
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear chromium complexes with [OSSO]-type ligands in the copolymerization of epoxides with CO2 and phthalic anhydride†
Fatemeh
Niknam
a,
Alina
Denk
b,
Antonio
Buonerba
 a,
Bernhard
Rieger
b,
Alfonso
Grassi
a and
Carmine
Capacchione
*a
a,
Bernhard
Rieger
b,
Alfonso
Grassi
a and
Carmine
Capacchione
*a
aDipartimento di Chimica e Biologia “Adolfo Zambelli”, Università degli Studi di Salerno, Via Giovanni Paolo II, 84084 Fisciano, SA, Italy. E-mail: ccapacchione@unisa.it
bWACKER-Lehrstuhl für Makromolekulare Chemie, Zentralinstitut für Katalyseforschung (CRC), Technische Universitat München, Lichtenbergstraße 4, 85747 Garching, Germany
First published on 11th July 2023
Abstract
In this study, a new family of dinuclear chromium complexes (1–3) containing bis-thioether-diphenolate ligands has been introduced for the binary copolymerization of carbon dioxide and epoxides and ternary copolymerization of the latter with phthalic anhydride. Complex 1 (0.1 mol%) in combination with (bis(triphenylphosphine)iminium chloride) (PPNCl, 0.5 mol) at 45 °C and 20 bar of CO2 showed high regioselective copolymerization to polypropylene carbonate (PPC) with conversion (up to 91%) and selectivity (up to 95%). The data presented in this work have consistently shown that complex 1 displayed higher catalytic activity in copolymerizing epoxides with CO2 than complexes 2, 3, and the analogous mononuclear 4. At the same time, complex 1 showed good catalytic properties in the terpolymerization of epoxides/CO2/phthalic anhydride among the tested complexes 2 and 4. In the case of cyclohexene oxide and vinylcyclohexene oxide, a selectivity of more than 99% towards polycyclohexene carbonate (PCHC) and polyvinylcyclohexene carbonate (PVCHC) with a TOF as high as 41 h−1 in the poly(ester-block-carbonate) was observed. Notably, a conversion higher than 99% towards the polyester block was also observed for all the studied epoxides. A bimetallic intramolecular cooperative mechanism was proposed for the copolymerization of propylene oxide and CO2 based on the first-order dependence with respect to complex 1 by the kinetic investigations.
Introduction
Polymeric materials are ubiquitous in our world because of their unique chemical and physical properties and low cost compared to other structural materials. For decades, the sole motivation for synthesizing new polymers was obtaining a more performing material for a given application. Only recently, the growing evidence that the accumulation of plastics in the environment causes severe risks for living organisms, with serious consequences also for the food chain and, therefore, human health,1,2 has shifted the attention of the academic and industrial communities toward the design and synthesis of polymers that have a lesser impact on ecosystems.3,4In this scenario, aliphatic polyesters5–8 and polycarbonates9–12 have attracted much attention due to the possibility of being hydrolyzed at their end of life and being built up from renewable resources starting from cyclic esters, cyclic anhydrides, and epoxides derived from biomass and using CO2 as a building block.13–16 As a matter of fact, the ring-opening copolymerization (ROCOP) of the epoxides can be performed either with CO2, giving the corresponding polycarbonates, or with cyclic anhydrides, which gives polyester chains.17,18
More recently, attention has shifted to the terpolymerization of epoxides with CO2 and cyclic anhydrides for the possibility of obtaining block copolymers with polycarbonate and polyester segments having different microstructural features and therefore offering a wider range of chemical and physical properties.19–21
In this field, dinuclear metal complexes have shown, in many cases, unique features in terms of activity and selectivity compared to their mononuclear counterparts.22–28 In particular, the work of the Williams research group using homodinuclear29 and heterodinuclear30,31 complexes supported by the Robson-type cyclic ligand has shown excellent behaviour in the terpolymerization of epoxides with CO2 and cyclic anhydrides. In all cases, the key to the good performance of these catalysts lies in the cooperativity between the two metal centres, which allows the lowering of the barriers of the coordination/insertion steps during the copolymerization process.
[OSSO]-type metal complexes have been widely used as catalysts in many polymerization reactions,32 and more tightly Fe(III)33–39 and Cr(III)40–42 complexes have a high potential for the copolymerization of various epoxides with CO2. Recently, we have also developed a Fe(III) dinuclear complex exhibiting good PO/CO2 coupling performances, with kinetic data showing the cooperativity of both metal centers in the catalytic process.43
Here we report on the synthesis of three new dinuclear Cr(III) complexes (1–3) based on the [OSSO]-type ligand framework and their behaviour in copolymerizing various epoxides with CO2 and their terpolymerization with phthalic anhydride.
