
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding structure–activity relationships: iron(II) complexes of “Legacy Guanidines” as catalysts for the synthesis of polylactide†
Christian
Conrads
 ,
Lisa
Burkart
,
Sven
Soerensen
,
Sandra
Noichl
,
Yasemin
Kara
,
Joshua
Heck
,
Alexander
Hoffmann
and
Sonja
Herres-Pawlis
*
,
Lisa
Burkart
,
Sven
Soerensen
,
Sandra
Noichl
,
Yasemin
Kara
,
Joshua
Heck
,
Alexander
Hoffmann
and
Sonja
Herres-Pawlis
*
Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany. E-mail: sonja.herres-pawlis@ac.rwth-aachen.de
First published on 18th September 2023
Abstract
In this work, eight novel iron(II) chloride complexes of well-known bisguanidine and N,N hybrid guanidine ligands are presented. Their activity in the synthesis of polylactide via a ring-opening polymerization was investigated under industrially relevant conditions with low catalyst loadings in the lactide melt. The conversion was monitored by in situ Raman spectroscopy to evaluate the reaction kinetics. The catalysts were investigated regarding their polymerization activity as well as their ability to maintain their polymerization activity over time. The most promising catalyst [Fe(TMGepy)Cl2] (C6) polymerizes L-lactide at monomer-to-initiator ratios of 1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and higher with a rate constant of propagation similar to the until now most active robust iron catalysts. Experiments on the influence of a co-initiator were carried out. Additionally, the experimental observations were further underlined with theoretical calculations explaining the stability and activity of the catalysts. Iron guanidines with rather simple ligands demonstrate a great potential for large-scale application in the industrial process. Finally, initial tests on the application of the compounds in the methanolysis of polylactide were conducted.
:1 and higher with a rate constant of propagation similar to the until now most active robust iron catalysts. Experiments on the influence of a co-initiator were carried out. Additionally, the experimental observations were further underlined with theoretical calculations explaining the stability and activity of the catalysts. Iron guanidines with rather simple ligands demonstrate a great potential for large-scale application in the industrial process. Finally, initial tests on the application of the compounds in the methanolysis of polylactide were conducted.
Introduction
Polylactic acid or polylactide (PLA) has been the focus of many research groups in the last two decades because it is bio-based as well as biodegradable and features properties resembling conventional plastics.1–3 Moreover, it serves as an excellent example of a plastic with a large potential for the circular economy offering various options for recycling, including mechanical and chemical recycling.4,5 The preparation of PLA via the ring-opening polymerization (ROP) of lactide is well-established and has been discussed in numerous publications following anionic,6 cationic,7 organocatalytic8 and coordination–insertion9 mechanisms. The latter takes a special role because it exhibits a high tolerance towards impurities and allows a controlled polymerization behavior. Complexes of various transition and main group metals were successfully applied as catalysts including complexes of Mg,10 Al,11 Fe,12–18 Zn,19–21 Zr,22 La23,24 and many more.However, only a rather small number of publications focused on catalysts that are appropriate for the industrially-relevant melt polymerization of lactide. Besides a sufficient activity, this requires an inexpensive and straightforward preparation of the metal complex, a sufficient stability towards impurities and temperature as well as control over the molar mass and dispersity.25 Additionally, the use of non-toxic metal catalysts is desirable instead of the commonly used tin(II) octoate (Sn(Oct)2).26 Numerous examples of robust guanidine catalysts based on bio-compatible iron or zinc metal ions were published by our group.27–30 Guanidines are strong organic bases and excellent neutral N donor ligands coordinating via the imine nitrogen atom. The most common guanidine units are the peralkylated tetramethylguanidine (TMG) and dimethylethyleneguanidine (DMEG) units (see Fig. 1A). The library of already published guanidine containing ligands is versatile.31,32 The ligands vary in the number of guanidine units, additional donor units (e.g. pyridines, quinolines, esters), the distance between donor units and the type of the ligand backbone which can be either aromatic or aliphatic (see Fig. 1B and C).
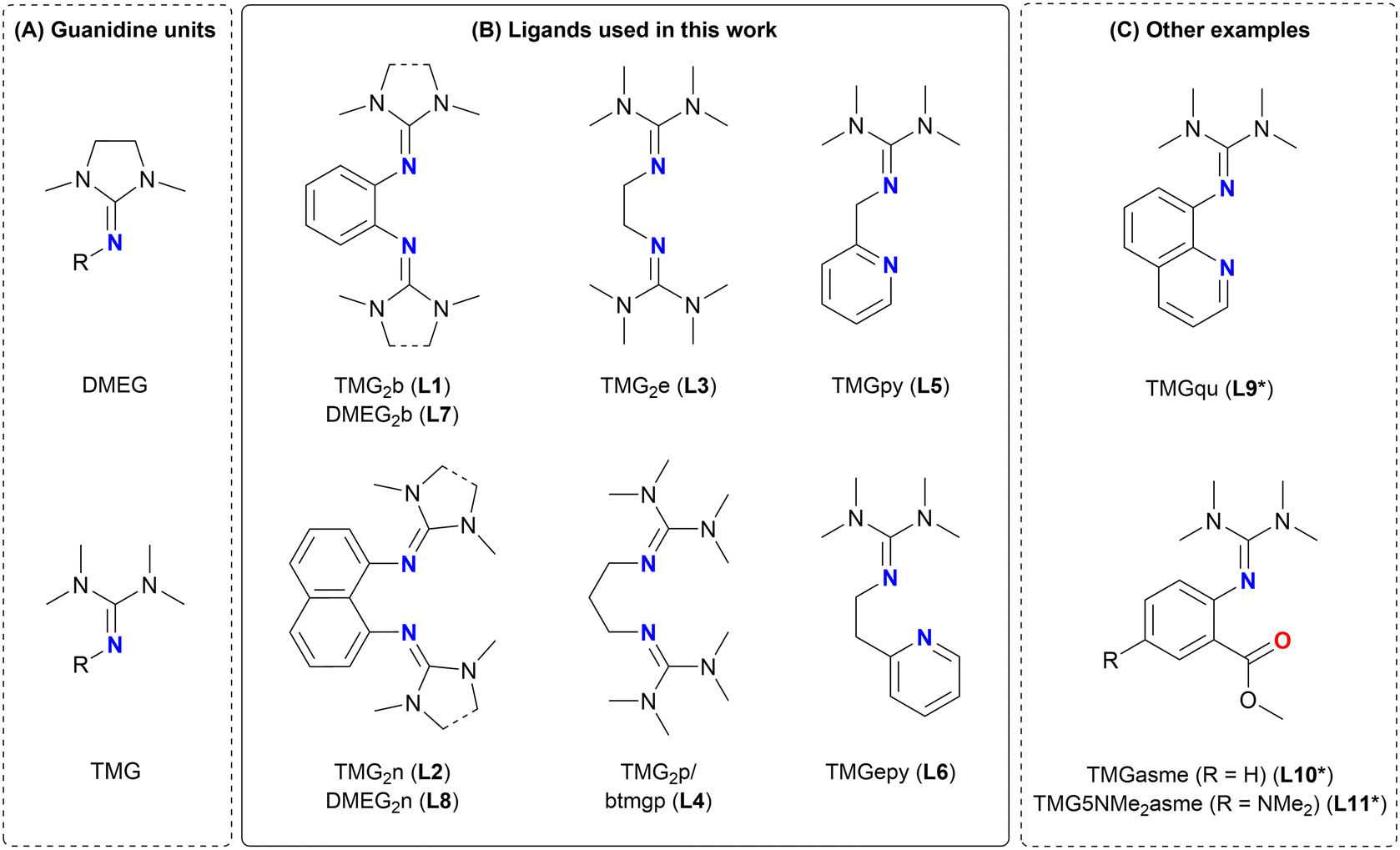 | ||
| Fig. 1 (A) Structures of the DMEG and TMG units. (B) Structures of literature-known bisguanidine and N,N hybrid guanidine ligands used in this work: L1: TMG2b;45L2: TMG2n;46L3: TMG2e;35L4: TMG2p/btmgp;33,35L5: TMGpy;49L6: TMGepy;50L7: DMEG2b;47L8: DMEG2n.48 (C) Other examples of N,N and N,O hybrid guanidine ligands: L9*: TMGqu;49L10*: TMGasme;38L11*: TMG5NMe2asme.39,40 | ||
The interest in multidentate guanidine ligands dates back to the year 2000, when the first examples of bisguanidine ligands such as propylene-bridged TMG2p (L4, also known as btmgp)33–35 and the ethylene-bridged TMG2e (L3)35 were presented. In the field of lactide polymerization first attempts were made with zinc complexes of bisguanidine and N,N hybrid guanidine ligands such as DMEG2e,36 the guanidine–pyridine hybrid ligands TMGpy (L5)/DMEGpy37 and the guanidine–quinoline hybrid ligands TMGqu (L9*)/DMEGqu.37 These complexes proved to be active in the bulk polymerization of lactide, however, on a rather large time scale of several hours to days exhibiting a significantly lower activity than the industrially-applied tin(II) octoate. A major step towards catching up tin(II) octoate was made when the first zinc chloride and zinc bromide complexes with N,O hybrid guanidine ligands (TMGasme (L10*)/DMEGasme, see Fig. 1C) were introduced to the lactide polymerization.38 The ligand system was later further optimized by systematical substitution at the aromatic backbone yielding the ligand TMG5NMe2asme (L11*).39 In 2019, tin(II) octoate was finally beaten for the first time by a N,O hybrid guanidine complex, however, not by a zinc complex but the iron complex [Fe(TMG5NMe2asme)Cl2].40 It was found that this complex, exhibiting a rate constant of propagation that is about one order of magnitude higher than for tin(II) octoate, is able to polymerize lactide following pseudo first order kinetics and the coordination–insertion mechanism. In the following years it was demonstrated that because of the iron center, these complexes are also able to catalyze the atom transfer radical polymerization (ATRP) and are active in a combination of ROP and ATRP yielding novel copolymers.41 Moreover, they can be used in the copolymerization of lactide with other cyclic esters, e.g. ε-caprolactone.42 Initially studied for the zinc TMGasme complexes,43 the complex [Fe(TMG5NMe2asme)Cl2] was further investigated in the alcoholysis and aminolysis as a chemical recycling method for PLA.44 The catalyst depolymerizes PLA to methyl lactate (MeLA) in the presence of methanol at 60 °C within 24 h completely.
Overall, these thoroughly investigated iron(II) TMGasme complexes reveal the potential of iron guanidine complexes and these promising results are the motivation for this work. Although guanidine metal complexes have been a relevant topic for years, iron guanidine complexes were only investigated scarcely in the field of ROP. The present study can therefore draw from the whole library of known guanidine ligands. Aiming at inexpensive starting materials and a straightforward synthesis as well as a versatile ligand architecture, this work reports eight novel iron(II) chloride complexes C1–C8 based on literature-known bisguanidine and N,N hybrid guanidine ligands L1–L8 (see Fig. 1B). Their catalytic activity and polymerization behavior in the ROP of lactide are evaluated under industrially relevant conditions to identify the most promising catalysts which are then studied in more detail. Findings are further supported by density functional theory (DFT) calculations. Moreover, a selection of catalysts is applied in the methanolysis of PLA. Finally, the experimental observations are related in order to deepen the understanding of the connection between structural motives and the polymerization activity and stability of the catalysts.
