
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CeO2-supported Ni and Co catalysts prepared by a solution combustion method for H2 production from glycerol: the effect of fuel/oxidizer ratio and oxygen excess†
Anna N.
Matveyeva
 *a,
Shamil O.
Omarov
a,
Marianna A.
Gavrilova
ab,
Andrey D.
Trofimuk
c,
Johan
Wärnå
d and
Dmitry Yu.
Murzin
*d
*a,
Shamil O.
Omarov
a,
Marianna A.
Gavrilova
ab,
Andrey D.
Trofimuk
c,
Johan
Wärnå
d and
Dmitry Yu.
Murzin
*d
aLaboratory of Materials and Processes for Hydrogen Energy, Ioffe Institute, Politekhnicheskaya ul. 28, 194021 St. Petersburg, Russia. E-mail: anna.matveyeva@mail.ioffe.ru
bDepartment of Physical Chemistry, St. Petersburg State Institute of Technology (Technical University), Moskovskiy pr. 26, 190013 St. Petersburg, Russia
cLaboratory of Physics for Cluster Structures and Colloid Physics Laboratory, Ioffe Institute, Politekhnicheskaya ul. 28, 194021 St. Petersburg, Russia
dLaboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University, Henriksgatan 2, 20500 Turku/Åbo, Finland. E-mail: dmurzin@abo.fi
First published on 3rd August 2023
Abstract
Glycerol is a promising raw material for obtaining various chemicals and fuels, including H2. Nevertheless, the development of cost-effective and stable catalysts with high activity and selectivity for H2 production from glycerol under mild conditions has been a serious problem for their practical use. In this work, a series of CeO2-supported Ni and Co catalysts with the content of Ni and Co as metal(II) oxides being 30 wt% was prepared by solution combustion synthesis via changing the fuel/oxidizer ratio (φ = 0.7–3), the fuel type (glycine, urea), and the oxygen excess. Thus, various species of Ni and Co and their interactions with CeO2 were obtained, which affected the reduction sequence, defectiveness, textural characteristics, and activity in steam and aqueous-phase reforming of glycerol. To study the characteristics of the obtained samples, instrumental methods such as XRD, DTA-TGA, low-temperature N2 physisorption, SEM-EDX, H2-TPR, and H2- and CO2-TPD, as well as Raman spectroscopy, were used. The results demonstrated that the Ni–Ce–O system synthesized with a high glycine-to-oxidizer ratio can successfully compete with catalysts based on noble metals (Pt/γ-Al2O3) in aqueous-phase reforming of glycerol.
1. Introduction
One of the consequences of global population growth and industrialization is the depletion of fossil fuels, the use of which has also exacerbated global warming and climate change.1 Among the alternatives to fossil sources, hydrogen (H2) has emerged as an energy vector of the future.2 Hydrogen is predicted to be the main source of 90% of the world's energy by 2080.3 One of the effective and promising methods of hydrogen production is steam (SR) and aqueous-phase reforming (APR) of glycerol.4 The latter is a by-product of the growing biodiesel industry, as a result of which the glycerol production has increased significantly in the few recent years, saturating the market and causing a free fall in prices.5,6 In this context, it is important to manage excess glycerol to avoid waste problems and adverse effects on biodiesel industrial development.7APR of glycerol compared to steam reforming is more preferable for hydrogen production in terms of side reactions, as well as more thermodynamically favorable conditions (200–260 °C; 20–50 bar) for the water–gas shift (WGS) reaction.3,8 The latter is especially important in the case of subsequent use of hydrogen in fuel cells that are sensitive to CO.9 In addition, APR of glycerol is an alternative to SR when glycerol is already obtained dissolved in H2O, in particular, in upstream biomass processing.
Steam reforming, which proceeds at higher temperatures and lower pressures, in contrast, imposes more stringent requirements regarding the thermal stability of the catalyst, especially in the presence of steam, as well as resistance to carburization. The latter issue is one of the main causes of catalyst deactivation, together with metal particle sintering.
In the few recent years, various combinations of catalysts and supports have been reported in the literature about reforming processes of glycerol. Among them, catalysts based on transition metals, in particular Ni and Co, are more preferable due to low costs and wide availability.
Ni-containing catalysts are known to have a high capability for C–C, C–H and O–H bond cleavage10 and can be easily dispersed on a range of supports. A number of nickel catalysts supported on Al2O3,11–20 CeO2,17,20–37 MgO,21,22,30 TiO2,21,22,38 SiO2,13,38–40 and ZrO2 (ref. 13, 38, and 41–44) have been studied in reforming processes of several renewable feeds, including methanol,9 ethanol,14–17,24,26,27,32–34,37,39,41 and glycerol.11–13,20–23,25,28–31,35,36,38,40,42–44 Al2O3 and CeO2 as supports are considered effective in terms of high H2 production and lower coke formation.17 However, according to ref. 23, Ni supported on CeO2 has a higher stability in glycerol steam reforming compared to Ni/Al2O3, despite having a significantly lower specific surface area. A comparative study of nickel catalyst deactivation on Al2O3, CeO2, ZnO, MgO, ZrO2, and SiO2 in low-temperature steam reforming of ethanol showed the following decreasing order of activity: CeO2 > Al2O3 > ZrO2.45
The excellent stability of Ni/CeO2 has been related to accelerated carbon removal from the metal surface due to the mobility of oxygen ions and formation of oxygen vacancies in the support.46 Recent studies of Ni/CeO2 systems in reforming processes are related to controlling the functional properties of catalysts through changing the morphology,32,33,36 preparation methods,24,28,34,35 active component content,24 promoters,26,47 and additives.27,48,49
Although Co-based catalysts supported on CeO2 have not been studied for glycerol reforming as extensively as nickel-based catalysts,50 they have also been reported to demonstrate excellent ethanol steam reforming activity.34,37,51–58 It has been found that Co/CeO2 exhibited optimum hydrogen yield and stability compared to Fe-, Ni-, and Cu-based catalysts.37 Recent studies of Co/CeO2 systems in reforming processes are associated with the modification of the support and/or active phase,56 and changing the cobalt content58 and its dispersion,34 or the CeO2 crystal facet59 to fine-tune the local structure of the surface.
It is well known that material preparation methods have a strong effect on the structural, textural, and catalytic properties. For Ni and Co-based catalyst preparation, the incipient wetness impregnation22,24,28,32,35–37,52,53,57 and wet impregnation21,25,29,49 methods are mainly used. However, these methods have limitations in the synthesis of a catalyst with a high content of the active component, at which it is difficult to achieve high metal dispersion. A small particle size with a uniform distribution of active components can also be achieved by the co-precipitation method even at a high metal content.15,58 The disadvantages of this method include a long duration and the overall complexity, careful control of the synthesis conditions, and precipitation and washing completeness. In addition, the precipitation methods are characterized by a significant consumption of reagents and large amounts of wastewater.
Therefore, other alternative methods for the formation of inorganic materials, such as the solution combustion synthesis (SCS) method, are becoming more widespread. SCS involves the rapid heating of a solution containing certain amounts of an oxidizing agent (metal precursors, often nitrates) and an organic fuel such as urea, glycine or citric acid. The combustion technique allows obtaining very fine crystalline oxide powders of high purity with a homogeneous, non-agglomerated and multicomponent composition.60 Some studies show that catalysts prepared using this method are comparable with catalysts obtained by classical methods, for example Ni-based supported catalysts for ethanol decomposition and reforming.26,27,31,61–63 In addition, a high conversion of glycerol (30%) was obtained for the 20 wt% Ni-based catalyst on CeO2 synthesized by the self-propagation combustion method compared to those by co-precipitation or wet impregnation due to the formation of smaller nickel crystallites.29
Fuel usually plays a central role in optimizing material properties because it often performs the triple function of a reducing and a complexation agent, and a microstructural template.64,65 However, the characteristics of the final powder obtained by the SCS method also depend on other synthesis parameters, such as the fuel-to-oxidizer ratio (φ), metal precursor type, chemical composition, pH, heating method, etc. Subsequently, the main drawback of this method is the difficulties in defining the optimal fuel-to-oxidizer ratio. The amount of fuel calculated directly from the stoichiometric ratio for combustion results frequently in a product of an undesired composition.66
There are several studies on Ni–Ce and Co–Ce oxide systems prepared by the SCS method, focused on the effect of Sn and Zr,26 NaCl,67 and colloidal SiO2 (ref. 68 and 69) (using glycine fuel), Ni loading (0–8.5 wt%, urea;31 3–68 wt%, hydrous hydrazine;70 3.1–15.6 wt%, urea71), Co loading (1–10 wt%, glycine69), the fuel-to-oxidizer ratio (φ = 0.5–3, hydrous hydrazine70), and the fuel type (glycine and hydrous hydrazine at φ = 2;70 oxalyldihydrazide, glycerol, carbohydrazide, urea at φ = 1 (ref. 71)) on their physical, chemical, and catalytic properties in various processes. However, no attention was paid to the features of their formation upon varying the ratio of glycine and urea to the oxidizing agent and their influence on the catalytic behavior during glycerol reforming. The ratio of fuel/oxidizer is one of the most important parameters affecting not only the phase composition, but also the structure, defectiveness, and related properties of the materials. Therefore, the present study aims to comparatively explore CeO2-supported Ni and Co catalysts prepared by varying the SCS parameters, such as the fuel type (glycine, urea), the fuel-to-oxidizer ratio, and the oxygen excess, in both steam and aqueous-phase reforming of glycerol.
2. Experimental
2.1. Reagents
Ni(NO3)2·6H2O (99.2%, Lenreactiv, Russia), Co(NO3)2·6H2O (97%, Lenreactiv, Russia), Ce(NO3)3·6H2O (99.8%, Chemcraft, Russia), ammonium nitrate (98.5%, Russia), glycine (99.2%, Lenreactiv, Russia), urea (99.8%, Lenreactiv, Russia), AlOOH (Catapal, Sasol), H2PtCl6 (37 wt% Pt), and glycerol (99.3%, Lenreactiv, Russia) were used.2.2. Preparation
An appropriate amount of glycine (or urea) and the corresponding metal nitrates were dissolved in DI water (1 mL per 1 g of the starting materials) in a wide glass beaker (250 mL) to obtain 1 g of NiO/CeO2 or CoO/CeO2. The theoretical content of nickel and cobalt calculated as NiO and CoO was 30 wt%. In some syntheses, 1 g of NH4NO3 was additionally used per 1 g of NiO/CeO2. The fuel-to-oxidizer ratio (φ) was taken from 0.7 to 3 and calculated via the reducing and oxidizing valence states (RV and OV, respectively) according to ref. 65 and 72:| φ = (−1)∑(coeff.·RV of fuel)/∑(coeff.·OV of nitrate), | (1) |
The methodology for calculating the reducing and oxidizing valence states can be found in ref. 73. By selecting the ratio between reagents, the value of φ can be changed. For example, the reaction equation occurring with glycine at φ = 1 (−1·9·100/(−15·36 + (−10)·36) can be represented as:
| 36Ce(NO3)3(aq) + 36Ni(NO3)2(aq) + 100C2H5NO2 + 9O2 → 36(NiO·CeO2) + 200CO2 + 140N2 + (250H2O), | (2) |
The resulting aqueous solution of fuel with salts was heated on an electric plate (1 kW) to boiling. It was burned with the formation of a solid powder, after complete evaporation of H2O. To avoid oxidation of the resulting combustion products, the beaker was covered with a Petri dish immediately after the end of combustion (denoted with *). Two or three batches of each catalyst were synthesized. Reaction equations for each synthesis can be found in the ESI.†
A platinum–alumina (Pt/Al2O3) catalyst was synthesized by impregnation of γ-Al2O3 (prepared by calcining AlOOH at 700 °C) with an excess of H2PtCl6 solution to obtain 1 wt% Pt. After impregnation, the sample was dried and heat-treated at 260 °C (according to ref. 74) in an oven. The catalyst was reduced in a reactor at 280 °C prior to experiments as discussed below.
