Reactivity and stability synergism directed by the electron transfer between polyoxometalates and metal–organic frameworks†
Received
25th April 2023
, Accepted 17th July 2023
First published on 3rd August 2023
Abstract
The synergism between polyoxometalates (POM) and Cu(II) ions in homogeneous aerobic thiol oxidative deodorization has been realized in a more utilitarian heterogeneous catalyst: a multi-electron-capable POM captured in the pores of a metal–organic framework (MOF), HKUST-1 (POM@HKUST). The synergism between POM and the Cu(II) nodes in the MOF depends on the type of POM. Phosphovanadomolybdates, PVxMo12−xO40(3+x)− (x = 1–3) (PVMo) but not transition-metal-substituted polytungstates PXW11 (X = V, Co, Zn and Co) result in POM@MOF materials that exhibit synergy relative to the individual structural components, the POM or MOF alone, not only for reactivity as in the case for the analogous homogeneous catalysts, but also for catalyst structural stability. The PVMo@HKUST-catalyzed reaction proceeds to essentially 100% conversion and the material is recoverable and unchanged based on FTIR spectroscopy, powder XRD data and other observations after reaction. The PXW11@HKUST materials produce only limited conversions and decompose to white powders after reaction. X-ray photoelectron spectroscopy reveals that all the Cu(II) sites in the HKUST-1 become Cu(I) sites that are stable in air. Further kinetics studies show that PVMo undergoes fast multielectron transfer with intermediate Cu/RSH complexes, while PXW11 show far slower and limited electron transfer ability with these Cu/RSH complexes. Limited electron transfer between Cu nodes and the encapsulated POM units not only hinders reactivity but also leads to MOF framework distortion and subsequent decomposition induced by the reduction of Cu(II) to Cu(I) sites in the framework.
Introduction
POM@MOF composites comprising polyoxometalates (POMs) residing in metal–organic framework (MOF) pores1–4 have been reported as catalysts for oxidative, electrocatalytic, photocatalytic and other reactions.3–6 A central point of interest is that they can combine the attractive properties of POMs and also those of MOFs. POMs have been investigated as catalysts for years,7–10 resulting from one or more of their following attributes: strong acidity,11,12 facile redox chemistry,13–16 photoactivity17–22 and multielectron transfer ability.17,23,24 POMs in conventional solid phases, not pseudo-liquid phases,25 have small surface areas but are soluble in aqueous and many organic solvents. Thus, the immobilization of POMs on porous materials, including polymers,26 covalent organic frameworks (COFs),27 zeolites28 and MOFs,29–32 is attractive because it converts these homogeneous catalysts into more utilitarian heterogeneous ones with substantial surface area. Among all POM supports, MOFs, such as MIL-101,33–35 MOF-199 (HKUST-1),4,36–38 NU-1000,39–42 UIO-66 (ref. 43–46) and ZIF-8 (ref. 47–49) are highly attractive host materials because they offer high porosity and sufficient pore (cage) size to encapsulate POMs, including Keggin,37 Wells–Dawson43 and sandwich structural families,50 as guest materials forming POM@MOF composites.
Recent work in our group51,52 reveals strong synergism effect between Cu(II) ion and phosphovanadomolybdates, PVxMo12−xO40(3+x)− (x = 1–6) (PVMo) for catalysis of air-based oxidative removal of odorous thiols, facilitated by the redox buffering effect of PVMo. Moreover, the redox buffering and synergism are only operative for PVMo and not phosphovanadotungstates, PVxW12−xO40(3+x)− (x = 1, 3, 6) (PVW). Triggered by this, we sought formulation of a solid and thus more useful catalyst for aerobic thiol deodorization by combining Cu(II) and POM together as a heterogeneous catalyst. Here, we use the POM, HKUST-1 with Cu-containing nodes to bring Cu(II) and reversible multi-electron POM centers into proximity (POM@HKUST, Fig. 1). HKUST-1, in addition to providing well-ligated Cu(II), contains large pores that accommodate Keggin POMs with their accompanying counter-cations in the smaller pores. Various Keggin POMs have been incorporated into HKUST-1 for oxidation,36,53–59 esterification,60,61 hydrolysis37 and other organocatalytic reactions.62 Many efforts have been made to increase the catalytic activity such as tuning the nanocrystal size,53 modifying the morphology,60 designing the hierarchical structure57 and regulating the pore size.63 Incorporating Keggin POMs into HKUST-1 not only increases the thermal64 and hydrolytic36 stability of the MOF but also increases the reactivity of the POM with a marked synergy between POM and MOF framework.36,53–55 However, almost all the synergism reported previously is due to the MOF framework confinement effect that enhances the absorption, selectivity, and conversion of organic substrates, while POM is the real catalytic center of the reaction.53–55 In this work, we show that the aerobic thiol oxidation mechanism involves both the Cu(II) nodes in the MOF framework and multi-electron redox processes of the POM polyanion. In addition, we show that synergistic electron transfer between MOF and POM maintains the overall POM@MOF framework.