Results and discussion
The three new dinuclear [OSSO]-type chromium complexes 1–3 (Scheme 1) were synthesized in THF solution by reaction of the sodium salt of the corresponding pro-ligands with CrCl3(THF)3. All the complexes have been isolated in good yield (94–98%) as brownish microcrystalline solids.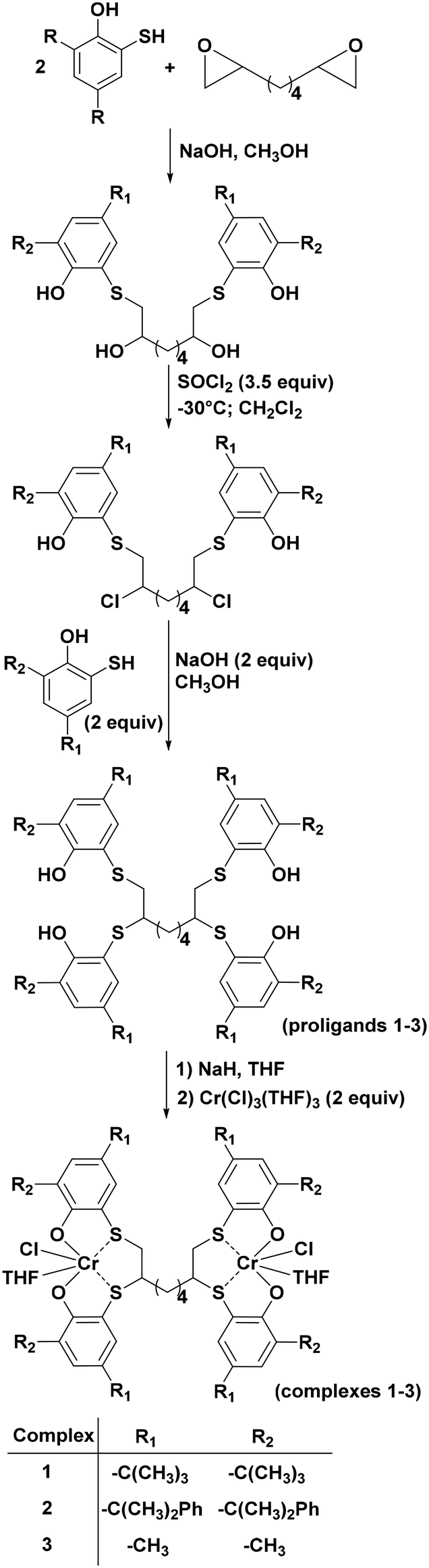 | ||
| Scheme 1 Synthetic strategy of complexes 1–3. | ||
The elemental analysis of the chromium compounds was in good agreement with obtaining of the desired products. Furthermore, complexes 1–3 were also characterized by MALDI-TOF spectrometry and FT-IR (see Fig. S7–S14 of ESI†).
In addition, the Evans method, which gives indirect access to the magnetic susceptibility of paramagnetic compounds in solution, confirmed the effective magnetic moments (μeff) for 1–3 in dichloromethane-d2 in the range between −10 and 25 °C. These values are higher than the calculated value for one isolated high spin (HS) Cr(III) centre (3.88 μB) and lower than the expected value for two isolated chromium-(III) HS nuclei (5.48 μB), suggesting an antiferromagnetic interaction between the two metal centres (see Fig. S15–S17 of ESI†), confirming the formation of dinuclear complexes stable in solution.42,44
Copolymerization of epoxides with CO2
The data relative to the copolymerization of propylene oxide with CO2 in the presence of 1 activated by different co-catalysts, including bis(triphenylphosphine)iminium chloride (PPNCl), tetrabutylammonium bromide (TBAB), tetrabutylammonium chloride (TBAC), tetrabutylammonium azide (TBAN3) and 4-dimethylaminopyridine (DMAP), are reported in Table 1.| Entrya | Co-cat/cat [molar ratio] | Co-cat | Conv.b [mol%] | PCb,c [mol%] | TOFd [h−1] | M w [kDa] | PDIe |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: PO = 0.014 mol; PO/complex 1 molar ratio = 1000; reaction time = 24 h; temperature = 45 °C; p CO2 = 20 bar; co-catalyst (PPNCl (bis(triphenylphosphine)iminium chloride); TBAB = tetrabutylammonium bromide; TBAC = tetrabutylammonium chloride; TBAN3 = tetrabutylammonium azide; DMAP = 4-dimethylaminopyridine). b Determined by 1H NMR in CDCl3. c Selectivity as mole percentage of polycarbonate linkage to polycarbonate + cyclic carbonate units. d TOF = (molepoxide reacted)/(molcat × time). e Determined by gel permeation chromatography (GPC) with respect to polystyrene standards. | |||||||
| 1 | 0.25 | PPNCl | 80 | 90 | 33 | 10.0 | 1.3 |
| 2 | 0.50 | PPNCl | 91 | 95 | 38 | 12.9 | 1.4 |
| 3 | 1.00 | PPNCl | 93 | 93 | 39 | 12.5 | 1.6 |
| 4 | 0.50 | TBAC | 67 | 46 | 28 | 6.3 | 1.3 |
| 5 | 1.00 | TBAC | 87 | 93 | 36 | 8.1 | 1.2 |
| 6 | 0.50 | TBAB | 22 | 81 | 9 | — | — |
| 7 | 1.00 | TBAB | 42 | 3 | 18 | — | — |
| 8 | 0.50 | TBAN3 | 71 | 89 | 30 | 17.4 | 1.4 |
| 9 | 1.00 | TBAN3 | 75 | 55 | 31 | 6.9 | 1.2 |
| 10 | 0.50 | DMAP | 40 | 0 | 17 | — | — |
Initially, we conducted experiments to find the optimized conditions for copolymerizing propylene oxide (PO) with CO2. Concerning the co-catalysts, the combination of nucleophilicity, steric hindrance, and leaving ability play an essential role in the selectivity and activity of the catalytic system.45,46 As observed for the mononuclear chromium complex 4,40 the best catalytic activity and selectivity were obtained using PPNCl in a substoichiometric amount. TBAC, TBAB, and DMAP are less selective in producing polypropylene carbonate, and in particular, the use of DMAP results in the exclusive formation of propylene carbonate (entry 10, Table 1).
The optimized conditions to produce polypropylene carbonate were found for catalyst loading (0.1 mol%) with 0.5 mol of PPNCl at reaction temperature (45 °C) at 20 bar of CO2, reaching high conversion (up to 91%) and selectivity (up to 95%) to PPC with a TOF up to 38 h−1 (entry 2, Table 1). For comparison, mono- and dinuclear salphen-type chromium complexes TOFs were found to be 61 h−1 and 49 h−1, respectively toward formation of PPC under higher catalyst loading and CO2 pressure.26 As expected, higher temperature and lower catalyst loading resulted in the production of predominantly propylene carbonate (see Table S1 of ESI†).
