J. M. Schouten, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2000, 28, 253 CrossRef CAS.
- E. Aneggi, C. de Leitenburg and A. Trovarelli, Inorganics, 2020, 8, 34 CrossRef CAS.
- E. Aneggi, V. Rico-Perez, C. de Leitenburg, S. Maschio, L. Soler, J. Llorca and A. Trovarelli, Angew. Chem., Int. Ed., 2015, 54, 14040 CrossRef CAS PubMed.
- S. Liu, X. Wu, W. Liu, W. Chen, R. Ran, M. Li and D. Weng, J. Catal., 2016, 337, 188 CrossRef CAS.
- A. Cooper, T. E. Davies, D. J. Morgan, S. Golunski and S. H. Taylor, Catalysts, 2020, 10, 294 CrossRef CAS.
- M. Montaña, M. S. Leguizamón Aparicio, M. A. Ocsachoque, M. B. Navas, I. de C. L. Barros, E. Rodriguez-Castellón, M. L. Casella and I. D. Lick, Catalysts, 2019, 9, 297 CrossRef.
- M. Chernykh, N. Mikheeva, V. Zaikovskii, M. Salaev, L. F. Liotta and G. Mamontov, Catalysts, 2020, 10, 580 CrossRef CAS.
- K. Shimizu, H. Kawachi and A. Satsuma, Appl. Catal., B, 2010, 96, 169 CrossRef CAS.
- D. K. Sharma and C. C. Giri, J. Power Technol., 2016, 96, 157 Search PubMed.
- Z. Yang, L. Zhang, J. Peng and M. Guo, Int. J. Green Energy, 2015, 12, 1046 CrossRef CAS.
- J. Rodríguez-Fernández, F. Oliva and R. A. Vázquez, Energy Fuels, 2011, 25, 2039 CrossRef.
- E. M. Iwanek (nee Wilczkowska), L. F. Liotta, S. Williams, L. Hu, H. Ju, G. Pantaleo, Z. Kaszkur, D. W. Kirk, W. Patkowski and M. Gliński, Catalysts, 2022, 12, 524 CrossRef.
- E. Iwanek (Wilczkowska), M. Gliński, A. Siwiec, S. Siennicka, M. Zybert and Z. Kaszkur, Catalysts, 2022, 12, 974 CrossRef.
- M. Machida, Y. Murata, K. Kishikawa, D. Zhang and K. Ikeue, Chem. Mater., 2008, 20, 4489 CrossRef CAS.
- W. Zhang, X. Niu, L. Chen, F. Yuan and Y. Zhu, Sci. Rep., 2016, 6, 29062 CrossRef CAS PubMed.
- Y. Wei, Z. Zhao, J. Jiao, J. Liu, A. Duan and G. Jiang, J. Rare Earths, 2014, 32, 124 CrossRef CAS.
- S. Bensaid, N. Russo and D. Fino, Catal. Today, 2013, 216, 57 CrossRef CAS.
- A. Bueno-Lopez, Appl. Catal., B, 2014, 146, 1 CrossRef CAS.
- P. T. Aragao Campos, J. P. Vieira Lima, D. R. de Queiroz Silva, C. F. Oliveira, S. C. Loureiro Dias and J. Alves Dias, RSC Adv., 2020, 10, 27428 RSC.
- C. J. Howard, R. J. Hill and B. E. Reichert, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 44, 116 CrossRef.
- P. Duwez, F. Odell and F. H. Brown, J. Am. Chem. Soc., 1952, 35, 107 CAS.
- M. Han, X. Tang, H. Yin and S. Peng, J. Power Sources, 2007, 165, 757 CrossRef CAS.
- T. H. Etsell and S. N. Flengas, Chem. Rev., 1970, 70, 339 CrossRef CAS.
- M. V. Swain, R. C. Garvie and R. J. Hannink, J. Am. Chem. Soc., 1983, 66, 358 CAS.
- M. Zieliński, Z. Kaszkur, W. Juszczyk and J. Sobczak, Sci. Rep., 2023, 13, 1469 CrossRef PubMed.
- G. S. A. M. Theunissen, A. J. A. Winnubst and A. J. Burggraaf, J. Mater. Sci., 1992, 27, 5057 CrossRef CAS.
- A. C. Simonsen, F. Yubero and S. Tougaard, Surf. Sci., 1999, 436, 149 CrossRef.
- J. F. Weaver and G. B. Hoflund, Chem. Mater., 1994, 6, 1693 CrossRef CAS.
- P. Norby, R. Dinnebier and A. N. Fitch, Inorg. Chem., 2002, 41, 3628 CrossRef CAS PubMed.
- B. Pettinger, X. Bao, I. Wilcock, M. Muhler, R. Schlögl and G. Ertl, Angew. Chem., Int. Ed. Engl., 1994, 33, 85–86 CrossRef.
- J. M. Saber, J. L. Falconer and L. F. Brown, Fuel, 1986, 65, 1356–1359 CrossRef CAS.
- C. A. Mims and J. K. Pabst, Fuel, 1983, 62, 176–179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: DSC-TGA-MS results of uncatalyzed soot combustion; Fig. S2: DTA-MS results of Ag2O with m/z signals 18, 32 and 44. See DOI: https://doi.org/10.1039/d3cy01183f |
| This journal is © The Royal Society of Chemistry 2023 |