
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the performance for palladium catalysed tert-butyl hydroperoxide-mediated Wacker-type oxidation of alkenes†
Matthew N.
Blair
 a,
Meadhbh
Murray-Williams
a,
Calum
Maguire
a,
Clare L.
Brown
a,
Qun
Cao
a,
Hongxin
Chai
a,
Yitong
Li
a,
Róisín L.
O'Hagan
a,
Paul
Dingwall
a,
Panagiotis
Manesiotis
a,
Catherine L.
Lyall
b,
John P.
Lowe
b,
Ulrich
Hintermair
c,
Peter C.
Knipe
a and
Mark J.
Muldoon
*a
a,
Meadhbh
Murray-Williams
a,
Calum
Maguire
a,
Clare L.
Brown
a,
Qun
Cao
a,
Hongxin
Chai
a,
Yitong
Li
a,
Róisín L.
O'Hagan
a,
Paul
Dingwall
a,
Panagiotis
Manesiotis
a,
Catherine L.
Lyall
b,
John P.
Lowe
b,
Ulrich
Hintermair
c,
Peter C.
Knipe
a and
Mark J.
Muldoon
*a
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, UK. E-mail: m.j.muldoon@qub.ac.uk
bDepartment of Chemistry/Dynamic Reaction Monitoring Facility, University of Bath, UK
cDepartment of Chemistry/Dynamic Reaction Monitoring Facility/Institute for Sustainability, University of Bath, UK
First published on 28th September 2023
Abstract
This work examines the palladium(II) catalysed oxidation of terminal alkenes to their corresponding methyl ketones using tert-butyl hydroperoxide (TBHP) as the oxidant. The study aimed to reduce catalyst loadings and to understand some of the factors which are important in the design of more effective methods. A series of ligands based around 2-(2-pyridyl)benzoxazole (PBO) were studied and a new dicationic catalyst was developed which can operate more efficiently than previously reported catalysts. The choice of solvent system was also found to have a significant impact on catalyst performance. In the case of oct-1-en-3-yl acetate, a model substrate for a challenging class of substrates (protected allylic alcohols), it was found that using 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), as part of a solvent mixture, greatly improved the reaction; enabling shorter reaction times and lower catalyst loadings.
Introduction
The oxidation of a terminal alkene to a methyl ketone is a very useful transformation in organic synthesis, one which could be used for the preparation of a range of fine chemicals and medicinal compounds. The most common methods for this transformation use a simple Pd(II) salt, water as the nucleophile, molecular oxygen as the terminal oxidant and an electron transfer mediator to facilitate the oxidation of the Pd(0) intermediate.1–6 The application of “Wacker-type” reactions for organic synthesis have been studied since the 1960s but there is still significant room for improvement, with methods often having problems such as high catalyst loadings (i.e. low turnover number (TON)), slow reaction kinetics and low product selectivity to the desired product. These challenges have inspired researchers to continue to study this important transformation. For example, in recent years, it has been shown that first-row transition metal catalysts can perform Wacker-type oxidation reactions,7 with iron based systems showing particular promise.8–13In the case of Pd based methods, an alternative approach to aerobic systems is to use peroxides to mediate these reactions. Peroxide-mediated Wacker reactions have been studied less than their aerobic counterparts, but some of the work to-date indicates they have the potential to address some of the limitations of aerobic systems. In addition, implementing peroxide based reactions is attractive to some users, as it does not require pressurised gas systems. This can make it more accessible to some research chemists or even those in production. This is supported by the fact that there are more examples of peroxide based oxidation reactions in the production of pharmaceuticals compared to aerobic methods.14
In 1980, the oxidation of alkenes to ketones by Pd(II) salts with H2O2 or tert-butyl hydroperoxide (TBHP) was reported by research groups led by Tsuji15 and Mimoun.16,17 The synthetic utility and mechanistic understanding of TBHP systems were later expanded, when the Sigman group examined such systems.18,19 They developed a method which utilised quinoline-2-oxazoline (quinox) as a ligand and generated a dicationic Pd complex in situ using Ag[SbF6] (Fig. 1A). Their studies gave valuable mechanistic insights,18,20 which support the catalytic cycle illustrated in Fig. 1B. Their studies also demonstrated that the catalyst was suitable for a wide-range of substrates, delivering excellent yields and high selectivity to the desired ketone, even for substrates which are typically very challenging for aerobic methods.19,21–23 This is further illustrated by others who have used the quinox/TBHP method in target-orientated synthesis.24–27
