
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolated PdO sites on SiO2-supported NiO nanoparticles as active sites for allylic alcohol selective oxidation†‡
Aleksandra
Ziarko
a,
Thomas J. A.
Slater
 b,
Mark A.
Isaacs
cd,
Lee J.
Durndell
e and
Christopher M. A.
Parlett
*fghi
b,
Mark A.
Isaacs
cd,
Lee J.
Durndell
e and
Christopher M. A.
Parlett
*fghi
aDepartment of Chemical Engineering and Applied Chemistry, Aston University, Birmingham, B4 7ET, UK
bSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
cDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
dHarwellXPS, Research Complex at Harwell, RAL, Harwell Campus, Didcot, OX11 0FA, UK
eSchool of Geography, Earth and Environmental Sciences, University of Plymouth, Plymouth, PL4 8AA, UK
fDepartment of Chemical Engineering, University of Manchester, Manchester, M13 9PL, UK. E-mail: christopher.parlett@manchester.ac.uk
gUniversity of Manchester at Harwell, Diamond Light Source, Rutherford Appleton Laboratory, Didcot, Oxfordshire OX11 0DE, UK
hDiamond Light Source, Rutherford Appleton Laboratory, Didcot, Oxfordshire OX11 0DE, UK
iUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Oxfordshire OX11 0FA, UK
First published on 12th May 2023
Abstract
Catalytic allylic alcohol oxidation to aldehydes is an industrial process that necessitates chemoselectivity. Surface PdO (on Pd) enables this transformation but does not represent optimal metal utilisation. Here we report a facile synthesis route to produce isolated surface PdO catalytic sites on an earth-abundant metal (NiO) for cinnamyl alcohol oxidation.
Palladium nanoparticles (NP) are ubiquitous within heterogeneous catalysis due to their activity within a broad spectrum of catalytic applications, including C–C coupling,1 hydrogenation,2 and oxidation.3 The selective oxidation (selox) of alcohols to their carbonyl derivatives, a vital conversion within the fine chemical industry, is one application where Pd based catalysts represent some of the most active reported systems.3 The product from the oxidation of an allylic alcohol, e.g. cinnamaldehyde, finds application in the food industry as it confers the aroma and flavours of cinnamon,4 can act as a food preservative,5 and is a precursor to an artificial sweetener.6 However, allylic alcohol selox requires chemoselectivity, specifically targeting the alcohol functionality within the multifunctional substrate. Such control over reaction selectivity enhances process sustainability through optimal desired product yield by minimising unwanted side product formation, which is critical to simplifying product isolation, a key factor for industrial chemical production.
Supported Pd NPs on various common support materials have been investigated for alcohol selox,7–10 with high surface area silica supports, comprising three-dimensional pore architectures or hierarchical bimodal porosity, reported for allylic alcohols.11,12 These supports facilitate high metal dispersions, with deposited NPs exhibiting high activity through the increased generation of surface PdO, the identified active species. This active site has been further confirmed through complementary operando studies.13 Optimal formation of surface PdO, and therefore efficient Pd utilisation, is critical to escalating overall selox performance while simultaneously enhancing the economics and sustainability of such catalytic systems.
While the miniaturisation of NPs to the few nm regime results in an ever-increasing surface-to-bulk ratio, even at such minute sizes optimal utilisation of the metal component is not realised until the diameter reaches 1 nm as, until this size, a significant amount of the metal is inaccessible within the bulk of the NP.14 Therefore, there is considerable scope for further development. Single atom catalysts, where isolated metal sites are supported on carbon or oxide supports,15 represent one approach, with core-shell structures a second.16,17 The latter looks to replace the metal beneath the surface of the NP with a second one, ideally utilising a cheaper, more earth-abundant metal as the core material.18 Such structures based on Ni core/Pd shells have been prepared via two-step reductions,18,19 co-reduction,20,21 sonication,22 and ligand assisted,23 with applications in fuel cells,18 C–H bond arylation,20 and coupling and reduction reactions.19,21–23 In contrast, NiO core PdO shell materials, the focus of less interest to date, can be produced via a two-step polyol reduction for methane oxidation.24
Here we report the development of highly dispersed PdO sites on silica support NiO NPs for the selox of cinnamyl alcohol produced via a galvanic displacement mechanism. The resulting catalyst displays efficient utilisation of Pd, which results in high performance and selectivity towards the desired product. To confirm the nature of active species and elucidate the deactivation pathway, operando X-ray absorption spectroscopy (XAS) has been employed.