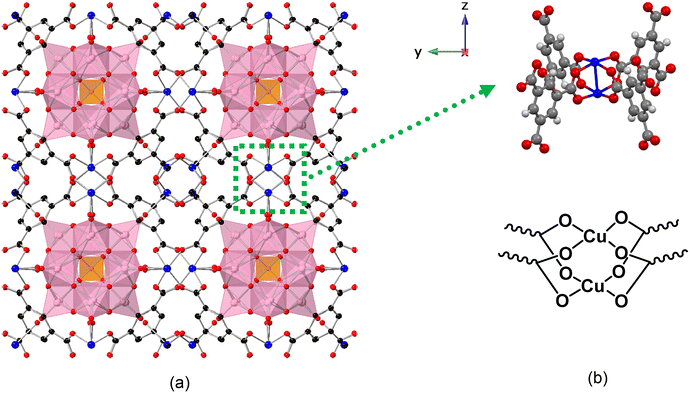 |
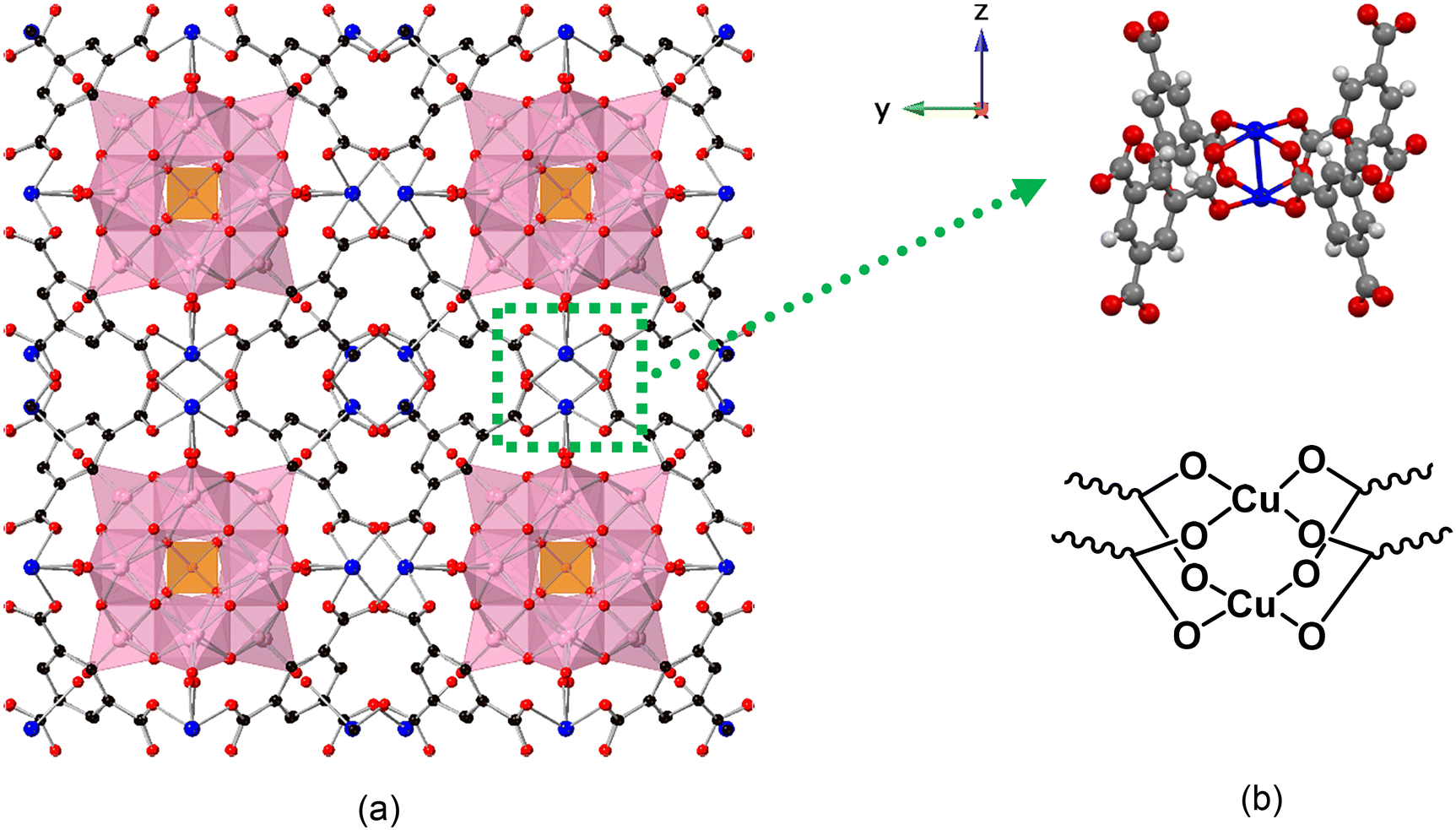
| | Fig. 1 (a) X-ray single-crystal structure of PV2Mo10@HKUST [100] direction. The purple polyhedral represent the MoO6 (or VO6) and orange tetrahedra represent the PO4 units in the POM. The HKUST-1 framework is represented in ball-and-stick form. C is black, O is red, Cu is blue. The tetramethylammonium (TMA) cations reside in the smaller pores and resolve as an average of 5 per POM unit (omitted for clarity). (b) The ball and stick and chemical formula of di-copper node. | |
Results and discussion
POM@MOF materials and characterization
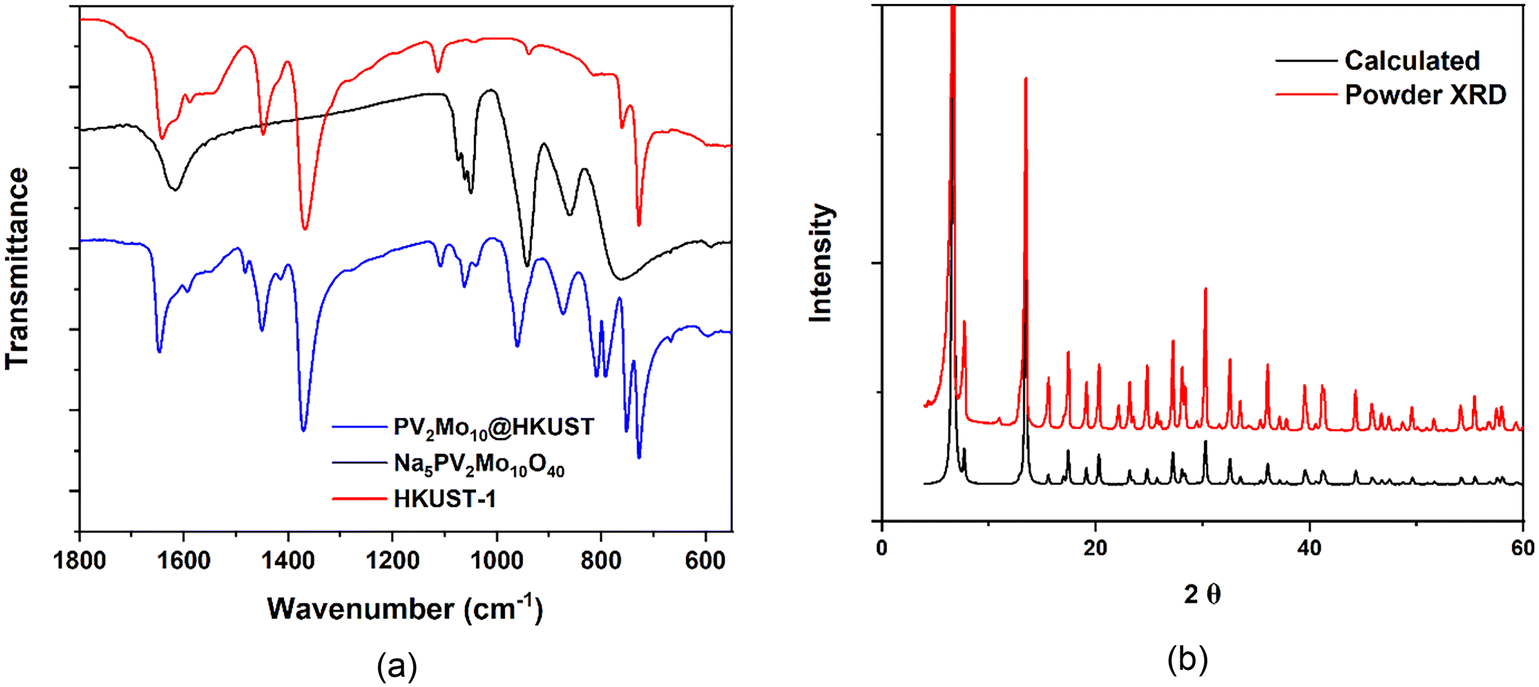
PVxMo12−xO40(3+x)− (x = 0–3) (PVMo) were incorporated into the HKUST-1 using the well-established hydrothermal synthetic method;37,38 however, the synthesis of POM@HKUST with PVMo with x > 3 using the same synthetic method failed. This likely reflects the decreasing hydrolytic stability of PVMo as the number of vanadium centers in the polyanion increases. We note that there are other synthetic methods to synthesize POM@HKUST materials.53,61,65,66 This paper does not explore various synthetic methods but only focuses on the hydrothermal approach. Thus, in this work, PVMo@HKUST (x = 0–3, PVMo11@HKUST, PV2Mo10@HKUST and PV3Mo9@HKUST) are discussed. The phase purity of PVMo@HKUST were established by FT-IR spectra and powder X-ray diffraction (PXRD) patterns (Fig. 2a and b and S1 and S2†). The PXRD patterns were compared with the calculated patterns from the single crystal X-ray diffraction of PV2Mo10@HKUST (Fig. 1, S3 and Table S1†). In Keggin crystal structures, the PO4 tetrahedron is disordered due to crystal symmetry, resulting in 8 positions for the mixed arrangement of the 4 O atoms. However, our study showed that if the crystals were treated as being chiral, F23, the PV2Mo10 anion resolves into a more ordered form with the tetrahedral arrangement resolved into a single component. This resolved PV2Mo10 anion was found to be packed into the pores of HKUST-1, with the terminal oxygen pointing towards the di-copper nodes. Furthermore, the crystals were found to be racemically twinned, resolving the Keggin structure. Each twin domain contained only one enantiomer, but due to the 50% twinning, the crystal had equal proportions of domains with each enantiomer. We determined that PV3Mo9@HKUST is also chiral and isostructural with PV2Mo10@HKUST (see ESI† for more details.) For the PVW, only x = 0, 1 can be inserted into HKUST-1 (PW12@HKUST and PVW11@HKUST). This likely derives from the fact that unlike the corresponding Mo-based Keggin POMs, the Keggin polytungstates are more rigid and far less hydrolytically labile. In addition, we succeeded in incorporating a series of transition-metal-substituted polytungstates (PXW11, X = Cu, Co and Ni) into HKUST-1 (PXW@HKUST: PCuW11@HKUST, PCoW11@HKUST and PNiW11@HKUST). The PXRD patterns of PVW11@HKUST, PCoW11@HKUST and PNiW11@HKUST were compared with the calculated pattern from the single crystal data of PCuW11@HKUST from previous work (Fig. S5†).36 Interestingly, PCuW11@HKUST and PVW11@HKUST single crystals do not show the ordered packed POM and chiral space groups. FT-IR and PXRD and establish the phase purity of these materials Fig. S4 and S5.† The incorporated transition metal, M, in the POM was established using SEM-EDX (Fig. S6–S8†). The SEM images show that POM@HKUST materials are large bulk crystals. EDX shows clearly that the POM elements, O, P, and W, including the transition metals, V, Co and Ni, are present in these crystalline materials. Nitrogen isotherms of PV2Mo10@HKUST and PCoW11@HKUST (Fig. S24†) show the specific surface areas are 619.2 and 413.5 m2 g−1, respectively. Comparing with the HKUST-1 alone, 1264 m2 g−1,36 the smaller specific surface area can attribute to the successful insertion of the POMs.
