
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the mechanism of benzimidazole synthesis via copper-catalysed intramolecular N-arylation†
Xiaodong
Jin‡
 ,
Yongjie
Lin
and
Robert P.
Davies
*
,
Yongjie
Lin
and
Robert P.
Davies
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: r.davies@imperial.ac.uk
First published on 28th November 2023
Abstract
Copper(I)-catalysed intramolecular Ullmann N-arylation has been widely used to synthesise benzimidazoles. However, the possible intermediates on the catalytic pathway and the role of the ancillary ligands in these systems have been seldom studied and are currently not fully understood, especially when compared to comparable intermolecular Ullmann reactions. Accordingly, this work explores the copper(I)-intramolecular N-arylation of 1,2-dimethylbenzimidazole, reporting on the solid-state structures of several copper(I) species with the reaction substrate and product. In addition, kinetic profiling using bis(tetra-n-butylphosphonium) malonate as a soluble base has been carried out, notably revealing negligible catalyst deactivation but significant catalyst inhibition. Based on these results an improved catalytic protocol using sub-mol% catalyst loading has been developed.
Introduction
Benzimidazoles are an important class of heterocycles comprising of benzene-fused imidazole scaffolds. They are often biologically active and therefore play an important role in pharmaceutical research.1–3 Benzimidazoles are one of the most commonly encountered nitrogen heterocycles in U.S. FDA approved drugs, and their derivatives have shown activity for a wide range of applications including antiulcers, anthelmintics, antivirals, anticancer drugs, antimicrobials, analgesics and anti-inflammatory agents.4The synthesis of benzimidazoles has therefore attracted great interest over the years.5,6 The two most common strategies reported for the preparation of substituted benzimidazoles are condensation and intramolecular C–N cross coupling (Scheme 1). The condensation method forms two new C–N bonds by reacting 1,2-diaminoarenes with carboxylic acids7–9 or aldehydes/oxidants.10–12 However, this long-standing protocol has some drawbacks, including the use of strong acids and high reaction temperatures. The alternative newer intramolecular cross-coupling method employs amidines as starting materials and can operate under milder conditions. The key ring-forming step is a metal-catalysed intramolecular C–N cross coupling using N′-(2-halogenphenyl)-N-substituted-ethanimidamide. Many different combinations of pre-catalyst and auxiliary ligand have been employed for these intramolecular cyclizations, including Pd-phosphine catalysts,13 Cu2O or CuI,14,15 CuO nanoparticles,16 and even transition-metal-free conditions (for example, KOH mediated couplings in DMSO at 120 °C).17
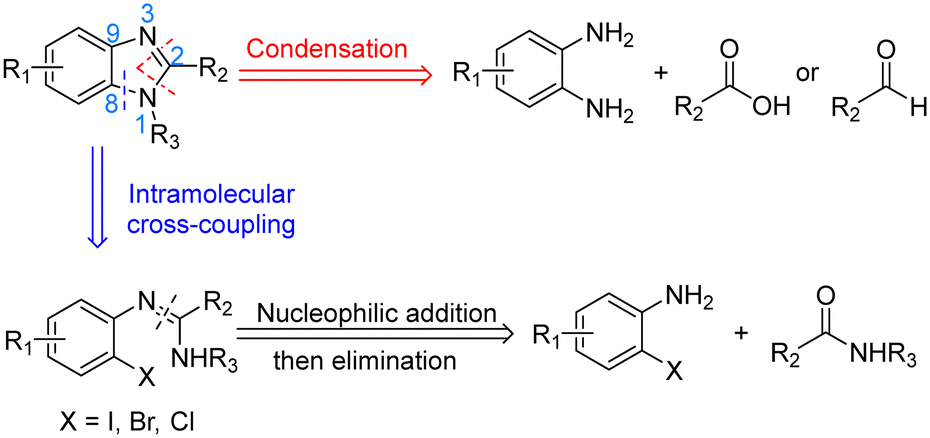 | ||
| Scheme 1 Retrosynthetic synthesis of benzimidazoles. | ||
Recent advances in intermolecular copper-catalysed N-arylation (including the introduction of ancillary ligands) have led to higher yields, improved reliability, lower catalyst loadings, and milder reaction conditions.18,19 In addition, it has permitted the use of more challenging substrates such as aryl chlorides. Over recent years, studies on the mechanism of the intermolecular N-arylation (Ullmann amination) reaction by our group and others has led to a much improved understanding of these systems.20–26 However, to date, mechanistic studies on related intramolecular Ullmann reactions are rare, and advances in the understanding and methodology for intermolecular coupling has yet to be fully transitioned to these intramolecular systems.
Therefore, in order to build an improved mechanistic understanding of copper-catalysed intramolecular N-arylation and to identify any differences with the intermolecular systems, a series of reactivity and mechanism studies have been carried out in this work for the synthetically important preparation of benzimidazoles. The solid-state structures of several copper(I) species derived from the reaction starting materials have been obtained. These structures reveal a range of potential coordination modes adopted by the substrate and auxiliary ligand (where present) with the copper(I) centre. The incorporation of both the aryl halide and amine functionalities within the same substrate permits additional insight not often accessible using intermolecular systems.
Detailed kinetic profiling using reaction progress kinetic analysis (RPKA) methodology27,28 has also been carried out to study the potential catalyst inhibition and deactivation pathways which may occur during the catalytic process, and to determine the rate dependence on the concentration of reactants. Based on these studies, an improved protocol capable of operating at sub-mol% copper catalyst loadings has been developed.
Results and discussion
TBPM promoted copper(I)-catalysed intramolecular N-arylation
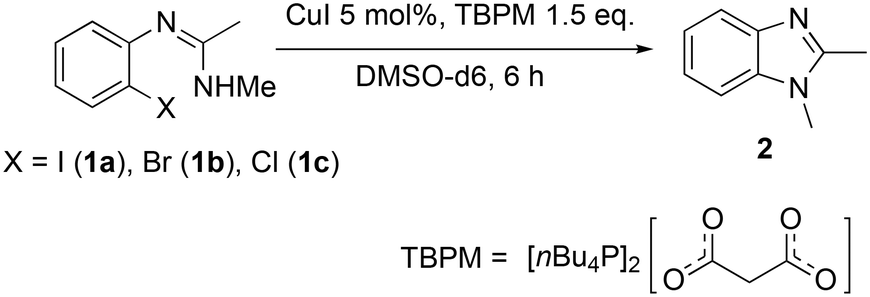
The copper(I)-catalysed synthesis of 1,2-dimethylbenzimidazole (2) via intramolecular C–N cross-coupling of N′-(2-halogenphenyl)-N-methylethanimidamide (1a–1c) was selected as a model reaction for these studies. The soluble ionic base n-tetrabutylphosphonium malonate (TBPM) has previously been reported in the literature to promote copper-catalysed inter-molecular N-arylation reactions,23,25,29 and was employed here as the base. Significantly, the excellent solubility of TBPM in the reaction mixture precludes any base mass transfer effects. In terms of substrate reactivity, aryl iodide (1a) was observed to be the most reactive with self-cyclisation occurring at room temperature (38% yield) even without a catalyst, and over 99% yield of the cyclised product was obtained with the addition of 5 mol% CuI (Table 1, entries 1 and 2). The aryl bromide 1b also gave near quantitative conversions in the presence of the catalyst, though no background reaction without a catalyst was observed in this case (entries 3 and 4). The less reactive aryl chloride 1c required both the copper catalyst and high reaction temperatures (130 °C) to give satisfactory yields (entries 5 and 6).|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | X = | CuI | Temp. (°C) | Yield (%) |
| Reaction yield was measured by 1H NMR using naphthalene as internal standard. | |||||
| 1 | 1a | I | — | r.t. | 38 |
| 2 | 1a | I | 5% | r.t. | >99 |
| 3 | 1b | Br | — | r.t. | 0 |
| 4 | 1b | Br | 5% | r.t. | >99 |
| 5 | 1c | Cl | — | 130 | 0 |
| 6 | 1c | Cl | 5% | 130 | 80 |
Copper(I) complexes: isolation and characterisation
In order to investigate the different complexes that might be present in the reaction mixture, a study of the solid-state structures of potential copper containing intermediates was carried out. Single crystals were obtained by reacting aryl halides (1a–1d) with mesitylcopper(I) followed by the recrystallisation of the resultant copper(I) complexes from toluene/hexane (Scheme 2).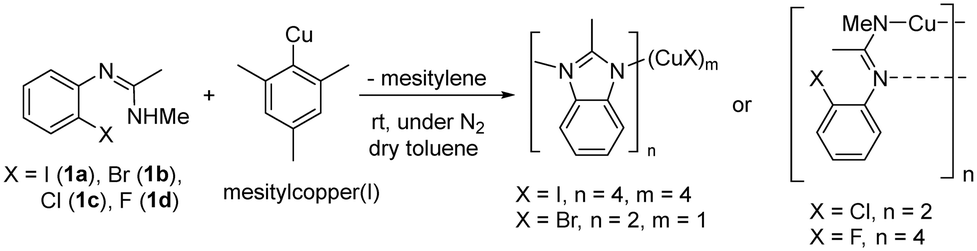 | ||
| Scheme 2 Synthesis of new copper(I) complexes in this work. | ||
For the more reactive aryl iodide (1a) and bromide (1b), intramolecular amination occurred readily giving complexes in which the imine group of the cyclised benzimidazole product 2 coordinates to a copper(I) centre. However, the less reactive aryl chloride (1c) and fluoride (1d) remained uncyclized with the negative charge on the substrate delocalised over the imidazole N–C–N unit.
