Catalytic hydrogenative dechlorination reaction for efficient synthesis of a key intermediate of SDHI fungicides under continuous-flow conditions†
Received
8th February 2023
, Accepted 16th April 2023
First published on 20th April 2023
Abstract
Selective hydrodechlorination reactions under continuous-flow conditions for the synthesis of an SDHI fungicide intermediate were investigated using originally developed heterogeneous Pd catalysts. Under optimal reaction conditions accompanying the use of tetrabutylammonium acetate, the desired hydrodechlorination reaction from a chlorodifluoromethyl-substituted pyrazole derivative proceeded selectively to afford the product quantitatively at 1.5 h−1 of the SVmol value. We tried to access the roles of the present binary support by a series of characterization techniques and revealed that the carbon support generates catalytic activity to the supported Pd species and Ca3(PO4)2 provides redox activity on the Pd through electron donation.
Introduction
One of the sustainable development goals, no hunger (G2), still relies on the use of highly efficient pesticides in required places and in required amounts. Considering efficient and practical contributions from the standpoint of the synthetic organic chemistry field, the design and implementation of on-demand and on-site production of active ingredients in an environmentally benign manner are of great importance. The demand for Succinate Dehydrogenase Inhibitor (SDHI) fungicides is increasing in the global market for agrochemicals; indeed, third-generation SDHIs have attracted a lot of attention due to their wide controlling spectrum against various pathogenic fungi.1 3-Difluoromethy-1-methyl pyrazole (DFMMP, 1) is a common unit of several commercialized SDHI fungicides, such as bixafen, sedaxane, isopyrazam, and fluxapyroxad, and is recognized as an important intermediate for the dissemination of these agrochemicals,2 and cost-efficient methods of constructing the difluoromethyl pyrazole unit are key to their efficient supply (Fig. 1). Several routes to DFMMP were already known in the literature, and these routes can be summarized in the usage of two headstream feedstocks for installation of the difluoromethyl (DFM) group (Fig. 1). Tetrafluoroethene (TFE) is one of the most useful and straightforward feedstocks of the DFM group, and it or its derivatives, such as ethyl 1,1,2,2-tetrafluoroethylether (2a)3,4 or 1,1,2,2-tetrafluoroethyl-N,N-dialkylamines 2b,5 are suitably converted to the corresponding vinylidene ketoester derivatives offering the corresponding pyrazole after cyclization.6 TFE is a common gaseous chemical used in the industrial preparation of fluoropolymers. However, its use is associated with a risk of explosion especially in the presence of oxygen, and thus specialized facilities are generally recommended to manage it. An alternative to the TFE-based route is the use of 1,2,2-trichloro-1,1-difluorethane (HCFC-122, 4),7 a by-product of the fluorocarbon industry, and post-modification of its derivatives into the DFM group. It is less expensive and can be easily converted into chlorodifluoroacetic acid derivatives 3 through photooxidation. To complete the synthesis of DFMMP from the given 3-chlorodifluoromethyl pyrazole (CDFMMP 5), efficient hydrodechlorination is required; however, previous research indicated its difficulty when utilizing the usual hydrogenation technologies due to the high resistance of the C(sp3)–Cl bond to reduction conditions.8 As a useful method from the latest literature, Braun et al. reported catalytic hydrogenation using Pd/C as a catalyst. This protocol provided the target DFMMP in 97% yield at 110 °C in the presence of K2CO3 as a base, but a high hydrogen pressure of 11 bar (1.1 MPa) is required and this may be affected by the production of over-hydrogenation products in a small amount.2,9
 |
| | Fig. 1 SDHI fungicides and synthesis of the common intermediate DFMMP. | |
Recently, we reported the continuous-flow synthesis of pyrazole derivatives including DFMMP through vinylidene ketoester synthesis with heterogeneous catalysts and successive cyclization with methylhydrazine.9 To brush-up this protocol for the cost-effective and safe production of DFMMP, we planned to develop continuous-flow dechlorination reactions of CDFMMP through catalytic hydrogenation using palladium-based heterogeneous catalysts supported on an activated carbon/calcium phosphate composite.10 It is expected that high mass-transfer and gas-distribution efficiencies will realize efficient C(sp3)–Cl bond cleavage by catalytic hydrogenation and suppress the co-production of undesirable over-hydrogenation products. Previous attempts to achieve this transformation mainly relied on catalysis by Pd/C and sometimes on the use of external modifiers. Our strategy for designing heterogeneous catalysts for continuous-flow reactions with packed-bed reactors is to utilize the chemical and physical characters of solid supports behaving as a static phase in reactors, and we envision that the chemical features of the support materials, carbon and calcium phosphate, possessing completely different chemical characters, would independently affect the catalytic features of palladium. Here we describe our efforts to achieve this transformation and elucidate the important factors which confer high activity and stability on Pd under continuous-flow conditions. Hydrodechlorination reactions are becoming increasingly important in environmental catalysis, especially in recent years for the treatment of halogenated plastics. Our research is expected to contribute not only to the specific organic synthesis field like pesticide synthesis, but also to the above fields in the future.11
