
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatial imaging of catalyst poisoning with SO2 on Pt/C PEFC electrocatalyst by operando Pt LIII-edge XAFS-CT imaging†
Hirosuke
Matsui
 *a,
Koshin
Sato
a,
Naoko
Isobe
a,
Gabor
Samjeské
a,
Tomoya
Uruga
b and
Mizuki
Tada
*a
*a,
Koshin
Sato
a,
Naoko
Isobe
a,
Gabor
Samjeské
a,
Tomoya
Uruga
b and
Mizuki
Tada
*a
aDepartment of Chemistry, Graduate School of Science & Research Center for Materials Science (RCMS) & Integrated Research Consortium on Chemical Science (IRCCS) & Institute for Advanced Study, Nagoya University, Furo, Chikusa, Nagoya, Aichi 464-8602, Japan. E-mail: matsui.hirosuke.x1@f.mail.nagoya-u.ac.jp; tada.mizuki.u6@f.mail.nagoya-u.ac.jp
bJapan Synchrotron Radiation Research Center, SPring-8, Koto, Sayo, Hyogo 679-5198, Japan
First published on 10th May 2023
Abstract
Sulfur dioxide (SO2), which is one of the air contaminants, causes drastic power loss of Pt electrocatalysts in polymer electrolyte fuel cells (PEFCs). Herein, we report spatial imaging of catalyst poisoning with SO2 on a Pt/C PEFC cathode electrocatalyst in a membrane electrode assembly (MEA) using operando Pt LIII-edge X-ray absorption fine structure (XAFS)-computed tomography (CT) imaging. The operando XAFS clearly suggested that SO2 is strongly adsorbed on active sites on the Pt cathode catalyst surface without the significant aggregation of Pt catalyst particles. However, it was found that subsequent electrochemical treatment at 1.0 V under 20% O2/N2 to remove the adsorbed S species induced not only the removal of S poisoning but also the degradative aggregation of the Pt electrocatalyst. The 3D imaging of the distribution and chemical states of the Pt electrocatalyst showed the degradative manner of the Pt electrocatalyst by the recovery process after the catalyst poisoning with SO2.
Polymer electrolyte fuel cell (PEFC) is a clean and highly efficient energy conversion device, and its practical application for fuel cell vehicles (FCVs) has been developed.1 FCVs require PEFC operations under various practical conditions, and catalyst poisoning at the PEFC cathode by air contaminants, such as SOx and NOx, is one of the essential issues to overcome by the further development of PEFC.2–6 The strong adsorption of S-containing compounds on Pt electrocatalysts often causes a rapid and drastic power loss of PEFC membrane electrode assembly (MEA).7–9
The poisoning of electrocatalysts with SO2 has been investigated by applying electrochemical and spectroscopic techniques. Even at a trace concentration of less than 1 ppm SO2, the total power loss of PEFC leached over 30% after 20 h of SO2 exposure.10 The cyclic voltammetry (CV) of an S-adsorbed Pt/C cathode electrocatalyst showed a significant decrease in the electrochemical active surface area (ECSA). The configurations of adsorbed S species on Pt surfaces in aqueous media were systematically characterized by applying the in situ IR and ATR-SEIRAS techniques.11,12 The analysis of S contaminants on the Pt surface was also conducted by in situ S K-edge XANES analysis13–16 and near-ambient pressure HAXPES analysis.17,18 However, the variation of adsorbed S species makes it difficult to regulate the reactions and removal of adsorbed S species on Pt electrodes for the recovery of electrocatalyst activity.
The electro-oxidation and reduction of adsorbed S species on Pt electrodes have been previously reported to partially recover the activity of poisoned Pt surfaces.19 Moreover, CV cycling,20,21 chronoamperometry (CA),22 and CA cycling11 processes achieved a 70–95% recovery rate of ECSA and power density relative to the original states. DFT calculations suggested that the oxidation and release of adsorbed SO2 proceeded on on-top and bridge Pt sites, but it was difficult to remove strongly coordinated S species even at high potential (>1.5 V).11 The lack of knowledge of the local structures of SO2-poisoned Pt electrocatalysts in practical PEFC MEAs prevents the investigation of the recovery processes of the SO2-poisoned Pt electrocatalysts under PEFC operating conditions.
In this study, we report changes in the local structures of a Pt cathode electrocatalyst in a PEFC MEA by catalyst poisoning with SO2 and subsequent electrochemical catalyst recovering processes by performing an operando X-ray absorption fine structure (XAFS) analysis. The operando X-ray spectroimaging combining XAFS spectroscopy and computed tomography (CT) imaging, which is called XAFS-CT, successfully provided three-dimensional (3D) maps of Pt local structure changes for catalyst poisoning and recovery with SO2 inside the MEA for the first time.
SO2, which is a serious air contaminant, is a strong catalyst poisoner. Changes in the electrochemical performance of a typical MEA with a Pt/C cathode electrocatalyst were investigated. A commercially available MEA with a 50 wt% Pt/C (TEC10E50E, TKK) cathode electrocatalyst and a 50 wt% Ru/C (TECRu(ONLY)50E, TKK) anode electrocatalyst was conditioned by 150 cycles of IV scan and used for consecutive experiments (hereinafter, denoted as fresh MEA) (ESI† 1).
