
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling synergistic effects in bi-metallic catalysts: deceleration of palladium–gold nanoparticle coarsening in the hydrogenation of cinnamaldehyde†
Jose
Pinto
 ab,
Andreas
Weilhard
*a,
Luke T.
Norman
a,
Rhys W.
Lodge
a,
David M.
Rogers
a,
Aitor
Gual
c,
Israel
Cano
d,
Andrei N.
Khlobystov
a,
Peter
Licence
*b and
Jesum
Alves Fernandes
*a
ab,
Andreas
Weilhard
*a,
Luke T.
Norman
a,
Rhys W.
Lodge
a,
David M.
Rogers
a,
Aitor
Gual
c,
Israel
Cano
d,
Andrei N.
Khlobystov
a,
Peter
Licence
*b and
Jesum
Alves Fernandes
*a
aSchool of Chemistry, University of Nottingham, NG7 2RD, Nottingham, UK. E-mail: Andreas.Weilhard1@nottingham.ac.uk; Jesum.AlvesFernandes@nottingham.ac.uk
bGSK Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, NG7 2TU, Nottingham, UK. E-mail: Peter.Licence@nottingham.ac.uk
cCTQC-Eurecat-UTQ, C/Marcel·lí Domingo 2, Building N5, 43007 Tarragona, Spain
dDepartment of Inorganic Chemistry, Faculty of Chemistry, Universidad Complutense de Madrid, 28040 Madrid, Spain
First published on 2nd June 2023
Abstract
In this work, we demonstrate that the synergistic effect of PdAu nanoparticles (NPs) in hydrogenation reactions is not only related to high activity but also to their stability when compared to Pd mono-metallic NPs. To demonstrate this, a series of mono- and bi-metallic NPs: Pd, Pd0.75Au0.25, Pd0.5Au0.5, Pd0.25Au0.75 and Au in ionic liquid [C4C1Im][NTf2] have been fabricated via a magnetron sputtering process. Bi-metallic NPs possess external shells enriched with Pd atoms that interact with [NTf2]− of the ionic liquid resulting in enhanced catalytic performance in hydrogenation of cinnamaldehyde compared to their mono-metallic counterparts. This is ascribed to their higher stability over 24 h of reaction, whilst the catalytic activity and selectivity are comparable for both catalysts. Using a bespoke kinetic model for in situ catalyst deactivation investigations and electron microscopy imaging at the nanoscale, we have shown that PdAu has a deactivation rate constant of 0.13 h−1, compared to 0.33 h−1 for Pd NPs, leaving 60% and 40% of available sites after the reaction, respectively. Beyond that, the kinetic model demonstrates that the reaction product has a strong stabilizing factor for bimetallic NPs against coarsening and deactivation, which is not the case for Pd NPs. In summary, our kinetic model enables the evaluation of the catalyst performance over the entire chemical reaction space, probing the contribution of each individual component of the reaction mixture and allowing the design of high-performance catalysts.
Introduction
Catalysis plays a vital role in mitigating hazards arising from environmental pollution and climate change.1 Indeed, 80% of industrial chemical transformations are catalytic, where the catalysts are expected to perform well in three key parameters: activity, selectivity and stability.2,3 In order to maintain a high catalytic activity and selectivity, the stability is paramount.4 However, whilst parameters influencing catalyst activity and selectivity have been widely investigated, catalyst deactivation pathways,5–7 determining the long-term performance, are poorly understood.8 The deactivation mechanisms may be affected by an array of parameters, such as reaction medium and conditions, supports/ligands and composition of the catalyst.9–11An excellent model reaction for the investigation of catalyst stability, selectivity and activity is the selective hydrogenation of cinnamaldehyde (CAL) to hydrocinnamaldehyde (HCAL). HCAL is employed as an important building block for the synthesis of important pharmaceuticals;12 however, the hydrogenation of CAL can result in a wide range of products, including cinnamyl alcohol (COL) and hydrocinnamyl alcohol (HCOL).13 Due to the competing selectivity, extensive research has been undertaken to obtain an active and selective catalyst for this transformation. In general, Ni14 and Pd15,16 based catalysts are found to be more active and selective towards HCAL compared to Pt,17 Ru,18,19 and Au,16 although a substantial effect can also be attributed to the employed supports and ligands.20–23 Another important aspect is the synergistic effect observed in bi-metallic systems leading to an increase of the activity and selectivity for this reaction either by facilitating the adsorption and activation of reactants and therefore enhancing the reaction rate,24–26 or by forming electrophilic sites, thus tuning the selectivity.27,28 In this regard, PdAu nanoparticles (NPs) represent an attractive catalyst as the H2 activation can be facilitated due to the synergistic effects of Pd and Au species.24–26 Moreover, PdAu NPs have found widespread application not only in the selective hydrogenation of CAL,29–31 but also in other catalytic hydrogenations32–34 and oxidations.35–39 However, mechanisms of mono- and bi-metallic catalyst deactivation remain largely unknown due to the lack of in situ deactivation kinetic measurements combined with local-probe analysis (e.g. electron microscopy) of the catalyst evolution.
In this work we developed a kinetic model describing mono- and bimetallic catalyst deactivation, by correlating transmission electron microscopy (TEM) imaging of catalytic Pd and PdAu NPs with reaction kinetics measurements. Direct growth of Pd and PdAu NPs in [C4C1Im][NTf2] ionic liquid (IL) via the magnetron sputtering approach provided 2.0–2.8 nm highly active catalytic centres with a core–shell structure for the bi-metallic NPs where Pd atoms are on the surface due to interactions with [NTf2]−. In the reaction of hydrogenation of CAL, the activity and selectivity of Pd0.5Au0.5 and Pd NPs were found to be similar but with stability significantly higher in the bi-metallic case due to a slower decrease of their available sites linked to a slower rate of coarsening of Pd0.5Au0.5 NPs as compared to Pd NPs. Interactions of the reaction product (HCAL) with the catalyst surface has been identified as the key factor for bi-metallic catalyst stabilisation during the reaction.