Dendritic mesoporous silica nanospheres were prepared by the method of Shen et al.25 using cyclohexane as the organic phase to produce a single-generation architecture. The pore templating agent was removed by calcination at 600 °C, with the solid sample denoted SiO2. The subsequent SiO2 support was impregnated using an ethylene glycol assist protocol with an aqueous solution of nickel nitrate and ethylene glycol in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ratio with Ni adjusted to achieve a nominal 5 wt% loading.26 The sample was dried at 50 °C before calcination at 400 °C. The resulting sample is denoted NiO/SiO2. Pd deposition onto the NiO/SiO2 was achieved by stirring the solid in a 1 mM Ammonium tetrachloropalladate(II) aqueous solution for 6 hours before recovering by centrifuge and drying at 100 °C (note the absence of a reduction step) with the sample denoted NiO@PdO/SiO2. The final catalyst was characterised by various techniques and screened for cinnamyl alcohol selox conducted under 1 Bar bubbling O2 at 80 °C. Full experimental details can be found in the ESI.†
:1 mol ratio with Ni adjusted to achieve a nominal 5 wt% loading.26 The sample was dried at 50 °C before calcination at 400 °C. The resulting sample is denoted NiO/SiO2. Pd deposition onto the NiO/SiO2 was achieved by stirring the solid in a 1 mM Ammonium tetrachloropalladate(II) aqueous solution for 6 hours before recovering by centrifuge and drying at 100 °C (note the absence of a reduction step) with the sample denoted NiO@PdO/SiO2. The final catalyst was characterised by various techniques and screened for cinnamyl alcohol selox conducted under 1 Bar bubbling O2 at 80 °C. Full experimental details can be found in the ESI.†
Porous silica nanospheres were confirmed by scanning transmission electron microscopy (STEM) and N2 isotherms (Fig. S1 and S2†), with average particle sizes and BJH pore diameters of 124 nm and 3.2 nm, respectively, and a BET surface area of 340 m2 g−1 (Table S1†). Imaging clearly reveals the radial pore architecture, which maximises mesopore openings.25 Ethylene glycol-assisted impregnation of Ni nitrate at a nominal 5 wt% loading, and subsequent thermal processing in air, yielded highly dispersed NiO nanoparticles, confirmed by X-ray diffraction (XRD), with average particle sizes of 2.2 nm and 2.3 nm by XRD and STEM, respectively (Fig. S3 and S4†). Uniform dispersion of NiO throughout the support (and the absence of large sintered Ni species) confirms the efficient utilisation of the mesopore skeleton, with over 93% of the NiO nanoparticles being under 3 nm, the average mesopore size (Table S1†). A significant drop in mesopore volume and BET surface area (Fig. S2 and Table S1†) is further evidence of these species residing within the mesopores. X-ray fluorescence (XRF) gave a Ni loading of 4.95 wt%, close to the nominal loading, with static hydrogen chemisorption evaluating Ni dispersion at 28%.
The substantial difference in isoelectric point of SiO2 and NiO, occurring at pH ∼427 and ∼8.2,28 respectively, allowed for control over the final location of the Pd sites,29 with the aim to prevent depositing Pd on the silica, which due to its weakly interacting nature results in poor metal stability against sintering during thermal processing. At pH 7, above its isoelectric point, SiO2 presents a negative charged surface with the opposite true for NiO. Therefore, the anionic Pd salt should result in mutually exclusive deposition onto the NiO with repulsion from the silica surface. Analysis of the deposition solution, post 6 h contact time with the NiO/SiO2 solid, indicated a ∼10% uptake of Pd, corresponding to a 0.38 wt% Pd loading. Analysis of the deposition solution revealed leaching of an equal molar Ni content. Hence, the process of incorporating Pd into our catalyst occurs via galvanic displacement of Ni by Pd,30,31 due to the difference in standard electrode potentials.31 XRF of the final solid catalyst NiO@PdO/SiO2 gave a Pd loading of 0.4 ± 0.06 wt% and a Ni loading of 4.72 ± 0.25 wt%, a decline of 0.23 wt%, which correspond to a bulk Ni:Pd ratio of 22.6, and a surface ratio of only 6.3.
STEM imaging, shown in Fig. 1 and S5,† shows the retainment of the support architecture after Pd deposition, with high-resolution imaging showing a comparable metal particle size to the parent NiO/SiO2 material. Fourier transform of the STEM image confirms these metal particles to be the parent NiO NPs, with no evidence for the co-existence of Pd NPs on the silica support. Furthermore, STEM-EDX confirms both the co-location of both Pd and Ni within single particles deposited on the SiO2 support and the absence of isolated Pd particles on the silica support.
