
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New prodigiosin derivatives – chemoenzymatic synthesis and physiological evaluation against cisplatin-resistant cancer cells†
Tim Moritz
Weber
 a,
Alexandra
Leyens
b,
Lena
Berning
c,
Björn
Stork
c and
Jörg
Pietruszka
*ab
a,
Alexandra
Leyens
b,
Lena
Berning
c,
Björn
Stork
c and
Jörg
Pietruszka
*ab
aInstitute of Bioorganic Chemistry, Heinrich Heine University Düsseldorf, Wilhelm-Johnen-Straße, 52428 Jülich, Germany
bInstitute of Bio- and Geosciences (IBG-1): Bioorganic Chemistry, Jülich Research Centre, Wilhelm-Johnen-Straße, 52428 Jülich, Germany. E-mail: j.pietruszka@fz-juelich.de
cInstitute of Molecular Medicine I, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany
First published on 21st September 2023
Abstract
Prodigiosin and its derivatives from the prodiginine family are a natural class of secondary metabolite alkaloids of bacterial origin. They are well known for multifarious biological activities against a broad range of bacteria, pathogenic fungi, parasites, and several cancer cell lines. Biosynthesis of natural derivatives is based on a converging route with a final ATP- and enzyme-dependent condensation reaction between the bipyrrole precursor MBC and miscellaneously substituted monopyrroles. Although these ligating enzymes have been recognised for promiscuity regarding monopyrroles, minor studies were exerted to investigate promiscuity for MBC derivatives. To overcome the current lack of structural knowledge, we synthesised six 5′-n-alkyl derivatives of MBC and validated their suitability for condensation with monopyrroles by the ligating enzymes PigC, TreaP, and TamQ to probe their active site experimentally. Moreover, chemically synthesised prodiginines with 5-n-alkylation on the A-ring were subjected to systematic cell viability screening with the urothelial cancer cell lines RT-112 (cisplatin-sensitive) and RT-112res (cisplatin-resistant) to fathom the effect of electron-donating substituents on cytotoxicity. Alongside an overall broad acceptance of short- and medium-chain alkylated MBC derivatives by the enzymes PigC, TreaP, and TamQ, we identified the A-ring substituted prodiginines with methyl substituents as superior anticancer agents against cisplatin-resistant RT-112res after 72 h (15.7–18.8 nM) compared to prodigiosin (41.1 nM) and the former phase II clinical candidate obatoclax mesylate (36.0 nM).
Introduction
Prodigiosin (1) is a deep red coloured natural pigment of bacterial origin and eponym for the generic family term of prodiginine alkaloids. During the last 70 years, the family of achiral and chiral prodiginines of natural and synthetic provenance has constantly grown and has been first under investigation for structural elucidation, but then more and more become of interest for the development of total synthesis strategies and for its manifold biological activities.1–11 Common for all prodiginines is a congeneric conjugated scaffold of A-, B-, and C-ring pyrroles and decorations on the ring systems. Prodigiosin (1) was once identified due to its striking colourful appearance,11 but nowadays its derivatives and their cytotoxic properties have raised scientific and clinical attention.12,13 The basic tripyrrolic core structure is able to bind divalent metal cations to induce oxidative stress. When binding CuII, prodigiosin was shown to efficiently cause copper-promoted single- and double-strand breaks of DNA.14–17 Moreover, protonated prodiginines are allowed to passively diffuse across biologic membranes, symporting the chloride counter ion. The co-transport implicates uncoupling and depletion of the proton gradient that is essential to acquire energy in the form of adenosine triphosphate (ATP) or, more generally, to maintain the proton motive force and associated biological processes.15,18–22 Albeit prodigiosin and prodiginine alkaloids exhibit a broad range of activities against bacteria, protozoa, pathogenic fungi, plants, and nematodes,23–26 prodiginines were well recognised for their activity against the malaria parasite Plasmodium falciparum.27–29 Furthermore, prodiginines feature anti-tumour activity against several human cancer cell lines by induction of apoptosis, showing relatively low effects against non-malignant tissue.30–34 The diversity of reported biophysical properties and biological activities adumbrate the great challenge, which cells are facing after prodigiosin (1) treatment. Presumably, the relatively low doses being necessary to observe lethal biological activity are a consequence of blurring multidimensional modes of action.Biosynthesis and condensing enzymes
With an increasing number of natural prodiginines and related natural products, the quantity of known bacterial gene clusters involved in their biosynthesis has constantly risen. To date, numerous clusters among diverse bacterial species, such as pig (Serratia marcescens, prodigiosin 1),35red (Streptomyces coelicolor, undecylprodigiosin and streptorubin B),36trea (Pseudoalteromonas citrea, tambjamine MYP1 2),37 and mar (marine Streptomyces sp., marineosin A) have been identified by genome mining and analyses of mutant strains (Fig. 1, bottom).38–42 Despite the structural diversity of prodiginine-related natural products, their biosynthesis resorts to clustered genes, encoding for non-ribosomal peptide synthetases (NRPS) of type II.36,43 Using the synthesis of prodigiosin (1) as representative example, the assembly employs a convergent route with two key precursors, namely the bipyrrole part (4-methoxy-2,2′-bipyrrole-5-carbaldehyde, MBC 3a) and the monopyrrole part (2-methyl-3-amylpyrrole, MAP 4a), which are consolidated in an ATP-catalysed condensation reaction to give the conjugated tripyrrole scaffold of prodigiosin (1) (Fig. 1, top).35,44–46 For further in-depth elaboration on (bio)synthesis of MBC, MAP, the included enzymatic steps, and prodiginine-related natural products, consultation of the review from Hu et al. is recommended.47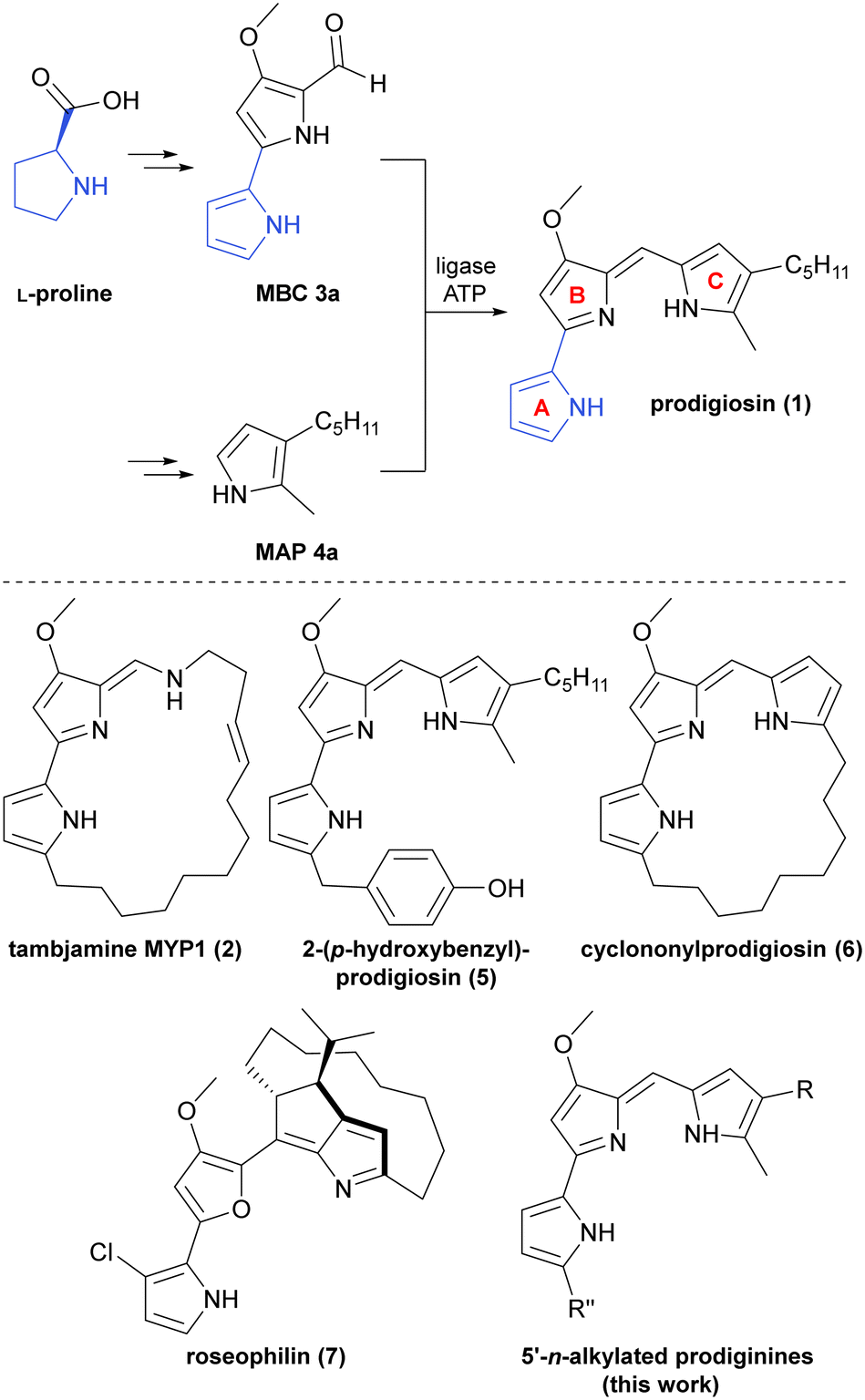 | ||
| Fig. 1 Top: Shortened biosynthesis of prodigiosin (1) by the related pig genes from S. marcescens. The final condensation step of MBC 3 and MAP 4a is catalysed by a ligase (here PigC) in an ATP-consuming manner. The fate of atoms from the initial precursor L-proline in the intermediate MBC 3 and the natural product prodigiosin (1) is highlighted in blue. Rings (A-, B-, and C-) of the prodiginine scaffold are accentuated in red. Bottom: Examples from the class of prodigiosin alkaloids and connatural natural products, illustrating the structural diversity. | ||
The enzymes, being responsible for the C–C-bond forming condensation reaction of monopyrroles and MBC, belong to the class of ligases (EC 6.4). Here, the enzymes PigC, RedH, TreaP, and MarH from afore mentioned gene clusters take on the role of ligases in the corresponding pathways. Unfortunately, crystallisation attempts of these enzymes did not lead to fruitful insights into structure-based mechanistic peculiarities. And yet, approximations of the structure were achieved by homology modelling and docking studies,48,49 while mechanistic intricacies were elucidated by kinetic studies.50
A-ring modifications of prodiginine-related natural products
The known natural prodiginine and connatural tambjamine derivatives featuring aliphatic substitutions on the A-ring are consistently belonging to the class of macrocycles, except the oddly protruding 2-(p-hydroxybenzyl)prodigiosin (5) from the marine bacterium Pseudoalteromonas rubra (Fig. 1, bottom).51 The formation of macrocyclic prodiginines is explicitly owing to late stage oxidative carbocyclisation of long chain alkyl chains on the prodiginine C-ring, including a carbon-centred radical intermediate.52,53 Because of this cyclisation, either formation of C- to A-ring (a) or C- to B-ring (b) bridging macrocycles is observed or formation of C-ring internal chiral cycles (c) catalysed. Prominent examples for those enzymes are the RedG homologue MarG involved in marineosin biosynthesis (b) or the Rieske oxygenase RedG in the making of streptorubin B (c).53For the C- to A-ring bridged carbomacrocycle cyclononylprodigiosin (6) the biosynthetic non gene cluster with the RedG-homologue NonG has been assigned (a).54 However, the upstream condensation of bipyrrole and monopyrrole exclusively relies on utilisation of MBC as bipyrrole condensation partner, meaning that no naturally occurring A-ring substituted MBC derivatives have been found to date. In case of A-ring modified 2-(p-hydroxybenzyl)prodigiosin (5), no proposal on biosynthesis and related genes, based on experimental evidence, has been published.55 Another well-known example of a prodigiosin-related natural product is roseophilin (7) from Streptomyces griseoviridis, which shows 3-chlorination of the A-ring, substitution of the B-ring pyrrolyl moiety by furyl and a chiral macrocyclic C-ring.
