
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbonic anhydrase mimics with rationally designed active sites for fine-tuned catalytic activity and selectivity in ester hydrolysis†
Foroogh
Bahrami
and
Yan
Zhao
 *
*
Department of Chemistry, Iowa State University, Ames, Iowa 50011-3111, USA. E-mail: zhaoy@iastate.edu; Fax: +1 515 294 0105; Tel: +1 515 294 5845
First published on 22nd August 2023
Abstract
Numerous hydrolytic enzymes utilize zinc as a cofactor for catalysis. We here report water-soluble polymeric nanoparticles with zinc ions in active sites and a nearby base as a mimic of carbonic anhydrase (CA). Their pKa of 6.3–6.4 for zinc-bound water is lower than the 6.8–7.3 value for natural enzymes, which allows the catalyst to hydrolyze nonactivated alkyl esters under neutral conditions—a long sought-after goal for artificial esterases. The size and shape of the active site can be rationally tuned through a template used in molecular imprinting. Subtle structural changes in the template, including shifting an ethyl group by one C–N bond and removal of a methylene group, correlate directly with catalytic activity. A catalyst can be made to be highly specific or have broad substrate specificity through modular synthesis of templates.
Introduction
Over 1000 enzymes use zinc as a cofactor,1,2 often for the hydrolysis of important biomolecules such as esters, peptides, and phosphate esters.3,4 To better understand catalytic mechanisms, researchers prepared numerous small-molecule zinc complexes as active site models for these enzymes, which have been thoroughly reviewed.5–8 These studies reveal three particular challenges in duplicating the catalytic feats of zinc enzymes. First, zinc ions in a catalyst need to be accessible to reactants (substrates and water) to be active but an exposed zinc cation complexed with a small organic molecule ligand can easily dimerize and lose its activity.9 Second, the products of hydrolysis in esters or phosphodiesters—e.g., anionic carboxylates or phosphates—are generally better ligands for zinc than the substrates and thus frequently prevent the catalysts from turning over. Third, the protein framework in an enzyme is not an innocent bystander in the catalysis but plays important roles, including in the binding of substrates and release of products, as well as other functions, such as mediation of proton transfers.More recently, zinc-based artificial enzymes have been constructed using peptides.9–19 Peptide-based materials have the advantage of using similar amino acids as natural enzymes to coordinate with zinc thus closely mimicking the biological ligand environment. In addition, they readily afford water-solubility and can protect the zinc center by sterics to eliminate metal center dimerization. Total turnover numbers (TONs), however, continue to be low in most reported cases (e.g., <50 and often <20),9–19 suggesting that product inhibition remains a challenge.
Zinc as a Lewis acid lowers the pKa of water significantly, from 15.7 to 9.0 in [Zn(OH2)6]2+.7 An enzyme active site can reduce the value further. Carbonic anhydrase (CA, EC 4.2.1.1), for example, has a pKa value of 6.8–7.3 for metal-bound water.20 A low pKa value means a significant percentage of zinc-bound water is deprotonated under physiological conditions. In contrast, most synthetic mimics of zinc enzymes, whether built on small organic molecules7 or peptide platforms,9–14 have higher pKa values for zinc bound water, typically in the range of 8 to 10. This limits their activity as a synthetic esterase, as the secondary rate constant for ester hydrolysis by hydroxide is 107–11 times faster than that by water.21
In this work, we report a method to create synthetic mimics of CA with an even lower pKa value than their natural counterparts. The reduced pKa allows the synthetic catalyst to hydrolyze esters under neutral conditions. A TON in the hundreds was obtained with little slowdown of the catalyst at higher substrate conversion. In the literature, highly activated substrates such as p-nitrophenyl esters have been the predominant substrates for traditional CA mimics.9–19,22 It has been demonstrated by Menger and co-workers that activated esters magnify the catalytic effects of synthetic catalysts and the effects tend to disappear even when a simple ethyl group replaces the p-nitrophenyl leaving group on the ester.23 In our catalysts, nonactivated alkyl esters are readily hydrolyzed, with high substrate selectivity or broad specificity, depending on how the active site is constructed.
Results and discussion
Our CA mimics were prepared through molecular imprinting. The technique generally creates imprinted binding sites in a cross-linked polymer network from template molecules and functional monomers (FMs).24–26 Molecularly imprinted polymers (MIPs) are useful for a wide range of applications including sensing, imaging, and biotechnology.27–29 MIP-based synthetic esterases have also been reported, although they also generally hydrolyze only activated esters.30–34In our case, molecular imprinting is performed in the mixed micelles of 1 containing the appropriate template, FM, divinyl benzene (DVB) as a radical cross-linker, and an oil-soluble radical initiator (Scheme 1).35 Briefly, the click reaction with diazide 2 affords a surface-cross-linked micelle (SCM) with some residual alkynes on the surface (Scheme 1, step a). Another round of the click reaction with monoazide 3 installs a layer of hydrophilic ligands on the surface of the SCM (step b). In step c, free radical polymerization cross-links the SCM core around the template, affording the so-called molecularly imprinted nanoparticles (MINPs). The templates are removed by precipitation of the nanoparticles in acetone and solvent washing (see the ESI† for details).
