
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring CO2 hydrogenation to methanol at a CuZn–ZrO2 interface via DFT calculations†
Aku
Lempelto
 ,
Lars
Gell
,
Toni
Kiljunen
and
Karoliina
Honkala
*
,
Lars
Gell
,
Toni
Kiljunen
and
Karoliina
Honkala
*
Department of Chemistry, University of Jyväskylä, Nanoscience Center, P.O. Box 35, FI-40014, Finland. E-mail: karoliina.honkala@jyu.fi
First published on 21st June 2023
Abstract
Multi-component heterogeneous catalysts are among the top candidates for converting greenhouse gases into valuable compounds. Combinations of Cu, Zn, and ZrO2 (CZZ) have emerged as promisingly efficient catalysts for CO2 hydrogenation to methanol. To explore the catalytic mechanism, density functional theory (DFT) calculations and the energetic span model (ESM) were used to study CO2 conversion routes to methanol on CuZn–ZrO2 interfaces with a varying Zn content. Our results demonstrate that the presence of Zn sites at the interface improves CO2 binding. However, the adsorption and activation energies are insensitive to Zn concentration. The calculations also show that the hydrogenation of adsorbate oxygen atoms at the interface is kinetically more favourable and requires hydrogen spillover from the metal to the zirconia. This leads to barriers that are lower than those reported on interface or metal-only sites in previous literature. While DFT calculations alone are unable to identify which one of the competing pathways is more favourable, the ESM model predicts that the carboxyl pathway has a higher turnover frequency than the formate route. Our findings also show the importance of considering effects such as hydrogen spillover which take place at a metal-oxide interface when modelling complex catalytic environments.
1 Introduction
Transforming CO2 into value-added products such as methanol, hydrocarbon fuels, and other platform chemicals has gained attention as an attractive approach to reduce the negative impact carbon dioxide has on the climate.1–4 Using renewable energy and sustainable hydrogen would allow us to establish a circular economy based on carbon recycling.1,2,5–8 However, due to the stable nature of CO2, highly active, selective, and deactivation-resistant catalysts are required to make large-scale adaptation feasible.5 Various catalytic systems with different combinations of active metal and supporting metal oxide have been prepared and characterised to maximise catalytic activity and selectivity for CO2 conversion to methanol (CTM):3,9–18| CO2(g) + 3H2(g) → CH3OH(g) + H2O(g). | (R1) |
Oxide-supported Cu nanoparticles have been extensively studied for catalysing CTM due to their promising methanol selectivity, which can be upwards of 60%.3,19 Several experimental3,20–24 and computational3,21,23–26 studies have associated the catalytic activity with active sites at the metal-oxide interface. The type of oxide support can substantially influence the activity and selectivity of the catalyst.3,4,24 Common choices for suitable oxides include zinc oxide ZnO, zirconia ZrO2, titania TiO2, and alumina Al2O3. For example, the ternary system of Cu/ZnO/Al2O3 is already an industrially established CTM catalyst, but it displays relatively low conversion, typically below 20%, leading to efforts to develop more selective catalysts in addition to the continuous search for increased activity.3,6,9,27 To this end, zirconia (ZrO2) has been suggested as an alternative oxide support due to increased turnover rates and selectivity of ZrO2-supported Cu.3,24 Additionally, zirconia has been reported to offer enhanced thermal and mechanical stability and to prevent Cu particles from sintering and thus to hinder the deactivation of the catalyst.3,24,28–33 Adding ZnO as another oxide into Cu/ZrO2 has demonstrated higher methanol production rates and total conversion percentages than either Cu/ZnO or Cu/ZrO2, often reaching a 20% conversion whereas a lower 5 to 10 percent conversion is typical for binary systems.3,4,30,34
The precise function of ZnO in Cu/ZnO/ZrO2 (CZZ) is still under debate.33 The ZnO component is suggested to be able to temporarily store hydrogen,33 and it may also act as an additional structural modifier, enhancing Cu dispersion and increasing its surface area.30 The actual oxidation state of Zn and the mechanism of catalytic promoting remain somewhat controversial,3,9–11,13,20,22,23,35–39 and the question of whether or not a CuZn-alloy is involved as the active phase remains open. It is known that the strong interaction between the components may lead to partial ZnO coverage of a Cu surface.11,20 Under certain conditions the ZnO component can partially reduce into metallic Zn which has two possible outcomes: either forming oxygen vacancies or creating surface alloys of Cu and Zn.9,10,13,40,41 The CuZn alloys formed at defect and edge sites of metal particles can then be partially reduced by the adsorbates and serve as the active sites of Cu/ZnO catalysts.9 Based on a combination of in situ analyses, it has been determined that the extent of the alloying varies largely based on the exact temperatures, the presence of CO2 or other gaseous species i.e. the reducing quality of the conditions.13,22,23,41,42 Therefore the significance of the alloying remains controversial. A bulk alloy may oxidise and separate into Cu and oxidised Zn,22 or simply lose its ability to act as an efficient catalyst.42 Instead, an oxidised phase of Zn in the form of Zn formates has been suggested to be the active intermediate species that lead to methanol formation.22,36 While the discussion is often centered around bulk alloying, the presence of metallic Zn impurities or dilute alloys at the metal-oxide interface acting as the active sites demand investigation.