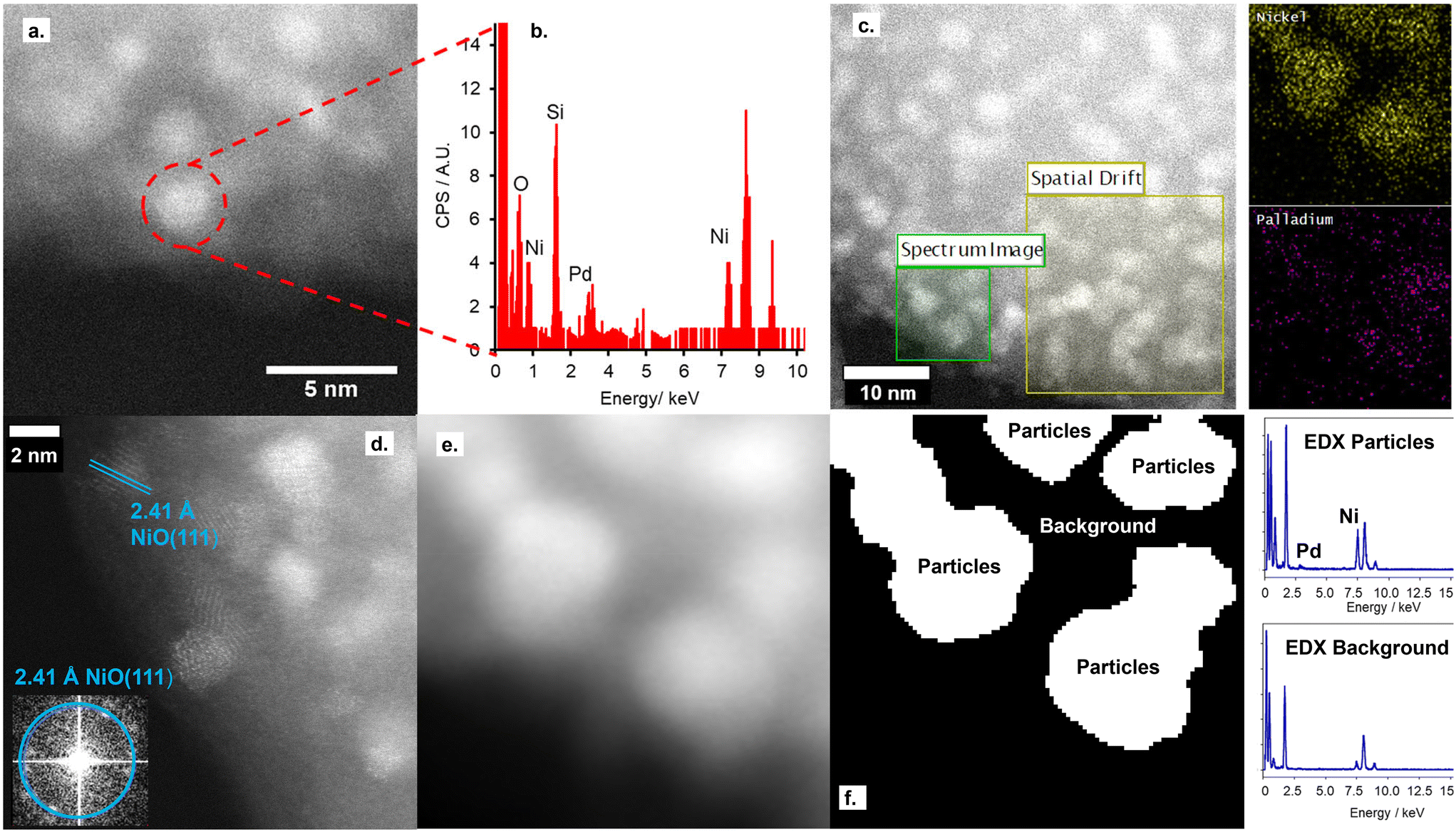 | ||
| Fig. 1 a. High-angle annular dark field (HAADF)-STEM of NiO@PdO/SiO2, b. selected area energy dispersive X-ray (EDX) spectrum of highlighted nanoparticles, c. EDX mapping of NiO@PdO/SiO2, d. atomic resolution imaging of NiO@PdO/SiO2 with Fourier transform of the STEM image (inset), and e. and f. HAADF-STEM imaging and applied particle and background mask for EDX spectra subtractions with corresponding EDX spectra. | ||
Nitrogen porosimetry isotherms and XRD patterns (Fig. S2 and S3, and Table S1†) are equivalent to the parent NiO/SiO2, further evidence for the absence of Pd NPs. While a decrease in the reduction temperature of NiO@PdO/SiO2 relative to NiO/SiO2, Fig. S6,† is indicative of the co-location of Pd and Ni within a single nanoparticle.32 The Pd species on the NiO NP surface facilitates this shift due to the lower reduction temperature of PdO. If PdO presents solely as a second isolated species, no change in NiO reduction temperature would be observed.