Results and discussion
System development under batch and continuous-flow conditions
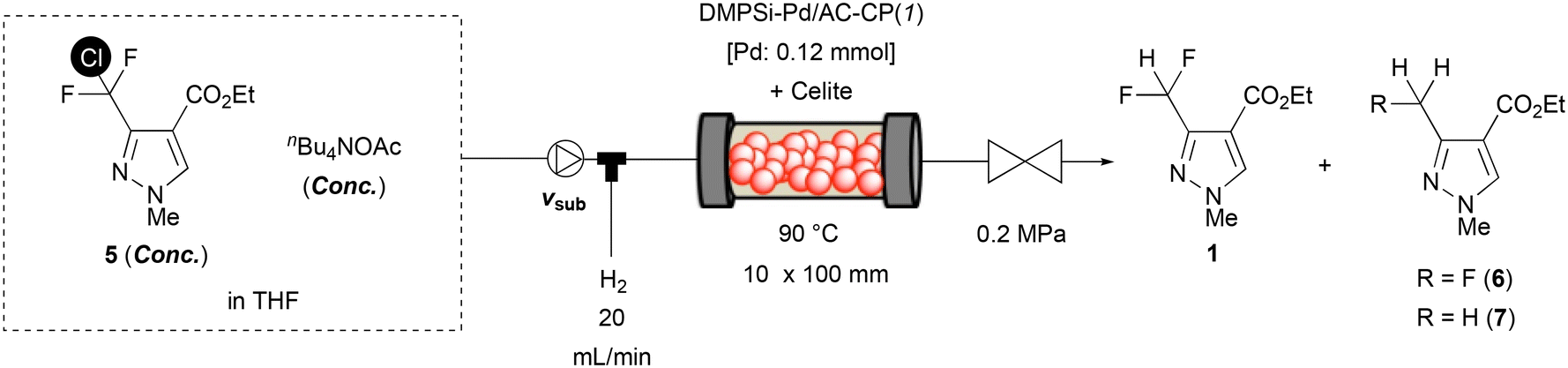
Since our originally developed polysilane-modified palladium on activated carbon/calcium phosphate composite catalyst showed potential activity for the hydrogenation of the aromatic C(sp2)–Cl bond under continuous-flow hydrogenation conditions,10a we started our investigation with the benchmark catalyst, DMPSi-Pd/AC-CP(3), whose binary support material was composed of activated carbon (AC)/calcium phosphate (CP) = 3/1, under batch conditions (Table 1). When the starting material, CDFMMP 5, was treated as a THF solution with the benchmark catalyst under 1.0 MPa of hydrogen pressure, the given conditions resulted in very low conversion of 5. On the other hand, the addition of 1.0 equiv. of an inorganic base such as Cs2CO3 progressed the reaction and gave the desired dechlorinated product 1 in moderate yield with significant amounts of over-hydrogenated products 6 and 7 (run 2). Thus, we examined various base conditions to attain higher selectivity of the desired product 1 against defluorinated products. A constraining condition should be considered that the base must be dissolved in the reaction medium to conduct the reaction under flow conditions. The addition of water to THF (THF/H2O = 9/1) dissolved these inorganic bases; however, a significant loss of conversion was observed. A smaller amount of water in THF or the use of ethanol improved the conversion; however, in comparison to the reaction without the addition of water, the selectivity decreased (runs 3–5). An organic base such as triethylamine provided a homogeneous reaction solution, but was ineffective for the progress of the reaction (run 6). We then focused on quaternary ammonium salt soluble in THF and found a fruitful result in the reaction with nBu4NOAc, although the conversion of 5 under standard conditions was moderate, and the desired product 1 was obtained in 40% GC yield with 98% selectivity, suggesting defluorinated products 6 and 7 were significantly suppressed (run 7).
Table 1 Initial investigations on solvent and base in batch
|
|
| Run |
Base |
Solv. |
Conv. [%] |
Yields [%] |
|
1
|
6
|
7
|
|
The reaction was performed at 100 °C.
THF/H2O = 9/1.
10 equiv. of H2O was utilized.
|
| 1 |
Without |
THFa |
10 |
5 |
<1 |
5 |
| 2 |
Cs2CO3 |
THF |
>99 |
90 |
5 |
5 |
| 3 |
Cs2CO3 |
THF/H2Ob |
24 |
10 |
3 |
11 |
| 4 |
Cs2CO3 |
THF/H2Oc |
>99 |
77 |
10 |
13 |
| 5 |
Cs2CO3 |
EtOH |
>99 |
46 |
15 |
40 |
| 6 |
Et3N |
THF |
<1 |
Trace |
Trace |
Trace |
| 7 |
n
Bu4NOAc |
THF |
41 |
40 |
1 |
<1 |
With a promising additive, we tried to build the flow hydrodechlorination system with a catalyst-embedded column-type reactor. For a typical reaction set-up, a stainless 10 (id) × 50 (L) mm column equipped with stainless filters on both column ends was used as a plug-flow reactor. On top of the reactor, a tube-in-tube type column head was mounted for independent feeding of liquid (inside) and hydrogen gas (outside). The catalyst was diluted with Celite, a diatomaceous filtering agent, and was packed inside the column reactor; then, the other column end was sealed with an appropriate one-way column cap. A reactant stream containing CDFMMP 5 and nBu4NOAc pumped by a plunger pump was connected to the liquid inlet of the column head, and mass-flow controlled hydrogen gas was supplied from the gas inlet. A characteristic gas–liquid slug flow flowed out from the bottom column cap and the outlet tube was connected to an appropriate back pressure regulator to maintain the pressure of the reactor system at 0.1–0.6 MPa(G) measured at the liquid flow inlet. Initial investigations on column temperature, back pressure, and hydrogen flow rate under continuous-flow conditions are summarized in Fig. 2. Back pressure had less effect than column temperature, and these gave an optimal 79% yield under 0.2 MPa(G) back pressure at 90 °C or 0.6 MPa(G) at 110 °C. Hydrogen flow (20 mL min−1) gave an almost similar yield to 10 mL min−1, whereas a 30 mL min−1 flow rate resulted in a lowering of the yield of 1. It is worth noting that the complete suppression of over-hydrogenation under the continuous-flow conditions is due to an efficient mass-transfer effect. Although several conditions exhibited similar results of 77–79% yields, we decided that 90 °C column temperature, 10 or 20 mL min−1 hydrogen flow, and 0.2 MPa(G) of back pressure were the optimal conditions at 10 h−1 of substance amount (mole)-based space velocity (SVmol).‡ It should be noted that 10 mL min−1 hydrogen flow enabled stable continuous-flow without unexpected clogging.
