
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the performance of Cu catalysts for the reverse water–gas shift reaction using N-doped CNT–ZnO composite as support†
Ana Rita
Querido
 ab,
Liliana P. L.
Gonçalves
abc,
Yury V.
Kolen'ko
c,
M. Fernando R.
Pereira
ab and
O. Salomé G. P.
Soares
*ab
ab,
Liliana P. L.
Gonçalves
abc,
Yury V.
Kolen'ko
c,
M. Fernando R.
Pereira
ab and
O. Salomé G. P.
Soares
*ab
aLSRE-LCM – Laboratory of Separation and Reaction Engineering, Laboratory of Catalysis and Materials, Faculty of Engineering, University of Porto, Rua Dr. Roberto Frias, 4200-465, Porto, Portugal. E-mail: salome.soares@fe.up.pt
bALiCE – Associate Laboratory in Chemical Engineering, Faculty of Engineering, University of Porto, Rua Dr. Roberto Frias, 4200-465, Porto, Portugal
cInternational Iberian Nanotechnology Laboratory, Avenida Mestre José Veiga s/n, 4715-330 Braga, Portugal
First published on 17th May 2023
Abstract
The reverse water–gas shift reaction (RWGS) allows the conversion of CO2 to CO which, mixed with H2, forms syngas, the feedstock of most chemicals and synthetic fuels production. Consequently, it is crucial to develop efficient catalysts for this reaction. To further the development of RWGS catalysts, Cu-based catalysts supported on pristine CNTs and on composites of pristine and functionalized CNTs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZnO were prepared. ZnO's presence in the catalyst's structure proved to be beneficial, as the CO2 conversion and CO yield reached 49.0% whereas the catalysts supported on pristine CNTs only achieved a CO2 conversion and CO yield of 17.6%, at a temperature of 600 °C. The N-doping of CNTs further improved the CO2 conversion and CO yield to 54.8%, remaining stable at least for 93 h.
:ZnO were prepared. ZnO's presence in the catalyst's structure proved to be beneficial, as the CO2 conversion and CO yield reached 49.0% whereas the catalysts supported on pristine CNTs only achieved a CO2 conversion and CO yield of 17.6%, at a temperature of 600 °C. The N-doping of CNTs further improved the CO2 conversion and CO yield to 54.8%, remaining stable at least for 93 h.
Introduction
The reverse water–gas shift (RWGS) reaction has drawn interest from researchers as a possible route to reduce harmful CO2 emissions. This reaction enables the conversion of CO2 to CO, a gas used as an intermediary in CO2 hydrogenation reactions. Since CO is more reactive than CO2, processes such as methanol synthesis (CAMERE process), Fischer–Tropsch synthesis or Cativa acetic acid synthesis, still use syngas (mixture of CO and H2) as feedstock. Therefore, the RWGS reaction can be used as an intermediate step in the production of many chemicals.1,2 Another possible application of this reaction is its use to couple CO2 with alkylene oxide to form chemicals, such as ethylene glycol or styrene. With the RWGS reaction, it is possible to overturn the thermodynamic equilibrium constraints observed in the direct thermal cracking process.2 This reaction can help the enhancement of the carbon recycling system.3The RWGS reaction (eqn (1)) is endothermic, and consequently, thermodynamically favourable at higher temperatures (ΔH25°C = +41.1 kJ mol−1) while not being pressure dependent. However, CO2 methanation (eqn (2)) may occur under the most favourable RWGS conditions due to excessive hydrogenation at ambient pressure. Methanation is an exothermic (ΔH25°C = −165.0 kJ mol−1) and pressure dependent reaction.2,4
| CO2 + H2 ↔ CO + H2O ΔH25°C = +41.1 kJ mol−1 | (1) |
| CO2 + 4H2 ↔ CH4 + 2H2O ΔH25°C = −165.0 kJ mol−1 | (2) |
For the active phase of the catalysts, two different possibilities have been studied: platinum group metals and non-platinum group metals. The active phase of the catalyst can affect the catalytic results, as the electron properties of d-orbital holes of the metal NPs influences the adsorption of the reactants.6
The platinum group metals have an incomplete d-orbital, which allows better adsorption of reactants, and an excellent resistance to oxidation and corrosion that converts them into an attractive option for the active phase of RWGS reaction catalysts. Since they have a high capacity to dissociate H2, and adsorb the reaction intermediates, noble metals like palladium (Pd) and rhodium (Rd) have been put to the test.4 Nevertheless, these types of metals have a higher cost than most metals, and more importantly, their reserves are limited, which is a disadvantage.4,6
For the non-platinum group metals, copper (Cu), nickel (Ni) and iron (Fe) have also been studied as active phases for the RWGS reaction.7 Cu-based catalysts are in the lead because they present excellent CO2 conversion and CO selectivity and are less expensive than the platinum group metals.3,6 Catalysts with Cu NPs have also demonstrated an extremely low selectivity to methane, and they have been able to present CO2 conversion at relatively low temperatures (165 °C). One of the main disadvantages of Cu-based catalysts is the need of a higher H2/CO2 ratio, as this type of catalyst is not capable of dissociating CO2 without H2.5 A high dispersion of the Cu metal NPs over the support is indispensable for a high catalytic performance, due to the increase of active sites.8
A low RWGS reaction temperature is fundamental to reduce the production costs of the syngas. This stimulates the search for RWGS catalysts that present lower selectivity to the unwanted methane while offering a better overall performance towards syngas. Nevertheless, the reaction temperatures used in the catalytic experiments are still high, and therefore, the catalysts for the RWGS reaction should be able to sustain the high temperatures needed to obtain high CO2 conversions.4 Cu-based catalysts might suffer deactivation by sintering or reoxidation.3,7 It is possible to revert this deactivation and increase the stability of the catalyst by adding a promoter, such as Fe, as Chen et al. demonstrated.9
Regarding the supporting material, nanostructured metal oxide supports are able to improve the metal NPs dispersion, thus decreasing the probability of deactivation of the catalyst by sintering; however, for this, carbon materials might be a more interesting material as they present generally higher specific surface areas than metal oxides.10–12 Additionally, metal oxide supports are also able to improve the CO2 activation on the surface of the catalyst, as the metal NPs on the metal oxide support interface are more easily reduced.2 Therefore, the support is also expected to influence the CO2 conversion and selectivity to CO of the catalyst.5,10 Nevertheless, excessive CO2 hydrogenation may also lead to the formation of methane. The most used metal oxide supports are ZnO, Al2O3, CeO2, SiO2 and ZrO2.2,6,10,13