With the optimized conditions in hand (0.1 mol% of complex, 0.5 mol of PPNCl, 45 °C, and 20 bar of CO2), we decided to compare, under the same reaction conditions, the catalytic activity of the complexes 1–3 and the mononuclear complex 4 (Scheme 2).
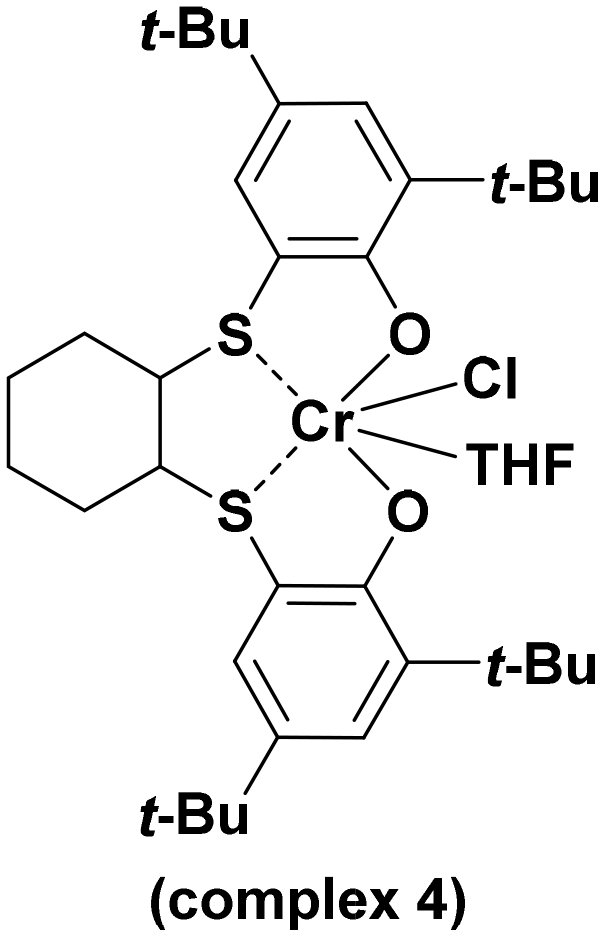 | ||
| Scheme 2 Structure of complex 4. | ||
The results are summarised in Table 2. Cyclohexene oxide (CHO) and vinylcyclohexene oxide (VCHO) activated by 1 both showed complete selectivity for the polycarbonate and higher activity, reaching a TOF of 40 h−1 in the case of VCHO at 80 °C (entry 9, Table 2). 1-Hexene oxide (HO) was also converted to the corresponding polycarbonate with good activity and selectivity, notably showing a higher molecular weight among the obtained polycarbonates (Mw = 23.4 kDa) (entry 13, Table 2).
| Entrya | Cat | Epoxide | T [°C] | Conv.b [mol%] | PCb,c [mol%] | TOFd [h−1] | M w [kDa] | PDIe |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: epoxide = 0.014 mol; epoxide/complex molar ratio = 1000; reaction time = 24 h; p CO2 = 20 bar; PPNCl/complex molar ratio = 0.5. b Determined by 1H NMR in CDCl3. c Selectivity as mole percentage of polycarbonate linkage to polycarbonate + cyclic carbonate units. d TOF = (molepoxide reacted)/(molcat × time). e Determined by GPC with respect to polystyrene standards. f PPNCl/complex 4 molar ratio = 1.0. | ||||||||
| 1 | 1 | PO | 45 | 91 | 95 | 38 | 12.9 | 1.4 |
| 2 | 2 | PO | 45 | 37 | 90 | 15 | 7.3 | 1.2 |
| 3 | 3 | PO | 45 | 20 | 54 | 23 | — | — |
| 4 | 4 | PO | 45 | 44 | 88 | 18 | 10.7 | 1.5 |
| 5 | 1 | CHO | 80 | 89 | >99 | 37 | 10.1 | 1.2 |
| 6 | 2 | CHO | 80 | 83 | >99 | 34 | 7.4 | 1.2 |
| 7 | 3 | CHO | 80 | 40 | >99 | 17 | 5.3 | 1.3 |
| 8 | 4 | CHO | 80 | 63 | >99 | 26 | 6.1 | 1.6 |
| 9 | 1 | VCHO | 80 | 97 | >99 | 40 | 14.2 | 1.4 |
| 10 | 2 | VCHO | 80 | 85 | >99 | 35 | 11.8 | 1.4 |
| 11 | 3 | VCHO | 80 | 76 | >99 | 32 | 7.1 | 1.5 |
| 12f | 4 | VCHO | 80 | 94 | >99 | 39 | 13.8 | 1.9 |
| 13 | 1 | HO | 45 | 79 | 74 | 33 | 23.4 | 1.3 |
| 14 | 2 | HO | 45 | 45 | 0 | 19 | — | — |
| 15 | 3 | HO | 45 | 5 | 0 | 2 | — | — |
| 16 | 4 | HO | 45 | 33 | 80 | 14 | 16.8 | 1.3 |
It is worth noting that complex 1 is the most active and selective in the copolymerization of all the investigated substrates (PO, CHO, VCHO, and HO) with the maximum molecular weight among all the other copolymerization products obtained in the presence of complexes 2 and 3 (entries 1, 5, 9, and 13, Table 2). The more sterically congested complex 2 was less active and selective (entries 2, 6, 10, 14, Table 2), and complex 3, bearing the less bulky methyl substituents, was less chemo-selective, giving a mixture of PPC and cyclic propylene carbonate (entry 3, Table 2) and the exclusive formation of the cyclic product in the case of HO (entry 15, Table 2).