The fresh MEA was treated at 0.4 V by supplying 4 ppm of SO2 diluted with humidified 20% O2/N2 (for electrochemical experiments) or 4 ppm of SO2 diluted with humidified N2 (for operando XAFS measurements to avoid sample damage during X-ray irradiation in the presence of SO2) to the PEFC cathode side (Fig. S1 and S2†). The CVs of the SO2-poisoned MEAs treated with N2 or 20% O2/N2 showed that the difference in the cathode atmosphere (N2 or 20% O2/N2) during the SO2-poisoning process was irrelevant to the CVs of the Pt catalyst (Fig. S3(a)†). The time course plot of the cell current at 0.4 V under 20% O2/N2 showed a significant loss of the cell current against the SO2-exposure time (Fig. S3(b)†). The current loss reached a steady state after 120 min at approximately 50–60% to that of the fresh state. The error ranges of the current decreases were estimated to be within 10% throughout the different batches of similar experiments, indicating that the catalyst poisoning treatment with SO2 was reproduced. As reported in a previous study,23 we increased the SO2 concentration, i.e., to 25 ppm or 50 ppm of SO2 in 20% O2/N2, and the deactivation of the Pt electrocatalyst was quickly completed within 30 min, as shown in Fig. S3(b).† The CV profiles of the Pt catalyst after the SO2-poisoning treatment with various SO2 concentrations (4–50 ppm) were similar, suggesting that the SO2 concentrations affected the catalyst deactivation rate but were irrelevant to the degree of catalyst poisoning (Fig. S3(c)†).
The performance of the MEA before and after the catalyst poisoning with 4 ppm of SO2 was evaluated by applying electrochemical CV and IV measurements. The ECSA of the fresh MEA was 62.5 m2 gPt−1, and the max power density was estimated to be 0.44 W cm−2; these values are similar to our previous report (Fig. 1(a and b)).24 After the SO2 treatment at 0.4 V with 4 ppm of SO2 in 20% O2/N2 at the cathode for 4 h, the hydrogen adsorption and desorption peaks in CV (0.05–0.4 V) almost disappeared, and the ECSA of the SO2-poisoned MEA was only 6.7 m2 gPt−1, which was 11% of that of the fresh MEA (Fig. 1(a)). The Pt oxidation peak above 0.6 V in the CV anodic scan (positive scan) disappeared, and a new peak appeared above 0.8 V, which was attributed to the oxidation of adsorbed S species.25
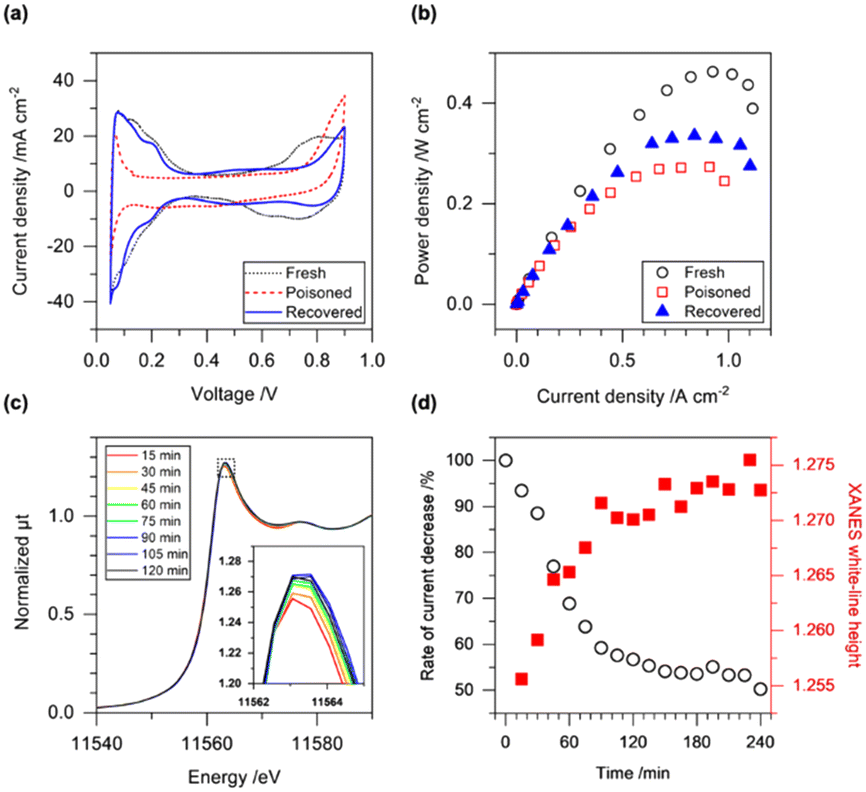 | ||
| Fig. 1 (a) CV profiles and (b) power plots of PEFC MEA after initial conditioning (fresh), after catalyst poisoning with SO2 (poisoned, 0.4 V, 4 ppm of SO2/20% O2/N2 for 4 h), and after electrochemical recovery process (recovered, 1.0 V, 20% O2/N2 for 1 h). (c) Operando time-resolved Pt LIII-edge XANES spectra during the poisoning process with SO2 at 0.4 V. The inset indicates the expanded image around the white-line peak top. (d) Time-course plots of current decrease during the poisoning process with SO2 (left axis, white circle) and Pt LIII-edge XANES white-line height (right axis, red square). | ||
A potential sweep to high voltage causes the oxidation of S absorbates on Pt, and changes in the shapes of the CVs were clearly observed for various potential sweep ranges after the catalyst poisoning with SO2 (Fig. S4†). The hydrogen adsorption and desorption peaks reappeared after the potential sweep greater than 1.0 V (Fig. S4(b)†), and the electrochemical operation procedure with the potential sweep can be used for the recovery procedure of Pt electrocatalyst performance under PEFC operating conditions. Several electrochemical protocols have been reported to recover electrochemical performance after catalyst poisoning with SO2 (Table S1†). For example, the CA cycles (3 cycles) in the range of 0.4–1.5 V for a rotating disk working electrode with a Pt/C electrocatalyst achieved a significant recovery of the SO2-poisoned Pt electrocatalyst (94%),11 and adsorbed S species were reported to be oxidized and desorbed by reactions with water and/or oxygen to form sulfate.