Interestingly, carbon materials have emerged as possible supports for catalysts for the RWGS reaction. Amorphous carbon materials, such as activated carbon (AC), have been used to prepare catalysts for diverse reactions, and nowadays, other relatively new carbon materials, such as carbon nanotubes (CNTs), have also been studied.14,15 Carbon materials have a hydrophobic nature, resulting in high water stability and a great resistance to structure changes by acids, bases, or high temperatures (an important property for RWGS reaction catalysts). Furthermore, these materials also facilitate the reduction of the active phases. Another advantage that makes carbon materials attractive to the chemical industry is their reduced cost.16 Moreover, the carbon materials surface can be modified to improve their reactivity and, as a result, their catalytic performance. It is possible to add functional groups (O-groups) or even replace some carbon atoms with other heteroatoms (e.g., N, S, P and B).14,17,18
In our previous studies, it was demonstrated that the surface functionalization of carbon materials can improve CO2 conversion due to an increase in the active phase dispersion and CO2 adsorption capacity.19 In a methanation investigation, catalysts supported on N-doped AC improved CO2 conversion from 54% to 70% when compared to catalysts supported by pristine AC, whereas catalysts supported by oxidized AC improved CO2 conversion from 54% to 62%.19 Another methanation study revealed that Ni-based catalysts supported on CNTs provide an even better CO2 conversion of 78.5%, confirming the benefit of having CNTs as a catalyst support.20 An additional former work on the methanation reaction, consisted in evaluating if catalysts supported on carbon materials and metal oxide composites could further improve CO2 conversion. The catalyst supported on a composite of carbon–CeO2 achieved a higher CO2 conversion, at a lower reaction temperature, than the catalyst supported on a carbon material. In this work, it was concluded that compositing carbon materials with metal oxide can provide a high-performing catalyst while also allowing a decrease in the usage of expensive and rare metal oxides.21
In this work, the possibility of taking advantage of the carbon materials properties as support for Cu-based catalysts for the RWGS reaction was evaluated. A preliminary analysis demonstrated that the catalytic performance of Cu-based catalysts supported on CNTs outperformed those supported on AC. Thereafter, the addition of ZnO to the composition of the CNTs supporting material was assessed via preparation of CNTs:ZnO composite, followed by an evaluation of the influence of the functionalization of the surface of the CNTs in the performance of the catalysts.
Materials and methods
Materials
The following materials and reactants were used: activated carbon (AC) NORIT GAC 1240 (Cabot), carbon nanotubes (CNTs) (Nanocyl 3100, 90%), carborundum (SiC, VWR Chemicals, 34%), distilled water (H2O), ultrapure water obtained from a Milli-Q Advantage A10 equipment, melamine (C3H6N6, Sigma Aldrich, 99%), nitric acid (HNO3, Supelco, 65%), copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, Emsure, 99.5%), and zinc oxide (ZnO, VP AdNano, 34%).Functionalization of carbon supporting materials
To obtain O-doped CNTs (CNT–O), 4 g of commercial CNTs were placed in a round bottom flask with 0.300 dm3 of a 7 mol dm−3 solution of HNO3. The mixture was heated until its boiling point and refluxed for 180 min. Afterwards, the mixture was washed with distilled water until it reached neutral pH, and the resultant product was dried overnight at 100 °C.To obtain N-doped CNTs (CNT–N), 0.60 g of commercial CNTs and 0.39 g of melamine were co-ball-milled for 4 h at a frequency of 15 vibrations per second. Then, the mixture obtained was annealed at 600 °C for 1 h, under a N2 flow of 100 cm3 min−1.
Preparation of composite supporting materials
A composite of CNTs and ZnO, with a proportion of 90:10 (wt%), in which 90 wt% regards to CNTs and 10 wt% to ZnO, was prepared. To obtain the composite, the materials were co-ball-milled (Retsch MM 200) at a frequency of 20 vibrations per second for 30 min.
Catalysts preparation
The catalysts were prepared by incipient wetness impregnation (IWI). To prepare the catalysts, 0.50 g of the supporting material (AC, CNT, CNT–O, CNT–N, ZnO, CNT–ZnO, CNT–O–ZnO, or CNT–N–ZnO) was placed under ultrasonic vibration, and an aqueous solution of Cu(NO3)2·3H2O was added, to achieve the intended amount of metal content, 15 wt% of Cu. The samples were left under ultrasonic vibration for 90 min and then dried at 100 °C overnight.After determining the reduction temperature of each catalyst, using H2 temperature programmed reduction (H2-TPR), the catalysts were reduced. The catalysts were placed in a quartz reactor and heated, at a rate of 10 °C min−1 under a N2 flowrate of 100 cm3 min−1, until the desired reduction temperature. After 1 h at the reduction temperature, the gas was changed to H2 (under the same flowrate), and the catalysts were reduced for 3 h.
Characterization
The resultant catalysts and their supports were characterized by N2 physisorption at −196 °C, elemental analysis (EA), temperature programmed desorption (TPD), hydrogen temperature programmed reduction (H2-TPR), powder X-ray diffraction analysis (XRD), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). Additional information can be found in the ESI† material.Catalytic experiments
The catalytic experiments for the reverse water–gas shift reaction were carried out in a Microactivity XS15 reactor (PID Eng & Tech), in a fixed bed quartz reactor. The resulting gaseous products were analysed using a GC 1000 gas chromatograph (DANI) equipped with a thermal conductivity detector (TCD) and a GS-CarbonPLOT capillary column. He was used as carrier gas while N2 was used as internal standard.For the experiments, 100 mg of the catalyst was mixed with SiC and placed into a fixed bed quartz reactor with internal diameter of 1 cm. An in situ reduction was performed for every catalyst with a 40 cm3 min−1 flowrate of H2 for 30 min, at 600 °C and 1 bar. Then, after a decrease of the reactor's temperature to 100 °C under a 50 cm3 min−1 flowrate of He, the reactor was fed with 10 cm3 min−1 of CO2, 40 cm3 min−1 of H2, and 50 cm3 min−1 of He, at 1 bar (GHSV = 60000 cm3 g−1 h−1). A temperature ramping from 100 °C to 600 °C at 5 °C min−1 was performed to assess each catalyst's performance at different temperatures and at 1 bar.