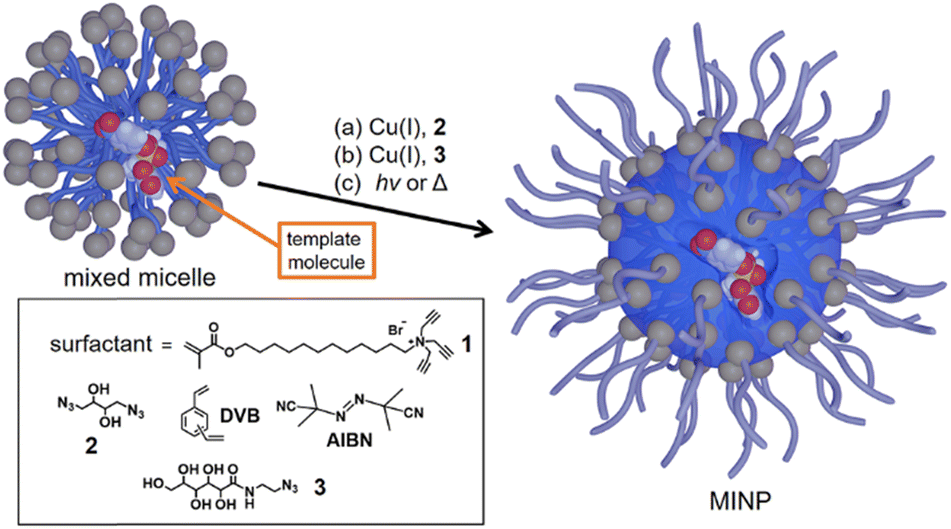 | ||
| Scheme 1 General procedure to prepare molecularly imprinted nanoparticles (MINPs) through templated polymerization in cross-linked micelles. | ||
Nanoconfinement of the imprinting within the micelle is highly beneficial to imprinting,36 with the imprint/nonimprint ratio (i.e., the imprinting factor) reaching up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for certain templates (e.g., peptides).37 The high fidelity of the imprinting enables MINPs to distinguish subtle structural changes in the guest binding, including the addition,36 removal,36 and shift38 of a single methyl (or methylene) group.
000 for certain templates (e.g., peptides).37 The high fidelity of the imprinting enables MINPs to distinguish subtle structural changes in the guest binding, including the addition,36 removal,36 and shift38 of a single methyl (or methylene) group.
Our strategy to lower the pKa of zinc-bound water in this work is to introduce a basic group precisely near the water, much like histidine 64 in human CA II.39,40 To create such a feature in the MINPs, we designed and synthesized template 4a, which contains a (magenta colored) basic nitrogen that coordinates to the zinc-containing FM 5a to form the template–FM complex. Amine–zinc complexation has been used in our first-generation zinc esterase.41 However, without a nearby base to help the proton transfer, the resulting CA mimic could hydrolyze nothing but activated esters.
Synthesis of 4a was achieved through reductive amination between aldehyde 6 and amine 7a. Modularity of the synthesis enabled us to prepare a range of templates 4b–e from the corresponding amine derivatives 7b–e. As the template is tuned in size and shape in this way, the imprinted pocket and the final active site can be fine-tuned as well.
Both 4a and 5a have a polymerizable vinyl group. Hence, the template–FM complex would be copolymerized into the micellar core during free radical polymerization. The template contains an ortho-nitrobenzyl ester linkage, which can be cleaved cleanly inside the nanoparticle.42 The resulting MINP(4a·5a)-COOH has a carboxylic acid in the imprinted site, which was activated with N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) and amidated42 with amine-derived 8 to afford MINP(4a·5a + 8), which is our CA-mimicking catalyst.
It should be mentioned that amine 8 is designed intentionally to match the dimension of the blue substructure of the template, as highlighted in yellow in Scheme 2. The idea is to position the pyridyl nitrogen near the zinc metal in the final MINP(4a·5a + 8), with a gap defined by the magenta-colored CH2NH moiety in the template. This gap in the final catalyst will be used to accommodate a water molecule, which is hypothesized to coordinate to zinc and form a hydrogen-bond to the pyridyl nitrogen, hence doubly activated.
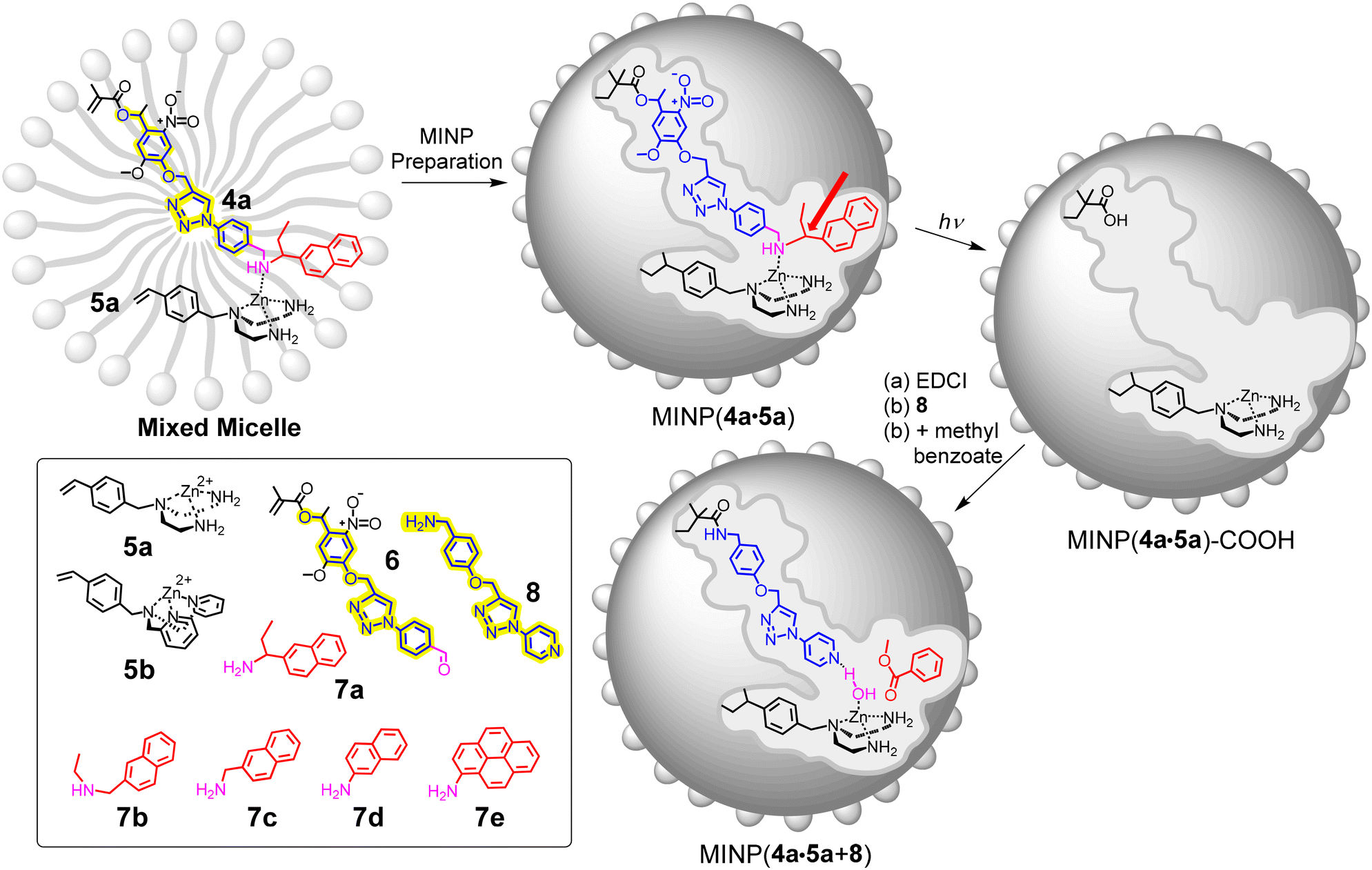 | ||
| Scheme 2 Preparation of molecularly imprinted nanoparticles for hydrolysis of methyl benzoate. The surface ligands (3) are omitted for clarity. | ||