Although the chlorination pattern implies utilisation of a chlorinated MBC analogue, comparable to the biosynthesis of pyoluteurin,56 an adequate precursor and identification of the responsible halogenating enzyme is still missing. Late stage halogenation of dechlororoseophilin could be plausible, too. Indeed, the lack in discovery of substituted A-ring MBC derivatives from natural sources is not surprising, as the A-ring is built from the amino acid L-proline (cf.Fig. 1, top). Within the prodigiosin pig biosynthesis cluster of S. marcescens and undecylprodigiosin red gene cluster in S. coelicolor, the early steps of transformation include PigI/RedM-catalysed activation of proline as thioester and the subsequent oxidation of the pyrrolidine core by PigI/RedA to give acylated 1H-pyrrole.35,36,47 In other words, a prerequisite for natural A-ring variation on MBC derivatives would be the acceptance of alternative canonic amino acids by PigI/RedA during the initial steps in biosynthesis or functionalisation of the A-ring pyrrole in successive steps. Nevertheless, no experimental proof has been furnished for existence of this kind of A-ring substituted MBC so far.
Shifting the focus from the bipyrrole MBC 3a to the monopyrrole unit, promiscuity of condensing enzymes, exemplified by PigC from S. marcescens, is well-known for monopyrroles with cyclic and acyclic aliphatic side chains and the C-ring moiety of prodiginines.57–59 Contemporaneously, little evidence was provided in the context of MBC derivatives being suitable substrates, yet. Haynes et al. attempted investigation of RedH-catalysed condensation of a dechlororoseophilin-inspired MBC analogue in combination with 2-undecylpyrrole, but did not observe any activity based on product formation. Since their study was conducted as feeding experiment with the mutant strain S. coelicolor W39, limited phase transfer across the bacterial membrane or lacking eligibility as RedH substrate were deduced as potential cause.60 Chawrai et al. pioneered demonstration of accepted aryl substituted A-ring derivatives of MBC (e.g. thienyl-, furyl-, phenyl-) by PigC,46 but since then, no studies were performed beyond that. Even though broad accessibility of A-ring substituted prodiginines by biosynthetic methods is not given from a today's perspective, those derivatives can be assessed by means of total synthesis. Melvin et al. attributed a negative effect on copper-promoted DNA cleavage, when the A-ring was either substituted with electron-withdrawing substituents in 5-position or replaced by aryl residues other than pyrrolyl.15 Similar effects were observed by D'Alessio et al. with focus on cytotoxicity in extensive structure–activity-relationship studies (SAR).61 At the same time, both studies denoted positive effects by adding electron-donating alkyl substitutions in 5-position of the A-ring,15,61 providing substantial evidence of value to further analyse the consequences of A-ring associated alkylations. In the present study, we aim at expanding the knowledge about acceptance of A-ring n-alkylated MBC derivatives by prodiginine ligases to probe the active site of investigated enzymes experimentally and systematically infer the effects on prodiginine cytotoxicity against cisplatin-sensitive and -resistant urothelial carcinoma cell lines.
Results and discussion
Synthesis of prodiginine precursors
Prodiginine ligases exploit the bipyrrole MBC 3a and derivatives of 1H-monopyrroles as substrates for their ATP-catalysed reaction mechanism.62,63 Thus, the chemical synthesis of monopyrroles was approached using the Trofimov procedure, which allows conversion of oximes to 2,3-dialkylated 1H-pyrroles.57,64 Starting from commercially available ketones, namely octan-2-one (8a), hexan-2-one (8b), and decan-2-one (8c), the respective oximes 9a–c were synthesised in yields of ≥98% by refluxing the ketones with hydroxylamine hydrochloride and sodium acetate in EtOH/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Fig. 2). As conversions were near quantitative, no further purification was needed and the E/Z mixtures could be used straightaway for the subsequent Trofimov reaction. The latter allows cyclisation of alkylated oximes in the presence of 1,2-dichloroethane under superbasic reaction conditions that can be obtained by mixing DMSO, ground potassium hydroxide and a small amount of H2O.64 The electron-rich pyrroles are highly sensitive towards oxygen and light. Consequently, in addition to inert reaction conditions we identified utilisation of thoroughly degassed and dried DMSO as key requirement for a reliable outcome of the reaction. Only with degassed solvent, the application of reaction temperatures of 90–100 °C over a prolonged period led to a minimal amount of polymerisation during the reaction and yielded 52–54% of pyrroles 4a–c (Fig. 2).
:1) (Fig. 2). As conversions were near quantitative, no further purification was needed and the E/Z mixtures could be used straightaway for the subsequent Trofimov reaction. The latter allows cyclisation of alkylated oximes in the presence of 1,2-dichloroethane under superbasic reaction conditions that can be obtained by mixing DMSO, ground potassium hydroxide and a small amount of H2O.64 The electron-rich pyrroles are highly sensitive towards oxygen and light. Consequently, in addition to inert reaction conditions we identified utilisation of thoroughly degassed and dried DMSO as key requirement for a reliable outcome of the reaction. Only with degassed solvent, the application of reaction temperatures of 90–100 °C over a prolonged period led to a minimal amount of polymerisation during the reaction and yielded 52–54% of pyrroles 4a–c (Fig. 2).
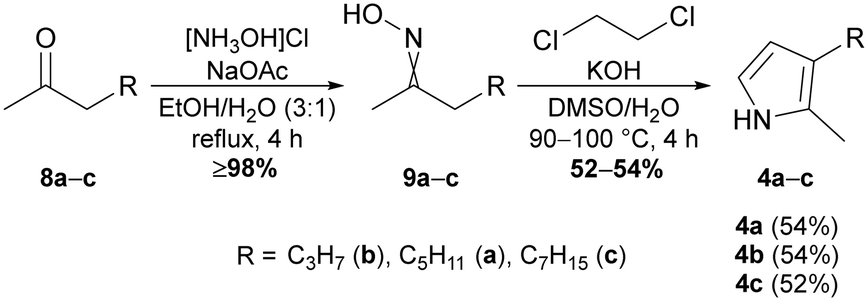 | ||
| Fig. 2 Synthesis scheme of 2,3-dialkylated 1H-pyrroles 4a–c from ketones 8a–c and intermediate oximes 9a–cvia Trofimov pyrrole synthesis. | ||
Apparently, the use of such synthetic schemes that enable both, use of the products as substrates for chemical synthesis of reference compounds and as potential substrates for ligating enzymes are inevitable for our approach. Based on the biomimetic two-step sequence devised by Dairi et al. for gaining access to the natural prodigiosin and tambjamine precursor 4-methoxy-2,2′-bipyrrole-5-carboxaldehyde (3a, MBC),65 we identified an adapted route to make 5′-n-alkylated derivatives of MBC accessible. Although other positions for MBC alkylation might be of interest as well, we focused on the 5′-position for this fundamental study. The devised synthetic route is predicated on the haloformylation of 4-methoxy-3-pyrrolin-2-one to give bromide 10, followed by a palladium-catalysed Suzuki–Miyaura cross-coupling of bromide 10 with 5-n-alkylated pyrrole-2-boronic acids. The fundament for the desired boronic acids were 2-alkylated N-Boc-protected 1H-pyrroles, which were afforded by a consecutive Friedel–Crafts acylation of 1H-pyrrole with acyl chlorides 11c–g under usage of aluminium trichloride as Lewis acid.66 Reduction of the 2-acylation products 12c–g with sodium borohydride in iPrOH and final N-protection of the 2-alkyl-pyrroles 13b–g with Boc-anhydride yielded the target compounds 14b–g (Fig. 3).1 In this reaction sequence, we were able to reduce the purification effort due to usage of crude reaction products and exclusive purification of the Boc-protected products (14b–g) by ordinary flash column chromatography. By this means, the N-Boc-2-alkylated pyrroles were synthesised in yields of 43–59%.
 | ||
| Fig. 3 Synthesis scheme of 2-n-alkylated N-Boc-pyrroles 14b–g. 2-Methyl-1H-pyrrole (13b) was purchased from commercial suppliers. Intermediates were not purified and the yield over three steps determined after final purification of Boc-protected 2-alkylated pyrroles. For further information regarding systematic nomenclature, please see ref. 67. | ||
Since boronic acids are more reactive than their related boronic acid pinacol or MIDA esters,68 we aimed for the synthesis of free pyrrole-2-boronic acids from N-Boc-2-alkylated pyrroles for the cross-coupling reaction with bromide 10. Admittedly, 2-substituted boronic acids of N-heterocycles, such as pyrroles or pyridines, are delicate and significantly lacking storage stability.69 Thus, utilisation of crude boronic acids from pyrrole borylation reactions was considered more reliable rather than isolation of pure boronic acids. Indeed, the crude products of the borylation sequence, which comprises deprotonation of pyrroles 14b–g with lithium tetramethylpiperidide, borylation with trimethyl borate and lastly deprotection of the methyl ester with aqueous hydrochloric acid were ideal starting materials. Catalysed by palladium acetate, SPhos-assisted Suzuki–Miyaura cross-coupling in degassed n-butanol/H2O (4:1) and with potassium phosphate as base allowed C–C coupling between pyrrole-2-boronic acids 15b–g and bromide 10 under mild reaction conditions (Fig. 4). The reactions typically gave mixtures of Boc-protected and deprotected carbaldehydes. Full deprotection of crude reaction was rendered by refluxing in 2,2,2-trifluoroethanol to provide the desired products after filtration. Resulting from this sequence, the 5′-n-alkylated MBC derivatives 3b–g were prepared in 52–80% yield (Fig. 4). Unsubstituted MBC 3a was synthesised according to the published literature procedure.65