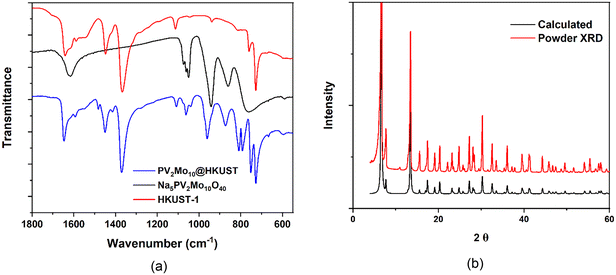 |
| | Fig. 2 (a) FT-IR of Na5PV2Mo10O40, HKUST-1 and PV2Mo10@HKUST; (b) PV2Mo10@HKUST calculated XRD pattern from single-crystal refinement data compare with the experimental powder XRD pattern. | |
POM leaching and solvent selection
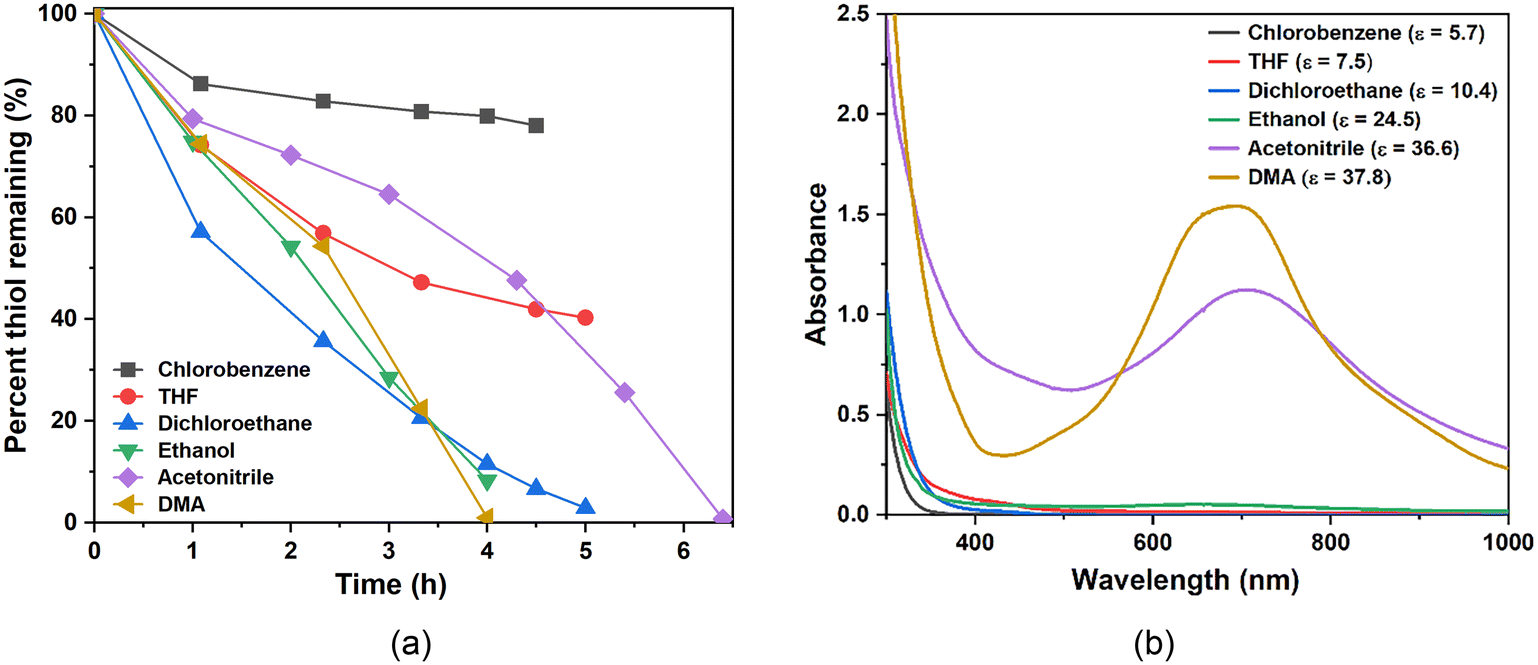
The leaching of POM from MOF pores during the reaction is a potential issue for POM@MOF materials.3,4Fig. 3a shows the thiol (RSH) oxidation reactivity of PV2Mo10@HKUST in different organic solvents (see thiol oxidation section in ESI†). Auto-catalytic behavior is seen in acetonitrile (AN) and dimethylacetamide (DMA). This is because POM is leached into the solution during reaction in these polar POM-solubilizing solvents, and the dominant mode of catalysis switches from heterogeneous to homogeneous by POM in solution. To further address POM leaching from MOF pores in these catalytic studies, we developed a colorimetric method to detect whether POM is extracted into the solution. Supernatants were filtered after the oxidation reaction, then SnCl2, a strong reducing agent, was added to the Ar-purged, filtered solution. Since Mo(VI)–Mo(V) intervalence charge transfer band has very high extinction coefficient around 600–800 nm,67 even a trace amount of POM that leaks into the solution is rapidly reduced and quantified by UV-vis spectroscopy. Fig. 3b shows the UV-vis results. When ethanol is used as a solvent, a slight absorbance due to reduced POM in solution is observed; when AN and DMA are used, large absorbances are observed. When solvents less polar than ethanol are used, no absorbances around 600–800 nm are generated. Here we use dielectric constant (ε) to differentiate and quantify the polarity of the organic solvents. The reactivity increases as ε increases (Fig. 3b), however, when ε is too large (ethanol, 24.6), the POM starts to leach from the MOF pores. As a consequence, we chose dichloroethane for all the catalytic studies.