Another point worth mentioning is the amphiphilicity of FM 5a, which contains a metal ion and two primary amino groups. Amphiphilic structures prefer to stay near the surfactant/water interface in a micelle, so that the polar groups/ions are exposed to water for solvation, while the hydrophobic groups are inserted into the micelles. This strategy of “hydrophilic anchoring” is important to the removal of the template after imprinting43 and expected to facilitate mass transfer during catalysis.
Our synthesis overall is designed to afford an active site in the cross-linked micellar core that is tunable in size and shape. The zinc ion and the nearby pyridine are positioned to work cooperatively, similar to natural CA, producing a metal-bound hydroxide for nucleophilic attack on the ester substrate.44 Synthesis and characterization of the MINP catalysts followed previously established procedures41,42 and are reported in the ESI.†
As shown in Fig. 1a, MINP(4a·5a + 8) is indeed a powerful catalyst for the benchmarking substrate, p-nitrophenyl acetate (PNPA). When 1000 equiv of the substrate is mixed with the MINP catalyst, a TON of 474 is obtained at 186 min. Meanwhile, the reaction in a buffer and in the presence of nonimprinted nanoparticles (NINPs) prepared without templates proceeded minimally. The high TON, as well as the small slowdown of the reaction over time, suggests minimal product inhibition in the catalysis. We attributed the success to the location of the active site in the hydrophobic core of the cross-linked micelle. The ester substrate is significantly more hydrophobic than the hydrolyzed products (acetic acid and p-nitrophenol) and should have no problem outcompeting the latter in binding with the catalyst.
 | ||
| Fig. 1 (a) The amount of p-nitrophenol formed in PNPA hydrolysis catalyzed by MINP(4a·5a + 8) in a 25 mM HEPES buffer (pH 7) at 40 °C in comparison to those in the buffer and in the presence of NINPs. The amount is calculated based on an extinction coefficient of ε400 = 0.0091 μM−1 cm−1. [PNPA] = 100 μM. [MINP] = 0.1 μM. (b) Michaelis–Menten plot of the hydrolysis of PNPA by MINP(4a·5a + 8) in a 25 mM HEPES buffer at 40 °C and pH 7.0. [MINP] = 8 μM. | ||
Fig. 1b shows the Michaelis–Menten plot for PNPA hydrolysis catalyzed by MINP(4a·5a + 8) in a 25 mm HEPEs buffer (pH 7) at 25 °C. The kcat value is 0.032 ± 0.002 s−1 and the KM value is 0.165 ± 0.002 mM−1, affording a catalytic efficiency of kcat/KM = 194 M−1 s−1. The rate acceleration (kcat/kuncat) is 7.4 × 104, based on the reported kuncat value of 4.3 × 10−7 s−1.45Table 1 shows a comparison between our MINP catalysts with several artificial Zn metalloenzymes reported in the literature. Both the catalytic efficiency and TON generally show a considerable improvement despite the lower pH used. The catalyst displays a higher activity than human CA B but is less efficient than human CA C at the same pH (entries 11–12).