We investigated the oxidation of the S poisoning species at 1.0 V under 20% O2/N2 flow at the cathode for 1 h, which was used as a typical activity recovery procedure for SO2-poisoned Pt electrocatalysts. The CV profile of the SO2-poisoned MEA after the recovery treatment showed a significant recovery of the hydrogen desorption/adsorption peak, and the recovery ratio reached 80% of the fresh MEA (Fig. 1(a)). Notably, the Pt surface after the recovery treatment showed a distinct peak in the CV at 0.2 V, which may indicate the rearrangement of the Pt surface structure by etching with S adsorbates.26 The new peak observed above 0.8 V in the anodic scan of the CV was attributed to the oxidation of S species adsorbed on the Pt surface19 and became smaller compared to that after the SO2 treatment, suggesting the partial desorption of the S adsorbates from the Pt surface (Fig. 1(a)). Although the Pt oxidation peak in the anodic scan was still small compared to the fresh MEA, the max power density of the MEA obtained from the IV profile was recovered to be 77% of the fresh MEA (Fig. 1(b)). Thus, the electrochemical treatment at 1.0 V led to the partial recovery of the redox properties of the SO2-poisoned Pt electrocatalyst although its recovery ratio was imperfect.
Changes in the local structures (valence state and local coordination) of the SO2-poisoned and partially recovered Pt catalysts in the MEA were characterized by operando Pt LIII-edge XAFS analysis. Under PEFC operating conditions with large amounts of water and fuels, hard X-ray XAFS enables the elucidation of the local structures of Pt electrocatalysts in PEFC MEAs.27 The operando Pt LIII-edge X-ray absorption near edge structure (XANES) spectra were recorded every 15 min during the SO2-poisoning treatment at 0.4 V under a supply of 4 ppm SO2 in N2 at the cathode (Fig. 1(c)). After the exposure of SO2, a slight increase and positive shift in the Pt LIII-edge white-line peak were observed. They were almost steady after 120 min, and the time course of the cell current decrease was also steady after 120 min, in agreement with the trend in the changes in the Pt LIII-edge XANES spectra (Fig. 1(d)). Hence, the changes in the Pt LIII-edge XANES spectra were consistent with the SO2 poisoning of the Pt electrocatalyst.
We investigated the local structure of the SO2-poisoned Pt electrocatalyst and that after the recovery process by the curve-fitting analysis of the operando Pt LIII-edge XANES and extended X-ray absorption fine structure (EXAFS) spectra measured at cell voltages of 0.4 V and 1.0 V. The XANES spectra of the fresh MEA at 0.4 V and 1.0 V showed clear differences in the white-line region of the Pt LIII-edge spectra, indicating the high redox sensitivity of the Pt surface in the fresh MEA (Fig. 2(a-1)). The k3-weighted EXAFS-Fourier transform (FT) of the fresh MEA at 1.0 V was successfully fitted with the 2 shells of Pt–Pt and Pt–O bonds; the coordination numbers (CNs) of the Pt–Pt and Pt–O bonds were estimated to be 8.0 ± 0.7 and 0.9 ± 0.3, respectively (Fig. 2(b-1) and Table S2†). The CNs at 0.4 V were 8.6 ± 0.8 and 0.1 ± 0.2, respectively, indicating that the Pt–O bonds were negligible on the Pt catalyst surface at 0.4 V. These results agree with our previous results,28 and the CN of the Pt–O bond (0.9 ± 0.3) at 1.0 V suggested a high redox response to the oxidation of the Pt catalyst surface in fresh MEA.
 | ||
| Fig. 2 (a) Operando Pt LIII-edge XANES spectra and (b) k3-weighted EXAFS-Fourier transforms (black line) with curve-fitting results (red dotted line) of the Pt/C cathode electrocatalyst in the PEFC MEA measured at the cell voltages of 0.4 V and 1.0 V. (1) After the initial conditioning (fresh), (2) after the catalyst poisoning with SO2 (poisoned), and (3) after the subsequent electrochemical recovery process (recovered). | ||
After treatment with SO2, both the XANES and EXAFS spectra significantly changed, as shown in Fig. 2(a-2 and b-2). The difference in Pt LIII-edge XANES white-line heights at 0.4 V was small between the MEA before and after the SO2 treatment (Fig. 2(a-2)). Moreover, the Pt LIII-edge XANES white-line height at 1.0 V after the SO2 treatment was smaller than that of the fresh MEA at 1.0 V. A previous Pt LIII-edge EXAFS study on an S-contaminated Pt/C electrocatalyst in an aqueous half cell reported the existence of two-scattering shells of Pt–Pt and Pt–O bonds without Pt–S bonds;29 however, our curve-fitting analysis of the recorded EXAFS-FTs after the treatment with SO2 suggested the formation of Pt–S bond at 0.229 nm, whose CN was found to be 0.6 ± 0.3 at 0.4 V. The CN of Pt–Pt was observed to be 8.5 ± 1.1, which was similar to that of the fresh MEA (8.6 ± 0.8). Accompanied by the appearance of the Pt–S bond, the CN of Pt–O at 1.0 V (0.6 ± 0.2) became relatively smaller compared to that of the fresh MEA (0.9 ± 0.3) (Fig. 2(b-2) and S5, and Table S3†). It was reported that S adsorption on Pt proceeds under the formation of a single S overlayer without the dissociation of Pt–Pt bonds.30 A  R45-S structure,31 a c(2 × 2)-S structure on Pt(100),32 and a
R45-S structure,31 a c(2 × 2)-S structure on Pt(100),32 and a 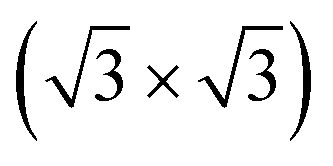 R30-S structure on Pt(111)33 were proposed as S-adsorbed Pt surfaces. The curve-fitting results of the EXAFS-FTs agree with the reported results and indicate that the surface of the Pt electrocatalyst was covered by S absorbates by the SO2 exposure.