In computational studies, a pristine Cu facet such as Cu(111) or Cu(211) has typically been chosen as the model to represent the Cu-containing catalyst system.9,27,43 While simple to work with, these models obviously omit the effects of the supporting oxide. For example, when CO2 is electronically activated upon adsorption, it takes a bent shape with an O–C–O angle of 123°, as if sp2 hybridised. However, both at and stepped Cu surfaces bind CO2 only in a linear fashion even though physisorption energies as large as −0.56 eV and −0.71 eV have been reported on Cu(111)9,27,43,44 and Cu(211),9,45,46 respectively. On the other hand, the copper-oxide interface has been reported to activate CO2, which bends upon adsorption and the adsorption energy varies in the range of −0.4–1.8 eV, depending on the specific structure of the interface.25,44,47 Therefore, binary systems where a metal cluster or nanorod is supported on an oxide slab are a common alternative to a purely metallic model.25,44,47,48 In a recent study combining experiments and calculations, a single Cu atom catalyst on ZrO2 was found to promote CTM with near 100% selectivity whereas additional active sites at larger Cu clusters and particles were shown to diminish this efficiency.14
As the precise role of the Zn promoter in CTM is unsettled,35,41 the choice of how it should be included in computational models has varied. The density functional theory (DFT) studies on CZZ catalysts conducted so far have only considered one or two of the three components. For example, a CZZ system was recently modelled21 by depositing a small ZrO2 cluster on a ZnO(11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) surface to better understand the catalytic behavior on oxide interfaces. Very recently, an inverse ZrZn2O3/Cu(111) system was used to model ZrO2/Cu and ZnO/Cu interfaces.49 Purely metallic Cu or CuZn models have also been used to mimic active sites at facets and edges of nanoparticles.9,23,27,46,50 Even though a CuZn(211) surface is unable to activate CO2 and the intermediates bound to it are thermodynamically less stable than the gas-phase reagents,9 the hydrogenation intermediates are nevertheless more strongly bound to an alloyed CuZn surface than to a pure Cu surface.9
0) surface to better understand the catalytic behavior on oxide interfaces. Very recently, an inverse ZrZn2O3/Cu(111) system was used to model ZrO2/Cu and ZnO/Cu interfaces.49 Purely metallic Cu or CuZn models have also been used to mimic active sites at facets and edges of nanoparticles.9,23,27,46,50 Even though a CuZn(211) surface is unable to activate CO2 and the intermediates bound to it are thermodynamically less stable than the gas-phase reagents,9 the hydrogenation intermediates are nevertheless more strongly bound to an alloyed CuZn surface than to a pure Cu surface.9
While the reaction mechanism of CTM has been studied for a variety of catalyst compositions, the views are not unanimous on the identity of the key intermediates, the main reaction pathway, and an active catalyst domain.3,19,24 Generally, the plausible pathways have been narrowed down to two competing ones: one converting CO2 to a formate (HCOO) which then reacts onwards to methanol, and the other, where CO2 first breaks down to carbon monoxide (CO) through a reverse water–gas shift (RWGS) reaction and then hydrogenates to methanol via a formaldehyde intermediate. Different interpretations of computational and experimental results have led to differing opinions on the dominant reaction route. For example, a formate species bound to the zirconia surface has been both computationally and experimentally determined to be highly stable and could be considered a strong thermodynamic sink.25,28,44,48 Therefore, formates have been suggested to accumulate at the zirconia and poison the active sites rather than acting as key intermediates in CTM. However, the barrier for HCOO conversion to dioxymethylene (H2COO) has been computed to be only 0.66 eV on a ZrO2/ZnO interface.21 This finding is supported by in situ diffuse reflectance infrared spectroscopy measurements, which have shown that the CZZ system can quickly convert formate to a methoxy (CH3O) species.21,25
Another key feature for a hydrogenation catalyst is the ability to efficiently dissociate molecular hydrogen, which, in a Cu-based system, is assumed to be take place on the metal component, where hydrogen readily adsorbs dissociatively.4,9,21,51–53 Hydrogen spillover from the metal to the ZrO2 surface may take place and promote a zirconia-bound hydrogen to participate in the CTM reactions.51,54,55 However, the efficiency and mechanism of the spillover on irreducible oxides, such as zirconia, has been a subject of debate56–58 and the importance of the kinetic control of the spillover step is unclear.
In this work, we employ DFT calculations to examine the intermediate and transition states for the branched reaction network of CO2 CTM by first discussing the reactant properties at the interface using supported CuZn nanorod models, then outlining the formate and RWGS route specifics, combining them to form the products, and finally evaluating the kinetic aspects of the catalytic cycle. We model the active interface by constructing mixed CuZn structures with varying concentration of Zn atoms at the Cu-monoclinic ZrO2(![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11) interface mimicking a diluted interface nanoalloy. The energetic span model is used to identify rate-controlling intermediates and to draw a simplified comparison between the optimal kinetics of competing mechanisms.
11) interface mimicking a diluted interface nanoalloy. The energetic span model is used to identify rate-controlling intermediates and to draw a simplified comparison between the optimal kinetics of competing mechanisms.
2 Computational methods
DFT calculations were carried out using the BEEF-vdW exchange–correlation functional59 in the projector-augmented wave (PAW)60 formalism as implemented in the GPAW61 package. The core electrons of all elements were described in the frozen-core approximation. A maximum spacing of 0.20 Å was used for the real-space grid basis, and the reciprocal space was sampled at the Γ point. A Hubbard U correction62 of 2.0 eV, determined using a self-consistent linear response method detailed in ref. 63, was applied to the d-orbitals of the zirconium atoms. A higher value of 4 eV is common in literature64–67 but the difference is not unusual as the value of the U parameter is very sensitive to the specific DFT implementation used. The geometry optimisations were performed using the fast inertial relaxation engine (FIRE) algorithm as implemented in the atomic simulation environment (ASE).68,69 During optimisations, the bottom layer of the ZrO2 slab was kept frozen in its initial bulk geometry. All other atoms were allowed to relax until the maximum residual force was reduced below 0.02 eV Å−1. The transition state searches were carried out using the climbing image nudged elastic band (CI-NEB)70,71 method where the maximum residual force was set to 0.05 eV Å−1 which keeps the computational cost feasible. Hydrogenation reactions were carried out such that the H atom was initially placed either on the metal side of the interface or on the oxide side, close to the molecule to be hydrogenated. The transition states were confirmed by calculating the vibrational modes using the Frederiksen method72 and verifying that only one mode with an imaginary frequency exists along the reaction coordinate. Partial charges on atoms were analysed with the Bader method73 using code written by Tang et al.74For the metal-oxide interface, we utilise the oxide-supported metal nanorod concept consisting of a Cu rod with some edge Cu atoms replaced with Zn atoms at different concentrations (see Fig. 1 for the structures and denominations). The Cu–(m-ZrO2(11)) interface model was adopted from our previous study,47 where the length of the nanorod is eight atoms and the thickness three atomic layers. A (111) plane is exposed towards the interface. The zirconia surface is described by a two-layer-thick slab model, built from a 3 × 2 m-ZrO2(11) supercell with periodic boundary conditions in the horizontal directions. While a Cu rod was found to be more stable on a tetragonal zirconia surface,47 the monoclinic variety is thermodynamically favoured under reaction conditions75 and has shown activity towards CO2 activation and CTM in experiments.34,76 Three different Zn concentrations were examined by replacing every fourth Cu atom, 3/4 of Cu atoms, or all Cu atoms on the nanorod edge with Zn. These models were named the Zn-dilute, the Zn-rich, and the Zn interface, respectively. This represents situations where Zn centres are dispersed to a varying degree, such as the case where atomic layer deposition has been used to prepare the catalyst.34 In this instance we have chosen to place the Zn centers along the active interface instead of a more homogeneous alloying with Cu, allowing the use of a pure Cu interface model as a reference, assisting interpretation.
 | ||
| Fig. 1 The four metal/ZrO2 interface systems with varying amounts of Zn in the Cu nanorod. A: Cu interface, B: Zn-dilute interface, C: Zn-rich interface, D: Zn interface. Red: O, white: H, dark grey: C, purple: Zn, turquoise: Zr, orange: Cu. | ||
The simulation cell measures 20.67 × 14.79 × 24.0 in Å with angles of 90°/90°/116.5°. This unit cell size results in a minor compressive strain of −1.02% for the Cu atoms along the direction of the nanorod.47 For the Zn interface, the strain is −4.2% as a result of the longer, 2.69 Å, Zn–Zn bulk distance. The strains experienced by the other interfaces are less clearly defined but are likely between the −1.02% of Cu and −4.2% of the Zn interface. A strain of this magnitude at the Cu interface47 was found to enhance CO2 binding by up to 0.4 eV. On the other hand, a computed lattice constant of 2.91 Å has been reported for a bulk β-CuZn,77 corresponding to a shorter Cu–Zn distance of 2.52 Å. This could mean that the compressive strain of the Zn-dilute interface is slightly lower than that of the Cu interface. All three doped interfaces have a Zn atom at the site that binds CO2 and many further reaction intermediates the strongest, so that they all measure the impact of Zn against the performance of the pure Cu interface. The relative strength of the adsorption at this site is likely due to a lower coordination caused by the interaction between the nanoparticle and the oxide. The models represent cases where the Zn atoms exist as single atom centers dispersed at the metallic interface or as more conjugated assemblies that span several Zn centers. The energy ΔEdef by which CO2 adsorption deforms the interface was calculated as
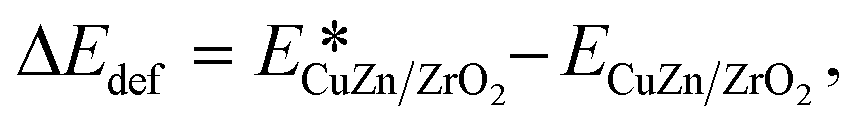 | (1) |
 is the energy of a CuZn/ZrO2 configuration after removal of CO2 from an optimised adsorption geometry and ECuZn/ZrO2 is the energy of the optimal interface without any adsorbate.
is the energy of a CuZn/ZrO2 configuration after removal of CO2 from an optimised adsorption geometry and ECuZn/ZrO2 is the energy of the optimal interface without any adsorbate.