| Entry | Catalysts | pH | pKa | k cat/Km (M−1 s−1) | TON |
|---|---|---|---|---|---|
| a The data for the artificial zinc enzymes reported in the literature were obtained at 22–25 °C for PNPA, unless otherwise indicated. b The peptide assembly upon ageing becomes more active, giving a kcat/KM value of 355 M−1 s−1 after 10 days at 37 °C in a pH 8 buffer and 1 mM ZnCl2.16 | |||||
| 1 | MINP(4a·5a + 8) | 7 | 6.2 | 194 | >474 |
| 2 | Modified TRI peptide-Zn (ref. 9) | 8 | 8.8 | 3 | >10 |
| 3 | MID1-Zn (ref. 10) | 8 | 8.2 | 180 | >50 |
| 4 | Ac-IHIHIQI-CONH2 (ref. 12) | 8 | 9.3 | 62 | >20 |
| 5 | A104AB3 (ref. 13) | 9 | 9.0 | 32 | — |
| 6 | CC-Hept–Cys–His–Glu (ref. 14) | 8 | 9.0 | 18 | >12 |
| 7 | VK2H (ref. 15) | 9 | — | 19 | — |
| 8 | Ac-IHIHIYI-NH2 at 37 °C (ref. 16) | 8 | — | 138b | — |
| 9 | F–Zn assembly18 | 7 | — | 11 | — |
| 10 | VFFAHH assembly19 | 7.4 | — | 1.7 | — |
| 11 | Human CA B20 | 7 | 7.3 | 150 | — |
| 12 | Human CA C20 | 7 | 6.8 | 1670 | — |
Efficient hydrolysis of PNPA at pH 7 suggests successful activation of water under neutral conditions. To gain additional mechanistic insights into the catalysis, we used linear free energy relationships.46,47 For aryl ester hydrolysis, the amount of negative charge buildup on the phenol oxygen depends on the type of nucleophile involved in the hydrolysis.48,49 When the carbonyl is attacked by a strong, anionic oxygen-based nucleophile such as hydroxide (Scheme 3a), the reaction constant (ρ) is typically 1–1.2. Nucleophilic attack by a water molecule in general base catalysis (Scheme 3b) affords much less negative charge on the phenol oxygen, giving a lower ρ value of 0.5–0.7. As shown in Fig. 2, hydrolysis of a series of p-substituted phenyl acetates by MINP(4a·5a + 8) follows a linear relationship to the Hammett substituent constants and reveals a ρ value of 1.08 in a pH 7 buffer. Thus, a (metal-bound) hydroxide is indeed generated in the active site of the catalyst under neutral conditions.
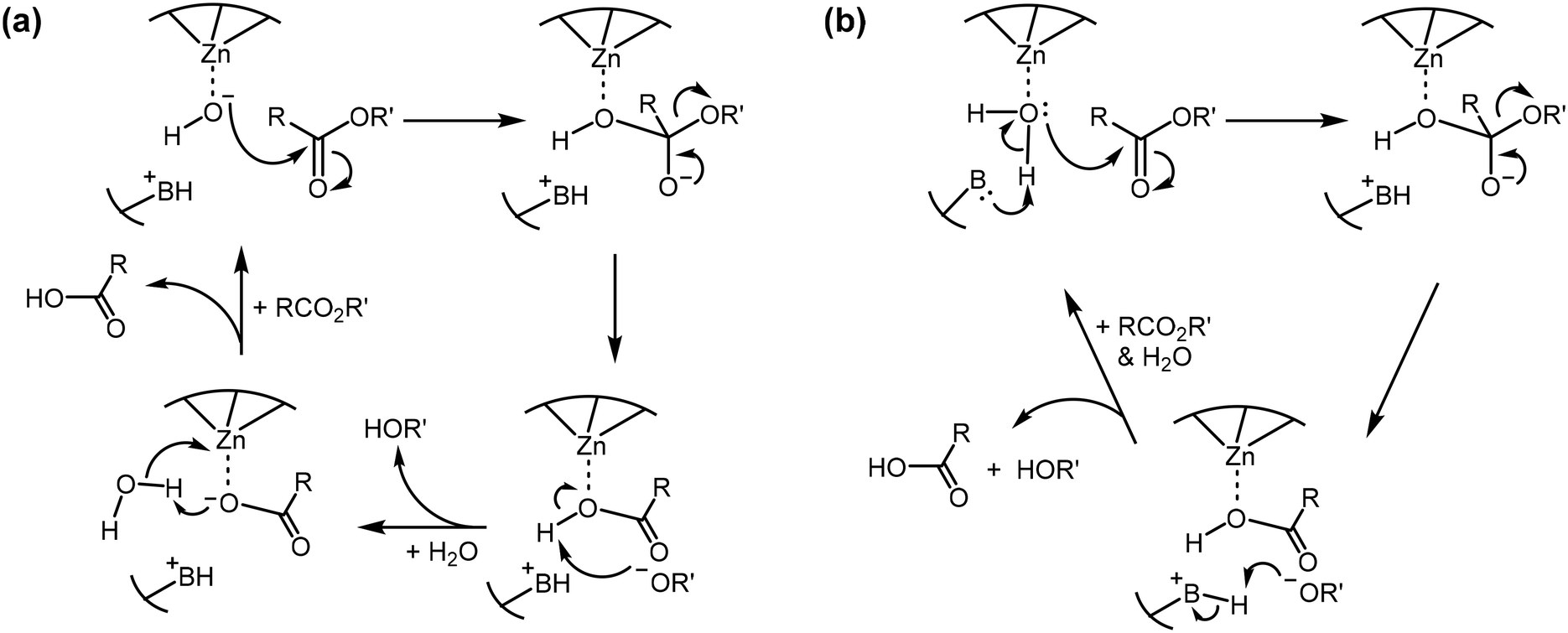 | ||
| Scheme 3 Comparison between a potential (a) nucleophilic attack of ester by an active site hydroxide and (b) general base-catalyzed attack of ester by an active site water molecule. | ||
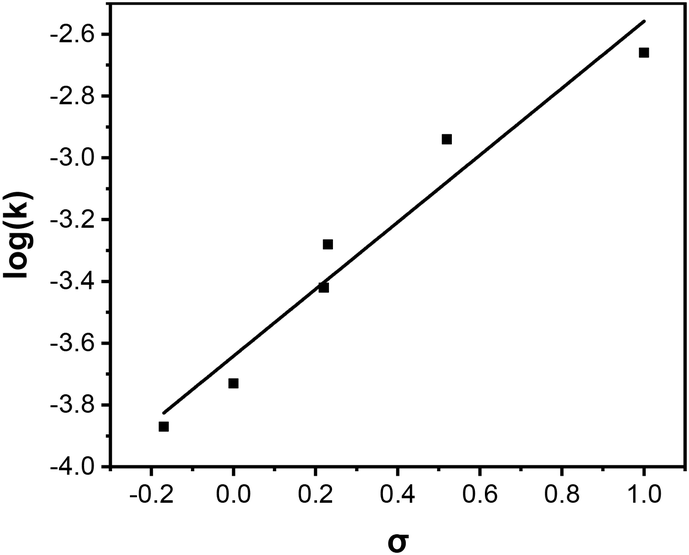 | ||
| Fig. 2 Hammett σ–ρ correlation in the hydrolysis of para-substituted-phenyl hexanoates catalyzed by MINP(4a·5a + 8). Reaction rates were measured in a 25 mM HEPES buffer (pH 7) at 25 °C. [Ester] = 50 μM. [Catalyst] = 15 μM. σ values: p-NO2, 1.00; p-CH3CO, 0.52; p-Cl, 0.23; p-CHO, 0.22; p-H, 0.00; p-CH3, −0.17. | ||