Ex situ Pd 3d XPS and Pd K-edge XAS were deployed to evaluate the nature of the deposited Pd species (Fig. S6 and S7†). XPS identifies the surface Pd as Pd(II), with a peak maxima at 336.4 eV. Comparison of XAS spectra to plausible reference standards likewise indicates Pd to present in its 2+ oxidation state as PdO, based on edge position and the X-ray absorption near edge spectroscopy (XANES) region, with quantification via linear combination fitting (LCF) confirming this assignment. Optimal LCF returns a composition of ∼90% PdO, with the remainder Pd metal (Fig. S7†). This demonstrates Pd deposition does not occur via simple adsorption of the Pd salt precursor ((NH4)2PdCl4) nor PdCl2, i.e. consistent with a galvanic displacement mechanism. The extended X-ray absorption fine structure (EXAFS) region probes local geometry, specifically the first few coordination shells, with coordination numbers (CN) indicative of species size (and therefore dispersion) when analysing species in the size regime below a few nm in diameter.33 Pd K-edge EXAFS of NiO@PdO/SiO2 fits well to Bulk PdO (Fig. 2, S7, and Table S2†), albeit with a significantly reduced CN in the second and third shells and quality of the fit in this region. This shrink in CN is consistent with highly dispersed species, while the poor fit and large increase in Debye Waller values reflects greater structural disorder,34 likely due to these second and third shells comprising a mixture of Ni and Pd scatters from NiO and PdO with subtle differences in distance. The former is consistent with isolated Pd sites, i.e., single atom catalysts of PdO, also apparent by STEM (Fig. 1d, S5e and f†). Furthermore, the absence of a scatter at 2.62 and 2.74 Å rules out the presence of bimetallic Ni-Pd and metallic Pd.35,36
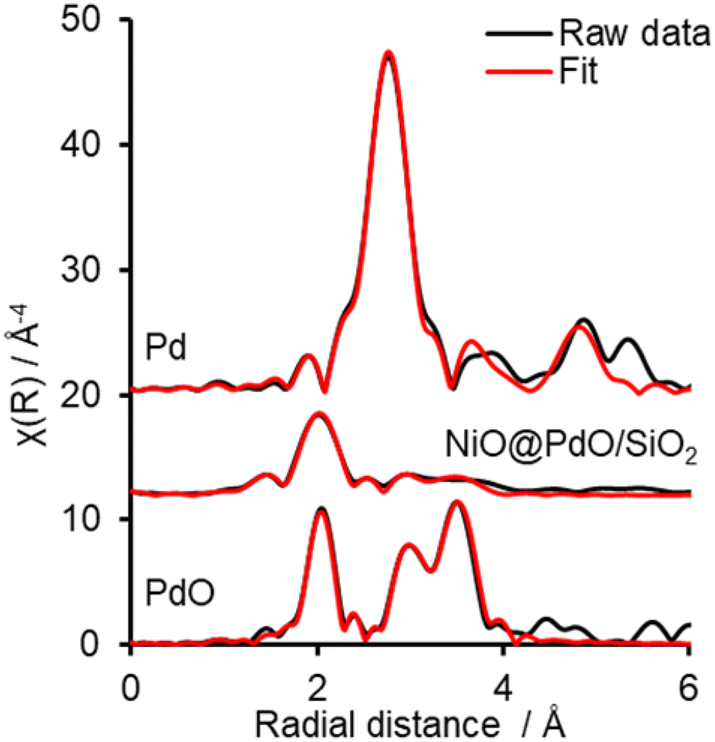 | ||
| Fig. 2 EXAFS Fourier transforms and fits for PdO and Pd references and NiO@PdO/SiO2. | ||
Complementary Ni K-edge XAS confirms the nature of the Ni species. Comparison for XAS spectra, Fig. S8,† confirms NiO as the species in our catalyst, however, overlaying the XANES region shows a more intense white line for NiO@PdO/SiO2 relative to NiO (with the opposite observed at the Pd K edge). As Ni K-edge white line intensity inversely correlates to the degree of occupancy of the Ni 3d orbitals,37 this increase in intensity reflects a greater degree of unoccupancy in the Ni 3d orbitals, due to the great electronegativity of Pd (2.2 vs. 1.9), which is consistent with the slightly lower than expected binding energy reported by XPS. This electron transfer between the two metals further confirms direct interaction, i.e., co-location within a single NP, while simultaneously accounting for the fit returning a small degree of Pd metal in the LCF analysis of the Pd K edge XANES. The presence of Pd metal is inconsistent with the EXAFS data and arises in the LCF due to the slight decrease in the white line of NiO@PdO/SiO2 compared to PdO. The Ni K edge EXAFS region fits to bulk NiO (Fig. S8 and Table S3†), again with a decline in the second shell CN, but to a lesser extent than in Pd, as expected given the NiO NPs size. However, a satisfactory fit requires a second oxide scatter at a significantly shorter scatter distance. The O scatter at 1.996 Å, a decrease of ∼0.09 Å from the bulk value, is again accounted for by surface Pd incorporation with NiO and resulting surface crystal strain induced by the larger ionic radii of Pd.