Reaction of aryl iodide 1a with mesitylcopper led to the formation of crystals which were identified, using single crystal X-ray diffraction, as the tetramer Cu4I424 (Fig. 1a). Each copper(I) centre forms bonds with either two or three bridging iodide anions and one nitrogen (imine) atom. Cu–N distances are 1.957(3) and 1.980(3) Å. This gives rise to a four rung CuI twisted ladder structure in which the copper atoms adopt a close to planar rhombic Cu4 arrangement. Closer inspection of the benzimidazole groups within the copper complex reveals them to stack in an almost head-to-tail confirmation (offset 156°). Adjacent imidazole rings stack directly on one another with a distance between the centroids of 3.536 and 3.614 Å. A search of the literature shows benzimidazole-ligated copper(I) iodide complexes to have been reported with monomeric, dimeric, tetrameric, and polymeric CuI units. However, only a small number of tetrameric clusters are known, all of which contain cubic Cu4I4 units based around a tetrahedral arrangement of copper atoms (Fig. 1b).30–32 The unique twisted ladder cluster in Cu4I424 maybe due to the stabilisation obtained from the π-stacking combined with the steric requirements of the methyl group on the carbon of the imidazole ring.
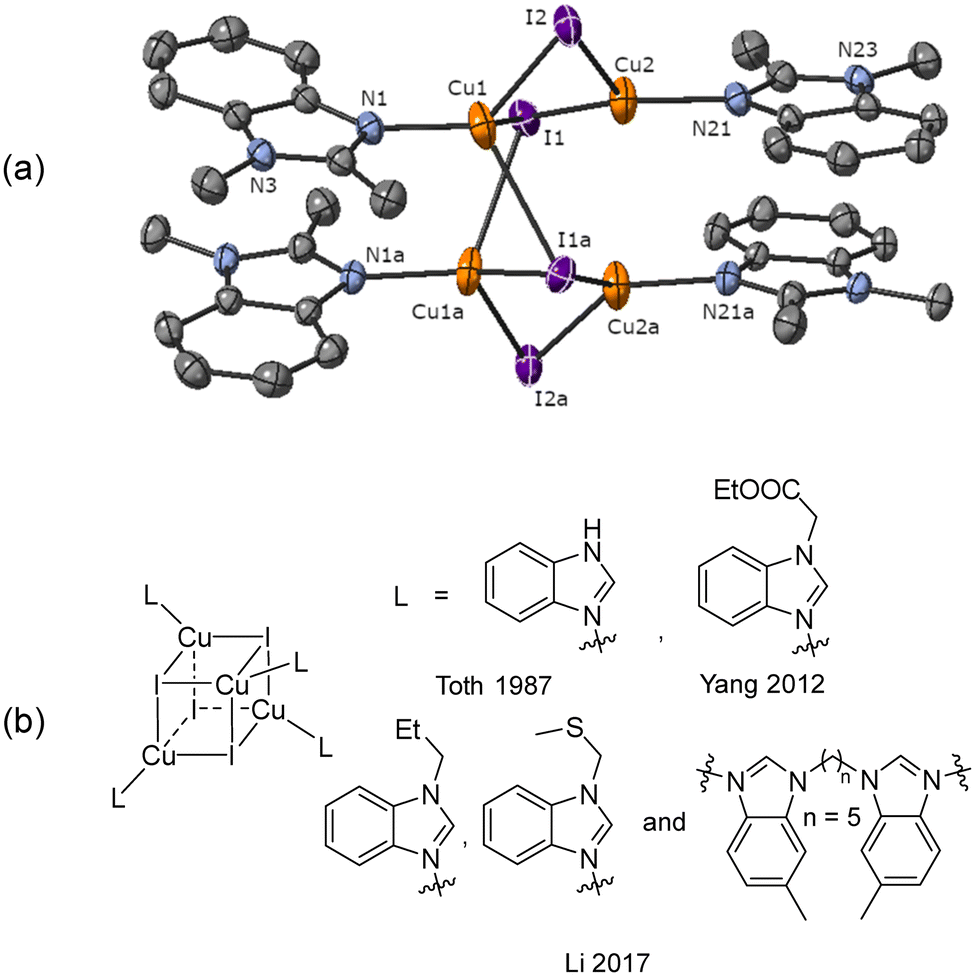 | ||
| Fig. 1 (a) Solid-state structure of Cu4I424; thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (b) Benzimidazole ligated Cu4I4 complexes reported in the literature.30–32 | ||
Similar to 1a, intramolecular amination also occurred when aryl bromide 1b was reacted with mesitylcopper(I). However, in this case, a different aggregation state was observed in the solid state with just one CuBr unit coordinating to two molecules of 2 (Fig. 2a). The CuBr22 complex has comparable Cu–N bond distances (1.9718(18) and 1.9853(18) Å) to those in the Cu4I424 tetramer. Two other examples of benzimidazole ligated copper(I) bromide complexes have previously been reported in the literature (Fig. 2b).33,34
 | ||
| Fig. 2 (a) Solid-state structure of CuBr22; thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (b) Benzimidazole ligated CuBr complexes reported in literature.33,34 | ||
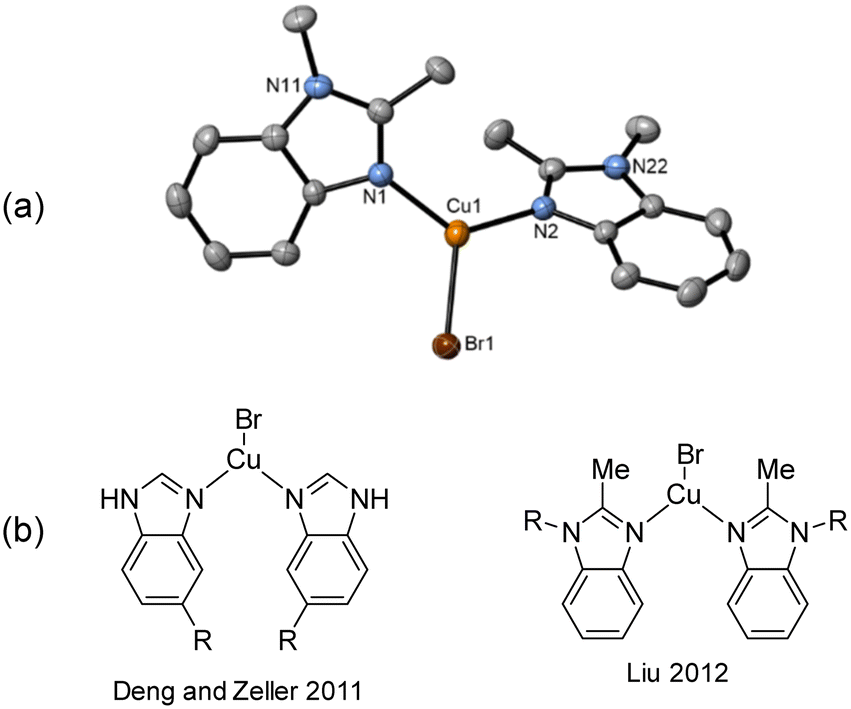
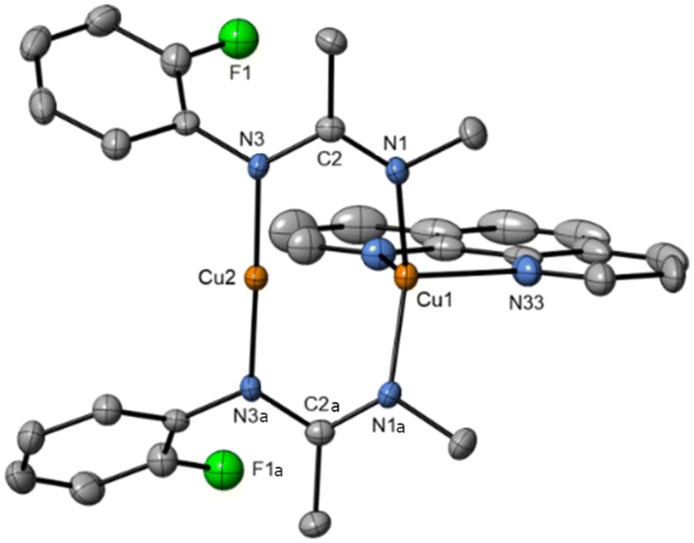
Due to the stronger aryl-halide bond in 1c, intramolecular amination did not proceed in the reaction of mesitylcopper(I) with 1c at room temperature. X-ray diffraction studies instead revealed the formation of the dimeric structure 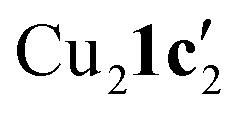 (where 1c′ is deprotonated 1c) comprising of a nearly planar Cu2N4C2 8-membered ring (Cu⋯Cu distance 2.441 Å). The secondary amine group in 1c has been deprotonated by mesitylcopper(I) to give a Cu–N bond. The Cu–N bonds in
(where 1c′ is deprotonated 1c) comprising of a nearly planar Cu2N4C2 8-membered ring (Cu⋯Cu distance 2.441 Å). The secondary amine group in 1c has been deprotonated by mesitylcopper(I) to give a Cu–N bond. The Cu–N bonds in 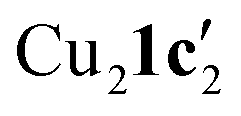 are shorter (mean 1.882°) than those in either Cu4I424 or CuBr22. Furthermore, the C1–N1 and C1–N11 bond distances (1.344(6) and 1.314(6) Å) are indicative of at least partial delocalization of the negative charge over the amidinate units. Although several copper(I) complexes containing planar Cu2N4C2 8-membered rings have been reported in the literature,35–37 these differ from
are shorter (mean 1.882°) than those in either Cu4I424 or CuBr22. Furthermore, the C1–N1 and C1–N11 bond distances (1.344(6) and 1.314(6) Å) are indicative of at least partial delocalization of the negative charge over the amidinate units. Although several copper(I) complexes containing planar Cu2N4C2 8-membered rings have been reported in the literature,35–37 these differ from 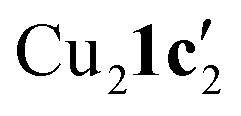 by virtue of being cationic, with the copper centers ligated by neutral bridging ligands (Fig. 3b).