 |
| | Fig. 2 System development under continuous-flow conditions. Note: unless otherwise indicated in bars (A) and (B), the reactions were conducted with 10 mL min−1 H2 flow. | |
Improvement in yield and productivity in the continuous-flow hydrodechlorination
To examine the catalyst's stability, an extended-time experiment was conducted (Fig. 3). Here the SVmol value was set to 10 h−1 and the hydrogen flow was controlled to 10 mL min−1. After a 5 h incubation period, the yield of desired product 1 reached 69%, which was maintained for more than 13 h without significant loss of yield. The upper x-axis indicates the ratio of a cumulative amount of CDFMMP 5 provided to a reactor to an amount of Pd inside the reactor; no drop in yield during the period tested suggested that the catalyst's activity was maintained until at least 200 equiv. of the substrate to Pd were supplied. The average yield from 5.6 to 18.3 h was calculated to be 69%; based on this value, the turnover number of Pd in this period was estimated to be 96, suggesting the present catalytic flow system possessed sufficient stability. The investigation was further continued to access high conversion conditions with lowered SVmol value (Table 2). Beforehand, we confirmed that the use of a larger reactor with the same Pd loading by using a double amount of Celite provided no advantage (run 2). Since we noted that one analogue of the benchmark catalyst, whose binary support material was composed of AC/CP = 1/1, gave a slightly better yield than the original AC/CP = 3/1 catalyst, we reset the standard catalyst to DMPSi-Pd/AC-CP(1) (run 3). By lowering the SVmol value to 5 h−1 with a halving of flow rate of substrate 5, the yield of product 1 was improved to 88% (run 6), whereas the same effect obtained by halving the concentration of 5 or the use of a double amount of Pd resulted in no improvement (runs 4 and 5). The SVmol value was further reduced to 2.5 or 1.5 h−1 by reducing substrate concentration while keeping nBu4NOAc concentration. The yield of 1 finally reached >99% using a 0.03 M solution containing 3 equiv. of nBu4NOAc.§ Worth noting in the present reaction was the effect of acetic acid. We confirmed the negative effect of acetic acid, a by-product derived from nBu4NOAc, in batch control experiments: a hydrodechlorination reaction with DMPSi-Pd/AC-CP(1) for 3 h with 1.0 equiv. of nBu4NOAc and the co-existence of 1.0 equiv. of acetic acid resulted in only 17% conversion and 16% yield, whereas 45% conversion with 43% yield of 1 was attained in the absence of additional acetic acid (Table 3, exp. 4). Clearly, co-production of acetic acid suppressed the progress of the reaction. A constant liquidity of solutes under continuous flow effectively excluded the deposition of acetic acid and played a definitive role in reaction completion. To check Pd leaking from the heterogeneous catalyst system, an outcome solution after continuous operation for 5 hours under the high-yield conditions of run 8 in Table 2 was recovered and converted into a sample for ICP-AES measurement. The amount of Pd from the sample was under the detection limit (UDL), and no leaking of Pd into the homogeneous phase was confirmed.
 |
| | Fig. 3 Extended-time operation at SVmol = 10 h−1. Conditions: catalyst; DMPSi-Pd/AC-CP(3), Pd = 0.12 mmol, packed in 10 × 100 mm stainless column with Celite as a dilutant: substrate solution; 5 (0.1 M), nBu4OAc (0.12 M) in THF, 0.2 mL min−1: H2 flow; 10 mL min−1: column temp.; 90 °C: back pressure; 0.2 MPa(G). | |
Table 2 Optimization studies for high-yielding conditions
|
|
| Run |
v
sub [mL min−1] |
5 [M] |
n
Bu4NOAc [M] |
SVmol [h−1] |
Conv.a [%] |
Yield, 1a,b [%] |
Yield, 6 + 7a [%] |
|
Determined by GC analysis.
Estimated TOF [h−1] value.
DMPSi-Pd/AC-CP(3) was used.
10 × 200 mm stainless column was used with a double amount of Celite.
The amount of Pd was increased to 0.24 mmol.
|
| 1c |
0.2 |
0.10 |
0.12 |
10 |
80 |
77 (7.7) |
3 |
| 2c,d |
0.2 |
0.10 |
0.12 |
10 |
79 |
76 |
3 |
| 3 |
0.2 |
0.10 |
0.12 |
10 |
84 |
81 |
3 |
| 4e |
0.2 |
0.10 |
0.12 |
5 |
81 |
76 |
5 |
| 5 |
0.2 |
0.05 |
0.06 |
5 |
78 |
74 |
4 |
| 6 |
0.1 |
0.10 |
0.12 |
5 |
91 |
88 |
3 |
| 7 |
0.1 |
0.05 |
0.10 |
2.5 |
96 |
93 |
3 |
| 8 |
0.1 |
0.03 |
0.09 |
1.5 |
>99 |
>99 |
Trace |
Table 3 Comparison of catalytic activity in batch
|

|
| Exp. |
Catalyst |
Additive |
Time [h] |
Yielda [%] |
|
Determined by GC analysis.
Estimated TOF [h−1] value.
1.0 equiv. of AcOH to 5 was added.
500 mg mmol5−1 of Ca3(PO4)2 was used. Note: exp. 4 was discussed in the earlier part.