Results and discussion
Pd, Au and PdAu NPs were prepared in [C4C1Im][NTf2] by the magnetron sputtering process. A co-deposition mode, where Pd and Au species were sputtered simultaneously from two targets, was employed for PdAu NP preparation (Fig. 1a). The PdAu NP composition was finely tuned by varying the power applied on each target (see details in the ESI†), thus achieving the desired metal ratio in the catalyst, as verified by inductively coupled plasma atomic emission spectroscopy (ICP-OES) (Fig. S4 and Table S1†). The size distributions of Pd, Au and PdAu NPs were evaluated directly in IL, without the use of solvent, via bright-field TEM imaging (Fig. 1b and S5†). | ||
| Fig. 1 The magnetron sputtering process, and structural and chemical characterisation of Pd and PdAu NPs. (a) Scheme of Pd and Au magnetron sputtering co-deposition in [C4C1Im][NTf2]. (b) Transmission electron microscopy (TEM) imaging of Pd0.5Au0.5 NPs in [C4C1Im][NTf2] shows the homogeneous NP size distribution generated by the magnetron sputtering process. A complete set of TEM images is available in the ESI† (Fig. S5). (c) X-ray photoelectron spectroscopy (XPS) spectra of Pd, Au and PdAu NPs in [C4C1Im][NTf2]. (d) Pd K-edge and Au L-edge X-ray absorption near edge structure (XANES). (e) Pd K-edge Fourier transform of k2-weighted extended X-ray absorption fine structure (EXAFS) spectra of PdAu NPs. (f) Schematic showing the formation of PdAu NPs with Au-core and Pd-shell as the proportion of Pd increases. | ||
Bright-field TEM imaging revealed a similar average NP size for Pd and Pd0.75Au0.25 and Pd0.5Au0.5 NPs, from ca. 1.98 to 2.18 nm, with a slightly larger NP size increase for high Au NP content Pd0.25Au0.75 and Au NPs from ca. 2.35 to 2.84 nm. This behaviour can be associated with the weaker interaction of [NTf2]− with Au atoms compared to Pd atoms, thus leading to a larger Au NP average size.40–42
X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) provided insights into the interactions between Pd–Au atoms within NPs and metal atoms with ionic liquid. The XPS spectra of monometallic Pd and Au NPs in [C4C1Im][NTf2] show two distinct electronic environments: the metallic Pd 3d5/2 at 336.1 eV and Au 4f7/2 at 84.3 eV ascribed to atoms in a metallic environment in the NP core, and another set of peaks at 338.4 eV for Pd 3d5/2 and 85.7 eV for Au 4f7/2 corresponding to surface atoms interacting with ionic liquid (Fig. 1c).43 For PdAu NPs in [C4C1Im][NTf2], XPS detects significant changes in the electronic environment of the metals as the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au atomic ratio is varied (Fig. 1c). As the Pd concentration increases in the Au matrix (Au → Pd0.25Au0.75 → Pd0.5Au0.5), the intensity of the peak associated with surface Au atoms interacting with ionic liquid (ca. 85.7 eV) decreases and completely disappears for Pd0.75Au0.25. Additionally, the Au peak at 84.3 eV, attributed to Au NP core metal atoms, showed a significant shift towards a lower binding energy as the Pd concentration increased (ca. 0.5 eV shift for Pd0.75Au0.25), while no significant changes were observed for Pd peaks.44 These XPS measurements demonstrate the interaction between Pd and Au atoms within NPs, thus confirming the formation of bi-metallic PdAu NPs, as well as suggesting that the surface of NP is enriched with Pd atoms.
:Au atomic ratio is varied (Fig. 1c). As the Pd concentration increases in the Au matrix (Au → Pd0.25Au0.75 → Pd0.5Au0.5), the intensity of the peak associated with surface Au atoms interacting with ionic liquid (ca. 85.7 eV) decreases and completely disappears for Pd0.75Au0.25. Additionally, the Au peak at 84.3 eV, attributed to Au NP core metal atoms, showed a significant shift towards a lower binding energy as the Pd concentration increased (ca. 0.5 eV shift for Pd0.75Au0.25), while no significant changes were observed for Pd peaks.44 These XPS measurements demonstrate the interaction between Pd and Au atoms within NPs, thus confirming the formation of bi-metallic PdAu NPs, as well as suggesting that the surface of NP is enriched with Pd atoms.
X-ray absorption near edge structure (XANES) analysis for Pd and Au NPs displayed a similar adsorption edge to metal foil but with a higher white line (Hw) intensity, which can be assigned to the Pd and Au surface atoms interacting with the IL (Fig. 1d).43 For PdAu NPs, the Hw intensity of the Pd K-edge is higher than that for Pd NPs, whereas for the Au L-edge, the Hw intensity peak of PdAu NPs is lower than that of Au NPs, which corroborates the XPS results.44 Extended X-ray absorption fine structure (EXAFS) measurements of Pd NPs show a peak at 1.5 Å associated with the Pd–IL interaction and another peak at 2.5 Å ascribed to the Pd–Pd bond (Fig. 1e). As the Au concentration increases in the Pd matrix, a peak for longer bond distances is observed at 2.7 Å, which is attributed to the Pd–Au bond.43 Additionally, in PdAu NPs the peak intensity associated with the Pd–Pd bond (ca. 2.5 Å) is dramatically decreased with respect to the Pd–IL peak (ca. 1.5 Å), which indicates the tendency of Pd atoms to be on the NP surface in the presence of Au. Interestingly, the formation of Pd shell/Au core NPs contrasts the thermodynamically most stable alloy configuration for these bimetallic nanoparticles.45 However, it has been shown that the interaction with strongly coordinating molecules can lead to the segregation of Pd from the nanoparticle surface.45 Similarly, the interaction with [NTf2]− could lead to the segregation of Pd atoms from the NP surface, especially considering that during the initial metal NP formation in IL by magnetron sputtering dynamic clusters are formed, as we recently demonstrated.43 The dynamic metal clusters are most likely to be thermodynamically unstable and kinetically reactive, thus small differences between Pd and Au in coordination strengths with [NTf2]− may lead to the segregation of Pd atoms to the NP surface. Furthermore, XPS and XAS data are consistent with a core–shell model in which Au atoms are preferentially located in the NP core and Pd atoms on the NP surface (Fig. 1f).46,47
Pd, Au and PdAu NPs in [C4C1Im][NTf2] were tested in the hydrogenation of CAL. Under the optimised reaction conditions, all NPs containing Pd showed high selectivity towards HCAL (>90%, Fig. 2a). Our results are comparable to PdAu NPs supported on high surface area mesoporous silica48 (however, it must be noted that the catalyst support and reaction conditions may have a significant influence on this reaction).29–31,49 Furthermore, Pd0.75Au0.25 and Pd0.5Au0.5 NPs exhibited higher conversions after 24 h than Pd and Pd0.25Au0.75 NPs (Fig. 2a, entries 3, 2 and 1, 4, 5).24–26 Au NPs were inactive under the assayed reaction conditions, indicating that Pd is crucial for the catalytic turnover in this transformation (Fig. 2a, entries 4, 5).