A graph theory-based energetic span model78,79 (ESM) for complex reaction networks was utilised to assess the catalytic cycles. By summing together pairs of intermediate and transition state energies, it is possible to estimate turnover frequency of each mechanism,
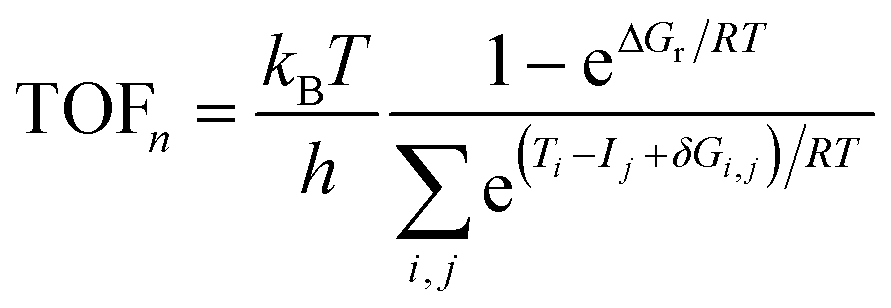 | (2) |
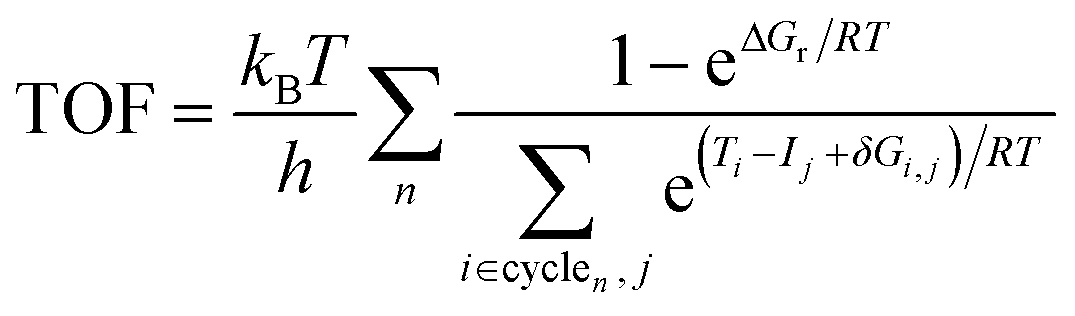 | (3) |
 | (4) |
 | (5) |
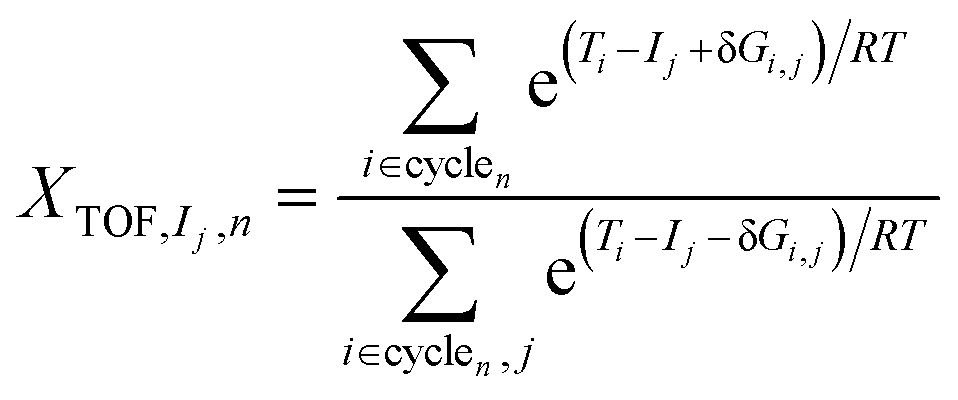 | (6) |
 | (7) |
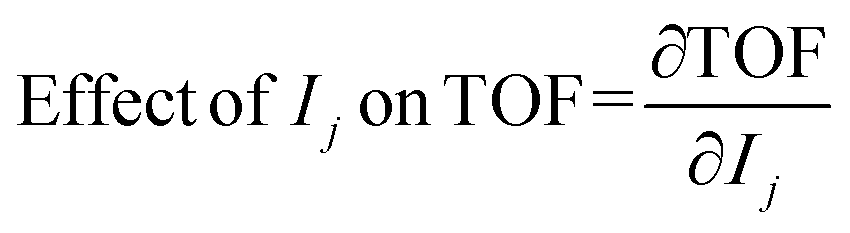 | (8) |
The ESM analysis was originally developed for studying homogeneous catalysis78 but it has also been successfully applied for heterogeneous systems including CO2 hydrogenation to methanol on Cu(111).81 The basic assumptions of the ESM include that: (i) Eyring's transition state theory is valid, (ii) a steady-state regime is applicable, and (iii) the intermediates undergo fast relaxation to the thermodynamic equilibrium described by the Boltzmann distribution.78 While the reaction kinetics for heterogeneous catalyst systems is often studied via microkinetic modelling or kinetic Monte Carlo simulations,23,26–28,48,49,82 the ESM offers a simplified way to estimate which pathway is optimal. We used the gTOFfee software,81,83 which was slightly modified to improve the performance for the present reaction network. Additionally, an extension was made to the code for calculating the degrees of TOF control, see ESI† for details.