R30-S structure on Pt(111)33 were proposed as S-adsorbed Pt surfaces. The curve-fitting results of the EXAFS-FTs agree with the reported results and indicate that the surface of the Pt electrocatalyst was covered by S absorbates by the SO2 exposure.
The subsequent electrochemical recovery process at 1.0 V in 20% O2/N2 at the cathode for 1 h resulted in further changes in the local structure of the Pt electrocatalyst. The XANES spectrum of the recovered MEA measured at 0.4 V was similar to that of the fresh MEA, but its white-line height at 1.0 V was slightly smaller than that of the fresh MEA, as shown in Fig. 2(a-3). The curve-fitting analysis of the EXAFS-FTs after the recovery process showed a negligible contribution of the Pt–S bond (0.0 ± 0.2) at both 0.4 V and 1.0 V, implying that most of the S absorbates were released from the Pt catalyst surface (Fig. 2(b-3) and S5, and Table S4†). Notably, increases in the Pt–Pt CNs (9.0–9.1 at 0.4 V; 8.5 at 1.0 V) were observed after the recovery process, suggesting the aggregation of the Pt electrocatalyst by the recovery process.
We measured the transmission electron microscopy (TEM) images of four Pt samples collected from MEAs similarly prepared and treated (fresh; after the catalyst poisoning with SO2; after the SO2 treatment and the subsequent electrochemical recovery process; and after the electrochemical recovery process without the SO2 treatment), the results of which are illustrated in Fig. 3. The average particle sizes of the Pt electrocatalysts were estimated to be 2.6 ± 0.6 nm (fresh), 2.7 ± 0.5 nm (after the catalyst poisoning with SO2), 3.4 ± 0.9 nm (after the SO2 treatment and the subsequent electrochemical recovery process), and 2.8 ± 0.7 nm (after the electrochemical recovery process without the SO2 treatment). The fact that the growth of the Pt particle size was observed for the sample after both SO2 treatment and the subsequent electrochemical recovery process clearly suggested that the subsequent electrochemical treatment for the recovery of electrocatalyst activity caused the aggregation of the Pt electrocatalyst in the MEA. Exposure to SO2 itself did not result in irreversible aggregation of the Pt electrocatalyst. However, the subsequent electrochemical treatment induced not only the removal of the adsorbed S species (poisoner) but also the aggregation of the Pt catalyst particles in the PEFC cell.
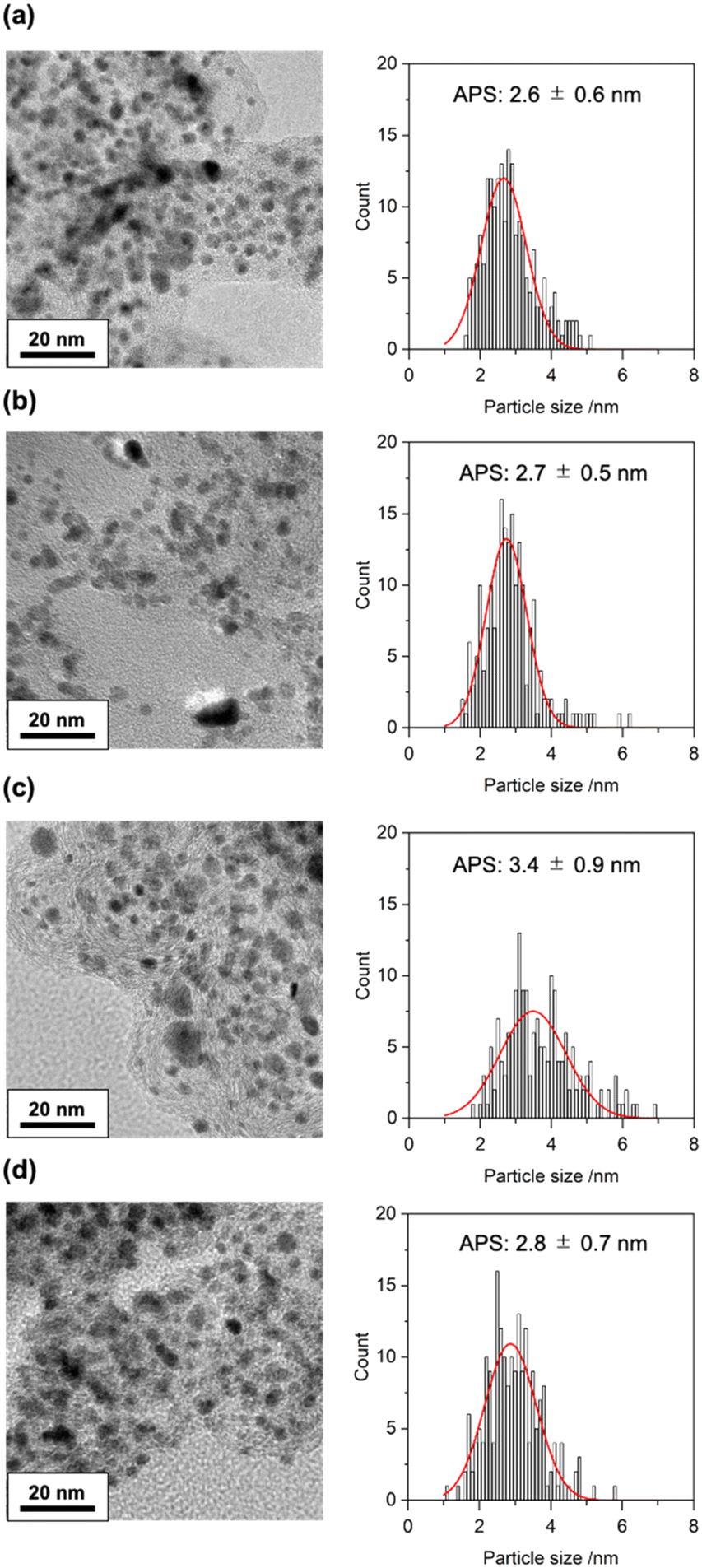 | ||
| Fig. 3 TEM images and particle size distributions of the Pt electrocatalysts in the MEAs (a) after the conditioning (fresh), (b) after the poisoning with 4 ppm of SO2 balanced with 20% O2/N2 at 0.4 V for 4 h, (c) after the subsequent electrochemical recovery treatment in 20% O2/N2 at 1.0 V for 1 h for the SO2-poisoned MEA, and (d) after the electrochemical treatment in 20% O2/N2 at 1.0 V for 1 h for the fresh MEA. Red curves indicate Gaussian fitting curves to estimate the average particle size (APS; peak top) and its error range (full-width half maximum). | ||