To study the stability of the best-performing catalyst, a catalytic experiment was conducted for an extended time-on-stream (TOS) at a CO2 conversion below thermodynamic equilibrium.
CO2 conversion (XCO2) was determined by eqn (3), while selectivity to CO (SCO), selectivity to CH4 (SCH4) and CO yield (YCO) were calculated by eqn (4)–(6), respectively.
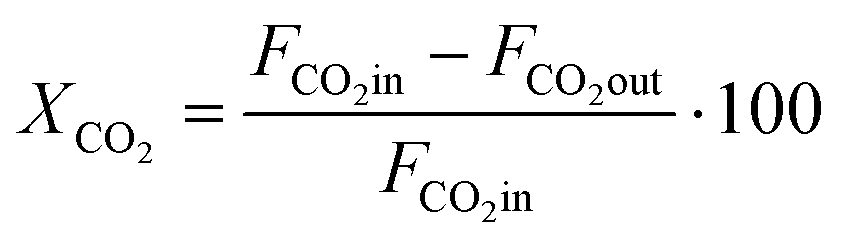 | (3) |
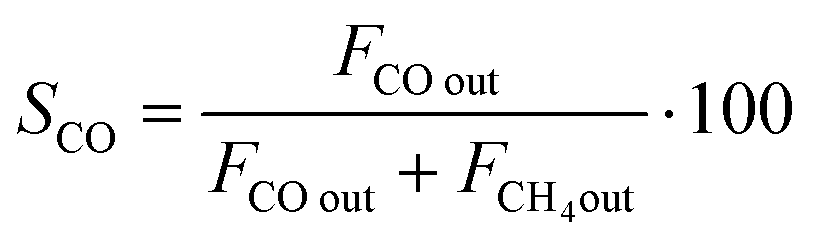 | (4) |
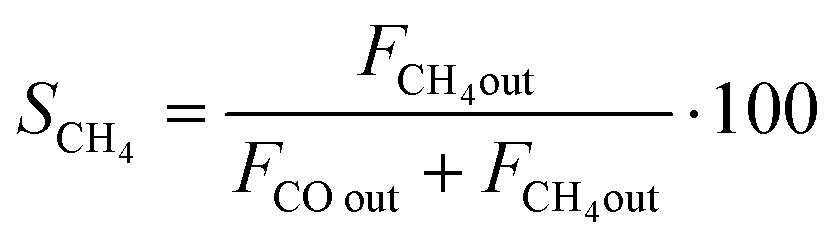 | (5) |
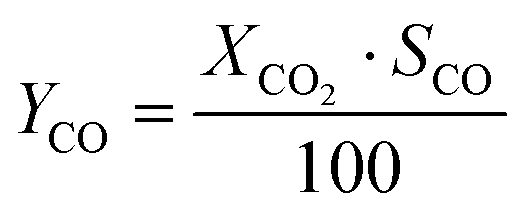 | (6) |
Results and discussion
Catalytic experiments
Firstly, preliminary catalytic experiments were conducted to find out which carbon supporting material, AC or CNT, could provide better catalytic properties in the RWGS reaction (Fig. S1†). It was established that the Cu-based catalyst supported on CNT, 15 wt%, demonstrates higher conversion (XCO2 = 17.6% at T = 600 °C) than the Cu-based catalyst supported on AC, also 15 wt%, (XCO2 = 10.4% at T = 600 °C). Notably, both catalysts display a complete selectivity to carbon monoxide (SCO = 100%).To evaluate if adding ZnO to CNT supported catalyst system could improve its catalytic properties, a Cu-based catalyst supported on a composite CNT–ZnO material (proportion of 90:10 wt%) was prepared through a co-ball milling. Fig. 1 shows the catalytic results obtained for the prepared catalysts on composite support. Notably, the catalyst Cu/CNT–ZnO demonstrates a better catalytic properties than either Cu/CNT or Cu/ZnO reference catalysts,22 reaching XCO2 = 49.0%. At the same time, this catalyst exhibits a small production of methane by-product in the 525–575 °C temperature range.
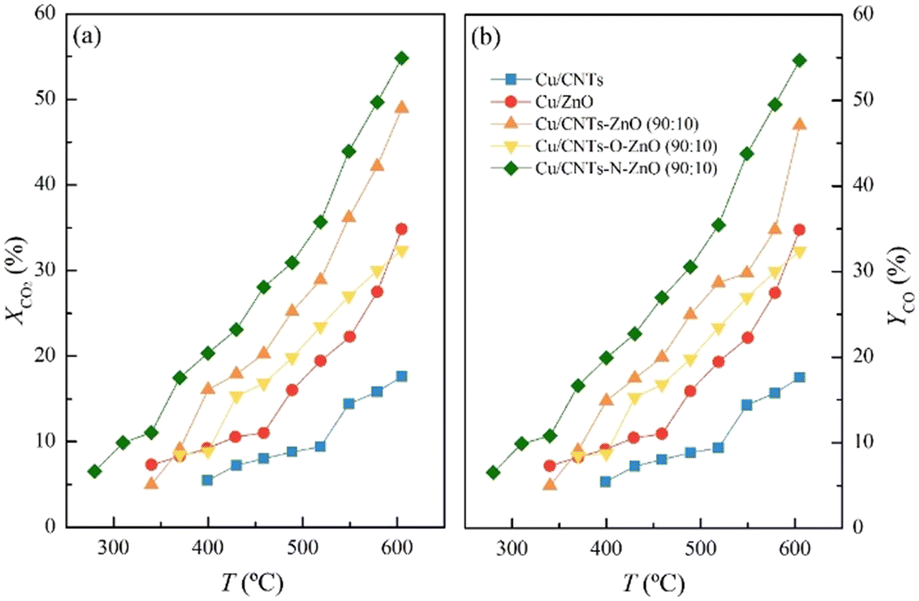 | ||
| Fig. 1 Comparison of the XCO2 (a) and YCO (b) as a function of the RWGS reaction temperature for Cu-based catalysts supported on different composite materials. Reaction conditions: P = 1 bar; GHSV = 60000 cm3 g−1 h−1; CO2:H2 (V:V) = 1:4. | ||
Considering that Cu catalyst supported on the composite CNT–ZnO material achieves a better CO2 conversion and CO yield than the reference catalysts, new catalysts, supported on functionalized CNT composites were prepared. Two different functionalization treatments were performed on CNT: N-doping (CNT–N) and O-doping (CNT–O).