3 Results and discussion
3.1 CO2 and H2 activation
The CO2 adsorption properties of the interfaces were determined first since the CO2 reduction pathways start with the adsorption of the reactants on the catalyst surface. All the interface atoms were first considered as potential sites for the CO2 adsorption. Subsequent hydrogenation steps are then performed for the most stable CO2 adsorption geometry, which is similar to the one for the Cu interface.47 CO2 binds to the CuZn–ZrO2 interface in a conformation where the carbon atom resides on top of a Zn atom (C–Zn bond length 2.1 Å) and the two oxygen atoms bind on two Zr cations close to the interface (see Fig. S6†). Upon adsorption, CO2 takes a bent shape, which resembles a carbonate anion25,47 and indicates the activation of the molecule with a partial charge of 1.3|e|.47 The interaction of CO2 with the CuZn–ZrO2 interface leads to a local deformation of the rod such that the metal atom in contact with the C atom is pulled out from the (111) plane.The CO2 adsorption is exothermic by −1.17 eV, −1.13 eV, and −1.30 eV at the Zn-dilute, Zn-rich and Zn interfaces, respectively. The Cu interface exhibits significantly weaker binding with an adsorption energy of −0.64 eV.47 The difference can be rationalised by examining the energy penalty of deformation ΔEdef, calculated using eqn (1), which is +1.7 eV for the Cu interface and +1.1 eV for the Zn-dilute interface. The ΔEdef is consistent with the difference in adsorption energies: the stronger binding at the Zn-containing interface is due to smaller deformation energy. No energy penalty is seen for the Zn-rich and Zn interfaces, as their intrinsic strain already favours a deformed structure. CO2 adsorption at the Zn-rich interface is a kinetically activated process with a barrier of 0.16 eV, which is 0.22 eV lower than the barrier computed for the Cu interface.47 Similarly to the more negative adsorption energies, the lower barrier is likely due to the increased mobility of the Zn. Adsorption of CO2 on the low-coordinated oxygen atoms of the ZrO2 surface, without the involvement of the interface, is also possible in a trigonal carbonate-like geometry where the carbon atom of the CO2 binds the lattice oxygen and one of its oxygens rests on a Zr cation. With an adsorption energy of −0.59 eV, this binding is likely in competition with the adsorption at the interface. In addition to adsorption, we considered dissociative adsorption of CO2 into CO and O, as studied previously.84–86 However, the reaction is endothermic with a barrier of 1.7 eV, see ESI† section 1.1 for details, and therefore this pathway was omitted from further evaluation.
The dissociative adsorption of H2 was considered at the CuZn–ZrO2 interface at various positions. The dissociation can be homolytic, where both H atoms adsorb on Cu/Zn sites on the rod and have similar small charges of ca. −0.3 e, regardless of if they are in the vicinity of a Zn site. Alternatively, it can happen across the interface either homolytically, where one H binds on the surface of the rod and the other on top of a Zr, or heterolytically, where a hydride binds to the surface of the metal rod and a proton to an oxide anion. These sites are illustrated in Fig. S7.† Hydrides and protons on the zirconia have charges of −0.6 e and +0.6 e, respectively. The heterolytic dissociation is endothermic by ca. +1 eV and has a kinetic barrier of 1.4 eV, while the homolytic path on the rod is endothermic only by +0.4 eV with a slightly smaller activation energy of 1.1 eV. The spillover of hydrogen from the metal to the oxide surface is endothermic by ca. +0.6 eV depending slightly on the interface site. The kinetic barrier for hydride migration from the metal to the Zr on-top site of the oxide is 0.8 eV. However, the presence of CO2 or further reaction intermediates can stabilise the oxide-bound H, up to the point where it becomes thermodynamically favourable compared to the metal-bound H atom.
3.2 The formate and RWGS routes
The two most commonly proposed CTM reaction mechanisms are considered here: the formate pathway with intermediates labeled Fi, and the RWGS pathway with labels Ri, Fig. 2 displaying the detailed reaction network. The elementary steps along the formate and RWGS pathways were examined at both the Zn-dilute and Zn-rich interfaces, as well as examining selected steps at the Zn interface. We highlight the differences between the systems when they are relevant but in many cases they behave very similarly. In these situations, we use the Zn-dilute interface as an example. The computed adsorption and activation energies for all interfaces are summarised in the ESI† in Table S2 and the corresponding atomic structures are shown in Fig. S8–S13.† | ||
| Fig. 2 The CO2 the two most common reaction routes for the CTM reaction network. The formate route is given in yellow whereas the RWGS route is displayed in pink. Solid lines imply hydrogenation and dashed lines stand for dissociation or ad-/desorption of the intermediate. Less favourable intermediates/paths are more faintly coloured. | ||
 | ||
| Fig. 3 Initial, transition and final state geometries for formate formation at the Zn-dilute interface. Light red: lattice O, dark red: adsorbate O, white: H, dark grey: C, purple: Zn, turquoise: Zr, orange: Cu. | ||
12) interface.26 The transition state geometry is also similar with a C–H distance of 1.58 Å. A study of a zirconia-supported Cu38 cluster reported a slightly higher barrier of ca. +0.7 eV (ref. 25) still suggesting a fast interconversion between the HCOO and H2COO. Interestingly, at a model ZrO2/ZnO interface,21 the reaction energy for H2COO formation is exothermic by −1.27 eV, but the activation energy of +0.66 eV is close to our values. Low activation energies reported for Cu/ZnO, Cu/ZrO2, and ZnO/ZrO2 interfaces are in strong contrast to the high activation energies on bare Cu surfaces. For example, the barrier on a Cu(111) surface has been reported to be 1.59 eV27 or 0.97 eV.43 On a stepped Cu(533) surface, the activation energy was determined to be 1.42 eV. Consequently, several computational studies9,23,27,28,45,46 on metal surfaces and inverse oxide-on-metal models have suggested that HCOO hydrogenation leads to formic acid (HCOOH) instead. In these cases, an oxygen atom of the adsorbate and the reacting H are connected to the same component, e.g. the metal surface. Thus it is sensible that the formation of an O–H bond to create formic acid is more facile than the reaction with the carbon atom of the formate, which points away from the surface. We find, however, that on CuZn/ZrO2 the reaction to HCOOH is thermodynamically and kinetically significantly less favourable than the reaction to H2COO with an activation energy that is over 1 eV higher, as apparent in Fig. S2 and S4 (see ESI† section 1.2 for the full discussion). The difference can be explained simply by the fact that the structure of a metal-oxide interface is able to bring the reacting H and the carbon centre of the HCOO species closer together to create a more favourable pathway.
 | ||
| Fig. 4 Initial, transition and final state geometries for HCOO hydrogenation to H2COO, at the Zn-dilute interface. Light red: lattice O, dark red: adsorbate O, white: H, dark grey: C, purple: Zn, turquoise: Zr, orange: Cu. | ||
The next step on is the dissociation of H2COOH into hydroxyl (OH) and formaldehyde (H2CO). This reaction shows significant energetic variation depending on the interface, being practically thermoneutral at the Zn-dilute and Zn interfaces, and slightly endothermic by +0.2 eV at the Zn-rich interface. At the Cu interface, the reaction is more endothermic by ∼+0.4 eV, due to the weaker adsorption of the formaldehyde. The activation barriers are in the order of +0.2 to +0.5 eV. A previous study80 found the reaction and activation energies on a Zn-decorated Cu(211) surface to be similarly slightly endothermic with a low barrier. When the C–O bond is broken, the OH part remains bound to a Zr-top site whereas the H2CO (F06), while still attached to an oxide cation via its oxygen, tilts toward the nearby interface and binds to a Zn via its C atom. The resulting C–Zn distance of 2.17 Å is similar to that of activated CO2. The Zn atom is again slightly pulled out from the ideal position and there is no significant energy difference regarding which of the two neighbouring Zr atoms the oxygen atom binds to. Alternatively, H2COOH may hydrogenate to methanediol H2COHOH (F07). However, we found the activation energy of +0.9 eV to be clearly higher than that of the dissociation. Therefore, the pathway was not considered further.