To further confirm the mechanism, we measured the kinetic solvent isotope effects for the PNPA hydrolysis. This is another way to distinguish a nucleophilic mechanism (by hydroxide) and general base catalysis (by the active site base).50–53 A nucleophilic attack by hydroxide does not involve O–H bond cleavage in the rate-determining step (Scheme 3a) and usually gives a kinetic solvent isotope effect of kH2O/kD2O ≈ 1. A general base mechanism is characterized by a cleavage of the water O–H bond in the rate-determining step (Scheme 3b), affording a primary solvent isotope effect of kH2O/kD2O = 2–3. In our case, the solvent isotope effect was found to be kH2O/kD2O = 1.23 at pH 7, confirming that a nucleophilic attack by hydroxide was responsible for the catalysis.54 During the measurement, the pD value was determined by adding 0.4 to the pH meter reading, since water and D2O have different dissociation constants.51
With a metal-bound hydroxide in the active site, our catalysts should be able to hydrolyze nonactivated esters. This represents a long sought-after goal in synthetic esterase.22,23,44Table 2 shows that our catalysts were able to hydrolyze methyl benzoate, a substrate that can also fit in the active site. The hydrolysis was performed at pH 7 and 40 °C for 4 h. The yield was determined by GC-MS and clearly depended on the structure of the template and thus the size and shape of the active site. The best catalyst was MINP(4a·5a + 8) (entry 1) and the NINP gave a negligible yield of <5% (entry 7).
| Entry | Catalyst | Yield (%) |
|---|---|---|
| a The conversion yields were determined by GC-MS analysis with p-xylene (600 μM) as the internal standard. [Methyl benzoate] = 600 μM. [MINP] = 15 μM. | ||
| 1 | MINP(4a·5a + 8) | 85 ± 2 |
| 2 | MINP(4b·5a + 8) | 72 ± 2 |
| 3 | MINP(4c·5a + 8) | 46 ± 1 |
| 4 | MINP(4d·5a + 8) | 34 ± 2 |
| 5 | MINP(4e·5a + 8) | <5 |
| 6 | MINP(4a·5b + 8) | 55 ± 2 |
| 7 | NINP | <5 |
Templates 4a and 4b (prepared from amines 7a and 7b, respectively) are isomers differing in the position of the ethyl group. Interestingly, shifting the ethyl from the carbon next to the (magenta-colored) nitrogen by 1 C–N bond length lowered the yield of hydrolysis from 85% to 72% (Table 2, entries 1 and 2). Thus, micellar imprinting could indeed faithfully reproduce structural features of the template in the imprinted site and impact the catalytic activity. To hydrolyze the ester efficiently, the catalyst needs to position the metal-bound hydroxide near the carbonyl of the substrate. Our design, as discussed above, is meant to place the metal-bound hydroxide at the former position of nitrogen in the template (Scheme 2). We designed the template so that the nitrogen-bonded secondary carbon (highlighted by the filled red arrow in the top middle structure in Scheme 2) on the template acts as a space holder for the carbonyl carbon, and the ethyl group more or less is the space holder for the methoxy of methyl benzoate. Although the design is simplistic, the fact that moving the ethyl by 1 C–N bond length made a significant impact on the catalytic hydrolysis suggests that this model is not without merit.