Results and discussion
Iron(II) guanidine complexes
The eight ligands L1–L8 were prepared following literature procedures in one step starting from the corresponding amine and the DMEG or TMG Vilsmeier reagent. Ligands L1,45L2,46L747 and L848 feature rigid aromatic backbones, while L335 and L433,35 possess an aliphatic backbone. L549 and L650 are guanidine–pyridine hybrid ligands. The complex syntheses were conducted under inert conditions with varying procedures depending on the complexes' solubility in acetonitrile (see Scheme 1). Especially complexes C2, C3 and C8 exhibit a straightforward preparation since they are hardly soluble in acetonitrile and precipitated as a powder upon formation. Complexes C1, C5, C6 and C7 were prepared by vapor diffusion of diethyl ether into a solution of the complex in acetonitrile and were obtained as crystalline solids. C4 was prepared by layering a solution of the complex in acetonitrile with diethyl ether. The molecular structures of complexes C1–C8 in the solid state were determined by single-crystal X-ray diffraction (see Fig. 2). Suitable single-crystals were either obtained directly from the crystallization via vapor diffusion (C1, C5, C6 and C7) or were prepared by recrystallization from acetonitrile (C2, C3, C4, C8). Key geometric data of all complexes is summarized in Table 1. | ||
| Scheme 1 Preparation of the iron(II) guanidine complexes starting from FeCl2 and ligands L1–L8 (gua = guanidine, py = pyridine). C5 is an exception forming a complex cation with an iron center that is fivefold coordinated by two TMGpy (L5) ligands and one chlorido ligand. | ||
 | ||
| Fig. 2 Molecular structures of complexes C1–C8 in the solid state. Hydrogen atoms and the non-coordinating chloride ion in C5 were omitted for clarity. | ||
| Complex | Fe–Ngua [Å] | Fe–Ngua,py [Å] | Ngua–Fe–Npy/gua [°] | ρ | τ 4 /τ5c |
|---|---|---|---|---|---|
a Degree of delocalization within the guanidine moiety: ρ = 2a/(b + c) with a = d(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ngua) and b, c = d(C–Namine).48
b
τ
4 = [360° − (α + β)]/141°.51α and β are the largest angles in a complex with a fourfold coordinated metal center.
c
τ
5 = (β − α)/60°.52α and β are the largest angles in a complex with a fivefold coordinated metal center with β ≥ α.
d Two complex molecules are found in the asymmetric unit. Ngua) and b, c = d(C–Namine).48
b
τ
4 = [360° − (α + β)]/141°.51α and β are the largest angles in a complex with a fourfold coordinated metal center.
c
τ
5 = (β − α)/60°.52α and β are the largest angles in a complex with a fivefold coordinated metal center with β ≥ α.
d Two complex molecules are found in the asymmetric unit.
|
|||||
| C1 | 2.093(1) | 2.068(1) | 79.9(1) | 0.98/0.98 | 0.89 |
| C2 | 2.051(2) | 2.052(2) | 86.7(1) | 0.98/0.99 | 0.90 |
| C3 | 2.084(1) | 2.084(1) | 81.9(1) | 0.96/0.96 | 0.73 |
| C4 | 2.055(2) | 2.054(1) | 93.4(1) | 0.97/0.97 | 0.81 |
| C5 | 2.092(3)/2.072(2) | 2.218(3)/2.213(3) | 76.9(1)/75.8(1) | 0.98/0.96 | 0.68 |
| C6 | 2.021(3) | 2.120(3) | 94.5(2) | 0.98 | 0.85 |
| C7 | 2.076(3) | 2.073(3) | 79.8(2) | 0.99/1.01 | 0.88 |
| 2.073(3) | 2.073(3) | 80.3(2) | 0.97/0.97 | 0.88 | |
| C8 | 2.054(1) | 2.036(1) | 87.6(1) | 0.97/0.99 | 0.84 |
The degree of delocalization (ρ) with values close to 1 indicates that the CN double bond in all guanidine units of complexes C1–C8 is fully delocalized. Except for C5, all iron centers are fourfold coordinated by two chlorido ligands and one bidentate N,N ligand. Complexes C1–C4 and C6–C8 possess a distorted tetrahedral coordination geometry as indicated by the τ4 values,51 with C1 and C2 being closest to a tetrahedral geometry and C3 being most distorted with a τ4 value of 0.73. The bite angles of the N,N ligands in the fourfold coordinated complexes C1–C4 and C6–C8 deviate strongly from the ideal tetrahedral angle of 109.47° with the strongest deviation for the ligands with C2-spacers in their backbone (C1, C3, C7). Complex C5 is an exception: the metal center is fivefold coordinated by two TMGpy (L5) ligands and one chlorido ligand. The second chloride anion is non-coordinating. The τ5 value52 of 0.68 indicates a strongly distorted trigonal bipyramidal coordination geometry. Both pyridine donor units are found in the apical positions, while the bulky TMG donor units and the chlorido ligand are located in the equatorial positions.
In the bisguanidine complexes C2–C4 and C7, both Fe–Ngua bond lengths are equal. Only C1 and C8 exhibit a slight difference in their two iron-guanidine bond lengths. In C5 and C6, the Fe–NTMG bond lengths are clearly shorter than the Fe–Npy bond lengths indicating a stronger donation through the TMG unit. Comparing complex C1 with C7 which are only different in their guanidine unit (TMG or DMEG), one of the Fe–NTMG bond lengths in C1 is slightly longer than the Fe–NDMEG bond lengths in C7. One of the Fe–NDMEG bond lengths in C8 is slightly shorter than the Fe–NTMG bond lengths in C2. Complex C7 contains two independent complex molecules in the asymmetric unit with the major difference that the DMEG units are tilted into different directions.
All catalysts were examined by thermogravimetric analysis (TGA) at 150 °C for 1 h. Catalysts C2–C8 proved to be thermally stable. The supposed sample of C1 showed a mass loss of approximately 8% starting at about 125 °C. The IR spectra recorded before and after the TGA experiment differ due to loss of solvent molecules (see Fig. S11–S13†). Single-crystal X-ray diffraction data indicates that the acetonitrile solvate [Fe(TMG2b)Cl2]·1.5MeCN (C1·1.5MeCN) can also be obtained during crystallization. Due to the insufficient quality of the dataset, the structure of this acetonitrile solvate could not be solved properly. It was not possible to selectively crystallize one of the compounds.
Evaluation of polymerization activities
The melt polymerization of L-lactide with complexes C1–C8 was conducted in a batch reactor on an 8 g-scale monitored by in situ Raman spectroscopy. Recrystallized L-lactide was employed in order to improve the reproducibility of the kinetics experiments. All experiments were conducted in duplicate at a monomer-to-initiator ratio ([M]/[I]) of 1000:1 at 150 °C under argon as inert gas. No additional co-initiator (CoI) was applied because zinc and iron guanidine complexes are known to function as single-site catalysts initiating through their nucleophilic guanidine ligands.29,40 Polymerization times varied depending on the catalyst activity (see Table 2). For comparison, the polymerization was performed with anhydrous FeCl2 as catalyst and an additional control experiment without catalyst can be found in the ESI† (see Fig. S40).
| Cat. | t [min] | p [%] | k app [10−4 s−1] | M n,theo [kg mol−1] | M n [kg mol−1] | Đ |
|---|---|---|---|---|---|---|
|
a Conditions: bulk polymerization of recrystallized L-lactide, [M]/[I] = 1000:1, T = 150 °C, stirring speed: 260 rpm, experiments were conducted in duplicate. Conversion p, molar masses and dispersities are only given for one experiment per catalyst. All experiments are listed in the ESI.†
b Conversion determined by 1H NMR spectroscopy.
c Initial apparent rate constant kapp, for details and all plots see ESI.†
d
M
n,theo = [M]/[I]·M(LA)·p.
e Determined by GPC with THF as eluent and a conventional calibration using polystyrene standards. Molar masses were corrected by a correction factor of 0.58.24
f For this catalyst, the two different crystal structures C1 and C1·1.5MeCN were obtained. The [M]/[I] ratio was calculated assuming the structure of C1.
g Molar mass and dispersity were not determined because no precipitation from EtOH at 20 °C was observed.
|
||||||
| C1 | 300 | 11 | 0.0629 ± 0.0047 | 15.9 | n.d.g | n.d.g |
| C2 | 300 | 65 | 0.575 ± 0.073 | 93.7 | 25.0 | 1.5 |
| C3 | 60 | 56 | 9.63 ± 0.05 | 80.7 | 25.3 | 1.5 |
| C4 | 60 | 42 | 54.2 ± 14.1 | 60.5 | 14.1 | 1.5 |
| C5 | 60 | 41 | 2.35 ± 0.27 | 59.1 | 11.0 | 1.4 |
| C6 | 10 | 59 | 24.1 ± 0.2 | 85.0 | 47.3 | 1.4 |
| C7 | 300 | 7 | 0.0280 ± 0.0001 | 10.1 | n.d.g | n.d.g |
| C8 | 300 | 80 | 1.32 ± 0.02 | 115.3 | 45.7 | 1.5 |
| FeCl2 | 300 | 16 | 0.0786 ± 0.0085 | 23.1 | n.d.g | n.d.g |
Two factors must be discussed: the activity as described by the initial apparent rate constant kapp and the stability of the complex meaning the time the polymerization activity is maintained before the activity decreases. The kapp values were determined from the semilogarithmic plots for each catalyst (see Fig. 3, all plots can be found in the ESI†). The kapp values of C1–C8 differ drastically over four orders of magnitude (10−6–10−3 s−1). Therefore, the catalytic activity is compared with respect to the order of magnitude rather than the absolute kapp values.
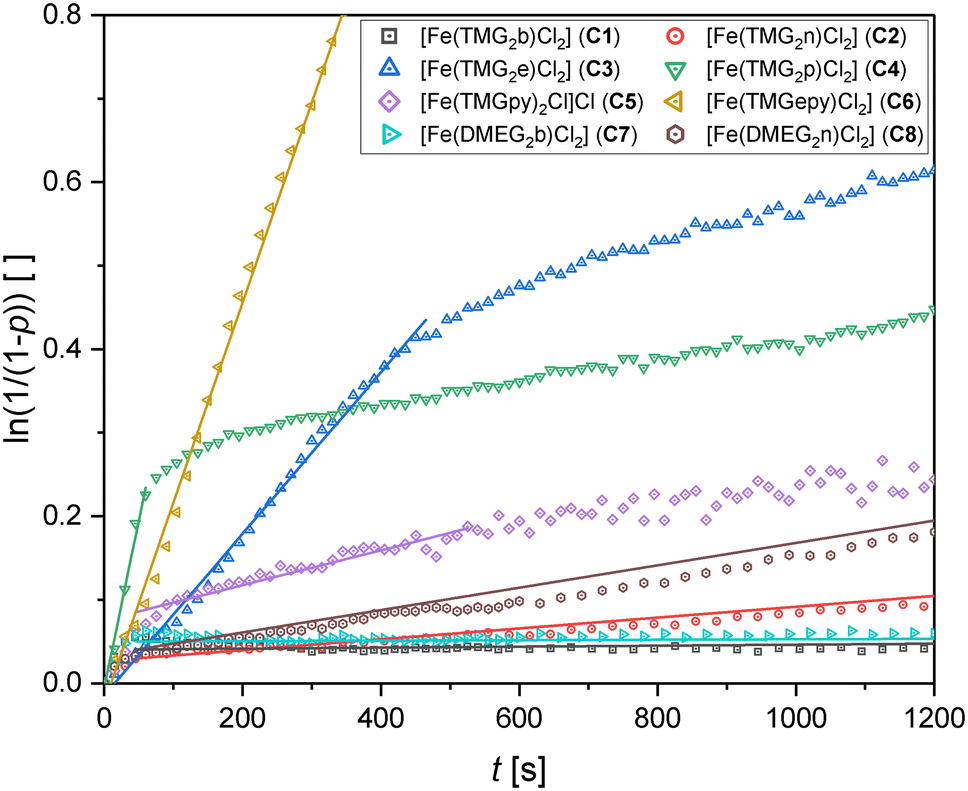 | ||
| Fig. 3 Pseudo first order semilogarithmic plots for complexes C1–C8 for the polymerization of recrystallized L-lactide (150 °C, [M]/[I] = 1000:1). Shown are the first 20 min of the experiments listed in Table 2. The plot for FeCl2 is omitted for clarity. All plots can be found in the ESI.† | ||
Since the selective formation of one of the species of C1 could not be controlled, the molar mass was assumed to be the molar mass of the acetonitrile-free species which may lead to slightly changed [M]/[I] ratios. This was, however, not further investigated because C1 as well as C7 exhibit very low polymerization activities comparable to FeCl2 with kapp values in the order of magnitude of 10−6 s−1. After 5 h, low conversions in the range of 10% were obtained and the product did not precipitate in ethanol meaning only short chains were formed. Due to the low activity and the comparably strong noise, no statement about the stability of the complexes can be made. The different guanidine units (TMG in C1 and DMEG in C7) do not seem to change the rate constant drastically. Because of the very low activity, these two iron complexes are not suitable as catalysts for the ROP of lactide.