The full potential energy diagram of CTM through the formate mechanism is shown in Fig. 5. The final steps from H2CO to methanol are shared between both the formate and the RWGS route.
 | ||
| Fig. 5 Potential energy diagrams of the formate route for CO2 hydrogenation to methanol. Dissociative adsorption steps of hydrogens are not included as they are assumed to be available. | ||
 | ||
| Fig. 6 CO2 hydrogenation to COOH at the Zn-dilute interface. Light red: lattice O, dark red: adsorbate O, white: H, dark grey: C, purple: Zn, turquoise: Zr, orange: Cu. | ||
The COOH is thermodynamically less stable by ca. 1.5 eV compared to the HCOO intermediate but the activation energy for COOH formation is 0.5 eV lower. A similar 1.4 eV difference in adsorption was found at a Cu38/ZrO2 interface.25 However, in that case the H reacted directly from the Cu38 cluster which corresponds to an activation energy of around +2 eV which is 1.2 eV higher than that of the formate formation. These activation energies and their differences are in line with our results but the case where H reacts from the oxide was not previously included.
In some previous studies, the formation of HCO has had a different character compared to our CuZn/ZrO2 results. Namely, on a Cu(111) surface,27 the reaction is endothermic by +0.78 eV and has an activation energy of almost +1 eV. Furthermore, on an inverse ZnO/Cu model, the reaction is endothermic by +0.39 eV and the barrier is +0.88 eV.23 These differences point to HCO adsorbing less strongly to Cu surfaces, and to the stabilising effect of the interface.
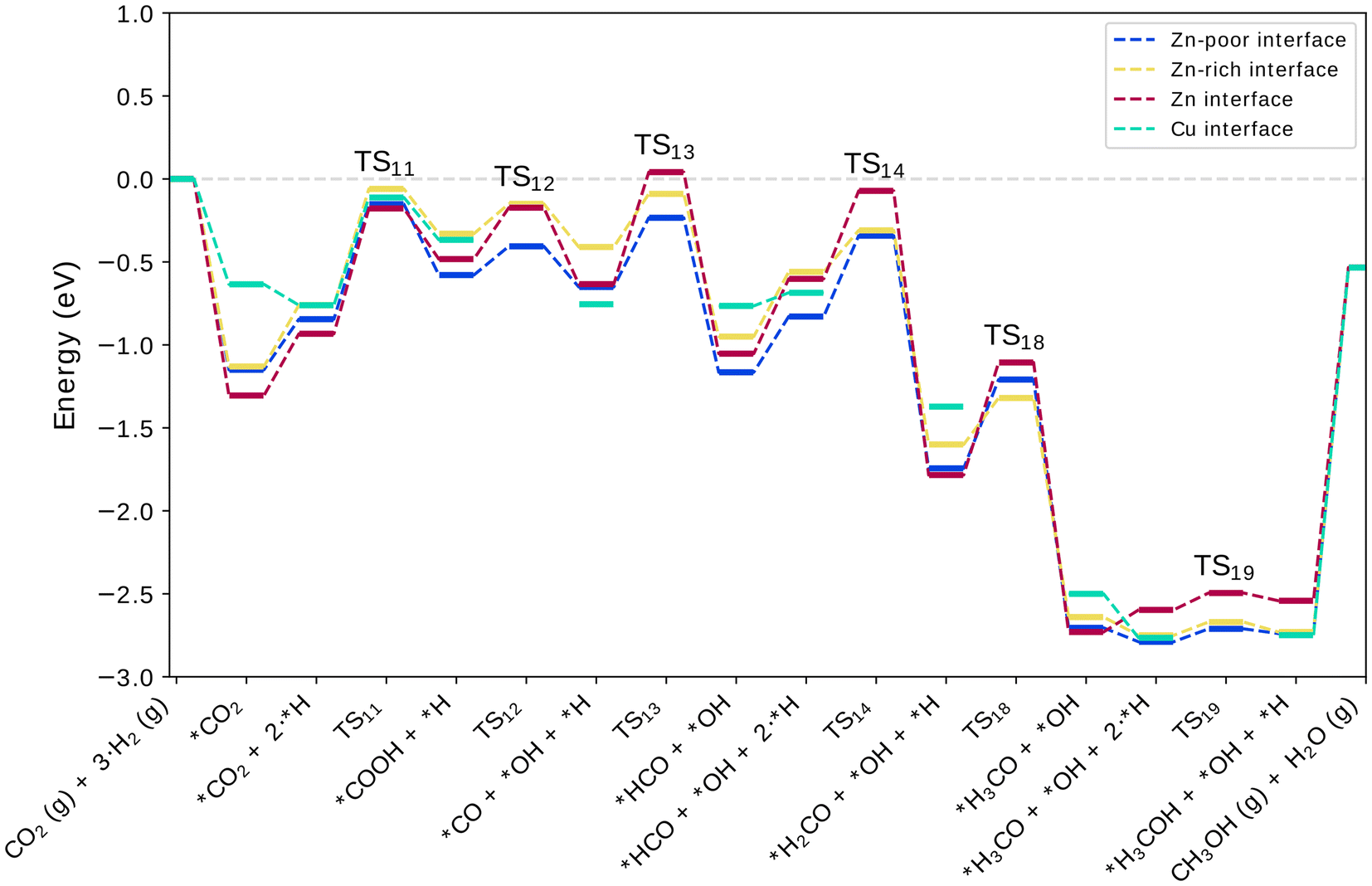 | ||
| Fig. 7 Potential energy diagrams of the RWGS path. States from H2CO forward include an OH on the zirconia surface. | ||
3.3 Formaldehyde hydrogenation to methanol
The formate and RWGS pathways discussed above merge after the formation of formaldehyde (H2CO). This species is stable, as the adsorption energy of H2CO relative to formaldehyde in the gas phase is −1.4 eV. Next, we address two different ways to progress from H2CO onward by considering the options of removing and keeping the ZrO2-bound OH group produced during the previous reaction steps. This is done to investigate the influence of OH on reaction energetics, as it could have a co-catalysing effect via hydrogen bonds or due to its Lewis acidity. The removal of the OH from the zirconia can take place via H2O formation and desorption, which is endothermic by +0.7 eV on average. A previous study48 showed no kinetic barriers for the dissociative adsorption of water on ZrO2 and therefore its desorption likely does not have a kinetic barrier either. For brevity, these two routes are shown in the PES diagrams of the previously discussed formate and RWGS pathways. The case where OH has been removed is shown in the diagram of the formate route (Fig. 5) whereas the OH is kept on the surface at the end of the RWGS path in Fig. 7.Upon hydrogenation to methoxy (H3CO), the bond between the product and the metal at the interface is broken and the methoxy binds solely to ZrO2via its O atom. The reaction is exothermic by −1 eV and requires an activation energy of 0.4–0.6 eV. In this case, the influence of the ZrO2-bound OH is rather insignificant as the reaction and activation energies are similar to those without OH. In previous studies, the reaction was practically thermoneutral on Cu(111)27 and exothermic by −0.67 eV on an inverse ZnO/Cu system.23 The kinetic barriers at the CuZn interfaces are higher than those found on the Cu(111) and inverse ZnO/Cu surfaces where in both cases an activation energy ∼+0.2 eV was reported. This may simply be due to more favourable adsorption and transition state geometries. We also studied the alternative pathway forward via the formation and hydrogenation of hydroxymethyl (H2COH) but found that it is significantly less favourable due to its relative instability and high activation barriers as seen in Fig. S2–S5.† Discussion of this pathway can be found in the ESI† section 1.3.