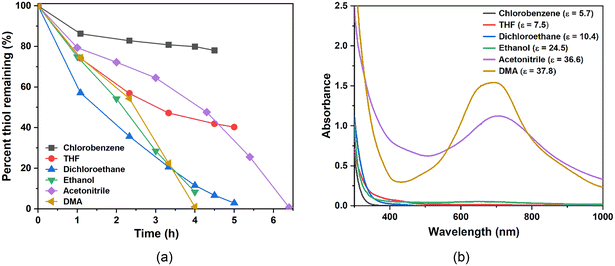 |
| | Fig. 3 (a) RSH consumption catalyzed by PV2Mo10@HKUST in different solvents. Conditions: dichloroethane (5 mL), 2-mercaptoethanol (28.6 mM), catalyst (0.774 mM), reaction at 50 °C. (b) UV-vis absorbance of the reduced supernatant solution after addition of SnCl2 under argon after reaction. | |
Activity and stability synergism
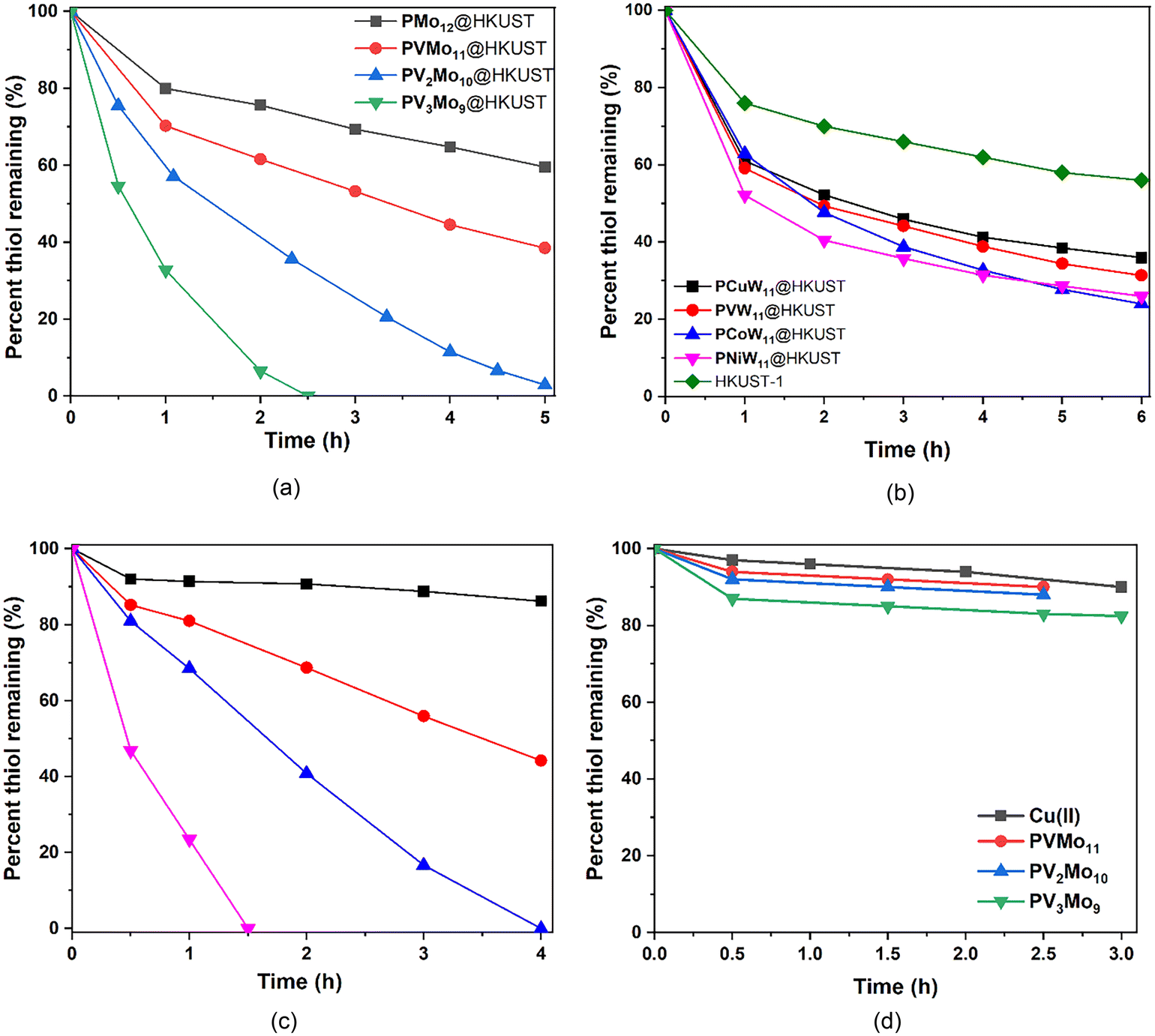
A core thrust of this study is to determine whether the synergism between PVMo and Cu(II) in homogeneous solution can still exist while both parts are confined without free diffusion. The aerobic oxidation of RSH catalyzed homogeneously by PVMo and Cu(II) in dichloroethane (30% acetonitrile for POM solubility) was evaluated (Fig. 4c and d). The same results were obtained in pure acetonitrile,51,52i.e. catalytic activity increases with increasing number of V centers in the POM. Notably, neither Cu(II) ion nor POM alone (PVMo, x = 1–3, but not x = 0) have significant activity which proves a synergy exists between these two catalytic components. The conversion and turnover frequency (TOF) data for all POM@HKUST catalyzing aerobic RSH oxidation are summarized in Table S2.† The consumption curves of PVMo@HKUST (x = 0–3) are shown in Fig. 4a. The catalytic activity, as for the homogeneous PVMo/Cu catalytic systems, increases with increasing number of vanadium centers in PVMo. Nearly 100% conversion is achieved in 3 h, 6 h and 15 h by PV3Mo9@HKUST, PV2Mo10@HKUST and PVMo11@HKUST, respectively. PMo12@HKUST does not achieve 100% after 24 h and HKUST-1 alone converts only 45% after 20 h (Fig. 4b). PXW@HKUST (X = V, Cu, Co and Ni) have much lower catalytic activity than PVMo@HKUST, Fig. 4b. The biphasic thiol consumption kinetics in the polytungstate@HKUST systems reflects initial rapid uptake of the thiol into the POM@MOF pores, followed continued but slower thiol uptake that parallels the time-dependent decomposition of the polytungstate@HKUST systems (see discussion below). Changing the transition metal in the POM does not have a significant effect on activity. PXW@HKUST do not achieve 100% conversion after 24 h. A homogeneous aerobic catalytic RSH oxidation investigation reveals no synergistic effect between the phosphotungstate POMs and Cu(II) ion (Fig. S9†). The same Keggin POMs were incorporated into the Cu-free large-pore MIL-101 MOFs (Fig. S10†). POM@MIL-101 materials have been widely studied.33,34,68–71 The hydrothermal synthesis described in literature has been used in this work (see ESI† for synthesis detail).72 Fig. S11† gives the FT-IR of all POM@MIL-101 materials that are consistent with the insertion of POMs into the MIL-101 pores.68,73 The resulting POM@MIL-101 binary materials have very little activity implicating a direct role of the HKUST-1 Cu(II) centers in catalytic aerobic thiol oxidation.
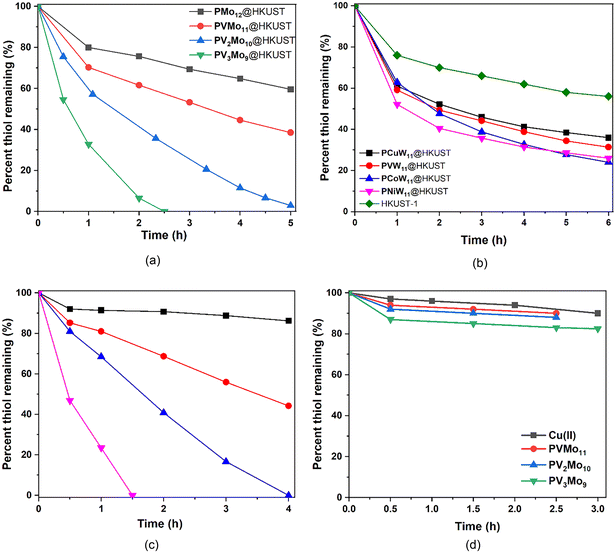 |
| | Fig. 4 RSH (28.6 mM) consumption catalyzed by (a) PVMo@HKUST (n = 0–3) (20 mg); (b) HKUST-1 alone (20 mg) and by PXW@HKUST (X = Cu, V, Ni and Co) (23 mg) in dichloroethane (5 mL) at 50 °C under air. (c) Aerobic RSH (30 mM) oxidation catalyzed by POM (0.2 mM) with Cu(II) (1.0 mM) at room temperature in a dichloroethane/acetonitrile 30% (v/v) solvent system. Black: H3PMo12O40 (PMo12); red: H4PVMo11O40 (PVMo11); blue: H5PV2Mo10O40 (PV2Mo10); pink: H6PV3Mo9O40 (PV3Mo9). (d) Aerobic RSH (30 mM) oxidation activity in the presence of Cu(II) (0.5 mM) alone or POM (0.5 mM) alone. | |
Interestingly, the Cu–POM synergy is not limited to catalytic activity: it also extends to catalyst stability. PVMo@HKUST are stable during the oxidation reactions (Fig. S12† compares spectra before and after 37 equivalents of thiol are converted). Fig. 5a and b show that both the FT-IR spectra and PXRD patterns of PV3Mo9@HKUST are the same before and after reaction consistent with structural integrity of both the POM and MOF components. In contrast, the FT-IR of HKUST-1 shows that this MOF is destroyed after oxidation (Fig. 5c). Thus, there is a dramatic oxidative stabilization of the MOF framework when these Keggin POMs are incorporated into the MOF pores. Significantly, all the PXW@HKUST materials decompose to white powders after reaction (Fig. S13 and S14†). Fig. 5d shows the FT-IR of PVW11@HKUST. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches (1644 cm−1) and C–O stretches (1368 cm−1) shift indicating changes in the MOF structures. This POM stabilization of the POM@MOF framework during the oxidation reaction, is POM selective: only phosphomolybdates but not phosphotungstates facilitate MOF stabilization. These data collectively indicate that the reactivity synergism between Cu(II), the MOF framework and the POM is crucial for stability of these hybrid materials.