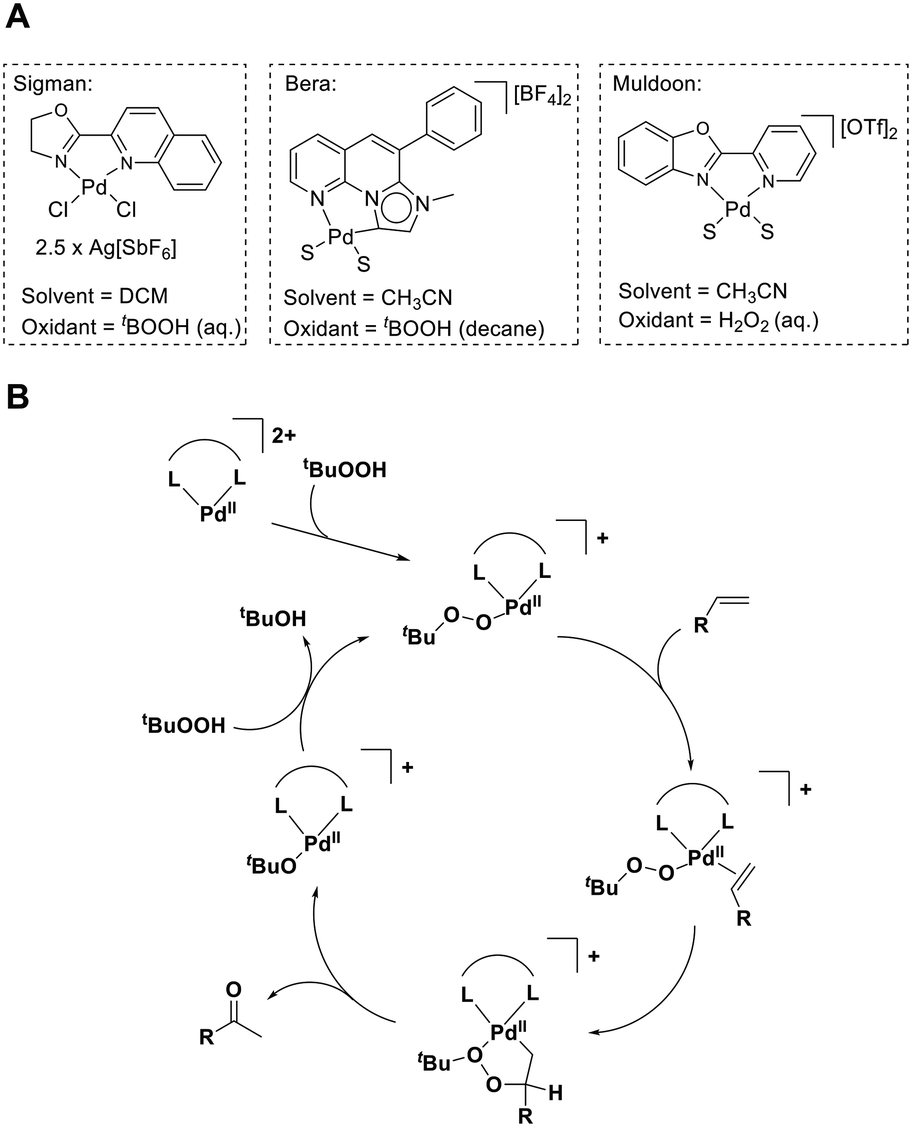 | ||
| Fig. 1 A Examples of previous dicationic palladium catalyst systems for peroxide-mediated Wacker-type oxidations. In the case of isolated complexes, S = CH3CN. B Postulated mechanism for TBHP-mediated oxidation by a dicationic Pd(II) complex with a bidentate ligand (L⌢L). | ||
There are several important characteristics that are needed to produce effective catalysts for these Wacker-type reactions. Sigman's studies showed that generating dicationic complexes (using weakly coordinating anions) enables activity under mild conditions,20 likely because such complexes improve the electrophilic activation of metal bound alkenes.28 The electronic asymmetric nature of the quinox ligand was also shown to play a key role in the efficiency of these TBHP-mediated reactions.20 More recently, Bera and co-workers reported the use of an isolated dicationic Pd(II) complex which employed an annelated pyridyl mesoionic carbene as an electronically asymmetric ligand (Fig. 1A).29 We have an interest in Pd(II) catalysed oxidation reactions and have reported new catalysts for aerobic30 and hydrogen peroxide-mediated31 Wacker-type reactions using dicationic Pd(II) complexes. In these previous Wacker studies we explored ligands based on the 2-(2-pyridyl)benzoxazole (PBO) type core. Using H2O2, the PBO-based catalyst (Fig. 1A) was only effective for styrene type substrates. We carried out mechanistic studies in collaboration with Waymouth and co-workers, and found there were mechanistic differences between the H2O2 and TBHP-mediated reactions.32 It was found that with H2O2, two pathways to the product were possible. One pathway which was the same as TBHP, with the product formed via a 1,2-hydride shift (as illustrated in Fig. 1B) and another (which dominates) involving a palladium enolate intermediate. During these studies, we observed that our [Pd(PBO)(MeCN)2][OTf]2 catalyst could employ TBHP as an oxidant, and for a number of reasons, we were interested in exploring this class of ligand further for TBHP based methods. The evidence suggests that TBHP systems enable faster rates and a wide substrate scope, and previous studies20 (discussed more below) have also shown that ligand structure can have a significant influence on reaction rate.
Despite the significant progress, there are still challenges remaining in this area of Wacker-type oxidations. As is often the case with oxidation reactions, it would be beneficial if the catalyst loadings required could be reduced. Catalyst loadings of 5–10 mol% are common4,5 and for some substrates, 20 mol% may be required.26,27 When compared to Pd cross-coupling reactions, which can often use ppm or even ppb levels of catalyst,33 it is clear that there is still significant room for improvement in terms of catalyst stability. Not only is Pd an expensive metal, but the ligand contributes significantly to the cost and sustainability of an industrial process.34 The elegant mechanistic studies by Sigman and co-workers gave valuable design principles, when it showed that the 4-CF3-quinox ligand delivered the best initial rates.20 A drawback is that this 4-CF3-quinox ligand is obtained in low yield via a 5-step synthesis. Additionally, this ligand did not improve catalyst stability and the catalyst TON was similar to the catalyst with the standard quinox ligand. In contrast to substituted quinox-type ligands, it is possible to make a range of functionalised PBO based ligands in a straightforward manner. As Sigman and co-workers have extensively explored substrate scope widely for these TBHP-mediated reactions,19,21–23 we have focused on the development of the catalyst and reaction conditions. We have selected two challenging model substrates, namely 1-octene and oct-1-en-3-yl acetate, for investigation. Aliphatic substrates such as 1-octene are less reactive compared to styrene type substrates and the oxidation reaction has to compete with isomerisation of the double bond. In the case of oct-1-en-3-yl acetate, this protected allylic alcohol often reacts more slowly and can have product selectivity issues, as a result of the alkene being close to a functional group which can coordinate to the Pd.