The complexes C2 and C8 containing a naphthalene backbone show a better activity with kapp values in the magnitude of 10−5 s−1 for C2 and 10−4 s−1 for C8, respectively. Interestingly, for these two complexes only a slight decay of activity is observed after more than 1 h. Although they exhibit a mediocre activity, they follow pseudo first order kinetics. Comparing C2 and C8, the change of the TMG groups to DMEG groups leads to a different activity, however, this effect is not as pronounced as for a change of the ligand backbone. The different activity is also reflected by the conversions after 5 h and the molar masses. Although these values show fluctuations, a general trend is observable. While C2 reaches conversions of up to 65% and an Mn of 25.0 kg mol−1, C8 polymerizes the lactide to a conversion of 80% with an Mn of up to 45.7 kg mol−1.
Compared to these four examples with aromatic ligand backbones, complexes C3–C6 bearing aliphatic ligand backbones with two guanidine units (C3, C4) or a combination of one guanidine unit and a pyridine unit (C5, C6) show significantly higher initial kapp values. C4 is the most active catalyst in the range of the complexes investigated in this work. However, it is also the complex with the shortest span of high activity of merely 1 min. Therefore, the experiment was repeated with sublimated L-lactide in order to evaluate the influence of lactide purity (see Fig. S52†). However, this did not change the polymerization rate significantly nor the short period of high activity. C3 also shows a decay of activity but significantly later than C4 with an initial kapp value similar to the second most active iron guanidine catalyst [Fe(TMGasme)Cl2] and the industrially-applied Sn(Oct)2 under comparable conditions (see Table 3 entries 1 and 3).40C5 being the only five-coordinate complex in this study exhibits a rather mediocre activity with a curve-like semilogarithmic plot. Also, the polymers produced by this complex show the strongest deviation from the theoretical molar masses. This is likely due to the two ligand molecules per complex molecule that can function as an initiator and lead to a larger number of polymer chains compared to the other complexes containing only one ligand molecule per complex molecule. Although for all three complexes C3–C5 the polymerization rate decreases over the course of the polymerization, a complete decay of activity was not observed. The final kapp values at the end of the polymerization time of 1 h are still in the range of 1 × 10−4 s−1 for C3–C5 which is significantly higher than for FeCl2. This indicates that a species different from the original iron complex is further supporting polymerization. This decrease of kapp during the polymerization process was also observed before for the zinc TMGasme systems and was attributed to the guanidine ligand functioning as an initiator for new growing polymer chains.53
| No. | Catalyst | [M]/[I] | t [h] | p [%] | k app [10−4 s−1] | M n [kg mol−1] | Đ | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Conditions: recrystallized L-lactide, 150 °C, same setup as in this work. b Conditions: rac-lactide, 150 °C, same setup as in this work. c rac-Lactide, 150 °C, trz: triazolylidene N-heterocyclic carbene ligand.18 d rac-Lactide, 150 °C. e This complex was not evaluated in the polymerization of lactide. | ||||||||
| 1a | [Fe(TMGasme)Cl2] | 1000/1 | 0.5 | 66 | 7.18 | 96.7 | 1.3 | Rittinghaus et al. 201940 |
| 2a | [Fe(TMG5NMe2asme)Cl2] | 1000/1 | 1.6 min | 73 | 37.52 | 147.0 | 1.6 | Rittinghaus et al. 201940 |
| 3a | Sn(Oct)2 | 1000/1 | 25 min | 69 | 9.88 | 168.0 | 1.9 | Rittinghaus et al. 201940 |
| 4b | [Zn((R,R)-DMEG2(1,2)ch)2](OTf)2·THF | 1250/1 | 7.4 min | 89 | 225 | 83.6 | 1.6 | Hermann et al. 202029 |
| 5c | [FeCp(CO)(trz)I] | 1000/1 | 2 | 91 | 53.9 ± 4.1 | 21 | 2.5 | Nylund et al. 202218 |
| 6d | [Zn(TMG2b)Cl2] | 500/1 | 48 | 80 | 7.0 | 2.09 | Vieira 201355 | |
| 7e | [Zn(TMG2n)Cl2] | — | — | — | — | — | Reinmuth et al. 200954 | |
| 8d | [Zn(TMG2e)Cl2] | 500/1 | 24 | 87 | 45.0 (Mw) | 1.9 | Börner 200956 | |
| 9d | [Zn(TMG2p)Cl2] | 500/1 | 24 | 71 | 26.0 (Mw) | 1.7 | Börner 200956 | |
| 10d | [Zn(TMGpy)Cl2] | 1000/1 | 24 | 67 | 62.0 (Mw) | 2.32 | Börner et al. 200937 | |
| 11d | [Zn(TMGepy)Cl2] | 500/1 | 18 | 87 | 0.26 | 19.0 | 1.69 | Vieira 201355 |
| 12d | [Zn(DMEG2b)Cl2]·MeCN | 500/1 | 48 | 67 | 4.0 | 1.79 | Vieira 2013;55 Roquette et al. 201157 | |
The iron(II) N,N hybrid guanidine complex C6 combines a high polymerization activity with stability. The experiments performed at an [M]/[I] ratio of 1000:1 lead to high conversions and no deactivation behavior was observed for this catalyst. After 10 min, the reaction had to be stopped due to the high viscosity causing fluctuations of the stirring speed. Conversions in the range of 60% and polymers with Mn values higher than 40 kg mol−1 could be obtained. Thus, this complex is the most promising catalyst in the scope of the herein investigated ROP catalysts and was therefore also investigated in detail (see below). In comparison to Sn(Oct)2 and other robust iron catalysts that have been investigated under industrially relevant conditions (see Table 3 entries 1–3 and 5) the catalyst can keep up regarding the order of magnitude of kapp at an [M]/[I] ratio of 1000:1. However, it yields polymers with lower molar masses than several of the other examples. Possibilities to improve the molar masses and the dispersities are discussed below. The zinc guanidine complex [Zn((R,R)-DMEG2(1,2)ch)2](OTf)2·THF from a former publication is however still surpassing the activity by an order of magnitude (see Table 3 entry 4).
As already observed for the complexes [Zn(TMG5NMe2asme)Cl2] and [Fe(TMG5NMe2asme)Cl2] in previous studies, exchanging the metal ion can lead to a drastically changed polymerization activity.38,40 Therefore, an intriguing comparison are the activities of the herein presented iron(II) guanidine complexes and the zinc chloride analogs that were investigated in the early stages of the research on metal guanidine catalysts for the ROP of lactide. The zinc chloride analogs of C1–C7 are known and were, except of the C2-analog,54 evaluated for the lactide polymerization (see Table 3 entries 6–12). Although the polymerization method in these early publications deviates considerably from the polymerization applied in this work the polymerization results for the six complexes [Zn(TMG2b)Cl2],55 [Zn(TMG2e)Cl2],56 [Zn(TMG2p)Cl2],56 [Zn(TMGpy)Cl2],37 [Zn(TMGepy)Cl2]55 and [Zn(DMEG2b)Cl2]·MeCN55,57 illustrate which enormous effect the replacement of a zinc to an iron center can have. While this is not obvious for the zinc chloride analogs of C1 and C7 (see Table 3 entries 6 and 12) because C1 and C7 show a low activity by themselves, the difference becomes apparent for the other iron complexes C3–C6. The polymerizations with the zinc complexes had to be conducted for several hours up to days at 150 °C and were carried out with higher catalyst concentrations ([M]/[I] = 500:1) in order to achieve high conversions. However, the long polymerization times were accompanied by broad molar mass distributions.
To have a more comparable result, the polymerization with C3 was repeated using a [M]/[I] ratio of 500:1 (see Table 4 entry 1). After merely 1 h, a conversion of 80% was obtained while the zinc chloride analog requires 24 h for a conversion of 87% (see Table 3 entry 8). Moreover, [Zn(TMGepy)Cl2], the zinc analog of C6, exhibits a much decreased activity with a kapp value of merely 2.6 × 10−5 s−1 at an [M]/[I] ratio of 500:155 while C6 exhibits a kapp value that is two orders of magnitude higher at an even lower catalyst concentration ([M]/[I] = 1000:1, see Table 2). This comparison again highlights the potential of iron-based catalysts for the ROP of lactide.
| No. | Cat. | CoI/ additive | [M]/[I]/[CoI] | t [min] | p [%] | k app [10−4 s−1] | M n,theo [kg mol−1] | M n [kg mol−1] | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: bulk polymerization of recrystallized L-lactide, T = 150 °C, stirring speed: 260 rpm. b Conversion determined by 1H NMR spectroscopy. c Initial apparent rate constant kapp, for details and all plots see ESI.† d M n,theo = [M]/([CoI] + [C3/C6])·M(LA)·p. FeCl2 was not considered as a co-initiator. e Determined by GPC with THF as eluent and a conventional calibration using polystyrene standards. Molar masses were corrected by a correction factor of 0.58.24 | |||||||||
| 1 | C3 | — | 500/1/— | 60 | 80 | 14.8 | 57.7 | 46.2 | 1.4 |
| 2 | C3 | — | 1000/1/— | 60 | 56 | 9.67 | 80.7 | 25.3 | 1.5 |
| 3 | FeCl2 | TMG2e (L3) | 1000/1/— | 60 | 25 | 1.22 | 36.0 | 3.0 | 1.2 |
| 4 | C3 | pMeBnOH | 1000/1/1 | 60 | 63 | 10.1 | 45.4 | 33.7 | 1.2 |
| 5 | C3 | FeCl2 | 1000/1/1 | 60 | 76 | 8.79 | 109.5 | 67.7 | 1.5 |
| 6 | C3 | TMG2e (L3) | 1000/1/1 | 60 | 45 | 19.8 | 64.9 | 8.1 | 1.4 |
| 7 | C6 | — | 2000/1/— | 60 | 44 | 8.47 | 126.8 | 29.9 | 1.5 |
| 8 | C6 | pMeBnOH | 2000/1/1 | 60 | 68 | 17.8 | 98.0 | 68.3 | 1.4 |
| 9 | C6 | pMeBnOH | 2000/1/4 | 60 | 75 | 27.8 | 43.2 | 48.7 | 1.3 |
| 10 | C6 | pMeBnOH | 5000/1/5 | 60 | 34 | 6.02 | 40.8 | 26.9 | 1.2 |
| 11 | C6 | FeCl2 | 2000/1/1 | 60 | 65 | 11.0 | 187.4 | 68.5 | 1.4 |
Experiments on extending the duration of high activity with [Fe(TMG2e)Cl2] (C3)
In order to gain deeper insight into the interplay of the iron center and the ligand and to extent the duration of high activity, additional experiments with C3 were conducted (see Fig. 4 and Table 4 entries 1–6). This catalyst was chosen because of its comparably high activity, its straightforward preparation and the solid ligand TMG2e (L3). Firstly, the in situ formation of the catalyst was tested by adding solid FeCl2 and L3 to the L-lactide (see Table 4 entry 3). However, the resulting kapp value of 1.22 × 10−4 s−1 measures only one eighth compared to the iron(II) complex C3 ([M]/[I] = 1000:1, see Table 4 entry 2). Given the fact that FeCl2 and L3 on their own exhibit very low kapp values in the magnitude of 10−6 s−1 for [M]/[I] = 1000:1 (see Table 2 and Fig. S45†), this result shows that the activity must be caused by the complex C3. Moreover, the determined kapp value is similar to the kapp value obtained for the complex C3 after the decrease of activity. Therefore, small proportions of complex might be formed in situ in the lactide melt and/or the same species forms that forms after the decay of activity in the polymerization with C3.