3.4 Potential energy landscape
Fig. 8 shows the potential energy profiles for the formate and RWGS pathways. At the Zn-dilute interface, the formate pathway exhibits lower energies and thus appears thermodynamically more stable as compared to the RWGS pathway. However, accessibility of the formate pathway is limited by the major barrier (TS01) for HCOO formation which is energetically well in line with previous computational studies that have considered CuZn surfaces or Cu cluster models.23,25 Despite the strong binding of formate, it does not severely poison the CuZn/ZrO2 interface. This is because it readily reacts onward to H2COO with a barrier around +0.4 eV. After a further hydrogenation to H2COOH, the reaction proceeds via decomposition into H2CO and a surface-bound OH group, which is removed to the gas phase as water. According to activation energies, the formation of H2COH is favoured over H3CO. However, the activation energy required for the final hydrogenation step from H2COH to methanol is considerably larger than that from methoxy, which in turn favours the path through the H3CO intermediate. Therefore, the methoxy intermediate is likely the dominant one, similar to what previous computational studies have suggested on both cluster and surface models.23,25,27 In general, intermediates along the formate pathway tend to be slightly more stable at the Zn interface than the Zn-dilute and the Zn-rich interfaces (see Fig. 5). Overall, the variation in energy is modest with the largest difference being 0.25 eV in the case of the formate species. | ||
| Fig. 8 Potential energy diagram of both the formate and the RWGS paths (Zn-dilute interface). States from H2CO forward include an OH on the zirconia surface. | ||
Along the RWGS pathway, the activation energies are in general lower than those of the formate route. The carboxyl intermediate readily dissociates into OH and CO. At the interface, CO prefers to bind to a Cu site over a geometrically equivalent Zn site and can easily diffuse to a neighbouring Cu or even onto the ZrO2 surface with no energy penalty. CO converts to HCO and H2CO at a low energy cost. From this point, the reactions proceed similarly to the formate pathway. Again, the nearly non-existent barrier from H3CO to methanol would suggest that the methoxy intermediate is dominant over H2COH. In the case of the RWGS pathway, we often see the Zn-dilute interface with the most stable intermediates. The largest difference occurs for the COOH species with 0.27 eV energy difference between the Zn-dilute and Zn-rich interface. A possible explanation for these small trends between different Zn concentrations could lie in the increased mobility of Zn atoms going from Zn-dilute to the full-Zn interface.
The CuZn interfaces bind several intermediates stronger than the Cu interface does, including CO2, COOH, HCO, HCOH, H2CO, and H2COH. The commonality between these intermediates is that they are bound at the interface, connected to the metal component via a Zn–C bond, and most of them are found along the RWGS pathway. Zn centers bind these adsorbates, on average, 0.4 eV stronger than geometrically equivalent adsorption sites at the Cu interface. The important exception to this is carbon monoxide, CO, which is adsorbed 0.1 to 0.3 eV stronger to Cu sites. We also note that a hydrogen placed in the immediate vicinity of the Zn center weakens the adsorption of all aforementioned intermediates and brings the adsorption energies roughly 0.3 eV closer to those at the Cu interface. Intermediates that preferably bind to the support are largely unaffected by the presence of Zn and its concentration.
3.5 Energetic span analysis
The results given by the energetic span model allow us to compare the competing formate and RWGS mechanisms. We note that the predicted turnover frequencies are unlikely directly comparable to experimental numbers.78,79,81 They are, however, representative of the relative kinetics of the two pathways. The analysis was done separately for the Zn-dilute and Zn-rich interfaces. The relative energies in Table S2 were used as inputs and the model was run at 500 K. To obtain reliable results and to avoid problems with coverage effects, we limit the ESM analysis to competing pathways with the same number of atoms. Therefore the model can not be used to assess if the presence of OH groups speeds up latter reaction steps and they are left out of the following discussion. The endothermicity of the CO production and release also prevents its examination using the computational code of Garay-Ruiz and Bo81 as an endothermic (endergonic) cycle would lead to a negative TOF.78,79 Nevertheless, CO is observed in most real-world CTM systems.3,19 The production of CO is likely controlled, to some extent, by its endothermic nature and the fact that typical reaction conditions include high pressures.In the terminology of the ESM, the formate and RWGS pathways are competing catalytic cycles. Both pathways are combined and presented as a network graph in Fig. 9a), labelling each intermediate state as a node and each transition state as a line. Starting from the left by adsorption of CO2, the network is simplified and categorised by ignoring side branches or processes such as CO desorption. The four cycles given in Fig. 9b) feature the mechanisms with the highest turnover frequencies. The analogous Fig. S14 in the ESI† represents the Zn-rich interface. Cycles 1 and 2 follow the RWGS pathway, while Cycles 3 and 4 progress along the formate route. Furthermore, cycles 1 and 3 progress via the H2COH intermediate while cycles 2 and 4 go through the formation of H3CO instead.
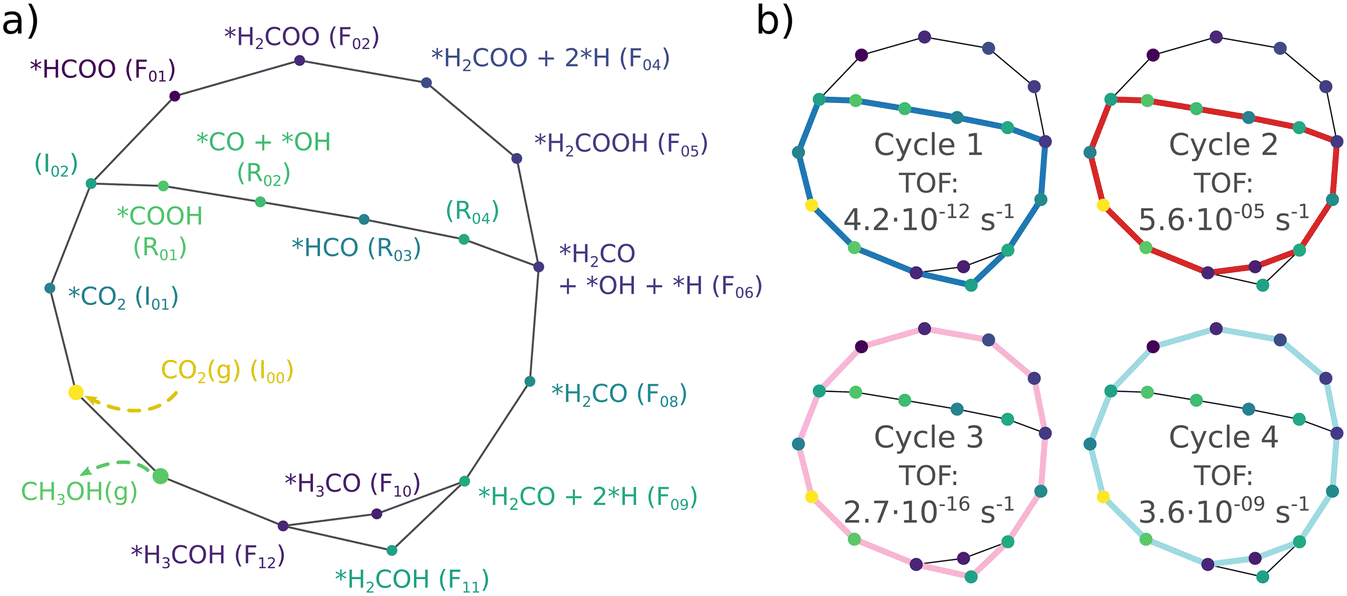 | ||
| Fig. 9 a) The simplified network used in the energetic span analysis. b) The catalytic cycles in the simplified network with their corresponding turnover frequencies on the Zn-dilute interface. | ||
Cycle 2, i.e. the RWGS route with methanol formed from H3CO, yields the highest TOF of the four cycles. Accordingly, the energetic span is lowest, being 1.89 eV at the Zn-dilute interface and 1.81 eV at the Zn-rich. The corresponding calculated turnover frequencies are 5.6 × 10−5 s−1 and 2.9 × 10−4 s−1, respectively, showing a relatively high degree of sensitivity to the change in δE. The next highest TOF belongs to cycle 4 representing the formate/H3CO pathway where the effective energetic span is 0.48 eV higher. Cycles 1 and 3 which go through H2COH have to pass a very high transition state, which becomes TOF-determining, and thus have significantly higher energetic spans and lower turnover frequencies.