Results and discussion
We began our studies with 1-octene and Fig. 2, shows the ligands that were evaluated in our study. There was significant variation in performance among the ligands, in terms of both rate and TON, but it was found that the best performance was obtained with 5-CF3-PBO (see Table S1, and Fig. S3 and S5† and later for further details and discussion). Fig. 3 illustrates the preparation of the dicationic catalysts with this with 5-CF3-PBO ligand. This ligand can be synthesized in one-step from inexpensive commercially available starting materials. The synthesis of 2-aryl(benzo)oxazoles is well developed, as such compounds are of importance in medicinal chemistry.35 In the case of 5-CF3-PBO we utilised conditions based on previous reports for the Pd catalyzed C–H bond arylation of benzoxazole.36,37 The dicationic Pd oxidation complexes can be prepared in situ using the Pd chloride complex and silver salts. This is most effective when performed as a two-step procedure, involving the initial preparation and isolation of the PdCl2 complex. A number of dicationic PBO complexes can also be prepared via the acidic route, as illustrated in Fig. 3 for the 5-CF3-PBO ligand. We were unable to isolate a dicationic complex with quinox. As discussed, later, the use of silver salts to generate dicationic complexes often results in the same catalytic performance, but isolated complexes (resulting in fully homogeneous systems) offer advantages in system analysis and investigating potential issues such as aggregation. In addition, there are examples in catalysis, where silver salts have played multiple and sometimes unexpected roles in catalytic reactions.38,39 | ||
| Fig. 2 Ligands examined in this study of TBHP-mediated Wacker-type oxidation, highlighting Sigman's quinox ligand and our best performing ligand, 5-CF3-PBO. | ||
 | ||
| Fig. 3 Synthesis of the 5-CF3-PBO ligand and associated dicationic Pd catalysts. | ||
The electronic asymmetric nature of the 5-CF3-PBO ligand was confirmed by obtaining an X-ray crystal structure of the dicationic Pd(II) complex with bis(trifluoromethanesulfonyl)imide ([(CF3SO2)2N]−/[Tf2N]−) anions, as shown in Fig. 4. Although attempts to crystalise the trifluoromethanesulfonate ([CF3SO3]−/[OTf]−) complex were unsuccessful, both complexes displayed similar performance in reactions (see Fig. 9 and S8†). The trifluoromethanesulfonate complex was primarily used in most catalyst studies herein, owing to its lower cost, compared to bis(trifluoromethanesulfonyl)imide.
 | ||
| Fig. 4 Single crystal X-ray structure of [Pd(5-CF3-PBO)(MeCN)2][Tf2N]2 (CCDC deposition #2094910). The [Tf2N]− counter-ions are omitted for clarity. Distances are indicated in Å. Displacement ellipsoids are drawn at the 50% probability level. | ||
Our investigation initially employed similar reaction conditions as Sigman and co-workers, utilising dichloromethane (DCM) as solvent for the oxidation of 1-octene (Fig. S1 and S4†). Our results were in-line with their observations, in terms of quinox performance and also the fact that different ligands could deliver different initial rates, but ultimately result in similar final TONs (Fig. S3†). As we began to vary the conditions, we observed greater differences in catalyst performance. As [SbF6]− is known to readily hydrolyse,40 we sought a more stable anion. This was important not only for developing catalysts with higher TONs, but to also deliver practical advantages in terms of general handling and storage. Both [Tf2N]− and [OTf]− are stable to water and it is possible to make isolated complexes as well as generating them in situ. For these initial 1-octene studies, the isolated catalysts gave higher yields and selectivity than those generated with silver salts (Fig. S6 and S7†). In terms of the selectivity, the other products were mainly internal isomers and small quantities of internal ketones, as a result of oxidation of these isomers. It is worth noting that there are aerobic methods available which enable the anti-Markovnikov aldehyde product to be produced as the major product.41 However, when TBHP is used as an oxidant, the selectivity to the methyl ketone tends to be very high, with no anti-Markovnikov aldehyde formed in some cases.19,21,22,42
The biggest improvement in catalyst performance was obtained when we examined different solvents (Table S2 and Fig. S9†). As seen in Fig. 5, the best results were obtained in α,α,α-trifluorotoluene (TFT), where 1 mol% loading of our 5-CF3-PBO catalyst was employed.
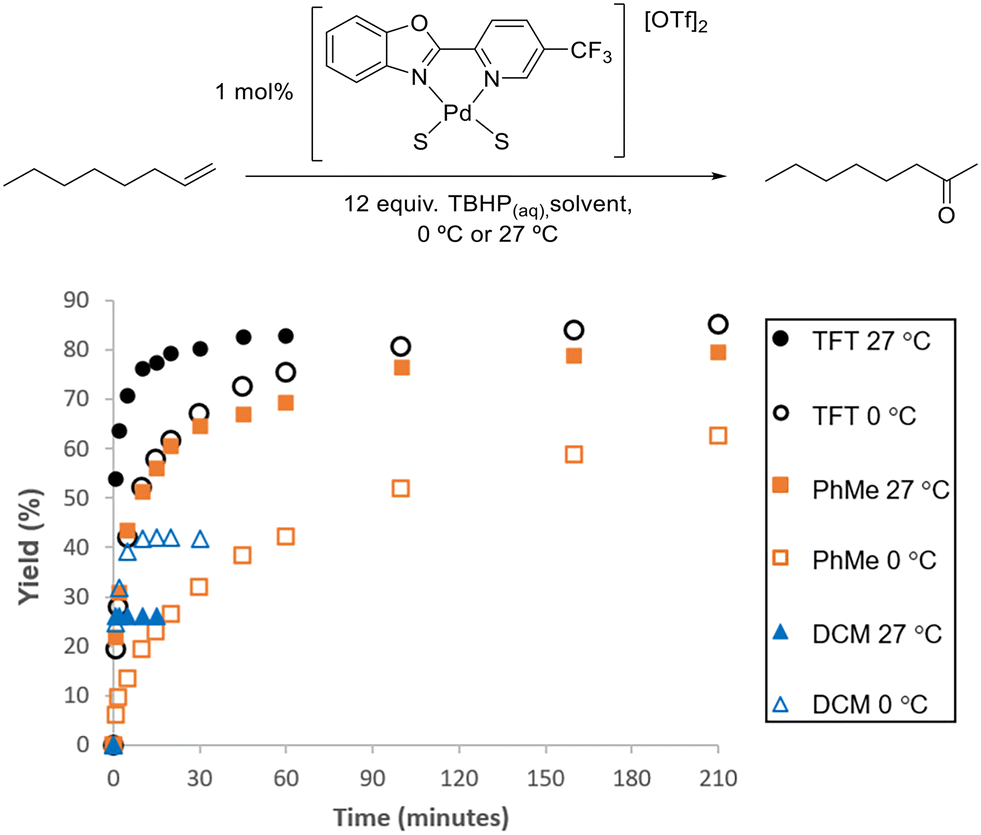 | ||
| Fig. 5 Comparison of TFT, toluene and DCM at 0 °C and 27 °C using 1 mol% [(5-CF3-PBO)Pd(S) 2][OTf]2 catalyst and aqueous TBHP. Plots are an average of two reactions. | ||
In DCM, the reaction proceeded rapidly, but the catalyst became deactivated in a similarly rapid manner, with no activity observed after 1 min, with a yellow solid precipitated from the initially homogeneous solution. The higher yield obtained at 0 °C indicates that this lower temperature slows down the catalyst deactivation. For this class of substrate, Sigman and co-workers started reactions at 0 °C and allowed them to warm to room temperature. Fig. 5 illustrates that the use of TFT results in the reactivity being maintained for longer and leads to an excellent yield. Toluene performed relatively well, and chlorobenzene was also a suitable solvent (Fig. S19†). The rate of reaction for this substrate is particularly fast in TFT and due to the exothermic nature of the reaction, further studies with this substrate were conducted at 0 °C (as detailed in the ESI,† with Fig. S17–S19 and S28–S30).