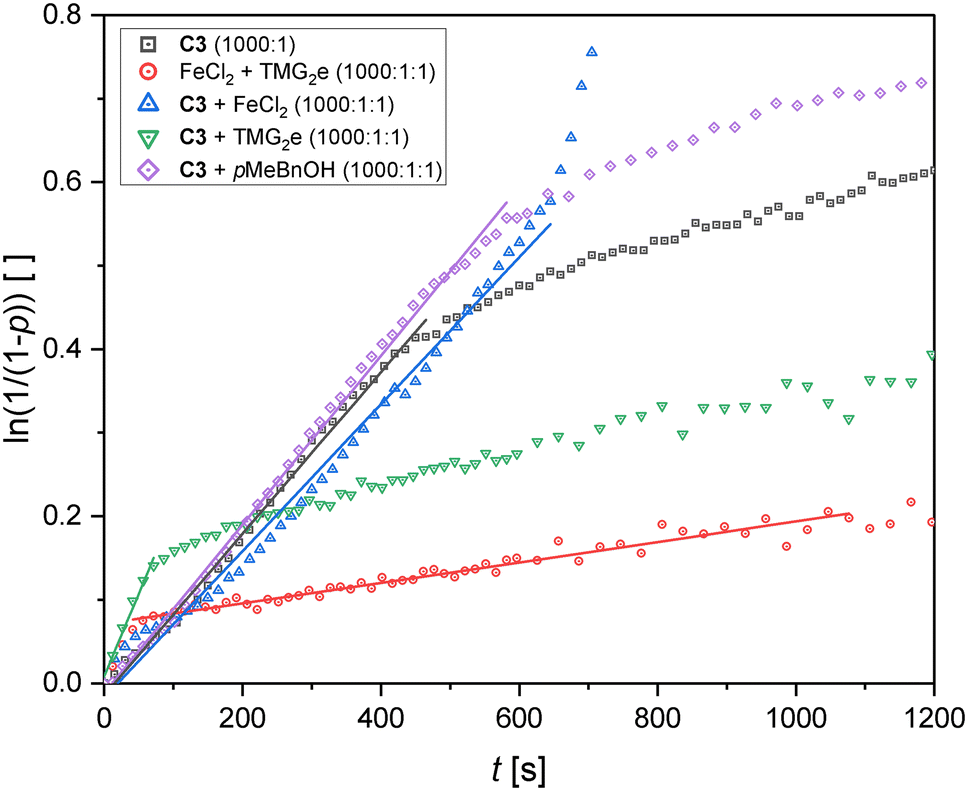 | ||
| Fig. 4 Pseudo first order semilogarithmic plots for the polymerization of recrystallized L-lactide with C3 and various additives. For comparison, the polymerization without additive is depicted ([M]/[I] = 1000:1). Shown are the first 20 min of the experiments. Full plots can be found in the ESI.† | ||
Since the addition of a co-initiating alcohol is common in literature, an additional polymerization experiment with 1 equivalent of p-methyl benzyl alcohol (pMeBnOH) was performed ([M]/[I]/[pMeBnOH] = 1000:1:1, see Table 4 entry 4). Indeed, the addition of alcohol leads to a modest improvement with a slightly increased conversion and a higher molar mass with a lower dispersity after 1 h at 150 °C. Surprisingly, the addition of 1 equivalent of ligand L3 does not result in a better polymerization performance (see Table 4 entry 6). In the first minute, the rate is increased compared to the experiment without additional ligand. However, afterwards the rate is significantly lower. Therefore, additional ligand might function as a co-initiator to some extent but at the same time it acts as a deactivator probably by coordination to the iron center. However, such a species could not be isolated since in crystallization experiments with 2 equivalents of L3 only C3 was obtained. The addition of 1 equivalent of FeCl2 to the polymerization mixture leads to the highest conversion after 1 h (76%) which indicates that the oxidation of iron(II) might be a problem and can be partially suppressed by additional iron(II) salt (see Table 4 entry 5). The molar mass is also significantly increased to 67.7 kg mol−1 compared to 25.3 kg mol−1 for the experiment without FeCl2. However, the addition of FeCl2 does not lead to a higher initial kapp, the high activity is merely maintained for a longer time.
In-detail investigation of the polymerization behavior of [Fe(TMGepy)Cl2] (C6)
Since C6 showed the most promising activity with high conversions within minutes, the polymerization behavior of this ROP catalyst was investigated further. First the [M]/[I] ratio was varied at 150 °C between 1000:1 and 5000:1 in order to determine the concentration-independent rate constant of propagation kp. In this context, it was also found that at [M]/[I] ratios higher than 1000:1 a decay of activity is also observed for C6 (see Fig. S47–S51†). The determined initial kapp values were plotted against the catalyst concentration [I] and a regression line was determined showing that the reaction is well in accordance with a pseudo first order mechanism (see Fig. 5). The slope of the regression line provides a kp value of (0.365 ± 0.004) L mol−1 s−1. This value is in the same order of magnitude as for [Fe(TMG5NMe2asme)Cl2] being one of the to-date most active robust iron catalysts with a kp value of (0.554 ± 0.02) L mol−1 s−1.40 However, the one-step preparation of L6 proves to be straightforward and more efficient compared to the four-step synthesis of TMG5NMe2asme (L11*).
 | ||
| Fig. 5 Plot of kappvs. the catalyst concentration [I] for the determination of kp for C6 at 150 °C. The end points were determined in duplicate ([M]/[I] = 1000:1 and 5000:1). The error bars show the standard deviation of the two experiments. | ||
Increasing the polymerization temperature to 180 °C leads to a doubling of the initial kapp value (39.8 × 10−4 s−1 for 180 °C vs. (24.1 ± 0.2) × 10−4 s−1 for 150 °C with [M]/[I] = 1000:1, see Fig. S55†). Lowering the temperature to 135 °C has an even more drastic effect with a conversion of only 16% after 10 min compared to 59% for 150 °C (see Fig. S54†). This highlights the importance of an appropriate polymerization temperature when applying the presented iron(II) guanidine catalysts. Additional experiments were performed with sublimated L-lactide and technical-grade rac-lactide with an [M]/[I] ratio of 2000:1 at 150 °C (see Fig. S57 and S58†). With increasing degree of purification, the kapp value rises from 6.89 × 10−4 s−1 (technical grade rac-lactide) to 1.14 × 10−3 s−1 (sublimated L-lactide) which is likely due to the purity.
In order to lower the catalyst loading, experiments with the co-initiator pMeBnOH were conducted (see Fig. 6 and Table 4 entries 8–10). Adding pMeBnOH generally leads to higher initial kapp values, lower dispersities and can improve the molar masses at high [M]/[I] ratios. This is most pronounced for experiments with very low catalyst concentrations. While for the experiment without co-initiator at [M]/[I] = 5000/1 no polymer could be precipitated from ethanol (see Table S7†), for [M]/[I]/[pMeBnOH] = 5000/1/5 polymer with an Mn of 26.9 kg mol−1 and a dispersity of 1.2 was obtained (see Fig. 6 and Table 4 entry 10). This illustrates that an alcohol such as pMeBnOH can be used as an inexpensive instrument to both lower the catalyst loading as well as improve the polymerization rate and polymer properties. The co-initiator is functioning as a nucleophile that initiates new growing chains. These observations are also in accordance with a former study on a zinc N,O hybrid guanidine complex.53 Analogous to C3, an experiment with the addition of 1 equivalent of FeCl2 was conducted ([M]/[I]/[FeCl2] = 2000:1:1, see Fig. 6 and Table 4 entry 11). Again, a longer linear behavior was observed and a significantly higher molar mass was obtained after 1 h at 150 °C compared to the experiment without FeCl2 (see Table 4 entry 7).
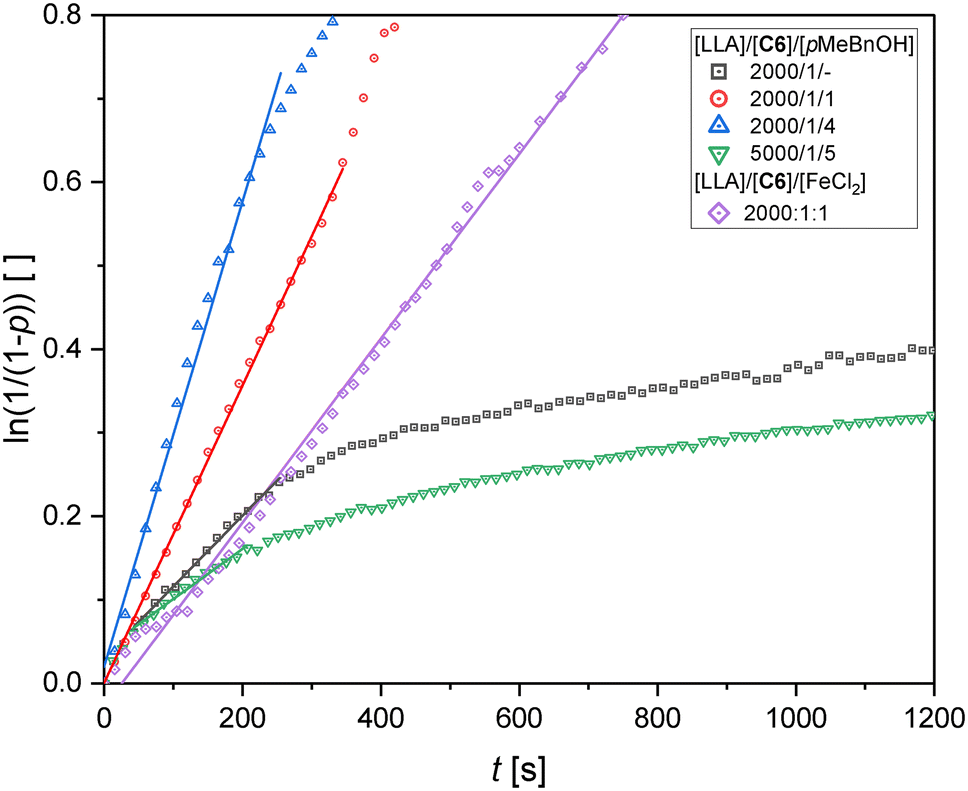 | ||
| Fig. 6 Pseudo first order semilogarithmic plots for the polymerization with C6 and co-initiator pMeBnOH as well as the polymerization with C6 and FeCl2. For comparison, the polymerization without co-initiator is depicted ([M]/[I] = 2000:1). Shown are the first 20 min of the experiments. Full plots can be found in the ESI.† | ||
MALDI-TOF mass spectra of polymer produced with C6 with an [M]/[I] ratio of 100:1 showed that the ligand TMGepy (L6) can be an end group (see Fig. S66†). This highlights the underlying coordination–insertion mechanism and the nucleophilicity of guanidine ligands. Additionally, in an experiment where pMeBnOH was applied as co-initiator ([M]/[I]/[pMeBnOH] = 100:1:1, see Fig. S67†), co-initiator was also found as end group. These observations are in accordance with the polymerization with other zinc and iron guanidine catalysts.38,40
The thermal properties of the poly-L-lactide produced by C6 with and without co-initiator were investigated by DSC (see Table S10†). The resulting glass transition temperatures in the range of 60 °C and melting peaks in the range of 170 °C are consistent with literature values for poly-L-lactide.2
DFT calculations
To understand the very different polymerization activities of C1–C8, DFT calculations were conducted (see Table 5). The functional TPSSh58,59 and the basis set def2-TZVP60–62 with a solvent model for MeCN using the polarizable continuum model (PCM) and an empirical dispersion correction using the D3 version of Grimme's dispersion with Becke–Johnson damping (GD3BJ)63–65 were applied. The optimized structures are in good accordance with the experimentally determined solid state structures as indicated by the low root mean-square deviations (RMSD values) and the comparison of the characteristic bond lengths and structure parameters (see Tables S14 and S15†). All calculations were carried out for the high-spin state (quintet) since the Fe–N bond lengths are in the typical range of high-spin iron complexes.| Complex | NBO charges [e units] | Charge-transfer energies [kcal mol−1] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe | Ngua,1 | Ngua,2/py/qu | Cl1 | Cl2 | Ngua,1 → Fe | Ngua,2/py/qu → Fe | ∅(N → Fe) | Cl1 → Fe | Cl2 → Fe | ∅(Cl → Fe) | |
| a Further details and calculated values as well as RMSD values can be found in Table S14.† b Calculation was performed for the complex cation [Fe(TMGpy)2Cl]+ (C5#). | |||||||||||
| C1 | 1.25 | −0.69 | −0.69 | −0.74 | −0.75 | 42.0 | 42.1 | 42.1 | 93.3 | 85.5 | 89.4 |
| C2 | 1.24 | −0.71 | −0.70 | −0.72 | −0.75 | 47.1 | 47.4 | 47.3 | 100.2 | 83.8 | 92.0 |
| C3 | 1.24 | −0.69 | −0.69 | −0.75 | −0.75 | 49.1 | 49.1 | 49.1 | 84.8 | 84.8 | 84.8 |
| C4 | 1.28 | −0.70 | −0.70 | −0.75 | −0.76 | 43.3 | 43.2 | 43.3 | 83.3 | 80.4 | 81.9 |
| C5# | 1.30 | −0.68/−0.68 | −0.52/−0.52 | −0.72 | — | 47.7/47.2 | 40.0/39.9 | 43.9/43.6 | 95.4 | — | 95.4 |
| C6 | 1.27 | −0.71 | −0.54 | −0.73 | −0.77 | 52.3 | 41.4 | 46.9 | 95.4 | 76.0 | 85.7 |
| C7 | 1.24 | −0.71 | −0.71 | −0.74 | −0.74 | 38.8 | 39.0 | 38.9 | 90.3 | 90.3 | 90.3 |
| C8 | 1.23 | −0.71 | −0.71 | −0.72 | −0.75 | 44.8 | 47.4 | 46.1 | 97.5 | 86.8 | 92.2 |
| C9* | 1.26 | −0.69 | −0.51 | −0.73 | −0.74 | 41.2 | 42.7 | 42.0 | 96.7 | 88.5 | 92.6 |
The structural optimization calculations were followed by natural bond orbital (NBO) calculations.66,67 The NBO charges of the iron centers in complexes C1–C8 only cover a small range between +1.23 and +1.30. Moreover, the NBO charges do not match the polymerization activities, and therefore, the Lewis acidity of the metal center cannot be the only factor influencing the catalytic activity. However, the charge-transfer energies between the N and Cl donor atoms, respectively, and the iron center can contribute to an explanation of the different activities and also stabilities. The complex cation C5# is only scarcely considered in the following discussion because of its significantly different structure, and therefore, a very limited comparability to the other complexes C1–C4 and C6–C8.