A comparison with the mononuclear complex 4 also revealed that complex 1 is significantly more active and selective toward polycarbonates in the copolymerization of PO and CHO with CO2 (see entries 1, 4, 5, and 8 in Table 2). It is worth noting that only complexes 1 and 4 containing tert-butyl groups showed selectivity toward poly(1-hexene carbonate) (PHC), in which complex 1 showed again higher activity (see entries 13 and 16 of Table 2).
The two-dimensional 13C{1H} NMR spectrum was recorded to provide better insight into the purified resulting copolymer microstructural features (see Fig. S19–S23 of ESI†), the only peak at δ = 154.3 ppm attributed to the carbonyl group in the case of PPC revealed a high regioselective copolymerization in which the nucleophile attacks the sterically less hindered carbon atom to open the epoxide ring, resulting in head-to-tail (HT) arrangements without regioerrors.47 In the case of poly(cyclohexene) carbonate (PCHC) and poly(vinylcyclohexene) carbonate (PVCHC), the 13C{1H}NMR analysis is in agreement with the formation of stereoirregular polymer chains (see Fig. S22 of ESI†).48
Terpolymerization of epoxides/phthalic anhydride with CO2
The good performances obtained in the copolymerization of the tested epoxides towards the formation of polycarbonates prompted us to investigate the terpolymerization of epoxide/CO2/phthalic anhydride (PA) (Table 3). Complexes 1, 2, and 4 were examined under optimized conditions (0.1 mol% of complex, 0.5 mol of PPNCl, epoxide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PA = 10:1, mol/mol, and 20 bar of CO2). The data are shown in Table 3. The reported conversions were calculated by comparing the intensity of the 1H signals of the reagents (PO, CHO, VCHO, and HO) with those of the methine protons of polycarbonates (at δ = 4.98 ppm for PPC, δ = 4.60 ppm for PCHC, δ = 4.77 ppm for PVCHC, and δ = 4.89 ppm for PHC) and of the cyclic propylene carbonate (δ = 4.99–4.82 ppm). From the data in Table 3, complex 1 showed the highest activity in the epoxide/CO2/PA terpolymerization. Notably, for all the studied epoxides, a conversion higher than 99% towards the polyester block was observed due to the absence of signals belonging to unreacted PA in the 1H NMR spectrum of the resulting crude product.
:PA = 10:1, mol/mol, and 20 bar of CO2). The data are shown in Table 3. The reported conversions were calculated by comparing the intensity of the 1H signals of the reagents (PO, CHO, VCHO, and HO) with those of the methine protons of polycarbonates (at δ = 4.98 ppm for PPC, δ = 4.60 ppm for PCHC, δ = 4.77 ppm for PVCHC, and δ = 4.89 ppm for PHC) and of the cyclic propylene carbonate (δ = 4.99–4.82 ppm). From the data in Table 3, complex 1 showed the highest activity in the epoxide/CO2/PA terpolymerization. Notably, for all the studied epoxides, a conversion higher than 99% towards the polyester block was observed due to the absence of signals belonging to unreacted PA in the 1H NMR spectrum of the resulting crude product.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Cat | Epoxide | T [°C] | Conv.b,c [mol] | PCb,d [mol%] | TOFe [h−1] | M w [kDa] | PDIf |
|
a Reaction conditions: epoxide = 0.014 mol; epoxide/complex molar ratio = 1000; PPNCl/complex molar ratio = 0.5; reaction time = 24 h; epoxide:PA molar ratio = 10:1; p CO2 = 20 bar; co-catalyst = PPNCl.
b Determined by 1H NMR in CDCl3.
c The ester-selectivity in the polyester segment/block >99%.
d Selectivity as mole percentage of polycarbonate linkage to polycarbonate + cyclic carbonate units.
e TOF = (molepoxide reacted)/(molcat × time).
f Determined by GPC with respect to polystyrene standards.