Combining XAS and STEM results, supported by the additional characterisation, advocates Pd presents as highly dispersed PdO species solely on the NiO NPs. These PdO species comprise isolated single atoms and small 2-dimensional surface species comprising a few Pd atoms, which reside at the surface, i.e., flat, rather than as islands on the NiO surface, due to the galvanic displacement mechanism. Given the high Ni:Pd surface ratio, it is not plausible to form a complete PdO shell encapsulating the NiO NP, which is also apparent from the STEM images, but it can be considered a partial shell covering a NiO core.
The catalytic performance of NiO@PdO/SiO2 was evaluated for cinnamyl alcohol selox to cinnamaldehyde, via an oxidative dehydrogenation mechanism,38 under mild conditions (O2 5 ml min−1 at 1 bar and 80 °C), as shown in Fig. 3 and S9.† It should be pointed out that these reactions were conducted at high substrate:catalyst active site ratios of ∼10800 (and ∼325 for the NiO/SiO2) to facilitate kinetics studies at low conversion and in an attempt to induce deactivation over a reasonable time frame. Conversion and selectivity are dominated by the presence of Pd in NiO@PdO/SiO2, with ∼98% of the initial activity, 20995 mmol h−1 gpd−1vs. 390 mmol h−1 gsurfaceNi−1, accounted for by the Pd sites. These mass normalised activities illustrate a significant enhancement over comparable Pd loading on silica supports, ∼8000 mmol h−1 gpd−1.11,12 Furthermore, the presence of Pd imparts a selectivity of ∼80% to the desired aldehyde compared to only 25% over NiO/SiO2, determined after 30 minutes. NiO/SiO2 shows greater selectivity to 3-phenylpropanoic acid (hydrogenation and over oxidation) and propylbenzene (hydrodeoxygenation and hydrogenation), with a selectivity of 40 and 33%, respectively. In part, the oxidative dehydrogenation mechanism, specifically the liberation of hydrogen onto the catalyst surface, is likely partially responsible for the formation of hydrogenation and hydrodeoxygenation by-products.39 A turnover frequency (TOF) for NiO@PdO/SiO2, based on all Pd being accessible for catalysis, of 2234 h−1 is comparable to previous reports for Pd/mesoporous SiO2, ∼5500 h−1 at 90 °C (10 °C higher than this study), for which surface PdO was confirmed as the active species.35 Additionally, the performance of NiO@PdO/SiO2 under identical reaction conditions to our previous studies (8.4 mmol alcohol, 25 mg catalysts, 90 °C and static air) is comparable to one of the superior systems, i.e. 0.46 wt% Pd/KIT-6, albeit using only half the mass of catalyst. Conversions and selectivity at 24 h are ∼70 and 77%, respectively, which compares to 76 and 59% for 0.46 wt% Pd/KIT-6.35 These comparable performances, specifically TOFs, are further confirmation that PdO is the active species responsible here, which is further verified through subjecting NiO@PdO/SiO2 to a reduction pre-treatment under flowing H2 at 600 °C. This significantly impacts cinnamaldehyde production (Fig. S9a†), with NiO@PdO/SiO2red displaying a ∼20-fold drop, resulting in comparable selective oxidation performance to that of the NiO/SiO2. We attribute this to the reduction of the isolated PdO species to Pd (Fig. S6†). The reduced Pd species displays a binding energy of 334.7 eV, a decrease relative to bulk metal due to the electronegativity differences discussed earlier.