by virtue of being cationic, with the copper centers ligated by neutral bridging ligands (Fig. 3b).
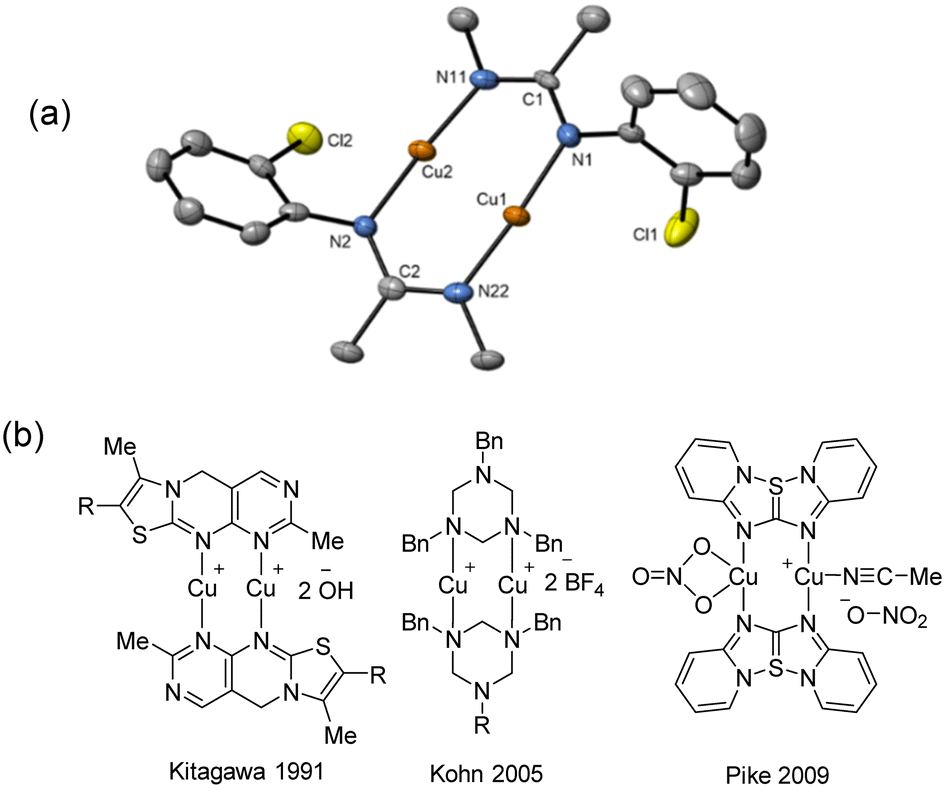 | ||
Fig. 3 (a) Solid-state structure of one of the two independent molecules in 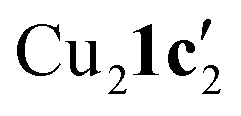 ; thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (b) Copper(I) complexes containing planar Cu2N4C2 8-membered rings.35–37 ; thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (b) Copper(I) complexes containing planar Cu2N4C2 8-membered rings.35–37 | ||
When aryl fluoride 1d was employed in the reaction with mesitylcopper(I), a copper complex with a tetrameric aggregation state was observed (Fig. 4a). 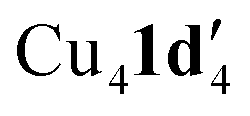 (where 1d′ is deprotonated 1d) contains a folded Cu4N4C4 16-membered ring with a central distorted rhombic Cu4 core (Fig. 4b). Similar to
(where 1d′ is deprotonated 1d) contains a folded Cu4N4C4 16-membered ring with a central distorted rhombic Cu4 core (Fig. 4b). Similar to 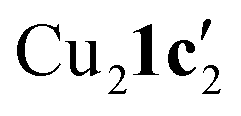 , the negative charge is delocalized within the N–C–N units with, for example, C1–N1 and C1–N11 bond distances of 1.343(5) Å and 1.313(5) Å respectively. Similar copper(I) complexes containing 16-membered Cu4N8C4 (ref. 38) or Cu4N12 (ref. 39–43) rings, planar Cu4 arrangements, and close to linear N–Cu–N coordination have been reported in the literature (Fig. 4c).
, the negative charge is delocalized within the N–C–N units with, for example, C1–N1 and C1–N11 bond distances of 1.343(5) Å and 1.313(5) Å respectively. Similar copper(I) complexes containing 16-membered Cu4N8C4 (ref. 38) or Cu4N12 (ref. 39–43) rings, planar Cu4 arrangements, and close to linear N–Cu–N coordination have been reported in the literature (Fig. 4c).
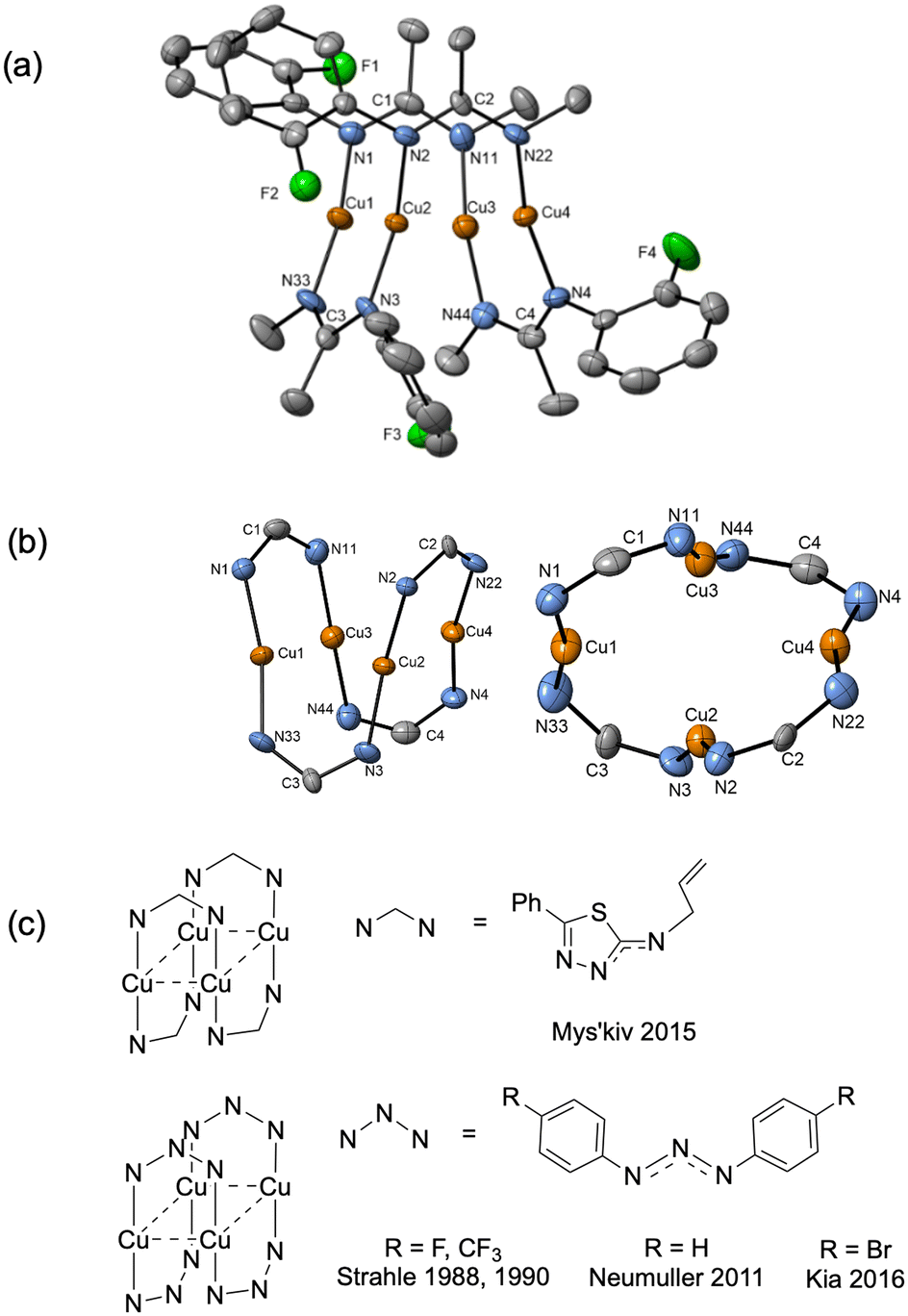 | ||
Fig. 4 (a) Solid-state structures of 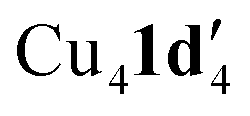 ; thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (b) Different views of the Cu4N4C4 16-membered ring. (c) Copper(I) complexes containing a 16-membered ring and N–Cu–N coordination.38–43 ; thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. (b) Different views of the Cu4N4C4 16-membered ring. (c) Copper(I) complexes containing a 16-membered ring and N–Cu–N coordination.38–43 | ||
Given that the addition of 1,10-phenanthroline (L1) has been shown to increase product yield in copper catalysed benzoxazole synthesis,44 the interaction of L1 with 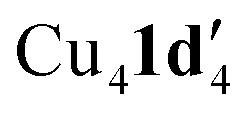 was also investigated. Thus, reaction of 1d with mesitylcopper(I) in the presence of L1 gave crystals which were characterized by X-ray diffraction as Cu2(1d′)2(L1) (Fig. 5). The addition of L1 therefore leads to a dimeric complex containing an 8-membered Cu2N4C2 ring. This contrasts to the tetramer
was also investigated. Thus, reaction of 1d with mesitylcopper(I) in the presence of L1 gave crystals which were characterized by X-ray diffraction as Cu2(1d′)2(L1) (Fig. 5). The addition of L1 therefore leads to a dimeric complex containing an 8-membered Cu2N4C2 ring. This contrasts to the tetramer 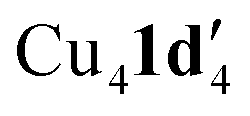 observed without the presence of L1 (Fig. 4a). The structure of Cu2(1d′)2(L1) also differs from the dimeric structure of
observed without the presence of L1 (Fig. 4a). The structure of Cu2(1d′)2(L1) also differs from the dimeric structure of 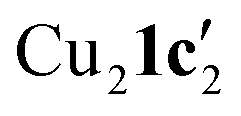 (Fig. 3a) as the two aryl halides adopt a head-to-head arrangement in Cu2(1d′)2(L1), rather than the head-to-tail arrangement in