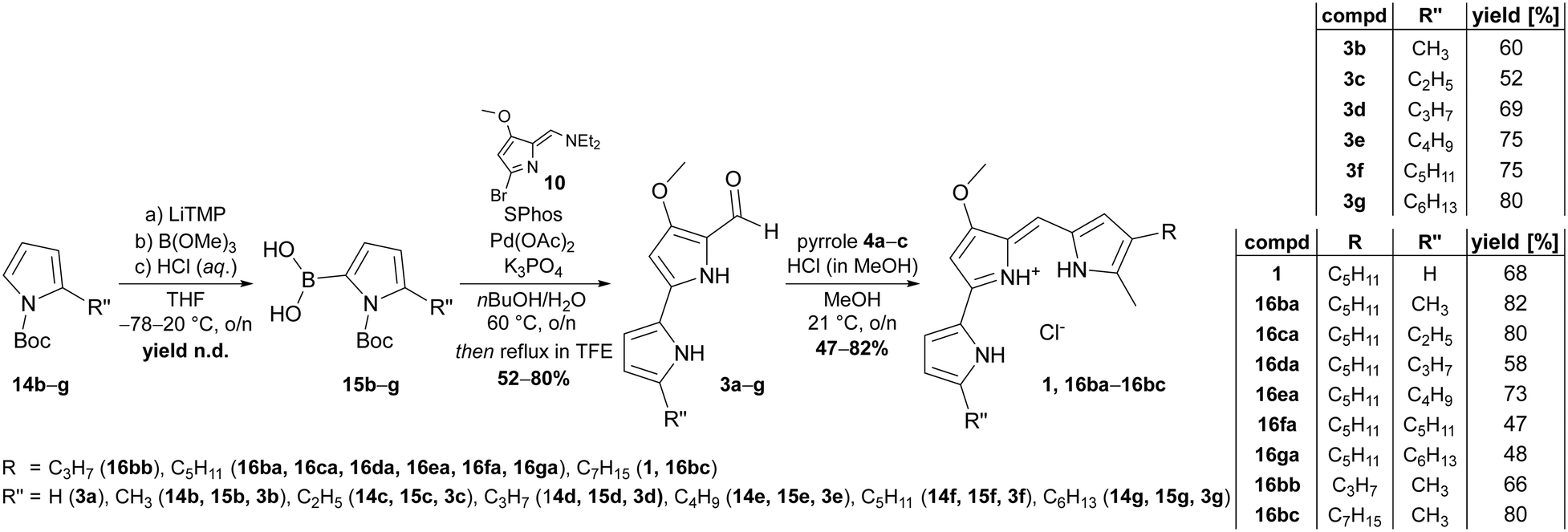 | ||
| Fig. 4 Synthesis scheme of n-alkylated A-ring derivatives of MBC (3b–g) and prodigiosin (16ba–bc) from N-Boc-pyrrole-2-boronic acid precursors (15b–g). Substituted prodiginines 16 were numbered in a modular fashion (16R′′R), including the substitution pattern of the MBC derivative (3a–g, R′′) and the monopyrrole (4a–c, R). For example, prodiginine 16ba was synthesised from MBC 3b and pyrrole 4a, prodiginine 16bb was synthesised from MBC 3b and pyrrole 4b. Abbreviations: n.d. – not determined; compd – compound; LiTMP – lithium 2,2,6,6-tetramethylpiperidide; SPhos – 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl; TFE – 2,2,2-trifluoroethanol. | ||
Synthesis of prodiginines
Although the alkylated MBC derivatives were primarily synthesised for systematic characterisation of the prodigiosin ligases substrate scope, they were also eligible for chemical prodiginine synthesis. Therefore, carbaldehydes 3a–g and pyrroles 4a–c were subjected to a condensation reaction under acid catalysis with hydrochloric acid to yield the corresponding prodiginines 16ba–bc in yields of 47–82% (Fig. 4). The hydrochlorides of prodigiosin (1) and novel prodiginines 16ba–bc generated thereby could be used as synthetic references for enzymatic condensation reactions, prodiginine quantification, toxicity screening, and were accessible in purities of up to 99.9%.Absorption spectra and molar extinction coefficients
Prodiginines are well known to display solvent-dependent absorption properties, which greatly vary with pH.70 To ensure that the molecule of interest is uniformly protonated, extinction coefficients and absorption spectra are typically recorded in acidified ethanol in which the purple pigment prodigiosin exhibits an absorption maximum at 535 nm.70,71 By adding a 5-n-alkyl substitution on the A-ring, the absorption maximum was increased to 545–547 nm, noticeable in a visible small shift from pink to purple in solution. With pentyl-substitution on the C-ring, variation in A-ring substitution greatly enhanced the molar extinction coefficient with increasing chain length, based on the inductive electron-donating effect of the attached alkyl substituents (Fig. S2A, Table S2, ESI†). For prodigiosin (1) a molar extinction coefficient of 139800 M−1 cm−1 (535 nm) was reported in the literature.71 The A-ring methyl-derivative (16ba), however, already presented an extinction coefficient of 163233 M−1 cm−1 (546 nm), followed by the ethyl- and propyl-derivative with 177092 M−1 cm−1 (16ca, 546 nm) and 181692 M−1 cm−1 (16da, 546 nm), respectively. From butyl- to hexyl-substitutions 16ea–ga, no further increase of significance was observed and the molar extinction coefficients seemed to reach a plateau around 190000 M−1 cm−1 at 547 nm (Fig. S2A, ESI†). Interestingly, molar extinction coefficients at 535 nm ranged from 126508 M−1 cm−1 to 138200 M−1 cm−1, showing less chain length-dependent behaviour and solely fluctuations in the magnitude of standard deviations (Fig. S2B, ESI†). With constant methyl-substitution on the A-ring and variation in C-ring chain length, a linear correlation was observable at 535 nm for prodiginines 16ba, 16bb, and 16bc allowing approximation of molar extinction coefficients (Table S3, ESI†) based on the experimental coefficients and the slope of linear regression (Fig. S2C, ESI†). In this way, we were able to quantify even prodiginines whose references had not been synthesised by chemical means.
Prodigiosin ligase substrate scope for A-ring substituted MBC
Chawrai et al. showed that prodigiosin derivatives with alternative aromatic A-rings are accepted by the ligase PigC from S. marcescens. Enzymatic activity was still present when the A-ring pyrrole moiety was either substituted by 2-phenyl-, 2-thienyl-, 2-furyl-, or 2-indolyl-residues. No activity was seen for napthyl- and biphenyl-substitution instead.46 Their findings prompted us to investigate alkyl-substituted pyrrole derivatives more thoroughly, as they were not characterised as suited substrates for PigC and, moreover, with TreaP and TamQ two new ligating enzymes from Pseudoalteromonadaceae were found since then.59In our experiment, we tested the three ligases PigC, TreaP, and TamQ for their substrate acceptance of A-ring substituted alkyl-derivatives 3b–g of MBC and MBC 3a itself and quantified the extracted prodiginines in LC-MS measurements with the corresponding extinction coefficients, which had been determined experimentally or by approximation (vide supra). As we used Escherichia coli cell lysate after expression of the corresponding genes rather than purified enzymes, E. coli harbouring the pET28a(+) empty vector was used as control. Unusual for this experimental setup in comparison to earlier studies on prodiginine ligases was the two-dimensional testing, varying MBC derivatives and monopyrroles at the same time (Fig. 5). We would like to emphasise that quantification was normalised to the amount of cells and not to the amount of enzyme being used in the experiment. Thus, differences in expression efficiencies for the analysed ligases might have an impact on absolute prodiginine titres but should not affect the relative acceptance of A-ring substituted MBC derivatives for each enzyme (Fig. 6A). Firstly, we validated whether prodiginines are readily formed in a catalyst-free environment, containing pyrroles, MBC, ATP, and buffer. It was found that no background reaction takes place (control reactions 1–6, ESI,† Fig. S111–S116) and that the ligating enzymes are needed for catalysis of the condensation reaction. Secondly, we performed reactions with enzyme containing cell lysates and ATP in buffer but without pyrroles and MBC to exclude formation of prodiginines from cell metabolites within the lysate. Again, no prodiginines were traced by coupled LC-MS measurements, proving that all four components (ligating enzyme, MBC, pyrrole, and ATP) are needed for their formation (negative controls, ESI,† Fig. S107–S110).
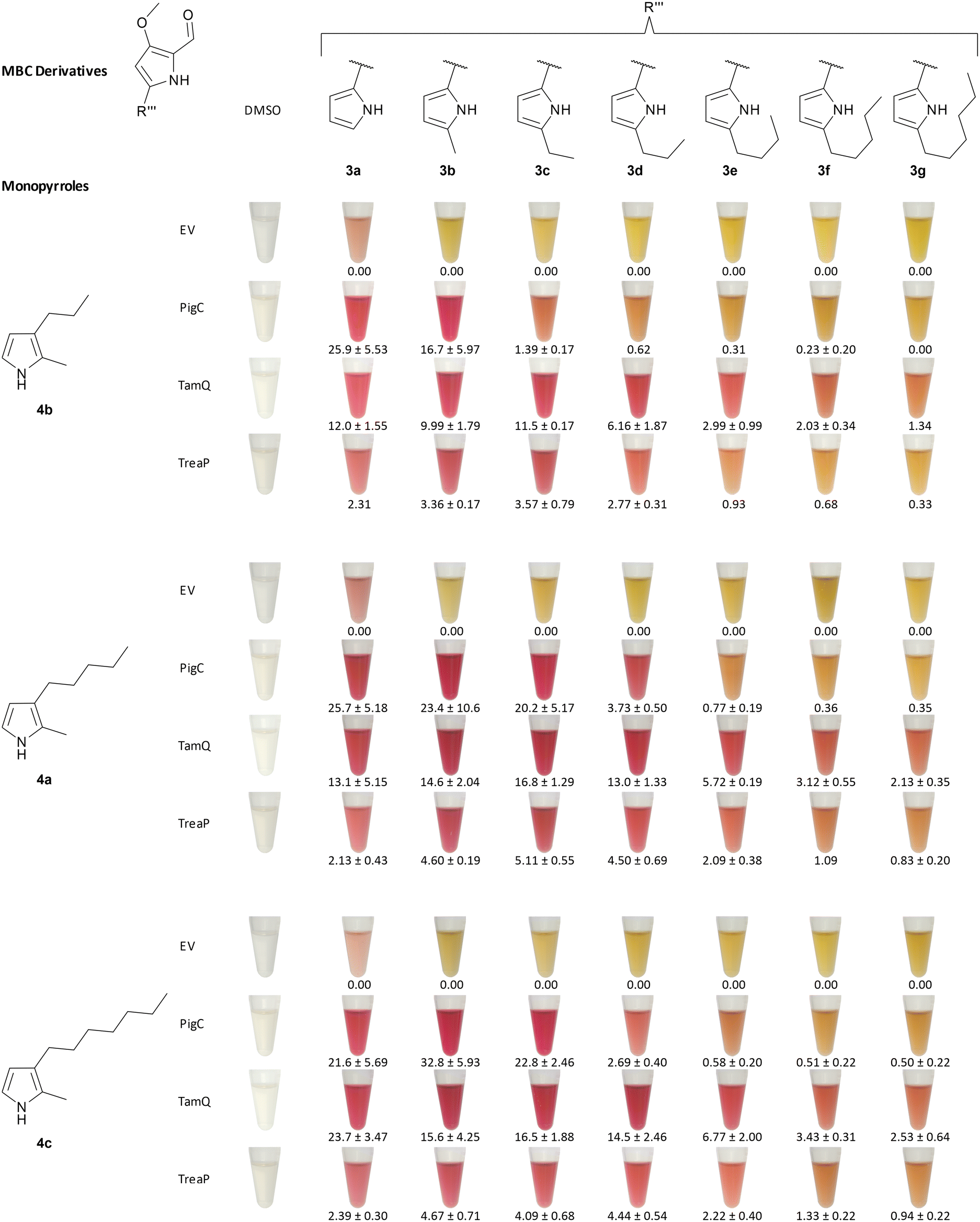 | ||
| Fig. 5 Substrate spectrum of the prodiginine ligases PigC, TamQ, and TreaP for combinations of monopyrroles 4a–c, MBC 3a and 5-n-alkylated MBC derivatives 3b–g. Cell lysates of E. coli BL21 with corresponding ligases in KPi buffer were treated with pyrroles (1 mM), MBC (1 mM), and ATP (1.25 mM) and incubated at 30 °C for 4 h. Cell lysate of E. coli BL21 pET28a(+) was used as negative control. A red colouration of the solutions indicates the formation of products. Photographic record was employed to document the organic extracts in acidic ethanol. Product titres (mg L−1) were determined by LC-MS measurements and are given below the corresponding tubes. Abbreviations: EV – empty vector. | ||
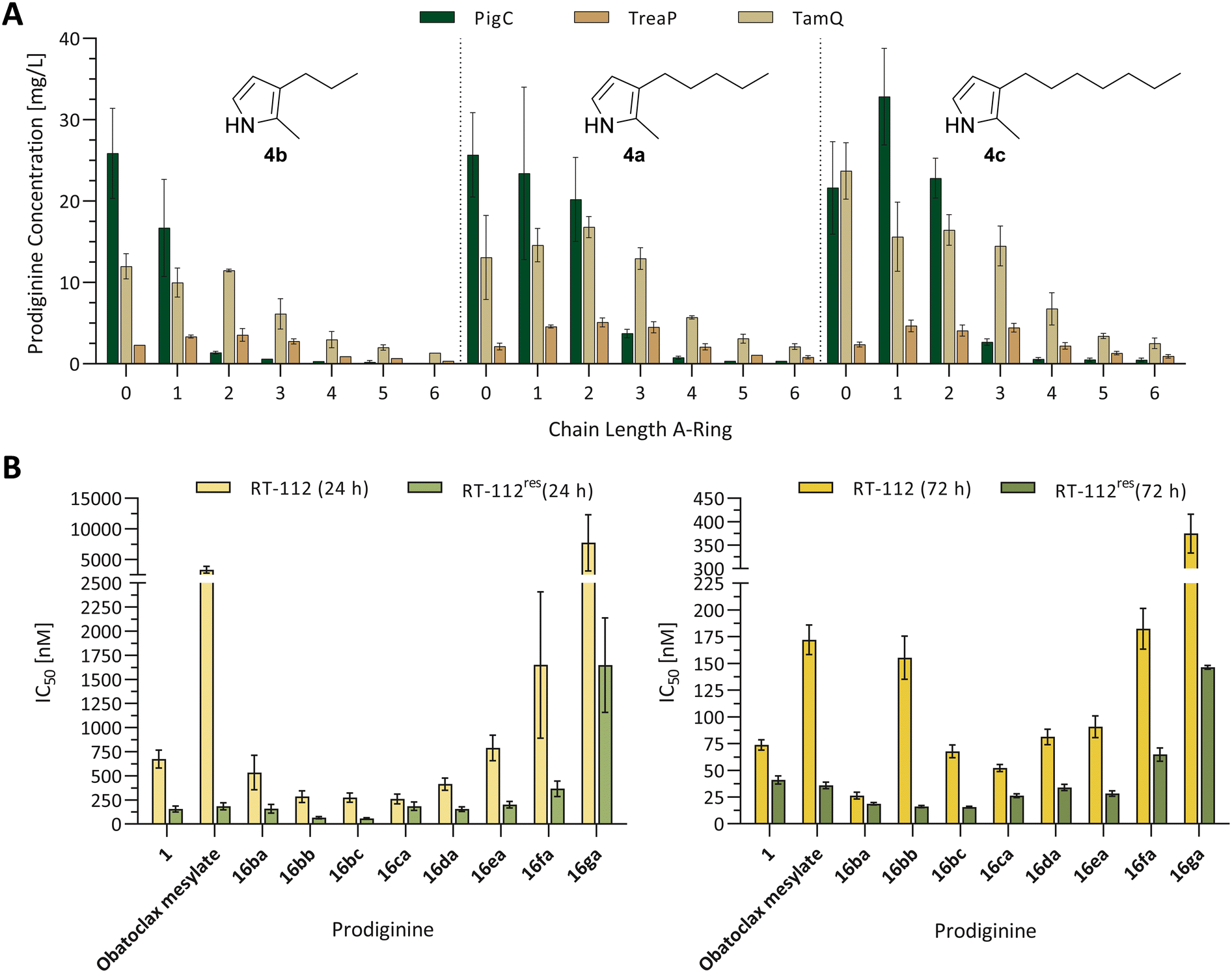 | ||
| Fig. 6 (A) Quantification of prodiginines from in vitro reactions with the prodiginine ligases PigC, TamQ, and TreaP with varying chain lengths on the A- and C-ring. Methanolic extracts of biocatalytic reactions between monopyrroles 4a–c and 5-alkyl MBC derivatives 3a–g were subjected to coupled LC-MS measurements and the amount of substance quantified from the corresponding UV absorption at 535 nm and the prodiginine extinction coefficients. The chain length of alkylated MBC is depicted on the x-axis of the bar diagram, the used monopyrrole is shown in the diagram. Since all values for the empty vector control were zero, the data is not shown, but plotted raw data can be found in the supporting information (Fig. S3†). Each experiment was performed in triplicates and the standard error is given. (B) Cell viability of urothelial cancer cell lines RT-112 (cisplatin sensitive) and RT-112res (cisplatin resistant) in the presence of prodigiosin (1), obatoclax mesylate, and chemically synthesised prodiginines (16ba–ga) after 24 h (left) and 72 h (right). For improved reading and resolution of data sets, the scales of ordinates for the 24 h and 72 h diagrams were adjusted. Experiments were performed in triplicates and cell survival was analysed using the MTT assay. Error bars are representing the 95% confidence interval. Plotted raw data and dose–response fits for determination of IC50 values can be found in the supporting information (plots and fits: Fig. S117–S119, fitted IC50 values: Table S4†). | ||