2.3. Characterization
XRD analysis was done on a Rigaku SmartLab 3 diffractometer (Japan) equipped with a 1D DteX250 detector (Rigaku, Japan) and Ni-filter (Rigaku, Japan) at λ = 1.54056 Å, 30 mA, 40 kV, a 4° min−1 scan speed, and a 0.01° step width. The diffraction data were analyzed by the Rietveld method. The refinement procedure and software are similar to those given in ref. 69. The structural parameters for CeO2 (no. 193169), NiO (no. 184918), Ni (no. 426960), CoO (no. 245319), Co3O4 (no. 290720) and Co (no. 622442) with the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m were taken using the ICDD PDF-2 database.
m were taken using the ICDD PDF-2 database.
N2-physisorption data were obtained on Quantachrome's Autosorb-6iSA (USA) and Micromeritics ASAP 2020 (USA) units. The samples were preliminarily degassed at 200–250 °C under vacuum. The specific surface area, the total pore volume, and the average pore diameter were determined using the BET equation, while the distribution of pore size was obtained by the NLDFT method.
SEM was carried out on a TESCAN VEGA 3 SBH microscope (Czech Republic) equipped with an INCAx-act EDS detector (Oxford Instruments).
H2-TPR, H2-TPD, and CO2-TPD were performed on a SOLO Chemosorb (Russia) equipped with a TCD (thermal conductivity detector). In H2-TPR tests, ca. 30 mg of powder without pretreatment was reduced from 80–100 to 620–720 °C at a 10 °C min−1 rate under 20 mL min−1 of 10 vol% H2/Ar (99.998 vol% purity). 2-Propanol cooled in liquid N2 was used to trap water vapor.
For H2-TPD, ca. 100 mg of a sample was first reduced in situ with a H2/Ar flow (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 vol%, 40 mL min−1) at a ramp rate of 20 °C min−1 to 450 °C and 10 °C min−1 to 500 °C and held for 30 min. Then, it was cooled to 60 °C and saturated with H2 (H2/Ar flow, 50:50 vol%, 20 mL min−1) for 20 min. After removing the physically adsorbed H2 by purging with Ar (20 mL min−1), the sample was heated to 700–800 °C at a ramping rate of 10 °C min−1. The nickel dispersion (DNi, %), specific metal surface area (A, m2 g−1 Ni), and the average Ni diameter (d, nm) were calculated by assuming a stoichiometric ratio of H/Ni using the following equations:75
:50 vol%, 40 mL min−1) at a ramp rate of 20 °C min−1 to 450 °C and 10 °C min−1 to 500 °C and held for 30 min. Then, it was cooled to 60 °C and saturated with H2 (H2/Ar flow, 50:50 vol%, 20 mL min−1) for 20 min. After removing the physically adsorbed H2 by purging with Ar (20 mL min−1), the sample was heated to 700–800 °C at a ramping rate of 10 °C min−1. The nickel dispersion (DNi, %), specific metal surface area (A, m2 g−1 Ni), and the average Ni diameter (d, nm) were calculated by assuming a stoichiometric ratio of H/Ni using the following equations:75
 | (3) |
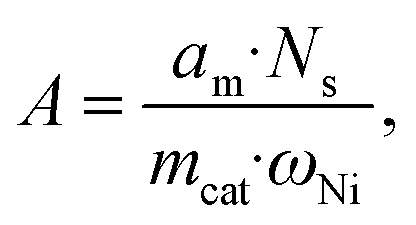 | (4) |
 | (5) |
For CO2-TPD, 100–200 mg of a sample was first reduced in situ with a H2/He flow (50:50 vol%, 40 mL min−1) at a ramp rate of 20 °C min−1 to 450 °C and 10 °C min−1 to 520 °C and held for 30 min. Then, it was cooled to 110 °C and saturated with CO2 (CO2/He flow, 10:90 vol%, 20 mL min−1) for 20 min. After removing the physically adsorbed CO2 by purging with He (20 mL min−1), the sample was heated to 600 °C at a ramping rate of 10 °C min−1.
Raman patterns were obtained using an NTEGRA Spectra spectrometer (“NT-MDT”, Russia) equipped with a 532.01 nm diode laser, a 100× objective and a 1800 lines per mm grating. The measurements were carried out at room temperature. To prevent sample heating, a beam with a power density <50 W cm−2 was used. Spectra were recorded at several random points, with each graph representing the average spectrum for a sample.
A simultaneous thermal analysis was done on a Shimadzu DTG-60A analyzer (Japan) by heating a sample in static air to 800 °C with a ramp rate of 10 °C min−1.
2.4. Catalytic tests
:1; total liquid flow 7.2–16.6 mL h−1). The resulting gaseous products were accumulated in a gas holder for sampling for 5–7 min with fixation of the volume of the formed gas. The duration of the tests did not exceed 2 h. The gaseous products were analyzed on a Shimadzu GC-2010 Plus chromatograph (Japan) with a TCD and RT-Msieve 5A and Rt-Q-BOND capillary columns. The heating program was similar to a program used in previous studies.31,42
The glycerol conversion to gaseous products (XGgas, %), hydrogen yield (Y(H2), %), and selectivity to H2, CO2, CO, CH4, C2H4, C2H6, and C3H8 (S(i), mol%) were calculated according to the following equations:
 | (6) |
 | (7) |
 | (8) |
 | (9) |
The deactivation constant (kd) was determined by fitting the dependence of hydrogen yield on TOS in accordance with the equation:
| a = ate−kdt + a∞ = (a0 − a∞)e−kdt + a∞, | (10) |
The hydrogen formation rate (mol H2 per mol Ni per s) was also calculated:
 | (11) |
The weight hourly space velocity (WHSV, h−1) was calculated as:
| WHSV = Vlq·ρlq/mcat, | (12) |
:water = 10:90 wt%, total liquid flow rate 3–9.8 mL h−1). Detailed test conditions are given in the corresponding figure captions. The gas sampling and analysis procedure is similar to the test procedure for glycerol steam reforming.
3. Results and discussion
3.1. Ce–Ni–O: synthesis features and phase composition
Fig. 1 presents the visual course of the combustion process of glycine–nitrate solutions and the macromorphology of the obtained Ce–Ni-containing materials. Under fuel deficit conditions (φ = 0.72), the thermal decomposition of nitrates dominates, as evidenced by the release of a brown gas. The most pronounced ignition is observed at the stoichiometric fuel-to-oxidizer ratio (φ = 1), which, however, entails the greatest entrainment of powder particles from the reaction zone. The powders remaining in the beaker and outside were analyzed separately using simultaneous thermal analysis (Fig. 2). Due to the fact that the combustion mode is disturbed, the escaped particles are characterized by large weight losses during heating in air compared to the sample remaining in the beaker (Fig. 2a). | ||
| Fig. 1 The course of the glycine–nitrate solution combustion and macromorphology of the Ce–Ni–O systems obtained at various fuel-to-oxidizer ratios. φ = 2* – the beaker was covered with a Petri dish immediately after completion of combustion; φ = 1.6 – ammonium nitrate was additionally used, while maintaining the glycine-to-nitrate ratio equal to 2. | ||
 | ||
| Fig. 2 Total weight change (a) and weight gain (b) during heating in air for the as-prepared Ce–Ni–O systems produced using different conditions. | ||
It should be noted that with greater contact of the solid products with oxygen (in the case of particle entrainment, φ = 1.25–1.4), the color of the samples changes from black to brown, which can indicate the formation of metallic nickel during combustion and its further oxidation. This assumption is supported by the results shown earlier in ref. 77–80, where metallic Ni is easily formed from nickel nitrate under fuel rich conditions without any post-reduction treatment. According to ref. 78 and 80, the formation of metallic Ni is triggered by the reducing action of nitrogen oxides and NH3 or CO produced by the fuel (urea, glycine, citric acid, hexamethylenetetramine) decomposition. However, there are no data for the formation of nickel during combustion of a mixture of nickel and cerium nitrates with any fuel, which can be explained by calcination of the freshly prepared samples.29,31,67,68,70,71 In ref. 62, the NiO phase was established as the only combustion product of nickel nitrate with fuel on a porous support in air, regardless of the φ value, which varied between one and three.
An increase in the sample weight during heating in air also indicates the interactions of metal Ni particles with oxygen (Fig. 2b). To minimize these interactions, experiments were carried out by covering the beaker with a Petri dish immediately after the end of combustion, as well as with the addition of NH4NO3. The results show that abundant gas evolution when using NH4NO3 does not prevent the oxidation of the samples. The most effective approach from this point of view is to cover the beaker, judging by the rich black color (Fig. 1, φ = 2*) and the increase in weight gain when heated in air (Fig. 2b). Minimizing oxygen access in the literature was carried out in various ways, for example, by using a beaker with a perforated rubber plug81 or conducting an experiment in a completely inert environment.62 According to the time-resolved XRD experiments performed in ref. 62, allowing the monitoring of the evolution of phase formation during combustion of Ni(NO3)2 + glycine + NH4NO3 + SiO2 mixture (φ = 3) in air, it turned out that during the first reaction stage, only the Ni phase is detected in the combustion front. In this case, oxygen does not influence the reaction in the combustion front, but it oxidizes nickel later in the post-combustion zone. Therefore, minimizing the time of contact with oxygen after the completion of combustion led to the results described above. It should be noted that the combustion mechanism in the absence of SiO2 is different.78
The carbamide–nitrate solution burns without entrainment of sample particles (Fig. 3). In addition, the macromorphology of the solid products corresponds to that of sintered powder particles, in agreement with literature data. For pure NiO, it has also been previously shown that due to poorer interactions of urea with the metal cations, more spherical particles, harder agglomerates, and a higher residual carbon content and ability to sinter were obtained compared to those with glycine.79
 | ||
| Fig. 3 The carbamide–nitrate solution combustion synthesis and macromorphology of the Ce–Ni–O systems obtained at various fuel-to-oxidizer ratios. | ||
At a urea-to-oxidizer ratio equal to 2, the weight gain is 3.2 wt%, which is the largest among all synthesized Ce–Ni–O systems (Fig. 2b). The total weight change in this case is 0.6 wt%, whereas that at φ = 1 and 3 is ca. −6 wt%. It is also important to note that the total weight loss in all cases does not exceed 10.3 wt%, which indicates the efficient course of the combustion process, without the need for further calcination of the samples.
XRD patterns of the obtained Ce–Ni–O systems are presented in Fig. 4. The main reflections correspond to oxide phases of cerium and nickel. After stoichiometric combustion with glycine, in addition to the oxide phases, the presence of metallic nickel is also observed. The metallic phase is also preserved at other fuel-to-oxidizer ratios by limiting the access of oxygen after the completion of combustion.62 It should be noted that at 2θ = 25.7° (Fig. 4c), a low-intensity reflection peak appears, corresponding to the orthorhombic phase of CeNiO3 (111), the presence of which in trace amounts was typical for most samples. Heat treatment at 700 °C for 6 h did not lead to a change in the intensity of this peak; thus, it cannot be related to the residual fuel or products of its decomposition. Previously, CeNiO3 was reported to be active in dry methane reforming (DRM) to produce syngas.82 However, during operation, deactivation of the catalyst caused by coke deposits was observed.
 | ||
| Fig. 4 XRD patterns of Ce–Ni oxide systems obtained by combustion of glycine–nitrate (a and c) and carbamide–nitrate (b) solutions at different fuel-to-oxidizer ratios (φ) (where o-CeNiO3 is orthorhombic CeNiO3 perovskite (Materials Project mp-777024), c-CeO2 – cubic ceria (JCPDS card no. 01-075-8371), NiO – JCPDS card no. 00-044-1159, Ni – JCPDS no. 00-004-0850, * samples were obtained with covering after the completion of the combustion). | ||
In the literature, CeNiO3 was synthesized via the co-precipitation method,83 electrospinning with calcination,84 the soft-templated sol–gel method with post-calcination,85 pulsed laser deposition,86 and the self-combustion method.82,87 However, in most cases, a mixture of oxides of Ce and Ni is taken as Ce nickelite due to the similarity of most of their reflections.