O stretches (1644 cm−1) and C–O stretches (1368 cm−1) shift indicating changes in the MOF structures. This POM stabilization of the POM@MOF framework during the oxidation reaction, is POM selective: only phosphomolybdates but not phosphotungstates facilitate MOF stabilization. These data collectively indicate that the reactivity synergism between Cu(II), the MOF framework and the POM is crucial for stability of these hybrid materials.
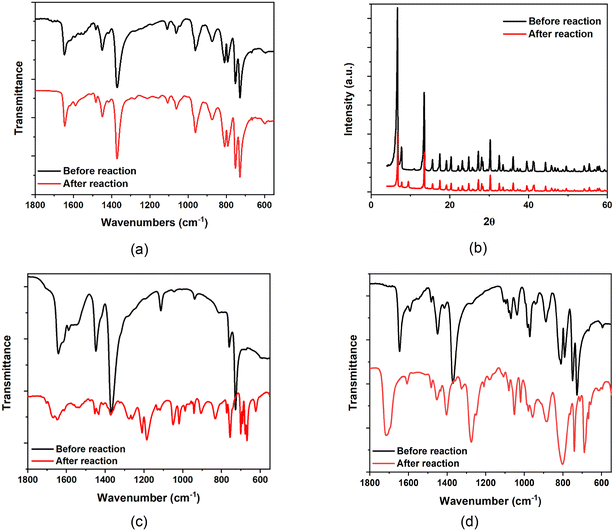 |
| | Fig. 5 (a) FT-IR and (b) PXRD spectra of PV3Mo9@HKUST before and after reaction. FT-IR spectra of (c) HKUST-1 and (d) PVW11@HKUST before and after reaction. | |
We turn to X-ray photoelectron spectroscopy (XPS) to further probe decomposition of the PXW@HKUST materials. The spectra of POM@HKUST before and after reaction were collected. Fig. 6a shows the Cu 2p spectra of PV3Mo9@HKUST. Peaks at 935.1 eV and 955.1 eV belong to Cu2+ 2p3/2 and 2p1/2, and are accompanied by satellite peaks at 944.5 and 940.5 eV.74–76 There are no detectable peaks due to Cu1+ indicating all Cu has been reoxidized back to its initial oxidation state after reaction. PCuW11@HKUST before reaction shows Cu2+ 2p peaks, and Cu1+ 2p3/2 and 2p1/2 peaks at 933.0 and 952.9 eV, respectively. This is consistent with charge transfer between Cu2+ nodes and POM.75–80 Here, we found that under the hydrothermal synthesis conditions, the transition-metal-substituted phosphotungstates prefer to undergo electron transfer with HKUST-1 (Fig. S16†) than PVMo (Fig. S15†). Since even perfect HKUST-1 crystals have Cu1+ defects,79 all the POM@MOF materials may contain low percents of Cu1+ in their nodes. Moreover, Fig. 6b shows that after reaction, the Cu2+ 2p peak and corresponding satellite peaks disappear, and only Cu1+ 2p peaks are evident. This indicates that the HKUST-1 framework breaks down and generates a Cu1+-based structure that is stable when exposed to ambient air. This also directly implicates a redox role for the Cu centers in the MOF nodes. Its structural distortion results from a lack of reoxidation of Cu1+ to Cu2+.
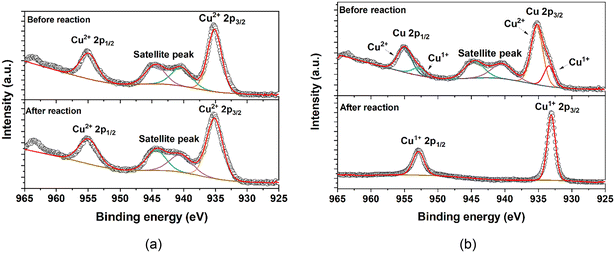 |
| | Fig. 6 Cu 2p XPS spectra of POM@MOF before and after reaction: (a) PV3Mo9@HKUST; (b) PCuW11@HKUST. | |
The mechanism of RSH oxidation catalyzed by the homogeneous PVMo/Cu system was recently reported.51 The heterogeneous POM@MOF requires a modified mechanism relative to freely-diffusing copper and phosphovanadomolybdate due to the confinement of Cu(II) and POM in their respective locations. The crystal structure of POM@MOF is given in Fig. 1 and S3.† Each POM is in proximity, on average, of two di-copper(II) groups. Cu, on average, binds two RSH and forms a polymeric network,81,82 eqn (1). The CuII(RSH)2 complex is a strong reducing agent, and when POM is present, the former reduces the latter by one electron to form CuI(RS˙)2 which then decomposes to CuI and RSSR products.51 In the POM@MOF, each di-copper(II) site reduces the proximal POM by two electrons and generates a di-copper(I) site (Fig. 7, eqn (2)). One possibility for the reoxidation process is that the di-copper(I) reacts with O2 forming a peroxo-CuII intermediate, then the peroxo group is reduced to water after receiving two electrons from a reduced POM (eqn (3) and (4))83 However, for the PVW11 and PXW11@HKUST materials, the decomposition to stable CuI sites indicates a lack of electron transfer between the POM and Cu nodes. Cu alone can catalyze the RSH oxidation in alkaline aqueous conditions,84–88 and the mechanism is given in eqn (5). Without a POM catalyst, the di-copper sites may result in CuI/RSH complexes.81,82 The XPS data suggest that the CuI/RSH complex is inert to O2. The XPS survey spectra of PCuW11@HKUST after RSH oxidation shown in Fig. S17,† compared to PV3Mo9@HKUST after reaction, clearly indicates a S 2p peak at 163.5 eV. For PCuW11@HKUST, the calculated atomic content (%) of Cu and S is 6.29 and 7.35 respectively. This ratio is close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the slightly larger amount of S could be due to the trapped unreacted RSH and/or product RSSR in the MOF pores. The XPS results are consistent with the stable CuI site being a di-CuIRSH complex.
:1, and the slightly larger amount of S could be due to the trapped unreacted RSH and/or product RSSR in the MOF pores. The XPS results are consistent with the stable CuI site being a di-CuIRSH complex.