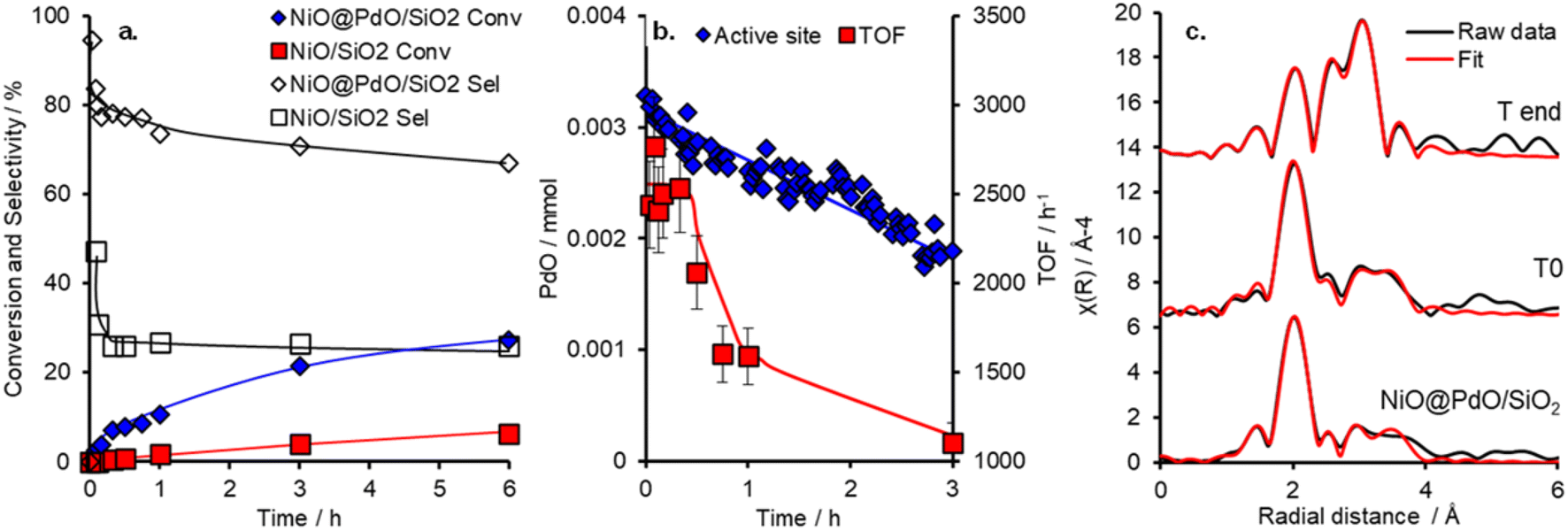 | ||
| Fig. 3 a. Selox reaction and selectivity profiles for NiO@PdO/SiO2 and NiO/SiO2, b. evolution of catalytic PdO content and instantaneous TOFs derived from LCF of operando Pd K edge XAS during cinnamyl alcohol selox, and c. FT EXAFS and fit of ex situ and operando NiO@PdO/SiO2 at T0 and 6 hours. | ||
Deactivation of NiO@PdO/SiO2 is apparent after the initial 30 minutes, with the mechanism behind this and its reversibility key to the recyclability of the catalytic system (which is part of the focus of the operando studies discussed below). We suspected that the deactivation was due to PdO reduction due to the oxidative dehydrogenation mechanism of alcohol oxidation over Pd sites.38 If this was the case and the only source of deactivation, it should be easily reversed. Catalyst recycle tests showed that washing and drying in air (16 hours at 120 °C) facilitated the complete recovery of the initial performance of the freshly prepared catalyst for two recycles (Fig. S9†).