Consistent with the above design hypothesis, removing the ethyl from the template altogether has an even larger negative effect on the catalysis, as MINP(4c·5a + 8) gave an even lower yield of 46% in the hydrolysis of methyl benzoate (Table 2, entry 3). Removing the methylene group near the naphthyl ring in the template lowered the yield further to 34% (entry 4). Apparently, micellar imprinting was able to translate the structural information from the template to the catalytic active site very well, even changes such as shifting the ethyl group by one bond length or removal of a methylene group.
Template 4e has a larger pyrenyl group than the naphthyl group in 4d. The seemingly “harmless” change practically erased the catalytic effects of MINP(4e·5a + 8) altogether (Table 2, entry 5). How could this happen? One likely reason is the positioning of the carbonyl. In our cross-linked polymeric nanoparticle, the position of the zinc complex is expected to be largely fixed by the polymerization, not only by the styrenyl group of the FM but also by the core-cross-linking around the template–FM complex. Since the active site hydroxide is anticipated to be fixed in position between the zinc ion and the pyridyl nitrogen, the nucleophile is held rigidly in the active site. For a naphthyl-shaped active site in MINP(4a–e·5a + 8), the long and narrow imprinted site restricts the orientation of the (long and narrow) methyl benzoate substrate, thus proving a better chance for the carbonyl to be placed near the nucleophilic hydroxide. It is most likely for this reason that even small structural changes in the template translate to large effects in the catalysis. On the other hand, a pyrenyl group is much wider than a naphthyl ring. The chance of orienting the substrate in a catalytically productive fashion is much reduced. If the carbonyl cannot be positioned near the fixed hydroxide, even if both the nucleophile and the electrophile exist in the same pocket, they could be prevented from reacting with each other.
Fig. 3 shows the pH profile for the catalytic hydrolysis of methyl benzoate. The reaction is nearly negligible at pH 5, rapidly increases over pH 6–7, and plateaus over pH 7–10 for both MINP(4a·5a + 8) and MINP(4a·5b + 8). The constant yield over a three-orders-of-magnitude change in the external hydroxide concentration over pH 7–10 indicates that a hydroxide in the active site is responsible for the hydrolysis. Nonlinear least squares curving fitting of the data affords a pKa value of 6.4 for MINP(4a·5a + 8) and 6.3 for MINP(4a·5b + 8).55 These numbers (for zinc-bound water) are even lower than that of CA (6.8–7.3, see Table 1, entries 11–12).20 FM 5b is more hydrophobic than 5a and has a different ligand environment. We expect its hydrophobicity would alter the position of the template–FM complex in the micelle and, in turn, the location of the active site. The lower activity of MINP(4a·5b + 8) is likely caused by a slower binding of the substrate and/or release of the products as the active site moves deeper into the micellar core. It is also possible that the charged, anionic tetrahedral transition state of the ester hydrolysis is less stable in a more hydrophobic environment.
 | ||
| Fig. 3 Hydrolysis of methyl benzoate as a function of pH by MINP(4a·5a + 8) (blue data points) and MINP(4a·5b + 8) (red data points). The smooth curves were obtained by nonlinear least-squares curve fitting of the data to the equation, y = a/(1 + 10pKa−pH), in which y is the yield of the catalyzed reaction and a is the maximum yield obtained under the reaction conditions. [Methyl benzoate] = 600 μM. [MINP] = 15 μM. Buffers: MES for pH 5.0–6.5, HEPES for pH 7.0–8.5, CHES for pH 9.0–10. | ||
MINP(4a·5a + 8), our best synthetic esterase, has significant selectivity in its hydrolysis. Table 3 shows the hydrolytic yields of a series of ester substrates. Methyl benzoate and phenyl acetate swap the positions of their carbonyl and ester oxygen in the structure. Since an aryl ester with a better leaving group has a higher intrinsic reactivity than an alkyl ester, the experimentally identical yields of the two might be caused by better positioning of the carbonyl in methyl benzoate, offset by the better leaving group of phenyl acetate. The active site excludes nearly everything larger than these two esters; ethyl, butyl, hexyl, and phenyl benzoates all gave little or no reaction at all under the same conditions (Table 3, entries 3–6).
| Entry | Ester substrate | % Yield | |
|---|---|---|---|
| MINP(4a·5a + 8) | MINP(4f·5a + 8) | ||
| a The conversion yields were determined by GC-MS analysis with p-xylene (600 μM) as the internal standard. [Substrate] = 600 μM. [MINP] = 15 μM. | |||
| 1 | Methyl benzoate | 85 ± 2 | 71 ± 2 |
| 2 | Phenyl acetate | 83 ± 3 | 88 ± 2 |
| 3 | Ethyl benzoate | 9 ± 1 | 70 ± 2 |
| 4 | Butyl benzoate | 5 ± 2 | 76 ± 1 |
| 5 | Hexyl benzoate | 0 | 89 ± 2 |
| 6 | Phenyl benzoate | 0 | 91 ± 2 |
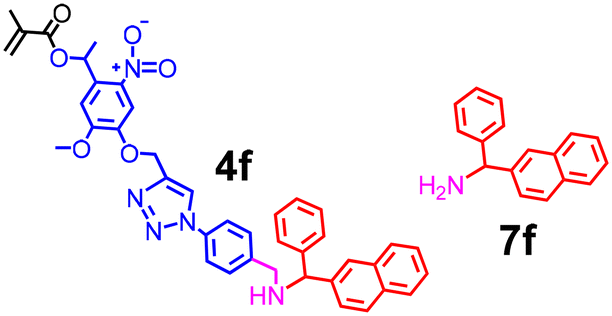
It is good that our synthetic catalysts can be made to distinguish small structural changes in the substrate. This is similar to many enzymes that have high substrate specificity. With the modular synthesis of the template and the facile catalyst preparation, we could also make our catalyst “promiscuous”. By simply changing the ethyl group of 4a to a phenyl group in 4f (prepared from 6 and 7f), we enlarged the part of the imprinted site that is hypothesized to accommodate the methoxy group of methyl benzoate (Scheme 2). Consistent with this postulation, MINP(4f·5a + 8) can hydrolyze all the esters in Table 3 nearly indiscriminately. The small differences in the yields are likely caused by the differences in the binding affinity of the substrates, the turnover frequency, and/or other factors discussed above such as intrinsic reactivity and positioning of the reactive groups. Regardless of the exact reasons, the formerly unreactive substrates (hexyl benzoate and phenyl benzoate) with MINP(4a·5a + 8) become the most reactive with MINP(4f·5a + 8), highlighting the tunability of our catalysts for their substrates.