Fig. 1 shows the catalytic results obtained for the Cu catalysts supported on functionalized CNT and ZnO composite materials. Remarkably, the catalyst supported on CNT–N–ZnO surpasses the performance of Cu/CNT–ZnO, achieving a XCO2 = 54.8% at 600 °C. For this catalyst, a small amount of CH4 by-product production between 340 and 600 °C (<1%) was recorded. On the other hand, the Cu/CNT–O–ZnO catalyst, with O-doped CNTs, achieves a lower CO2 conversion than that of Cu/CNT–ZnO with a XCO2 = 32.4% at 600 °C and CO selectivity equal to 100%. The catalytic results of CO2 conversion for all these catalysts have an associated error of 3%, and for CO selectivity an associated error of 1%.
Finally, the stability over long time-on-stream (TOS) of the best-performing catalyst, namely Cu/CNT–N–ZnO, was evaluated (Fig. 2). The sample remains stable for at least 93 h under the studied reaction conditions, exhibiting high CO2 conversion and complete CO selectivity (SCO = 100%) over this time span.
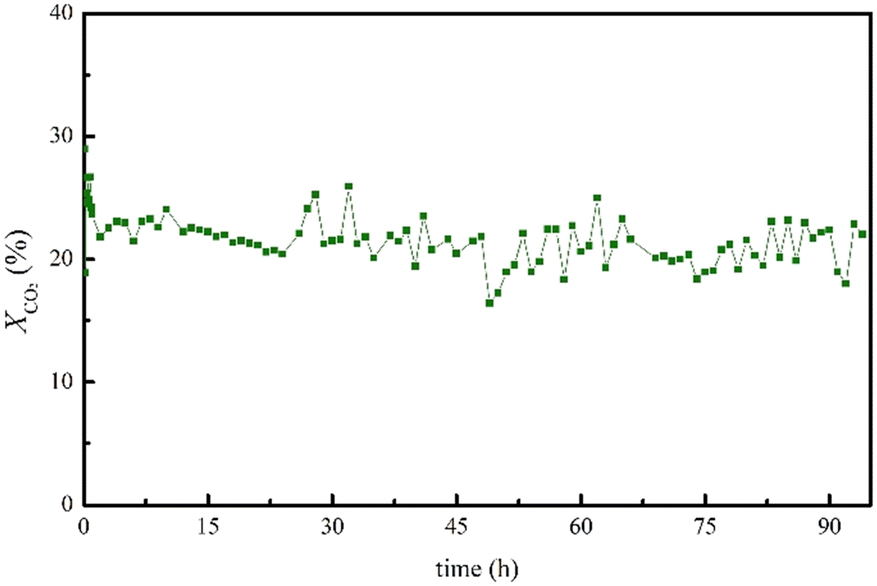 | ||
| Fig. 2 TOS results in terms of XCO2 and YCO over 93 h for the best-performing Cu/CNT–N–ZnO catalyst. Reaction conditions: T = 500 °C; P = 1 bar; GHSV = 60000 cm3 g−1 h−1; CO2:H2 (V:V) = 1:4. | ||
Characterization
Throughout the catalytic experiments, it was possible to optimize the preparation of the catalysts for the RWGS reaction. Therefore, an extensive characterization of the prepared materials was performed to correlate them with the catalytic performance.Regarding the textural properties of the materials, the specific surface area (SBET), total pore volume (VP,P/P0 = 0.95) and micropore volume (Vmicro) of the synthesized catalysts and the respective supporting materials were determined through their N2 adsorption–desorption isotherms at −196 °C (Fig. S2–S4†) and the obtained values are summarized in Table S1.†
CNT materials isotherms' are classified as type II isotherms, and CNTs materials display a type H3 hysteresis, according to the IUPAC classification.23 ZnO exhibits behaviour comparable to that of CNTs, with a type II isotherm. In this instance, an H3 hysteresis may also be seen, although its loop is not closed.23 The catalysts developed maintain the isotherm type defined for the support itself, as the addition of the active phase to the support does not alter its nature. Relatively to the CNTs' isotherms, the large increase shown in N2 adsorption when P/P0 equals 0.9, points to a mostly mesoporous character for these materials. The observed hysteresis in the isotherms may be explained by empty spaces in the CNTs' entanglements that result in a mesoporous nature.20
For the functionalized CNT materials, different outcomes occurred over the value of SBET, depending on the treatment done on the support. The oxidation of CNTs resulted in an increase of SBET and VP,P/P0 = 0.95, which can be explained by the used oxidative treatment, which can open some of the CNT tips and create sidewall defects, increasing the adsorption area of the material.24 The N-doped CNTs, although submitted to a ball-milling treatment that decreases the entanglement of this material, displayed a slightly smaller SBET compared to pristine CNTs, which could have been caused by a blockage of some of the pores of the material by the N-groups that were added. As expected, the Vmicro was null for all CNTs samples, confirming the mesoporous nature of CNTs.
Regarding the CNTs–ZnO (90:10) composite, this material presents a higher SBET than pristine CNTs, which can be explained by the decrease in the CNTs' entanglement due to the ball-milling treatment used to prepare the composite.
It is possible to conclude that each catalyst had a lower SBET, Vmicro, and VP,P/P0 = 0.95 values than the respective supports alone, because the active phase particles add weight to the sample without a considerable increase in surface area.
Through elemental analysis (Table S2†), it was confirmed that CNTs' oxidation was successful, as there was an increase of over 3.1% of O compared to pristine CNTs. The introduction of N-groups to pristine CNTs was effective, with a 7.2% increase in N-content for CNTs–N.