Complexes C3, C4 and C6 turned out to be the most active catalysts with the highest kapp values in the experimental studies (see above). The DFT calculations show that these three complexes with an aliphatic ligand backbone exhibit lower Cl → Fe charge-transfer energies compared to the less active complexes C1, C2, C7 and C8 with an aromatic ligand backbone. To further illustrate this relation, the kapp values at [M]/[I] = 1000:1 were plotted versus the average charge-transfer energies of the two Fe–Cl bonds (see Fig. 7). A low Cl → Fe charge-transfer energy is probably essential for the lactide coordination since the peralkylated guanidine donor units are sterically demanding and to enable the coordination of a lactide molecule as well as the growing polymer chain, the cleavage of an Fe–Cl bond might be necessary.
 | ||
| Fig. 7 Plot of the experimental initial kapp values ([M]/[I] = 1000:1, 150 °C) vs. the average Cl → Fe charge transfer energy (ECT(Cl → Fe)) in complexes C1–C4 and C6–C8. The complex cation C5# was omitted because it is not comparable to the other complexes. The error bars are partially invisible due to too small error values. | ||
The second factor relevant for the overall catalyst performance is the catalysts' ability to maintain the controlled polymerization behavior following pseudo first order kinetics over time. This stability seems to be linked to the strength of the Fe–N bonds. Complexes C2, C3, C6 and C8 exhibit the highest values for the N → Fe charge-transfer energies. The strong Fe–Ngua bonds explain the much higher stabilities of C3 and C6 compared to complex C4, which possesses rather weak N–Fe bonds and looses its high activity already after the first minute of polymerization. Moreover, the comparatively high N → Fe charge-transfer energies explain the steady polymerization activity of complexes C2 and C8 based on ligands with naphthalene backbones. Additionally, the much stronger electron density donation through a guanidine donor compared to a pyridine donor becomes apparent in the calculated N → Fe charge-transfer energies as can be seen for the hybrid guanidine complexes C5 and C6. This might be an additional reason for the good activity of hybrid guanidine complexes: the guanidine is tightly bound to the metal center while the second donor unit, the pyridine unit, can be more easily replaced by an approaching lactide molecule.
The combination of both influences on the catalyst performance, the Cl → Fe and the N → Fe charge transfer-energies, explains the very low polymerization activities of complexes C1 and C7. In these complexes with an aromatic ligand backbone, the Ngua–Fe bonds are weakened in comparison to the other complexes, while the Fe–Cl bonds are strong leading to an activity in the order of magnitude of FeCl2. Therefore, these complexes unite the features that are opposite to the trends found for suitable polymerization catalysts. This is also true for the iron(II) guanidine–quinoline complex [Fe(TMGqu)Cl2] (C9*) that has been investigated in the polymerization of technical-grade rac-lactide before and showed a very low polymerization activity with a kapp value of 3 × 10−6 s−1 at an [M]/[I] ratio of 500:1.40 All DFT results considered, it can be concluded that for an active and stable catalyst a weak Fe–Cl bond as well as strong Fe–N bonds are required. An aliphatic ligand backbone is beneficial for a high activity. Complexes C3 and C6 combine these features explaining their outstanding polymerization performances. These findings will support the future design of iron(II) guanidine catalysts.
PLA depolymerization with the complexes C2–C6
Besides the research on the PLA synthesis via a ROP, the chemical recycling of PLA becomes more and more important in order to integrate the material into the circular economy approach. Therefore, and to investigate whether the trend found for the polymerization can be transferred to the depolymerization, initial tests using C2–C6 for the methanolysis of PLA were performed (see Table 6). Choosing the same conditions as in previous publications43,44 allows a comparison of C2–C6 with other catalysts: PLA was depolymerized at 60 °C using 1 mol% catalyst, 7.13 equivalents MeOH and THF as solvent. The conversion of internal methine groups (Xint), the selectivity towards methyl lactate (MeLA) (SMeLA) and the yield (YMeLA) were calculated. The apparent rate constants (kapp) were determined from the semilogarithmic plots assuming pseudo first order kinetics (see Fig. S68–S70†).68| Cat. | t [h] | X int [%] | S MeLA [%] | Y MeLA [%] | k app [min−1] |
|---|---|---|---|---|---|
| a Conditions: 250 mg PLA (bio-mi Ltd., Mn = 46.7 kg mol−1, Đ = 1.5), 1 mol% catalyst (regarding the PLA ester bonds), methanol (7.13 equiv.), THF (4 mL), T = 60 °C, stirring speed: 260 rpm. For every catalyst, only one experiment is shown. The results of the other experiments can be found in Table S13.† b The conversion of internal methine groups of PLA (Xint), the selectivity towards the product methyl lactate (SMeLa) and the yield of the product (YMeLa) were calculated from the 1H NMR spectrum according to literature.20 c Determined from the plot of ln([Int]0/[Int]t) vs. t. The average value with standard deviation is shown. d The depolymerization proceeds very slowly and the semilogarithmic plot is not in good accordance with a pseudo first order reaction. Therefore, kapp was not determined. e The depolymerization proceeds very rapidly. Therefore, the kapp value was not determined. | |||||
| C2 | 24 | 69 | 52 | 36 | n.d.d |
| C3 | 3 | 100 | 93 | 93 | 0.0264 ± 0.0023 |
| C4 | 3 | 99 | 82 | 81 | 0.0306 ± 0.0010 |
| C5 | 0.25 | 100 | 100 | 100 | n.d.e |
| C6 | 3 | 67 | 42 | 28 | 0.0050 ± 0.0005 |
As for the polymerization, C2 shows a low activity with a maximum conversion of only 69% after 24 h and the kinetics deviated from a pseudo first order reaction. Therefore, no kapp value was determined for this complex. C6 being the most promising ROP catalyst features a similar conversion (Xint = 67%) within 3 h reaching a methyl lactate yield of 28%. With a kapp of 0.0050 ± 0.0005 min−1 it has a comparable activity to [Fe(TMG5NMe2asme)Cl2] with a kapp of 0.0048 min−1 under the same reaction conditions.44 The complete depolymerization of PLA to methyl lactate could not be observed (see Fig. S70†). C3 and C4 are an order of magnitude faster with kapp values of 0.0264 ± 0.0023 min−1 and 0.0306 ± 0.0010 min−1, respectively (see Fig. S68 and S69†). High methyl lactate yields are reached within 3 h. Complex C5 features an extraordinary activity being able to convert PLA completely to methyl lactate within 15 min. Overall, these experiments allow no correlation of the catalytic activities of the investigated complexes for the polymerization of lactide and the methanolysis of PLA. Due to the promising first results, further in-detail research will be conducted concerning the depolymerization abilities of the presented iron(II) guanidine catalysts and the structure–activity relationships.
Conclusion
In this study, eight well-known bisguanidine and N,N hybridguanidine ligands were reactivated showing the potential of their iron(II) chloride complexes C1–C8. Although ligands with a straightforward topology were chosen, they cover a wide range of structural motives with varying distances between the donor units, two types of the guanidine donor units and a different second donor unit in case of two guanidine–pyridine hybrid complexes. Except for one five-coordinate complex (C5) all complexes are four-coordinate with a distorted tetrahedral coordination geometry.All complexes were evaluated in the melt polymerization of L-lactide under industrially relevant conditions revealing substantial differences in their activities and stabilities. However, they generally exhibit faster polymerization rates than their zinc chloride analogs that have been studied before. The complexes with aromatic ligand backbones (C1, C2, C7 and C8) show low to mediocre activities rendering them unsuited for the application as ROP catalysts. On the contrary, the bisguanidine complexes C3 and C4 with an aliphatic ligand backbone exhibit a high initial activity. For C4 this activity decreased drastically after the first minute, while C3 is able to maintain its high activity for a longer time. The high activity phase of C3 could be further extended by the addition of FeCl2 improving conversion and molar masses. The N,N hybrid guanidine complex C6 stood out with its ability to polymerize lactide within minutes to molar masses of more than 40 kg mol−1. The catalyst loading could be successfully lowered by the use of a co-initiating alcohol, yielding polymers with even higher molar masses and low dispersities.
DFT calculations suggested that the strength of the Fe–Cl bond in iron(II) chloride guanidine complexes might be one of the main reasons for a catalyst's activity. At the same time, these calculations allowed to explain the decay of activity over time for some complexes by their decreased strength of the Fe–N bonds.