|
| 1-1 |
DMPSi-Pd/AC-CP(1) |
wo |
3 |
43 (2.9b) |
| 4 |
DMPSi-Pd/AC-CP(1) |
AcOHc |
3 |
16 (1.1b) |
| 1-2 |
DMPSi-Pd/AC-CP(1) |
wo |
6 |
50 |
| 1-3 |
DMPSi-Pd/AC-CP(1) |
wo |
24 |
63 |
| 2-1 |
DMPSi-Pd/AC |
wo |
3 |
31 (2.1b) |
| 2-2 |
DMPSi-Pd/AC |
wo |
6 |
39 |
| 2-3 |
DMPSi-Pd/AC |
wo |
24 |
39 |
| 3-1 |
DMPSi-Pd/AC |
Ca3(PO4)2d |
3 |
43 |
| 3-2 |
DMPSi-Pd/AC |
Ca3(PO4)2d |
6 |
48 |
| 3-3 |
DMPSi-Pd/AC |
Ca3(PO4)2d |
24 |
60 |
Hydrodechlorination of pyrimidine derivative
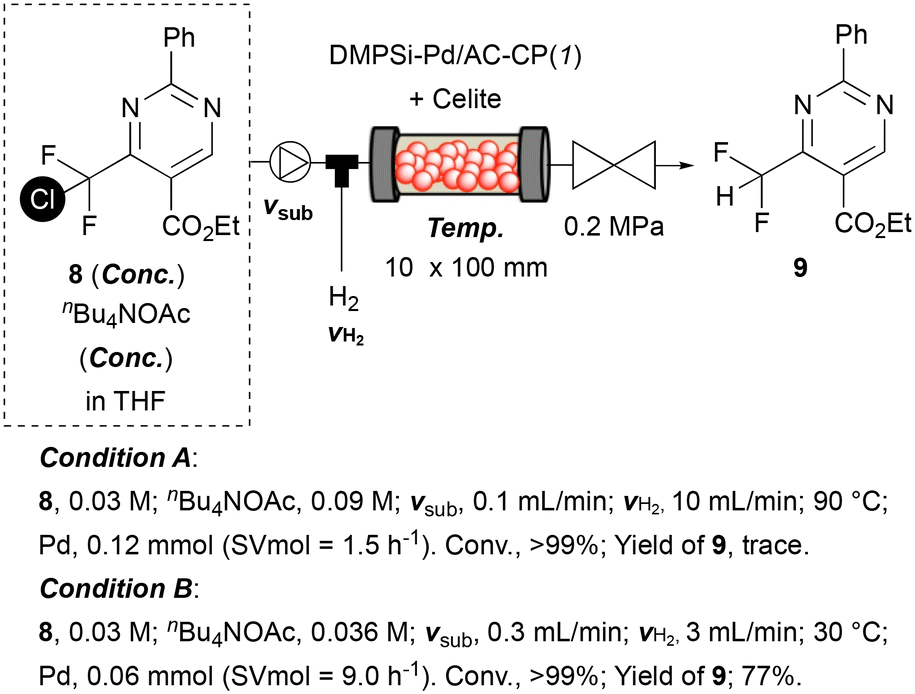
As a demonstration of the current protocol for the other series of difluoromethyl-substituted compounds, chlorodifluoromethyl-substituted pyrimidine 8 was subjected to standard reaction conditions.9,12 When the optimized continuous-flow conditions for the hydrodechlorination of pyrazole 5 were applied to pyrimidine 8, desired product 9 was obtained in a trace amount; instead by-products suggestive of over-hydrogenation were obtained. After several optimizations, increasing the SVmol value to 9.0 h−1 with a reduction of hydrogen flow to 3.0 mL min−1 gave the target difluoromethyl-substituted pyrimidine 9 in 77% isolated yield (Scheme 1; the optimization studies are shown in ESI†).
 |
| | Scheme 1 Continuous-flow hydrodechlorination of pyrimidine derivative 8. | |
Effect of supports and elucidation of the role of the binary support
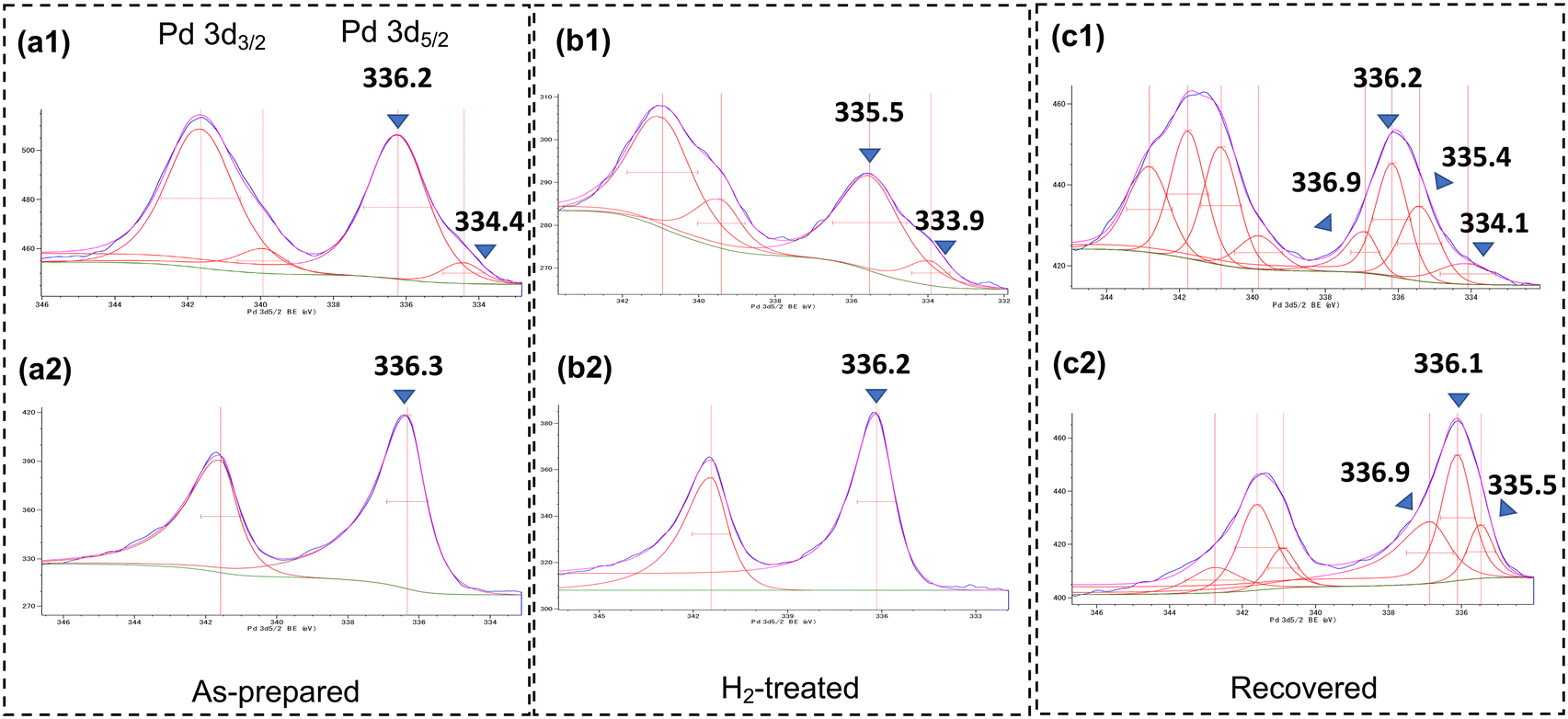
To determine the feasibilities and differences in performance of a commercially available heterogeneous Pd catalyst for this system, 5 wt% Pd/C provided by Fujifilm Wako Pure Chemical Co. was subjected to the catalyst bed. Pd/C also gave the target product with high selectivity; however, the yield was 57% compared to 81% for the use of DMPSi-Pd/AC-CP(1) under the same conditions, suggesting the superiority of our designed catalyst. We then examined the effect of a carbon–Ca3(PO4)2 binary support material. Fig. 4 displays the results of continuous-flow hydrodechlorination of CDFMMP 5 with different types of catalyst under particular conditions. A dimethylpolysilane-modified activated carbon-supported Pd catalyst (DMPSi-Pd/AC) prepared by us showed a yield between that obtained using DMPS-Pd/AC-CPs and that obtained using the commercially available 5 wt% Pd/C. DMPSi-Pd/CP surprisingly showed poor catalytic activity, indicating the necessity for Pd species on carbon to generate catalytic activity. Al2O3 and SiO2 supported catalysts also showed low activity. To elucidate the roles of calcium phosphate (CP) on the continuous-flow reaction outcome from the viewpoint of a carbon-supported Pd environment, we adopted XPS analyses of the catalysts (Fig. 5). Surface Pd species of bench-stocked DMPSi-Pd/AC-CP(1) and DMPSi-Pd/AC catalysts were mainly PdO whose bond energies were 336.2–336.3 eV (Fig. 5a1 and b1).13 To understand a terminal Pd state under H2 flow conditions, the two catalysts were treated with 1.0 MPa of H2 at 50 °C in THF in individual batch reactors. The PdO-like species on DMPSi-Pd/AC-CP(1) in the initial state were fully reduced to Pd0, whereas there was no significant change in the PdO-like species on DMPSi-Pd/AC (Fig. 5a2 and b2). The significant difference in redox activity will be one of the key features in the catalytic superiority of the composite catalyst. In addition to this redox behaviour, it was found that DMPSi-Pd/AC-CP(1) possessed another characteristic Pd0 species whose bond energy was 333.9–334.4. Relatively low bond energy compared with usual bulk Pd0 suggested electron donation from the phosphate to Pd0. We next compared the behaviour of Pd species by XPS analyses before and after batch catalytic reactions. The used catalysts DMPSi-Pd/AC-CP(1)_used and