During the first experiments, the most prominent enzyme PigC showed high acceptance of methyl- and unsubstituted MBC (3b and 3a) in combination with the short-chain monopyrrole 4b, whereat the natural precursor MBC 3a turned out to be the favoured bipyrrole, yielding 25.9 mg L−1versus 16.7 mg L−1 of prodiginine extracts for methyl-MBC 3b. Other MBC derivatives were not converted to the related prodiginines in noteworthy amounts. In contrast, TamQ, which is naturally involved in tambjamine biosynthesis, apparently converted all derivatives including butyl-MBC 3e, but not pentyl- and hexyl-MBC 3f and 3g. Here, the obtained product titres of prodiginines derived from short-chain MBC derivatives 3a, 3b, and 3c were rather similar in a range from 10–12 mg L−1. The acceptance of MBC derivatives for TreaP was limited to C0–C3-substituted MBC 3a–d and resulted in considerably lower titres (<3.6 mg L−1) than PigC or TamQ. However, TreaP showed highest relative product concentrations for the ethyl-substituted MBC 3c. With increasing chain length of the monopyrrole all enzymes appeared to possess greater substrate promiscuity and conversions. For both, the medium- and long-chain monopyrroles 4a and 4c, the MBC substrate spectrum for PigC from S. marcescens was expanded to propyl-MBC 3d but also to higher prodiginine titres. Again, unsubstituted MBC 3a was apparently converted best, but only for the medium-chain monopyrrole 4a at 25.7 mg L−1. With the long-chain monopyrrole 4c, the non-natural methyl-MBC derivative 3b was identified as ideal partner for condensation, providing the related product in a titre of 32.8 mg L−1. Supported by previous findings regarding the enzymes TamQ and TreaP,59 either preferentially converted long-chain monopyrroles to the corresponding prodiginines, at which TamQ in total exhibited the broadest acceptance of 5-alkyl-substituted MBC derivatives. Even though MBC 3a is the common prodiginine and tambjamine precursor in nature for all three enzymes under investigation, TreaP and TamQ shared a similar trend of MBC derivative promiscuity for the medium-chain monopyrrole 4a, showing highest product titres for ethyl-MBC 3c with 5.1 mg L−1 and 16.8 mg L−1, respectively. The long-chain monopyrrole 4c was best accepted in combination with methyl-MBC 3b by PigC, providing the highest prodiginine product titre of 32.8 mg L−1 in our experiment.
From the contingent of tested enzymes, PigC was in fact the least versatile enzyme, demonstrated by tight restraints in terms of tolerating alkyl-substitution on the MBC part. In contrast, TamQ was identified as most profitable catalyst for substitutions on the monopyrrole and the A-ring of MBC. It is hypothesised that multiple reasons contribute to the observed acceptance pattern of combinations for monopyrroles and MBC. As the monopyrrole serves as nucleophile in the proposed mechanism (devised for PigC), reviewed by Hu et al.,47 it is believed that shorter chain length may result in a higher degree of freedoms and flexibility of side chains, less tight binding of the pyrrole core within the enzyme pocket, potentially leading to decreased or depleted reaction rates. In addition, short chain pyrroles are lacking additional activation, which is contributed by the electron-donating effect of alkyl-chains. Significance of the latter can be seen from the drastic effect of increasing chain-length on prodiginine extinction coefficients. For the alkyl-chain variation on MBC derivatives, we assume steric reasons as principal force behind the lack of conversion with increased number of methylene groups within the alkyl chain. Electronic contributions are judged rather implausible, as the condensation chemistry between monopyrrole and MBC is taking place at the distal part of the MBC derivatives, namely the carbaldehyde.
Evaluation of cytotoxicity for A-ring substituted prodiginines
To assess the impact of 5-n-alkyl substitutions on cytotoxic properties of chemically synthesised prodiginines, the urothelial carcinoma cell lines RT-112 (cisplatin sensitive) and RT-112res (cisplatin resistant) were selected for evaluation.72–74 Cisplatin-based therapy is the standard of care for this kind of carcinomas, however, miscellaneous mechanisms are involved in emerging resistance against cisplatin, such as autophagy and apoptosis.72,75–78 Prodigiosin (1) has been shown to display cytotoxic activity at nanomolar concentrations against both, cisplatin sensitive and resistant cell lines, and was thus judged as potential drug candidate for treatment of cisplatin resistant urothelial carcinomas.30 Thus, prodigiosin (1) and the former phase II clinical candidate obatoclax mesylate were selected as benchmark compounds.12,13,79,80 In order to assort the impact of A-ring associated alkyl substitutions on prodiginine derivatives, the two RT-112 cell lines were subjected to cell viability monitoring after 24 h and 72 h, using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (Fig. 6B, data and dose–response fits in the supporting information). For the lead compound prodigiosin (1), inhibitory concentrations (IC50) of 675 nM and 157 nM were traced after 24 h and were further reduced to 73.8 nM and 41.1 nM after 72 h for RT-112 and RT-112res, respectively. In contrast, obatoclax mesylate exhibited relatively low cytotoxic activity in RT-112 cells after 24 h of incubation. With 3327 nM against RT-112 and 184 nM against RT-112res after 24 h, obatoclax was acting similarly to the least cytotoxic long-chain derivatives 16ea–ga. With increased incubation time of 72 h, improved cytotoxicity of 172 nM (RT-112) and 36.0 nM (RT-112res) were achieved, judging obatoclax significantly less effective against RT-112 than prodigiosin (1) and of similar value against RT-112res.In the following, prodiginines with altered A-ring alkyl substitution, but constant C-ring substitution of prodigiosin (1) that originated from chemical synthesis were investigated for their cytotoxic effects. Simple addition of an electron donating methyl-group in prodiginine 16ba resulted in a strong increase of cytotoxicity against both cell lines. With a final IC50 of 26.4 nM for RT-112 and 18.8 nM for RT-112res after 72 h, an up to 2.8-fold increase in cytotoxicity was observed. However, IC50 values after 24 h initially resembled the cytotoxicity of prodigiosin (1) itself. Interestingly, further chain elongation on the A-ring of prodiginines 16ca–ga led to constant loss of biological activity with each step of elongation. Especially the transition from pentyl to hexyl substitution on the A-ring displayed a turning point in cytotoxicity and was accompanied by a significant loss of biological activity, independent from the cell line and time point of analysis. Aside from the effect of A-ring substitution with invariable C-ring substitution pattern of compounds 16ba–ga, we chose the most cytotoxic methyl-substituted prodiginine 16ba as starting point and scrutinised which effect a substitution in 4-position of the C-ring chain toward longer or shorter alkyl residues might have. All analysed prodiginines (16ba, 16bb, and 16bc) with methyl substitution on the A-ring appeared to have unspoiled high cytotoxicity against the cisplatin resistant subtype RT-112res after 72 h when the C-ring was modulated. With IC50 values ranging between 15.7 nM (16bc) and 18.8 nM (16ba), the impact was rather negligible. Nonetheless, the tested derivatives were shown to act differently after 24 h, as prodiginine 16ba with C5-chain on the C-ring revealed slightly higher inhibitory concentrations against both cell lines than observed for derivatives 16bb and 16bc. Summarising the presented data from Fig. 6B, an additional methyl group in 5-position of the A-ring boosted the cytotoxicity against both, cisplatin sensitive and resistant urothelial carcinoma cell lines. In addition to this increased performance, we found prodiginine 16ba to display the highest cytotoxicity against both cell lines after 72 h (RT-112: 26.4 nM, RT-112res: 18.8 nM). Concurrently, prodiginines 16bb and 16bc were outstandingly fast in unleashing their cytotoxic potential, having substantial improvements of toxicity after 24 h in comparison to derivative 16ba, the mother compound prodigiosin (1) and the former phase II clinical candidate obatoclax mesylate. The tripyrrole core structure of prodigiosin (1) and its substituted clinical relative obatoclax is well known to inhibit the cellular process of autophagy,30,57,81,82 which is essential to recycle or degrade cell organelles or anomalous proteins (misfolded, aggregated, or damaged).30 At the same time, prodigiosin (1) was shown to induce apoptosis, an ensemble of signalling pathways of programmed cell death.33,34,83 In the past, the mechanistic cause for autophagy inhibition and induction of apoptosis in prodigiosin-treated cancer cell lines was accounted to the capability of H+/Cl− symport across the membranes of lysosomes or other eukaryotic cell compartments and the cell cycle arrest.18,20,21,84