From the point of view of thermodynamic stability, CeNiO3 has a positive convex hull energy (Ehull), signifying its instability, with an absolute value of ca. 89 meV per atom, which indicates the release of energy upon decomposition to the stable states CeO2 and NiO.88 Nevertheless, a structure exhibiting E > 0 does not necessarily mean that the structure cannot exist in nature. There are several examples of metastable structures at 0 K that either become stable at higher temperatures or exist as a kinetically-trapped state. Thus, as far as is known, pure cerium nickelite has not yet been obtained. Samples containing CeNiO3 were synthesized by pulsed laser deposition86 and the SCS method.82,87
The quantitative phase composition, the average crystallite size (D), and unit cell parameters for the Ce–Ni oxide systems were determined by the Rietveld refinement method; the results are presented in Tables 1 and S1† (all data). The phase composition was corrected taking into account the weight gain during heating of the sample in air, which corresponds to the oxidation of metallic nickel and amorphous NiO depending on the stoichiometry and content of crystalline nickel oxide.
| N | Fuel | Covering | n(AN)/n(MeN) | φ | Phase composition according to Rietveld/corrected,b wt% | ||
|---|---|---|---|---|---|---|---|
| CeO2 | NiO | Ni | |||||
| Note:a The sample contains traces of the CeNiO3 phase.b Taking into account amorphous nickel oxide and TGA results; n(AN) is the number of moles of ammonium nitrate; n(MeN) is the number of moles of cerium and nickel nitrates. | |||||||
| 1 | Glycine | − | — | 0.72 | 79/67 | 21/33 | 0 |
| 2a | − | — | 1 | 71/67 | 24/32 | 5/1 | |
| 3-1a | − | — | 1.25 | 70/67 | 30/33 | 0 | |
| 5-1a | − | — | 1.5 | 72/67 | 21/31 | 7/2 | |
| 5-2a | + | — | 1.5 | 75/67 | 9/29 | 16/4 | |
| 6-1 | − | — | 2 | 73/67 | 27/33 | 0 | |
| 6-2 | + | — | 2 | 79/67 | 0/29 | 21/4 | |
| 8a | − | 1.54 | 1.6 | 70/67 | 23/32 | 7/1 | |
| 10 | Urea | + | — | 1 | 77/67 | 23/33 | 0/0 |
| 11a | + | — | 2 | 71/67 | 11/21 | 18/12 | |
| 12a | + | — | 3 | 71/67 | 19/no data | 10/no data | |
As expected, covering makes it possible to preserve a larger fraction of metal in the samples (N. 5-1 and 5-2, Table 1). In addition, in this case, the content of amorphous nickel oxide increases from 6–10 to 20–32 wt%, the reason being that the heat released during interactions of nickel with oxygen promotes the crystallization of amorphous nickel oxide. The reproducibility of the phase composition of the samples with the use of covering is shown in Fig. S1.†
Certain correlations between the sizes of Ni, NiO, and CeO2 crystallites, the unit cell parameter of CeO2, and φ were found for the samples prepared using glycine (Fig. 5). The average size of CeO2 crystallites ranges from 4.5 to 35 nm, NiO – from 4.7 to 29 nm, and Ni – from 4.8 to 19 nm, the maximum values of which are obtained at a stoichiometric ratio of glycine and oxidizer. Therefore, the smallest crystals in this study within the investigated range of fuel-to-oxidizer ratios are obtained at φ < 1 and φ ≥ 2. The use of urea as fuel led to similar regularities; however, compared with glycine, the maximum crystallite size of CeO2 was obtained at φ = 2 (Table S1†). Apparently additional studies are required to elucidate the general validity of these observations.
 | ||
| Fig. 5 Dependence of the Ni, NiO, and CeO2 crystallite sizes and CeO2 cell parameter on the fuel (glycine)-to-oxidizer ratio (φ). Blue dots correspond to nickel. | ||
As described previously,89 the crystallite growth occurs due to higher flame temperatures achieved in the reactions with higher fuel-to-oxidizer ratios. Subsequently, smaller crystallite sizes can be related to the increasing release of gases that remove heat from the reaction.65 Because the amount of energy released during combustion depends on the initial fuel and reagents, as well as the resulting products, the maximum crystallite size can be obtained at various values of φ. For example, the dependence of the crystallite size on φ with an extremum close to 2 was previously shown for α-alumina,90 CuFe2O4,91 and (Y1−mCem)2SiO5.92 Considering that the molar enthalpy of urea formation is less than that of glycine,89 it can be supposed that the maximum temperature for urea is reached at a higher fuel-to-oxidizer ratio.
The cell parameter of CeO2 at a glycine-to-oxidizer ratio of 1–1.6 turned out to be lower than 5.419 Å, which indicates the incorporation of nickel ions into the CeO2 lattice and formation of a solid solution. In addition, lattice distortions may indicate the formation of new crystalline phases, including cerium nickelite.
The average sizes of crystallites of all phases turned out to be the largest for the samples containing the intense CeNiO3 (111) reflection in the XRD pattern.
It has been proven that steam and aqueous phase reforming of alcohols are structure-sensitive reactions, i.e., their rate, normalized to the number of exposed metal surface atoms, changes with the particle size.93–95 However, there is no apparent agreement whether smaller of bigger particles are more beneficial for hydrogen production.34,96,97
3.2. Ce–Co–O: synthesis features and phase composition
The synthesis of the cobalt-containing systems, as well as Ce–Ni–O, proceeds with a sparkling flame (Fig. 6, left). As a result, the powder is carried away from the reaction zone, especially at the stoichiometric fuel-to-oxidizer ratio (φ = 1). The abundant gas evolution leads to the formation of fluffy powders that are black at an increased fuel content (φ = 1.5–2). The obtained materials gain weight during heating in air (Fig. 6, left), with the largest weight gain obtained for φ = 2. | ||
| Fig. 6 The glycine–nitrate solution combustion synthesis and macromorphology of the Ce–Co–O systems obtained at various fuel-to-oxidizer ratios and with covering (left, where Δm and WG – the total weight change and the weight gain, respectively). XRD patterns of Ce–Co oxide systems obtained by combustion of glycine–nitrate solutions at different fuel-to-oxidizer ratios (right, where c-CeO2 – cubic ceria (JCPDS card no. 01-075-8371); Co3O4 – JCPDS card no. 00-042-1467; CoO – JCPDS card no. 00-048-1719; Co – JCPDS card no. 00-015-0806). | ||
According to the XRD data, the obtained materials consist mainly of CoO and CeO2 phases (Fig. 6, right). In addition, there are also XRD reflections related to Co and Co3O4. The data for the phase composition, the average crystallite size, and the CeO2 unit cell parameter for Ce–Co oxide systems are given in Table 2.
| φ | Phase composition according to Rietveld/corrected,a wt% | Co (TGA),c wt% | D(CeO2), nm | a(CeO2), Å | D(CoO), nm | D(Co3O4), nm | D(Co), nm | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CeO2 | CoO | Co3O4 | Am. Co oxideb | Co | |||||||
| Note:a Taking into account amorphous cobalt oxide and TGA results.b The amount of amorphous cobalt oxide is equal to the difference between the total amount (according to stoichiometry, no more than 33 wt%) and crystalline cobalt oxide (determined by the Rietveld method) with metallic Co.c Calculated from the weight gain of the sample during heating, equal to the mass of oxygen involved in the oxidation of cobalt according to the equation 3Co + 2O2 = Co3O4; φ is the fuel-to-oxidizer ratio; the ratio of the weighted (Rwp) and expected (Re) R-factors was 0.97–0.98, which characterizes the goodness of fit (if the squared value is equal to one or constant, the refinement procedure is completed); “—” not possible to determine using the Rietveld refinement. | |||||||||||
| 1 | 73/67 | 27/27 | 0/0 | 6 | 0/0 | 0.1 | 24.0 | 5.4094 | 28.1 | 0 | 0 |
| 1.5 | 72/67 | 8/8 | 14/14 | 9 | 6/2 | 2.1 | 21.1 | 5.4119 | 10.6 | 8.9 | 8.2 |
| 2 | 100/67 | 0/0 | 0/0 | 26 | 0/7 | 7.2 | 9.8 | 5.4161 | — | 0 | — |
The phase composition is also corrected for the increase in mass during heating, which corresponds to the oxidation of Co rather than CoO to Co3O4, because oxidation occurred between 145 and 310 °C,98 and amorphous cobalt oxide based on the content of crystalline CoO, Co3O4, and metallic Co. With increasing fuel-to-oxidizer ratio, the fraction of both metal and amorphous cobalt oxide increases. In this case, the average size of CeO2 crystallites decreases, while the lattice parameter increases. In general, for this system, as well as for Ce–Ni–O, the formation of solid solutions at φ = 1–1.5 is typical.
It is noteworthy that the size of the crystalline phases in the Ce–Ni–O and Ce–Co–O systems is approximately the same under the same experimental conditions, which can be explained by the close values of the formation enthalpies for metal nitrates. Compared to the data presented in ref. 99, where combustion of a mixture of cobalt nitrate and glycine was carried out, the crystallites of cobalt and cobalt oxides are smaller in size.
3.3. Structural and textural properties
According to SEM micrographs, the Ce–Ni- and Ce–Co-containing materials obtained by the SCS method using glycine as a fuel are identical and have a typical foamy structure (Fig. 7a–d). Unlike that of glycine, the morphology of the powders obtained by combustion with urea is represented by agglomerates consisting of rounded aggregates (Fig. 7e and f). The formation of such agglomerates for a Ni–CeO2 system was also achieved in ref. 31 and 71. It was found that glycine, being able to interact very well with metal cations, led to more intense combustion, giving a branched wool structure with softer agglomerates compared to urea during the preparation of nickel oxide powders.79 The data for the elemental composition indicate an excess of nickel with respect to cerium (Ni:Ce (N. 3-2) = 53.5:46.5 at%), which means a slight deviation of the nickel oxide content from the targeted one (ca. 32.9 wt% instead of 30%). Based on the arithmetic average content of nickel oxide in four samples (Table S2†), 33 wt% NiO was taken for calculations of the phase composition.
 | ||
| Fig. 7 SEM micrographs of Ce–Ni(Co)–O systems obtained by the SCS method using different fuels (with covering). | ||
The results of studying the porous structure of the as-prepared samples are shown in Fig. S2† and Table 3. With increasing φ, the specific surface area for the Ce–Ni–O systems varies from 8 to 43 m2 g−1 with a minimum at φ = 1, and the pore size changes from 13 to 54 nm with a maximum for the same sample. The observed effect can be explained by the influence of the CeO2 crystallite size, which decreases proportionally with the increase in the specific surface area. The porous characteristics for Ni- and Co-containing systems with the highest content of the amorphous phase (φ = 2*) turned out to be identical. The specific surface area of these systems is ca. 26 and 20 m2 g−1, respectively, with a total pore volume of 0.1 cm3 g−1 and an average pore diameter of 13–15 nm. According to ref. 31, amorphous oxides of Ni and Co can fill the intergranular space of CeO2, thereby blocking small pores with the largest surface.
| Fuel, additive, φ | Ni, wt% | S BET (m2 g−1)/∑Vp (cm3 g−1)/dp (nm) | D(Ni), nm | D(CeO2), nm | Methodc | Ref. |
|---|---|---|---|---|---|---|
| Note:a After reduction.b For NiO.c Determination method of crystallite sizes.d Instead of cerium nitrate, ammonium cerium nitrate was used. | ||||||
| Glycine, 0.72 | 26.3 | 43/0.24/22.5 | — | 6.5 | XRD | This work |
| Glycine, 1 | 26.3 | 8/0.11/53.7 | 18.7 | 35.2 | XRD | This work |
| Glycine, 2* | 26.7 | 26/0.09/13.3 | 4.8 | 9.5 | XRD | This work |
| Glycine, NaCl, 1–1.1 | 46.3 | 77/0.18/9.9 | 7.2a | 11.2a | XRD | 67 |
| Glycine, 1 | 15.1 | 21/0.06/12.6 | 19.2a | 10.4 | XRD/CO chemisorption | 100 |
| Glycine, 2 | 5.8 | 12/—/13.2 | 16.2a | — | TEM | 70 |
| Glycine, 0.7 | 5.4 | 7/0.05/— | 22.3a | 40.8a | XRD | 68 |
| Urea, 0.95 | 2.25–2.34 | 72/0.05/3.0 | 2.4a | 10.8 | XRD/TEM | 31 |
| Urea, 0.95 | 8.46–8.71 | 73/0.05/3.0 | — | 9.5 | XRD | 31 |
| Urea, 1d | 7.8 | 49/—/— | 5.5b | 8.6 | XRD | 71 |
| Urea, 1 | 7.8 | 14/—/— | 5.6b | 12.9 | XRD | 71 |
| Urea, 1 | 19.6 | <10/0.01/18.5 | 18.8 ± 1a | — | XRD | 29 |
| Hydrous hydrazine, 0.5 | 5.8 | 12/—/14.8 | 10.3a | — | TEM | 70 |
| Hydrous hydrazine, 2 | 23.6 | 14/—/15.2 | 23.1–23.9a | — | XRD/TEM | 70 |
The characteristics of the systems obtained in this study were compared with the characteristics of Ni/CeO2 previously synthesized by the solution combustion synthesis method (Table 3).