The formation of insoluble, inactive μ-hydroxy bridged dimers has previously been proposed as a possible pathway for catalyst deactivation.20 Our studies support this hypothesis, as higher yield of ketones could be achieved when reactions were conducted in anhydrous conditions, using organic solvent extracted TBHP. In the case of TFT, when using 0.5 mol% catalyst 0 °C, we could obtain 80% yield of the ketone using the anhydrous conditions, compared to 56% yield with aqueous TBHP (Fig. S17†). The reaction profile in Fig. S19† indicates there is still catalyst deactivation, but it is lessened; as discussed later, it is possible that TBHP also cause aggregation. Anhydrous TBHP is available commercially in decane, but we found its performance to be inferior in comparison to TBHP we had extracted into TFT (Fig. S18†). We found that the quinox systems did not operate in anhydrous conditions (Fig. S51†), which is in agreement with previous observations by Sigman and co-workers.20
We carried out diffusion-ordered spectroscopy (DOSY) NMR using the CF3 group on the ligand as a diagnostic handle (ESI†). We studied a post-reaction mixture when the reaction was carried out in toluene (Fig. S13†) and also examined 19F DOSY of the isolated [Pd(5-CF3-PBO)(S)2][OTf]2 complex in wet and anhydrous acetonitrile (Fig. S25 and S27†). These studies indicated that the complex/catalyst does aggregate in the presence of water. It is worth noting in the recent work by Bera and co-workers, that utilising aqueous TBHP resulted in precipitation of a black solid, which did not occur when they used TBHP in decane, with acetonitrile as the bulk solvent.29 It is also worth highlighting again, the earlier studies by Mimoun.16 These non-catalytic studies used pre-formed peroxidic complexes. Their studies predominantly employed anhydrous conditions and the introduction of water was found to lead to the precipitation of metallic palladium. Such precipitation of metallic palladium could be due to water acting as a nucleophile, and this pathway would proceed via a Pd(0) intermediate which would readily aggregate and precipitate if not quickly re-oxidised by O2 or an electron transfer mediator. Mimoun also found that internal alkenes led to the formation of π-allyl complexes which could precipitate from the reaction mixture.16 However, our findings indicated that addition of an internal alkene (4-octene) did not affect the catalytic performance for the oxidation of 1-octene (Fig. S30†).
Furthermore, little evidence of product inhibition on the oxidation of 1-octene was observed by adding 2-butanone and t-butanol (Fig. S29 and S30†). Taken as a whole, these data suggest that aggregation due to water is the major issue causing catalyst deactivation.
In terms of performance for aliphatic alkenes, our system compares favorably to the prior work. With aqueous TBHP, we can obtain high yields (82–85%) in short reaction times at 0 or 27 °C using 1 mol% catalyst (Fig. 5). Under anhydrous conditions, 80% yield can be achieved at 0 °C with 0.5 mol% catalyst (Fig. S17†). In the Sigman system, simple aliphatic alkenes could be oxidised to 86% yield in 20 min (0 °C to room temperature) using 2 mol% catalyst.19 In the recent Bera studies, it was found that aliphatic alkenes required 5 mol% catalyst and temperatures of 70 °C to obtain high yields in 24 h.29 Given that TBHP can pose thermal hazards,43 it seems preferable to have catalysts that can effectively operate at lower temperatures.
We next wanted to examine a particularly challenging model substrate; oct-1-en-3-yl acetate. Oxidation of protected allylic alcohols give acyloin products which are valuable building blocks.44 Aerobic conditions often lead to a mixture of products (e.g. ketone and aldehyde), but the Sigman method enables the oxidation of such substrates with high selectivity to the ketone product.19 In the case of oct-1-en-3-yl acetate, Sigman and co-workers reported that their quinox/TBHP system could deliver the product in 89% yield, with 5 mol% loading of the Pd catalyst and a reaction time of 20 h.19 We postulated that this substrate was slower (compared to unfunctionalized alkenes) due to the acetoxy group coordinating to the Pd, and that this could be addressed by the addition of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP). HFIP has some unique solvent properties and solvent effects,45–49 and has previously been used to prevent product inhibition of a catalyst.50 It has a strong hydrogen-donating ability, and a weak hydrogen bond accepting ability/nucleophilicity. This is an ideal combination for our system, as it can interact strongly with Lewis basic species but not with our dicationic Pd(II) catalysts. The interactions between HFIP and the substrate and the product were examined by 1H NMR and Job plot analysis found that HFIP interacted with both (Fig. 6 and ESI†).
 | ||
| Fig. 6 Job plots demonstrating the hydrogen bonding between HFIP and oct-1-en-3-yl acetate (top) and 2-oxooctan-3-yl acetate (bottom) in CDCl3 (the HFIP hydroxy proton shift was monitored). | ||
In the case of the substrate, there was a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hydrogen bond adduct with HFIP, and for the product it was a mixture of 1:1 and 1:2. The Job plot findings are in good agreement with similar studies which have shown that HFIP interacts with Lewis basic functionalities such as carbonyls and ethers.50–52 In our studies, we postulate that HFIP interactions with the oct-1-en-3-yl acetate substrate will reduce its ability to chelate to the metal center. In addition, interacting with the product will reduce its ability to inhibit the catalyst (Fig. S32†). We initially examined this substrate without HFIP, using both aqueous TBHP and anhydrous conditions. We found that reaction rate was significantly slower than for 1-octene (as expected), and the rates were similar in both the wet and anhydrous reactions. The final yield was higher using the anhydrous conditions which again supports the theory that water leads to an increase in catalyst deactivation.