DMPSi-Pd/AC_used showed somewhat broad XPS peaks of both 3d3/2 and 3d5/2 compared to those of the fresh catalysts (Fig. 5a3 and b3). Careful deconvolution suggested that both used catalysts possessed at least three kinds of Pd species, corresponding to bulk-like Pd0, PdO-like Pd2+, and the other electron-deficient Pd2+. Pd0-occupancy in DMPSi-Pd/AC-CP(1)_used was quite a bit higher than that in DMPSi-Pd/AC_used, again indicating active redox behaviour. The electron-rich Pd0 species in the former remained in the used catalyst. These significant differences in peak appearance and in redox activity would be provided by electron donation from a phosphate anion and be one of the key features in the catalytic superiority of the composite catalyst.
 |
| | Fig. 4 Effect of supports on activity. Note: yield, red bars; selectivity of 1, blue bars; 10 mL min−1 of hydrogen flow was utilized for the reaction with DMPSi-Pd/CP. | |
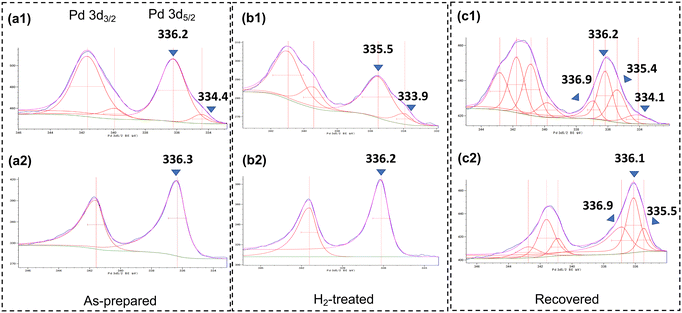 |
| | Fig. 5 Comparison of DMPSi-Pd/AC-CP(1) and DMPSi-Pd/AC in XPS analyses. Note: (a1 and a2) as-prepared sample; (b1 and b2) after H2-treatment at 50 °C for 12 h in THF; (c1 and c2) after batch reactions for 3 h under the standard conditions (DMPSi-PD/AC-CP(1)_used and DMPSi-PD/AC_used). The upper row (a1–c1): Pd 3d3/2 and 3d5/2 of DMPSi-Pd/AC-CP(1); the bottom row (a2–c2): Pd 3d3/2 and 3d5/2 of DMPSi-Pd/AC. | |
Scanning transmission electron microscope (STEM) and energy dispersive X-ray spectroscopy (EDX) analyses were conducted to visualize the location of Pd species and support components (Fig. 6). In the DMPSi-Pd/AC-CP(1) catalyst, 3–5 nm well-dispersed Pd nanoparticles were observed over the support material. EDX elementary mappings of carbon and elements from calcium phosphate clearly indicated the layered structure of the AC-CP interface, and suggested that Pd particles were selectively deposited on carbon as well. These would reflect the order of addition in the preparation procedure of this material. The DMPSi-Pd/AC catalyst also gave similar photography, matching the results in XPS initial state analyses. From the EDX mapping image of Ca and P, we assumed that the calcium phosphate layer does not exist separately from the activated carbon layer, but is partially dispersed and spread over the carbon layer, and the Pd species on carbon would be coordinated by phosphate.14 An additional series of batch reactions pursuing the reaction profile of the following three conditions supported the above considerations (Table 3): exp. 1 and exp. 2 are the time courses of batch hydrodechlorination reactions of CDFMMP 5 catalyzed by DMPSi-Pd/AC-CP(1) or DMPSi-Pd/AC, and exp. 3 is that catalyzed by the latter with the co-existence of Ca3(PO4)2. In the case of the DMPSi-Pd/AC-catalyzed reaction without additives, exp. 2, the reaction almost stopped after 6 h when the yield reached 40%. In contrast, the yield of the product gradually increased up to 24 hours in the DMPSi-Pd/AC-CP(1) case, despite some sluggish reaction progress. The reaction profile of exp. 3 was almost identical to that of exp. 1, suggesting that externally added Ca3(PO4)2 could also act like a Ca3(PO4)2 component internally present in the composite-supported catalyst. The XPS spectra of the recovered solid materials from exp. 3, which might contain the catalyst and Ca3(PO4)2, were similar to the residue of exp. 1, including the specific electron-rich Pd0 peak shown in the previous part. All these results suggest that Ca3(PO4)2, whether it was included internally as a component of the binary support or added externally, interacted with Pd species on the carbon support and may contribute their redox activity and stability in catalytic turnover.