|
||||||||
| 1 | 1 | PO | 45 | 62 | 88 | 29 | 12.3 | 1.2 |
| 2 | 2 | PO | 45 | 55 | 94 | 25 | 13.0 | 1.6 |
| 3 | 4 | PO | 45 | 60 | 88 | 29 | 19.0 | 1.4 |
| 4 | 1 | CHO | 80 | >99 | >99 | 41 | 11.0 | 1.3 |
| 5 | 2 | CHO | 80 | 77 | >99 | 32 | 9.6 | 1.2 |
| 6 | 4 | CHO | 80 | 81 | >99 | 34 | 10.5 | 1.2 |
| 7 | 1 | VCHO | 80 | >99 | >99 | 41 | 12.0 | 1.3 |
| 8 | 2 | VCHO | 80 | 89 | >99 | 37 | 12.7 | 1.4 |
| 9 | 4 | VCHO | 80 | 77 | >99 | 32 | 11.6 | 1.3 |
| 10 | 1 | HO | 45 | 67 | 93 | 30 | 8.5 | 1.0 |
| 11 | 2 | HO | 45 | 0 | 0 | 0 | — | — |
| 12 | 4 | HO | 45 | 58 | 91 | 26 | 9.0 | 1.1 |
The 1H and 13C{1H} NMR spectra do not show polycarbonate sequences in the polyester segments suggesting the formation of diblock poly(ester-block-carbonate) copolymers through the faster reaction of epoxide/anhydride coupling and, after the complete conversion of the anhydride, the carbonate block was grown by CO2/epoxide alternating insertion.49,50
Data from Table 3 can be compared with the data in Table 2, which show sensibly lower conversion in terpolymerization toward PPC and PHC activated by complex 1 (see entries 1 and 10, Table 3). In contrast, CHO and VCHO have been converted more than 99% (TOF of 41 h−1) to their related polycarbonates (entries 4, and 7, Table 3). A negligible amount of polyether (peak at δ = 3.45 ppm in the 1H NMR spectrum) was observed in the terpolymerization reaction of CHO/PA with CO2 (see Fig. S27 of ESI†). DOSY NMR spectroscopy for purified poly(ester-block-cyclohexene carbonate) (PE-co-PCHC) and poly(ester-block-vinylcyclohexene carbonate) (PE-co-PVCHC) was carried out (see Fig. S28 and S29 of ESI†). It is apparent that all signals show the same diffusion coefficient, indicating the existence of a genuine copolymer. Interestingly, in the 13C{1H} NMR spectra of terpolymers, the chemical shifts and patterns of the carbonyl groups in the backbone of polycarbonates are similar to those observed for copolymers. This finding agrees with our earlier observation for polypropylene carbonate, which shows that the PO/CO2 terpolymerization with PA is also regioselective (see Fig. S24–S26 of ESI†). The thermal properties of the poly(ester-block-carbonate)s show that the presence of a semi-aromatic polyester block due to the presence of PA in the polymer chain causes an increase in the Tg values compared to the corresponding polycarbonates. As already reported for these diblock copolymers, the phases are completely miscible; therefore, only one value for Tg was observed (see Fig. S18 of ESI†).51
Kinetic studies
The kinetic investigations for 1 and 4 with combinations of PPNCl as catalytic systems were performed for a more detailed understanding of the copolymerization process towards the formation of PPC. In situ attenuate total reflection infrared (ATR-IR) spectroscopy provides a valuable tool for monitoring the reaction progress by increasing the carbonyl stretching bond (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1750 cm−1) (see Fig. S39–S46 of the ESI†). The solution of PO/CH2Cl2/PPNCl was prepared as the reaction medium to guarantee the homogeneity of the system during the experiment. The reaction order with respect to 1 and 4 was determined by performing a series of experiments varying the concentration of 1 (in the range from 2.26 to 9.03 mM) and of 4 (in the range from 3.76 to 18.05 mM) while keeping the concentration of PPNCl constant at 2.26 mM (see Tables S2 and S3 of ESI†). The values of initial velocities vo were obtained from the slope of the initial straight line at low conversion of the epoxide. The double logarithmic plot of the initial rate (vo) versus the concentration of the complexes gave orders of 0.98 and 1.94, very close to first-order and second-order dependences with respect to complexes 1 and 4, respectively (see Fig. 1).
O = 1750 cm−1) (see Fig. S39–S46 of the ESI†). The solution of PO/CH2Cl2/PPNCl was prepared as the reaction medium to guarantee the homogeneity of the system during the experiment. The reaction order with respect to 1 and 4 was determined by performing a series of experiments varying the concentration of 1 (in the range from 2.26 to 9.03 mM) and of 4 (in the range from 3.76 to 18.05 mM) while keeping the concentration of PPNCl constant at 2.26 mM (see Tables S2 and S3 of ESI†). The values of initial velocities vo were obtained from the slope of the initial straight line at low conversion of the epoxide. The double logarithmic plot of the initial rate (vo) versus the concentration of the complexes gave orders of 0.98 and 1.94, very close to first-order and second-order dependences with respect to complexes 1 and 4, respectively (see Fig. 1).
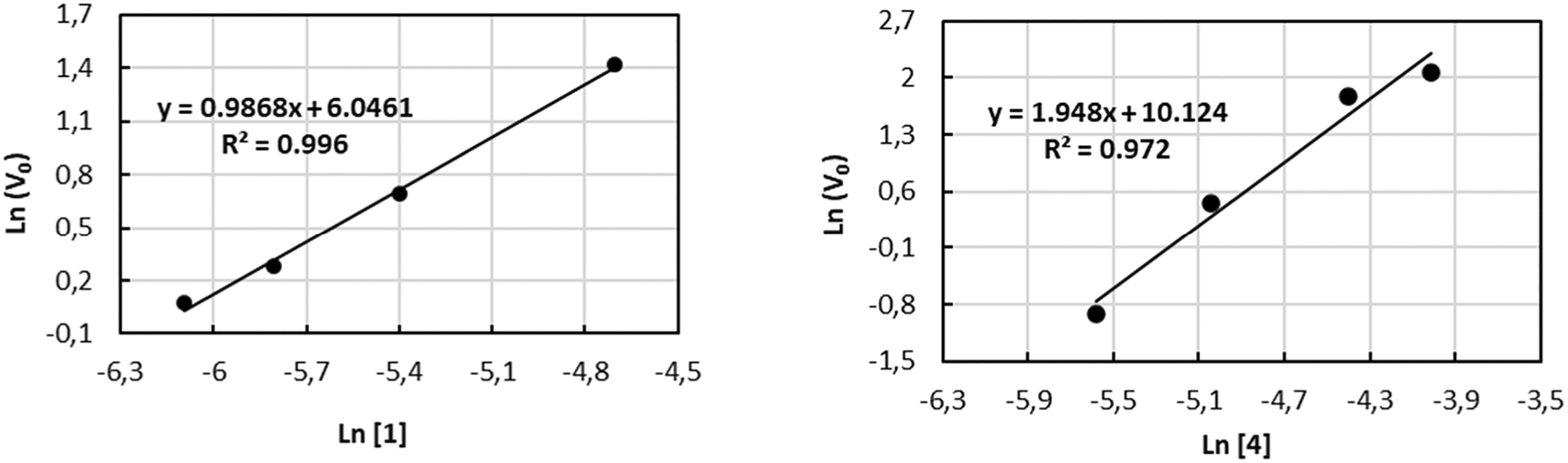 | ||
| Fig. 1 Plot of ln(v0) for the formation of PPC versus: (left) ln[1], (right) ln[4]. | ||
These data support the involvement of two metal centres during the polymerization process, as already observed in other dinuclear systems,52,53 in which the presence of two vicinal metal centers allows coordination and ring opening of the incoming monomer (epoxide, CO2) and the chain growth on the second metal center. The higher activity and selectivity observed for dinuclear complex 1vis-à-vis complex 4 are therefore a consequence of a cooperative mechanism due to the proximity of the two metal centres.