:1 hydrogen bond adduct with HFIP, and for the product it was a mixture of 1:1 and 1:2. The Job plot findings are in good agreement with similar studies which have shown that HFIP interacts with Lewis basic functionalities such as carbonyls and ethers.50–52 In our studies, we postulate that HFIP interactions with the oct-1-en-3-yl acetate substrate will reduce its ability to chelate to the metal center. In addition, interacting with the product will reduce its ability to inhibit the catalyst (Fig. S32†). We initially examined this substrate without HFIP, using both aqueous TBHP and anhydrous conditions. We found that reaction rate was significantly slower than for 1-octene (as expected), and the rates were similar in both the wet and anhydrous reactions. The final yield was higher using the anhydrous conditions which again supports the theory that water leads to an increase in catalyst deactivation.
When HFIP was added to catalytic reactions with 1 mol% [Pd(5-CF3-PBO)(S)2][OTf]2 it led to a significant improvement in rate (Fig. 7 and S37† (for wet conditions)). With 10 equiv. of HFIP added to the anhydrous TFT system, 84% yield of the ketone product was obtained in just 2 hours. HFIP also appears to improve catalyst stability (see later for more discussion on this). In 4 hours, 81% yield of product could be obtained with 0.5 mol% catalyst. With longer reactions times, it was found that just 0.25 mol% catalyst could deliver high yields (Fig. S41†). This is a lower catalyst loading than we could achieve with 1-octene, but we found that HFIP was not suitable for this substrate due double bond migration. This is in agreement with previous studies, which have used Pd(OAc)2 and HFIP for the isomerisation of allylbenzenes.53
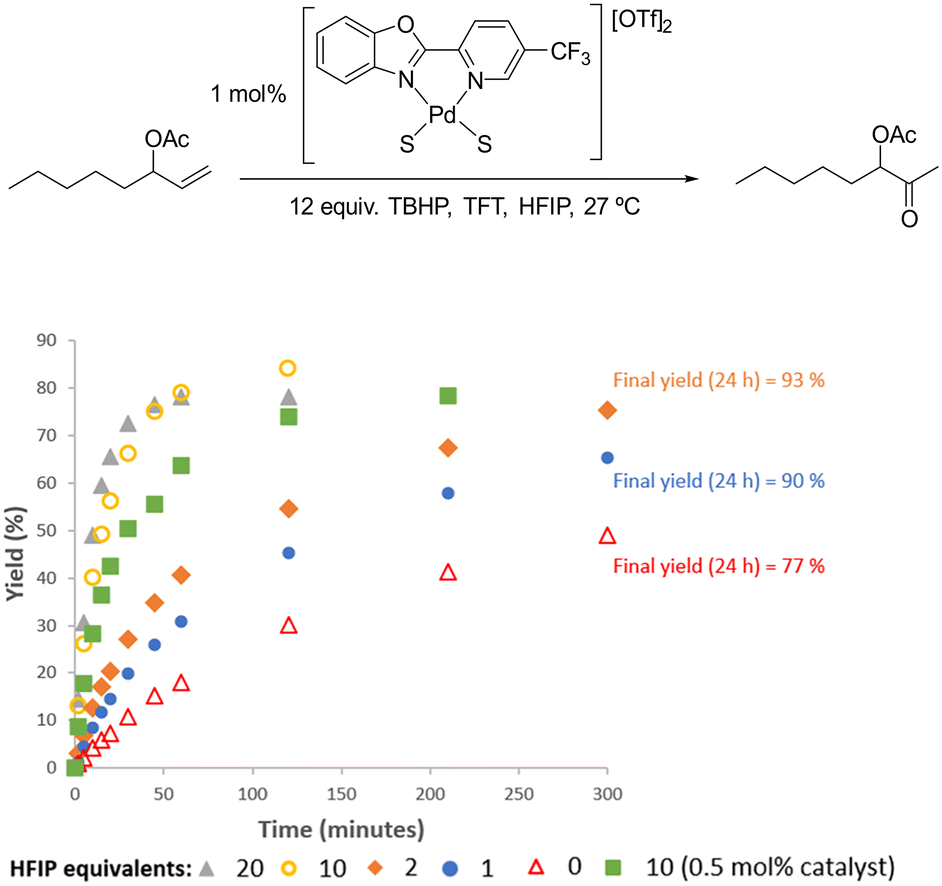 | ||
| Fig. 7 Addition of different equivalents of HFIP for the Wacker oxidation of oct-1-en-3-yl acetate under anhydrous conditions. Each plot an average of 2 reactions. | ||
When we discovered that we could use 0.25 mol% catalyst with the anhydrous/HFIP conditions, we decided to study a number of catalysts again at this loading. We thought these conditions could give further insights into the influence of ligand structure (see Fig. S43, S45 and S46†). These experiments indicated that electronic factors affect the rate and that steric factors can improve catalyst stability. The 5-CF3-PBO ligand was still found to deliver the best performance. It is worth noting that for other reactions, ligands containing a pyridine with a CF3 group at position 5, have also had the best performance.54,55 In the case of TBHP-mediated Wacker-type oxidations, the electronic benefits of electron withdrawing groups on our ligands is in-line with the previous ligand studies by Sigman and co-workers.20 Although most of these catalysts had TONs of over 200, not all could deliver complete conversion, suggesting that there is still some catalyst deactivation over time. This is also supported by 19F NMR DOSY studies in anhydrous PhCl (Fig. S23†). It is plausible that the TBHP could also lead to some aggregation, by acting as a bridging ligand. Mimoun and co-workers reported tetrameric structures of the type [RCO2PdOO-t-Bu]4, formed from the treatment of the corresponding Pd(O2CR)2 salts with TBHP (see Fig. S47†).16 In our studies, it was found that 5-ethyl-PBO had a slower initial rate but resulted in good final yield, which may be due the ethyl group reducing the rate of the competing catalyst aggregation.