Conclusions
Nature is an unlimited source of inspiration for chemists when it comes to designing highly active and selective catalysts. It is encouraging that micellar imprinting is able to faithfully reproduce functionalized active sites from templates in water-soluble polymeric nanoparticles. The most important finding in this work is that accurate positing of the catalytic groups (base and zinc ion) is able to duplicate features previously only observed in natural enzymes such as a greatly reduced pKa value for zinc-bound water. This allows metal-bound hydroxides to be generated under neutral or even slightly acidic conditions (Fig. 3). As a result, nonactivated esters that formerly represented unreachable substrates for synthetic esterases22 become readily hydrolyzed under neutral pH. The overall hydrophobicity of the active site also minimizes product inhibition and enables our synthetic esterase to turn over hundreds of substrates with little slowdown at high substrate conversions (Fig. 1a). MINPs are robust cross-linked polymeric nanoparticles and can tolerate high temperatures and organic solvents (e.g., ionic liquids and DMSO mixture at high temperatures).56,57 These features make it straightforward to recycle the catalysts.Another important finding is that the catalytic activity of the molecularly imprinted catalysts can be readily controlled by modularly synthesized templates. The direct correlation between the structural features of the template and the observed catalytic activity and selectivity means that rational design of synthetic enzymes is feasible. It is remarkable that moving the ethyl group by 1 C–N bond length or removal of a single methylene group can change the activity of the catalyst significantly.
Ester as a functional group is found in fats, oils, polyesters, and polycarbonates which exist in great quantities on Earth. Although they can be hydrolyzed under strongly acidic and basic conditions, the process generates large amounts of waste if performed on an industrial scale. For challenging applications such as plastic recycling, it is desirable to have robust synthetic catalysts that can selectively cleave target ester bonds in the presence of similar structures. Doing so under mild conditions is even more desirable.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NIGMS (R01GM138427) for financial support for this research.Notes and references
- D. S. Auld, in Encylopedia of Inorganic Chemistry, ed. R. B. King, Wiley, New York, 2nd edn, 2007, pp. 5885–5927 Search PubMed.
- B. L. Vallee and D. S. Auld, Acc. Chem. Res., 1993, 26, 543–551 CrossRef CAS.
- B. L. Vallee and D. S. Auld, Biochemistry, 1990, 29, 5647–5659 CrossRef CAS PubMed.
- J. E. Coleman, Curr. Opin. Chem. Biol., 1998, 2, 222–234 CrossRef CAS PubMed.
- E. Kimura, H. Hashimoto and T. Koike, J. Am. Chem. Soc., 1996, 118, 10963–10970 CrossRef CAS.
- H. Vahrenkamp, Acc. Chem. Res., 1999, 32, 589–596 CrossRef CAS.
- G. Parkin, Chem. Rev., 2004, 104, 699–767 CrossRef CAS PubMed.
- D. Desbouis, I. P. Troitsky, M. J. Belousoff, L. Spiccia and B. Graham, Coord. Chem. Rev., 2012, 256, 897–937 CrossRef CAS.
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118–123 CrossRef CAS PubMed.
- B. S. Der, D. R. Edwards and B. Kuhlman, Biochemistry, 2012, 51, 3933–3940 CrossRef CAS PubMed.
- M. L. Zastrow and V. L. Pecoraro, J. Am. Chem. Soc., 2013, 135, 5895–5903 CrossRef CAS PubMed.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stohr, T. A. Smith, X. Z. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- W. J. Song and F. A. Tezcan, Science, 2014, 346, 1525–1528 CrossRef CAS PubMed.
- A. J. Burton, A. R. Thomson, W. M. Dawson, R. L. Brady and D. N. Woolfson, Nat. Chem., 2016, 8, 837–844 CrossRef CAS PubMed.
- C. Zhang, R. Shafi, A. Lampel, D. MacPherson, C. G. Pappas, V. Narang, T. Wang, C. Maldarelli and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 14511–14515 CrossRef CAS PubMed.
- Z. S. Al-Garawi, B. A. McIntosh, D. Neill-Hall, A. A. Hatimy, S. M. Sweet, M. C. Bagley and L. C. Serpell, Nanoscale, 2017, 9, 10773–10783 RSC.
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. E. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285–1288 CrossRef CAS PubMed.