 |
| | Fig. 6 STEM and EDX images of DMPSi-Pd/AC-CP(1). | |
Assumed mechanism
The assumed mechanism of the current reaction is illustrated in Fig. 7 based on previous reports.15 The surface Pd0 species I of the activated catalyst would insert into a C–Cl bond of CDFMMP 5 to give Pd2+ intermediate II, and subsequently transform to intermediate III by salt metathesis, which has more nucleophilic acetate anions than intermediate II. A surface hydride species on a Pd particle would move to the reaction centre and give Pd hydride intermediate IV with liberation of AcOH. Finally, a hydrogenated compound forms by reductive elimination, along with the regeneration of Pd0 species. As the reaction progresses and acetic acid concentration increases, conversion from intermediate III to IV should be counteracted by its reverse reaction, and this might be a reason for reaction blunting at 50% conversion in a batch system.
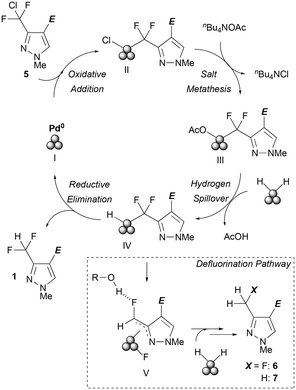 |
| | Fig. 7 Plausible mechanism for C–Cl and C–F bond cleavage. | |
In the course of our investigations, we found fast defluorination in the hydrodechlorination reaction of 1 to give a fully hydrogenated product when the reactions were conducted using Cs2CO3 as a base in aqueous media or ethanol. In contrast, the corresponding 3-CF3 (10) derivatives or DFMMP 1 were completely inert under the present reaction conditions and no hydrodefluorination proceeded (Scheme 2). According to the bond energy of C(sp3)–Cl (∼70 kcal mol−1)16 and C(sp3)–F (>105 kcal mol−1)17 bonds, the low reactivities of 1 and 10 were reasonable and over-defluorination of 5 may happen through migratory insertion of a Pd centre to an adjacent C–F bond giving intermediate V. It is also supposed that protic solvents assisted C–F bond activation by hydrogen bonding.18
 |
| | Scheme 2 Trials of hydrodefluorination reaction. | |
Conclusions
In conclusion, we investigated selective hydrodechlorination reactions under continuous-flow conditions using originally developed heterogeneous Pd catalysts. Under optimal reaction conditions accompanied by the use of tetrabutylammonium acetate, the desired DFMMP, an important target as a common fungicide intermediate, was obtained quantitatively at 1.5 h−1 of SVmol value. At high-throughput condition of 10 h−1 of SVmol value, the present catalysis worked well to give the product in 81% yield. Through meticulous XPS studies of the catalysts, we could access the roles of the present binary support, and revealed that the carbon support generates catalytic activity to the supported Pd species and Ca3(PO4)2 provides redox activity to Pd through electron donation. Further improvement in catalytic activity and expanding the ability and utility of continuous-flow hydrogenation are now ongoing in our laboratory.
Experimental
General information
Nuclear magnetic resonance (NMR) spectra were recorded on a JEOL ECX-600 or ECA-500 spectrometer, operating at 600 MHz or 500 MHz for 1H, 150 MHz for 13C NMR in CDCl3 unless otherwise noted. Tetramethylsilane (TMS) served as the internal standard (δ = 0) for 1H NMR, CDCl3 (δ = 77.0) was used as the internal standard for 13C NMR. Gas chromatography (GC) was recorded on a Shimadzu GC-2030AF, 100 V spectrometer using an SH-Rtx-5 Amine column (30 m, 0.25 mm id, 0.25 μm df). Inductively coupled plasma-atomic emission spectrometry (ICP-AES) analysis was performed on Shimadzu ICPS-7510 equipment. STEM/EDS images were obtained using a JEOL JEM-2100F instrument operated at 200 kV. All STEM specimens were prepared by placing a catalyst directly on carbon-coated copper grids. X-ray photoelectron spectroscopy (XPS) was measured using JEOL JPS-9010MC equipment. High-pressure hydrogenation reactions under batch conditions were carried out using a Tokyo Rikakikai (EYELA) Co., Ltd ChemiStation™ (PPV-CTRL1). As apparatus for flow systems, a plunger pump (FLOM, UI-22-110P), a column heater (Tokyo Rikakikai Co., Ltd, LCR-1300), a back pressure regulator (DFC Co., Ltd, FC-BPV-100) and a stainless column with column ends were used. Preparative thin-layer chromatography (PTLC) was carried out using Wakogel B-5F from Wako Pure Chemical Industries, Ltd. High resolution mass spectra (HRMS) were recorded using a JEOL JMS-T100TD (DART) spectrometer. THF and toluene were purchased from Fujifilm Wako Pure Chemical Co. Ltd as dried solvents and were used directly. Other organic solvents used were commercially available dry solvents, which were distilled appropriately under an argon atmosphere or were stored over molecular sieves prior to use. Ethyl 3-(chlorodifluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (5) and ethyl 3-(trifluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (12) were prepared following the literature.9,19 Ethyl 4-(chlorodifluoromethyl)-2-phenylpyrimidine-5-carboxylate was prepared following the literature.9 Tetrabutylammonium acetate was purchased from Tokyo Chemical Industry Co., Ltd or Sigma-Aldrich Co. LLC and was used directly. Activated carbon and calcium phosphate were purchased from Fujifilm Wako Pure Chemical Co. Ltd and used directly. Dimethylpolysilane was purchased from Nippon Soda Co. Ltd. Polycarbosilane was purchased from JGC Catalysts and Chemicals Ltd.
Preparation of catalysts: DMPSi-Pd/supports
Dimethylpolysilane-modified Pd on support catalysts was prepared according to our previous work10a with small modifications. The following is a representative example of a DMPSi-Pd/AC catalyst; to a suspension of 4.5 g of activated carbon in toluene (50 mL), a THF (50 mL) solution of Pd(OAc)2 (112.4 mg, 0.5 mmol) was added dropwise. After stirring for 30 min, polycarbosilane (250 mg), a reductant, was added portionwise and after completion of addition, the mixture was heated to 80 °C for 1 h in an oil bath. The resulting mixture was cooled to room temperature, and dimethylpolysilane (0.5 g), toluene (25 mL), and MeOH (25 mL) were added to the mixture and stirred overnight at 80 °C. After completion of the above operation, the solution was then cooled to room temperature and a black solid material was filtered. This material was washed with acetone and water, and at 80 °C under reduced pressure for 15 h to give 0.1 mmol g−1 DMPSi-Pd/AC catalyst.