To shed further insight on the true nature of the Pd species responsible for the catalytic selox of alcohols and elucidate the reason for its on-stream deactivation, operando XAS was employed (Fig. S10†).40 An initial high and constant TOF (Fig. 3) is witnessed over the initial 30 minutes of the reaction, which corresponds to optimal PdO concentration within the system. A similar initial stability was also reported over Pd/Al-SBA-15.41 However, as the reaction proceeds, catalytic performance falls, both in activity and selectivity, which coincides with a steady decline in PdO active species and its metamorphosis into Pd(0). Upon completion of the reaction, the content of PdO had dropped by ∼44% from an initial 95% at T0 (Fig. S10†). EXAFS fitting of the catalyst before initiating the reaction (via the addition of the allylic alcohol), i.e., T0, closely matches the ex situ results (Fig. 3 and S11 and Table S4†). So, the PdO species is stable at reaction temperature and in the presence of solvent and O2. EXAFS analysis of the Pd species upon completion of the reaction, system quenched at room temperature, reveals a decrease in oxide species CN for first and second shells and the emergence of a new scatter at 2.8 Å, which is attributed to a Pd(0) and/or Ni(0), i.e. the formation of (bi)metallic species. This species is consistent with XANES analysis and corresponds to the reduction of the catalyst nanoparticle surface. However, a potential carbide phase, observed for Pd/Al-SBA-15, is not apparent.41 Thus, from the insight provided by this operando investigation, the reduction of the PdO active species is one contributor to catalyst deactivation. Nevertheless, if this were the sole factor, the instantaneous TOFs, based on PdO content at the corresponding time, would remain constant. Therefore, a second deactivation factor is also apparent. Given the mild treatment required for the spent catalyst to reactivate to its original performance, i.e. when freshly prepared, it is unlikely that sintering is driving this. A more plausible justification is that surface poisoning is the cause.
Conclusions
Here we report a facile way to improve the efficient utilisation of rare platinum group metals, specifically Pd, to produce allylic alcohol selox catalysts via the replacement of the Pd within the core of a nanoparticle with Ni, a more abundant and cheaper metal. The surface functionalisation via galvanic displacement of a NiO NP with isolated PdO species represents a highly efficient utilisation strategy with optimal accessibility and Pd dispersion, which in turn provide excellent catalytic activity and selectivity to produce desirable cinnamaldehyde. While catalyst deactivation via reduction is apparent, the reversal of this is facile.Author contributions
Aleksandra Ziarko: investigation, methodology, formal analysis. Thomas J. A. Slater: investigation, visualisation, formal analysis. Writing – original draft. Mark A. Isaacs: investigation, writing – original draft. Lee J. Durndell: investigation, writing – original draft. Christopher M. A. Parlett: conceptualisation, investigation, methodology, formal analysis, methodology, writing – original draft, data curation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Diamond Light Source for time on beamlines B18 under proposals SP21795-1 and SP15151 and ePSIC (instrument E01) under proposal mg22736. C. A. M. P. would like to thank the Research Complex for access and support to these facilities and equipment. In addition, the UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC grant EP/R027129/1.References
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- G. Vile, D. Albani, N. Almora-Barrios, N. Lopez and J. Perez-Ramirez, ChemCatChem, 2016, 8, 21–33 CrossRef CAS.
- C. P. Vinod, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2011, 86, 161–171 CrossRef CAS.
- M. Friedman, N. Kozukue and L. A. Harden, J. Agric. Food Chem., 2000, 48, 5702–5709 CrossRef CAS PubMed.
- S. Qu, K. L. Yang, L. Chen, M. Liu, Q. R. Geng, X. N. He, Y. X. Li, Y. G. Liu and J. Tian, Front. Microbiol., 2019, 10, 2895 CrossRef PubMed.
- C. T. Evans, C. Choma, W. Peterson and M. Misawa, Biotechnol. Bioeng., 1987, 30, 1067–1072 CrossRef CAS PubMed.
- S. E. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- C. Chen, X. L. Fang, B. H. Wu, L. J. Huang and N. F. Zheng, ChemCatChem, 2012, 4, 1578–1586 CrossRef CAS.
- L. Saputra, T. Kojima, T. Hara, N. Ichikuni and S. Shimazu, Mol. Catal., 2018, 453, 132–138 CrossRef CAS.
- S. Z. Ding, M. Ganesh, Y. L. Jiao, X. X. Ou, M. A. Isaacs, M. S'Ari, A. T. Lopez, X. L. Fan and C. M. A. Parlett, R. Soc. Open Sci., 2021, 8, 211086 CrossRef CAS PubMed.
- C. M. A. Parlett, D. W. Bruce, N. S. Hondow, A. F. Lee and K. Wilson, ACS Catal., 2011, 1, 636–640 CrossRef CAS.
- C. M. A. Parlett, P. Keshwalla, S. G. Wainwright, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, ACS Catal., 2013, 3, 2122–2129 CrossRef CAS.
- A. F. Lee, C. V. Ellis, J. N. Naughton, M. A. Newton, C. M. A. Parlett and K. Wilson, J. Am. Chem. Soc., 2011, 133, 5724–5727 CrossRef CAS PubMed.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- H. B. Zhang, G. G. Liu, L. Shi and J. H. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- J. G. Ruiz-Montoya, L. M. S. Nunes, A. M. Baena-Moncada, G. Tremiliosi and J. C. Morales-Gomero, Int. J. Hydrogen Energy, 2021, 46, 23670–23681 CrossRef CAS.