Conclusions
This study set out to report the synthesis of a new series of dinuclear Cr(III) complexes (1–3), supported by bis-thioether-diphenolate ligand and their catalytic activity in combination with PPNCl as a co-catalyst to promote the reaction of epoxides with CO2. It was observed that the activity of complexes mainly relied on the nature of the substituents present in the phenolate group. Complex 1 with tert-butyl substitutions displayed good activity and selectivity for the formation of polycarbonates starting from the corresponding epoxides as compared to complexes 2, 3, and the analogous mononuclear 4. In the case of PO, a high conversion (up to 91%, with a TOF up to 38 h−1) to polypropylene carbonate with negligible production of the cyclic carbonate during a regioselective copolymerization was observed. 1 showed good catalytic properties in the terpolymerization of epoxide/CO2/phthalic anhydride among all the other tested complexes 2 and 4. In the case of CHO and VCHO, a selectivity of more than 99% towards PCHC and PVCHC with a TOF as high as 41 h−1 in the poly(ester-block-carbonate) was observed. The kinetic investigation supported the occurrence of the bimetallic intramolecular and bimetallic intermolecular ring-opening mechanisms by observation of the first-order and second-order dependences with respect to the complexes 1 and 4, respectively.Experimental part
General considerations
All synthesizes containing air- and moisture-sensitive compounds were performed under a dry nitrogen atmosphere using standard Schlenk techniques and the Jacomex glovebox. All chemicals and solvents were purchased from Sigma Aldrich or TCI. THF (99%) was refluxed for 72 hours over sodium and sodium benzophenone ketyls and distilled before use for moisture- and oxygen-sensitive reactions. Monomers were dried over calcium hydride, distilled, and stored over activated 4 Å molecular sieves prior to polymerization.NMR (1H, 13C, Dosy, and HSQC) measurements were recorded on Bruker Avance spectrometers (300, 400, or 600 MHz). Chemical shifts δ were reported in ppm and referenced to tetramethylsilane (TMS), and calibrated to the residual 1H or 13C signal of the deuterated solvent. Deuterated solvents were obtained from Aldrich and dried over a 3 Å molecular sieve.
Measurements of effective magnetic moments (μeff) were performed on Bruker Avance spectrometers (300 and 600 MHz) in deuterated solvent (dichloromethane-d2) using a 5 mm Wilmad coaxial insert NMR tube.33 The effective magnetic moment (μeff) was calculated from μeff = 8χgMwT. The χg (cm3 g−1) is the corrected molar susceptibility derived from χg = 3Δf/4πfoCMw + χo. Δf (Hz) is the shift in frequency of the residual proton signal of the solvent in the presence of the complex from the value of the pure solvent. C (mol cm−1) is concentration, Mw (g mol−1) is the molecular weight of the complex, fo (Hz) is the operating frequency of the spectrometer, and χo is the mass susceptibility of the pure solvent (−0.5486 × 10−6 cm3 g−1 for dichloromethane-d2). 4π/3 is the shape factor for a cylindrical sample in a superconducting magnet.
DSC was conducted on a DSC Q2000 instrument. 3–10 mg of the polymer was filled into a DSC aluminum pan and heated from −80 to 170 °C at a rate of 5 K min−1. The reported values were determined with TA Universal Analysis from the second heating cycle.
MALDI-TOF analysis was performed on a Waters Quattro Micro triple quadrupole mass spectrometer equipped with an electrospray ion source. Anthracene and 2,5-dihydroxybenzoic acid were used as matrixes.
GPC was performed on a Varian PL-GPC 50 equipped with a Deflection RI detector to obtain size exclusion chromatography (SEC) measurements. Polystyrene was used as a standard at 40 °C with THF (flow = 1 mL min−1) as an eluent.
FT-IR measurements were carried out on a Bruker Vertex 70 spectrometer equipped with a DTGS detector and a Ge/KBr beam splitter, analyzing the solid state as KBr disks. In situ IR measurements for kinetic investigations were performed under an argon atmosphere using an ATR IR Mettler Toledo system.