As can be seen, almost all samples differ primarily in nickel content. It was found that the samples prepared in this study at φ = 0.72 and 2*, the most dispersed phases of ceria and nickel, have the highest nickel content and the largest specific surface area among the Ni–CeO2 systems synthesized with glycine without additives. NaCl acts as a diluent and reduces the combustion temperature,89 resulting in an increase in the specific surface area and a decrease in the particle and crystallite sizes even for a highly loaded Ni-based system.67 However, in this case, sodium and chlorine impurities remain in the catalyst, requiring washing.
In the case of urea, the stoichiometric combustion (φ = 0.95–1) mainly leads to a highly developed specific surface area and a small crystallite size at a low nickel content (up to 10 wt%). Other less often used fuels (oxalyldihydrazide, glycerol, carbohydrazide,71 hydrous hydrazine70) result in worse textural properties of the materials.
3.4. Reducibility
Various forms of Ni2+ compounds, the sequence of their reduction, and the interaction type between the metal and CeO2 were determined by H2-TPR. Fig. 8a shows the reduction curves for the Ce–Ni oxide systems obtained at various fuel-to-oxidizer ratios, as well as with and without covering. With increasing φ, the main reduction peak shifts towards lower temperatures, which is explained by an increase in the fraction of amorphous NiO (compared to crystalline), a decrease in the degree of interaction between NiO and CeO2 particles, or an average crystallite size. It is known from previous studies31 that amorphous NiO is distributed in voids between CeO2 agglomerates, subsequently exhibiting poor self-interaction. The peak α shown in Fig. 8a can correspond to the reduction of amorphous nickel oxide, with an increase in the amount of which shifting the peak towards higher temperatures, while the β peak can be attributed to the reduction of fine NiO particles distributed over the CeO2 surface and is characterized by increased interactions with CeO2. | ||
| Fig. 8 H2-TPR curves of the Ce–Ni–O (a) and Ce–Co–O (b) systems, obtained by the SCS method at various fuel-to-oxidizer ratios (φ). | ||
At a stoichiometric fuel-to-oxidizer ratio, the largest NiO crystallites and a CeO2(Ni2+) solid solution are obtained, whose reduction is difficult from the topochemical point of view and occurs at high temperatures (γ and ε peaks, respectively). The high-temperature δ-peaks for the samples obtained at φ = 0.72 and 2 can be explained by the strong interactions of nickel nanoparticles with the ceria surface. Some examples of TPR data modelling assuming the presence of different nickel oxide species are presented in Fig. S3 and Table S3.† Ceria is also reduced in the process, as evidenced by the ratio of the amount of consumed hydrogen to metal (mol H2 per mol Ni, Table S4†) exceeding unity.
Fig. 8b shows the reduction curves for the Ce–Co oxide systems, whose behavior is similar to those of the Ni-containing systems. With increasing φ, the peaks shift towards lower temperatures due to an increase in the amorphous cobalt oxide fraction, a decrease in the interactions between CeO2 and cobalt oxide, and the average size of crystallites. The peaks at 295–334 °C can be associated with the reduction of amorphous cobalt oxide, as well as Co3+ at the contact point between Co3O4 and CeO2.101 According to the literature,102 large Co3O4 crystallites are often reduced to Co0 in one stage. Therefore, the peaks at 385–393 °C can be attributed to both the reduction of CoO particles, formed due to the reduction of Co3O4, and bulk CoO, which weakly interacts with CeO2.
The reduction of the largest CoO crystallites obtained at φ = 1 is difficult and occurs at higher temperatures. In addition, in this case, the reduction profile consists of peaks that have merged into one, which can be explained by the strong interactions of cobalt particles with the surface of cerium oxide. It is also known that cobalt ions can improve the reduction of bulk oxygen CeO2,103 which usually occurs at 800 °C. This phenomenon is interpreted as the transition of hydrogen from cobalt to cerium. The same promoting effect of cerium reduction was reported in the literature.104
Thus, the results of H2-TPR indicate that the most reactive, easily reduced oxides of Co and Ni, which act as active components in many heterogeneous catalytic processes, are obtained with an excess of fuel and covering with a Petri dish after completing combustion to minimize the interactions of solid products with oxygen.
3.5. Chemisorption properties and defectiveness of CeO2
The chemisorption properties and dispersion for various Ce–Ni–O systems were measured using H2- and CO2-TPD. The choice of Ni-containing systems instead of Ce–Co–O is due to a wide variety of conditions for their preparation. Similar dependencies can be expected for both of these systems.The spectra show a broad asymmetric H2 desorption peak resulting from the overlap of signals from metal particles of different diameters, surfaces, or positions (Fig. S4†). The Ce–Ni–O system obtained at stoichiometric φ has the smallest area of low-temperature peaks (<250 °C) associated with H2 weakly chemisorbed on the surface due to highly dispersed Ni and defects. Thus, the highest defect density of the samples can be expected with a deficit and excess of fuel (φ = 0.72 and 2). At the same time, at φ = 0.72, despite the high specific surface area, the dispersion (D) and specific metal surface area (A) are lower than those at φ = 2 (Table 4), which can be explained by the influence of the synthesis conditions. Furthermore, Raman spectroscopy was carried out to identify the differences in defectiveness.
| φ | H2, μmol g−1 | D, % | A, m2 per g Ni | d, nm | SDs/F2g | D2/F2g | D3/F2g | μmol CO2 per ga |
|---|---|---|---|---|---|---|---|---|
| Note:a According to CO2-TPD; D – dispersion; A – metal specific surface area; d – metal diameter; SDs – surface defects; D2 – extrinsic defects; D3 – oxygen vacancies; n/d – no data. | ||||||||
| 0.72 | 129.4 | 5.7 | 38.2 | 17.6 | 0.31 | 1.55 | 1.89 | 199 |
| 1 | 47.0 | 2.1 | 14.0 | 48.2 | 0.00 | 0.19 | 0.36 | 52 |
| 2 | 152.8 | 6.8 | 45.5 | 14.8 | 0.06 | 0.95 | 1.63 | n/d |
| 2* | 156.3 | 6.9 | 45.9 | 14.7 | 0.00 | 1.04 | 1.33 | 261 |
Basic catalyst sites play an important role in the reaction pathways and selectivity during GSR, facilitating the adsorption of CO2 generated during steam reforming of glycerol. According to CO2-TPD, the amount of adsorbed CO2 on the surface of reduced Ni–Ce–O catalysts depends substantially on φ with a minimum at φ = 1 (Table 4).
Raman spectra of the as-prepared Ce–Ni–O systems are presented in Fig. 9. It should be noted that the studied samples are generally homogeneous, but large particles (10–20 μm) are visible by optical microscopy. They have also been studied at several random points and their spectra are well reproduced (Fig. S5†). Each spectrum contains an intense band at 462 ± 2 cm−1 associated with the F2g mode (triply degenerate) of CeO2. According to ref. 105, the F2g band belongs to the symmetrical stretching vibration of CeO8 units.
 | ||
| Fig. 9 Raman spectra with deconvolution for Ce–Ni–O systems prepared at different φ values. | ||
In addition, for all samples, the D band phonon (or LO mode) in the region of 500–700 cm−1 is observed, indicating the presence of CeO2 bulk defects, which can be divided into three bands: 591 ± 2 (D1, Fig. 9, red area), 631 ± 2 (D2, Fig. 9, yellow area), and 553–554 (D3, Fig. 9, blue area). According to ref. 106, the D1 band is attributed to “internal” defects already present in pure ceria, for example, a Frenkel type oxygen vacancy generated by the migration of oxygen anions from tetrahedral sites to octahedral sites. The D2 band is related to “external” defects generated by the Ni addition, while the third component (D3) is usually assigned to oxygen vacancies associated with the presence of Ce3+ cations. The spectra also show broad signals from NiO at 419–424 cm−1 (TO mode) and 509–516 cm−1 (LO mode).107,108
It was found that only two samples (φ = 0.72 and 2) had Raman bands at ca. 224 cm−1, which is associated with surface defects (SDs), namely, the Ce–OH vibration.105 As noted in ref. 105, the highest intensity of this peak is observed for samples with smaller crystals and a larger specific surface area.
The defect amount in the four samples was estimated by calculating the ratios between the area of the bulk (D2, D3) or surface defects (SDs) and the F2g mode; these parameters, called D1/F2g, D2/F2g, and SDs/F2g, respectively, are summarized in Table 4. The obtained data confirm that a fuel deficiency and excess contribute to the formation of both bulk and surface defects. At the same time, for the sample obtained at φ = 2* (with covering), no surface defects were found, which can be explained by the presence of surface metal particles.
3.6. Activity in GSR
The initial screening of the catalysts was performed in glycerol steam reforming, which is due to the simplicity, as well as the possibility of selecting the most efficient catalysts for their subsequent study in glycerol aqueous-phase reforming. The samples that differ significantly in the dispersity and amorphization of the phases were selected. The same fuel (glycine) for synthesis was chosen to compare the different catalytic systems. Fig. 10a and b show the data for hydrogen yield as a function of time-on-stream for the selected compositions. | ||
| Fig. 10 Hydrogen yield versus time-on-stream for Co- (a) and Ni-containing (b) systems; and dependence of the rate of hydrogen formation on the glycine-to-oxidizer ratio (c). Conditions: mcat = 40 mg, Vlq(water + glycerol) = 7.3–7.9 mL h−1, V(N2) = 30 mL min−1, T = 520 °C. Because of a limited number of data points, the statistical analysis of the parameters, determined using eqn (10), was reliable just only in a few cases indicated by errors for the corresponding constants. In other cases, an exact approximation of the experimental points did not allow determination of the parameter errors. | ||
In terms of hydrogen yield, the nickel systems are more active than the cobalt systems. Both systems are characterized by the following general trends: the samples with the highest initial activity (time-on-stream of 15 min) undergo rapid deactivation in the first hour of testing. For them, in general, a higher value of the deactivation constant is obtained (Fig. 10a and b). This is explained by the partial encapsulation by carbonaceous deposits of metal Ni and Co nanoparticles localized on the surface and between CeOx aggregates.21 The lack of contact between the metal particles and the defective support, as well as the low process temperature, makes it difficult to remove coke during the reaction with steam. It should be noted that catalysts with this feature are characterized by increased residual activity (Fig. 10b, time-on-stream is more than 1 h), which exceeds the residual activity of the other systems studied in this work.