The ion pair of protonated prodigiosin and the chloride counter ion is lipophilic and tight enough to penetrate the membrane and diffuse across the permeability barrier.11 A possible explanation for the increased cytotoxicity of prodiginines with short-chain electron-donating alkyl substituents on the A-ring could be a tighter bond between the B-ring nitrogen atom and the proton from the hydrochloride complex, increasing the pKa value and facilitating the trespass of the charged complex through the membrane. A further increase of the A-ring chain length could potentially result in a more lipophilic character for the resulting prodiginine, raising the risk of functioning as membrane anchor and attaching derivatives to the membrane rather than diffusing across, thereby reducing the cytotoxic potential. In fact, our results regarding A-ring substitution are in agreement with earlier studies on alkyl substitution on the C-ring, where short-chain propyl substitution was characterised by drastically stronger autophagy inhibition than for the long-chain octyl variant.57
Conclusions
Prodiginine ligating enzymes are known to exhibit substrate promiscuity on C-ring monopyrroles and MBC derivatives with aromatic A-ring substitution. However, reliable structural information for these enzymes in free or substrate-bound state are still lacking. Whilst enzyme structures remain unclear, probing of the active site and testing of suitable substrate combinations is limited to means of trial and error. We were able to transfer the previously gained knowledge of enzyme promiscuity in a systematic screening on 5′-n-alkylated MBC derivatives, showing broad substrate acceptance for both key precursors of the convergent biosynthesis route. While PigC only accepted short-chained MBC derivatives, TamQ and TreaP displayed fewer restraints, also for longer alkyl chains. During cell viability screening of A-ring substituted prodiginines in cisplatin-sensitive and -resistant urothelial carcinoma cell lines, methyl-substituted derivatives 16ba, 16bb, and 16bc were cytotoxic at low nanomolar concentrations, and thereby up to 2.6-times more potent than prodigiosin (1) or obatoclax mesylate against the cisplatin-resistant cell line RT-112res after 72 h. Consequently, these derivatives are suggested as potential lead compounds for future structural optimisation in terms of cytotoxicity, drug administration and drug metabolism and could be potential candidates for treatment of cisplatin resistant carcinomas.Experimental
Synthetic procedures
:1. The reaction vessel is lowered into a pre-heated 60 °C bath and the orange solution is stirred under argon overnight. The solvent is evaporated, 50 mL of water are added to the slurry and the mixture is then extracted with EtOAc (3 × 50 mL). Combined organic phases are washed with brine (2 × 25 mL), dried over MgSO4, filtered over degreased cotton wool and the solvent is removed. The resulting solid is recovered by filtration and washed with cold distilled n-pentane. The green solid is refluxed for 6 h at 90 °C in 45 eq. 2,2,2-trifluoroethanol. Evaporation of the solvent and trituration of the remaining solid in n-pentane provides the product as green solid that is recovered by filtration and washed with distilled n-pentane and Et2O.
δ1H (300 MHz, DMSO) 1.19 (3 H, q, 7′-H), 2.53–2.65 (2 H, m, 6′-H), 3.67–3.92 (3 H, m, 10-H), 5.66–5.97 (1 H, m, 4′-H), 6.08–6.35 (1 H, m, 3-H), 6.49–6.78 (1 H, m, 3′-H), 9.26 (1 H, d, 7-H), 10.95 (1 H, s, 1′-NH), 11.27 (1 H, s, 1-NH). δ13C (76 MHz, DMSO) 13.7 (C-7′), 20.4 (C-6′), 57.7 (C-10), 90.4 (C-3), 106.2 (C-4′), 108.7 (C-3′), 117.2 (C-5), 122.0 (C-2′), 133.6 (C-2), 136.6 (C-5′), 158.8 (C-4), 171.1 (C-6). FT-IR (neat, cm−1): 3250, 3197, 2972, 2833, 1593, 1553, 1513, 1445, 1422, 1357, 1302, 1284, 1248, 1199, 1175, 1161, 1147, 1039, 1014, 990, 832, 766, 740, 698, 588, 484. Tm: 243.7–245.5 °C (n-pentane). HRMS-ESI (m/z): [M + H]+ calculated for C12H15N2O2 219.1128, found 219.1132. tR (LC-MS method): 7.93 min.
:10) + 1% triethylamine. δ1H (600 MHz, CDCl3) 0.90 (3 H, t, J = 6.7 Hz, 5′′-H), 1.28–1.38 (4 H, m, 3′′-H, 4′′-H), 1.54 (2 H, p, J = 7.4 Hz, 2′′-H), 2.19 (3 H, s, 1′-H), 2.39 (2 H, t, J = 7.7 Hz, 1′′-H), 6.02 (1 H, t, J = 2.8 Hz, 4-H), 6.60 (1 H, t, J = 2.7 Hz, 5-H), 7.70 (1 H, brs, 1-NH). δ13C (151 MHz, CDCl3) 11.2 (C-1′), 14.3 (C-5′′), 22.8 (C-4′′), 26.0 (C-1′′), 31.2 (C-2′′), 31.9 (C-3′′), 109.0 (C-4), 114.9 (C-5), 119.9 (C-3), 123.3 (C-2). FT-IR (neat, cm−1): 3376, 2956, 2923, 2854, 1464, 1444, 1378, 1246, 1107, 1064, 954, 901, 831, 771, 670, 551. HRMS-ESI (m/z): [M + H]+ calculated for C10H18N 152.1434, found 152.1434. tR (LC-MS method): 8.52 min.
:15) + 1% triethylamine. δ1H (600 MHz, CDCl3) 0.95 (3 H, t, J = 7.3 Hz, 3′′-H), 1.56 (2 H, h, J = 7.5 Hz, 2′′-H), 2.19 (3 H, s, 1′-H), 2.37 (2 H, t, J = 7.6 Hz, 1′′-H), 6.02 (1 H, t, J = 2.7 Hz, 4-H), 6.60 (1 H, t, J = 2.7 Hz, 5-H), 7.70 (1 H, brs, 1-NH). δ13C (151 MHz, CDCl3) 11.2 (C-1′), 14.2 (C-3′′), 24.6 (C-2′′), 28.2 (C-1′′), 109.0 (C-4), 114.9 (C-5), 119.7 (C-3), 123.4 (C-2). FT-IR (neat, cm−1): 3378, 2956, 2925, 2869, 1464, 1455, 1376, 1249, 1106, 1066, 955, 904, 889, 832, 801, 712, 663, 549. HRMS-ESI (m/z): [M + H]+ calculated for C8H14N 124.1121, found 124.1120. tR (LC-MS method): 7.90 min.
:15) + 1% triethylamine. δ1H (600 MHz, CDCl3) 0.89 (3 H, t, 7′′-H), 1.23–1.38 (8 H, m, 3′′-H, 4′′-H, 5′′-H, 6′′-H), 1.48–1.58 (2 H, m, 2′′-H), 2.19 (3 H, s, 1′-H), 2.39 (2 H, t, 1′′-H), 6.02 (1 H, t, J = 2.8 Hz, 4-H), 6.60 (1 H, t, J = 2.7 Hz, 5-H), 7.71 (1 H, brs, 1-NH). δ13C (151 MHz, CDCl3) 11.2 (C-1′), 14.3 (C-7′′), 22.8 (C-6′′), 26.1 (C-1′′), 29.4 (C-3′′), 29.7 (C-4′′), 31.5 (C-2′′), 32.1 (C-5′′), 109.0 (C-4), 114.9 (C-5), 119.9 (C-3), 123.3 (C-2). FT-IR (neat, cm−1): 3485, 3377, 2955, 2923, 2871, 1583, 1463, 1445, 1377, 1272, 1247, 1144, 1108, 1064, 953, 901, 831, 711, 668, 646, 634, 548, 485, 468. HRMS-ESI (m/z): [M + H]+ calculated for C12H22N 180.1747, found 180.1749. tR (LC-MS method): 9.01 min.
:1). The reaction mixture is then heated for 4 h to reflux. The mixture is cooled to room temperature and excess of EtOH is removed under reduced pressure. Water (100 mL) is added to the crude mixture and the resulting aqueous solution is extracted with EtOAc (3 × 150 mL). The combined organic layer is washed with saturated NaHCO3 (2 × 100 mL) and water (2 × 100 mL), dried over MgSO4, filtered over degreased cotton wool, and concentrated in vacuo. The oxime products are obtained in quantitative yields as volatile oils and used without further purification.
:27 (NMR). δ1H (600 MHz, CDCl3, E-isomer) 0.90 (3 H, t, J = 7.4 Hz, 6-H), 1.32 (2 H, h, J = 7.3 Hz, 5-H), 1.44–1.52 (2 H, m, 4-H), 1.87 (3 H, s, 1-H), 2.15–2.23 (2 H, m, 3-H), 9.09 (1 H, brs, 8-H). δ13C (151 MHz, CDCl3, E-isomer) 13.5 (C-1), 13.9 (C-6), 22.4 (C-5), 28.5 (C-4), 35.6 (C-3), 158.8 (C-2). δ1H (600 MHz, CDCl3, Z-isomer) 0.92 (3 H, t, J = 7.30 Hz, 6′-H), 1.36 (2 H, h, J = 7.4 Hz, 5′-H), 1.44–1.52 (2 H, m, 4′-H), 1.85 (3 H, s, 1′-H), 2.34–2.40 (2 H, m, 3′-H), 9.09 (1 H, brs, 8′-H). δ13C (151 MHz, CDCl3, Z-isomer) 14.0 (C-6′), 20.0 (C-1′), 23.0 (C-5′), 27.8 (C-4′), 28.5 (C-3′), 159.2 (C-2′). FT-IR (neat, cm−1): 3222, 3118, 2957, 2928, 2873, 2862, 1664, 1467, 1433, 1368, 1331, 1262, 1207, 1105, 1010, 951, 919, 878, 823, 742, 648, 613. HRMS-ESI (m/z): [M + H]+ calculated for C6H14NO 116.1070, found 116.1071.
:24 (NMR). δ1H (600 MHz, CDCl3, E-isomer) 0.871 (3 H, t, 10-H), 1.19–1.35 (10 H, m, 5-H, 6-H, 7-H, 8-H, 9-H), 1.49 (2 H, p, 4-H), 1.88 (3 H, s, 1-H), 2.14–2.20 (2 H, m, 3-H), 8.14 (1 H, brs, 12-H). δ13C (151 MHz, CDCl3, E-isomer) 159.0 (C-2), 35.9 (C-3), 32.0 (C-8), 29.5 (C-6), 29.33 (C-5), 29.32 (C-7), 26.4 (C-4), 22.8 (C-9), 14.2 (C-10), 13.5 (C-1). δ1H (600 MHz, CDCl3, Z-isomer) 0.874 (3 H, t, 10′-H), 1.19–1.35 (10 H, m, 5′-H, 6′-H, 7′-H, 8′-H, 9′-H), 1.49 (2 H, p, 4′-H), 1.86 (3 H, s, 1′-H), 2.33–2.38 (2 H, m, 3′-H), 8.14 (1 H, brs, 12′-H). δ13C (151 MHz, CDCl3, Z-isomer) 159.4 (C-2′), 32.0 (C-8′), 29.9 (C-6′), 29.5 (C-5′), 29.32 (C-7′), 28.7 (C-3′), 25.6 (C-4′), 22.8 (C-9′), 19.9 (C-1′), 14.2 (C-10′). FT-IR (neat, cm−1): 3223, 3117, 2954, 2924, 2855, 1664, 1464, 1458, 1377, 1368, 1258, 1217, 1168, 1113, 994, 956, 943, 830, 823, 724, 651, 614. HRMS-ESI (m/z): [M + H]+ calculated for C10H22NO 172.1696, found 172.1699.