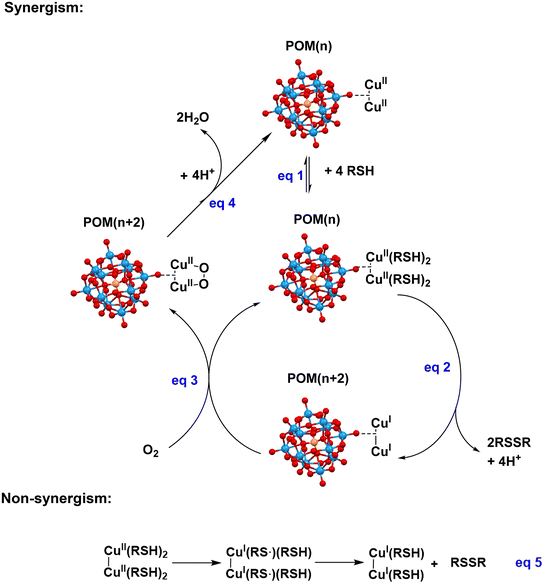 |
| | Fig. 7 Proposed mechanism of POM@MOF-catalyzed RSH aerobic oxidation. | |
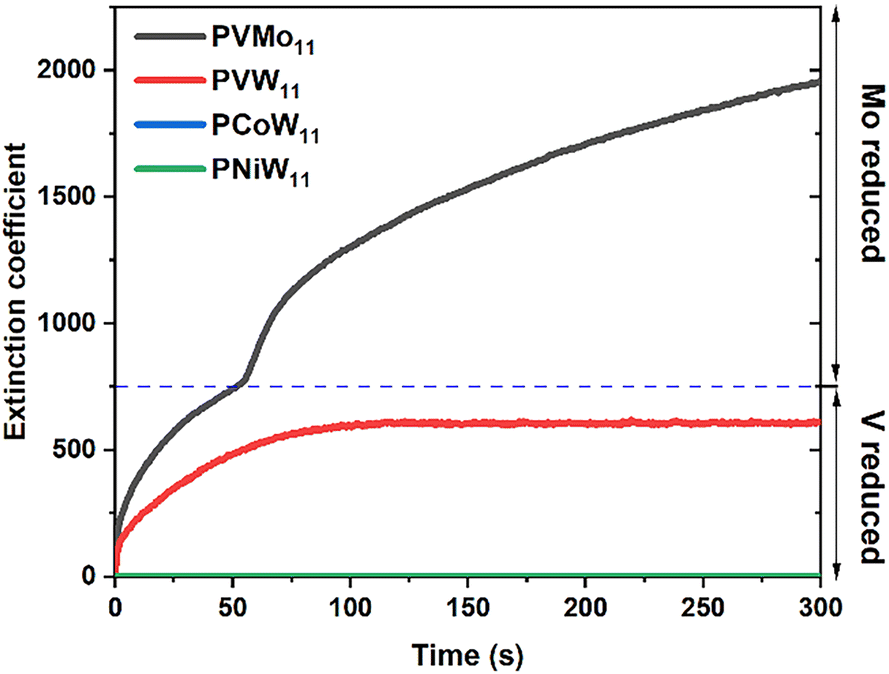
Homogeneous electron transfer between POM and the Cu/RSH complex can be studied by stopped-flow spectroscopy. Previous work52 in aqueous buffer and acetonitrile shows that PVMo (x = 1–6) can transfer multiple electrons involving both V and Mo atoms, depending on reaction conditions, while PVW (x = 1, 3 and 6) can only transfer one electron independent of reactant concentrations. Fig. 8 shows the stopped-flow kinetics of PVMo11 and PVW11 reduction in pH = 2 H2SO4/Na2SO4 buffer and PCoW11 and PNiW11 reduction in pH = 5 sodium acetate buffer by RSH catalyzed by μM quantities of Cu(II) under Ar (the pH changes according to the stability of POM). PVMo11 exhibits two-stage kinetics that involves the Mo redox couple, while PVW11 only accepts one electron on a V atom; the W centers are not involved. The Cu concentration dependence is given in Fig. S18.†PVMo11 is reduced by more than two electrons with increasing the Cu concentration, while PVW11 is only reduced by one electron independent of Cu concentration. The transition-metal-substituted polytungstates are not reduced by the Cu/RSH complex (Fig. 8). Calibration of the extinction coefficient based on the number of electrons was done by UV-vis spectra in conjunction with bulk electrolysis, Fig. S19.† Based on our proposed mechanism above, the POM should have multielectron transfer ability with di-copper thiol complexes. Therefore, the synergistic effects of the PVMo@HKUST materials derive from fast, multi-electron transfer between POM and the Cu nodes in the MOF.
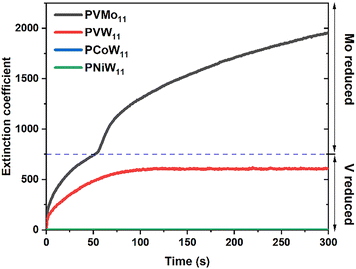 |
| | Fig. 8 Kinetics of 0.5 mM PVMo11 and PVW11 reduction by 25 mM RSH under Ar catalyzed by 2 μM Cu(II) in pH = 2 H2SO4/Na2SO4 buffer, and 0.5 mM PCoW11 and PNiW11 reduction by 25 mM RSH under Ar catalyzed by 10 μM Cu(II) in pH = 5 sodium acetate buffer. | |
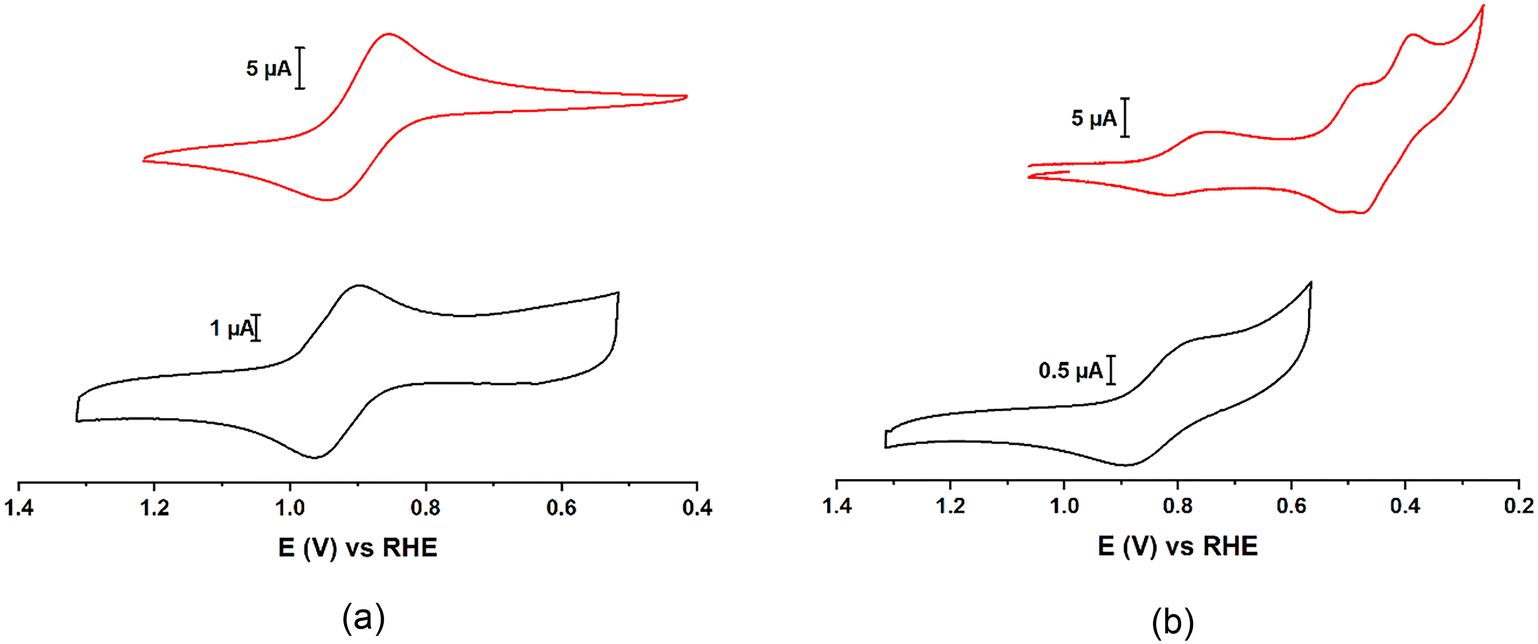
Cyclic voltammograms (CVs) show that PVMo11 and PVW11 have similar V redox potentials in both aqueous (Fig. 9, Table S5†) and acetonitrile solutions (Fig. S20, Table S4†). The Mo peaks have more positive potentials than the W peaks.89PVMo11@HKUST and PVW11@HKUST cyclic voltammograms (CVs) were collected by immobilizing the materials on glassy carbon working electrodes (Fig. 9). The observed V peaks in both POM@MOFs have potentials close to those of their homogeneous vanadium-containing POM counterparts.43,59,73 Thermodynamically, Mo centers are much easier to reduce than W centers in the same POM structure, thus the former centers are more likely to engage in redox processes than the latter. This agrees with the fact that PVMo can transfer multiple electrons including both V and Mo centers; whereas, PVW under identical experimental conditions, can only transfer one electron to a V center. Fig. S22† shows the CVs of PCoW11 and PNiW11 in pH = 5 acetate buffer. Both POMs have two redox peaks at negative potential (vs. RHE). Some transition-metal redox peaks overlap with W redox peaks consistent with a weaker oxidative ability than PVW11.90
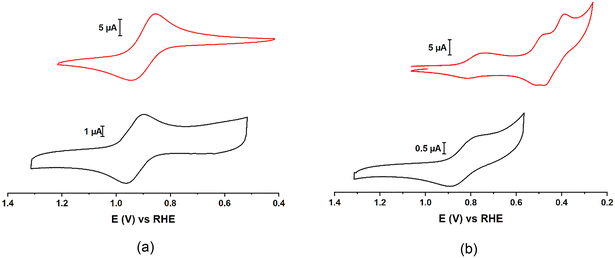 |
| | Fig. 9 a) Black: cyclic voltammograms (CV) of homogeneous 0.5 mM H3PVW11O40 (PVW11) on glassy carbon (GC) electrode. Red: CV of PVW11@HKUST immobilized on GC electrode. b) Black: CV of homogeneous 0.5 mM H3PVMo11O40 (PVMo11) on GC electrode. Red: CV of PVMo11@HKUST immobilized on GC electrode. Conditions: pH = 2 H2SO4/Na2SO4 buffer, 0.1 M KNO3, scan rate, v = 100 mV s−1, T = 298 K. The immobilization can be proved by the linear relationship of I vs. v in Fig. S22.† | |
Conclusions
In this work, we give compelling evidence of synergistic electron transfer between POM and MOF framework in host–guest PVMo@HKUST materials. The kinetic and thermodynamic results indicate that PXW11 materials have limited electron transfer ability with Cu, thus when all the Cu(II) is reduced to Cu(I) via eqn (2) (Fig. 7), no Cu(II) sites remain in the MOF. As a result, the catalytic aerobic thiol removal reaction (deodorization) has limited conversion. The Cu(I) sites in the MOF nodes cannot be reoxidized back to Cu(II) via eqn (3), leading to distortion of the MOF framework. Therefore, POM@MOF synergism regarding activity parallels this synergism for POM@MOF stability.