The heterogeneous structure and distribution of catalyst particles in a real MEA cause spatial differences in the reactivity of electrocatalysts in the MEA, and the operando 3D spectroimaging using XAFS-CT is a powerful tool for visualizing the spatial distribution of structural differences of PEFC electrocatalysts in MEAs.34–39 XAFS-CT imaging, which is the combination of XAFS spectroscopy and CT imaging, provides not only the 3D morphological image of a sample but also the 3D images of element-specific chemical states inside a material. As depicted in Fig. 2(a), the Pt LIII-edge XANES white-line height at 1.0 V was indicative of the catalyst poisoning with SO2. We investigated the operando XAFS-CT spectroimaging at the Pt LIII-edge to visualize the 3D degradation manner of the Pt electrocatalyst in the practical MEA by the catalyst poisoning with SO2 and the subsequent electrochemical recovery process with a voxel resolution of 1.3 μm.
The operando XAFS-CT images of the fresh MEA and the recovered MEA (after the catalyst poisoning with SO2 and the electrochemical recovery process) were measured at 1.0 V under N2 flow at the cathode. We recorded the 3D XAFS-CT data for the same view of the MEA for the fresh and recovered states using an on-site catalyst poisoning and recovery system. The operando measurement of one XAFS-CT dataset required at least 1.1 h of X-ray irradiation, and the sample damage was found to be serious in the presence of SO2. Hence, we skipped the operando XAFS-CT measurement of the SO2-poisoned MEA in the presence of SO2 and compared the XAFS-CT images of the fresh with those of the recovered states.
The reconstruction of the Pt LIII-edge XAFS-CT data was processed for three extracted parameters: (1) morphology obtained by the reconstruction of Pt LIII-edge XANES background intensity, (2) Pt density obtained by the reconstruction of the Pt LIII-edge jump, and (3) Pt valence state obtained by the reconstruction of the Pt LIII-edge white-line height. The first and second images show the structure and Pt catalyst distribution of the cathode catalyst layer in the PEFC MEA (Fig. 4(a and b) and S7(a and b)†). The morphology images of the fresh MEA and the recovered ones revealed significant expansion of cracks in the cathode catalyst layer (Fig. 4(a) and S7(a)†). It was reported that exposure to SO2 enhanced the formation of radical species for the 2e− ORR pathway,40,41 and treatments at high potential caused the rapid corrosion of carbon support.42 The observed changes in the 3D morphology images indicate the corrosion of carbon support in the catalyst layer.