Despite the excellent performance under the anhydrous conditions, we are aware that simply using commercial aqueous TBHP is clearly more convenient than having to prepare anhydrous TBHP solutions; an issue which only gets more complicated for larger scale applications.56 Fortunately, we found that with 20 equiv. of HFIP we could obtain high yields in short reaction times with 1 mol% catalyst using commercial aqueous TBHP (Fig. 8).
 | ||
| Fig. 8 The influence of solvent and fluorinated alcohols on the oxidation of oct-1-en-3-yl acetate. Yields are an average of two reactions. | ||
This method was found to be particularly robust, whereas the 0.25 mol% loading using anhydrous conditions was more susceptible to impurities in reagents (see page 79 of ESI† for more discussion). At 1 mol% catalyst loading, we found that there was no real benefit increasing to 30 equiv. of HFIP (Fig. 8) and using pure HFIP as the solvent was not suitable as lower yields were obtained (50% with 1 mol% catalyst) and the catalyst became inactive within the first 30 min. Chlorobenzene (PhCl) with 20 equiv. HFIP also delivered good performance, significantly better than toluene (PhMe), therefore PhCl offers an alternative to TFT (Fig. 8).
Although, our initial hypothesis on the role of HFIP was to prevent substrate and product inhibition of our catalyst, HFIP has the potential to influence the reaction in several other ways. The hydrogen donating ability of HFIP may slow down catalyst aggregation caused by water and/or TBHP, perhaps due to HFIP interacting with water and TBHP. For example, it has been shown that the addition HFIP can have a significant impact on the nucleophilicity of water.57 We carried out experiments that support that HFIP slows down catalyst aggregation. Typically, the catalyst is stirred in solution (with TBHP present) for 5 min before finally adding the alkene substrate. It was found that if the catalyst was stirred in solution for 3 hours prior to adding the substrate, the performance of the catalyst was better if HFIP was present during this 3 h incubation period (Fig. S40†).
There could also be an enhancement of the oxidant. It is known that HFIP can activate H2O2,45,46 and TBHP, as shown by Kühn and co-workers.58 They studied a fluorinated organomolybdenum complex for the epoxidation of alkenes using TBHP. In their study it was found that using HFIP as the solvent led to the best results. They carried out a control reaction with cyclooctene as the substrate and found that in the absence of the Mo catalyst, HFIP/TBHP could still produce epoxide. We did our own control experiments to examine if there was any oxidation activity without the palladium catalyst. In the case of 1-octene we used TBHP(aq.) with HFIP as the sole solvent and no epoxide was observed, however, terminal alkenes such as 1-octene are known to be more difficult to epoxidise than internal olefins such as cyclooctene. We also tested oct-1-en-3-yl acetate under optimal conditions (12 equiv. TBHP and 20 equiv. HFIP in TFT) and found there was no conversion of the substrate in the absence of a palladium complex. An additional possibility is that HFIP is oxidised to hexafluoroacetone (HFA). Hexafluoroacetone hydrate is a commercial reagent that can act as a catalytic oxidant when combined with H2O2,59 so in theory it is possible that HFA could be generated in situ and play a role as an active oxidant in our system. We therefore carried out a number of tests to explore this possibility. Although HFA is a gas at room temperature, the HFA hydrate has a boiling point over 100 °C, therefore we examined reactions which used aqueous TBHP. The 19F NMR analysis of post-reaction mixtures did not show any HFA signals (Fig. S54†). To check that small amounts of HFA were not playing a role, control experiments with one equivalent of HFA (hydrate) were carried out, and the catalyst performance was found to be similar to using pure TFT as the solvent (Fig. S55†). Furthermore, as shown in Fig. 8, we found that using perfluoro-tert-butyl alcohol (PFTB) instead of HFIP, could lead to improved performance, which suggests that the primary benefits are derived by the hydrogen bond donating ability of HFIP.
We also compared a variety of catalysts at 1 mol% catalyst loading with aqueous TBHP, as shown in Fig. 9.
 | ||
| Fig. 9 The influence of catalyst structure (and solvent) on the oxidation of oct-1-en-3-yl acetate. Yields are an average of two reactions. In the case of palladium chloride complexes, 2.5 mol% Ag[X] was used to make the dicationic catalyst in situ. | ||
With HFIP present we observed little difference in performance between the isolated catalyst and that generated in situ (something we had observed when examining 1-octene). It is possible that when HFIP is present it can interact with chloride ions,60 and reduce their ability to inhibit the Pd catalyst. It can also be seen that there is no real difference between [Tf2N]− and [OTf]−. PBO ligands could deliver better performance than quinox, although we found that HFIP also had a considerable positive influence on the quinox catalysts for this substrate (Fig. 9 and also Fig. S52 and S53†). These experiments have been carried out on a 0.9 mmol scale using GC with an internal standard for accurate analysis of reactions. In order to showcase the scalability of this system, a reaction was performed on a 7 mmol scale using 1 mol% [Pd(5-CF3-PBO)(S)2][OTf]2 and an isolated product yield of 93% (1.21 g, 6.5 mmol) was obtained.