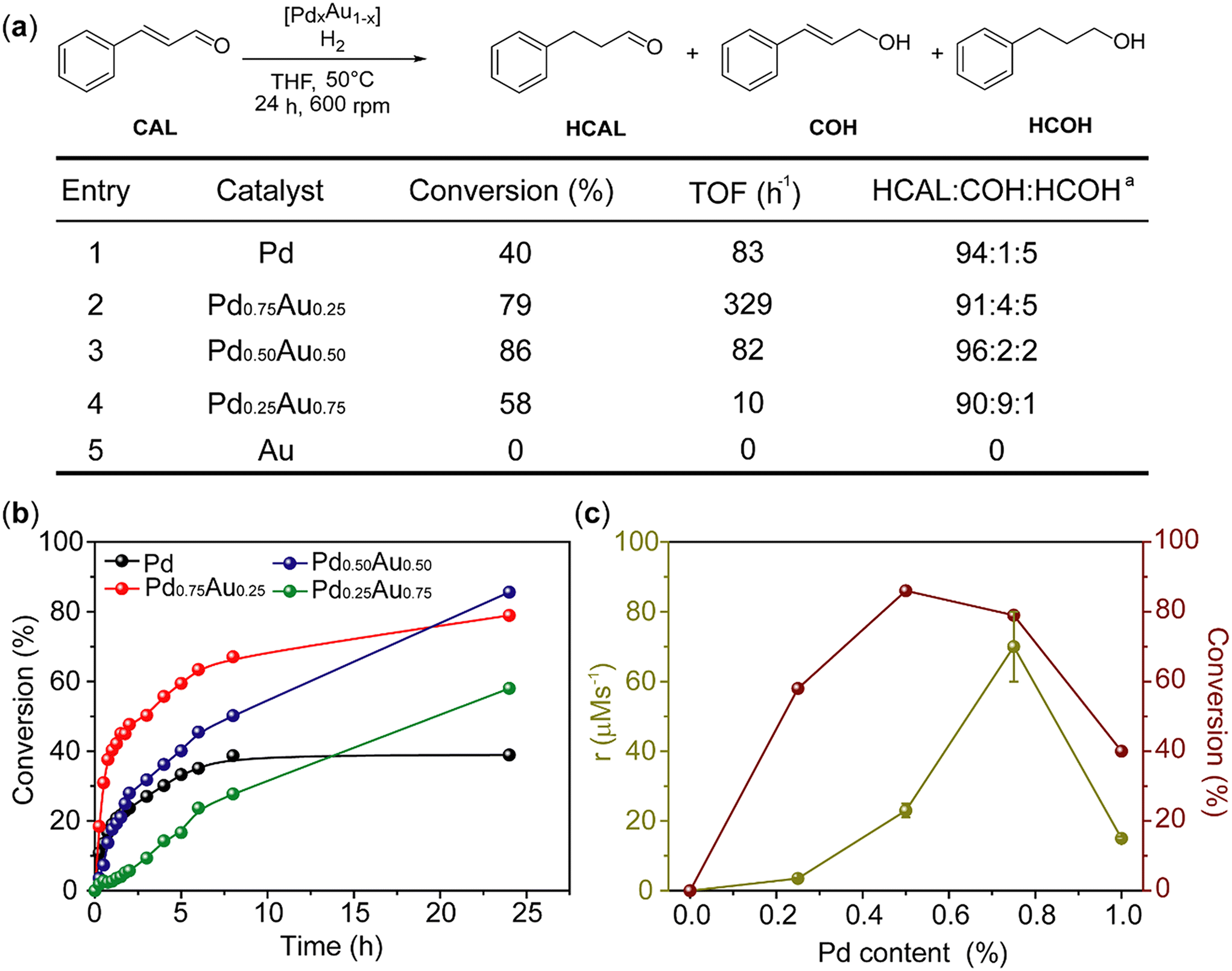 | ||
| Fig. 2 (a) Conversion, turnover frequency (TOF) and selectivity for Pd, Au and PdAu NPs. Reaction conditions: catalyst (Pd = 0.11 mg) in C4C1Im·NTf2 (130 mg), THF (10 mL), T = 50 °C, t = 24 h, P(H2) = 5 bar, CAL (0.5 M). aSelectivity towards HCAL. All reaction components were determined using gas chromatography and tridecane as an internal standard. (b) Reaction profiles for Pd, Au and PdAu NPs. At each point of the reaction the selectivity towards HCAL was >90%, thus the selectivity was omitted. (c) Comparison between achieved conversion after 24 h and the initial rate. The initial rate was determined by linear regression performed on the consumption of CAL. | ||
A more detailed analysis of the reaction profiles and rates provided insight into the superior catalytic performance of PdAu NPs compared to the Pd catalyst, both in terms of activity and stability (Fig. 2b and c). For example, for Pd NPs the reaction rate drops and thus the conversion has only a small increase after reaching ca. 40% conversion, whereas no such effect was observed for Pd0.5Au0.5 and Pd0.25Au0.75 NPs (Fig. 2b). Pd0.75Au0.25 NPs displayed the highest initial rate, achieving 70% conversion of CAL to HCAL after 8 h, but thereafter the conversion increased only slightly (Fig. 2b). Even though Pd0.75Au0.25 NPs exhibit much higher initial rates than Pd0.25Au0.75, Pd0.5Au0.5 and Pd NPs, the highest conversion after 24 h was found for Pd0.5Au0.5 NPs with a continuous increase in conversion over time (Fig. 2c). These results show that the correlation of the initial reaction rate (related to catalyst activity) with the overall conversion (related to catalyst activity and stability) is not always correct, thus the overall catalytic performance cannot be solely attributed to an enhancement of the initial reaction rates,24–26 and must include catalyst stability over an extended period of time as well as any changes in the reaction mechanism caused by the different catalysts.24 As these differences are most pronounced with Pd and Pd0.5Au0.5 NPs, these two catalysts were investigated in greater detail. TEM analysis of the catalysts after reaction was performed to determine the catalyst changes occurring during the reaction (Fig. 3).