In addition to the polymerization study, first experiments regarding the methanolysis of PLA were conducted with complexes C2–C6. Especially complex C5 depolymerizes PLA to methyl lactate within minutes demonstrating the potential of these compounds for a circular use of PLA. Overall, this work provides new impetus for the research on iron guanidine complexes as polymerization catalysts and illustrates that the combination of well-known and easily prepared ligands with other metal precursors can unlock a previously unknown potential. This brings non-toxic catalysts for bioplastics closer to industrial application.
Experimental
Materials
All manipulations involving air- and/or moisture-sensitive materials were conducted under nitrogen or argon as inert gas applying standard Schlenk-techniques or in a nitrogen-filled glovebox. The glassware was dried at 150 °C and assembled while hot. Solvents used for the preparation of the iron complexes were purchased in analytical grade and were purified and degassed before use according to standard procedures.69 Acetonitrile was distilled over CaH2 and degassed by three freeze–pump–thaw cycles. Diethyl ether was distilled over sodium and degassed by three freeze–pump–thaw cycles. Anhydrous iron(II) chloride (purity 99.5%) was purchased from Alfa Aesar. L-Lactide (Lumilact L polymer grade, stereochemical purity ≥99.5%, free acid ≤7 meq kg−1, water ≤0.03%) was donated by TotalEnergies Corbion and was recrystallized from toluene and stored in a nitrogen-filled glovebox at room temperature. p-Methyl benzyl alcohol (4-methyl benzyl alcohol, pMeBnOH) was purchased from TCI Deutschland and was recrystallized from toluene. PLA film (Mn = 46.7 kg mol−1, Đ = 1.5) for the depolymerization experiments was provided by bio-mi Ltd. (Matulji, Croatia). Methanol (Acros Organics, 99.8%, extra dry over molecular sieves) was degassed and stored over molecular sieves.The guanidine ligands TMG2b (L1),45 TMG2n (L2),46 TMG2e (L3),35 TMG2p (L4),35 TMGpy (L5),49 TMGepy (L6),50 DMEG2b (L7)47 and DMEG2n (L8)48 were synthesized according to literature starting from the respective amine and the tetramethyl or dimethylethylene Vilsmeier reagent.
Preparation of iron(II) chloride guanidine complexes
IR for species without MeCN (C1) (crystals that were used for SC-XRD): IR (ATR, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ) = 2936 (w, ν(CHaliph)), 2932 (w, ν(CHaliph)), 2878 (w, ν(CHaliph)), 2798 (w, ν(CHaliph)), 1577 (w), 1540 (vs, ν(CNgua)), 1534 (vs, ν(CNgua)), 1518 (vs, ν(CNgua)), 1481 (s), 1460 (m), 1446 (w), 1434 (w), 1417 (vs), 1406 (vs), 1392 (vs), 1334 (s), 1286 (w), 1271 (w), 1239 (m), 1210 (m), 1155 (vs), 1149 (vs), 1108 (w), 1065 (w), 1030 (vs), 948 (w), 931 (w), 869 (w), 837 (s), 817 (s), 798 (m), 770 (vs), 748 (vs), 710 (m), 637 (m), 624 (w), 566 (m), 508 (w) cm−1. IR for acetonitrile-containing species (C1·1.5MeCN) (before TGA): IR (ATR, ) = 2934 (w, ν(CHaliph)), 2873 (w, ν(CHaliph)), 2801 (vw, ν(CHaliph)), 1581 (w), 1557 (m), 1538 (s, ν(CNgua)), 1520 (vs, ν(CNgua)), 1484 (m), 1464 (m), 1448 (w), 1436 (w), 1420 (s), 1408 (vs), 1402 (vs), 1396 (vs), 1334 (m), 1292 (w), 1288 (w), 1272 (w), 1238 (w), 1212 (w), 1187 (vw), 1162 (m), 1155 (m), 1147 (m), 1114 (w), 1066 (w), 1058 (vw), 1039 (w), 1029 (s), 960 (vw), 937 (vw), 923 (w), 875 (w), 843 (m), 818 (s), 797 (w), 746 (vs), 704 (w), 631 (w), 623 (w), 577 (vw), 563 (m), 513 (w), 510 (w), 477 (vw) cm−1. HR-MS (ESI+, MeCN), m/z (%): isotopic distribution calculated for [C16H28Cl2FeN6]+ [M]+: 428.11432 (6) [C16H2835Cl2N654Fe]+, 429.11767 (1) [C1513CH2835Cl2N654Fe]+, 430.10964 (100) [C16H2835Cl2N6Fe]+, 431.11300 (17) [C1613CH2835Cl2N6Fe]+, 432.10670 (64) [C16H2835Cl37ClN6Fe]+, 433.11005 (11) [C1613CH2835Cl37ClN6Fe]+, 434.10374 (10) [C16H2837Cl2N6Fe]+, 435.10710 (2) [C1513CH2837Cl2N6Fe]+; found: 430.10920 (100), 431.11188 (21), 432.10638 (62); isotopic distribution calculated for [C16H28ClFeN6]+ [M–Cl]+: 393.14546 (6) [C16H2835Cl54FeN6]+, 395.14079 (100) [C16H2835ClFeN6]+, 396.14415 (17) [C1513CH2835ClFeN6]+, 397.13784 (32) [C16H2837ClFeN6]+; found: 393.14394 (8), 395.14037 (100), 396.14296 (20), 397.13758 (31).
) = 2936 (w, ν(CHaliph)), 2932 (w, ν(CHaliph)), 2878 (w, ν(CHaliph)), 2798 (w, ν(CHaliph)), 1577 (w), 1540 (vs, ν(CNgua)), 1534 (vs, ν(CNgua)), 1518 (vs, ν(CNgua)), 1481 (s), 1460 (m), 1446 (w), 1434 (w), 1417 (vs), 1406 (vs), 1392 (vs), 1334 (s), 1286 (w), 1271 (w), 1239 (m), 1210 (m), 1155 (vs), 1149 (vs), 1108 (w), 1065 (w), 1030 (vs), 948 (w), 931 (w), 869 (w), 837 (s), 817 (s), 798 (m), 770 (vs), 748 (vs), 710 (m), 637 (m), 624 (w), 566 (m), 508 (w) cm−1. IR for acetonitrile-containing species (C1·1.5MeCN) (before TGA): IR (ATR, ) = 2934 (w, ν(CHaliph)), 2873 (w, ν(CHaliph)), 2801 (vw, ν(CHaliph)), 1581 (w), 1557 (m), 1538 (s, ν(CNgua)), 1520 (vs, ν(CNgua)), 1484 (m), 1464 (m), 1448 (w), 1436 (w), 1420 (s), 1408 (vs), 1402 (vs), 1396 (vs), 1334 (m), 1292 (w), 1288 (w), 1272 (w), 1238 (w), 1212 (w), 1187 (vw), 1162 (m), 1155 (m), 1147 (m), 1114 (w), 1066 (w), 1058 (vw), 1039 (w), 1029 (s), 960 (vw), 937 (vw), 923 (w), 875 (w), 843 (m), 818 (s), 797 (w), 746 (vs), 704 (w), 631 (w), 623 (w), 577 (vw), 563 (m), 513 (w), 510 (w), 477 (vw) cm−1. HR-MS (ESI+, MeCN), m/z (%): isotopic distribution calculated for [C16H28Cl2FeN6]+ [M]+: 428.11432 (6) [C16H2835Cl2N654Fe]+, 429.11767 (1) [C1513CH2835Cl2N654Fe]+, 430.10964 (100) [C16H2835Cl2N6Fe]+, 431.11300 (17) [C1613CH2835Cl2N6Fe]+, 432.10670 (64) [C16H2835Cl37ClN6Fe]+, 433.11005 (11) [C1613CH2835Cl37ClN6Fe]+, 434.10374 (10) [C16H2837Cl2N6Fe]+, 435.10710 (2) [C1513CH2837Cl2N6Fe]+; found: 430.10920 (100), 431.11188 (21), 432.10638 (62); isotopic distribution calculated for [C16H28ClFeN6]+ [M–Cl]+: 393.14546 (6) [C16H2835Cl54FeN6]+, 395.14079 (100) [C16H2835ClFeN6]+, 396.14415 (17) [C1513CH2835ClFeN6]+, 397.13784 (32) [C16H2837ClFeN6]+; found: 393.14394 (8), 395.14037 (100), 396.14296 (20), 397.13758 (31).
IR (ATR, ) = 3009 (vw, ν(CHarom)), 2954 (w, ν(CHaliph)), 2924 (w, ν(CHaliph)), 2887 (w, ν(CHaliph)), 2862 (w, ν(CHaliph)), 2789 (vw, ν(CHaliph)), 1549 (s), 1538 (s), 1517 (vs, ν(CNgua)), 1503 (vs), 1463 (m), 1440 (w), 1415 (s), 1406 (s), 1398 (vs), 1378 (vs), 1343 (m), 1328 (s), 1278 (m), 1232 (m), 1176 (vw), 1159 (vs), 1145 (m), 1127 (w), 1115 (w), 1103 (w), 1064 (m), 1060 (m), 1017 (vs), 996 (vs), 976 (w), 959 (vw), 927 (w), 920 (w), 847 (vs), 807 (vs), 786 (s), 768 (vs), 754 (s), 691 (s), 667 (vw), 624 (m), 576 (vw), 545 (w), 526 (vw), 508 (m), 480 (w), 475 (w), 412 (vw), 410 (vw) cm−1. HR-MS (ESI+, MeCN), m/z (%): isotopic distribution calculated for [C20H30Cl2FeN6]+ [M]+: 478.12997 (6) [C20H3035Cl254FeN6]+, 479.13332 (1) [C1913CH3035Cl254FeN6]+, 480.12529 (100) [C20H3035Cl2FeN6]+, 481.12865 (22) [C1913CH3035Cl2FeN6]+, 482.12234 (64) [C20H3035Cl37ClFeN6]+, 483.12570 (14) [C1913CH3035Cl37ClFeN6]+, 484.11939 (10) [C20H3037Cl2FeN6]+, 485.12275 (2) [C1913CH3037Cl2FeN6]+, 486.12610 (<1) [C1813C2H3037Cl2FeN6]+; found: 478.13097 (6), 479.13409 (1), 480.12639 (100), 481.12914 (25), 482.12358 (63), 483.12621 (16), 484.12130 (11), 485.12349 (2), 486.12586 (<1). EA (C20H30Cl2FeN6): calculated C 49.92%; H 6.28%; N 17.46%. Found C 49.84%; H 6.16%; N 17.63%.
Additional information on the synthesis of the target compound and original analysis data files are available via the Chemotion repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-QUQGCQVELV-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
IR (ATR, ) = 3003 (vw), 2983 (vw, ν(CHaliph)), 2943 (w, ν(CHaliph)), 2893 (w, ν(CHaliph)), 2851 (w, ν(CHaliph)), 2822 (w, ν(CHaliph)), 2804 (vw), 1554 (vs, ν(CNgua)), 1521 (vs, ν(CNgua)), 1476 (m), 1462 (w), 1442 (w), 1424 (s), 1415 (m), 1408 (m), 1389 (vs), 1342 (m), 1326 (w), 1240 (m), 1210 (w), 1151 (s), 1123 (w), 1109 (w), 1079 (w), 1068 (s), 1043 (s), 1017 (w), 914 (w), 894 (s), 776 (w), 765 (s), 735 (w), 595 (w), 583 (vw), 578 (w), 553 (w), 500 (w), 493 (w), 489 (vw), 438 (vw), 432 (vw) cm−1. HR-MS (APCI+, MeCN), m/z (%): isotopic distribution calculated for [C12H28Cl2FeN6]+ [M]+: 380.11432 (6) [C12H2835Cl254FeN6]+, 382.10964 (100) [C12H2835Cl2FeN6]+, 383.11300 (13) [C1113CH2835Cl2FeN6]+, 384.10670 (64) [C12H2835Cl37ClFeN6]+, 385.11005 (8) [C1113CH2835Cl37ClFeN6]+, 386.10374 (10) [C12H2837Cl2FeN6]+, 387.10710 (1) [C1113CH2837Cl2FeN6]+; found: 382.10950 (100), 384.10689 (57). EA (C12H28Cl2FeN6): calculated C 37.62%; H 7.37%; N 21.93%. Found C 37.53%; H 7.22%; N 22.10%.