(Fig. 3a) as the two aryl halides adopt a head-to-head arrangement in Cu2(1d′)2(L1), rather than the head-to-tail arrangement in 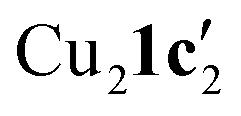 . The presence of L1 leads to a significant elongation of the Cu1–N1 bond (1.999(2) Å) when compared to the Cu2–N3 bond (1.859(2) Å). This can be attributed to the increased coordination number (four) of Cu1 compared to Cu2 (which is two coordinate). The delocalization of the negative charge within the N–C–N unit was also confirmed by the C–N bond distances (N1–C2 = 1.307(4) Å; C2–N3 = 1.352(4) Å).
. The presence of L1 leads to a significant elongation of the Cu1–N1 bond (1.999(2) Å) when compared to the Cu2–N3 bond (1.859(2) Å). This can be attributed to the increased coordination number (four) of Cu1 compared to Cu2 (which is two coordinate). The delocalization of the negative charge within the N–C–N unit was also confirmed by the C–N bond distances (N1–C2 = 1.307(4) Å; C2–N3 = 1.352(4) Å).
 | ||
| Fig. 5 Solid-state structure of Cu2(3d′)2(L2). Thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. | ||
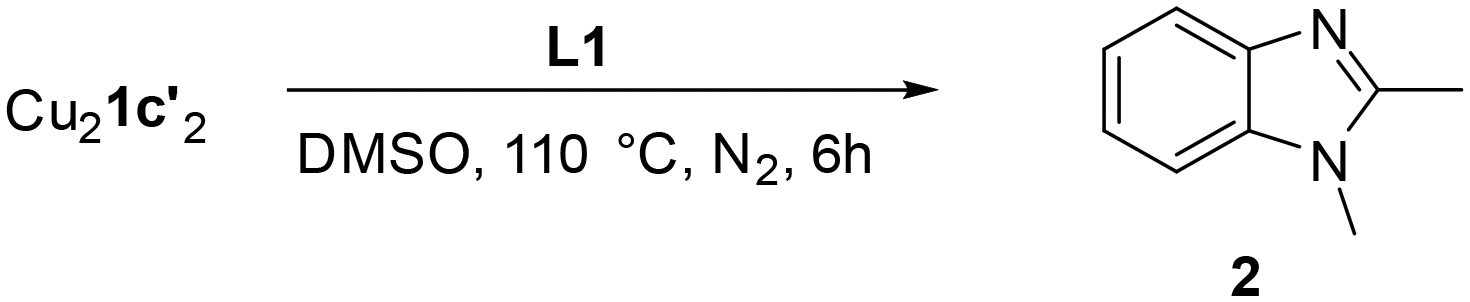
To confirm whether these copper(I) complexes are feasible reaction intermediates, the reactivity of 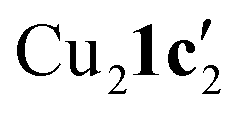 was studied by heating the complex, with or without L1, to 110 °C in DMSO (Table 2).
was studied by heating the complex, with or without L1, to 110 °C in DMSO (Table 2). 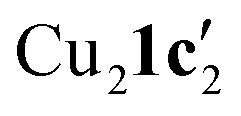 reacts at this higher temperature to give the desired cyclized benzimidazole product 2, supporting its viability as an intermediate on the reaction pathway. Furthermore, addition of L1 gave an improved yield for this reaction (see Fig. 11 and discussion later for a comparison of catalytic reaction rates with different ligands).
reacts at this higher temperature to give the desired cyclized benzimidazole product 2, supporting its viability as an intermediate on the reaction pathway. Furthermore, addition of L1 gave an improved yield for this reaction (see Fig. 11 and discussion later for a comparison of catalytic reaction rates with different ligands).
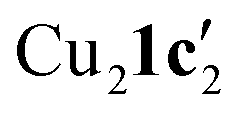 toward benzimidazole synthesis
toward benzimidazole synthesis
|
|
|||
|---|---|---|---|
| Entry | Substrate | L1 | 2 (%) |
| The reaction was carried out by heating the copper(I) complex (0.1 mmol) in DMSO (1 mL) with or without L1 (0.1 mmol). The yield of 2 was determined by 1H NMR using naphthalene as internal standard. | |||
| 1 |
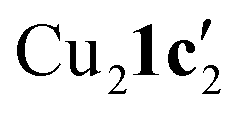
|
— | 65 |
| 2 |
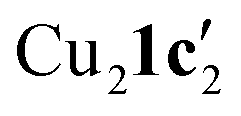
|
1 equiv. | 89 |
Overall, the solid-state structures of these copper(I) complexes furnish some novel insights into copper coordination modes and aggregation states with amidinate and imidazole substrates. In all the uncyclized products, it is interesting to note that the copper centers coordinate to the amidinate nitrogens and thus remain relatively distant from the aryl-halide bonds. If a similar motif were adopted under the catalytic reaction conditions it is likely that aryl-halide activation would require the involvement of an additional copper center for the intramolecular cyclization reaction to proceed. However, it is difficult to draw any direct parallels between the solid-state structures and catalyst speciation in the reaction due to the different conditions employed (solvent, presence of base, temperature etc.). Therefore, an alternative approach was also employed based on kinetic measurements of the actual catalytic system as detailed below.