A degree of turnover frequency control XTOF analysis (detailed in the ESI† section 3) confirms that the formate acts as a thermodynamic sink. The XTOF for HCOO is practically 1 in all unique cycles of the network (Fig. 9), indicating a large degree of turnover frequency control. Near the calculated formate energy, its effect on the TOF of cycle 2 (eqn (8)) is 1.2 × 10−3 s−1 eV−1. Note that, as described earlier, the presence of Zn sites at the interface does not affect the stability of the formate. Assigning the TOF-limiting transition states depends on the cycle. The transition state from H2COH to CH3OH (TS10), present only in cycles 1 and 3, has the highest degree of TOF control owing to its significantly high activation energy. In cycles 2 and 4 the transition state from H2CO to H3CO (TS07) takes dominance. In cycle 4, corresponding to a formate mechanism, the TS01 from CO2 to HCOO also has some TOF-controlling character with a  of 0.32. The influence of TS10 on cycle 1 (eqn (7)) is −9.0 × 10−11 s−1 eV−1 and the influence of TS07 on cycle 2 is −1.2 × 10−3 s−1 eV−1 near the calculated energies. Interestingly, all these elementary steps take place at the interface. Based on the energetic span analysis, we can conclude that the RWGS pathway is more favourable compared to the formate pathway, and that the last steps clearly involve the methoxy intermediate.
of 0.32. The influence of TS10 on cycle 1 (eqn (7)) is −9.0 × 10−11 s−1 eV−1 and the influence of TS07 on cycle 2 is −1.2 × 10−3 s−1 eV−1 near the calculated energies. Interestingly, all these elementary steps take place at the interface. Based on the energetic span analysis, we can conclude that the RWGS pathway is more favourable compared to the formate pathway, and that the last steps clearly involve the methoxy intermediate.
4 Conclusions
We have employed DFT calculations to obtain a thermodynamic and kinetic view of CO2 hydrogenation to methanol using a Cu/Zn/ZrO2 catalyst. To this end, we constructed a mixed CuZn/ZrO2 interface model using Cu nanorods with a varying Zn concentration at the interface. Our results show that intermediates binding to metal atoms at the interface adsorb stronger to Zn sites than to geometrically equivalent Cu sites. Enhanced binding is visible for all the studied Zn configurations and most pronounced on the interface where the Zn solute is the most dispersed. CO is the one exception to this trend as it preferentially binds to Cu sites. The addition of Zn at the interface has minimal effect on intermediates that do not bind to the metallic component, including some key intermediates such as the formate and methoxy species. Comparison between the four considered interface models shows that the reaction steps are not very sensitive to the concentration and arrangement of Zn at the interface. Zinc centers are active regardless of the identity of their surrounding atoms.A graph-based energetic span analysis can provide estimates for turnover frequencies to compare the competing mechanisms. Analysing the catalytic cycle as a whole allows us to identify the TOF-determining intermediates and transition states. The results support the RWGS route being the main CTM pathway. This can be explained by smaller reaction barriers along the RWGS mechanisms. Considering hydrogen spillover from the metal component to the support oxide is necessary as it facilitates the protonation of the surface-bound oxygen atoms of the intermediates. By including spillover, the barriers for the formation of several intermediates, notably carboxyl (COOH), become much lower than previously reported making the RWGS route more accessible.
Incorporating a Zn promoter into the interface structure selectively stabilises some intermediates, highlighting the importance of the effort to identify key intermediates and transition states. As Zn binds H less strongly, the role of the Cu is in hydrogen dissociation, storage, and spillover as well as enhancing CO binding. An increased understanding of promoter–adsorbate interactions allows tailoring of catalyst properties to influence adsorbate binding, which is important for selectivity and yield.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The work was funded by Academy of Finland (project 329977). The electronic structure calculations were made possible by computational resources provided by the CSC – IT Center for Science, Espoo, Finland (https://www.csc.fi/en/) and computer capacity from the Finnish Grid and Cloud Infrastructure (urn:nbn:fi:research-infras-2016072533). Dr Minttu Kauppinen is acknowledged for carefully reading the manuscript.References
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- K. Li and J. G. Chen, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS.
- S. Roy, A. Cherevotan and S. C. Peter, ACS Energy Lett., 2018, 3, 1938–1966 CrossRef CAS.
- B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y. G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- S. Zander, E. L. Kunkes, M. E. Schuster, J. Schumann, G. Weinberg, D. Teschner, N. Jacobsen, R. Schlögl and M. Behrens, Angew. Chem., Int. Ed., 2013, 52, 6536–6540 CrossRef CAS PubMed.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed.
- J. D. Grunwaldt, A. M. Molenbroek, N. Y. Topsøe, H. Topsøe and B. S. Clausen, J. Catal., 2000, 194, 452–460 CrossRef CAS.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nat. Catal., 2022, 5, 818–831 CrossRef CAS.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- A. Bavykina, I. Yarulina, A. J. Al Abdulghani, L. Gevers, M. N. Hedhili, X. Miao, A. R. Galilea, A. Pustovarenko, A. Dikhtiarenko, A. Cadiau, A. Aguilar-Tapia, J. L. Hazemann, S. M. Kozlov, S. Oud-Chikh, L. Cavallo and J. Gascon, ACS Catal., 2019, 9, 6910–6918 CrossRef CAS.
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2016, 55, 7968–7973 CrossRef CAS PubMed.
- M. R. Gogate and R. J. Davis, Catal. Commun., 2010, 11, 901–906 CrossRef CAS.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS PubMed.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Nat. Commun., 2019, 10, 1166 CrossRef PubMed.
- M. Zabilskiy, V. L. Sushkevich, D. Palagin, M. A. Newton, F. Krumeich and J. A. van Bokhoven, Nat. Commun., 2020, 11, 2409 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- N. Scotti, F. Bossola, F. Zaccheria and N. Ravasio, Catalysts, 2020, 10, 168 CrossRef CAS.
- K. Larmier, W.-C. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS PubMed.
- Q.-L. Tang, Q.-J. Hong and Z.-P. Liu, J. Catal., 2009, 263, 114–122 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- T. Witoon, J. Chalorngtham, P. Dumrongbunditkul, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2016, 293, 327–336 CrossRef CAS.
- Y. Nitta, O. Suwata, Y. Ikeda, Y. Okamoto and T. Imanaka, Catal. Lett., 1994, 26, 345–354 CrossRef CAS.