- P. Makam, S. S. R. K. C. Yamijala, K. Tao, L. J. W. Shimon, D. S. Eisenberg, M. R. Sawaya, B. M. Wong and E. Gazit, Nat. Catal., 2019, 2, 977–985 CrossRef CAS PubMed.
- A. Singh, J. P. Joseph, D. Gupta, C. Miglani, N. A. Mavlankar and A. Pal, Nanoscale, 2021, 13, 13401–13409 RSC.
- J. A. Verpoorte, S. Mehta and J. T. Edsall, J. Biol. Chem., 1967, 242, 4221–4229 CrossRef CAS PubMed.
- J. F. Kirsch and W. P. Jencks, J. Am. Chem. Soc., 1964, 86, 837–846 CrossRef CAS.
- M. D. Nothling, Z. Xiao, A. Bhaskaran, M. T. Blyth, C. W. Bennett, M. L. Coote and L. A. Connal, ACS Catal., 2019, 9, 168–187 CrossRef CAS.
- F. M. Menger and M. Ladika, J. Am. Chem. Soc., 1987, 109, 3145–3146 CrossRef CAS.
- G. Wulff, Chem. Rev., 2002, 102, 1–28 CrossRef CAS PubMed.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- J. Pan, W. Chen, Y. Ma and G. Pan, Chem. Soc. Rev., 2018, 47, 5574–5587 RSC.
- H. Zhang, Adv. Mater., 2020, 32, 1806328 CrossRef CAS PubMed.
- K. Haupt, P. X. Medina Rangel and B. T. S. Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef CAS PubMed.
- J.-Q. Liu and G. Wulff, J. Am. Chem. Soc., 2008, 130, 8044–8054 CrossRef CAS PubMed.
- G. Wulff and J. Liu, Acc. Chem. Res., 2012, 45, 239–247 CrossRef CAS PubMed.
- D. K. Robinson and K. Mosbach, J. Chem. Soc., Chem. Commun., 1989, 969–970 RSC.
- K. Ohkubo, Y. Urata, S. Hirota, Y. Honda, Y.-I. Fujishita and T. Sagawa, J. Mol. Catal., 1994, 93, 189–193 CrossRef CAS.
- B. Sellergren, R. N. Karmalkar and K. J. Shea, J. Org. Chem., 2000, 65, 4009–4027 CrossRef CAS PubMed.
- I. Bose and Y. Zhao, ACS Catal., 2022, 12, 3444–3451 CrossRef CAS PubMed.
- K. Chen and Y. Zhao, Org. Biomol. Chem., 2019, 17, 8611–8617 RSC.
- M. Zangiabadi and Y. Zhao, ACS Appl. Polym. Mater., 2020, 2, 3171–3180 CrossRef CAS.
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 2188–2191 CrossRef CAS PubMed.
- D. N. Silverman and R. McKenna, Acc. Chem. Res., 2007, 40, 669–675 CrossRef CAS PubMed.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946–1051 CrossRef CAS PubMed.
- M. Arifuzzaman and Y. Zhao, ACS Catal., 2018, 8, 8154–8161 CrossRef CAS.
- J. K. Awino and Y. Zhao, Chem. – Eur. J., 2015, 21, 655–661 CrossRef PubMed.
- J. K. Awino and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 12552–12555 CrossRef CAS PubMed.
- M. D. Arifuzzaman, I. Bose, F. Bahrami and Y. Zhao, Chem Catal., 2022, 2, 2049–2065 CrossRef CAS.
- W. P. Jencks and M. Gilchrist, J. Am. Chem. Soc., 1968, 90, 2622–2637 CrossRef CAS.
- L. P. Hammett, Physical organic chemistry; reaction rates, equilibria, and mechanisms, McGraw-Hill Book Company, Inc., New York, London, 1st edn, 1940 Search PubMed.
- A. Williams, Free energy relationships in organic and bio-organic chemistry, RSC, Cambridge, UK, 2003 Search PubMed.
- T. C. Bruice and S. J. Benkovic, J. Am. Chem. Soc., 1964, 86, 418–426 CrossRef CAS.
- J. F. Kirsch, W. Clewell and A. Simon, J. Org. Chem., 1968, 33, 127–132 CrossRef CAS.
- M. L. Bender, E. J. Pollock and M. C. Neveu, J. Am. Chem. Soc., 1962, 84, 595–599 CrossRef CAS.
- R. A. Gibbs, P. A. Benkovic, K. D. Janda, R. A. Lerner and S. J. Benkovic, J. Am. Chem. Soc., 1992, 114, 3528–3534 CrossRef CAS.
- W. P. Jencks and J. Carriuolo, J. Am. Chem. Soc., 1961, 83, 1743–1750 CrossRef CAS.
- D. Stefanidis and W. P. Jencks, J. Am. Chem. Soc., 1993, 115, 6045–6050 CrossRef CAS.
- The small solvent effect suggests that a small degree of proton transfer is involved in the rate-limiting step. It is possible that the real mechanism is somewhere in between Scheme 3a and b but much closer to the proposed nucleophilic attack.
- K. Al Adem, J. C. Ferreira, S. Fadl and W. M. Rabeh, J. Biol. Chem., 2023, 299, 102790 CrossRef CAS PubMed.
- X. Li, M. Zangiabadi and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 5172–5181 CrossRef CAS PubMed.
- M. Zangiabadi and Y. Zhao, J. Am. Chem. Soc., 2022, 144, 17110–17119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, syntheses, and characterization of catalysts. See DOI: https://doi.org/10.1039/d3cy00704a |
| This journal is © The Royal Society of Chemistry 2023 |