Conclusions
We have shown that 2-(2-pyridyl)benzoxazole type ligands can be used to deliver excellent catalyst performance for TBHP-mediated Wacker-type oxidation reactions. These ligands are be easily prepared and can be used to synthesize stable dicationic Pd(II) complexes which can be isolated. The choice of solvent and the presence of water was found to have a considerable effect on catalyst stability. A significant pathway for catalyst deactivation is aggregation, something which is promoted by water. The use of anhydrous conditions enabled higher TONs to be achieved, although our catalyst system still demonstrated comparatively excellent performance with commercial aqueous TBHP. For the challenging model substrate, oct-1-en-3-yl acetate, we have found that the addition of HFIP led to significant improvements in catalyst performance. Hydrogen bonding between HFIP and this substrate (and product) enables the Pd(II) catalyst to oxidise the substrate at faster rates. The HFIP also improves catalyst stability and allows lower catalyst loadings to be used, with 0.25 mol% being used under anhydrous conditions. With aqueous TBHP it can be carried out with 1 mol% in short reaction times. These results compare well with other reported methods for this substrate, which have used 5 mol% of their respective Pd catalyst.19,61,62 In the future, we will explore how such solvent effects can be applied to other catalysts,63 and functionalised molecules of relevance to target orientated organic synthesis.Author contributions
MNB: investigation, methodology, visualization, writing – original draft, writing – review & editing, MMW: investigation, methodology, writing – review & editing, CM: investigation, writing – review & editing, CLB: investigation, writing – review & editing, QC: investigation, methodology, writing – review & editing, HC: investigation, YL: investigation, RLH: investigation, writing – review & editing, PD: methodology, writing – review & editing, PM: methodology, investigation, writing – review & editing, CLL: methodology, writing – review & editing JPL: methodology, writing – review & editing, UH: methodology, resources, writing – review & editing, PCK: investigation, writing – review & editing, MJM: conceptualization, methodology, investigation, funding acquisition, resources, project administration, supervision, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Conor McGrann and Donal Moran of Queen's University's ASEP Analytical Services for ESI-MS and elemental analysis. For funding, we are grateful to the following: the Department for the Economy (DfE) Northern Ireland for studentships (M. Blair, M. Murray-Williams, C. Maguire), China Scholarship Council (No. 201506030041) (H. Chai) and the EPSRC/Catalysis Hub (EP/R027129/1) for postdoctoral funding (Q. Cao). The Royal Society for a University Research Fellowship (UF160458) (U. Hintermair), and EPSRC for funding the Dynamic Reaction Monitoring Facility at the University of Bath (EP/P001475/1).Notes and references
- J. Tsuji, Synthesis, 1984, 1984, 369–384 CrossRef.
- C. N. Cornell and M. S. Sigman, Inorg. Chem., 2007, 46, 1903–1909 CrossRef CAS PubMed.
- S. E. Mann, L. Benhamou and T. D. Sheppard, Synthesis, 2015, 47, 3079–3117 CrossRef CAS.
- T. V. Baiju, E. Gravel, E. Doris and I. N. Namboothiri, Tetrahedron Lett., 2016, 57, 3993–4000 CrossRef CAS.
- R. A. Fernandes, A. K. Jha and P. Kumar, Catal. Sci. Technol., 2020, 10, 7448–7470 RSC.
- O. Temkin, Kinet. Catal., 2020, 61, 663–720 CrossRef CAS.
- P. Rajeshwaran, J. Trouvé, K. Youssef and R. Gramage-Doria, Angew. Chem., Int. Ed., 2022, 61, e202211016 CrossRef CAS PubMed.
- B. Liu, F. Jin, T. Wang, X. Yuan and W. Han, Angew. Chem., Int. Ed., 2017, 56, 12712–12717 CrossRef CAS PubMed.
- F. Puls and H. J. Knölker, Angew. Chem., 2018, 130, 1236–1240 CrossRef.
- F. Puls, F. Seewald, V. Grinenko, H. H. Klauß and H. J. Knölker, Chem. – Eur. J., 2021, 27, 16776–16787 CrossRef CAS PubMed.
- F. Puls, P. Linke, O. Kataeva and H. J. Knölker, Angew. Chem., 2021, 133, 14202–14209 CrossRef.
- J. Trouvé, K. Youssef, S. Kasemthaveechok and R. Gramage-Doria, ACS Catal., 2023, 13, 4421–4432 CrossRef.
- T. Schuh, O. Kataeva and H.-J. Knölker, Chem. Sci., 2023, 14, 257–265 RSC.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- J. Tsuji, H. Nagashima and K. Hori, Chem. Lett., 1980, 9, 257–260 CrossRef.
- H. Mimoun, R. Charpentier, A. Mitschler, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1980, 102, 1047–1054 CrossRef CAS.
- M. Roussel and H. Mimoun, J. Org. Chem., 1980, 45, 5387–5390 CrossRef CAS.
- C. N. Cornell and M. S. Sigman, J. Am. Chem. Soc., 2005, 127, 2796–2797 CrossRef CAS PubMed.
- B. W. Michel, A. M. Camelio, C. N. Cornell and M. S. Sigman, J. Am. Chem. Soc., 2009, 131, 6076–6077 CrossRef CAS PubMed.
- B. W. Michel, L. D. Steffens and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 8317–8325 CrossRef CAS PubMed.
- B. W. Michel, J. R. McCombs, A. Winkler and M. S. Sigman, Angew. Chem., 2010, 122, 7470–7473 CrossRef.
- J. R. McCombs, B. W. Michel and M. S. Sigman, J. Org. Chem., 2011, 76, 3609–3613 CrossRef CAS PubMed.
- R. J. DeLuca, J. L. Edwards, L. D. Steffens, B. W. Michel, X. Qiao, C. Zhu, S. P. Cook and M. S. Sigman, J. Org. Chem., 2013, 78, 1682–1686 CrossRef CAS PubMed.
- A. Kreuzer, S. Kerres, T. Ertl, H. Rücker, S. Amslinger and O. Reiser, Org. Lett., 2013, 15, 3420–3423 CrossRef CAS PubMed.
- F. Perez, A. R. Waldeck and M. J. Krische, Angew. Chem., 2016, 128, 5133–5136 CrossRef.
- R. J. Smith and B. C. Hawkins, Eur. J. Org. Chem., 2019, 2019, 6847–6854 CrossRef CAS.
- P. Steib and B. Breit, Chem. – Eur. J., 2019, 25, 3532–3535 CrossRef CAS PubMed.
- C. Hahn, Chem. – Eur. J., 2004, 10, 5888–5899 CrossRef CAS PubMed.