 | ||
| Fig. 3 TEM images and histograms of Pd and Pd0.5Au0.5 NPs before and after the reaction (later denoted by AR). (a and b) Pd and Pd-AR NP TEM images, respectively, show drastic changes in NP size and dispersion in IL after the reaction. (c) Pd and Pd-AR NPs displayed a mean diameter size of 2.0 ± 0.7 and 5.4 ± 1.4 nm, respectively. Low TEM image magnification inset in the figure (b) highlights the broad Pd NPs dispersion after the reaction. (d and e) Pd0.5Au0.5 and Pd0.5Au0.5-AR NP TEM images, respectively, show only a slight change in NP size and dispersion in IL after the reaction, which is highlighted in the inset in the figure (e). (f) Pd0.5Au0.5 and Pd0.5Au0.5-AR NPs display a mean diameter size of 2.2 ± 0.8 and 3.3 ± 0.9 nm, respectively. | ||
TEM analysis of Pd NPs showed a significant increase in NP size after the reaction with the mean diameter changing from 2.0 to 5.46 nm, whereas the size distribution of Pd0.5Au0.5 NPs changed only slightly with the mean diameter before and after the reaction from 2.2 to 3.3 nm, respectively (Fig. 3). Significant coarsening of Pd NPs during the reaction leads to reduction of the surface area available for catalysis, whilst Pd0.5Au0.5 NPs changing only slightly under the same reaction conditions and retain much of their surface area. Indeed, a more detailed analysis of the size distribution shows that for Pd0.5Au0.5 NPs 61% of surface sites after the reaction remain available for catalysis, whilst in Pd NPs only 36% of initially active sites remain available. This suggests that the long-term catalyst stability (rather than activity) is the main factor discriminating the performance of Pd and Pd0.5Au0.5 NPs.
To reveal how the compounds of the reaction mixture (THF, H2, CAL, HCAL) affect the catalyst stability, Pd and Pd0.5Au0.5 NPs were exposed to the individual components for an incubation period (τ) prior to the reaction initiation (Fig. 4).
 | ||
| Fig. 4 Dependence of the relative concentration of total available sites [ST] on the incubation time τ for (a) Pd NPs and (b) Pd0.5Au0.5 NPs in the presence of only THF (black), in the presence of 0.5 M HCAL (green), in the presence of 0.5 M CAL (red), and under 5 bar H2 (blue). In all experiments, it was assumed that the catalyst deactivation takes place only during the incubation period, thus the initial rates are not affected by further catalyst deactivation. The relative concentration of available sites was estimated using the fraction of the initial rate after the incubation period and the initial rate at τ = 0 (see ESI† for further details). | ||
For the first set of experiments, Pd and Pd0.5Au0.5 NPs were subjected to incubation periods, without addition of reagents/products (only THF solvent), prior to initiating the reaction (Fig. 4). This led to an exponential decay of the initial reaction rates as a function of pre-reaction incubation time, suggesting a first order dependence for the catalyst deactivation (Fig. 4, S7 and S8†) measured as the total concentration of available sites [ST]. Comparing Pd and Pd0.5Au0.5 NPs, a much faster deactivation rate for Pd NPs was observed, which is consistent with extensive Pd NP coarsening shown by TEM analysis (Fig. 3). The presence of reagents or product(s) in the pre-reaction exposure of catalysts retarded deactivation (e.g. increased their stability) for both catalysts, which can be ascribed to catalyst surface stabilisation due to adsorption of reagents or products (Fig. 4). For example, the presence of H2 considerably slowed the deactivation of Pd NPs during the incubation period as compared to pure solvent, whereas the addition of CAL and HCAL had a negligible effect on Pd NPs. For Pd0.5Au0.5 NPs, under the same conditions, the presence of HCAL had the greatest effect on the catalyst during the incubation period, with effects of H2 and CAL being less pronounced. It is important to highlight that these results were obtained using the standard reaction conditions assayed in this work (Fig. 3a).
The experimental results presented in Fig. 4 strongly indicate a pseudo-first order mechanism for the catalyst deactivation due to the particle coarsening (hence leading to reduced surface area), which is retarded by both reagent and product adsorption on the catalysts. By considering the quasi-equilibria taking place during the incubation periods, eqn (1) can describe the concentration of available sites ([ST]τ) as a function of time τ (see ESI† for further details), where [ST]0 is the total concentration of initial available sites, k5 is the rate constant for the catalyst deactivation, and K1 and K4 are the equilibrium constants for the adsorption of CAL and HCAL on the catalyst surface, respectively. The adsorption equilibrium constant for H2 (K2) is multiplied by the Henry constant to give Kp which allows the use of the hydrogen pressure P(H2) in the equation:
 | (1) |
As expected, Pd0.5Au0.5 NPs exhibit a significantly lower rate constant for the catalyst deactivation than Pd NPs (Table 1). For Pd NPs, CAL showed a stronger effect on their stability compared to H2 (Table 1), which was an unexpected outcome based on the results shown in Fig. 4 explained by the much higher concentration of H2 than that of CAL in the reaction conditions (Fig. 4), whereas in the quasi-equilibrium constant calculation, the concentrations of reagents and the product were normalised (Table 1). Furthermore, these results were confirmed by DFT calculations (see details in the ESI†), which showed that the computed Gibbs free energy for adsorption of CAL and H2 adsorbed on a Pd19 cluster are −36.76 and −4.87 kcal mol−1, respectively, in line with the experimental quasi equilibrium constants (Table 1). For Pd0.5Au0.5 NPs, HCAL had the largest adsorption equilibrium constant (Table 1), following a similar trend as that observed under the reaction conditions (Fig. 4), which underpins the strong effect of HCAL on Pd0.5Au0.5 NP stabilisation.
| Constant | Pd NPs | Pd0.5Au0.5 NPs |
|---|---|---|
| Catalyst deactivation rate constant (k5/h−1) | 0.33 | 0.13 |
| CAL adsorption equilibrium constant (K1/M−1) | 2.75 | 0.54 |
| H2 adsorption equilibrium constant (Kp/bar−1) | 1.30 | 0.70 |
| HCAL adsorption equilibrium constant (K4/M−1) | 0.25 | 6.36 |
Using the kinetic parameters in Table 1, the relative number of catalyst available sites was calculated and correlated with TEM results (Fig. 4) providing deep insights into the mechanism of the catalyst deactivation process (Fig. 5).