- Y. F. Li, L. J. Xiao, F. F. Liu, Y. S. Dou, S. M. Liu, Y. Fan, G. Cheng, W. Song and J. L. Zhou, J. Nanopart. Res., 2019, 21, 28 CrossRef.
- M. G. Hosseini, R. Mahmoodi and V. Daneshvari-Esfahlan, Energy, 2018, 161, 1074–1084 CrossRef CAS.
- Z. P. Dong, X. D. Le, C. X. Dong, W. Zhang, X. L. Li and J. T. Ma, Appl. Catal., B, 2015, 162, 372–380 CrossRef CAS.
- H. Kilic, M. Turgut, M. S. Yilmaz, O. Dalkilic and O. Metin, ACS Sustainable Chem. Eng., 2018, 6, 11433–11440 CrossRef CAS.
- S. P. Jian and Y. W. Li, Chin. J. Catal., 2016, 37, 91–97 CrossRef CAS.
- N. Ghanbari, S. J. Hoseini and M. Bahrami, Ultrason. Sonochem., 2017, 39, 467–477 CrossRef CAS PubMed.
- Z. J. Wei, D. Thushara, X. H. Li, Z. Y. Zhang, Y. X. Liu and X. Y. Lu, Catal. Commun., 2017, 98, 61–65 CrossRef CAS.
- X. L. Zou, Z. B. Rui and H. B. Ji, ACS Catal., 2017, 7, 1615–1625 CrossRef CAS.
- D. K. Shen, J. P. Yang, X. M. Li, L. Zhou, R. Y. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Y. Zhao, Nano Lett., 2014, 14, 923–932 CrossRef CAS PubMed.
- C. M. A. Parlett, A. Aydin, L. J. Durndell, L. Frattini, M. A. Isaacs, A. F. Lee, X. T. Liu, L. Olivi, R. Trofimovaite, K. Wilson and C. F. Wu, Catal. Commun., 2017, 91, 76–79 CrossRef CAS.
- L. Jiao and J. R. Regalbuto, J. Catal., 2008, 260, 342–350 CrossRef CAS.
- T. Mahmood, M. T. Saddique, A. Naeem, P. Westerhoff, S. Mustafa and A. Alum, Ind. Eng. Chem. Res., 2011, 50, 10017–10023 CrossRef CAS.
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 CrossRef CAS PubMed.
- X. Tai, B. Wu, J. Bao, W. Qu, L. Zhao and Z. Wang, Mater. Today Sustain., 2022, 18, 100140 CrossRef.
- S. M. Alia, Y. S. Yan and B. S. Pivovar, Catal. Sci. Technol., 2014, 4, 3589–3600 RSC.
- R. Camposeco, M. Hinojosa-Reyes, S. Castillo, N. Nava and R. Zanella, Environ. Sci. Pollut. Res., 2021, 28, 10734–10748 CrossRef CAS PubMed.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- P. Fornasini and R. Grisenti, J. Synchrotron Radiat., 2015, 22, 1242–1257 CrossRef CAS PubMed.
- C. M. A. Parlett, D. W. Bruce, N. S. Hondow, M. A. Newton, A. F. Lee and K. Wilson, ChemCatChem, 2013, 5, 939–950 CrossRef CAS.
- T. T. Huynh, M. C. Tsai, C. J. Pan, W. N. Su, T. S. Chan, J. F. Lee and B. J. Hwang, Electrochem. Commun., 2019, 101, 93–98 CrossRef CAS.
- D. J. Morris and P. Zhang, Chem.: Methods, 2021, 1, 162–172 CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef CAS.
- L. J. Durndell, C. M. A. Parlett, N. S. Hondow, K. Wilson and A. F. Lee, Nanoscale, 2013, 5, 5412–5419 RSC.
- L. Negahdar, C. M. A. Parlett, M. A. Isaacs, A. M. Beale, K. Wilson and A. F. Lee, Catal. Sci. Technol., 2020, 10, 5362–5385 RSC.
- C. M. A. Parlett, L. J. Durndell, A. Machado, G. Cibin, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, Catal. Today, 2014, 229, 46–55 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00198a |
| ‡ The data underpinning the publication are available from the University of Manchester repository. See https://doi.org/10.48420/c.6647102. |
| This journal is © The Royal Society of Chemistry 2023 |