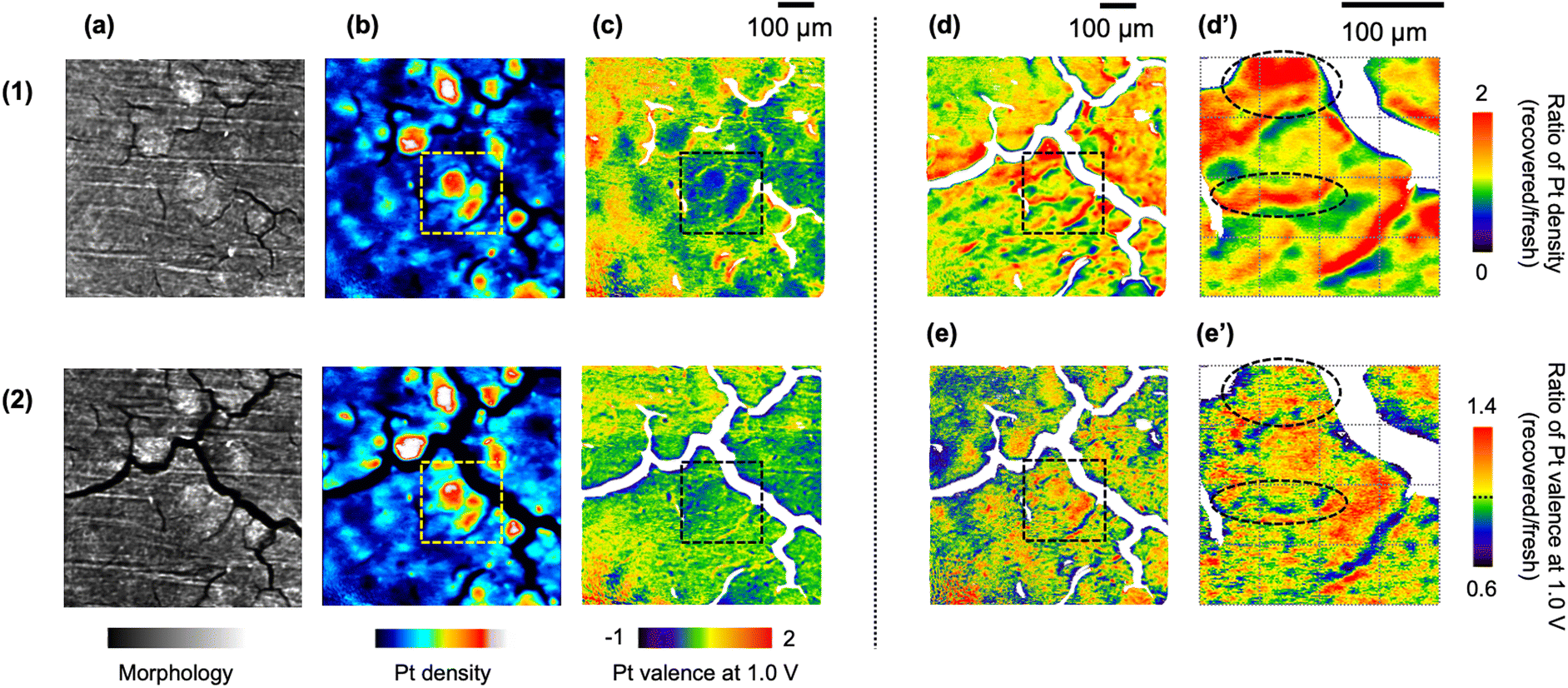 | ||
| Fig. 4 Cross-sectional images at the center of the cathode catalyst layer (Z = 20 μm). (a) Morphology (the XANES background), (b) Pt density (the Pt LIII-edge jump), and (c) Pt valence at the cell voltage of 1.0 V (the normalized Pt LIII-edge XANES white-line height) of the same MEA for (1) the initial conditioning (fresh) and (2) after the catalyst poisoning with SO2 and the subsequent electrochemical recovery treatment. (d) The ratio of Pt density estimated by (b-2)/(b-1). (e) The ratio of Pt valence at 1.0 V estimated by (c-2)/(c-1). (d′ and e′) Enlarged views of dotted square areas in (d) and (e), respectively. The field-of-view and voxel size of the XAFS-CT imaging were 665.6 μm × 665.6 μm × 665.6 μm and 1.3 μm × 1.3 μm × 1.3 μm, respectively. | ||
Regarding the pattern of the crack structure in the cathode catalyst layer, the distribution of the Pt electrocatalyst was clearly visualized on the Pt density images (Fig. 4(b) and S7(b)†). The catalyst poisoning and recovery processes maintained the heterogeneity of the Pt catalyst distribution, but significant changes in Pt density were also observed after the catalyst poisoning and recovery treatments. The ratio of Pt density, which was defined as (Pt density image of the recovered MEA; Fig. 4(b-2))/(that of the fresh MEA; Fig. 4(b-1)), was calculated, as shown in Fig. 4(d) and S7(d),† suggesting spatial changes in the Pt distribution by sequential processes. The image of the ratio of Pt density directly showed the spatial position of the Pt aggregation in the cathode catalyst layer, which was confirmed by Pt LIII-edge EXAFS and TEM analyses. We similarly calculated the image of the ratio of the Pt valence at 1.0 V, as shown in Fig. 4(e) and S7(e).† The enlarged views (dashed squares in Fig. 4(d) and (e)) showed spatial variations in Pt migration and Pt valence changes (Fig. 4(d′ and e′)). Areas circled by dotted lines in Fig. 4(e′) with an increase or decrease in the Pt valence at 1.0 V were observed as parts with an increase in the Pt density illustrated in Fig. 4(d′) by sequential processes. Thus, 3D XAFS-CT imaging clearly demonstrated a heterogeneous degradation manner in the MEA.