Preparation of catalyst; DMPSi-Pd/AC-CP
Dimethylpolysilane-modified Pd on composite support catalysts was also prepared according to our previous work10a with slight modifications. The following is a representative example of a DMPSi-Pd/AC-CP(3) catalyst; to a suspension of 3.38 g of activated carbon in toluene (50 mL), a THF (50 mL) solution of Pd(OAc)2 (112.4 mg, 0.5 mmol) was added dropwise. After stirring for 30 min, polycarbosilane (250 mg), a reductant, was added portionwise and after completion of addition, the mixture was heated to 80 °C for 1 h in an oil bath. The resulting mixture was cooled to room temperature. Ca3(PO4)2 (1.13 g) and dimethylpolysilane (0.5 g) were successively added in this order at room temperature with an interval of 30 minutes; then, toluene (25 mL) and MeOH (25 mL) were added to the mixture and stirred overnight at 80 °C. After completion of the above operation, the solution was then cooled to room temperature and a black solid material was filtered. This material was washed with acetone and water, and at 80 °C under reduced pressure for 15 h to give 0.1 mmol g−1 DMPSi-Pd/AC-CP catalyst.
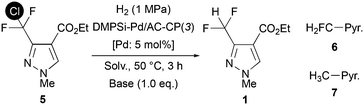
Hydodechlorination of CDFMMP 5 under batch conditions
A mixture of ethyl 3-(chlorodifluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (1: CDFMMP) (23.2 mg, 0.10 mmol), DMPSi-Pd/AC-CP(3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (51.0 mg, Pd: 0.005 mmol) and tetrabutylammonium acetate (30.1 mg, 0.10 mmol) in THF (5 mL) was put in a stainless autoclave reactor. Then, the reactor was set to ChemiStation™ and filled with 1 MPa of H2. After stirring at 50 °C for 3 h at 900 rpm, the reaction mixture was opened to the air and vacuum filtrated with Celite, and washed with EtOH. The mixture was then analyzed by GC using decane as an internal standard. After that, the solvent of the collected sample was removed under reduced pressure and the residue was purified by preparative TLC (n-hexane/EtOAc = 2/1) to give ethyl 3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (1: DFMMP) (7.2 mg, 35% yield) as a white solid.
:1) (51.0 mg, Pd: 0.005 mmol) and tetrabutylammonium acetate (30.1 mg, 0.10 mmol) in THF (5 mL) was put in a stainless autoclave reactor. Then, the reactor was set to ChemiStation™ and filled with 1 MPa of H2. After stirring at 50 °C for 3 h at 900 rpm, the reaction mixture was opened to the air and vacuum filtrated with Celite, and washed with EtOH. The mixture was then analyzed by GC using decane as an internal standard. After that, the solvent of the collected sample was removed under reduced pressure and the residue was purified by preparative TLC (n-hexane/EtOAc = 2/1) to give ethyl 3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (1: DFMMP) (7.2 mg, 35% yield) as a white solid.
Hydodechlorination of CDFMMP 5 under continuous-flow conditions
DMPSi-Pd/AC-CP(1) (0.1 mmol g−1, 1.20 g) was mixed well with Celite 545® (3.0 g) in a screw vial and filled a 10 mm (id) × 100 mm (L) stainless pipe with stainless filters on the both column ends. On top of the reactor, a tube-in-tube type column head was mounted for independent feeding of liquid (inside) and hydrogen gas (outside). The other column end was sealed with an appropriate one-way column cap. Before starting the reactions, liquid (solvent) and hydrogen gas flow were stabilized at the required column temperature, gas flow, and back pressure for at least 1 h. A substrate THF solution of required concentration of substrate 5 containing tetrabutylammonium acetate was then provided at 0.2 mL min−1 flow rate. After stabilization of the continuous-flow conditions, the resulting solution (1.0 mL) was collected and the yields of products were analyzed by GC using decane as an internal standard.
Ethyl 3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (1)9
White solid. 1H NMR (600 MHz, CDCl3): δ = 1.35 (t, J = 7.2 Hz, 3H), 3.97 (s, 3H), 4.32 (q, J = 7.1 Hz, 2H), 7.10 (t, JH–F = 54.0, 1H), 7.90 (s, 1H). 13C NMR (150 MHz, CDCl3): δ = 14.2, 39.7, 60.7, 109.3 (t, JC–F = 237 Hz), 113.4, 134.9, 146.3, 161.8.
Ethyl 3-(fluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (6)
Pale yellow oil. 1H NMR (500 MHz, CDCl3): δ = 1.26 (t, J = 7.1 Hz, 3H), 3.84 (s, 3H), 4.21 (q, J = 7.0 Hz, 2H), 5.51 (d, JH–F = 47.6 Hz, 2H), 7.80 (s, 1H). 13C NMR (150 MHz, CDCl3): δ = 14.0, 39.1, 60.0, 75.9, 112.8, 134.7, 148.3 (d, JC–F = 19 Hz), 162.3. IR (neat, cm−1): 3138, 2983, 1705, 1545, 1255, 1101, 977, 781, 764. HRMS (DART): calcd for C8H12FN2O2 [M + H]+ 187.08773, found 187.08797.
Ethyl 1,3-dimethyl-1H-pyrazole-4-carboxylate (7)9
White solid. 1H NMR (600 MHz, CDCl3): δ = 1.34 (t, J = 6.9 Hz, 3H), 2.45 (s, 3H), 4.27 (q, J = 7.1 Hz, 2H), 7.79 (s, 1H). 13C NMR (150 MHz, CDCl3): δ = 13.4, 14.4, 39.0, 59.8, 112.4, 134.6, 151.3, 163.7.