Comparison of systems in terms of the hydrogen formation rate r(H2), at which the influence of the deactivation is minimal, shows its extreme dependence on φ (Fig. 10c) with minima at φ = 1 and 1.5. Results in the Co–Ni series are in line with the experimental and theoretical work on methane steam reforming.109 Namely an increase of activity depending on the metal can be related to the energy of oxygen adsorption on the surface.
The highest initial rate of hydrogen formation was shown by those Ce–Ni–O and Ce–Co–O systems, which are characterized by a high dispersion of Ni and Co nanoparticles, which is provided by moderate combustion conditions with a deficit or excess of fuel (φ < 1 and φ ≥ 2), and the increased dispersion and defectiveness of CeO2 in combination with an increased specific surface area. At the same time, the Ce–Ni(Co)–O samples with a large crystallite size (φ = 1–1.5) are characterized by a low initial hydrogen formation rate, including the Ce–Ni–O sample obtained at φ = 1 containing the largest amount of perovskite o-CeNiO3.
The experiments also showed that the covering that prevents the oxidation of fine Ni particles makes it possible to exclude the reductive activation stage of the sample with hydrogen directly in the catalytic reactor before testing without a loss of activity and stability.
The Ce–Ni–O systems obtained at φ = 0.72, 2, and 2* have a hydrogen yield close to the highest thermodynamically possible value for these conditions (88.5% or 6.2 mol mol−1). The calculation of the Mears and Weisz–Prater criteria demonstrated that during experiments, there were no internal and external diffusion limitations. To establish the maximum productivity of these samples, additional experiments were carried out with a lower catalyst loading, as well as a high flow of the feedstock. Fig. S6† shows the yield of glycerol to gaseous products and the integral rates of hydrogen formation r(H2) and glycerol transformation r(gly) as a function of WHSV. The rates have been calculated at 4 min of time on stream, and thus the dependence of the integral rates on WHSV is related to a non-zero order reaction rather than deactivation. Subsequently, the turnover frequency (TOF) was calculated, showing an increase of TOF in the presence of surface defects (Table 5), which is consistent with the literature.
| Catalyst | Synthesis method | Ni loading, wt% | d(Ni)XRD or TEM, nm | d(Ni)Chem, nm | SDs/F2g | T, °C | TOFH2, s−1 | TOFgly, s−1 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 2500 h−1 | 60 h−1 | 2500 h−1 | 40–60 h−1 | |||||||
| Note: SCS – solution combustion synthesis; WI – wet impregnation; d(Ni)XRD or TEM – the mean diameter of Ni particles calculated from XRD or TEM; d(Ni)Chem – the mean diameter of Ni particles calculated from chemisorption data; SDs – surface defects (Table 4). | ||||||||||
| Ni/CeO2φ = 0.72 | SCS | 26.3 | 6.4 (NiO) | 17.6 | 0.31 | 520 | 10.9 | 1.4 | 2.1 | 0.33 |
| Ni/CeO2φ = 2 | SCS | 26.3 | 6.1 (NiO) | 14.8 | 0.06 | 520 | 11.5 | 1.4 | 2.1 | 0.19 |
| Ni/CeO2φ = 2* | SCS | 26.7 | 4.8 | 14.7 | 0.00 | 520 | 9.0 | 1.1 | 1.7 | 0.26 |
| Ni/CeO2 (ref. 31) | SCS | 6.8 | 2.6 | — | 0.47 | 550 | — | — | — | 0.06 |
| Ni/CeO2 (ref. 22) | WI | 11.6 | — | 16.5 | — | 600 | — | — | — | 0.31 |
The surface anion vacancies of CeO2 are the activation sites for additional water molecules involved in the catalytic cycle. TOFGly was also found to increase with increasing Ni particle size at a constant Ni content. Similar regularities were obtained in ref. 42 and 96. These results suggest that steam reforming is a structure sensitive reaction. Comparison of TOF values shows that the catalysts obtained in this work have the highest values among other Ni/CeO2 systems (Table 5 and data presented in ref. 31). These values are obtained at a relatively low reforming temperature.
3.7. Selectivity in GSR
The series of performed tests also made it possible to compare the synthesized systems in terms of selectivity to the main (H2, CO2, CO) and side (C1–C3 hydrocarbons) reaction products. The specified selectivity values do not take into account coke formation. Systems containing Ni and Co are generally characterized by a positive slope of the S(H2)–XGgas dependence (Fig. 11a), which corresponds to the consecutive reactions of H2 formation. A simultaneous decrease of S(H2) and XGgas is associated with a lower contribution of WGS or an increase in the reverse WGS (RWGS, CO + H2O ↔ CO2 + H2). This is also evidenced by a decrease in S(CO) and an increase in S(CO2) with elevation of XGgas (Fig. 11b and c). Extrapolation of S(CO2) and S(CO) to close to zero glycerol conversion shows that these products are formed directly from glycerol as a result of two different reactions – steam reforming and thermal decomposition, respectively:110| C3H8O3 + 3H2O = 3CO2 + 7H2, ΔG0298 = −48.85 kJ mol−1 | (13) |
| C3H8O3 = 3CO + 4H2, ΔG0298 = 37.04 kJ mol−1 | (14) |
 | ||
| Fig. 11 Dependencies of selectivity to the main gaseous products (a–c), and H2/CO2 (d) and H2/CO (e) ratios on glycerol conversion into gaseous products (XGgas). Conditions: mcat = 40 mg, Vlq(water + glycerol) = 7.3–7.9 mL h−1, V(N2) = 30 mL min−1, T = 520 °C. | ||
The most active Ce–Ni–O catalysts exhibited a higher S(CO2) and, correspondingly, a lower S(CO) at the same XGgas compared to Ce–Co–O. Therefore, the Ce–Ni–O systems obtained at φ = 0.72 and 2 make it possible to achieve the highest H2/CO ratio (Fig. 11e), closest to the stoichiometric one, with complete conversion of glycerol into CO2 and H2 (H2/CO2 = 2.33, Fig. 11d). For these Ce–Ni–O systems (φ = 0.72 and 2), an increased basicity is observed, which affects CO2 adsorption and, consequently, the H2/CO2 ratio. However, in addition to basicity, with a change in φ, other properties also change, such as defectiveness, dispersion, and crystallite size, affecting the activity and selectivity. Therefore, the selectivity cannot be attributed solely to basicity.
There is a very small amount of data in the literature for steam glycerol reforming on cobalt–ceria systems. All available results of the activity of various cobalt-based systems are presented in Table 6. Reaction conditions significantly affect activity and selectivity. The experimental conditions closest to those in this work were used in ref. 111 for 15Co/Al2O3, which demonstrated a significantly low glycerol conversion into gaseous products and H2 selectivity compared to 26.3Co/CeO2 synthesized by SCS. Data comparison at approximately the same reaction temperature (500–525 °C) shows that 9.2 wt% Co on Ca–HTlc has a higher hydrogen yield due to lower WHSV.116 However, the samples obtained in this work demonstrated a rather high activity and selectivity upon a nonproportional elevation of WHSV, pointing out a higher productivity.
| Ref. | Catalyst | Synthesis method | T, °C | WHSV, h−1 | TOS, min |
|
Y(H2), % | S(H2), % |
|---|---|---|---|---|---|---|---|---|
| Note:a GHSV, mL g−1 h−1.b Supercritical water reforming of glycerol at 25 MPa.c Ni and Co species in the cave of the support; “≈” – values were calculated from the data presented in the literature; WI – wetness impregnation; CP – co-precipitation; P – precipitation; UWI – ultrasonic wetness impregnation. | ||||||||
| 50 | 15Co/CeO2 | WI | 500 | ≈2.7 | — | 100* | — | 92 |
| 111 | 15Co/Al2O3 | WI | 500 | 50000a |
— | ≈12 | ≈77 | ≈64 |
| 112 | 10Co/YSZ | WI | 525 | 6.45 | 230 | 94*b | 53b | — |
| 10.8 | 87*b | 36b | — | |||||
| 113 | 10Co/HAp | WI | 650 | ≈22 | 60 | 92* | — | 64 |
| 10Co–10Ce/HAp | 96* | — | 63 | |||||
| 114 | 7Co/SBA-15 | WI | 600 | 7.7 | ≈60/600 | ≈62*/56* | — | ≈91/92 |
| 7Co–8.5Ce/SBA-15 | ≈60/600 | ≈70*/64* | — | 100/100 | ||||
| 115 | La0.7Ce0.3CoO3 | CP | 500 | ≈5.1 | 60 | ≈90 | ≈51 | ≈56 |
| 116 | 9.2Co–Ca–HTlc | P | 500 | 5600a | 120 | ≈86* | ≈86 | — |
| 117 | 5Ni–5Co/CNTsc | UWI | 400 | ≈41 | 480 | 96* | — | 87 |
| This work | 26.3Co/CeO2 | SCS, φ = 2* | 520 | 197 (≈63000a) |
15 | 58 | 52 | 89 |
| 90 | 36 | 31 | 84 | |||||

For a more detailed understanding of the differences in the catalytic action of the synthesized systems, the S–XGgas dependencies for by-products were also analyzed (Fig. 12). The presence of a slope in the graphs of S–X dependencies for ethane, ethylene, and propane indicates that they are formed directly from glycerol as a result of consecutive dehydration and hydrogenation reactions of intermediate products. The decrease in selectivity to C2–C3 products indicates the possible occurrence of their reforming as well, being apparently more prominent with increasing glycerol conversion. Ni-containing systems are characterized by the lowest selectivity for these products. At the same time, for Ce–Ni–O (φ = 0.72 and 2), an increased S(CH4) is observed, which is apparently formed mainly as a result of CO and/or CO2 hydrogenation. This is evidenced by a drop in S(H2) and S(CO2), a stop in the decrease in S(CO) at XGgas > 50%, and a growth of S(H2) at the same time to diminish S(CH4) and XGgas. Moreover, a non-zero S(CH4) for a near-zero XGgas indicates the existence of some additional route for the formation of methane directly from glycerol.
 | ||
| Fig. 12 Dependencies of selectivity to by-products on glycerol conversion into gaseous products. | ||
Based on the obtained results and the literature data,42,118 a network of the main reactions in the gas phase can be proposed as shown in Fig. 13. The scheme is divided into three main paths of formation: (1) methane from glycerol and syngas or via RWGS; (2) C2-gases from ethanol; (3) C3-gases from 2-propanol.
 | ||
| Fig. 13 The reaction network in GSR. | ||
The spent catalysts were examined by simultaneous thermal analysis (Fig. S7†) and XRD (Fig. S8†) to determine the amount and type of coke deposits, as well as changes in the phase composition and crystallinity. The obtained thermograms for the Ce–Ni systems clearly show two exothermic peaks in the temperature ranges of 200–400 and 400–600 °C. The exothermic effect of nickel oxidation is also superimposed on the first peak, since an increase in mass occurs in this range. According to the literature,119,120 amorphous coke burns up to 550 °C, while graphite-like and/or filiform coke burns out at higher temperatures. At the same time, the presence of amorphous coke is observed for almost all samples (except for Ce–Ni–O obtained at φ = 1.5*), and “heavy” coke is observed for samples with a high dispersion of Ni nanoparticles (φ = 0.72, 2, 2*), which is consistent with their highest activity in glycerol steam reforming. For the sample obtained at φ = 0.72, the highest content of coke was observed due to a higher specific surface area. At the same time, according to the hydrogen yield, the maximum coke formation was not reached for all samples, since there was no sharp activity drop depending on TOS (Fig. 10). The exo-effects of the oxidation of Co and CoO are observed for all spent Ce–Co samples, except for Ce–Co at φ = 2, for which amorphous coke burns out at 277 °C.
XRD patterns of the spent catalytic systems (Fig. S8†) indicate the transition of cobalt and nickel oxides to a metallic form, which, as a rule, do not undergo strong enlargement during testing (with the exception of the most amorphous systems), as well as maintaining the size of cerium oxide crystallites.