- S. Saha, S. Yadav, N. U. D. Reshi, I. Dutta, S. Kunnikuruvan and J. K. Bera, ACS Catal., 2020, 10, 11385–11393 CrossRef CAS.
- H. Chai, Q. Cao, L. M. Dornan, N. L. Hughes, C. L. Brown, P. Nockemann, J. Li and M. J. Muldoon, Eur. J. Inorg. Chem., 2017, 2017, 5604–5608 CrossRef CAS.
- Q. Cao, D. S. Bailie, R. Fu and M. J. Muldoon, Green Chem., 2015, 17, 2750–2757 RSC.
- K. L. Walker, L. M. Dornan, R. N. Zare, R. M. Waymouth and M. J. Muldoon, J. Am. Chem. Soc., 2017, 139, 12495–12503 CrossRef CAS PubMed.
- D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602–625 CrossRef CAS.
- J. D. Hayler, D. K. Leahy and E. M. Simmons, Organometallics, 2018, 38, 36–46 CrossRef.
- C. S. Demmer and L. Bunch, Eur. J. Med. Chem., 2015, 97, 778–785 CrossRef CAS PubMed.
- F. Derridj, J. Roger, F. Geneste, S. Djebbar and H. Doucet, J. Organomet. Chem., 2009, 694, 455–465 CrossRef CAS.
- X.-B. Shen, Y. Zhang, W.-X. Chen, Z.-K. Xiao, T.-T. Hu and L.-X. Shao, Org. Lett., 2014, 16, 1984–1987 CrossRef CAS PubMed.
- T. Bhattacharya, S. Dutta and D. Maiti, ACS Catal., 2021, 11, 9702–9714 CrossRef CAS.
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed.
- W. A. Mazeika and H. Neumann, Inorg. Chem., 1966, 5, 309–311 CrossRef CAS.
- J. J. Dong, W. R. Browne and B. L. Feringa, Angew. Chem., Int. Ed., 2015, 54, 734–744 CrossRef CAS PubMed.
- J. A. Wright, M. J. Gaunt and J. B. Spencer, Chem. – Eur. J., 2006, 12, 949–955 CrossRef CAS PubMed.
- Y.-W. Wang and C.-M. Shu, Ind. Eng. Chem. Res., 2010, 49, 8959–8968 CrossRef CAS.
- R. Kommera, V. R. Kasireddy, V. R. Ghojala, M. Bekkam, P. Rebelli and J. R. Yerrabelly, Synlett, 2018, 29, 1076–1078 CrossRef CAS.
- I. Colomer, A. E. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 1–12 CrossRef.
- X. D. An and J. Xiao, Chem. Rec., 2020, 20, 142–161 CrossRef CAS PubMed.
- V. Pozhydaiev, M. Power, V. Gandon, J. Moran and D. Lebœuf, Chem. Commun., 2020, 56, 11548–11564 RSC.
- H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. Koehn, V. M. Norwood IV and J. Aubé, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- T. Bhattacharya, A. Ghosh and D. Maiti, Chem. Sci., 2021, 12, 3857–3870 RSC.
- H. F. Motiwala, C. Fehl, S.-W. Li, E. Hirt, P. Porubsky and J. Aubé, J. Am. Chem. Soc., 2013, 135, 9000–9009 CrossRef CAS PubMed.
- E. Gaster, S. Kozuch and D. Pappo, Angew. Chem., Int. Ed., 2017, 56, 5912–5915 CrossRef CAS PubMed.
- A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed.
- N. Nishiwaki, R. Kamimura, K. Shono, T. Kawakami, K. Nakayama, K. Nishino, T. Nakayama, K. Takahashi, A. Nakamura and T. Hosokawa, Tetrahedron Lett., 2010, 51, 3590–3592 CrossRef CAS.
- D. Marcos-Atanes, C. Vidal, C. D. Navo, F. Peccati, G. Jiménez-Osés and J. L. Mascareñas, Angew. Chem., Int. Ed., 2023, 62, e202214510 CrossRef CAS PubMed.
- G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC.
- B. Li, S. M. Guinness, S. Hoagland, M. Fichtner, H. Kim, S. Li, R. J. Maguire, J. C. McWilliams, J. Mustakis and J. Raggon, Org. Process Res. Dev., 2018, 22, 707–720 CrossRef CAS.
- J. Ammer and H. Mayr, J. Phys. Org. Chem., 2013, 26, 59–63 CrossRef CAS.
- S. A. Hauser, M. Cokoja, M. Drees and F. E. Kühn, J. Mol. Catal. A: Chem., 2012, 363, 237–244 CrossRef.
- P. A. Ganeshpure and W. Adam, Synthesis, 1996, 1996, 179–188 CrossRef.
- L. Wang, Q. Yuan, W. Cao, J. Han, X. Zhou, S. Liu and X.-B. Wang, J. Phys. Chem. A, 2020, 124, 2036–2045 CrossRef CAS PubMed.
- D. A. Chaudhari and R. A. Fernandes, J. Org. Chem., 2016, 81, 2113–2121 CrossRef CAS PubMed.
- R. A. Fernandes and D. A. Chaudhari, J. Org. Chem., 2014, 79, 5787–5793 CrossRef CAS PubMed.
- While this manuscript was in preparation, Zou and co-workers reported the TBHP-mediated Wacker oxidation using palladium acetate complexes with 2-(1H-indazol-1-yl)quinoline as the ligand. In this study, they utilised HFIP as the sole/bulk solvent, having found it gave superior performance to the other solvents tested (methanol and ethanol), but they did not study solvent effects in detail. This report is quite different from our study in a number of ways. In terms of HFIP, we found using HFIP as the bulk solvent led to reduced performance. In addition, the substrates studied were mostly styrene-type derivatives, and the conditions used (2 mol% catalyst, 12 h, 60 °C), indicate that this is significantly less active in comparison to our system. S. Zhang, J. Zhang and H. Zou, Org. Lett., 2023, 25, 1850–1855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental sections, supplementary data, and additional discussion. CCDC 2094910. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy01046e |
| This journal is © The Royal Society of Chemistry 2023 |