Additional information on the synthesis of the target compound and original analysis data files are available via the Chemotion repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-CMAKKIUQDS-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
IR (ATR, ) = 3003 (vw), 2926 (w), 2911 (w, ν(CHaliph)), 2894 (w, ν(CHaliph)), 2856 (w, ν(CHaliph)), 1613 (vw), 1545 (vs, ν(CNgua)), 1532 (vs, ν(CNgua)), 1477 (vw), 1465 (vw), 1440 (vw), 1421 (m), 1404 (vw), 1393 (s), 1360 (w), 1354 (w), 1342 (w), 1236 (w), 1195 (vw), 1167 (w), 1160 (w), 1151 (w), 1145 (w), 1120 (vw), 1100 (vw), 1083 (w), 1074 (vw), 1067 (vw), 1058 (vw), 1034 (m), 939 (m), 915 (w), 901 (vw), 834 (w), 774 (s), 765 (w), 724 (vw), 715 (vw), 576 (w), 525 (vw), 506 (w) cm−1. HR-MS (ESI+, MeCN), m/z (%): calculated for [C13H30Cl2FeN6]+ [M]+: 396.12529, not found; only an adduct of the ligand L4 was found: 271.26151 (100) [C13H31N6]+ [L4 + H]+. EA (C13H30Cl2FeN6): calculated C 39.31%; H 7.61%; N 21.16%. Found C 39.29%; H 7.29%; N 21.49%.
Additional information on the synthesis of the target compound and original analysis data files are available via the Chemotion repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-OCIRWSBUTV-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
IR (ATR, ) = 3016 (vw, ν(CHarom)), 2983 (vw), 2947 (vw, ν(CHaliph)), 2890 (vw, ν(CHaliph)), 2874 (vw, ν(CHaliph)), 2832 (vw), 1602 (w), 1577 (w), 1570 (m), 1536 (vs, ν(CNgua)), 1477 (m), 1463 (w), 1453 (w), 1437 (m), 1427 (m), 1421 (w), 1408 (w), 1393 (vs), 1370 (m), 1291 (w), 1257 (vw), 1234 (w), 1158 (s), 1147 (w), 1111 (vw), 1096 (vw), 1069 (w), 1063 (w), 1056 (w), 1019 (w), 1012 (w), 1009 (w), 981 (vw), 914 (w), 896 (vw), 856 (vw), 846 (vw), 802 (m), 777 (vs), 744 (vw), 727 (w), 643 (w), 618 (w), 579 (w), 576 (w), 553 (vw), 486 (vw) cm−1. HR-MS (APCI+, MeOH), m/z (%): isotopic distribution calculated for [C22H36Cl2FeN8]+ [M]+: 536.18306 (6) [C22H3635Cl254FeN8]+, 537.18642 (2) [C2113CH3635Cl254FeN8]+, 538.17839 (100) [C22H3635Cl2FeN8]+, 539.18175 (24) [C2113CH3635Cl2FeN8]+, 540.17544 (64) [C22H3635Cl37ClFeN8]+, 541.17880 (15) [C2113CH3635Cl37ClFeN8]+, 542.17249 (10) [C22H3637Cl2FeN8]+, 543.17585 (2) [C2113CH3637Cl2FeN8]+, 544.17920 (<1) [C2013C2H3637Cl2FeN8]+; found: 538.1787 (100), 539.1831 (8), 540.1761 (47). EA (C22H36Cl2FeN8): calculated C 48.99%; H 6.73%; N 20.78%. Found C 48.73%; H 6.66%; N 21.16%.
Additional information on the synthesis of the target compound and original analysis data files are available via the Chemotion repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-YBCYHBWFSB-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
IR (ATR, ) = 3061 (vw, ν(CHarom)), 3010 (vw, ν(CHarom)), 2927 (w, ν(CHaliph)), 2900 (w, ν(CHaliph)), 2875 (w, ν(CHaliph)), 2845 (w), 2806 (vw), 1603 (w), 1537 (vs, ν(CNgua)), 1485 (m), 1480 (m), 1459 (w), 1452 (w), 1442 (s), 1423 (s), 1416 (w), 1405 (s), 1395 (vs), 1360 (w), 1345 (s), 1305 (w), 1258 (w), 1238 (w), 1230 (w), 1219 (w), 1161 (m), 1143 (m), 1115 (w), 1110 (w), 1073 (w), 1062 (w), 1054 (w), 1027 (w), 1017 (vw), 984 (m), 969 (w), 907 (w), 876 (w), 797 (w), 781 (vs), 765 (s), 748 (w), 716 (w), 646 (w), 590 (w), 581 (w), 511 (m), 458 (m), 417 (w) cm−1. HR-MS (APCI+, MeOH), m/z (%): isotopic distribution calculated for [C12H20Cl2FeN4]+ [M]+: 344.04557 (6) [C12H2035Cl254FeN4]+, 345.04892 (<1) [C1113CH2035Cl254FeN4]+, 346.04090 (100) [C12H2035Cl2FeN4]+, 347.04425 (13) [C1113CH2035Cl2FeN4]+, 348.03795 (64) [C12H2035Cl37ClFeN4]+, 349.04130 (8) [C1113CH2035Cl37ClFeN4]+, 350.03500 (10) [C12H2037Cl2FeN4]+, 351.03835 (1) [C1113CH2037Cl2FeN4]+; found: 346.04117 (100), 347.04587 (23), 348.03838 (63), 349.04296 (14), 350.03576 (7). EA (C12H20Cl2FeN4): calculated C 41.53%; H 5.81%; N 16.14%. Found C 41.54%; H 6.05%; N 16.40%.
Additional information on the synthesis of the target compound and original analysis data files are available via the Chemotion repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-SUGFTNYFKC-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
IR (ATR, ) = 2955 (w, ν(CHaliph)), 2928 (w, ν(CHaliph)), 2888 (w), 2870 (w, ν(CHaliph)), 1588 (m), 1547 (vs, ν(CNgua)), 1528 (vs, ν(CNgua)), 1479 (vs), 1461 (m), 1459 (m), 1450 (m), 1412 (vs), 1378 (vs), 1292 (s), 1281 (vs), 1235 (w), 1202 (vw), 1187 (vw), 1161 (vw), 1114 (w), 1079 (vw), 1036 (vs), 976 (m), 939 (vw), 887 (m), 856 (w), 811 (m), 761 (vs), 739 (vs), 705 (m), 663 (vw), 646 (w), 604 (w), 586 (w), 557 (m), 499 (w), 478 (w), 471 (w), 447 (vw), 428 (vw), 417 (vw) cm−1. HR-MS (APCI+, MeCN/MeOH), m/z (%): isotopic distribution calculated for [C16H24Cl2FeN6]+ [M]+: 424.08302 (6) [C16H2435Cl254FeN6]+, 425.08637 (1) [C1513CH2435Cl254FeN6]+, 426.07834 (100) [C16H2435Cl2FeN6]+, 427.08170 (17) [C1513CH2435Cl2FeN6]+, 428.07539 (64) [C16H2435Cl37ClFeN6]+, 429.07875 (11) [C1513CH2435Cl37ClFeN6]+, 430.07244 (10) [C16H2437Cl2FeN6]+, 431.07580 (2) [C1513CH2437Cl2FeN6]+; found: 424.08303 (6), 426.07859 (100), 427.08201 (23), 428.07576 (61), 429.07910 (13), 430.07363 (10). EA (C16H24Cl2FeN6): calculated C 44.99%; H 5.66%; N 19.67%. Found C 44.88%; H 5.63%; N 19.81%.
Additional information on the synthesis of the target compound and original analysis data files are available via the Chemotion repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-LGNUUDHCCJ-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
IR (ATR, ) = 2951 (vw, ν(CHaliph)), 2890 (w, ν(CHaliph)), 2868 (w, ν(CHaliph)), 2797 (vw, ν(CHaliph)), 1583 (w), 1544 (vs, ν(CNgua)), 1505 (w), 1489 (w), 1477 (m), 1463 (w), 1448 (w), 1427 (w), 1408 (s), 1387 (w), 1373 (w), 1342 (w), 1322 (w), 1294 (vs), 1239 (w), 1209 (w), 1164 (w), 1139 (w), 1127 (w), 1089 (w), 1060 (w), 1025 (m), 1009 (s), 977 (m), 969 (m), 921 (vw), 893 (vw), 852 (m), 823 (w), 810 (m), 783 (s), 770 (vs), 703 (w), 651 (w), 635 (m), 603 (w), 595 (w), 533 (m), 500 (s), 472 (m), 451 (vw), 435 (w), 431 (w), 423 (w), 414 (w) cm−1. HR-MS (ESI+, MeCN), m/z (%): isotopic distribution calculated for [C20H26Cl2FeN6]+ [M]+: 474.0987 (6) [C20H2635Cl254FeN6]+, 475.1020 (1) [C1913CH2635Cl254FeN6]+, 476.0940 (100) [C20H2635Cl2FeN6]+, 477.0973 (22) [C1913CH2635Cl2FeN6]+, 478.0910 (64) [C20H2635Cl37ClFeN6]+, 479.0944 (14) [C1913CH2635Cl37ClFeN6]+, 480.0881 (10) [C20H2637Cl2FeN6]+, 481.0914 (2) [C1913CH2637Cl2FeN6]+; found: 474.0975 (2), 476.0936 (100), 477.0964 (21), 478.0912 (68), 479.0933 (13), 480.0890 (9). EA (C20H26Cl2FeN6): calculated C 50.34%; H 5.49%; N 17.61%. Found C 49.98%; H 5.21%; N 17.90%.
Additional information on the synthesis of the target compound and original analysis data files are available via the Chemotion repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-CHWOIVIIAO-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
Bulk polymerization of L-lactide
Inside a nitrogen-filled glovebox, the catalyst, L-lactide (LLA) and, if appropriate, the co-initiator p-methyl benzyl alcohol (pMeBnOH), FeCl2 or the ligand TMG2e (L3) were homogenized in a mortar for 10 min. The mixture was transferred to a vial and removed from the glovebox. The used steel reactor was pre-heated under vacuum to the desired reaction temperature and flooded with argon gas for three times over the course of 1 h. The polymerization mixture was transferred to the reactor under a counter stream of argon. As soon as the reactor was closed, the mechanical stirrer was started and the acquisition of Raman spectra was initiated. After the desired reaction time, the stirrer and heating were turned off and the reactor was opened. An NMR sample for the determination of the conversion was taken. The residual polymer melt was distributed to two vials. The first fraction was used as a retention sample and the second fraction was dissolved in DCM (3–5 mL depending on the viscosity), precipitated from EtOH at 20 °C and the volatile compounds were removed under reduced pressure. The precipitated polymer was subjected to GPC analysis in order to determine the molar mass and the molar mass distribution.Methanolysis of polylactide
For the depolymerization experiments, PLA (0.25 g, amount of ester linkages nEL = 3.5 mmol, 1.0 eq.), the catalyst (0.035 mmol, 1 mol%), and absolute THF (4.0 mL) were added to a Young-type Schlenk tube inside a nitrogen-filled glovebox. The PLA and the catalyst were dissolved using an external heat source and placed in an oil bath at 60 °C. The reaction was started by addition of dry MeOH (1.00 mL, 24.7 mmol, 7.13 eq., THF/MeOH = 4:1) and stirred at 260 rpm. Samples were analyzed by 1H NMR spectroscopy in CDCl3.