3.8. Activity and selectivity in GAPR
Based on the results obtained in the previous section, it was decided to conduct further studies of the most active Ce–Ni–O (φ = 2*) and Ce–Co–O (φ = 2*) systems and compare them with a platinum–alumina catalyst in aqueous-phase reforming of glycerol. Fig. 14 displays the glycerol conversion and the yield of hydrogen as a function of time-on-stream for the studied systems. In all cases, an increase in conversion was observed in the first 3–6 hours of testing, which is associated with the filling of the entire system with generated gaseous products and a large set-up volume (a separator volume of 100 mL). A similar effect was observed in ref. 74.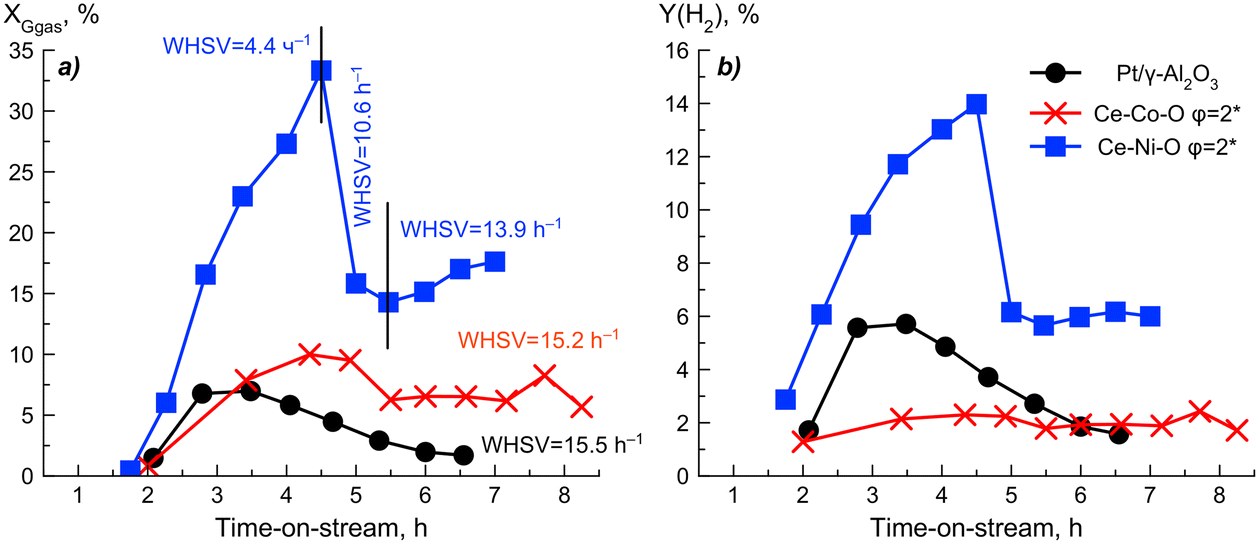 | ||
| Fig. 14 Glycerol conversion into gaseous products (a) and hydrogen yield (b) over time-on-stream. Conditions for Pt-containing catalysts: Tred = 280 °C, mcat = 200 mg, Vlq = 3 mL h−1, WHSV = 15.5 h−1. Conditions for Ce–Ni–O: Tred = 280 °C, mcat = 668 mg, Vlq = 3, 7.2 mL h−1, WHSV = 4.4, 10.6 h−1; mcat = 635 mg, Vlq = 9.8 mL h−1, WHSV = 13.9 h−1. Conditions for Ce–Co–O: Tred = 330 °C, mcat = 635 mg, Vlq = 9.8 mL h−1, WHSV = 15.2 h−1. | ||
It was found that the Ce–Ni–O system displays a significantly higher glycerol conversion and hydrogen yield compared to Pt/Al2O3. In general, the activity of the studied systems during aqueous-phase reforming increases in the same order as that during steam reforming of glycerol, namely: Co < Ni. A feature of Ce–Co–O compared to Ce–Ni–O, as well as Pt/Al2O3, is a low hydrogen yield at a relatively high conversion of glycerol into gaseous products. The Ce–Ni-containing system makes it possible to achieve a hydrogen yield comparable to that of Pt/Al2O3 at a close WHSV = 13.9–15.5 h−1 without undergoing significant deactivation. In addition, this sample does not reach the maximum rate of hydrogen formation at T = 231 °C (Fig. 15).
 | ||
| Fig. 15 Dependence of the H2 formation rate and hydrogen yield on the WHSV of the glycerol–water mixture for the Ce–Ni–O sample (φ = 2*). | ||
Additional information about the process, as well as on differences between the systems, is provided by the analysis of the S–XGgas dependencies (Fig. 16): as a general trend, a higher conversion (catalyst activity) at a close WHSV = 13.9–15.5 h−1 leads to more prominent side reactions: RWGS and CO and/or CO2 methanation. This is evidenced by the increase of S(CO) and S(CH4) (Fig. 16c and d), the decrease in S(H2) and S(CO2) (Fig. 16a and b), and the H2/CO ratio (Fig. 16e). In this case, the direct reaction of glycerol with water with the formation of H2 and CO2 (S(CO2) → 100%, S(CO) → 0% at XGgas → 0%) prevails, in contrast to glycerol steam reforming. The different character of the S–XGgas dependencies during testing of Ce–Ni–O with a lower WHSV = 4.4 h−1 is apparently associated with the non-stationary composition of the gas mixture at the outlet due to the filling of the setup volume with the gaseous products.
 | ||
| Fig. 16 Dependence of selectivity for all gas reaction products (a–d, g and h) and H2/CO (e) and H2/CO2 (f) ratios on glycerol conversion into gaseous products. | ||
The presence of methane, ethane, and propane in the reaction products causes a lower H2/CO2 ratio (Fig. 16f) for the Ce–Ni–O and Ce–Co–O catalysts compared to the stoichiometric one due to the occurrence of side reactions of dehydration, including liquid products, and subsequent hydrogenation.42,121 The systems under study are characterized by a higher selectivity to C2 hydrocarbons in GAPR compared to that in GSR (Fig. 16g and 12), which is related to the occurrence of decarbonylation leading to additional formation of CO from the liquid products of glycerol dehydrogenation.42 A high selectivity to propane for Ce–Co–O in combination with a low S(H2) and H2/CO2 ratio due to hydrogenation of propene explains the lower hydrogen yield with an increased glycerol conversion into gaseous products (Fig. 16). It should also be noted that the increased selectivity to methane and C2 hydrocarbons for Ce–Ni–O and Ce–Co–O, obtained in glycerol steam reforming, was also seen in aqueous-phase reforming. It additionally shows the similarity of these processes.
The selected test conditions – temperature, pressure, and WHSV, made it possible to compare the catalyst performance with those in the literature (Table 7), which for the Ce–Ni–O sample (φ = 2*) was similar to or even surpassed those for some nickel catalysts prepared by precipitation or impregnation in glycerol conversion into gaseous products, the H2 yield and the rate of hydrogen formation, as well as selectivity to H2 and CO2. Such a result is ensured by both the high dispersion of nickel compared to other systems, despite the high content of Ni, and interactions of Ni with CeOx.
| This work | 122 | 30 | 123 | 124 | ||
|---|---|---|---|---|---|---|
| Synthesis method | Ce–Ni–O (φ = 2*), SCS | Ni/MgAl (hydrotalcite), co-precipitation | NiO/CeO2, co-precipitation | Ni/γ-Al2O3, impregnation | NiAl2O4, co-precipitation | |
| Ni0, nm | 4.8 (XRD) | 12.0 (TEM) | 5.3 (XRD) | — | 12.0 (TEM) | |
| Ni, wt% | 26.7 | 23.0 | 14.9 | 20.0 | 42.3 | |
| Conditions | ||||||
| T, °C | 231 | 250 | 250 | 225 | 235 | |
| P, bar | 34 | 35 | 20 | 23 | 35 | |
| WHSV, h−1 | 4.4 | 13.9 | 5.0 | 5.0 | 4.0 | 24.5 |
| Glycerol, wt% | 10.0 | 7.8 | 15.0 | 10.0 | 10.0 | |
| Process indicators | ||||||
| X Ggas, % | 33.3 | 17.6 | ≈16.0 | 17.3 | ≈44.0 (full) | ≈27.0 |
| Y(H2), % | 14.0 | 6.0 | 10.1 | — | — | ≈8.1 |
| r(H2), μmol mol−1 Ni per s | 0.343 | 0.438 | 0.088 | — | — | 0.154 |
| S(H2), % | 41.9 | 39.6 | 31.7 | 95.7 | ≈42.0 | ≈35.0 |
| S(CO2), % | 69.5 | 68.3 | 72.5 | 56.1 | — | 55.9 |
| S(CO), % | 4.2 | 8.0 | 3.5 | 36.8 | — | 1.70 |
| S(CH4), % | 24.8 | 20.9 | 24.1 | 7.1 | — | 42.4 |
| H2/CO2 | 1.41 | 1.35 | 1.02 | — | — | 1.80 |
Conclusions
Currently, glycerol represents an emerging renewable bio-derived source that can be used to produce H2 through steam (GSR) or aqueous-phase reforming (GAPR). A recent focus in catalytic reforming is improving catalysts and processes to maximize the catalyst lifetime and H2 selectivity. For this purpose, in the present work, new Ce–Ni–O and Ce–Co–O catalytic systems (the content of Ni and Co, calculated as metal(II) oxides, is 30 wt%) were obtained and characterized. The synthesis was performed in one step by the SCS (solution combustion synthesis) method using different fuels and amounts relative to the oxidizing agents (φ = 0.7–3). Due to an increased and decreased glycine-to-oxidizer ratio (φ < 1; φ ≥ 2), it was possible to avoid the formation of solid solutions characteristic of these systems and achieve the most dispersed phases, which is important, because glycerol reforming is a structure-sensitive reaction. It has been established for the first time that covering the beaker with a Petri dish immediately after the completion of the combustion makes it possible to retain a larger amount of the metal phase in the samples and to increase the fraction of amorphous Ni or Co oxides. Thus, by changing the fuel-to-oxidizer ratio and the excess of oxygen, various forms of Ni or Co and their interactions with CeO2 were obtained, which affected the reduction sequence, defectiveness, porous characteristics, and chemical reactivity.The synthesized materials were tested in glycerol steam reforming at atmospheric pressure and 520 °C. It has been shown that the Ce–Ni–O and Ce–Co–O systems characterized by a high crystallinity of phases (obtained at φ < 1 and φ ≥ 2) have the highest activity and H2 yield in GSR. The Ce–Ni–O systems, in contrast to Ce–Co–O, make it also possible to achieve the H2/CO2 ratio closest to the stoichiometric one (2.33) with complete conversion of glycerol into CO2 and H2. Prevention of the material oxidation during synthesis makes it possible to exclude the reductive activation stage of the samples before testing without a loss of activity and stability. On the other hand, according to Raman spectroscopy, this negatively affected the defective structure of cerium oxide.
Additional experiments carried out at increased WHSV for Ce–Ni–O, which was obtained with a fuel deficiency (φ = 0.72), resulted in a higher TOF due to a large number of surface defects. In addition, such a result can be associated with the largest Ni particle size among other systems according to H2 chemisorption. However, in terms of the metal dispersion and the amount of coke deposits, this sample is inferior to the others.
The activity of the studied systems (obtained at φ = 2 without a posteriori oxidation) in aqueous-phase reforming of glycerol (34 bar, 231 °C) increased in the same order as that in GSR, namely, Co < Ni. With the Ni-containing system, it was possible to achieve a hydrogen yield comparable to that of Pt/γ-Al2O3 at a close WHSV without undergoing noticeable deactivation. The glycerol conversion into gaseous products, the yield and rate of hydrogen formation, and the selectivity to H2 and CO2 were also comparable.