Temperature programmed desorption (TPD) (Table S3†) allowed the evaluation of the amount of surface oxygenated groups present on the pristine carbon materials, CNTs, and their oxidized counterparts, CNTs–O. The O-groups present on the material's surface, decompose into CO and CO2 at different temperatures. Pristine CNTs (Fig. S6†) emits a small amount of CO2 (Table S3†); therefore, it was concluded that this material presents small quantities of acid carboxylic and lactone groups on its surface. Pristine CNTs also releases a small amount of CO (Table S3†), in a similar temperature range, hence it was concluded that CNTs materials contain phenol, carbonyl, ether and quinone groups.25,26
The material CNTs–O (Fig. S7†) exhibits an increase in the emission of both CO2 and CO (Table S3†) when compared to pristine CNTs. This result was expected since it went through an oxidation treatment with HNO3 to increase the number of O-containing surface groups. It was concluded that this material presents all the groups mentioned before: carboxylic acid, anhydrides and lactone groups (CO2 release); anhydrides, phenol, carbonyl, ether and quinone groups (CO release). However, it should be emphasised once more that in the material CNTs–O, there is a larger amount of O-groups, particularly the carboxylic acids, quinones, and carbonyl groups, giving the substance an acidic nature as it was intended.25,26
H2 temperature programmed reduction (H2-TPR) was used to study the reducibility of the prepared catalysts, as well as to determine the reduction temperature of each catalyst. Comparing the catalysts supported on pristine CNTs and ZnO with the same amount of Cu (15 wt%) (Fig. S8†), it becomes clearer that ZnO, as a support, decreases the reducibility of the catalyst, with its H2 consumption beginning later, after 300 °C. The catalyst supported on CNTs is more easily reduced, presenting H2 consumption at 250 °C. Therefore, the supports can be arranged by their reducibility as: CNTs > ZnO. The reference catalyst, Cu/ZnO, presents two peaks for two different types of Cu species: a peak at higher temperatures that corresponds to Cu species with stronger interactions with the support, and a peak at lower temperatures corresponding to Cu species with weaker ones. Cu/CNTs only features a single peak at a lower temperature than both Cu/ZnO peaks, suggesting a weaker interaction from all Cu species with pristine CNTs, when compared to ZnO. It was therefore concluded that Cu/ZnO undergoes stronger metal–support interactions overall than Cu/CNTs and, consequently, features smaller particle sizes, which contributes to the better catalytic performance of Cu/ZnO.
Regarding the catalysts supported on CNTs:ZnO composites (Fig. S8†), both Cu/CNTs–ZnO (90:10) and Cu/CNTs–O–ZnO (90:10) catalysts show a single peak at a similar temperature as Cu/CNTs, which again suggests a weaker interaction from all Cu species with the supports CNTs–ZnO (90:10) and CNTs–O–ZnO (90:10). However, Cu/CNTs–N–ZnO (90:10) displays two peaks similarly to the reference catalyst: a peak at lower temperatures corresponding to Cu nanoparticles with weaker metal–support interaction and a peak at higher temperatures, which may stem from Cu nanoparticles with stronger interaction with the support or the interactions between H2 and the N-groups in the support. Therefore, H2-TPR analysis of the CNTs–N–ZnO (90:10) support (Fig. S9†) was conducted. Interestingly, the second peak of the H2-TPR profile of Cu/CNTs–N–ZnO (90:10) differs from the peak seen in the profile of the support, being of a slightly different shape and temperature. Therefore, this second peak at the higher temperature in the Cu/CNTs–N–ZnO (90:10) catalyst may stem from both the interaction of H2 with the N-groups and the presence of smaller Cu nanoparticles with stronger metal–support interaction. Both facts might be responsible for the higher activity of Cu/CNTs–N–ZnO when compared to the reference Cu/ZnO.
For the catalyst supported on pristine CNTs, the reduction temperature was determined as 250 °C, and for Cu/ZnO it was 300 °C. The catalysts supported on CNTs:ZnO composites were also reduced at 300 °C. These temperatures were selected considering the 180 min length of the reduction process. It is however important to note, that a further reduction pre-treatment at 600 °C was performed prior to the catalytic experiments, since the maximum temperature of these experiments was 600 °C.
X-rays diffraction (XRD) was used to identify the crystallites present in the catalysts prepared (Fig. S10 and S11†). For the Cu/CNTs catalyst and the catalysts supported on CNTs:ZnO composites, peaks were detected at 2θ = 43.2°, 2θ = 50.5°, and 2θ = 70.4°, for Cu(111), Cu(200) and Cu(220), respectively, in metallic Cu. For the catalysts Cu/CNTs, Cu/CNTs–ZnO (90:10) and Cu/CNTs–O–ZnO (90:10), CuO was also identified.27 Graphite, C, was present in all catalysts studied except Cu/ZnO, with a constant peak at 2θ = 26.6° that corresponds to C(002).28 In the catalysts with ZnO in their constitution, the crystallite form zincite was identified.29
Characterization after the stability test
X-ray photoelectron spectroscopy (XPS) was used to evaluate the chemical composition of the best performing catalyst (Cu/CNTs–N–ZnO (90:10)) before and after the long time on stream (TOS) stability experiment. Fig. 3 presents the high-resolution XPS data for the Zn 2p and Cu 2p regions for the CNTs–N–ZnO (90:10) supporting material and for the fresh and after TOS Cu/CNTs–N–ZnO (90:10) catalyst.