Data availability
Additional information on the synthesis and characterization of the presented iron(II) guanidine complexes is available via the Chemotion repository. The respective links are given in the Experimental section.Crystallographic data for complexes C1–C8 has been deposited at the Cambridge Crystallographic Data Centre (CCDC) as supplementary no. CCDC – 2278549 for C1, CCDC – 2278550 for C2, CCDC – 2278551 for C3, CCDC – 2278552 for C4, CCDC – 2278553 for C5, CCDC – 2278554 for C6, CCDC – 2278555 for C7 and CCDC – 2278556 for C8.
Original data of polymerization and depolymerization experiments as well as the MALDI-TOF-MS spectra are available via the RADAR4Chem repository by FIZ Karlsruhe – Leibniz-Institut für Informationsinfrastruktur and are published under an Open Access model (CC BY-NC-SA 4.0 Attribution-NonCommercial-Share Alike: DOI: https://doi.org/10.22000/1625).
Author contributions
CC: conceptualization; methodology, investigation, formal analysis and data curation for complex synthesis & characterization and polymerization experiments; visualization, writing – original draft. LB: methodology, investigation, formal analysis and data curation for depolymerization experiments; writing – review & editing. SS: investigation (complex synthesis & characterization, polymerization experiments), writing – review & editing. SN: investigation (complex synthesis & characterization, polymerization experiments), writing – review & editing. YK: investigation (depolymerization experiments), writing – review & editing. JH: investigation, formal analysis (conducted DFT calculations); writing – review & editing. AH: supervision, validation, data curation, writing – review & editing. SHP: conceptualization, supervision, funding acquisition, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Commission under Horizon 2020 Grant Agreement no. 953073 (UPLIFT project) for funding. Moreover, we would like to thank bio-mi Ltd. (Matulji, Croatia) for the supply with PLA and TotalEnergies Corbion (Gorinchem, Netherlands) for the donation of lactide. We thank the RWTH High Performance Computing Cluster for computing time. We acknowledge Christian Broschinski, Maverick Eilers and Kristofer Weinberg for experimental support. We thank Henrika Hüppe for performing single-crystal X-ray diffraction measurements. We are grateful to Brigitte Pütz for conducting mass spectrometry and Brigitte Jansen for conducting TGA measurements (both IAC, RWTH Aachen University). In addition, we would like to thank Prof. Dr. Andrij Pich and Marion Connolly (both DWI – Leibniz-Institut für Interaktive Materialien, Aachen) for the MALDI-TOF-MS measurements. Finally, we thank NFDI4Chem for support and funding of the repositories Chemotion repository and RADAR4Chem.References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- R. A. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, Applications, and End of Life, Wiley, Hoboken, NJ, USA, 2nd edn, 2022 Search PubMed.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- P. McKeown and M. D. Jones, Sustainable Chem., 2020, 1, 1–22 CrossRef.
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- H. R. Kricheldorf and I. Kreiser-Saunders, Makromol. Chem., 1990, 191, 1057–1066 CrossRef CAS.
- M. Baśko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071–7081 CrossRef.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- T. Rosen, J. Rajpurohit, S. Lipstman, V. Venditto and M. Kol, Chem. – Eur. J., 2020, 26, 17183–17189 CrossRef CAS PubMed.
- J. Payne, P. McKeown, G. Kociok-Köhn and M. D. Jones, Chem. Commun., 2020, 56, 7163–7166 RSC.
- P. Marin, M. J.-L. Tschan, F. Isnard, C. Robert, P. Haquette, X. Trivelli, L.-M. Chamoreau, V. Guérineau, I. del Rosal, L. Maron, V. Venditto and C. M. Thomas, Angew. Chem., Int. Ed., 2019, 58, 12585–12589 CrossRef CAS PubMed.
- B. B. Idage, S. B. Idage, A. S. Kasegaonkar and R. V. Jadhav, Mater. Sci. Eng., B, 2010, 168, 193–198 CrossRef CAS.
- K. R. Delle Chiaie, A. B. Biernesser, M. A. Ortuño, B. Dereli, D. A. Iovan, M. J. T. Wilding, B. Li, C. J. Cramer and J. A. Byers, Dalton Trans., 2017, 46, 12971–12980 RSC.
- S. Impemba, F. Della Monica, A. Grassi, C. Capacchione and S. Milione, ChemSusChem, 2020, 13, 141–145 CrossRef CAS PubMed.
- B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 4384–4393 CrossRef PubMed.
- J. A. Stewart, P. McKeown, O. J. Driscoll, M. F. Mahon, B. D. Ward and M. D. Jones, Macromolecules, 2019, 52, 5977–5984 CrossRef CAS.
- P. V. S. Nylund, B. Monney, C. Weder and M. Albrecht, Catal. Sci. Technol., 2022, 12, 996–1004 RSC.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- P. McKeown, L. A. Román-Ramírez, S. Bates, J. Wood and M. D. Jones, ChemSusChem, 2019, 12, 5233–5238 CrossRef CAS PubMed.
- J. Stewart, M. Fuchs, J. Payne, O. Driscoll, G. Kociok-Köhn, B. D. Ward, S. Herres-Pawlis and M. D. Jones, RSC Adv., 2022, 12, 1416–1424 RSC.
- A. Buchard, C. J. Chuck, M. G. Davidson, G. Gobius du Sart, M. D. Jones, S. N. McCormick and A. D. Russell, ACS Catal., 2023, 13, 2681–2695 CrossRef CAS PubMed.
- C. Hermans, W. Rong, T. P. Spaniol and J. Okuda, Dalton Trans., 2016, 45, 8127–8133 RSC.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS PubMed.
- S. Jacobsen, H. G. Fritz, P. Degée, P. Dubois and R. Jérôme, Polym. Eng. Sci., 1999, 39, 1311–1319 CrossRef CAS.
- P. M. Schäfer and S. Herres-Pawlis, ChemPlusChem, 2020, 85, 1044–1052 CrossRef PubMed.
- A. Hermann, T. Becker, M. A. Schäfer, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2022, 15, e202201075 CrossRef CAS PubMed.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2020, 59, 21778–21784 CrossRef CAS PubMed.
- M. Fuchs, P. M. Schäfer, W. Wagner, I. Krumm, M. Walbeck, R. Dietrich, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2023, 16, e202300192 CrossRef CAS PubMed.
- S. Herres-Pawlis, A. Neuba, O. Seewald, T. Seshadri, H. Egold, U. Flörke and G. Henkel, Eur. J. Org. Chem., 2005, 2005, 4879–4890 CrossRef.
- J. Stanek, T. Rösener, A. Metz, J. Mannsperger, A. Hoffmann and S. Herres-Pawlis, Guanidines as Reagents and Catalysts II, 2015, pp. 95–164, DOI:10.1007/7081_2015_173.
- S. Pohl, M. Harmjanz, J. Schneider, W. Saak and G. Henkel, J. Chem. Soc., Dalton Trans., 2000, 3473–3479, 10.1039/B002554M.
- S. Pohl, M. Harmjanz, J. Schneider, W. Saak and G. Henkel, Inorg. Chim. Acta, 2000, 311, 106–112 CrossRef CAS.
- H. Wittmann, A. Schorm and J. Sundermeyer, Z. Anorg. Allg. Chem., 2000, 626, 1583–1590 CrossRef CAS.
- J. Börner, S. Herres-Pawlis, U. Flörke and K. Huber, Eur. J. Inorg. Chem., 2007, 2007, 5645–5651 CrossRef.
- J. Börner, U. Flörke, K. Huber, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2009, 15, 2362–2376 CrossRef PubMed.
- P. M. Schäfer, M. Fuchs, A. Ohligschläger, R. Rittinghaus, P. McKeown, E. Akin, M. Schmidt, A. Hoffmann, M. A. Liauw, M. D. Jones and S. Herres-Pawlis, ChemSusChem, 2017, 10, 3547–3556 CrossRef PubMed.
- P. M. Schäfer, P. McKeown, M. Fuchs, R. D. Rittinghaus, A. Hermann, J. Henkel, S. Seidel, C. Roitzheim, A. N. Ksiazkiewicz, A. Hoffmann, A. Pich, M. D. Jones and S. Herres-Pawlis, Dalton Trans., 2019, 48, 6071–6082 RSC.
- R. D. Rittinghaus, P. M. Schäfer, P. Albrecht, C. Conrads, A. Hoffmann, A. N. Ksiazkiewicz, O. Bienemann, A. Pich and S. Herres-Pawlis, ChemSusChem, 2019, 12, 2161–2165 CrossRef CAS PubMed.
- R. D. Rittinghaus, A. Karabulut, A. Hoffmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2021, 60, 21795–21800 CrossRef CAS PubMed.
- R. D. Rittinghaus, J. Zenner, A. Pich, M. Kol and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2022, 61, e202112853 CrossRef CAS PubMed.
- M. Fuchs, M. Walbeck, E. Jagla, A. Hoffmann and S. Herres-Pawlis, ChemPlusChem, 2022, 87, e202200029 CrossRef CAS PubMed.
- L. Burkart, A. Eith, A. Hoffmann and S. Herres-Pawlis, Chem. – Asian J., 2023, 18, e202201195 CrossRef CAS PubMed.
- M. Kawahata, K. Yamaguchi, T. Ito and T. Ishikawa, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o3301–o3302 CrossRef CAS.
- V. Raab, J. Kipke, R. M. Gschwind and J. Sundermeyer, Chem. – Eur. J., 2002, 8, 1682–1693 CrossRef CAS PubMed.
- M. Kawahata, K. Yamaguchi and T. Ishikawa, Cryst. Growth Des., 2005, 5, 373–377 CrossRef CAS.
- V. Raab, K. Harms, J. Sundermeyer, B. Kovačević and Z. B. Maksić, J. Org. Chem., 2003, 68, 8790–8797 CrossRef CAS PubMed.
- A. Hoffmann, J. Börner, U. Flörke and S. Herres-Pawlis, Inorg. Chim. Acta, 2009, 362, 1185–1193 CrossRef CAS.
- R. Wortmann, A. Hoffmann, R. Haase, U. Flörke and S. Herres-Pawlis, Z. Anorg. Allg. Chem., 2009, 635, 64–69 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964, 10.1039/B617136B.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356, 10.1039/DT9840001349.
- N. Conen, M. Fuchs, A. Hoffmann, S. Herres-Pawlis and A. Jupke, Adv. Sustainable Syst., 2023, 7, 2200359 CrossRef CAS.
- M. Reinmuth, U. Wild, D. Rudolf, E. Kaifer, M. Enders, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2009, 2009, 4795–4808 CrossRef.
- I. d. S. Vieira, PhD Thesis, Technische Universität Dortmund, 2013 Search PubMed.
- J. Börner, PhD Thesis, Universität Paderborn, 2009 Search PubMed.
- P. Roquette, A. Maronna, M. Reinmuth, E. Kaifer, M. Enders and H.-J. Himmel, Inorg. Chem., 2011, 50, 1942–1955 CrossRef CAS PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- A. Hoffmann, R. Grunzke and S. Herres-Pawlis, J. Comput. Chem., 2014, 35, 1943–1950 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO (7.0), Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, USA, 2018 Search PubMed.
- L. A. Román-Ramírez, P. McKeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef.
- W. L. F. Armarego, Purification of laboratory chemicals, Part 1: Physical techniques, chemical techniques, organic chemicals, Butterworth-Heinemann, Amsterdam, Netherlands, 9th edn, 2022 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of analytical methods, crystallographic information, TGA results, experimental data for polymerization and depolymerization, DSC results, MALDI-TOF-MS results, DFT details. CCDC 2278551–2278549. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy01117h |
| This journal is © The Royal Society of Chemistry 2023 |