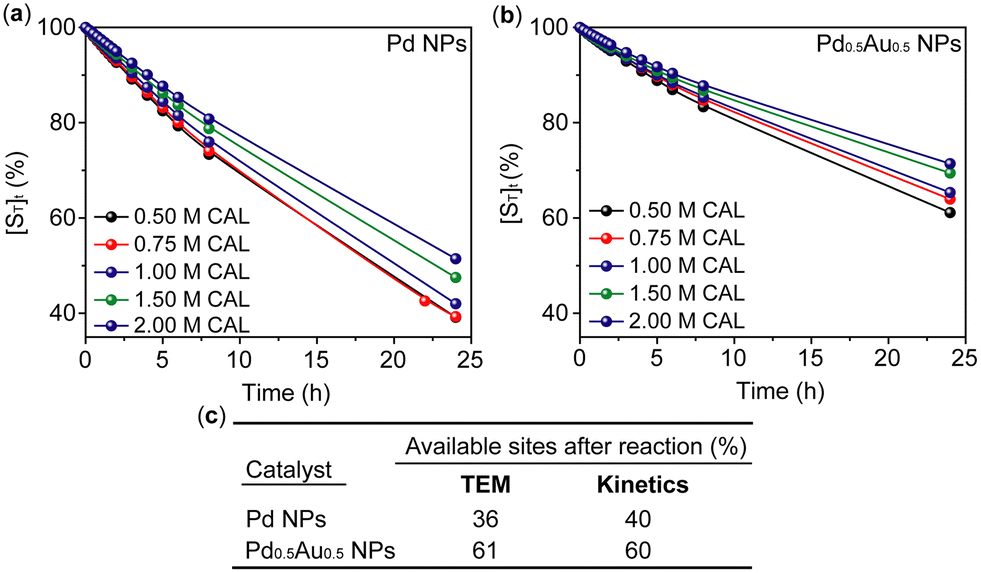 | ||
| Fig. 5 Calculated relative numbers of the catalyst available sites after the reaction using kinetic parameters and correlating them with TEM imaging. (a and b) Calculated kinetic traces of Pd and Pd0.5Au0.5 NPs for the loss of their available sites in different excess experiments, respectively (for original traces see Fig. S10†). (c) Comparison of Pd NPs and Pd0.5Au0.5 NPs for estimated available sites after the reaction determined by TEM and kinetic traces. | ||
Remarkably the loss of available sites calculated by the kinetic analysis (Fig. 5) for both Pd and Pd0.5Au0.5 NPs agrees with the TEM observations for the spent catalysts. For instance, Pd NPs increase in size by 3.5 nm during the reaction, whilst Pd0.5Au0.5 NPs increase only by 1.3 nm under the same conditions indicating 36% and 61% of remaining available sites, respectively, which correlates well with the kinetic data, emphasising the key difference in stability of mono- and bi-metallic catalysts (Fig. 5c).
In order to generate a complete kinetic model involving the catalytic transformation, the apparent rate orders were investigated using different excess experiments and analysed via the variable time normalization analysis (VTNA) method (detailed discussion in the ESI,† Fig. S10–S13).51–54 Therefore, with both the kinetic rate orders of reagents and product(s) (Fig. S11†) and the individual parameters influencing the catalyst deactivation in hand (Fig. 5), a complete picture of the catalytic transformation was established. As shown in Fig. 6a, the hydrogenation proceeds via L–H type transformation of CAL to HCAL, whilst the catalyst deactivates due to particle growth which is described as a pseudo first order. The relative number of active sites is utilised in conjunction with the rate orders determined from kinetics measurements (Fig. S10–S13†) to produce a complete normalisation of the kinetic traces (Fig. 6b and c). Note that the rate order for CAL changes for Pd NPs from 1 to −1, whilst Pd0.5Au0.5 NPs remaining +1 throughout the assayed reaction conditions. Furthermore, the unusual rate orders found for Pd NPs (Fig. S10–S13†) can be ascribed to the effects of the individual components (e.g. solvent, reactants and products) on the catalyst deactivation as only the order of +1 and −1 are present in the total normalisation of the kinetic traces (Fig. 6b). It is important to emphasise that it is the combination of complete normalisation of the kinetic traces (Fig. 6b and c) with TEM imaging of the catalyst that allows us to validate the kinetic model for in situ analysis of the catalyst deactivation process.