Reaction kinetic studies
In order determine the rate dependence in reactants as well as build a better understanding of the catalyst performance, the intramolecular N-arylation reaction was profiled using a superCRC calorimeter (see the ESI† for full details). Initial reaction calorimetry studies focused upon the rate dependence on catalyst concentration, with the order in the catalyst determined by comparing the graphical rate equations obtained using different catalyst concentrations.27 A positive non-integer order in [Cu]total was observed (Fig. 6a) with ‘best fit’ reaction rate plots obtained by normalising by [Cu]total1.35 (Fig. 6b). Non-integer orders in catalyst concentration have been used to infer catalyst deactivation or off-cycle equilibrium(s), whereas an order in [catalyst] greater than one can indicate catalyst inhibition or more than one catalytic species being involved in the rate determining step.45 This later scenario could fit with observations from the solid-state studies where the copper centre in the metalated aryl halide would seem to be sited too distant from the aryl-halide bond to permit oxidative addition at this centre, and hence an additional catalytic copper centre would be required for the rate limiting oxidative addition step. The following experiments explore this possibility in more detail. It is noteworthy that this behaviour therefore differs to that observed for intermolecular Ullmann aminations where generally first order reactions in [Cu] have been reported and where amide coordination/deprotonation and aryl halide activation occurs at the same copper centre.22,24,25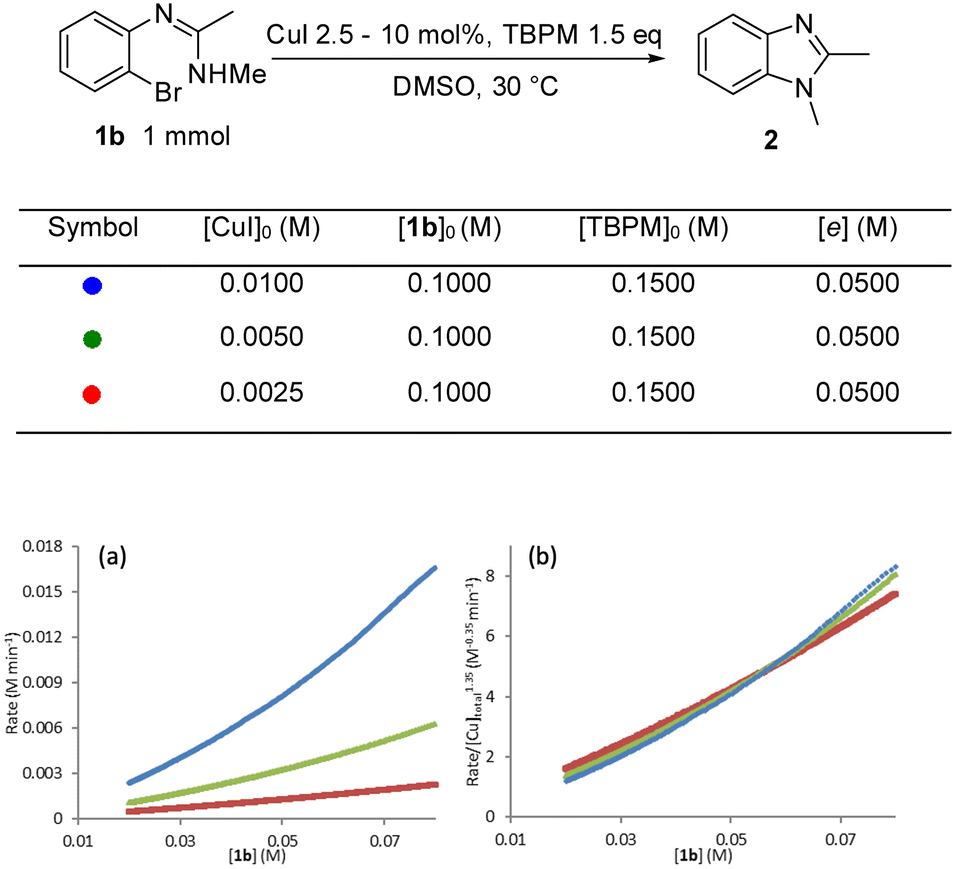 | ||
| Fig. 6 Rate dependence on [CuI]: (a) comparison of graphical rate equations using different [CuI]; (b) the reaction rate is normalized by [Cu]1.35 to show the best fit overlap. | ||
A “same excess” experiment was carried out to investigate potential catalyst deactivation and/or inhibition. This experiment involved reducing [TBPM]0 and [1b]0 in 0.0250 M increments, whilst keeping the excess [e] constant at 0.0500 (Fig. 7a). The marked lack of overlay of the graphical rate equations indicates that strong catalyst deactivation and/or inhibition processes are occurring. To distinguish between deactivation and inhibition, a “same excess” experiment was carried out with the addition of the three reaction products, namely 1,2-dimethylbenzimidazole (2), the mono-anion of malonic acid (MalH) and tetra-(n-butyl)phosphonium bromide (TBPB) (Fig. 7b) – note that in these experiments all reagents and additives are fully dissolved to give a homogeneous solution. All three graphical rate equations now overlayed one another very well, indicating that catalyst inhibition is occurring and any catalyst deactivation is negligible. This differs from our previous calorimetry study on intermolecular copper-catalysed N-arylation in which catalyst deactivation occurred and catalyst inhibition was negligible.36
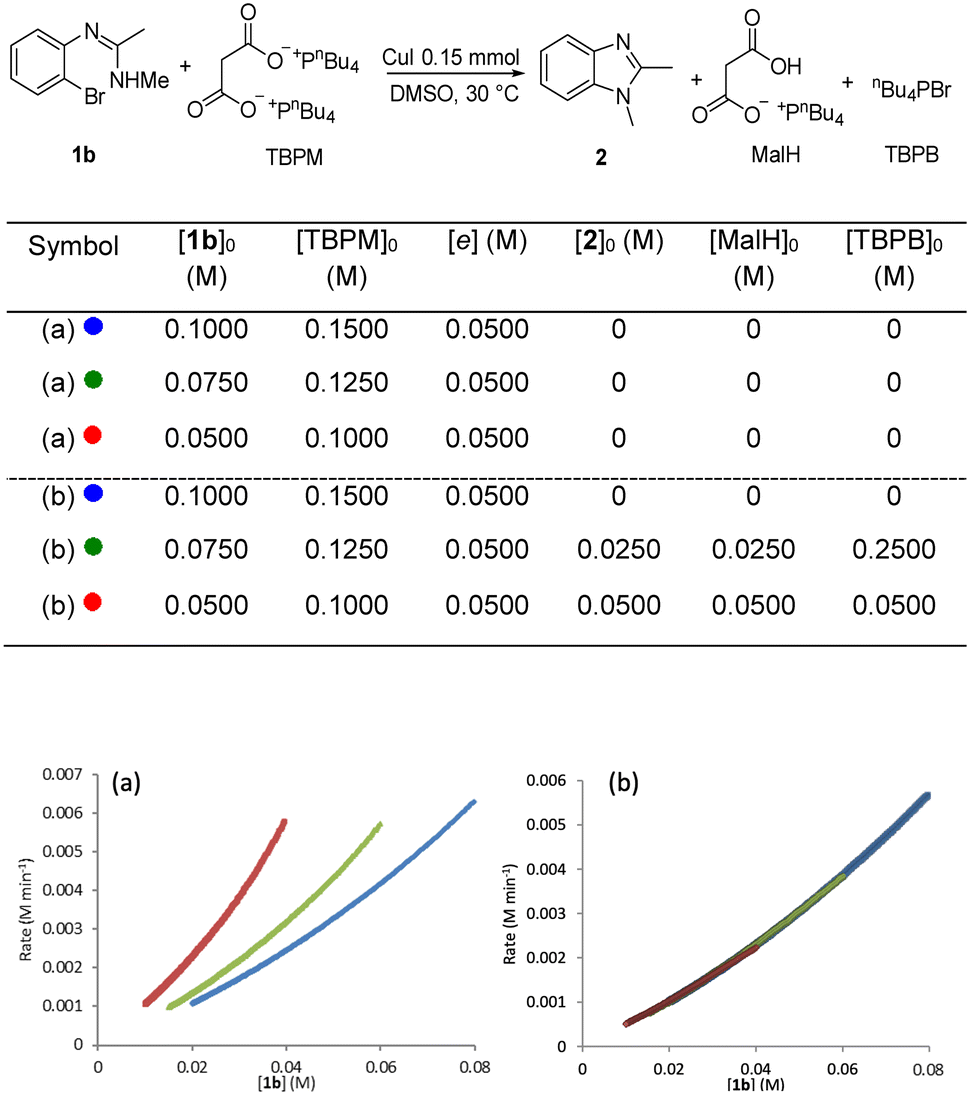 | ||
| Fig. 7 (a) “Same excess” experiment: graphical rate against [1b]; (b) “same excess” experiment with addition of corresponding amounts of 2, MalH and TBPB. | ||
As the initial inhibition experiment used addition of all products together ([2], [MalH] and [TBPB]), the inhibition cause by each component was also studied individually, starting with benzimidazole 2 (Fig. 8). This revealed a significant negative order in [2] which could be due to an off-cycle process involving coordination of 2 to the copper center. The predilection of 2 to coordinate to Cu(I) has already been observed in the solid-state studies (Fig. 1 and 2) and can be explained by the accessible exocyclic lone pair on the imidazole nitrogen which readily coordinates to the copper center. Inhibition experiments using the mono-anion of malonic acid (MalH) and tetra-(n-butyl)phosphonium bromide (TBPB) both showed less pronounced negative influences on the reaction rate (see the ESI† for plots), which can be rationalized respectively by the formation of an inactive diligated copper(I) complex with MalH and variations in solution conductivity with TBPB.