Acylation of 1H-pyrrole. The 2-acylation of 1H-pyrrole was performed according to the protocol of Ono et al.66 In a Schlenk flask under N2 atmosphere the acid chloride 11c–g (1.00 eq.) is dissolved in dry CH2Cl2 (3.74 mL mmol−1 acid chloride) and cooled to 0 °C. Aluminium trichloride (1.20 eq.) is added slowly to give a yellow suspension and the mixture is allowed to warm to 24 °C over 30 min. The solution is cooled to 0 °C again and a solution of 1H-pyrrole (1.10 eq.) in dry CH2Cl2 (0.42 mL mmol−1 acid chloride) is transferred slowly to the acid chloride solution. The reaction is stirred for further 1 h at 0 °C, then slowly thawed to 21 °C and stirred overnight. A saturated solution of NH4Cl is used at 0 °C for reaction quenching. The phases are separated, the aqueous phase subsequently extracted with CH2Cl2 (3 × 150 mL) and the combined organic phases washed with saturated NaHCO3 (2 × 150 mL) and brine (1 × 150 mL). Drying over MgSO4, filtration over degreased cotton wool and solvent removal in vacuo provides the crude acylation product 12c–g as brown oil, which is used without further purification and contains mostly 2-acylated 1H-pyrrole.
Reduction of acylated 1H-pyrrole. The reduction of 2-acylated 1H-pyrroles to 2-alkyl-1H-pyrroles was performed according to the protocol of Fürstner et al.1 Under N2 atmosphere sodium borohydride (2.80 eq.) is suspended in dry isopropyl alcohol (0.90 mL mmol−1 acylated pyrrole) and cooled to 0 °C. The crude acylated pyrrole 12c–g (1.00 eq.) is dissolved in dry isopropyl alcohol (3.15 mL mmol−1 acylated pyrrole), precooled to 0 °C and added to the sodium borohydride suspension at 0 °C. Afterwards, the solution is refluxed overnight at 99 °C. The reaction is quenched with water at 0 °C, additional water (300 mL) is added and the product extracted with MTBE (3 × 200 mL). Merged organic phases are washed with brine (2 × 200 mL), dried over MgSO4, and filtered over degreased cotton wool. After solvent removal, the oily brown crude product, which contains mostly 2-alkylated 1H-pyrrole 13c–g, is used without further purification.
Boc-protection of alkylated 1H-pyrrole. Boc-protection of 2-alkyl-1H-pyrroles was realised according to the protocol of Fürstner et al.1 In a Schlenk flask under N2 atmosphere crude alkylated 1H-pyrrole 13c–g or commercially available 2-methyl-1H-pyrrole (13b) (1.00 eq.) and DMAP (0.10 eq.) are dissolved in dry CH2Cl2 (1.42 mL mmol−1 alkylated pyrrole). A solution of Boc2O (1.20 eq.) in dry CH2Cl2 (2.37 mL mmol−1 alkylated pyrrole) is added to the prior solution at ambient temperature and the reaction is stirred overnight. The solvent is removed under reduced pressure and the residue purified by chromatography on silica using n-pentane/MTBE (99
:1). The Boc-protected 2-alkylated 1H-pyrroles 14b–g are afforded as colourless to yellow liquids.
:2), yielding 3.88 g (21.39 mmol, 80%) of a colourless liquid. δ1H (600 MHz, CDCl3) 1.59 (9 H, s, 10-H, 11-H, 12-H), 2.43 (3 H, d, J = 1.1 Hz, 1′-H), 5.92 (1 H, tt, J = 1.9 Hz, J = 1.0 Hz, 3-H), 6.06 (1 H, t, J = 3.3 Hz, 4-H), 7.18 (1 H, dd, J = 3.4 Hz, J = 1.8 Hz, 5). δ13C (151 MHz, CDCl3) 15.6 (C-1′), 28.2 (C-10, C-11, C-12), 83.4 (C-9), 110.0 (C-4), 111.9 (C-3), 120.7 (C-5), 131.7 (C-2), 149.9 (C-6). FT-IR (neat, cm−1): 2979, 2928, 1737, 1497, 1396, 1370, 1329, 1308, 1254, 1237, 1170, 1127, 1065, 983, 883, 851, 798, 772, 715, 676, 597, 552. HRMS-ESI (m/z): [M + H]+ calculated for C10H16NO2 182.1176, found 182.1176.
NMR data of crude boronic acids
:40) and 4% 7 N NH3 in MeOH to remove unconverted MBC precursor. Evaporation of the solvent and acidification with 1 M HCl in Et2O yields a purple film that is precipitated by repeated addition of petroleum ether. A deep purple amorphous solid is obtained.
Determination of molar extinction coefficients
Prodiginines were weighed in and dissolved in acidic EtOH (+ 4% 1 M HCl) to give a 10 mM solution. By means of serial 1:10 dilutions, the stock was diluted to 10 μM in acidic EtOH. Firstly, the absorption spectra of 10 μM solutions was collected on a Shimadzu UV-1800 UV Spectrophotometer, using the following instrument settings: 20 °C, 200–800 nm, 1.0 nm increment, Hellma Analytics quartz glass cuvette SUPRASIL QS (d = 10 mm light path length).
Secondly, from the diluted 10 μM stock, three concentrations were prepared in acidic EtOH, namely 1 μM, 3 μM and, 5 μM. The extinction was measured at 20 °C for each concentration at 535 nm, 545 nm and at the wavelength of maximum absorption, if the wavelength varied from the two aforementioned and the solvent background was subtracted on the instrument. The procedure was repeated three times for each compound. Resulting from the Beer–Lambert law (E = ε·c·d), the molar extinction E was plotted against the molar concentration c and the molar extinction coefficient ε determined from the slope of a linear regression curve using Origin 2019 (ESI†).
Biologic procedures
Chemically competent cells of E. coli BL21 (DE3) were transformed with of pET28a(+)-derived plasmids using the heat-shock protocol. In detail, 50 μL of cells were thawed on ice for 10 min and 1 μL of plasmid DNA was added. Incubation on ice for 30 min was followed by a heat shock at 42 °C for 30 s. The cells were again incubated on ice for 10 min and 700 μL lysogeny broth (LB) medium was added to allow cell proliferation at 37 °C for 1.5 h. Finally, the cells were plated on LB agar, containing 50 μg mL−1 kanamycin, and grown overnight at 37 °C.Protein production of prodiginine ligating enzymes
Single colonies of transformed E. coli BL21 (DE3) with pET28a(+) (empty vector, EV), pPigC_3, pET28a(+)::tamQ or pET28a(+)::treaP were used to inoculate 50 mL of LB media (50 μg mL−1 kanamycin) in a 250 mL Erlenmeyer flask and incubated overnight at 37 °C and 130 RPM. The precultures were used to inoculate 2 × 1 L of terrific broth (TB) media (50 μg mL−1 kanamycin) in 3 L baffled flasks to an optical density at 600 nm (OD600) of 0.05 and then cultivated at 37 °C and 130 RPM. At an OD600 of 0.6–0.9, the cultures were cooled at room temperature for 15 min and then induced by addition of 100 μM isopropyl β-D-1-thiogalactopyranoside (IPTG). The cultures were then transferred to 25 °C and incubated overnight at 130 RPM. Cells were harvested by centrifugation (15 min, 4.500 RPM, 4 °C), the supernatant discarded and dry pellets stored at −20 °C until further use.Cell lines and cell culture
RT-112 and RT-112res cells (kindly provided by Margaretha A. Skowron, Michèle J. Hoffmann, and Günter Niegisch; Department of Urology, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University Düsseldorf) were cultured in Dulbecco's modified Eagle medium (DMEM, Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS, Sigma-Aldrich), 4.5 g L−1D-glucose, 100 units mL−1 penicillin and 100 μg mL−1 streptomycin (Thermo Fisher Scientific). The cells were cultivated and treated at 37 °C and 5% CO2 in a humidified atmosphere. RT-112res cells have been previously described.73 Briefly, for the generation of this cisplatin-resistant cell line, RT-112 cells were treated with increasing dosages of cisplatin over several months. During cell cultivation 12 μg mL−1 cisplatin (NeoCorp, Pawtucket, RI, USA) was added to the media of RT-112res cells with every passage.Cell viability assay
Viability of RT-112 and RT-112res cells was measured using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. RT-122 and RT-112res cells were seeded in 96-well plates with a density of 2.5 × 103 or 5.0 × 103 cells per well, respectively. One day after seeding, cells were treated with prodigiosin (1) or derivatives 16ba–bc for 24 or 72 h. 5 μM staurosporine was used as a positive control and 0.1% DMSO (PanReac AppliChem, Darmstadt, Germany) was used as a solvent control. After the incubation time, thiazolyl blue was added to the cells and they were incubated at 37 °C and 5% CO2 in a humidified atmosphere for 45 min. After removal of the MTT-containing medium, 100 μL DMSO were added per well for extraction of the formazan. Absorbance was measured at 570 nm and 650 nm (reference) with a microplate reader (BioTek, Synergy Mx). After subtraction of the reference signal and the mean value of the positive control from each value, the mean of the absorbance of the solvent control was set as 100% and relative viability was calculated for each sample. All IC50 values were calculated using GraphPad Prism 7.01.In vitro assay with prodiginine ligating enzymes
Cells of E. coli BL21 (DE3) with the desired pET expression vector were resuspended after expression of the corresponding ligating enzyme in 50 mM potassium phosphate buffer, pH 7.0 (KPi) to a final mass concentration of 0.2 g mL−1 and cooled on ice. To disrupt the cells, sonication on ice was performed (40% amplitude, 3 × 5 min with 5 min rest on ice between each cycle, 0.5 s pulse and 0.5 s rest per pulse cycle).The 1H-pyrroles 4a–c, as well as the MBC derivatives 3a–g were freshly dissolved in DMSO to a final concentration of 20 mM. For ATP, a 62.5 mM stock solution in water was prepared from the disodium salt Na2-ATP × 3 H2O.
In a 1.5 mL reaction tube, 10 μL of 62.5 mM ATP, 25 μL of 20 mM MBC, and 25 μL of 20 mM pyrrole were mixed. Subsequently, the reaction was initiated by supplementation with 440 μL of homogenous sonicated cells in KPi buffer with a concentration of 0.20 g mL−1 (cell debris was not removed, as the enzymes are attached to the inner membrane). After carefully inverting the tubes, the reactions were incubated at 30 °C and 300 RPM for a total reaction time of 4 h. Afterwards, the analytical reactions were centrifuged (20300 rcf, 20 min, 4 °C) and the supernatant was disposed. Depending on the further utilisation of samples, two workup procedures were used as follows.
In vitro assay – workup for LC-MS measurements
The cell debris was resuspended in 400 μL of MeOH, however, the resuspension process was assisted by an ultrasound bath. Centrifugation (20300 rcf, 20 min, 4 °C) was used to remove cell debris and the supernatant transferred into a new reaction tube. MeOH was evaporated in a vacuum centrifuge at 45 °C and the residue was taken up in 200 μL water. Extraction with CH2Cl2 (2 × 200 μL) and successive evaporation of the solvent provided a residue, which was dissolved in 200 μL MeOH, filtered through a 0.45 μm syringe filter and subjected to LC-MS chromatography.