 | ||
| Fig. 6 (a) Schematic depiction of the catalytic cycle for the semi hydrogenation of CAL to HCAL. (b and c) A complete normalisation of the kinetic traces found by different excess experiments with CAL for Pd NPs and Pd0.5Au0.5NPs, respectively. | ||
Conclusion
Palladium-containing mono- and bi-metallic nanoparticles fabricated in ionic liquid using the magnetron sputtering method have been shown as highly effective catalysts for hydrogenation of cinnamaldehyde to hydrocinnamaldehyde taking place via the L–H mechanism. Comparison of reaction kinetics for Pd and Pd0.5Au0.5 NPs and correlation of the kinetic data with the particle size evolution during the reaction, identified the loss of surface area associated with the increase of the NP size as the main pathway for the catalyst deactivation. Bimetallic Pd0.5Au0.5 NPs have been shown to be significantly more stable than Pd NPs, with neither activity nor selectivity being hampered, and offering a wider optimal operation window for the selective hydrogenation of CAL. Importantly, bimetallic catalysts have demonstrated superior stability which was linked to stabilisation of catalyst particles against coarsening by the reaction product adsorbing on Pd0.5Au0.5 NPs. This work elucidates the synergistic effects emerging in bimetallic Pd–Au systems that can be effectively harnessed in hydrogenation reactions and opens new avenues for improving metal catalyst performance through innovative combination of kinetic tools with nanoscale imaging.Author contributions
J. P., A. W., I. C. and A. G. performed part of the ionic liquid synthesis and catalytic experiments. R. W. L., L. T. N. and A. N. K. conducted TEM measurements and analysis. J. P. performed the magnetron sputtering deposition of Pd species in ionic liquid and ICP-OES measurements. J. P. and P. L. performed XPS measurements and analysis. D. M. R. performed the DFT calculations. A. N. K., A. W., P. L. and J. A. F. designed the study, analysed the data and co-wrote the paper. All the authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors thank the University of Nottingham Advanced Molecular Materials Interdisciplinary Research Cluster, Propulsion Futures and Green Chemicals Beacons of Excellence for the financial support, the Nanoscale and Microscale Research Centre (nmRC) for access to material characterisation equipment. The authors acknowledge support of EPSRC Metal Atoms on Surfaces and Interfaces (MASI) for Sustainable Future programme grant (EP/V000055/1), EPSRC CDT in Sustainable Chemistry (EP/L015633/1) for the financial support. We are grateful for access to the University of Nottingham's Augusta HPC service. The authors acknowledge the Diamond Light Source and the UK Catalysis Hub for provision of beam time (proposal number SP19850-5 and through the Block Allocation Group (BAG) for Energy Materials under proposal sp17198), and Alan Chadwick and Giannantonio Cibin for assistance with the XAS measurements, and to MAX IV Laboratory for provision of beam time under proposal 20200213, and Stuart Ansell for assistance with the XAS measurements. The authors thank Mark Guyler and Richard Wilson (University of Nottingham) for the help with the magnetron sputtering and reactor laboratory setup.Notes and references
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488(7411), 294–303 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2(6), 65–81 CrossRef CAS.
- D. Murzin, Engineering catalysis, 2013 Search PubMed.
- N.-L. Michels, S. Mitchell and J. Pérez-Ramírez, Effects of Binders on the Performance of Shaped Hierarchical MFI Zeolites in Methanol-to-Hydrocarbons, ACS Catal., 2014, 4(8), 2409–2417 CrossRef CAS.
- G. Prieto, J. Zečević, H. Friedrich, K. P. de Jong and P. E. de Jongh, Towards stable catalysts by controlling collective properties of supported metal nanoparticles, Nat. Mater., 2013, 12(1), 34–39 CrossRef CAS PubMed.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, S. Dawson, X. Liu, L. Lu, K. Dymkowski, F. Fernandez-Alonso, S. Mukhopadhyay, E. K. Gibson, P. P. Wells, S. F. Parker, C. J. Kiely and G. J. Hutchings, Deactivation of a Single-Site Gold-on-Carbon Acetylene Hydrochlorination Catalyst: An X-ray Absorption and Inelastic Neutron Scattering Study, ACS Catal., 2018, 8(9), 8493–8505 CrossRef CAS.
- R. W. Lodge, G. A. Rance, M. W. Fay and A. N. Khlobystov, Movement of palladium nanoparticles in hollow graphitised nanofibres: the role of migration and coalescence in nanocatalyst sintering during the Suzuki–Miyaura reaction, Nanoscale, 2018, 10(40), 19046–19051 RSC.
- S. L. Scott, A Matter of Life(time) and Death, ACS Catal., 2018, 8(9), 8597–8599 CrossRef CAS.
- A. Kolpin, G. Jones, S. Jones, W. Zheng, J. Cookson, A. P. E. York, P. J. Collier and S. C. E. Tsang, Quantitative Differences in Sulfur Poisoning Phenomena over Ruthenium and Palladium: An Attempt To Deconvolute Geometric and Electronic Poisoning Effects Using Model Catalysts, ACS Catal., 2017, 7(1), 592–605 CrossRef CAS.
- C. Lancelot, V. V. Ordomsky, O. Stéphan, M. Sadeqzadeh, H. Karaca, M. Lacroix, D. Curulla-Ferré, F. Luck, P. Fongarland, A. Griboval-Constant and A. Y. Khodakov, Direct Evidence of Surface Oxidation of Cobalt Nanoparticles in Alumina-Supported Catalysts for Fischer–Tropsch Synthesis, ACS Catal., 2014, 4(12), 4510–4515 CrossRef CAS.
- J. Xie, H. M. Torres Galvis, A. C. J. Koeken, A. Kirilin, A. I. Dugulan, M. Ruitenbeek and K. P. de Jong, Size and Promoter Effects on Stability of Carbon-Nanofiber-Supported Iron-Based Fischer–Tropsch Catalysts, ACS Catal., 2016, 6(6), 4017–4024 CrossRef CAS PubMed.
- X. Lan and T. Wang, Highly Selective Catalysts for the Hydrogenation of Unsaturated Aldehydes: A Review, ACS Catal., 2020, 10(4), 2764–2790 CrossRef CAS.
- F. Delbecq and P. Sautet, Competitive C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO Adsorption of α-β-Unsaturated Aldehydes on Pt and Pd Surfaces in Relation with the Selectivity of Hydrogenation Reactions: A Theoretical Approach, J. Catal., 1995, 152(2), 217–236 CrossRef CAS.
C and CO Adsorption of α-β-Unsaturated Aldehydes on Pt and Pd Surfaces in Relation with the Selectivity of Hydrogenation Reactions: A Theoretical Approach, J. Catal., 1995, 152(2), 217–236 CrossRef CAS. - W. Lin, H. Cheng, L. He, Y. Yu and F. Zhao, High performance of Ir-promoted Ni/TiO2 catalyst toward the selective hydrogenation of cinnamaldehyde, J. Catal., 2013, 303, 110–116 CrossRef CAS.
- A. Cabiac, T. Cacciaguerra, P. Trens, R. Durand, G. Delahay, A. Medevielle, D. Plée and B. Coq, Influence of textural properties of activated carbons on Pd/carbon catalysts synthesis for cinnamaldehyde hydrogenation, Appl. Catal., A, 2008, 340(2), 229–235 CrossRef CAS.
- S. Fujiwara, N. Takanashi, R. Nishiyabu and Y. Kubo, Boronate microparticle-supported nano-palladium and nano-gold catalysts for chemoselective hydrogenation of cinnamaldehyde in environmentally preferable solvents, Green Chem., 2014, 16(6), 3230–3236 RSC.