Synthesis
Both the ligands 6,6′,6′′,6′′′-(pentane-1,2,4,5-tetrayltetrakis-(sulfanediyl)) tetrakis(2,4-bis(2-phenylpropan-2-yl)phenol), (L2), 6,6′,6′′,6′′′-(octane-1,2,7,8-tetrayltetrakis(sulfanediyl)) tetrakis(2,4-dimethylphenol), (L3), and the complexes (1–3) were synthesized according to the literature procedures.41The mixture of 2-mercapto-4,6-bis(2-phenylpropan-2-yl)phenol (16.0 mmol) and NaOH (16.0 mmol) in 70 ml of methanol was refluxed until complete dissolution of all reagents. After 20 minutes at 25 °C, compound c (8.0 mmol) was dissolved in the minimum amount of methanol and added dropwise into the mixture of solutions. The mixture was refluxed over night at 70 °C. After that, the solvent was evaporated, and 50 ml of water was added. The organic phase was extracted by diethyl ether (3 × 50 ml) and dried with MgSO4. The concentrated solution was precipitated from methanol in the fridge as a brownish solid. Yield 70%. 1H NMR (CDCl3): δ = 7.12–7.27 (m, 40H, arom. CH); 7.08 (s, 8H, Ar–H); 6.62 (br s, 2H, Ar–OH); 6.42 (br s, 2H, Ar–OH); 2.50 (br s, 6H), 1.67 (m, 28H, CH3 and (CH2)2-alkyl bridge); 1.61 (s, 28H, CH3 and (CH2)2-alkyl bridge); 13C{1H} NMR (CDCl3): δ 153.9; 153.0; 151.0; 150.8; 150.6; 142.4; 142.1; 135.1; 133.8; 132.2; 128.5; 128.4; 128.3; 127.1; 126.1; 126.0; 125.8; 125.7. Elemental analysis calcd. for C104H114O4S4 = C 80.26, H 7.38, O 4.11, S 8.24; found: C 80.46, H 7.33, O 4.16, S 8.34; MS: m/z = 1578.7 (L2 + Na)+, 1593.7 (L2 + K)+.
The mixture of 2-mercapto-4,6-dimethylphenol (16.0 mmol) and NaOH (16.0 mmol) in 70 ml of methanol was refluxed until complete dissolution of all reagents. After 20 minutes at 25 °C, compound c (8.0 mmol) was dissolved in the minimum amount of methanol and added dropwise into the mixture of solutions. The mixture was refluxed over night at 70 °C. After that, the solvent was evaporated, and 50 ml of water was added. The organic phase was extracted by diethyl ether (3 × 50 ml) and dried with MgSO4. The concentrated solution was washed with cold methanol and collected as a yellow, transparent oil. Yield 60%. 1H NMR (CDCl3): δ = 7.02 (s, 2H, Ar–H); 6.99 (s, 2H, Ar–H); 6.93 (s, 2H, Ar–H); 6.90 (s, 2H, Ar–H); 6.87 (br s, 2H, Ar–OH); 6.71 (br s, 2H, Ar–OH); 2.80 (m, 6H); 2.32 (s, 1H, CH-alkyl bridge); 2.25 (br s, 6H, 2CH3); 2.24 (br s, 2H, (CH2)2-alkyl bridge); 2.22 (br s, 6H, 2CH3); 2.19 (br s, 12H, 4CH3); 1.69 (m, 2H, CH2-alkyl bridge); 1.55 (m, 3H, CH2-alkyl bridge); 13C{1H} NMR (CDCl3): δ 154.1; 153.4; 134.5; 134.1; 133.7; 133.5; 132.1; 129.8; 129.6; 124.7; 117.6; 115.7; 50.56; 41.9; 33.5; 26.9; 20.7; 16.85. Elemental analysis calcd. for C40H50O4S4 = C 66.44, H 6.97, O 8.85, S 17.74; found: C 66.54, H, 6.90, O, 8.83, S, 17.77; MS: m/z = 745.2 (L3 + Na)+, 761.2 (L3 + K)+.
Polymerization and terpolymerization
The reaction vessels, needles, and 60 mL stainless steel reactor were dried in an oven at 140 °C for 24 hours before use. Complexes 1–3, PPNCl, and/or phthalic anhydride were weighed in the glovebox. The calculated amount of epoxides was taken under nitrogen flow and added to the vial containing complex and PPNCl. Then the mixture of solutions was injected into the reactor and fed with CO2. The catalytic reactions were run at an adjusted temperature for 24 hours. At the end of the reaction, the reactor was cooled with ice for 20 minutes, and then the CO2 was slowly released. A small portion of the reaction mixture was used for 1H NMR spectroscopy for the determination of conversion and selectivity. The crude polymer products were collected by dissolving them in a minimum amount of CH2Cl2 and washing them with methanol. After the consequent removal of the solvent, the precipitated polymers were dissolved in dichloromethane and then dried under vacuum for 24 hours.Kinetic investigations
The kinetic studies of polymerizations were performed with in situ monitoring using a React-IR Mettler-Toledo system. The 50 mL steel autoclave, equipped with a diamond window, a heating device, and mechanical stirring, was heated to 130 °C under vacuum overnight prior to polymerization. All chemicals were weighed in the glove box, stored in syringes, and rapidly transported to the reactor. The reaction was terminated when the signal of the carbonyl oxygen (1750 cm−1) was constant by adding dichloromethane and some drops of methanol.Author contributions
Fatemeh Niknam, investigation, methodology, synthesis, formal analysis, writing – original draft. Alina Denk, investigation, methodology. Antonio Buonerba, investigation, resources, writing – review & editing. Bernhard Rieger, visualization, resources, writing – review & editing. Alfonso Grassi, visualization, resources, writing – review & editing. Carmine Capacchione, conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial Support from Ministero dell'Università della Ricerca (MUR, Roma, PRIN 2017-CO2 as only source of carbons for monomers and polymers: a step forwards circular economy-CO2 only) and Università degli Studi di Salerno are gratefully acknowledged. Dr. Patrizia Iannece, Dr. Patrizia Oliva, Dr. Mariagrazia Napoli, and Dr. Ivano Immediata from Università degli Studi di Salerno for technical assistance are deeply acknowledged.References
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- H. Nakajima, P. Dijkstra and K. Loos, Polymers, 2017, 9, 523 CrossRef PubMed.
- D. M. Mitrano and M. Wagner, Nat. Rev. Mater., 2021, 7, 71–73 CrossRef.