The 3D imaging data contained the 8 million data sets of the structural parameters, and we prepared the kernel density plots of the Pt density and the Pt valence state at 1.0 V for the fresh and recovered MEAs (Fig. 5(a and b), respectively). The steep inclination of the kernel density plot of the Pt density (Fig. 5(a)) suggested the aggregation of the Pt electrocatalyst in the MEA after the catalyst poisoning with SO2 and subsequent recovering processes. H2O2 generation was reported to accelerate the degradation of Pt electrocatalyst on the SO2-poisoned Pt surface.40,41 The kernel density plot of the Pt valence at 1.0 V showed a gentle slope and wide variation, indicating that the Pt valence state at 1.0 V tended to be reduced in the recovered MEA (Fig. 5(b)).
 | ||
| Fig. 5 The kernel density plots of (a) the Pt density and (b) Pt valence state at 1.0 V for the fresh and recovered MEA. (c) The kernel density plot between the ratio of Pt density (Pt density of the recovered MEA/that of the fresh MEA) and the ratio of Pt valence at 1.0 V (Pt valence at 1.0 V of the recovered MEA/that of the fresh MEA). The kernel density map is a smoothed color density representation of a scatter plot based on kernel density estimation, a nonparametric method of probability distribution functions. The white dashed lines in (c) border the positive and negative trends and are divided into four quadrants (i–iv). (d) Ratio of voxels in the four quadrants (i–iv) to the total. (e) Spatial map of Pt species at the cathode center categorized in four quadrants (i; red, ii: green, iii; blue, and iv; yellow). | ||
The kernel density plot between the ratio of Pt density (Fig. 5(a)) and the ratio of Pt valence at 1.0 V (Fig. 5(b)) shows a relationship between changes in the two ratios of Pt density and Pt valence for the catalyst poisoning and recovery processes (Fig. 5(c)). The data appeared around the cross point of the two dashed lines depicted in Fig. 5(c), which separate four quadrants in the plot. For example, data in the first quadrant correspond to voxels whose Pt density increased, and Pt valence also increased during the recovery process. The numbers of voxels in the four quadrants differed, as shown in Fig. 5(d): the fractions of the first, second, third, and fourth quadrants were found to be 25.2%, 18.4%, 14.5%, and 41.9%, respectively. Most of the data were found in the fourth quadrant (the ratio of Pt density >1 and the ratio of Pt valence at 1.0 V < 1), corresponding to Pt aggregation with a loss of active surface sites. Thus, the cross-sectional maps of the location of the Pt species categorized in the four quadrants (Fig. 5(e) and S8†) were the spatial maps of catalyst degradation during the sequential catalyst poisoning/recovery processes.
In conclusion, we investigated the structural changes in the Pt cathode electrocatalyst in an MEA caused by catalyst poisoning with SO2 by operando Pt LIII-edge XAFS, TEM, and operando XAFS-CT imaging. SO2 acted as a serious poisoner of the Pt electrocatalyst, and S absorbates inhibited the coordination of oxygen on the Pt catalyst surface without the aggregation of the Pt catalyst particles in the MEA. However, the subsequent catalyst recovery process by an electrochemical treatment at 1.0 V under 20% O2/N2 at the cathode caused not only the removal of the S poisoner on the Pt surface but also the aggregation of the Pt catalyst particles. The operando XAFS-CT imaging and its statistical data analysis revealed the spatial variation in Pt electrocatalyst degradation in the MEA heterogeneously for the catalyst poisoning and recovery processes with SO2. The further development of catalyst recovery protocols at lower potential will be the key to achieving the full recovery of the SO2-poisoned MEA without the irreversible aggregation of the Pt electrocatalyst.
This research was supported by the New Energy and Industrial Technology Development Organization (NEDO) Program and the R-ing (Reaction Infography) World Research Unit (B-1) at Nagoya University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Debe, Nature, 2012, 486, 43 CrossRef PubMed.
- B. D. Gould, O. A. Baturina and K. E. Swider-Lyons, J. Power Sources, 2009, 188, 89 CrossRef CAS.
- O. A. Baturina and K. E. Swider-Lyons, J. Electrochem. Soc., 2009, 156, B1423 CrossRef CAS.
- Y. Zhai, G. Bender, K. Bethune and R. Rocheleau, J. Power Sources, 2014, 247, 40 CrossRef CAS.
- J. A. Prithi, B. S. Viswanath, N. Rajalakshmi and K. S. Dhathathreyem, Fuel Cells, 2017, 17, 308 CrossRef CAS.
- S. Tsuhima, K. Kaneko and S. Hirai, ECS Trans., 2010, 33, 1645 CrossRef.
- U. Misz, A. Talke, A. Heinzel and G. Konrad, Fuel Cells, 2016, 16, 444 CrossRef CAS.
- M. Angelo and J. St-Pierre, ECS Trans., 2014, 64, 773 CrossRef CAS.
- R. Mohtadi, W.-K. Lee and J. W. Van Zee, J. Power Sources, 2004, 138, 216 CrossRef CAS.
- Y. Zhai, G. Bender, S. Dorn, M. Angelo, K. Bethune and R. Rocheleau, ECS Trans., 2008, 16, 873 CrossRef CAS.
- Y. Liu, W. Zhang, G. Han, Y. Zhou, L. Li, F. Kong, Y. Gao, C. Du, J. Wang and L. Du, ACS Catal., 2021, 11, 9293 CrossRef CAS.
- I. Moraes, M. Weber and F. Nart, Electrochim. Acta, 1997, 42, 617 CrossRef CAS.
- O. Baturina, B. Gould, A. Korovina, Y. Garsanny, R. Stroman and P. Northrup, Langmuir, 2011, 27, 14930 CrossRef CAS PubMed.
- O. Baturina, B. Gould, P. Northrup and K. Swider-Lyons, Catal. Today, 2013, 205, 106 CrossRef CAS.
- K. Isegawa, T. Nagami, S. Jomori, M. Yoshida and H. Kondoh, Phys. Chem. Chem. Phys., 2016, 18, 25183 RSC.