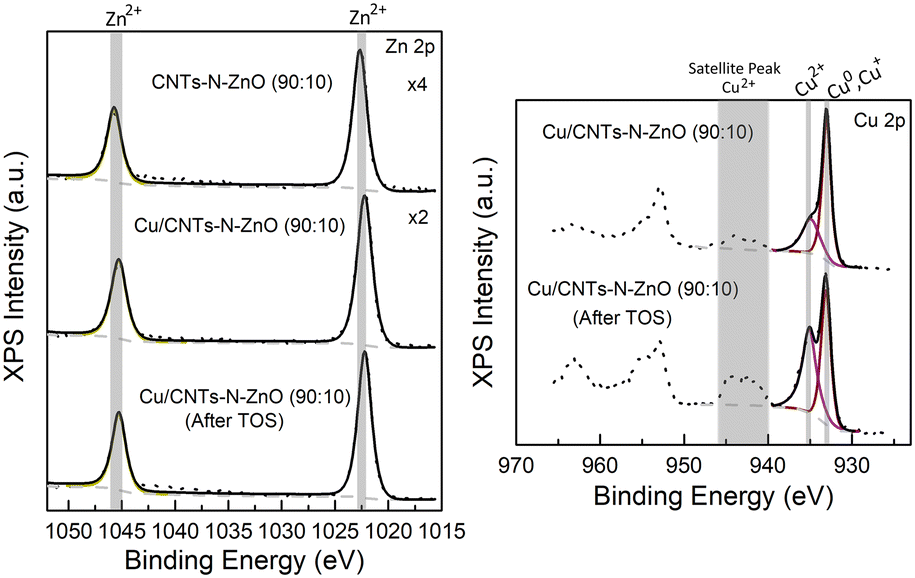 | ||
| Fig. 3 High-resolution XPS data for the Zn 2p and Cu 2p regions for the CNTs–N–ZnO (90:10) supporting material and for the fresh and after TOS Cu/CNTs–N–ZnO (90:10) catalyst. Symbols: raw data; black lines: overall fits; coloured lines: fits of individual components; dashed lines: background. | ||
The XPS spectra of Zn 2p of the support and the catalyst before and after TOS present two well defined peaks at 1022.4 ± 0.2 eV and 1045.4 ± 0.2 eV, which can be attributed to Zn 2p3/2 and Zn 2p1/2, respectively.30 The difference between the two peaks is 23 eV, which is characteristic of the Zn2+ valence state,30 confirming that Zn is present in the form of ZnO, as also demonstrated by XRD. The Zn phase does not suffer any modification either after the addition of Cu or under catalytic conditions, as observed by the lack of change in the binding energies of the peaks. Regarding the Cu 2p spectra, two main peaks and a satellite were identified in the Cu 2p3/2 region. The peak at 932.9 ± 0.04 eV can be attributed to Cu0 and/or Cu+, while the one at 934.6 ± 0.2 eV can be attributed to Cu2+.31 The Cu LMM spectra were also analysed, which indicated the presence of a mixture of Cu0, Cu+ and Cu2+ (Fig. S15†). Furthermore, the presence of the satellite peak further confirms the presence of Cu2+ in the sample. Interestingly, the relative intensity of the Cu2+ compared to Cu0/Cu+ increases after the long TOS experiment, which might indicate that the sample suffered some oxidation when exposed to the reaction conditions. It is worth to note that the limitations of the experimental protocols and sample handling should be considered. Namely, exposure to ambient air, after the reduction treatment, can partially oxidize the surfaces of transition-metal nanoparticles.
The C 1s (Fig. S12†), O 1s (Fig. S13†) and N 1s (Fig. S14†) regions of the spectra were also analysed and a brief discussion can be found in the ESI† material.19,30,32,33
Transmission electron microscopy (TEM) demonstrated that the Cu/CNTs–N–ZnO (90:10) sample is composed by agglomerates of nanoparticles, possibly ZnO, and small nanoparticles, presumably Cu and CuO, dispersed on the CNTs (Fig. 4). Interestingly, the sample after TOS demonstrated an apparent increase in nanoparticle size; however, the imaging contrast between the Cu and ZnO nanoparticles is very low; thus, it is difficult to differentiate the two types of nanoparticles in the TEM images. Therefore, it was not possible to obtain a clear particle size distribution.
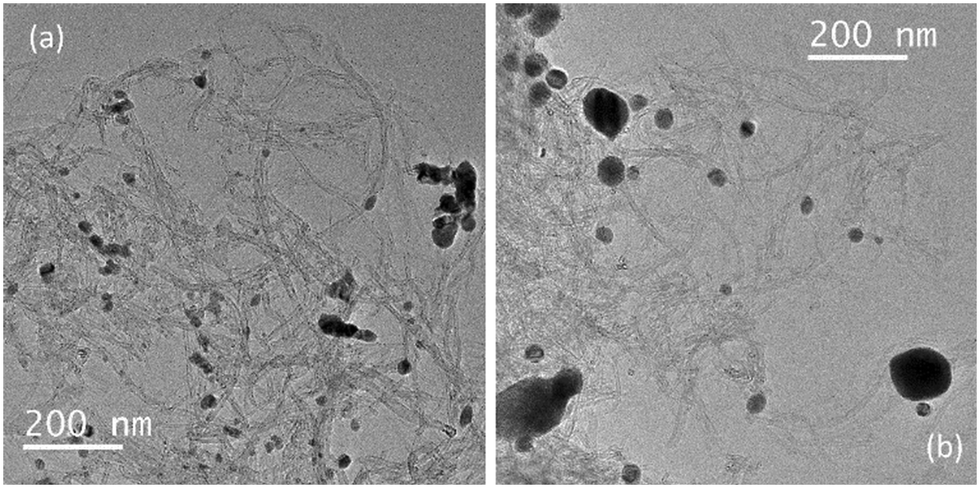 | ||
| Fig. 4 TEM image of the topmost performing catalyst Cu/CNTs–N–ZnO (90:10) before (a) and after (b) TOS. | ||
Discussion
In this work, a catalytic study of carbon materials as support for Cu-based catalysts for the RWGS reaction is presented. Firstly, Cu-based catalysts supported on pristine AC and CNTs were prepared, characterized, and tested to evaluate which carbon material could provide better catalytic performance.Through the first catalytic experiments (Fig. S1†), it was possible to conclude that Cu/CNTs outperformed Cu/AC in terms of CO2 conversion and CO yield, particularly in the higher reaction temperatures. This result can be explained by the CNTs' ability to adsorb H2 and transport electrons, and to the higher surface area of the catalyst as compared with Cu/AC, as indicated by the H2-TPR profile of this sample (Fig. S8†).14,16,17,34
Afterwards, to improve the catalytic performance, the possibility of adding ZnO to the catalysts was studied, through the preparation of Cu-based catalysts supported on a composite of CNT and ZnO prepared by ball milling (Fig. 1). Interestingly, the catalyst supported on a CNT:ZnO composite prepared by a ball milling procedure, Cu/CNTs–ZnO (90:10), achieved a superior catalytic performance not only compared to the Cu/CNTs catalyst but also to the reference catalyst Cu/ZnO, highlighting the benefit of taking advantage of the properties of both CNTs and ZnO to the catalyst's structure.
Finally, the functionalization of the CNT supports, with O-groups and N-groups, was assessed as a path to improve even further the catalytic performance (Fig. 1). The catalyst over the O-doped support, which contained carboxylic acid, lactone, anhydrides, phenol, carbonyl, ether and quinone groups in its surface as confirmed by the TPD analysis, achieved a lower conversion than that of supported on pristine CNT (although with CO selectivity equal to 100%). The idea of the O-groups functionalization would be to improve the metal nanoparticles' dispersion; however, the number of acidic groups present in the CNTs' surface, due to the oxidation treatment, can be the cause of the lower performance. As CO2 is considered a weak acid, its adsorption is facilitated by basic groups instead of the present acidic groups.14,17,18
Notably, N-doping of the surface of the CNTs, which allowed the introduction of quaternary ammonium, pyridinic, and pyrrolic groups to the catalyst as confirmed by the XPS analysis, led to an improvement in the CO2 conversion by 5.8% comparing to the Cu/CNTs–ZnO (90:10) catalyst, leading to the topmost performing catalyst of this study – Cu/CNTs–N–ZnO (90:10). The addition of N-groups to CNTs' surface possibly increases the CO2 adsorption capability and the overall performance of the catalyst, therefore, leading to a greater CO2 conversion and CO yield.35 This catalyst may also present a smaller particle size and larger active area when compared to the other catalysts prepared, since a strongest metal–support interaction was identified, from the H2-TPR analysis.