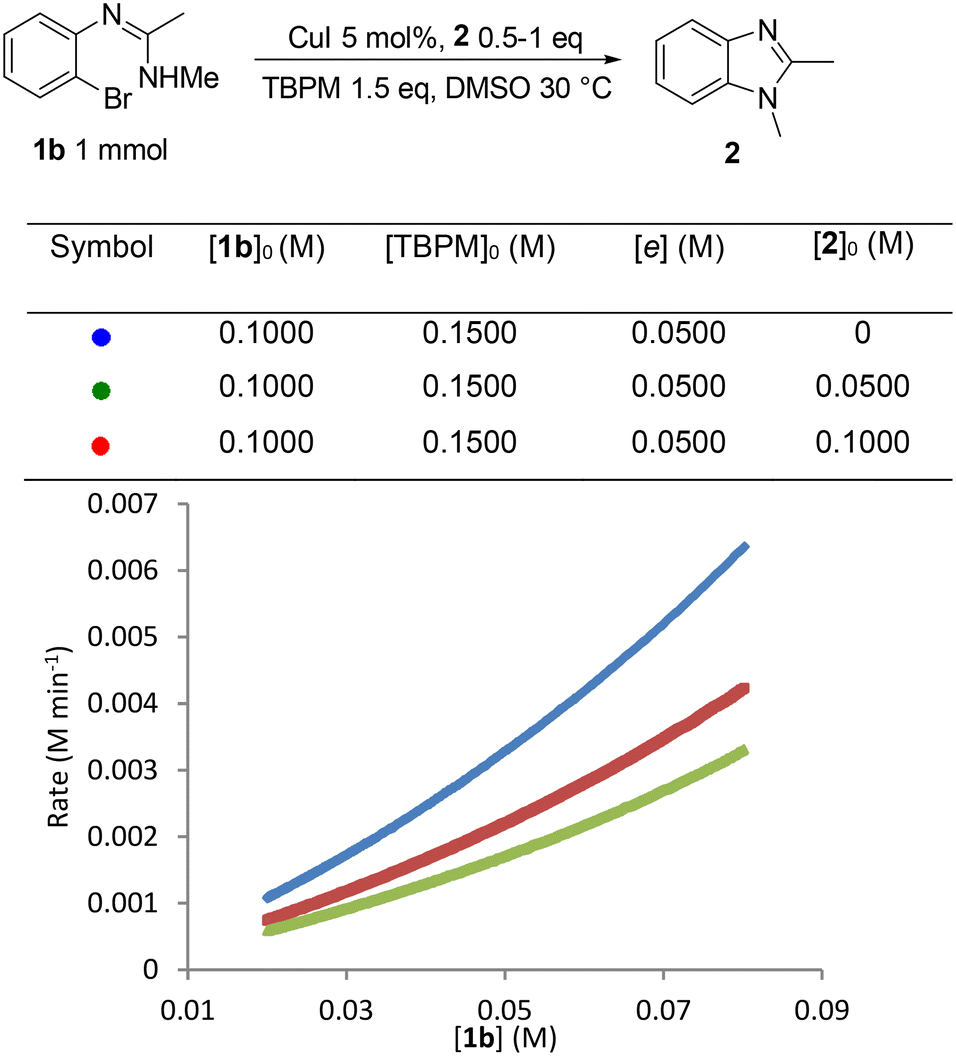 | ||
| Fig. 8 Catalyst inhibition experiment: graphical rate against [1b] with addition of 1,2-dimethylbenzimidazole 2. | ||
Different excess experiments28 were then carried out to determine the rate dependences on [1b] and [TBPM]. Fig. 9 shows the graphical equations for [1b]. The reaction rate can be normalized by [1b]1 indicating that the reaction is first order in the aryl bromide starting material (Fig. 9b).
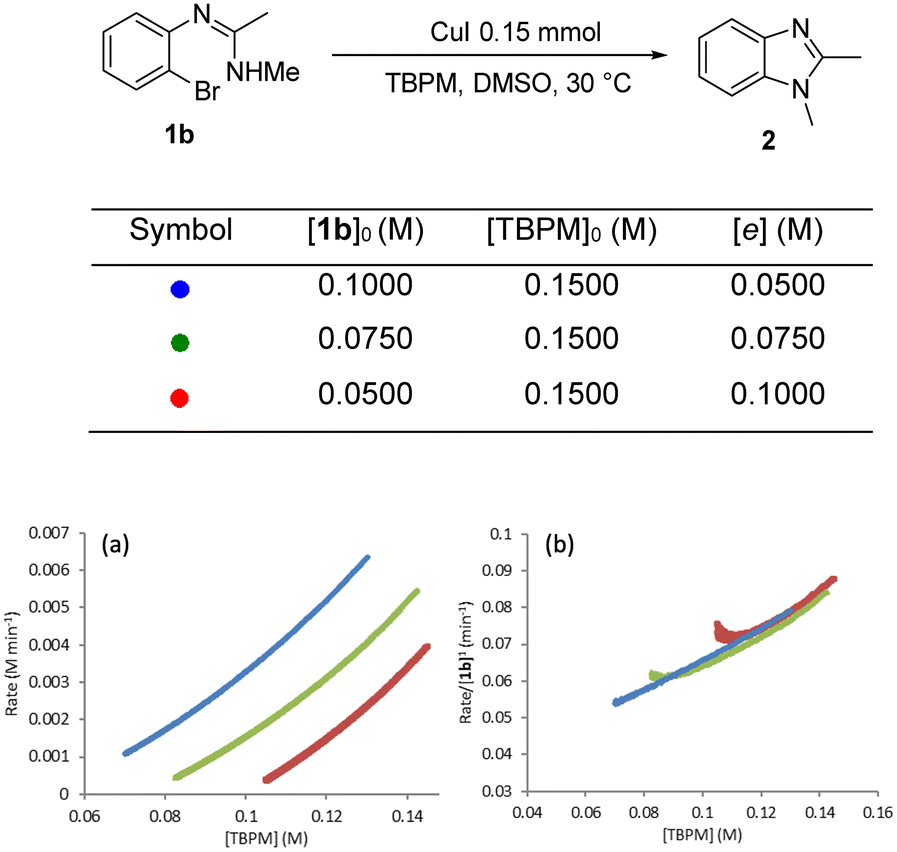 | ||
| Fig. 9 “Different excess” experiment: (a) graphical rate equation against [TBPM]; (b) normalised reaction rate by [1b]1 and overlapped. | ||
The rate dependence on [TBPM] was found to be approximately negative first order (Fig. 10). This shows that higher TBPM loading can inhibit the reaction to give a slower overall rate. A negative rate dependence on [TBPM] was also reported in TBPM promoted Cu(I)-catalysed inter-molecular N-arylation where this was rationalised by the formation of an inactive (off-cycle) di(malonate) copper(I) complex.25
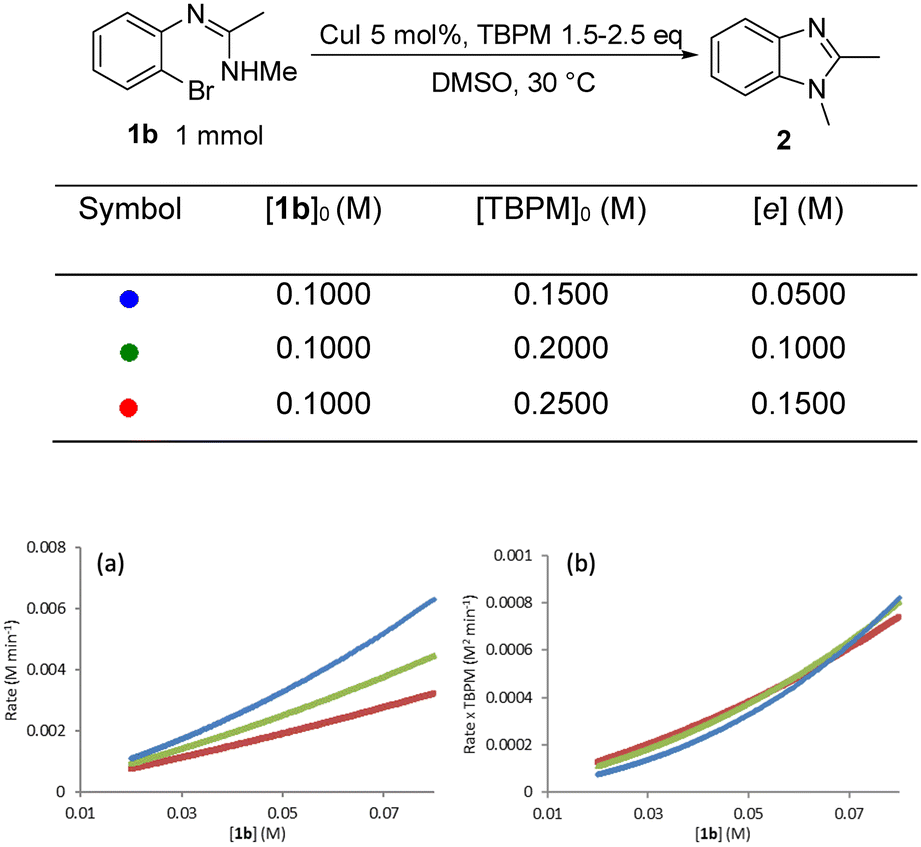 | ||
| Fig. 10 (a) “Different excess experiment”: graphical rate against [1b]; (b) normalized rate by [TBPM]−1. | ||
Based upon these kinetic studies and existing literature knowledge on related systems,23 we now put forward a postulated reaction mechanism (Scheme 3). The presence of two catalytic species in the rate limiting step, product inhibition, and a negative order in TBPM make this a challenging system to study, however some key salient similarities and differences can be observed between this intramolecular coupling and more widely studied intermolecular couplings.