In vitro assay – workup for photographic documentation
The cell debris was resuspended in 300 μL acidic EtOH (+ 4% 1 M HCl) and the resuspension process assisted by an ultrasound bath. Centrifugation (20300 rcf, 20 min, 4 °C) was used to remove cell debris and the supernatant was then transferred into a new 1.5 mL reaction tube for documentation (ESI†).
LC-MS parameters
Coupled LC-MS measurements were performed on a Thermo Scientific UltiMate 3000 UHPLC instrument with an Atlantis T3 3 μm, 3 × 100 mm column (Waters) and an ISQ-ES mass spectrometer. Sample volumes of 5 μL were injected at a temperature of 30 °C and a flow rate of 0.60 mL min−1. UV detection was realised at 535 nm via photo diode array detector. Gradient elution with MeOH + 0.1% formic acid (solvent A) and Millipore water + 0.1% formic acid (solvent B) allowed separation of prodiginines from MBC and pyrrole precursors. Elution profile: −5–0 min with 10% solvent A, 0–4 min with 10–60% solvent A, 4–6 min with 60–100% solvent A, 6–13 min with 100% solvent A. The following parameters were used for the ISQ-MS detection – mode: positive, vaporiser temperature: 282 °C, ITT temperature: 300 °C, source voltage positive ions: 3000 V, source voltage negative ions: −2000 V, sheath gas pressure: 49.9 psig, aux gas pressure: 5.7 psig, sweep gas pressure: 0.5 psig, mass area 10–1000, CID 20.Quantification of prodiginines from LC chromatograms
Based on the measured and approximated molar extinction coefficients for chemically synthesised prodiginine references, quantification of prodiginines in methanolic extracts from in vitro assays was performed. Therefore, eqn (1.2), devised by Torsi et al., was deployed.88 | (1.1) |
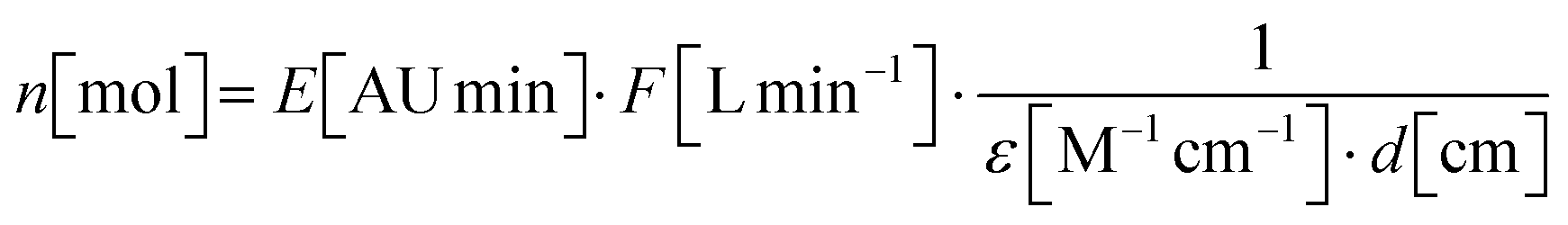 | (1.2) |
c: prodiginine concentration in the methanolic extract
n: mount of substance applied to chromatography
V inj: injected volume applied to chromatography
E: integrated extinction at 535 nm
F: chromatographic flow rate
ε: molar extinction coefficient at 535 nm
d: flow cell path length
The UV traces at 535 nm and the extracted ion chromatograms (EIC) of the appropriate m/z ratio for the proposed products were generated from the total ion chromatograms (TIC). EIC spectra were used to validate the estimated product masses and UV absorbance at 535 nm was utilised to calculate the corresponding peak areas by integration. For quantification of prodiginines from the methanolic extracts, the mean value of absorbance from triplicates was used to determine the amount of substance that had been injected to the chromatographic system (eqn (1.2)), based on the extinction coefficients, which had been determined experimentally or by approximation. From the results of eqn (1.2), the concentrations in methanolic extracts were calculated by eqn (1.1).
Abbreviations
| Compd | Compound |
| LiTMP | Lithium 2,2,6,6-tetramethylpiperidide |
| SPhos | 2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl |
| TFE | 2,2,2-Trifluoroethanol |
| tR | Retention time |
| LB | Lysogeny broth |
| TB | Terrific broth |
| KPi | Potassium phosphate |
| IC50 | Half maximal inhibitory concentration |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| o/n | Overnight |
Author contributions
Tim Moritz Weber: conceptualisation, methodology, investigation, writing – original draft, writing – review & editing, visualisation, supervision, project administration, funding acquisition; Alexandra Leyens: investigation; Lena Berning: investigation, visualisation, writing – original draft; Björn Stork: writing – review & editing, supervision, funding acquisition; Jörg Pietruszka: conceptualisation, writing – review & editing, supervision, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Jürgen Manchot Stiftung (scholarship to T. M. W.), the Deutsche Forschungsgemeinschaft DFG (GRK2158, project #270650915; to J. P. and B. S.) for funding as well as the Heinrich Heine University Düsseldorf and the Forschungszentrum Jülich GmbH for their ongoing support. We kindly acknowledge Birgit Henßen, Vera Ophoven, and Irene Küberl for their support in LC-MS data acquisition, chemical synthesis, and photo documentation, respectively. We thank Margaretha A. Skowron, Michèle J. Hoffmann, and Günter Niegisch (Department of Urology, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University Düsseldorf) for providing RT-112 and RT-112res cell lines.Notes and references
- A. Fürstner, J. Grabowski and C. W. Lehmann, J. Org. Chem., 1999, 64(22), 8275–8280 CrossRef PubMed.
- H. Rapoport and C. D. Willson, J. Am. Chem. Soc., 1962, 84(4), 630–635 CrossRef CAS.
- D. L. Boger and M. Patel, J. Org. Chem., 1988, 53(7), 1405–1415 CrossRef CAS.
- H. H. Wasserman and L. J. Lombardo, Tetrahedron Lett., 1989, 30(13), 1725–1728 CrossRef CAS.
- R. E. Johnson, T. de Rond, V. N. Lindsay, J. D. Keasling and R. Sarpong, Org. Lett., 2015, 17(14), 3474–3477 CrossRef CAS PubMed.
- H. H. Wasserman, G. C. Rodgers and D. D. Keith, J. Am. Chem. Soc., 1969, 91(5), 1263–1264 CrossRef CAS PubMed.
- M. D. Clift and R. J. Thomson, J. Am. Chem. Soc., 2009, 131(40), 14579–14583 CrossRef CAS PubMed.
- A. Fürstner, K. Radkowski and H. Peters, Angew. Chem., Int. Ed., 2005, 44(18), 2777–2781 CrossRef PubMed.
- A. Fürstner and H. Weintritt, J. Am. Chem. Soc., 1998, 120(12), 2817–2825 CrossRef.
- J. H. Frederich and P. G. Harran, J. Am. Chem. Soc., 2013, 135(10), 3788–3791 CrossRef CAS PubMed.
- A. Fürstner, Angew. Chem., Int. Ed., 2003, 42(31), 3582–3603 CrossRef PubMed.
- S. A. Parikh, H. Kantarjian, A. Schimmer, W. Walsh, E. Asatiani, K. El-Shami, E. Winton and S. Verstovsek, Clin. Lymphoma, Myeloma Leuk., 2010, 10(4), 285–289 CrossRef CAS PubMed.
- A. D. Schimmer, S. O'Brien, H. Kantarjian, J. Brandwein, B. D. Cheson, M. D. Minden, K. Yee, F. Ravandi, F. Giles and A. Schuh, Clin. Cancer Res., 2008, 14(24), 8295–8301 CrossRef CAS PubMed.
- A. Fürstner and E. J. Grabowski, ChemBioChem, 2001, 2(9), 706–709 CrossRef.
- M. S. Melvin, M. W. Calcutt, R. E. Noftle and R. A. Manderville, Chem. Res. Toxicol., 2002, 15(5), 742–748 Search PubMed.
- M. S. Melvin, J. T. Tomlinson, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Am. Chem. Soc., 2000, 122(26), 6333–6334 CrossRef CAS.
- M. S. Melvin, K. E. Wooton, C. C. Rich, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Inorg. Biochem., 2001, 87(3), 129–135 CrossRef CAS PubMed.
- H. Konno, H. Matsuya, M. Okamoto, T. Sato, Y. Tanaka, K. Yokoyama, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, J. Biochem., 1998, 124(3), 547–556 CrossRef CAS PubMed.
- H. Matsuya, M. Okamoto, T. Ochi, A. Nishikawa, S. Shimizu, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, Biochem. Pharmacol., 2000, 60(12), 1855–1863 CrossRef CAS PubMed.
- S. Ohkuma, T. Sato, M. Okamoto, H. Matsuya, K. Arai, T. Kataoka, K. Nagai and H. H. Wasserman, Biochem. J., 1998, 334(3), 731–741 CrossRef CAS PubMed.
- T. Sato, H. Konno, Y. Tanaka, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, J. Biol. Chem., 1998, 273(34), 21455–21462 CrossRef CAS PubMed.
- T. Kataoka, M. Muroi, S. Ohkuma, T. Waritani, J. Magae, A. Takatsuki, S. Kondo, M. Yamasaki and K. Nagai, FEBS Lett., 1995, 359(1), 53–59 CrossRef CAS PubMed.
- S. S. Habash, H. U. Brass, A. S. Klein, D. P. Klebl, T. M. Weber, T. Classen, J. Pietruszka, F. M. Grundler and A. S. S. Schleker, Front. Plant Sci., 2020, 11, 579807 CrossRef PubMed.
- J. Lapenda, P. Silva, M. Vicalvi, K. Sena and S. Nascimento, World J. Microbiol. Biotechnol., 2015, 31, 399–406 CrossRef CAS PubMed.
- D. C. Woodhams, B. C. LaBumbard, K. L. Barnhart, M. H. Becker, M. C. Bletz, L. A. Escobar, S. V. Flechas, M. E. Forman, A. A. Iannetta and M. D. Joyce, Microb. Ecol., 2018, 75, 1049–1062 CrossRef CAS PubMed.
- W. McRary, E. Beaver and E. Noble, Exp. Parasitol., 1953, 2(2), 125–128 CrossRef.
- H.-S. Kim, M. Hayashi, Y. Shibata, Y. Wataya, T. Mitamura, T. Horii, K. Kawauchi, H. Hirata, S. Tsuboi and Y. Moriyama, Biol. Pharm. Bull., 1999, 22(5), 532–534 CrossRef CAS PubMed.
- J. Lazaro, J. Nitcheu, R. Z. Predicala, G. C. Mangalindan, F. Nesslany, D. Marzin, G. P. Concepcion and B. Diquet, Nat. Toxins, 2002, 11(4), 367–377 CAS.
- M. Isaka, A. Jaturapat, J. Kramyu, M. Tanticharoen and Y. Thebtaranonth, Antimicrob. Agents Chemother., 2002, 46(4), 1112–1113 CrossRef CAS PubMed.
- L. Berning, D. Schlütermann, A. Friedrich, N. Berleth, Y. Sun, W. Wu, M. J. Mendiburo, J. Deitersen, H. U. Brass, M. A. Skowron, M. J. Hoffmann, G. Niegisch, J. Pietruszka and B. Stork, Molecules, 2021, 26(5), 1294 CrossRef CAS PubMed.
- C. Yamamoto, H. Takemoto, K. Kuno, D. Yamamoto, A. Tsubura, K. Kamata, H. Hirata, A. Yamamoto, H. Kano and T. Seki, Hepatology, 1999, 30(4), 894–902 CrossRef CAS PubMed.