As expected, the performance from all catalysts achieves its higher results at the highest temperature, 600 °C, due to the endothermic nature of the RWGS reaction. Notably, the results presented herein with the Cu/CNTs–N–ZnO (90:10) catalyst compare favourably with those reported in the literature for catalysts evaluated under similar conditions for reaction pressure and temperature (1 bar and 600 °C), but different WHSV, H2:CO2, and catalyst's mass. More details can be observed in ESI† material (Table S4). Chen et al.36 developed monometallic catalysts, 10% Cu/SiO2 and 0.3% Fe/SiO2, and achieved a 8.0% and 2.0% CO2 conversion for each, respectively, with CO selectivity equal to 100%. A bimetallic catalyst, 10:0.3 CuFe/SiO2, was also developed by Chen et al.,36 and while it achieved a CO selectivity equal to 100%, its CO2 conversion was 12%. They attributed this result to the formation of active sites between Cu and Fe, which contributes to an increase in CO2 conversion and catalysts stability, as Fe could prevent Cu from sintering at the highest temperatures. Ye et al.37 developed a Pd-based catalyst supported on SiO2, Pd/SiO2, and a bimetallic catalyst also supported on SiO2, PdIn/SiO2; the catalyst Pd/SiO2 demonstrated a CO2 conversion equal to 29% and a CO selectivity of 82%, whereas the catalyst PdIn/SiO2 achieved a CO2 conversion of 10% and CO selectivity of 100%. Per the authors, this result was attributed to a weaker CO adsorption on PdIn's surface, to a more energetically favoured CO hydrogenation on Pd′s surface, and to a weaker H2 dissociation on PdIn's surface. Liu et al.38 developed a bimetallic catalyst of Cu–Ni supported on Al2O3, and achieved a CO2 conversion of 28.7% and a CO selectivity of 79.7%. This result was attributed to the strong interaction between Cu and Ni, as Cu particles benefit CO2 adsorption and CO formation (higher CO2 conversion), and Ni particles favour H2 adsorption (higher selectivity).
The performance of the topmost performing catalyst – Cu/CNTs–N–ZnO (90:10) – was evaluated over a long time on stream (TOS) experiment for 93 h, to assess its stability (Fig. 2). This way, it would be possible to verify if this catalyst can nullify the problem observed for most Cu-based catalysts, deactivation by sintering at high temperatures. Notably, the catalyst displayed great stability over the 93 h of reaction. The catalyst characterization after TOS by XPS demonstrated that the ZnO phase did not suffer any modification in its chemical structures after TOS; however, Cu, which was present as metallic Cu and CuO, presented an increase in the relative intensity of Cu2+ phase as compared to Cu0 after TOS. Furthermore, the microstructure of the sample, analysed by TEM demonstrated an apparent increase in nanoparticle size; however, due to the low imaging contrast between the Cu and ZnO nanoparticles it was not possible to estimate the size variation.
Conclusions
Cu-based catalysts supported on CNTs were developed and evaluated for the RWGS reaction. The preliminary study conducted revealed that CNT-supported catalysts achieve a better overall catalytic performance when compared to AC-support present for the RWGS reaction, possibly due to the CNTs' capacity for H2 adsorption, electron transport, and the dispersion of the active phase via their mesoporous structure.Interestingly, the addition of ZnO to CNTs in the structure of the Cu-based catalysts – Cu/CNTs–ZnO (90:10) – resulted in an improvement of the catalytic performance when compared to both the Cu/CNTs and the Cu/ZnO reference catalyst. The N-doping of the CNTs in the CNTs:ZnO composite (Cu/CNTs–N–ZnO (90:10)) demonstrated to be beneficial for the catalytic performance, leading to the highest CO2 conversion accomplished, XCO2 = 54.8%, and a CO selectivity equal to 100%, at 600 °C. The catalyst's ability to adsorb CO2 and its overall performance were likely improved by adding N-groups to the surface of the support. Notably, this catalyst demonstrated excellent stability throughout 93 h of TOS.
The results presented herein with the Cu/CNTs–N–ZnO (90:10) catalyst compare favourably with those presented in the literature for catalysts under similar conditions. Therefore, this study provides proof that the catalytic performance of the RWGS reaction may benefit from combining the properties of carbon materials and metal oxide on composites, turning this approach an interesting toolbox for developing highly efficient catalysts for this reaction.
Author contributions
A. R. Querido: investigation, writing – original draft; Liliana P. L. Gonçalves: conceptualization, methodology, investigation, writing – original draft; Yury V. Kolen'ko: supervision, writing – review & editing; M. Fernando R. Pereira: supervision, writing – review & editing O. Salomé G. P. Soares: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by Move2LowC project (n. 46117), cofinanced by Programa Operacional Competitividade e Internacionalização (POCI); Programa Operacional Regional de Lisboa, Portugal 2020 and the European Union, through the European Regional Development Fund (ERDF), and by Project HyGreen&LowEmissions, reference NORTE-01-0145-FEDER-000077, Co-financed by the European Regional Development Fund (FEDER), through the North Portugal Regional Operational Programme (NORTE2020); LA/P/0045/2020 (ALiCE), UIDB/50020/2020 and UIDP/50020/2020 (LSRE-LCM), funded by national funds through FCT/MCTES (PIDDAC). O. S. G. P. S. acknowledges FCT funding under the Scientific Employment Stimulus – Institutional Call CEECINST/00049/2018.Notes and references
- A. M. Bahmanpour, M. Signorile and O. Kröcher, Appl. Catal., B, 2021, 295, 120319 CrossRef CAS.
- X. Chen, Y. Chen, C. Song, P. Ji, N. Wang, W. Wang and L. Cui, Front. Chem., 2020, 8, 1 CrossRef PubMed.