Author contributions
Xinlin Lu performed most of the experiments and drafted most of the article. Ting Cheng performed some of the electroanalytical experiments. Yurii V. Geletii oversaw the kinetics experiments and provided advice on the entire manuscript. John Bacsa performed the X-ray crystallography. Craig L. Hill oversaw all aspects of this study and did final editing of all documents.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors thank DTRA (Grant number W911NF-15-2-0107 through ARO).
References
- J. X. Liu, X. B. Zhang, Y. L. Li, S. L. Huang and G. Y. Yang, Coord. Chem. Rev., 2020, 414, 213260 CrossRef CAS.
- D. Y. Du, J. S. Qin, S. L. Li, Z. M. Su and Y. Q. Lan, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC.
- J. Sun, S. Abednatanzi, P. Van Der Voort, Y. Y. Liu and K. Leus, Catalysts, 2020, 10, 578 CrossRef CAS.
- M. Samaniyan, M. Mirzaei, R. Khajavian, H. Eshtiagh-Hosseini and C. Streb, ACS Catal., 2019, 9, 10174–10191 CrossRef CAS.
- M. Genovese and K. Lian, Curr. Opin. Solid State Mater. Sci., 2015, 19, 126–137 CrossRef CAS.
- Y. Liu, C. Tang, M. Cheng, M. Chen, S. Chen, L. Lei, Y. Chen, H. Yi, Y. Fu and L. Li, ACS Catal., 2021, 11, 13374–13396 CrossRef CAS.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS.
- C. L. Hill, L. Delannoy, D. C. Duncan, I. A. Weinstock, R. F. Renneke, R. S. Reiner, R. H. Atalla, J. W. Han, D. A. Hillesheim, R. Cao, T. M. Anderson, N. M. Okun, D. G. Musaev and Y. V. Geletii, C. R. Chim., 2007, 10, 305–312 CrossRef CAS.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 1997, 117, 151–158 CrossRef CAS.
- R. Neumann, Inorg. Chem., 2010, 49, 3594–3601 CrossRef CAS PubMed.
- I. A. Weinstock, R. E. Schreiber and R. Neumann, Chem. Rev., 2018, 118, 2680–2717 CrossRef CAS PubMed.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–218 CrossRef CAS PubMed.
- K. P. Sullivan, M. Wieliczko, M. Kim, Q. Yin, D. L. Collins-Wildman, A. K. Mehta, J. Bacsa, X. Lu, Y. V. Geletii and C. L. Hill, ACS Catal., 2018, 8, 11952–11959 CrossRef CAS.
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef CAS.
- T. Yamase, Chem. Rev., 1998, 98, 307–325 CrossRef CAS PubMed.
- E. Papaconstantinou, Chem. Soc. Rev., 1989, 18, 1–31 RSC.
- C. Streb, Dalton Trans., 2012, 41, 1651–1659 RSC.
- J. M. Cameron, D. J. Wales and G. N. Newton, Dalton Trans., 2018, 47, 5120–5136 RSC.
- M. Tao, Q. Yin, A. L. Kaledin, N. Uhlikova, X. Lu, T. Cheng, Y. S. Chen, T. Lian, Y. V. Geletii, D. G. Musaev, J. Bacsa and C. L. Hill, Inorg. Chem., 2022, 61, 6252–6262 CrossRef CAS PubMed.
- I. A. Weinstock, Chem. Rev., 1998, 98, 113–170 CrossRef CAS PubMed.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219–237 CrossRef CAS PubMed.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–217 CrossRef CAS PubMed.
- V. G. Snider and C. L. Hill, J. Hazard. Mater., 2023, 442, 130015 CrossRef CAS PubMed.
- H. Ma, B. Liu, B. Li, L. Zhang, Y. G. Li, H. Q. Tan, H. Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS PubMed.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 1–15 Search PubMed.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed.
- Q. L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- N. V. Maksimchuk, O. A. Kholdeeva, K. A. Kovalenko and V. P. Fedin, Isr. J. Chem., 2011, 51, 281–289 CrossRef CAS.
- J. Juan-Alcañiz, E. V. Ramos-Fernandez, U. Lafont, J. Gascon and F. Kapteijn, J. Catal., 2010, 269, 229–241 CrossRef.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed.
- C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 CrossRef CAS PubMed.
- L. Yang, H. Naruke and T. Yamase, Inorg. Chem. Commun., 2003, 6, 1020–1024 CrossRef CAS.
- S. Ahn, S. L. Nauert, C. T. Buru, M. Rimoldi, H. Choi, N. M. Schweitzer, J. T. Hupp, O. K. Farha and J. M. Notestein, J. Am. Chem. Soc., 2018, 140, 8535–8543 CrossRef CAS PubMed.
- C. T. Buru, A. E. Platero-Prats, D. G. Chica, M. G. Kanatzidis, K. W. Chapman and O. K. Farha, J. Mater. Chem. A, 2018, 6, 7389–7394 RSC.
- C. T. Buru, P. Li, B. L. Mehdi, A. Dohnalkova, A. E. Platero-Prats, N. D. Browning, K. W. Chapman, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 5174–5181 CrossRef CAS.
- C. T. Buru, M. C. Wasson and O. K. Farha, ACS Appl. Nano Mater., 2020, 3, 658–664 CrossRef CAS.
- W. Salomon, C. Roch-Marchal, P. Mialane, P. Rouschmeyer, C. Serre, M. Haouas, F. Taulelle, S. Yang, L. Ruhlmann and A. Dolbecq, Chem. Commun., 2015, 51, 2972–2975 RSC.
- W. Xie and F. Wan, Chem. Eng. J., 2019, 365, 40–50 CrossRef CAS.
- Y. L. Peng, J. Liu, H. F. Zhang, D. Luo and D. Li, Inorg. Chem. Front., 2018, 5, 1563–1569 RSC.
- L. Zeng, L. Xiao, Y. Long and X. Shi, J. Colloid Interface Sci., 2018, 516, 274–283 CrossRef CAS PubMed.