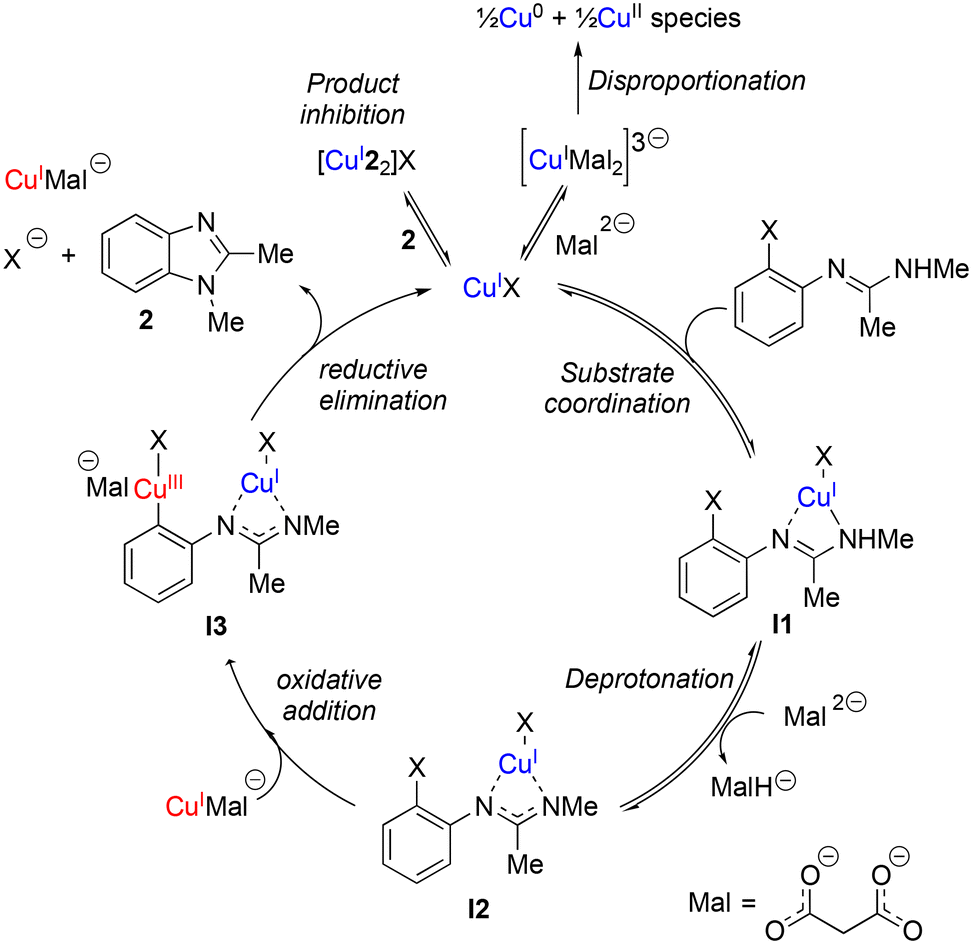 | ||
| Scheme 3 Proposed mechanism for TBPM promoted copper(I)-catalysed intramolecular N-arylation. | ||
Rate dependence on [ArBr] (1st order) and [TBPM] (approximately negative 1st order) are in agreement with the literature for intermolecular Ullmann reactions.23 Also the proposed oxidative addition rate limiting step which takes place at a copper center with a malonate XX-type ligand is the same. However a catalyst order greater than 1 is suggestive of two catalytic species reacting on a cycle and a ‘parent-offspring species mechanism’45 seems most probable. One copper centre aids with the deprotonation of the amidine unit (required due to the relative weakness of the TBPM base23) whilst the other copper centre facilitates the aryl halide oxidative addition step. Although all catalytic on-cycle species are shown in Scheme 3 as monomeric, aggregates similar to those observed in the X-ray diffraction studies cannot be ruled out.
Catalyst inhibition by the imidazole product (2) plays a significant role as the reaction progresses towards completion as revealed by the kinetic studies as shown in Scheme 3 as an off-cycle equilibrium. Additional equilibrium due to excess Mal coordination are also inhibitory to a lesser extent but are not shown in Scheme 3 for clarity.
Reaction optimisation
The addition of an ancillary ligand has previously been shown to help improve yields and reactions rates for Ullmann arylations when organic bases such as TBPM are used.24 However the use of organic bases with an ancillary ligand for intramolecular Ullmann arylations have not yet been investigated, hence TBPM promoted copper(I)-catalysed intramolecular N-arylation was also carried out in the presence of ancillary ligands. Unfortunately, none of the ligands studied (L1–L4) led to an improvement in the reaction rate compared to the ligandless system (Fig. 11). Moreover, increasing [L1] to 20 mol% resulted in an even lower reaction rate. Competition between the ancillary ligand and the malonate anion from TBPM for coordination to the metal center and the fact that malonate is already a good ligand for the reaction would explain these observations.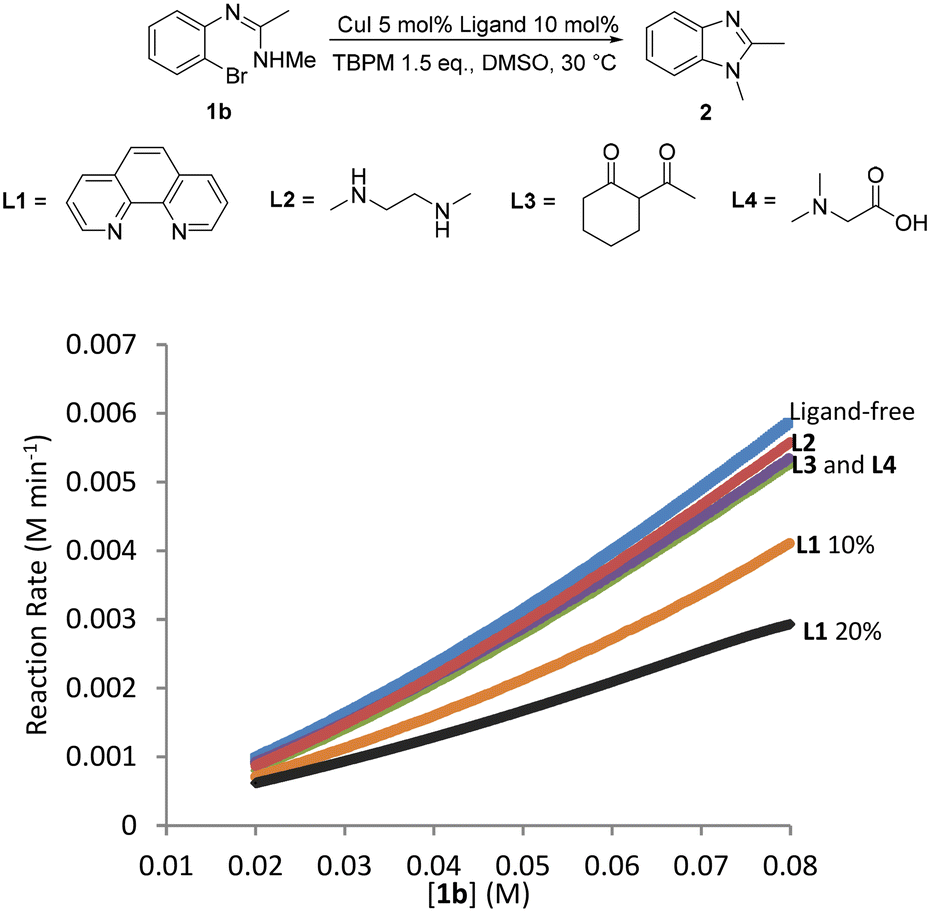 | ||
| Fig. 11 In situ ligand screening by calorimetry. | ||
Given the absence of any catalyst decomposition processes (as shown by the “same excess” experiments described above), attempts were made to lower the copper(I) loading to sub-mol% levels (Table 3). Pleasingly, it was shown that the reaction can proceed with catalyst loadings as low as 0.1 mol% CuI and still give satisfactory yields even at room temperature (entry 2). Over 99% yield can be achieved within 3 hours by increasing the reaction temperature to 110 °C (entry 5), although it should be noted that at this higher temperate the reaction can also proceed without a catalyst, although longer reaction times are required and the yield is significantly lower (entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | CuI (mol%) | Temp. (°C) | Time (h) | Yield (%) |
| 1 | — | 22 | 72 | n.r. |
| 2 | 0.1 | 22 | 72 | 91 |
| 3 | 1 | 22 | 72 | >99 |
| 4 | — | 110 | 24 | 63 |
| 5 | 0.1 | 110 | 3 | >99 |

Conclusion
New insights into the reactivity and mechanism of copper-catalysed intramolecular N-arylation for the synthesis of benzimidazoles have been obtained, including the solid-state structures of copper(I) complexes with the amidinate substrate and benzimidazole product. For aryl iodide and aryl bromide substrates, cyclisation occurred readily when they were treated with mesitylcopper(I) to give 1,2-dimetylbenzimidazole ligated copper(I) complexes Cu4I424 and CuBr22. The less reactive aryl chloride and fluoride substrates were found to form dimeric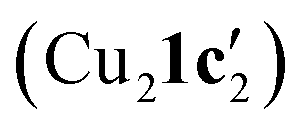 and tetrameric
and tetrameric 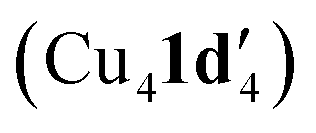 complexes respectively in which the aryl halide bond remained unreacted. Moreover, the solid-state structure of a 1,10-phenanthroline (L1) ligated intermediate (Cu2(1d′)2(L1)) was reported revealing how L1 can interact with just one of the metal centres.
complexes respectively in which the aryl halide bond remained unreacted. Moreover, the solid-state structure of a 1,10-phenanthroline (L1) ligated intermediate (Cu2(1d′)2(L1)) was reported revealing how L1 can interact with just one of the metal centres.