- A. M. Bahmanpour, F. Héroguel, M. Klllç, C. J. Baranowski, L. Artiglia, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, ACS Catal., 2019, 9, 6243 CrossRef CAS.
- X. Su, X. Yang, B. Zhao and Y. Huang, J. Energy Chem., 2017, 26, 854 CrossRef.
- D. Vovchok, C. Zhang, S. Hwang, L. Jiao, F. Zhang, Z. Liu, S. D. Senanayake and J. A. Rodriguez, ACS Catal., 2020, 10, 10216 CrossRef CAS.
- H. X. Liu, S. Q. Li, W. W. Wang, W. Z. Yu, W. J. Zhang, C. Ma and C. J. Jia, Nat. Commun., 2022, 13, 1 Search PubMed.
- C. Y. Chou, J. A. Loiland and R. F. Lobo, Catalysts, 2019, 9, 1 CrossRef CAS.
- X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y. W. Li, C. Shi and D. Ma, ACS Catal., 2017, 7, 912 CrossRef CAS.
- C. S. Chen, W. H. Cheng and S. S. Lin, Chem. Commun., 2001, 1770 RSC.
- M. Ronda-Lloret, S. Rico-Francés, A. Sepúlveda-Escribano and E. V. Ramos-Fernandez, Appl. Catal., A, 2018, 562, 28 CrossRef CAS.
- R. S. Oosthuizen and V. O. Nyamori, Platinum Met. Rev., 2011, 55, 154 CrossRef CAS.
- M. Bushell, S. Beauchemin, F. Kunc, D. Gardner, J. Ovens, F. Toll, D. Kennedy, K. Nguyen, D. Vladisavljevic, P. E. Rasmussen and L. J. Johnston, Nanomaterials, 2020, 10, 1 CrossRef PubMed.
- J. A. Loiland, M. J. Wulfers, N. S. Marinkovic and R. F. Lobo, Catal. Sci. Technol., 2016, 6, 5267 RSC.
- E. Furimsky, Carbons and Carbon Supported Catalysts in Hydroprocessing, The Royal Society of Chemistry, Ontario,Canada, 2008, vol. 1 Search PubMed.
- E. Furimsky, Ind. Eng. Chem. Res., 2020, 59, 15393 CrossRef CAS.
- P. Serp and B. Machado, Nanostructured Carbon Materials for Catalysis, The Royal Society of Chemistry, 2015 Search PubMed.
- E. Furimsky, Carbon Nanomaterials in Hydrogenation Catalysis, Royal Society of Chemistry, 2019 Search PubMed.
- P. Serp and J. L. Figueiredo, Carbon Materials for Catalysis, John Wiley & Sons, New Jersey, 2009 Search PubMed.
- L. P. L. Gonçalves, J. P. S. Sousa, O. S. G. P. Soares, O. Bondarchuk, O. I. Lebedev, Y. V. Kolen'ko and M. F. R. Pereira, Catal. Sci. Technol., 2020, 10, 7217 RSC.
- L. P. L. Gonçalves, M. Meledina, A. Meledin, D. Y. Petrovykh, J. P. S. Sousa, O. S. G. P. Soares, Y. V. Kolen'ko and M. F. R. Pereira, Carbon, 2022, 195, 35 CrossRef.
- L. P. L. Gonçalves, J. Mielby, O. S. G. P. Soares, J. P. S. Sousa, D. Y. Petrovykh, O. I. Lebedev, M. F. R. Pereira, S. Kegnæs and Y. V. Kolen'ko, Appl. Catal., B, 2022, 312, 121376 CrossRef.
- J. Wen, C. Huang, Y. Sun, L. Liang, Y. Zhang, Y. Zhang, M. Fu, J. Wu, L. Chen and D. Ye, Catalysts, 2020, 10, 533 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- R. S. Costa, O. S. G. P. Soares, R. Vilarinho, J. A. Moreira, M. F. R. Pereira, A. Pereira and C. Pereira, Carbon Trends, 2021, 5, 100137 CrossRef CAS.
- R. Rocha, O. Soares, J. Figueiredo and M. Pereira, C, 2016, 2, 17 Search PubMed.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379 CrossRef CAS.
- H. M. A. Hassan, M. S. Alhumaimess, I. H. Alsohaimi, S. K. Mohamed, O. F. Aldosari, T. S. Alraddadi and A. A. Essawy, Colloids Surf., A, 2022, 654, 130056 CrossRef CAS.
- Q. T. Ain, S. H. Haq, A. Alshammari, M. A. Al-Mutlaq and M. N. Anjum, Beilstein J. Nanotechnol., 2019, 10, 901 CrossRef CAS PubMed.
- A. K. Zak, R. Razali, W. H. A. Majid and M. Darroudi, Int. J. Nanomed., 2011, 6, 1399 Search PubMed.
- T. Wang, B. Jin, Z. Jiao, G. Lu, J. Ye and Y. Bi, J. Mater. Chem. A, 2014, 2, 15553 RSC.
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Ind. Eng. Chem. Res., 2019, 58, 9030 CrossRef CAS.
- J. L. Figueiredo and M. F. R. Pereira, Catal. Today, 2010, 150, 2 CrossRef CAS.
- L. P. L. Gonçalves, D. B. Christensen, M. Meledina, L. M. Salonen, D. Y. Petrovykh, E. Carbó-Argibay, J. P. S. Sousa, O. S. G. P. Soares, M. F. R. Pereira, S. Kegnæs and Y. V. Kolen'Ko, Catal. Sci. Technol., 2020, 10, 1991 RSC.
- I. U. Din, M. S. Shaharun, M. A. Alotaibi, A. I. Alharthi and A. Naeem, J. CO2 Util., 2019, 34, 20 CrossRef CAS.
- M. S. Shafeeyan, W. M. A. W. Daud, A. Houshmand and A. Shamiri, J. Anal. Appl. Pyrolysis, 2010, 89, 143 CrossRef CAS.
- C. S. Chen, W. H. Cheng and S. S. Lin, Appl. Catal., A, 2004, 257, 97 CrossRef CAS.
- J. Ye, Q. Ge and C. J. Liu, Chem. Eng. Sci., 2015, 135, 193 CrossRef CAS.
- Y. Liu and D. Liu, Int. J. Hydrogen Energy, 1999, 24, 351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00308f |
| This journal is © The Royal Society of Chemistry 2023 |