- R. Li, X. Ren, J. Zhao, X. Feng, X. Jiang, X. Fan, Z. Lin, X. Li, C. Hu and B. Wang, J. Mater. Chem. A, 2014, 2, 2168–2173 RSC.
- Q. Y. Li, L. Zhang, Y. X. Xu, Q. Li, H. Xue and H. Pang, ACS Sustainable Chem. Eng., 2019, 7, 5027–5033 CrossRef CAS.
- S. Mukhopadhyay, J. Debgupta, C. Singh, A. Kar and S. K. Das, Angew. Chem., Int. Ed., 2018, 57, 1918–1923 CrossRef CAS PubMed.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS PubMed.
- X. Lu, T. Cheng, Y. V. Geletii and C. L. Hill, Inorg. Chem., 2023, 5, 2404–2414 CrossRef PubMed.
- X. Lu, Y. V. Geletii, T. Cheng and C. L. Hill, Role of Multiple Vanadium Centers on Redox Buffering and Rates of Polyvanadomolybdate-Cu(II)-Catalyzed Aerobic Oxidations, Inorg. Chem., 2023, 62, 5822–5830 CrossRef CAS PubMed.
- Y. Liu, S. Liu, S. Liu, D. Liang, S. Li, Q. Tang, X. Wang, J. Miao, Z. Shi and Z. Zheng, ChemCatChem, 2013, 5, 3086–3091 CrossRef CAS.
- X. Zhong, Y. Lu, F. Luo, Y. Liu, X. Li and S. Liu, Chem. – Eur. J., 2018, 24, 3045–3051 CrossRef CAS PubMed.
- J. Zhu, P. C. Wang and M. Lu, Catal. Sci. Technol., 2015, 5, 3383–3393 RSC.
- J. Zhu, M. N. Shen, X. J. Zhao, P. C. Wang and M. Lu, ChemPlusChem, 2014, 79, 872–878 CrossRef CAS.
- X. Xu, S. Chen, Y. Chen, H. Sun, L. Song, W. He and X. Wang, Small, 2016, 12, 2982–2990 CrossRef CAS PubMed.
- H. Yang, J. Li, L. Wang, W. Dai, Y. Lv and S. Gao, Catal. Commun., 2013, 35, 101–104 CrossRef CAS.
- E. Rafiee and N. Nobakht, J. Mol. Catal. A: Chem., 2015, 398, 17–25 CrossRef CAS.
- Y. Liu, S. Liu, D. He, N. Li, Y. Ji, Z. Zheng, F. Luo, S. Liu, Z. Shi and C. Hu, J. Am. Chem. Soc., 2015, 137, 12697–12703 CrossRef CAS PubMed.
- L. H. Wee, S. R. Bajpe, N. Janssens, I. Hermans, K. Houthoofd, C. E. A. Kirschhock and J. A. Martens, Chem. Commun., 2010, 46, 8186–8188 RSC.
- Z. Wang and Q. Chen, Green Chem., 2016, 18, 5884–5889 RSC.
- L. H. Wee, C. Wiktor, S. Turner, W. Vanderlinden, N. Janssens, S. R. Bajpe, K. Houthoofd, G. Van Tendeloo, S. De Feyter, C. E. A. Kirschhock and J. A. Martens, J. Am. Chem. Soc., 2012, 134, 10911–10919 CrossRef CAS PubMed.
- D. Mustafa, E. Breynaert, S. R. Bajpe, J. A. Martens and C. E. A. Kirschhock, Chem. Commun., 2011, 47, 8037–8039 RSC.
- C. R. Wade and M. Dincă, Dalton Trans., 2012, 41, 7931–7938 RSC.
- S. R. Bajpe, C. E. A. Kirschhock, A. Aerts, E. Breynaert, G. Absillis, T. N. Parac-Vogt, L. Giebeler and J. A. Martens, Chem. – Eur. J., 2010, 16, 3926–3932 CrossRef CAS PubMed.
- E. A. Nagul, I. D. McKelvie, P. Worsfold and S. D. Kolev, Anal. Chim. Acta, 2015, 890, 60–82 CrossRef CAS PubMed.
- N. Maksimchuk, M. Timofeeva, M. Melgunov, A. Shmakov, Y. Chesalov, D. Dybtsev, V. Fedin and O. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS.
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Inorg. Chem., 2010, 49, 2920–2930 CrossRef CAS PubMed.
- N. V. N. Maksimchuk, M. N. Timofeeva, M. S. Melgunov, A. N. A. Shmakov, Y. A. Chesalov, D. N. D. Dybtsev, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS.
- N. V. N. Maksimchuk, M. N. Timofeeva, M. S. Melgunov, A. N. A. Shmakov, Y. A. Chesalov, D. N. D. Dybtsev, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS.
- L. Bromberg, Y. Diao, H. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef CAS.
- D. M. Fernandes, A. D. S. Barbosa, J. Pires, S. S. Balula, L. Cunha-Silva and C. Freire, ACS Appl. Mater. Interfaces, 2013, 5, 13382–13390 CrossRef CAS PubMed.
- M. Yin, C. K. Wu, Y. Lou, C. Burda, J. T. Koberstein, Y. Zhu and S. O'Brien, J. Am. Chem. Soc., 2005, 127, 9506–9511 CrossRef CAS PubMed.
- C. Chen, T. Wu, D. Yang, P. Zhang, H. Liu, Y. Yang, G. Yang and B. Han, Chem. Commun., 2018, 54, 5984–5987 RSC.
- T. Wei, M. Zhang, P. Wu, Y. J. Tang, S. L. Li, F. C. Shen, X. L. Wang, X. P. Zhou and Y. Q. Lan, Nano Energy, 2017, 34, 205–214 CrossRef CAS.
- P. Zhu, X. Yang, X. Li, N. Sheng, H. Zhang, G. Zhang and J. Sha, Dalton Trans., 2019, 49, 79–88 RSC.
- H. Liu, L. G. Gong, C. X. Wang, C. M. Wang, K. Yu and B. Bin Zhou, J. Mater. Chem. A, 2021, 9, 13161–13169 RSC.
- P. St. Petkov, G. N. Vayssilov, J. Liu, O. Shekhah, Y. Wang, C. Wöll and T. Heine, ChemPhysChem, 2012, 13, 2025–2029 CrossRef CAS PubMed.
- C. Chen, T. Wu, D. Yang, P. Zhang, H. Liu, Y. Yang, G. Yang and B. Han, Chem. Commun., 2018, 54, 5984–5987 RSC.
- A. D. Leu and D. A. Armstrong, J. Phys. Chem., 1986, 90, 1449–1454 CrossRef CAS.
- V. Vortisch, P. Kroneck and P. Hemmerich, J. Am. Chem. Soc., 1976, 98, 2821–2826 CrossRef CAS.
- R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS PubMed.
- G. A. Bagiyan, I. K. Koroleva, N. V. Soroka and A. V. Ufimtsev, Russ. Chem. Bull., 2003, 52, 1135–1141 CrossRef CAS.
- G. Scrivens, B. C. Gilbert and T. C. P. Lee, J. Chem. Soc., Perkin Trans. 2, 1995, 9, 955–963 RSC.
- M. G. Gantman, I. G. Tarkhanova and Y. G. Kolyagin, J. Sulfur Chem., 2016, 37, 501–514 CrossRef CAS.
- P. W. Albro, J. T. Corbett and J. L. Schroeder, J. Inorg. Biochem., 1986, 27, 191–203 CrossRef CAS PubMed.
- G. A. Bagiyan, I. K. Koroleva, N. V. Soroka and A. V. Ufimtsev, Kinet. Catal., 2004, 45, 372–380 CrossRef CAS.
- J. J. Altenau, M. T. Pope, R. A. Prados and H. So, Inorg. Chem., 1975, 14, 417–421 CrossRef CAS.
- J. H. Choi, J. K. Kim, D. R. Park, T. H. Kang, J. H. Song and I. K. Song, J. Mol. Catal. A: Chem., 2013, 371, 111–117 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
John
Bacsa
and
Craig L.
Hill
*,
John
Bacsa
and
Craig L.
Hill