- Q. Zhang, M. Song, Y. Xu, W. Wang, Z. Wang and L. Zhang, Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- K. M. Zia, A. Noreen, M. Zuber, S. Tabasum and M. Mujahid, Int. J. Biol. Macromol., 2016, 82, 1028–1040 CrossRef CAS PubMed.
- F. Siragusa, C. Detrembleur and B. Grignard, Polym. Chem., 2023, 14, 1164–1183 RSC.
- G. A. Bhat, M. Luo and D. J. Darensbourg, Green Chem., 2020, 22, 7707–7724 RSC.
- Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- R. D. Rittinghaus and S. Herres-Pawlis, Eur. J. Chem., 2023, 29, e202202222 CAS.
- S. Cui, J. Borgemenke, Z. Liu and Y. Li, J. CO2 Util., 2019, 34, 40–52 CrossRef CAS.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- X. Wang, Z. Huo, X. Xie, N. Shanaiah and R. Tong, Chem. – Asian J., 2023, 18, e202201147 CAS.
- G. L. Gregory, G. S. Sulley, J. Kimpel, M. Lagodzińska, L. Häfele, L. P. Carrodeguas and C. K. Williams, Angew. Chem., Int. Ed., 2022, 134, e202210748 CrossRef.
- D. H. Lamparelli and C. Capacchione, Catalysts, 2021, 11, 961 CrossRef CAS.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- S. Kissling, P. T. Altenbuchner, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, U. B. Seemann and B. Rieger, Eur. J. Chem., 2015, 21, 8148–8157 CrossRef CAS PubMed.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC.
- S. Klaus, S. I. Vagin, M. W. Lehenmeier, P. Deglmann, A. K. Brym and B. Rieger, Macromolecules, 2011, 44, 9508–9516 CrossRef CAS.
- Y. Liu, J.-Z. Guo, H.-W. Lu, H.-B. Wang and X.-B. Lu, Macromolecules, 2018, 51, 771–778 CrossRef CAS.
- F. de la Cruz-Martínez, M. M. de Sarasa Buchaca, J. Martínez, J. Tejeda, J. Fernández-Baeza, C. Alonso-Moreno, A. M. Rodríguez, J. A. Castro-Osma and A. Lara-Sánchez, Inorg. Chem., 2020, 59, 8412–8423 CrossRef PubMed.
- A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS PubMed.
- G. Rosetto, A. C. Deacy and C. K. Williams, Chem. Sci., 2021, 12, 12315–12325 RSC.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021, 60, 13372–13379 CrossRef CAS PubMed.
- V. Paradiso, V. Capaccio, D. H. Lamparelli and C. Capacchione, Coord. Chem. Rev., 2021, 429, 213644 CrossRef CAS.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- A. Buonerba, F. Della Monica, A. De Nisi, E. Luciano, S. Milione, A. Grassi, C. Capacchione and B. Rieger, Faraday Discuss., 2015, 183, 83–95 RSC.
- F. Della Monica, S. V. C. C. Vummaleti, A. Buonerba, A. D. Nisi, M. Monari, S. Milione, A. Grassi, L. Cavallo and C. Capacchione, Adv. Synth. Catal., 2016, 358, 3231–3243 CrossRef.
- F. Della Monica, M. Leone, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, Mol. Catal., 2018, 460, 46–52 CrossRef CAS.
- F. Della Monica, B. Maity, T. Pehl, A. Buonerba, A. De Nisi, M. Monari, A. Grassi, B. Rieger, L. Cavallo and C. Capacchione, ACS Catal., 2018, 8, 6882–6893 CrossRef CAS.
- F. Della Monica, A. Buonerba and C. Capacchione, Adv. Synth. Catal., 2019, 361, 265–282 CrossRef CAS.
- F. Della Monica, A. Buonerba, V. Paradiso, S. Milione, A. Grassi and C. Capacchione, Adv. Synth. Catal., 2019, 361, 283–288 CrossRef CAS.
- F. Niknam, V. Capaccio, M. Kleybolte, D. H. Lamparelli, M. Winnacker, G. Fiorani and C. Capacchione, ChemPlusChem, 2022, 87, e202200038 CAS.
- F. Della Monica, V. Paradiso, A. Grassi, S. Milione, L. Cavallo and C. Capacchione, Chem. – Eur. J., 2020, 26, 5347–5353 CrossRef CAS PubMed.
- I. Contento, D. H. Lamparelli, A. Buonerba, A. Grassi and C. Capacchione, J. CO2 Util., 2022, 66, 102276 CrossRef CAS.
- V. Paradiso, F. Della Monica, D. H. Lamparelli, S. D'Aniello, B. Rieger and C. Capacchione, Catal. Sci. Technol., 2021, 11, 4702–4707 RSC.
- A. M. Hollas, J. W. Ziller and A. F. Heyduk, Polyhedron, 2018, 143, 111–117 CrossRef CAS.
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 41141 CrossRef.
- R. Chiarcos, M. Laus, K. Sparnacci, R. Po, P. Biagini, I. Tritto, L. Boggioni and S. Losio, Eur. Polym. J., 2023, 192, 112058 CrossRef CAS.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed.
- G. Si, L. Zhang, B. Han, H. Zhang, X. Li and B. Liu, RSC Adv., 2016, 6, 22821–22826 RSC.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- J. Y. Jeon, S. C. Eo, J. K. Varghese and B. Y. Lee, Beilstein J. Org. Chem., 2014, 10, 1787–1795 CrossRef PubMed.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- M. R. Kember, F. Jutz, A. Buchard, A. J. White and C. K. Williams, J. Chem. Sci., 2012, 3, 1245–1255 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00832k |
| This journal is © The Royal Society of Chemistry 2023 |