- D. Yamamoto, Y. Kiyozuka, Y. Uemura, C. Yamamoto, H. Takemoto, H. Hirata, K. Tanaka, K. Hioki and A. Tsubura, J. Cancer Res. Clin. Oncol., 2000, 126, 191–197 CrossRef CAS PubMed.
- B. Montaner, S. Navarro, M. Piqué, M. Vilaseca, M. Martinell, E. Giralt, J. Gil and R. Pérez-Tomás, Br. J. Pharmacol., 2000, 131(3), 585–593 CrossRef CAS PubMed.
- B. Montaner and R. Pérez-Tomás, Life Sci., 2001, 68(17), 2025–2036 CrossRef CAS PubMed.
- A. K. Harris, N. R. Williamson, H. Slater, A. Cox, S. Abbasi, I. Foulds, H. T. Simonsen, F. J. Leeper and G. P. Salmond, Microbiology, 2004, 150(11), 3547–3560 CrossRef CAS PubMed.
- A. M. Cerdeño, M. J. Bibb and G. L. Challis, Chem. Biol., 2001, 8(8), 817–829 CrossRef PubMed.
- K. J. Picott, J. A. Deichert, E. M. deKemp, G. Schatte, F. Sauriol and A. C. Ross, MedChemComm, 2019, 10(3), 478–483 RSC.
- S. M. Salem, P. Kancharla, G. Florova, S. Gupta, W. Lu and K. A. Reynolds, J. Am. Chem. Soc., 2014, 136(12), 4565–4574 CrossRef CAS PubMed.
- X. Wang, T. Isbrandt, E. Ø. Christensen, J. Melchiorsen, T. O. Larsen, S.-D. Zhang and L. Gram, Microbiol. Spectrum, 2021, 9(2), e01171-21 CrossRef PubMed.
- N. L. Grenade, D. S. Chiriac, A. O. Pasternak, J. L. Babulic, B. E. Rowland, G. W. Howe and A. C. Ross, ACS Chem. Biol., 2023, 18(2), 223–229 CrossRef CAS PubMed.
- N. R. Williamson, H. T. Simonsen, R. A. Ahmed, G. Goldet, H. Slater, L. Woodley, F. J. Leeper and G. P. Salmond, Mol. Microbiol., 2005, 56(4), 971–989 CrossRef CAS PubMed.
- E. Coco, K. Narva and J. Feitelson, Mol. Genet. Genomics, 1991, 227, 28–32 CrossRef CAS PubMed.
- M. J. Jaremko, T. D. Davis, J. C. Corpuz and M. D. Burkart, Nat. Prod. Rep., 2020, 37(3), 355–379 RSC.
- H. Wasserman, C. Shaw, R. Sykes and R. Cushley, Tetrahedron Lett., 1974, 15(33), 2787–2790 CrossRef.
- H. Wasserman, R. Sykes, P. Peverada, C. Shaw, R. Cushley and S. Lipsky, J. Am. Chem. Soc., 1973, 95(20), 6874–6875 CrossRef CAS PubMed.
- S. R. Chawrai, N. R. Williamson, G. P. Salmond and F. J. Leeper, Chem. Commun., 2008, 1862–1864 RSC.
- D. X. Hu, D. M. Withall, G. L. Challis and R. J. Thomson, Chem. Rev., 2016, 116(14), 7818–7853 CrossRef CAS PubMed.
- Z. You, X. Liu, S. Zhang and Y. Wang, Biochem. Eng. J., 2018, 134, 1–11 CrossRef CAS.
- S. Brands, H. U. Brass, A. S. Klein, J. G. Sikkens, M. D. Davari, J. Pietruszka, A. J. Ruff and U. Schwaneberg, Catal. Sci. Technol., 2021, 11(8), 2805–2815 RSC.
- K. J. Picott, J. A. Deichert, E. M. DeKemp, V. Snieckus and A. C. Ross, ChemBioChem, 2020, 21(7), 1036–1042 CrossRef CAS PubMed.
- D. Fehér, R. S. Barlow, P. S. Lorenzo and T. K. Hemscheidt, J. Nat. Prod., 2008, 71(11), 1970–1972 CrossRef PubMed.
- D. M. Withall, S. W. Haynes and G. L. Challis, J. Am. Chem. Soc., 2015, 137(24), 7889–7897 CrossRef CAS PubMed.
- C. Perry, E. L. De los Santos, L. M. Alkhalaf and G. L. Challis, Nat. Prod. Rep., 2018, 35(7), 622–632 RSC.
- A. E. Silva, L. A. Guimarães, E. G. Ferreira, M. d. C. M. Torres, A. B. d. Silva, P. C. Branco, F. A. S. Oliveira, G. G. Silva, D. V. Wilke and E. R. Silveira, J. Braz. Chem. Soc., 2017, 28, 465–474 CAS.
- F. E. Sakai-Kawada, C. G. Ip, K. A. Hagiwara and J. D. Awaya, Front. Microbiol., 2019, 10, 1715 CrossRef PubMed.
- P. C. Dorrestein, E. Yeh, S. Garneau-Tsodikova, N. L. Kelleher and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(39), 13843–13848 CrossRef CAS PubMed.
- A. S. Klein, A. Domröse, P. Bongen, H. U. Brass, T. Classen, A. Loeschcke, T. Drepper, L. Laraia, S. Sievers and K.-E. Jaeger, ACS Synth. Biol., 2017, 6(9), 1757–1765 CrossRef CAS PubMed.
- A. S. Klein, H. U. C. Brass, D. P. Klebl, T. Classen, A. Loeschcke, T. Drepper, S. Sievers, K. E. Jaeger and J. Pietruszka, ChemBioChem, 2018, 19(14), 1545–1552 CrossRef CAS PubMed.
- H. U. Brass, A. S. Klein, S. Nyholt, T. Classen and J. Pietruszka, Adv. Synth. Catal., 2019, 361(11), 2659–2667 CrossRef CAS.
- S. W. Haynes, P. K. Sydor, A. E. Stanley, L. Song and G. L. Challis, Chem. Commun., 2008, 1865–1867 RSC.
- R. D'Alessio, A. Bargiotti, O. Carlini, F. Colotta, M. Ferrari, P. Gnocchi, A. Isetta, N. Mongelli, P. Motta and A. Rossi, J. Med. Chem., 2000, 43(13), 2557–2565 CrossRef PubMed.
- U. V. Santer and H. J. Vogel, Biochim. Biophys. Acta, 1956, 19(3), 578–579 CrossRef CAS PubMed.
- H. H. Wasserman, J. E. McKeon and U. V. Santer, Biochem. Biophys. Res. Commun., 1960, 3(2), 146–149 CrossRef CAS PubMed.
- B. Trofimov, A. Mikhaleva, A. Vasil'ev, S. Korostova and S. Shevchenko, Chem. Heterocycl. Compd., 1985, 21, 46–49 CrossRef.
- K. Dairi, S. Tripathy, G. Attardo and J.-F. Lavallée, Tetrahedron Lett., 2006, 47(15), 2605–2606 CrossRef CAS.
- T. Ono, D. Koga and Y. Hisaeda, Chem. Lett., 2017, 46(2), 260–262 CrossRef CAS.
- For the early steps in the synthesis of MBC derivatives (Fig. 3 and 4), the letter a was left out for acid chlorides, acyl pyrroles, alkyl pyrroles, Boc-protected alkyl pyrroles and Boc-protected pyrrole-2-boronic acids. As unsubstituted MBC 3a was synthesised by a different route (supporting information), omission allows systematic description and significantly enhances readability of the manuscript.
- A. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43(1), 412–443 RSC.
- D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131(20), 6961–6963 CrossRef CAS PubMed.
- R. Hubbard and C. Rimington, Biochem. J., 1950, 46(2), 220 CrossRef CAS PubMed.
- A. Domröse, A. S. Klein, J. Hage-Hülsmann, S. Thies, V. Svensson, T. Classen, J. Pietruszka, K.-E. Jaeger, T. Drepper and A. Loeschcke, Front. Microbiol., 2015, 6, 972 Search PubMed.
- D. Schlütermann, M. A. Skowron, N. Berleth, P. Böhler, J. Deitersen, F. Stuhldreier, N. Wallot-Hieke, W. Wu, C. Peter and M. J. Hoffmann, Urol. Oncol.: Semin. Orig. Invest., 2018, 36(4), 160 Search PubMed.
- M. A. Skowron, P. Petzsch, K. Hardt, N. Wagner, M. Beier, S. Stepanow, M. Drechsler, H. Rieder, K. Köhrer and G. Niegisch, Sci. Rep., 2019, 9(1), 14476 CrossRef PubMed.
- M. A. Skowron, G. Niegisch, G. Fritz, T. Arent, J. G. van Roermund, A. Romano, P. Albers, W. A. Schulz and M. J. Hoffmann, J. Exp. Clin. Cancer Res., 2015, 34(1), 1–15 CrossRef PubMed.
- K. Ma, S. Li, X. Huo, M. Guo, X. Du, C. Li, X. Liu, J. Lv and Z. Chen, Biochem. Biophys. Res. Commun., 2020, 533(3), 474–480 CrossRef CAS PubMed.
- M. A. Skowron, M. Melnikova, J. G. Van Roermund, A. Romano, P. Albers, J. Thomale, W. A. Schulz, G. Niegisch and M. J. Hoffmann, Int. J. Mol. Sci., 2018, 19(2), 590 CrossRef PubMed.
- S.-H. Chen and J.-Y. Chang, Int. J. Mol. Sci., 2019, 20(17), 4136–4156 CrossRef PubMed.
- R. M. Drayton and J. W. Catto, Expert Rev. Anticancer Ther., 2012, 12(2), 271–281 CrossRef CAS PubMed.
- S. M. O'Brien, D. F. Claxton, M. Crump, S. Faderl, T. Kipps, M. J. Keating, J. Viallet and B. D. Cheson, Blood, 2009, 113(2), 299–305 CrossRef PubMed.
- A. D. Schimmer, A. Raza, T. H. Carter, D. Claxton, H. Erba, D. J. DeAngelo, M. S. Tallman, C. Goard and G. Borthakur, PLoS One, 2014, 9(10), e108694 CrossRef PubMed.
- F. McCoy, J. Hurwitz, N. McTavish, I. Paul, C. Barnes, B. O'Hagan, K. Odrzywol, J. Murray, D. Longley and G. McKerr, Cell Death Dis., 2010, 1(12), e108–e108 CrossRef CAS PubMed.
- B. C. Koehler, A. Jassowicz, A.-L. Scherr, S. Lorenz, P. Radhakrishnan, N. Kautz, C. Elssner, J. Weiss, D. Jaeger and M. Schneider, BMC Cancer, 2015, 15, 1–11 CrossRef PubMed.
- S. Ji, R. Sun, K. Xu, Z. Man, J. Ji, Y. Pu, L. Yin, J. Zhang and Y. Pu, Toxicol. In Vitro, 2019, 60, 107–115 CrossRef CAS PubMed.
- W. Castillo-Ávila, M. Abal, S. Robine and R. Pérez-Tomás, Life Sci., 2005, 78(2), 121–127 CrossRef PubMed.
- X. Mo, T. D. Morgan, H. T. Ang and D. G. Hall, J. Am. Chem. Soc., 2018, 140(15), 5264–5271 CrossRef CAS PubMed.
- X.-C. Cai and B. B. Snider, J. Org. Chem., 2013, 78(23), 12161–12175 CrossRef CAS PubMed.
- D. L. Boger and M. Patel, Tetrahedron Lett., 1987, 28(22), 2499–2502 CrossRef CAS.
- G. Torsi, G. Chiavari, C. Laghi, A. Asmundsdottir, F. Fagioli and R. Vecchietti, J. Chromatogr. A, 1989, 482(1), 207–214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting spectra, chromatograms, dose response curves as well as synthesis are provided. See DOI: https://doi.org/10.1039/d3cy00913k |
| This journal is © The Royal Society of Chemistry 2023 |