Kinetic profiling of the intramolecular reaction was carried out using RPKA methods and in situ monitoring by a calorimeter. An organic ionic base (TBPM) was used in the reaction to avoid any base mass transfer effects and to allow comparison with similar intermolecular Ullmann couplings using this base.23 Several parallels were observed between the intramolecular and intermolecular Ullmann couplings, notably the same rate dependence on [ArBr] (1st order) and [TBPM] (approximately negative 1st order). However, several significant differences were also found. Most notably, the rate dependence study on catalyst concentration reveals a complex order in [Cu]total (1 < order < 2), suggestive of two catalytic species reacting on a cycle in a parent-offspring species mechanism. The solid-state structures of 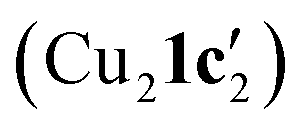 and
and 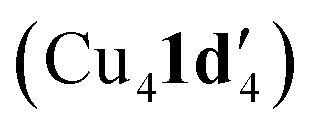 hint how one Cu centre might coordinate to the amidine functionality and aid its deprotonation, whilst the other facilitates activation of the aryl halide moiety via oxidative addition. Same excess experiments reveal high levels of catalyst inhibition by the product 2 which can be attributed to strong interactions between the readily accessible exocyclic nitrogen lone pair on benzimidazole and the copper centres – a similar motif was also observed in the solid-state structures of Cu4I424 and CuBr22. This highlights how small changes in the reaction components can have significant impact on the overall reaction kinetics. Based on these findings, we have been able to propose a new intramolecular sub-mol% copper(I)-catalysed process for the preparation of benzimidazoles. Further work is ongoing to further improve the scope of this reaction in terms of substrates and base.
hint how one Cu centre might coordinate to the amidine functionality and aid its deprotonation, whilst the other facilitates activation of the aryl halide moiety via oxidative addition. Same excess experiments reveal high levels of catalyst inhibition by the product 2 which can be attributed to strong interactions between the readily accessible exocyclic nitrogen lone pair on benzimidazole and the copper centres – a similar motif was also observed in the solid-state structures of Cu4I424 and CuBr22. This highlights how small changes in the reaction components can have significant impact on the overall reaction kinetics. Based on these findings, we have been able to propose a new intramolecular sub-mol% copper(I)-catalysed process for the preparation of benzimidazoles. Further work is ongoing to further improve the scope of this reaction in terms of substrates and base.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge Dr Andrew White for help with collecting and solving the crystal structure data.Notes and references
- B. Narasimhan, D. Sharma and P. Kumar, Med. Chem. Res., 2010, 21, 269–283 CrossRef.
- K. Shah, S. Chhabra, S. K. Shrivastava and P. Mishra, Med. Chem. Res., 2013, 22, 5077–5104 CrossRef CAS.
- Y. Bansal and O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208–6236 CrossRef CAS PubMed.
- R. Zou, K. R. Ayres, J. C. Drach and L. B. Townsend, J. Med. Chem., 1996, 39, 3477–3482 CrossRef CAS PubMed.
- M. Faheem, A. Rathaur, A. Pandey, V. Kumar Singh and A. K. Tiwari, ChemistrySelect, 2020, 5, 3981–3994 CrossRef CAS.
- V. A. S. Pardeshi, N. S. Chundawat, S. I. Pathan, P. Sukhwal, T. P. S. Chundawat and G. P. Singh, Synth. Commun., 2020, 51, 485–513 CrossRef.
- J. B. Wright, Chem. Rev., 1951, 48, 397–541 CrossRef CAS PubMed.
- S.-Y. Lin, Y. Isome, E. Stewart, J.-F. Liu, D. Yohannes and L. Yu, Tetrahedron Lett., 2006, 47, 2883–2886 CrossRef CAS.
- X. Wen, J. E. Bakali, R. Deprez-Poulain and B. Deprez, Tetrahedron Lett., 2012, 53, 2440–2443 CrossRef CAS.
- C. Mukhopadhyay and P. K. Tapaswi, Tetrahedron Lett., 2008, 49, 6237–6240 CrossRef CAS.
- M. Adharvana Chari, D. Shobha and T. Sasaki, Tetrahedron Lett., 2011, 52, 5575–5580 CrossRef CAS.
- S. M. Inamdar, V. K. More and S. K. Mandal, Tetrahedron Lett., 2013, 54, 579–583 CrossRef CAS.
- N. Zheng, K. W. Anderson, X. Huang, H. N. Nguyen and S. L. Buchwald, Angew. Chem., 2007, 119, 7653–7656 CrossRef.
- J. Peng, M. Ye, C. Zong, F. Hu, L. Feng, X. Wang, Y. Wang and C. Chen, J. Org. Chem., 2011, 76, 716–719 CrossRef CAS PubMed.
- K. Liubchak, K. Nazarenko and A. Tolmachev, Tetrahedron, 2012, 68, 2993–3000 CrossRef CAS.
- P. Saha, T. Ramana, N. Purkait, M. A. Ali, R. Paul and T. Punniyamurthy, J. Org. Chem., 2009, 74, 8719–8725 CrossRef CAS PubMed.
- H. Baars, A. Beyer, S. V. Kohlhepp and C. Bolm, Org. Lett., 2014, 16, 536–539 CrossRef CAS PubMed.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- E. Sperotto, G. P. van Klink, G. van Koten and J. G. de Vries, Dalton Trans., 2010, 39, 10338–10351 RSC.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- X. Ma and R. P. Davies, Adv. Synth. Catal., 2022, 364, 2023–2031 CrossRef CAS.
- Q. A. Lo, D. Sale, D. C. Braddock and R. P. Davies, Eur. J. Org. Chem., 2019, 2019, 1944–1951 CrossRef CAS.
- Q. A. Lo, D. Sale, D. C. Braddock and R. P. Davies, ACS Catal., 2017, 8, 101–109 CrossRef.
- S. Sung, D. Sale, D. C. Braddock, A. Armstrong, C. Brennan and R. P. Davies, ACS Catal., 2016, 6, 3965–3974 CrossRef CAS.
- S. Sung, D. C. Braddock, A. Armstrong, C. Brennan, D. Sale, A. J. White and R. P. Davies, Chemistry, 2015, 21, 7179–7192 CrossRef CAS PubMed.
- D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 10852–10866 CrossRef CAS PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- C. T. Yang, Y. Fu, Y. B. Huang, J. Yi, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed., 2009, 48, 7398–7401 CrossRef CAS PubMed.
- Y. Fang, W. Liu, S. J. Teat, G. Dey, Z. Shen, L. An, D. Yu, L. Wang, D. M. O'Carroll and J. Li, Adv. Funct. Mater., 2017, 27, 1603444 CrossRef.
- L. Yang and Z. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m796 CrossRef CAS PubMed.
- A. Toth, C. Floriani, A. Chiesi-Villa and C. Guastini, Inorg. Chem., 1987, 26, 3897–3902 CrossRef CAS.
- X. Wang, C. B. Liu, Y. S. Yan, S. T. Wang and L. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m153 CrossRef CAS PubMed.
- L. Ma, Y.-C. Qiu, G. Peng, J.-B. Cai, H. Deng and M. Zeller, CrystEngComm, 2011, 13, 3852 RSC.
- S. Kitagawa, S. Matsuyama, M. Munakata, N. Osawa and H. Masuda, J. Chem. Soc., Dalton Trans., 1991, 1717 RSC.
- R. D. Kohn, Z. Pan, M. Haufe and G. Kociok-Kohn, Dalton Trans., 2005, 2793–2797 RSC.
- A. Saxena, E. C. Dugan, J. Liaw, M. D. Dembo and R. D. Pike, Polyhedron, 2009, 28, 4017–4031 CrossRef CAS.
- Y. I. Slyvka, E. A. Goreshnik, B. R. Ardan, G. Veryasov, D. Morozov and M. G. Mys'kiv, J. Mol. Struct., 2015, 1086, 125–130 CrossRef CAS.
- E. Hartmann and J. Strähle, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 818–824 CrossRef CAS.
- E. Hartmann and J. Strähle, Z. Anorg. Allg. Chem., 1990, 583, 31–40 CrossRef CAS.
- M. Hörner, H. Fenner, W. Hiller and J. Beck, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 204–206 CrossRef.
- C. Lego and B. Neumüller, Z. Anorg. Allg. Chem., 2011, 637, 1784–1789 CrossRef CAS.
- R. Kia, Inorg. Chim. Acta, 2016, 446, 32–40 CrossRef CAS.
- G. Altenhoff and F. Glorius, Adv. Synth. Catal., 2004, 346, 1661–1664 CrossRef CAS.
- C. Alamillo-Ferrer, G. Hutchinson and J. Burés, Nat. Rev. Chem., 2022, 7, 26–34 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental and crystallographic details. CCDC 2263362 to 2263367. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy00767g |
| ‡ Current address: Department of Chemistry, Xi'an Jiaotong-Liverpool University, Suzhou 215123, China. |
| This journal is © The Royal Society of Chemistry 2023 |