- K. Isegawa, D. Kim and H. Kondoh, RSC Adv., 2018, 8, 38204 RSC.
- L. Yu, Y. Takagi, T. Nakamura, T. Sakata, T. Uruga, M. Tada, Y. Iwasawa, S. Masaoka and T. Yokoyama, J. Phys. Chem. C, 2019, 123, 603 CrossRef CAS.
- S. Chaveanghong, T. Nakamura, Y. Takagi, B. Cagnon, T. Uruga, M. Tada, Y. Iwasawa and T. Yokoyama, Phys. Chem. Chem. Phys., 2021, 23, 3866 RSC.
- T. Reshetenko, V. Laue, U. Krewer and K. Artyushkova, J. Power Sources, 2019, 438, 226949 CrossRef CAS.
- Y. Zhai, G. Bender, S. Dorn and R. Rocheleau, J. Electrochem. Soc., 2010, 157, B20 CrossRef CAS.
- Y. Fu, M. Hou, C. Du, Z. Shao and B. Yi, J. Power Sources, 2009, 187, 32 CrossRef.
- B. Gould, G. Bender, K. Bethune, S. Dorn, O. Baturina, R. Rocheleau and K. Swider-Lyons, J. Electrochem. Soc., 2010, 157, B1569 CrossRef CAS.
- S. Tsushima, K. Kaneko, H. Morioka and S. Hirai, J. Therm. Sci. Technol., 2012, 7, 619 CrossRef CAS.
- S. Ozawa, H. Matsui, N. Ishiguro, Y. Tan, N. Maejima, M. Taguchi, T. Uruga, O. Sekizawa, T. Sakata, K. Nagasawa, K. Higashi and M. Tada, J. Phys. Chem. C, 2018, 122, 14511 CrossRef CAS.
- A. Zolfaghari, F. Villiard, M. Chayer and G. Jerkiewicz, J. Alloys Compd., 1997, 253, 481 CrossRef.
- M. Foral and S. Langer, J. Electroanal. Chem. Interfacial Electrochem., 1988, 246, 193 CrossRef CAS.
- N. Ishiguro and M. Tada, Catal. Lett., 2018, 148, 1597 CrossRef CAS.
- H. Matsui, N. Ishiguro, Y. Tan, N. Maejima, Y. Muramoto, T. Uruga, K. Higashi, D. Nguyen, H. Dam, G. Samjeske and M. Tada, ChemNanoMat, 2022, 8, e202200008 CrossRef CAS.
- D. Ramaker, D. Gatewood, A. Korovina, Y. Garsany and K. Swider-Lyons, J. Phys. Chem. C, 2010, 114, 11886 CrossRef CAS.
- T. Fischer and S. Kelemen, J. Catal., 1978, 53, 24 CrossRef CAS.
- I. Villegas and M. J. Weaver, J. Electroanal. Chem., 1994, 373, 245 CrossRef CAS.
- H. Gutleben and E. Bechthold, Langmuir, 1989, 5, 990 CrossRef CAS.
- Y.-E. Sung, W. Chrzanowski, A. Zolfaghari, G. Jerkiewicz and A. Wieckowski, J. Am. Chem. Soc., 1997, 119, 194 CrossRef CAS.
- H. Matsui, N. Ishiguro, T. Uruga, O. Sekizawa, K. Higashi, N. Maejima and M. Tada, Angew. Chem., Int. Ed., 2017, 56, 9371 CrossRef CAS PubMed.
- H. Matsui, S. Takao, K. Higashi, T. Kaneko, G. Samjeske, T. Uruga, M. Tada and Y. Iwasawa, ACS Appl. Mater. Interfaces, 2022, 14, 6762 CrossRef CAS PubMed.
- J. Becher, D. F. Sanchez, D. E. Doronkin, D. Zengel, D. M. Meira, S. Pascarelli, J.-D. Grunwaldt and T. L. Sheppard, Nat. Catal., 2021, 4, 46 CrossRef CAS.
- D. F. Sanchez, J. Ihli, D. Zhang, T. Rohrbach, P. Zimmermann, J. Lee, C. N. Borca, N. Böhlen, D. Grolimund, J. A. van Bokhoven and M. Ranocchiari, Angew. Chem., Int. Ed., 2021, 60, 10032 CrossRef PubMed.
- J. E. Schmidt, R. Oord, W. Guo, J. D. Poplawsky and B. M. Weckhuysen, Nat. Commun., 2017, 8, 1666 CrossRef PubMed.
- J. Zhang, Q. Wang, S. Li, Z. Jiang, S. Tan, X. Wang, K. Zhang, Q. Yuan, S.-J. Lee, C. J. Titus, K. D. Irwin, D. Nordlund, J.-S. Lee, P. Pianetta, X. Yu, X. Xiao, X.-Q. Yang, E. Hu and Y. Liu, Nat. Commun., 2020, 11, 6342 CrossRef CAS PubMed.
- Y. Garsany, O. Baturina and K. Swider-Lyons, J. Electrochem. Soc., 2009, 156, B848 CrossRef CAS.
- T. Reshetenko, V. Laue, U. Krewer and K. Artyushkova, J. Power Sources, 2020, 458, 228032 CrossRef CAS.
- J. Wu, X. Yuan, J. Martin, H. Wang, H. Zhang, J. Shen, S. Wu and W. Merida, J. Power Sources, 2008, 184, 104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, electrochemistry, XAFS-CT analysis. See DOI: https://doi.org/10.1039/d3cy00241a |
| This journal is © The Royal Society of Chemistry 2023 |