- Y. Li, C.-H. Ge, J. Zhao and R.-X. Zhou, Influence of Preparation Modes on Pt–Ni/CNTs Catalysts Used in the Selective Hydrogenation of Cinnamaldehyde to Hydrocinnamaldehyde, Catal. Lett., 2008, 126(3), 280–285 CrossRef CAS.
- J. Hájek, N. Kumar, P. Mäki-Arvela, T. Salmi, D. Y. Murzin, I. Paseka, T. Heikkilä, E. Laine, P. Laukkanen and J. Väyrynen, Ruthenium-modified MCM-41 mesoporous molecular sieve and Y zeolite catalysts for selective hydrogenation of cinnamaldehyde, Appl. Catal., A, 2003, 251(2), 385–396 CrossRef.
- X. Ni, B. Zhang, C. Li, M. Pang, D. Su, C. T. Williams and C. Liang, Microwave-assisted green synthesis of uniform Ru nanoparticles supported on non-functional carbon nanotubes for cinnamaldehyde hydrogenation, Catal. Commun., 2012, 24, 65–69 CrossRef CAS.
- I. Cano, A. M. Chapman, A. Urakawa and P. W. N. M. van Leeuwen, Air-Stable Gold Nanoparticles Ligated by Secondary Phosphine Oxides for the Chemoselective Hydrogenation of Aldehydes: Crucial Role of the Ligand, J. Am. Chem. Soc., 2014, 136(6), 2520–2528 CrossRef CAS PubMed.
- Q. Wu, C. Zhang, M. Arai, B. Zhang, R. Shi, P. Wu, Z. Wang, Q. Liu, K. Liu, W. Lin, H. Cheng and F. Zhao, Pt/TiH2 Catalyst for Ionic Hydrogenation via Stored Hydrides in the Presence of Gaseous H2, ACS Catal., 2019, 9(7), 6425–6434 CrossRef CAS.
- S. Handjani, E. Marceau, J. Blanchard, J.-M. Krafft, M. Che, P. Mäki-Arvela, N. Kumar, J. Wärnå and D. Y. Murzin, Influence of the support composition and acidity on the catalytic properties of mesoporous SBA-15, Al-SBA-15, and Al2O3-supported Pt catalysts for cinnamaldehyde hydrogenation, J. Catal., 2011, 282(1), 228–236 CrossRef CAS.
- G. Kennedy, L. R. Baker and G. A. Somorjai, Selective Amplification of CO Bond Hydrogenation on Pt/TiO2: Catalytic Reaction and Sum-Frequency Generation Vibrational Spectroscopy Studies of Crotonaldehyde Hydrogenation, Angew. Chem., Int. Ed., 2014, 53(13), 3405–3408 CrossRef CAS PubMed.
- L. Luo, Z. Duan, H. Li, J. Kim, G. Henkelman and R. M. Crooks, Tunability of the Adsorbate Binding on Bimetallic Alloy Nanoparticles for the Optimization of Catalytic Hydrogenation, J. Am. Chem. Soc., 2017, 139(15), 5538–5546 CrossRef CAS PubMed.
- P. Kunal, H. Li, B. L. Dewing, L. Zhang, K. Jarvis, G. Henkelman and S. M. Humphrey, Microwave-Assisted Synthesis of PdxAu100–x Alloy Nanoparticles: A Combined Experimental and Theoretical Assessment of Synthetic and Compositional Effects upon Catalytic Reactivity, ACS Catal., 2016, 6(8), 4882–4893 CrossRef CAS.
- R. W. J. Scott, O. M. Wilson, S.-K. Oh, E. A. Kenik and R. M. Crooks, Bimetallic Palladium−Gold Dendrimer-Encapsulated Catalysts, J. Am. Chem. Soc., 2004, 126(47), 15583–15591 CrossRef CAS PubMed.
- K. Taniya, H. Jinno, M. Kishida, Y. Ichihashi and S. Nishiyama, Preparation of Sn-modified silica-coated Pt catalysts: A new PtSn bimetallic model catalyst for selective hydrogenation of crotonaldehyde, J. Catal., 2012, 288, 84–91 CrossRef CAS.
- Q. Wang, G. Wang, H. Xin, J. Liu, G. Xiong, P. Wu and X. Li, Sn-doped Pt catalyst supported on hierarchical porous ZSM-5 for the liquid-phase hydrogenation of cinnamaldehyde, Catal. Sci. Technol., 2019, 9(12), 3226–3237 RSC.
- P. Dash, N. A. Dehm and R. W. J. Scott, Bimetallic PdAu nanoparticles as hydrogenation catalysts in imidazolium ionic liquids, J. Mol. Catal. A: Chem., 2008, 286(1), 114–119 CrossRef CAS.
- S. Cattaneo, S. J. Freakley, D. J. Morgan, M. Sankar, N. Dimitratos and G. J. Hutchings, Cinnamaldehyde hydrogenation using Au–Pd catalysts prepared by sol immobilisation, Catal. Sci. Technol., 2018, 8(6), 1677–1685 RSC.
- T. Szumełda, A. Drelinkiewicz, R. Kosydar and J. Gurgul, Hydrogenation of cinnamaldehyde in the presence of PdAu/C catalysts prepared by the reverse “water-in-oil” microemulsion method, Appl. Catal., A, 2014, 487, 1–15 CrossRef.
- S. Seraj, P. Kunal, H. Li, G. Henkelman, S. M. Humphrey and C. J. Werth, PdAu Alloy Nanoparticle Catalysts: Effective Candidates for Nitrite Reduction in Water, ACS Catal., 2017, 7(5), 3268–3276 CrossRef CAS.
- M. Luneau, T. Shirman, A. C. Foucher, K. Duanmu, D. M. A. Verbart, P. Sautet, E. A. Stach, J. Aizenberg, R. J. Madix and C. M. Friend, Achieving High Selectivity for Alkyne Hydrogenation at High Conversions with Compositionally Optimized PdAu Nanoparticle Catalysts in Raspberry Colloid-Templated SiO2, ACS Catal., 2020, 10(1), 441–450 CrossRef CAS.