- D. Gasser and A. Baiker, Appl. Catal., 1989, 48, 279–294 CrossRef CAS.
- J. Weigel, R. A. Koeppel, A. Baiker and A. Wokaun, Langmuir, 1996, 12, 5319–5329 CrossRef CAS.
- I. Abbas, H. Kim, C. H. Shin, S. Yoon and K. D. Jung, Appl. Catal., B, 2019, 258, 117971 CrossRef CAS.
- A. Arandia, J. Yim, H. Warraich, E. Leppäkangas, R. Bes, A. Lempelto, L. Gell, H. Jiang, K. Meinander, T. Viinikainen, S. Huotari, K. Honkala and R. L. Puurunen, Appl. Catal., B, 2023, 321, 122046 CrossRef CAS.
- J. Sehested, J. Catal., 2019, 371, 368–375 CrossRef CAS.
- J. Nakamura, I. Nakamura, T. Uchijima, T. Watanabe and T. Fujitani, Stud. Surf. Sci. Catal., 1996, 101 B, 1389–1399 CrossRef.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 357, eaan8210 CrossRef PubMed.
- J. Nakamura, Y. Choi and T. Fujitani, Top. Catal., 2003, 22, 277–285 CrossRef CAS.
- J. Nakamura, T. Fujitani, S. Kuld, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2017, 357, eaan8074 CrossRef PubMed.
- N. Y. Topsøe and H. Topsøe, Top. Catal., 1999, 8, 267–270 CrossRef.
- P. Amann, B. Klötzer, D. Degerman, N. Köpfle, T. Götsch, P. Lömker, C. Rameshan, K. Ploner, D. Bikaljevic, H.-Y. Wang, M. Soldemo, M. Shipilin, C. M. Goodwin, J. Gladh, J. H. Stenlid, M. Börner, C. Schlueter and A. Nilsson, Science, 2022, 376, 603–608 CrossRef CAS PubMed.
- E. Frei, A. Gaur, H. Lichtenberg, L. Zwiener, M. Scherzer, F. Girgsdies, T. Lunkenbein and R. Schlögl, ChemCatChem, 2020, 12, 4029–4033 CrossRef CAS.
- D. Kopač, B. Likozar and M. Huš, Appl. Surf. Sci., 2019, 497, 143783 CrossRef.
- S. Polierer, J. Jelic, S. Pitter and F. Studt, J. Phys. Chem. C, 2019, 123, 26904–26911 CrossRef CAS.
- A. Cao, Z. Wang, H. Li, A. O. Elnabawy and J. K. Nørskov, J. Catal., 2021, 400, 325–331 CrossRef CAS.
- M. Kauppinen, A. Posada-Borbón and H. Grönbeck, J. Phys. Chem. C, 2022, 126, 15235–15246 CrossRef CAS.
- L. Gell, A. Lempelto, T. Kiljunen and K. Honkala, J. Chem. Phys., 2021, 154, 214707 CrossRef CAS PubMed.
- M. M. Kauppinen, M. M. Melander, A. S. Bazhenov and K. Honkala, ACS Catal., 2018, 8, 11633–11647 CrossRef CAS.
- B. C. Dharmalingam, A. V. Koushik, M. Mureddu, L. Atzori, S. Lai, A. Pettinau, N. S. Kaisare, P. Aghalayam and J. J. Varghese, Appl. Catal., B, 2023, 332, 122743 CrossRef CAS.
- X. Liu, J. Luo, H. Wang, L. Huang, S. Wang, S. Li, Z. Sun, F. Sun, Z. Jiang, S. Wei, W. Li and J. Lu, Angew. Chem., 2022, 61, e202202330 CAS.
- E. L. Fornero, A. L. Bonivardi and M. A. Baltanás, J. Catal., 2015, 330, 302–310 CrossRef CAS.
- J. M. Campbell and C. T. Campbell, Surf. Sci., 1991, 259, 1–17 CrossRef CAS.
- S. Sakong and A. Groß, Surf. Sci., 2003, 525, 107–118 CrossRef CAS.
- K. D. Jung and A. T. Bell, J. Catal., 2000, 193, 207–223 CrossRef CAS.
- M. D. Rhodes, K. A. Pokrovski and A. T. Bell, J. Catal., 2005, 233, 210–220 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- H. Y. T. Chen, S. Tosoni and G. Pacchioni, ACS Catal., 2015, 5, 5486–5495 CrossRef CAS.
- M. M. Bettahar, Catal. Rev.: Sci. Eng., 2022, 64, 87–125 CrossRef CAS.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- V. Korpelin, M. M. Melander and K. Honkala, J. Phys. Chem. C, 2022, 126, 933–945 CrossRef CAS.
- A. R. Puigdollers, F. Illas and G. Pacchioni, J. Phys. Chem. C, 2016, 120, 17604–17612 CrossRef.
- J.-H. Lan, L. Wang, S. Li, L.-Y. Yuan, Y.-X. Feng, W. Sun, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, J. Appl. Phys., 2013, 113, 183514 CrossRef.
- H. Koga, A. Hayashi, Y. Ato, K. Tada, S. Hosokawa, T. Tanaka and M. Okumura, Appl. Surf. Sci., 2020, 508, 145252 CrossRef CAS.
- O. A. Syzgantseva, M. Calatayud and C. Minot, J. Phys. Chem. C, 2012, 116, 6636–6644 CrossRef CAS.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Phys. Rev. Lett., 2006, 97, 170201 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- T. Frederiksen, M. Paulsson, M. Brandbyge and A.-P. Jauho, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 205413 CrossRef.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- M. Rühle, Adv. Mater., 1997, 9, 195–217 CrossRef.
- K. T. Jung and A. T. Bell, Catal. Lett., 2002, 80, 63–68 CrossRef CAS.
- J. S. Faulkner, Y. Wang and G. M. Stocks, Phys. Rev. Lett., 1998, 81, 1905–1908 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- S. Kozuch, ACS Catal., 2015, 5, 5242–5255 CrossRef CAS.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- D. Garay-Ruiz and C. Bo, ACS Catal., 2020, 10, 12627–12635 CrossRef CAS.
- H.-J. Chun, V. Apaja, A. Clayborne, K. Honkala and J. Greeley, ACS Catal., 2017, 7, 3869–3882 CrossRef CAS.
- D. Garay-Ruiz, gTOFfee, https://gitlab.com/dgarayr/gtoffee, (Feb. 2022) Search PubMed.
- A. Posada-Borbón, B. Hagman, A. Schaefer, C. Zhang, M. Shipilin, A. Hellman, J. Gustafson and H. Grönbeck, Surf. Sci., 2018, 675, 64–69 CrossRef.
- B. Hagman, A. Posada-Borbón, A. Schaefer, M. Shipilin, C. Zhang, L. R. Merte, A. Hellman, E. Lundgren, H. Grönbeck and J. Gustafson, J. Am. Chem. Soc., 2018, 140, 12974–12979 CrossRef CAS PubMed.
- B. Eren, R. S. Weatherup, N. Liakakos, G. A. Somorjai and M. Salmeron, J. Am. Chem. Soc., 2016, 138, 8207–8211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See ESI for the structures and energies of all adsorbed intermediates, potential energy diagrams, and details of the ESM analysis. See DOI: https://doi.org/10.1039/d3cy00549f |
| This journal is © The Royal Society of Chemistry 2023 |