In conclusion, the SCS-derived Ni/CeO2 systems have significant potential as catalysts because of a variety of tunable textural and surface properties. Undoubtedly, further research is required to optimize the synthesis conditions for each specific case.
Author contributions
ANM – conceptualization, investigation, visualization, and writing – original draft. SOO – formal analysis, investigation, and visualization. MAG – investigation. ADT – investigation. JW – formal analysis. DYM – conceptualization and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Russian Science Foundation (grant number 22-23-20094, https://rscf.ru/project/22-23-20094/, accessed on 18 June 2023) and the St. Petersburg Science Foundation (agreement number 26/2022 from 14 April 2022). The authors would like to thank MSc Maria Chebanenko (Ioffe Institute) and MSc Maria Serazhim (St. Petersburg State Institute of Technology (Technical University)) for performing N2 physisorption measurements.Notes and references
- A. G. Adeniyi and J. O. Ighalo, Chem. Pap., 2019, 73, 2619–2635 CrossRef CAS.
- P. J. Megía, A. J. Vizcaíno, J. A. Calles and A. Carrero, Energy Fuels, 2021, 35, 16403–16415 CrossRef.
- N. A. Roslan, S. Z. Abidin, A. Ideris and D. V. N. Vo, Int. J. Hydrogen Energy, 2020, 45, 18466–18489 CrossRef CAS.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- M. Anitha, S. K. Kamarudin and N. T. Kofli, Chem. Eng. J., 2016, 295, 119–130 CrossRef CAS.
- C. A. G. Quispe, C. J. R. Coronado and J. A. Carvalho, Renewable Sustainable Energy Rev., 2013, 27, 475–493 CrossRef CAS.
- Z. Pirzadi and F. Meshkani, Fuel, 2022, 329, 125044 CrossRef CAS.
- A. Fasolini, D. Cespi, T. Tabanelli, R. Cucciniello and F. Cavani, Catalysts, 2019, 9, 722 CrossRef CAS.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS.
- J. Chen, J. Sun and Y. Wang, Ind. Eng. Chem. Res., 2017, 56, 4627–4637 CrossRef CAS.
- K. N. Papageridis, G. Siakavelas, N. D. Charisiou, D. G. Avraam, L. Tzounis, K. Kousi and M. A. Goula, Fuel Process. Technol., 2016, 152, 156–175 CrossRef CAS.
- C. K. Cheng, S. Y. Foo and A. A. Adesina, Catal. Today, 2011, 178, 25–33 CrossRef CAS.
- N. D. Charisiou, K. N. Papageridis, G. Siakavelas, L. Tzounis, K. Kousi, M. A. Baker, S. J. Hinder, V. Sebastian, K. Polychronopoulou and M. A. Goula, Top. Catal., 2017, 60, 1226–1250 CrossRef CAS.
- A. Bshish, Z. Yaakob, A. Ebshish and F. H. Alhasan, J. Energy Resour. Technol., 2013, 136, 012601 CrossRef.
- T. Wang, H. Ma, L. Zeng, D. Li, H. Tian, S. Xiao and J. Gong, Nanoscale, 2016, 8, 10177–10187 RSC.
- O. Shtyka, Z. Dimitrova, R. Ciesielski, A. Kedziora, G. Mitukiewicz, J. Leyko, W. Maniukewicz, A. Czylkowska and T. Maniecki, React. Kinet., Mech. Catal., 2021, 132, 907–919 CrossRef CAS.
- S. Anil, S. Indraja, R. Singh, S. Appari and B. Roy, Int. J. Hydrogen Energy, 2022, 47, 8177–8213 CrossRef CAS.
- M. L. Dieuzeide, M. Jobbagy and N. Amadeo, Int. J. Hydrogen Energy, 2014, 39, 16976–16982 CrossRef CAS.
- E. A. Sanchez and R. A. Comelli, Int. J. Hydrogen Energy, 2014, 39, 8650–8655 CrossRef CAS.
- C. Italiano, K. Bizkarra, V. L. Barrio, J. F. Cambra, L. Pino and A. Vita, Int. J. Hydrogen Energy, 2019, 44, 14671–14682 CrossRef CAS.
- S. Adhikari, S. D. Fernando and A. Haryanto, Renewable Energy, 2008, 33, 1097–1100 CrossRef CAS.
- S. Adhikari, S. D. Fernando, S. D. Filip To, R. Mark Bricka, P. H. Steele and A. Haryanto, Energy Fuels, 2008, 22, 1220–1226 CrossRef CAS.
- K. K. Pant, R. Jain and S. Jain, Korean J. Chem. Eng., 2011, 28, 1859–1866 CrossRef CAS.
- E. V. Matus, A. S. Shlyakhtina, O. B. Sukhova, I. Z. Ismagilov, V. A. Ushakov, S. A. Yashnik, A. P. Nikitin, P. Bharali, M. A. Kerzhentsev and Z. R. Ismagilov, Kinet. Catal., 2019, 60, 221–230 CrossRef CAS.
- C. D. Dave and K. K. Pant, Renewable Energy, 2011, 36, 3195–3202 CrossRef CAS.
- A. K. Seriyala, S. Appari and B. Roy, Mater. Today: Proc., 2022, 76(Part 2), 279–288 Search PubMed.
- A. K. Seriyala, A. Rao, C. Leclerc, S. Appari and B. Roy, Int. J. Hydrogen Energy, 2023, 48, 15533–15554 CrossRef CAS.
- Y. Wang, S. Zhu, S. He, J. Lu, J. Liu, H. Lu, D. Song and Y. Luo, Nanomaterials, 2022, 12, 816 CrossRef CAS PubMed.
- R. L. Manfro, A. F. Da Costa, N. F. P. Ribeiro and M. M. V. M. Souza, Fuel Process. Technol., 2011, 92, 330–335 CrossRef CAS.
- H.-J. Lee, G. S. Shin and Y.-C. Kim, Korean J. Chem. Eng., 2015, 32, 1267–1272 CrossRef CAS.
- Sh. O. Omarov, K. D. Martinson, A. N. Matveyeva, M. I. Chebanenko, V. N. Nevedomskiy and V. I. Popkov, Fuel Process. Technol., 2022, 236, 107429 CrossRef CAS.
- D. G. Araiza, A. Gómez-Cortés and G. Díaz, Catal. Today, 2020, 349, 235–243 CrossRef CAS.
- F. Wang, L. Zhang, J. Deng, J. Zhang, B. Han, Y. Wang, Z. Li, H. Yu, W. Cai and Z. Deng, Fuel Process. Technol., 2019, 193, 94–101 CrossRef CAS.
- M. Greluk, W. Gac, M. Rotko, G. Słowik and S. Turczyniak-Surdacka, J. Catal., 2021, 393, 159–178 CrossRef CAS.
- B. Wang, Y. Xiong, Y. Han, J. Hong, Y. Zhang, J. Li, F. Jing and W. Chu, Appl. Catal., B, 2019, 249, 257–265 CrossRef CAS.
- Y. Wang, S. Zhu, J. Lu, S. He, H. Lu, D. Song, D. Chen and Y. Luo, Fuel Process. Technol., 2023, 243, 107677 CrossRef CAS.
- M. Konsolakis, Z. Ioakimidis, T. Kraia and G. E. Marnellos, Catalysts, 2016, 6, 39 CrossRef.
- I. Rossetti, A. Gallo, V. DalSanto, C. L. Bianchi, V. Nichele, M. Signoretto, E. Finocchio, G. Ramis and A. di Michele, ChemCatChem, 2013, 5, 294–306 CrossRef CAS.
- J. Vicente, J. Ereña, C. Montero, M. J. Azkoiti, J. Bilbao and A. G. Gayubo, Int. J. Hydrogen Energy, 2014, 39, 18820–18834 CrossRef CAS.
- A. Carrero, J. A. Calles, L. García-Moreno and A. J. Vizcaíno, Catalysts, 2017, 7, 55 CrossRef.
- V. Nichele, M. Signoretto, F. Pinna, F. Menegazzo, I. Rossetti, G. Cruciani, G. Cerrato and A. Di Michele, Appl. Catal., B, 2014, 150–151, 12–20 CrossRef CAS.
- Sh. O. Omarov, D. A. Sladkovskiy, K. D. Martinson, M. Peurla, A. Aho, D. Yu. Murzin and V. I. Popkov, Appl. Catal., A, 2021, 616, 118098 CrossRef CAS.
- N. D. Charisiou, G. Siakavelas, L. Tzounis, B. Dou, V. Sebastian, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, Int. J. Hydrogen Energy, 2020, 45, 10442–10460 CrossRef CAS.
- B. Jiang, L. Li, Z. Bian, Z. Li, Y. Sun, Z. Sun, D. Tang, S. Kawi, B. Dou and M. A. Goula, Int. J. Hydrogen Energy, 2018, 43, 13200–13211 CrossRef CAS.
- Y. C. Sharma, A. Kumar, R. Prasad and S. N. Upadhyay, Renewable Sustainable Energy Rev., 2017, 74, 89–103 CrossRef CAS.
- D. L. Trimm, Catal. Today, 1997, 37, 233–238 CrossRef CAS.
- Z. Xiao, Y. Li, F. Hou, C. Wu, L. Pan, J. Zou, L. Wang, X. Zhang, G. Liu and G. Li, Appl. Catal., B, 2019, 258, 117940 CrossRef CAS.
- Z. Xiao, C. Wu, L. Wang, J. Xu, Q. Zheng, L. Pan, J. Zou, X. Zhang and G. Li, Appl. Catal., B, 2021, 286, 119884 CrossRef CAS.
- M. D. Zhurka, A. A. Lemonidou and P. N. Kechagiopoulos, Catal. Today, 2021, 368, 161–172 CrossRef CAS.
- B. Zhang, X. Tang, Y. Li, Y. Xu and W. Shen, Int. J. Hydrogen Energy, 2007, 32, 2367–2373 CrossRef CAS.
- J. Llorca, N. Homs, J. Sales and P. Ramírez de la Piscina, J. Catal., 2002, 209, 306–317 CrossRef CAS.
- H. Song and U. S. Ozkan, J. Catal., 2009, 261, 66–74 CrossRef CAS.
- I. Ilgaz Soykal, H. Sohn and U. S. Ozkan, ACS Catal., 2012, 2, 2335–2348 CrossRef.
- Zs. Ferencz, A. Erdőhelyi, K. Baán, A. Oszkó, L. Óvári, Z. Kónya, C. Papp, H.-P. Steinrück and J. Kiss, ACS Catal., 2014, 4, 1205–1218 CrossRef CAS.
- E. Varga, Z. Ferencz, A. Oszkó, A. Erdohelyi and J. Kiss, J. Mol. Catal. A: Chem., 2015, 397, 127–133 CrossRef CAS.
- M. Greluk, M. Rotko and S. Turczyniak-Surdacka, Renewable Energy, 2020, 155, 378–395 CrossRef CAS.
- H. Wang, L. Zhang, M. Li, Y. Liu and X. Bai, J. Rare Earths, 2013, 31, 565–571 CrossRef CAS.
- M. Greluk, M. Rotko, G. Słowik and S. Turczyniak-Surdacka, J. Energy Inst., 2019, 92, 222–238 CrossRef CAS.
- R. Li, C. Liu, L. Li, J. Xu, J. Ma, J. Ni, J. Yan, J. Han, Y. Pan, Y. Liu and L. Lu, Fuel, 2023, 336, 126758 CrossRef CAS.
- C.-H. Yan, Z.-G. Yan, Y.-P. Du, J. Shen, C. Zhang and W. Feng, Handbook on the Physics and Chemistry of Rare Earths, 2011, vol. 41, pp. 275–472 Search PubMed.
- A. Cross, A. Kumar, E. E. Wolf and A. S. Mukasyan, Ind. Eng. Chem. Res., 2012, 51, 12004–12008 CrossRef CAS.