- H. Zhong, M. Iguchi, M. Chatterjee, T. Ishizaka, M. Kitta, Q. Xu and H. Kawanami, Interconversion between CO2 and HCOOH under Basic Conditions Catalyzed by PdAu Nanoparticles Supported by Amine-Functionalized Reduced Graphene Oxide as a Dual Catalyst, ACS Catal., 2018, 8(6), 5355–5362 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Solvent-Free Oxidation of Primary Alcohols to Aldehydes Using Au-Pd/TiO2 Catalysts, Science, 2006, 311(5759), 362–365 CrossRef CAS PubMed.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Catalytically highly active top gold atom on palladium nanocluster, Nat. Mater., 2012, 11(1), 49–52 CrossRef PubMed.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions, Science, 2017, 358(6360), 223–227 CrossRef CAS PubMed.
- X. Jin, K. Taniguchi, K. Yamaguchi and N. Mizuno, Au–Pd alloy nanoparticles supported on layered double hydroxide for heterogeneously catalyzed aerobic oxidative dehydrogenation of cyclohexanols and cyclohexanones to phenols, Chem. Sci., 2016, 7(8), 5371–5383 RSC.
- Z. Zhao, J. Arentz, L. A. Pretzer, P. Limpornpipat, J. M. Clomburg, R. Gonzalez, N. M. Schweitzer, T. Wu, J. T. Miller and M. S. Wong, Volcano-shape glycerol oxidation activity of palladium-decorated gold nanoparticles, Chem. Sci., 2014, 5(10), 3715–3728 RSC.
- L. Deng, M. T. Nguyen, J. Shi, Y.-t. R. Chau, T. Tokunaga, M. Kudo, S. Matsumura, N. Hashimoto and T. Yonezawa, Highly Correlated Size and Composition of Pt/Au Alloy Nanoparticles via Magnetron Sputtering onto Liquid, Langmuir, 2020, 36(12), 3004–3015 CrossRef CAS PubMed.
- M. Schrinner, S. Proch, Y. Mei, R. Kempe, N. Miyajima and M. Ballauff, Stable Bimetallic Gold–Platinum Nanoparticles Immobilized on Spherical Polyelectrolyte Brushes: Synthesis, Characterization, and Application for the Oxidation of Alcohols, Adv. Mater., 2008, 20(10), 1928–1933 CrossRef CAS.
- S. Suzuki, T. Suzuki, Y. Tomita, M. Hirano, K.-i. Okazaki, S. Kuwabata and T. Torimoto, Compositional control of AuPt nanoparticles synthesized in ionic liquids by the sputter deposition technique, CrystEngComm, 2012, 14(15), 4922–4926 RSC.
- I. Cano, A. Weilhard, C. Martin, J. Pinto, R. W. Lodge, A. R. Santos, G. A. Rance, E. H. Åhlgren, E. Jónsson, J. Yuan, Z. Y. Li, P. Licence, A. N. Khlobystov and J. Alves Fernandes, Blurring the boundary between homogenous and heterogeneous catalysis using palladium nanoclusters with dynamic surfaces, Nat. Commun., 2021, 12(1), 4965 CrossRef CAS PubMed.
- S. Marx and A. Baiker, Beneficial Interaction of Gold and Palladium in Bimetallic Catalysts for the Selective Oxidation of Benzyl Alcohol, J. Phys. Chem. C, 2009, 113(15), 6191–6201 CrossRef CAS.
- M. Mamatkulov, I. V. Yudanov, A. V. Bukhtiyarov, I. P. Prosvirin, V. I. Bukhtiyarov and K. M. Neyman, Pd Segregation on the Surface of Bimetallic PdAu Nanoparticles Induced by Low Coverage of Adsorbed CO, J. Phys. Chem. C, 2019, 123(13), 8037–8046 CrossRef CAS.
- Y. Yang, X. Shen and Y.-F. Han, Diffusion mechanisms of metal atoms in PdAu bimetallic catalyst under CO atmosphere based on ab initio molecular dynamics, Appl. Surf. Sci., 2019, 483, 991–1005 CrossRef CAS.
- P. S. West, R. L. Johnston, G. Barcaro and A. Fortunelli, The Effect of CO and H Chemisorption on the Chemical Ordering of Bimetallic Clusters, J. Phys. Chem. C, 2010, 114(46), 19678–19686 CrossRef CAS.
- X. Yang, D. Chen, S. Liao, H. Song, Y. Li, Z. Fu and Y. Su, High-performance Pd–Au bimetallic catalyst with mesoporous silica nanoparticles as support and its catalysis of cinnamaldehyde hydrogenation, J. Catal., 2012, 291, 36–43 CrossRef CAS.
- V. I. Pârvulescu, V. Pârvulescu, U. Endruschat, G. Filoti, F. E. Wagner, C. Kübel and R. Richards, Characterization and Catalytic-Hydrogenation Behavior of SiO2-Embedded Nanoscopic Pd, Au, and Pd–Au Alloy Colloids, Chem. – Eur. J., 2006, 12(8), 2343–2357 CrossRef PubMed.
- L. Luza, C. P. Rambor, A. Gual, J. Alves Fernandes, D. Eberhardt and J. Dupont, Revealing Hydrogenation Reaction Pathways on Naked Gold Nanoparticles, ACS Catal., 2017, 7(4), 2791–2799 CrossRef CAS.
- C. D. T. Nielsen and J. Burés, Visual kinetic analysis, Chem. Sci., 2019, 10(2), 348–353 RSC.
- J. Burés, Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles, Angew. Chem., Int. Ed., 2016, 55(52), 16084–16087 CrossRef PubMed.
- J. Burés, A Simple Graphical Method to Determine the Order in Catalyst, Angew. Chem., Int. Ed., 2016, 55(6), 2028–2031 CrossRef PubMed.
- A. Martínez-Carrión, M. G. Howlett, C. Alamillo-Ferrer, A. D. Clayton, R. A. Bourne, A. Codina, A. Vidal-Ferran, R. W. Adams and J. Burés, Kinetic Treatments for Catalyst Activation and Deactivation Processes based on Variable Time Normalization Analysis, Angew. Chem., Int. Ed., 2019, 58(30), 10189–10193 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00289f |
| This journal is © The Royal Society of Chemistry 2023 |