- A. Cross, S. Roslyakov, K. V. Manukyan, S. Rouvimov, A. S. Rogachev, D. Kovalev, E. E. Wolf and A. S. Mukasyan, J. Phys. Chem. C, 2014, 118, 26191–26198 CrossRef CAS.
- A. Kumar, A. Cross, K. Manukyan, R. R. Bhosale, L. J. P. van den Broeke, J. T. Miller, A. S. Mukasyan and E. E. Wolf, Chem. Eng. J., 2015, 278, 46–54 CrossRef CAS.
- F. Deganello, Mater. Today Proc., Elsevier Ltd, 2017, vol. 4, pp. 5507–5516 Search PubMed.
- F. Deganello and A. K. Tyagi, Prog. Cryst. Growth Charact. Mater., 2018, 64, 23–61 CrossRef CAS.
- B. D. Stojanovic, A. S. Dzunuzovic and N. I. Ilic, Magnetic, Ferroelectric, and Multiferroic Metal Oxides, Elsevier, 2018, pp. 333–359 Search PubMed.
- C. Han, Z. Cao, J. Yang, X. Lu, H. Liu, Z. Jin, Y. Zhang, S. Yang, X. Zheng and L. Wang, Crystals, 2022, 12, 702 CrossRef CAS.
- C. Zhang, R. Zhang, H. Liu, Q. Wei, D. Gong, L. Mo, H. Tao, S. Cui and L. Wang, Energies, 2020, 13, 5956 CrossRef CAS.
- L. Wang and H. Liu, Catal. Today, 2018, 316, 155–161 CrossRef CAS.
- W. Kang and A. Varma, Appl. Catal., B, 2018, 220, 409–416 CrossRef CAS.
- A. Vita, C. Italiano, C. Fabiano, M. Laganà and L. Pino, Mater. Chem. Phys., 2015, 163, 337–347 CrossRef CAS.
- A. N. Matveyeva, Sh. O. Omarov, M. A. Gavrilova, D. A. Sladkovskiy and D. Yu. Murzin, Materials, 2022, 15, 7970 CrossRef CAS PubMed.
- S. L. González-Cortés and F. E. Imbert, Appl. Catal., A, 2013, 452, 117–131 CrossRef.
- N. Luo, X. Fu, F. Cao, T. Xiao and P. P. Edwards, Fuel, 2008, 87, 3483–3489 CrossRef CAS.
- Handbook of Heterogeneous Catalysis. 2nd Edition (Second, Completely Revised and Enlarged), ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- Sh. O. Omarov, E. A. Vlasov, D. A. Sladkovskiy, K. V. Semikin, A. N. Matveyeva, S. P. Fedorov, G. V. Oganesyan and D. Yu. Murzin, Appl. Catal., B, 2018, 230, 246–259 CrossRef CAS.
- A. Kumar, E. E. Wolf and A. S. Mukasyan, AIChE J., 2011, 57, 2207–2214 CrossRef CAS.
- K. V. Manukyan, A. Cross, S. Roslyakov, S. Rouvimov, A. S. Rogachev, E. E. Wolf and A. S. Mukasyan, J. Phys. Chem. C, 2013, 117, 24417–24427 CrossRef CAS.
- S. Hadke, M. T. Kalimila, S. Rathkanthiwar, S. Gour, R. Sonkusare and A. Ballal, Ceram. Int., 2015, 41, 14949–14957 CrossRef CAS.
- A. Khort, K. Podbolotov, R. Serrano-García and Y. K. Gun'ko, J. Solid State Chem., 2017, 253, 270–276 CrossRef CAS.
- X. Wang, M. Qin, F. Fang, B. Jia, H. Wu, X. Qu and A. A. Volinsky, J. Alloys Compd., 2017, 719, 288–295 CrossRef CAS.
- N. Ahmad, F. Alharthi, M. Alam, R. Wahab, S. Manoharadas and B. Alrayes, Energies, 2021, 14, 2928 CrossRef CAS.
- M. P. Harikrishnan, A. J. C. Mary and A. C. Bose, Electrochim. Acta, 2020, 362, 137095 CrossRef CAS.
- Q. Hu, B. Yue, H. Shao, F. Yang, J. Wang, Y. Wang and J. Liu, J. Mater. Sci., 2020, 55, 8421–8434 CrossRef CAS.
- J. Chen, Z. He, G. Li, T. An, H. Shi and Y. Li, Appl. Catal., B, 2017, 209, 146–154 CrossRef CAS.
- H.-N. Barad, D. A. Keller, K. J. Rietwyk, A. Ginsburg, S. Tirosh, S. Meir, A. Y. Anderson and A. Zaban, ACS Comb. Sci., 2018, 20, 366–376 CrossRef CAS PubMed.
- N. Ahmad, R. Wahab, S. Manoharadas, B. F. Alrayes, M. Alam and F. A. Alharthi, Molecules, 2022, 27, 356 CrossRef CAS PubMed.
- G. Sai Gautam, E. B. Stechel and E. A. Carter, Chem. Mater., 2020, 32, 9964–9982 CrossRef CAS.
- E. Novitskaya, J. P. Kelly, S. Bhaduri and O. A. Graeve, Int. Mater. Rev., 2021, 66, 188–214 CrossRef CAS.
- C.-C. Chen and K.-T. Huang, J. Mater. Res., 2005, 20, 424–431 CrossRef CAS.
- N. M. Deraz, J. Alloys Compd., 2010, 501, 317–325 CrossRef CAS.
- E. J. Bosze, J. McKittrick and G. A. Hirata, Mater. Sci. Eng., B, 2003, 97, 265–274 CrossRef.
- G. Pipitone, G. Zoppi, R. Pirone and S. Bensaid, Int. J. Hydrogen Energy, 2022, 47, 151–180 CrossRef CAS.
- N. Palmeri, S. Cavallaro, V. Chiodo, S. Freni, F. Frusteri and J. C. J. Bart, Int. J. Hydrogen Energy, 2007, 32, 3335–3342 CrossRef CAS.
- M. Boudart, Adv. Catal., 1969, 20, 153–166 CAS.
- A. L. M. Da Silva, J. P. Den Breejen, L. V. Mattos, J. H. Bitter, K. P. De Jong and F. B. Noronha, J. Catal., 2014, 318, 67–74 CrossRef CAS.
- A. V. Kirilin, B. Hasse, A. V. Tokarev, L. M. Kustov, G. N. Baeva, G. O. Bragina, A. Yu. Stakheev, A.-R. Rautio, T. Salmi, B. J. M. Etzold, J.-P. Mikkola and D. Yu. Murzin, Catal. Sci. Technol., 2014, 4, 387–401 RSC.
- A. S. Lozhkomoev, A. V. Pervikov, S. O. Kazantsev, K. V. Suliz, R. V. Veselovskiy, A. A. Miller and M. I. Lerner, Nanomaterials, 2022, 12, 2523 CrossRef CAS PubMed.
- J. C. Toniolo, A. S. Takimi and C. P. Bergmann, Mater. Res. Bull., 2010, 45, 672–676 CrossRef CAS.
- C. Italiano, J. Llorca, L. Pino, M. Ferraro, V. Antonucci and A. Vita, Appl. Catal., B, 2020, 264, 118494 CrossRef CAS.
- F. Huang, C. Chen, F. Wang, B. Wang, L. Zhang, S. Lu and K. Li, Catal. Surv. Asia, 2017, 21, 143–149 CrossRef CAS.
- J. Y. Luo, M. Meng, X. Li, X. G. Li, Y. Q. Zha, T. D. Hu, Y. N. Xie and J. Zhang, J. Catal., 2008, 254, 310–324 CrossRef CAS.
- F. L. S. Carvalho, Y. J. O. Asencios, J. D. A. Bellido and E. M. Assaf, Fuel Process. Technol., 2016, 142, 182–191 CrossRef CAS.
- T. A. Maia, J. M. Assaf and E. M. Assaf, Fuel Process. Technol., 2014, 128, 134–145 CrossRef CAS.
- A. Filtschew, K. Hofmann and C. Hess, J. Phys. Chem. C, 2016, 120, 6694–6703 CrossRef CAS.
- E. Sartoretti, C. Novara, F. Giorgis, M. Piumetti, S. Bensaid, N. Russo and D. Fino, Sci. Rep., 2019, 9, 3875 CrossRef PubMed.
- X. Li, X. Liu, J. Hao, L. Li, Y. Gao, Y. Gu, Z. Cao and J. Liu, ACS Omega, 2022, 7, 24646–24655 CrossRef CAS PubMed.
- N. Mironova-Ulmane, A. Kuzmin, I. Sildos and M. Pärs, Open Phys., 2011, 9, 1096–1099 CrossRef CAS.
- G. Jones, J. G. Jakobsen, S. S. Shim, J. Kleis, M. P. Andersson, J. Rossmeisl, F. Abild-Pedersen, T. Bligaard, S. Helveg, B. Hinnemann, J. R. Rostrup-Nielsen, I. Chorkendorff, J. Sehested and J. K. Nørskov, J. Catal., 2008, 259, 147–160 CrossRef CAS.
- M. L. Dieuzeide and N. Amadeo, Chem. Eng. Technol., 2010, 33, 89–96 CrossRef CAS.
- C. K. Cheng, S. Y. Foo and A. A. Adesina, Catal. Commun., 2010, 12, 292–298 CrossRef CAS.
- T. Pairojpiriyakul, E. Croiset, W. Kiatkittipong, K. Kiatkittipong, A. Arpornwichanop and S. Assabumrungrat, Int. J. Hydrogen Energy, 2013, 38, 4368–4379 CrossRef CAS.
- J. Dobosz, M. Cichy, M. Zawadzki and T. Borowiecki, J. Energy Chem., 2018, 27, 404–412 CrossRef.
- A. Carrero, A. J. Vizcaíno, J. A. Calles and L. García-Moreno, J. Energy Chem., 2017, 26, 42–48 CrossRef.
- M. Surendar, T. V. Sagar, B. H. Babu, N. Lingaiah, K. S. R. Rao and P. S. S. Prasad, RSC Adv., 2015, 5, 45184–45193 RSC.
- S. A. Ghungrud and P. D. Vaidya, Int. J. Hydrogen Energy, 2020, 45, 9440–9450 CrossRef CAS.
- H. Zhou, S. Liu, F. Jing, S.-Z. Luo, J. Shen, Y. Pang and W. Chu, Ind. Eng. Chem. Res., 2020, 59, 17259–17268 CrossRef CAS.
- C. A. Schwengber, H. J. Alves, R. A. Schaffner, F. A. Da Silva, R. Sequinel, V. R. Bach and R. J. Ferracin, Renewable Sustainable Energy Rev., 2016, 58, 259–266 CrossRef CAS.
- C. K. S. Choong, Z. Zhong, L. Huang, Z. Wang, T. P. Ang, A. Borgna, J. Lin, L. Hong and L. Chen, Appl. Catal., A, 2011, 407, 145–154 CrossRef CAS.
- N. Charisiou, S. Douvartzides, G. Siakavelas, L. Tzounis, V. Sebastian, V. Stolojan, S. Hinder, M. Baker, K. Polychronopoulou and M. Goula, Catalysts, 2019, 9, 676 CrossRef CAS.
- A. J. Reynoso, J. L. Ayastuy, U. Iriarte-Velasco and M. A. Gutiérrez-Ortiz, J. Environ. Chem. Eng., 2022, 10, 107402 CrossRef CAS.
- R. L. Manfro, T. P. M. D. Pires, N. F. P. Ribeiro and M. M. V. M. Souza, Catal. Sci. Technol., 2013, 3, 1278–1287 RSC.
- S. H. Cho and D. J. Moon, J. Nanosci. Nanotechnol., 2011, 11, 7311–7314 CrossRef CAS PubMed.
- A. Morales-Marín, U. Iriarte-Velasco, M. Á. Gutiérrez-Ortiz and J. L. Ayastuy, Catalysts, 2022, 12, 668 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00854a |
| This journal is © The Royal Society of Chemistry 2023 |