Author contributions
A. D. provided initial trials on this subject. T. K. carried out all experiments and corrected all data. H. I. designed and directed this project. S. K. conceived and directed this project. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the research program on development of innovative technology grants (JPJ007097) from the Project of the Bio-oriented Technology Research Advancement Institution (BRAIN). We thank Dr. Tei Maki (JEOL Ltd) for STEM and EDX analyses.
Notes and references
- F. Giornal, S. Pazenok, L. Rodefld, N. Lui, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2013, 152, 2–11 CrossRef CAS.
- J. Jaunzems and M. Braun, Org. Process Res. Dev., 2014, 18, 1055–1059 CrossRef CAS.
-
(a)
A. Yamauchi, M. Tanaka, M. Shino and M. Koh, JP2008280305, 2008;
(b)
H. Aihara, W. Yokota, T. Yamakawa and K. Hirai, WO2006090778A1, 2006;
(c)
F. Giordano, T. Vettiger and J. G. Wiss, WO20081845257A1, 2008;
(d)
S. Pazenok, WO2009112157A1, 2009;
(e)
M. Dochnahl, M. Keil and R. Goetz, WO2011054733A1, 2011.
-
(a)
T. Zierke, V. Maywald, M. Rack, S. P. Smidt, M. Keil, B. Wolf and C. Koradin, WO2009133178A1, 2009;
(b)
S. Pazenok, L. Norbert and A. Neeff, WO2008022777A2, 2008.
- V. A. Petrov, S. Swearingen, W. Hong and W. C. Petersen, J. Fluorine Chem., 2001, 109, 25–31 CrossRef CAS.
-
(a)
R. Lantzsch, W. Joerges and S. Pazenok, WO2005042468A1, 2005;
(b)
R. Lantzsch, S. Pazenok and F. Memmel, WO2005044804A1, 2005;
(c) J. M. Kremsner, M. Rack, C. Pilger and O. C. Kappe, Tetrahedron Lett., 2009, 50, 3665–3668 CrossRef CAS;
(d)
O. Buisine and M. Dejoux, WO2012163905A1, 2012.
-
S. E. Jacobson and W. B. Ely, US5296640, 1994.
-
(a)
M. J. Braun and J. Jaunzems, WO2012010692A1, 2012;
(b) J. Xiong, Y. Ma, W. Yang and L. Zhong, J. Hazard. Mater., 2018, 355, 89–95 CrossRef CAS PubMed;
(c) J. Xiong and Y. Ma, Catalysts, 2019, 9, 77 CrossRef.
- A. Das, H. Ishitani and S. Kobayashi, Adv. Synth. Catal., 2019, 361, 5127–5132 CrossRef CAS.
-
(a) H. Ishitani, Y. Furiya and S. Kobayashi, Chem. – Asian J., 2020, 15, 1688–1691 CrossRef CAS PubMed;
(b) S. J. Miller, Y. Furiya, H. Ishitani and S. Kobayashi, Org. Process Res. Dev., 2021, 25, 192–198 CrossRef CAS;
(c) Y. Saito, K. Nishizawa, B. Laroche, H. Ishitani and S. Kobayashi, Angew. Chem., Int. Ed., 2022, e202115643 CAS.
-
(a) M. A. Keane, ChemCatChem, 2011, 3, 800–821 CrossRef CAS;
(b) Z. Peng, X. Wang, Z. Li, X. Chen, Y. Ding and J. Zhang, React. Chem. Eng., 2022, 7, 1827–1835 RSC.
-
(a)
H. Yamada, K. Tanaka, H. Adachi, S. Yamada and S. Shimoda, WO9408975A1, 1994;
(b)
M. Maue, I. Adelt, W. Gienke, M. Heil, P. Jeschke, B.-W. Krueger, F. A. Muehltrau, A. Sadau, K. Raming, U. Ebbinghaus-Kintscher, M. Adamczewski, A. Voerste, U. Goergens, T. Kapferer, M. W. Drewes and A. Becker, WO2010051926, 2010;
(c) T. T. Tung, S. B. Christensen and J. Nielsen, Chem. – Eur. J., 2017, 23, 18125–18128 CrossRef CAS PubMed;
(d) E. Zarenezhad, M. Farjam and A. Iraji, J. Mol. Struct., 2021, 1230, 129833 CrossRef CAS.
-
(a) C. Murali, M. S. Shashidhar and C. S. Gopinath, Tetrahedron, 2007, 63, 4149–4155 CrossRef CAS;
(b) C. J. Crawford, Y. Qiao, Y. Liu, D. Huang, W. Yna, P. H. Seeberger, S. Oscarson and S. Chen, Org. Process Res. Dev., 2021, 25, 1573–1578 CrossRef CAS PubMed.
-
(a) K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144–7145 CrossRef CAS;
(b) K. Mori, K. Yamaguchi, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2002, 124, 11572–11573 CrossRef CAS PubMed;
(c) G. Xu, J. Guo, Y. Zhang, Y. Fu, J. Chen, L. Ma and Q. Guo, ChemCatChem, 2015, 7, 2485–2492 CrossRef CAS.
-
(a) J. Moon and S. Lee, J. Organomet. Chem., 2009, 694, 473–477 CrossRef CAS;
(b) F.-L. Xue, J. Qi, P. Peng, G.-Z. Mo and Z.-Y. Wang, Lett. Org. Chem., 2013, 12, 64–79 Search PubMed.
- K. B. Wiberg and P. R. Rablen, J. Am. Chem. Soc., 1993, 115, 614–625 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
-
(a) P. A. Champagne, J. Pomarole, M.-É. Thérien, Y. Benhassine, S. Beaulieu, C. Y. Legault and J.-F. Paquin, Org. Lett., 2013, 15, 2210–2213 CrossRef CAS PubMed;
(b) P. A. Champagne, A. Saint-Martin, M. Drouin and J.-F. Paquin, Beilstein J. Org. Chem., 2013, 9, 2451–2456 CrossRef PubMed;
(c) P. A. Champagne, M. Drouin, C. Y. Legault, C. Audubert and J.-F. Paquin, J. Fluorine Chem., 2015, 171, 113–119 CrossRef CAS;
(d) N. S. Keddie, P. A. Champagne, J. Desroches, J.-F. Paquin and D. O'Hagan, Beilstein J. Org. Chem., 2018, 14, 106–113 CrossRef CAS PubMed;
(e) H.-J. Ai, X. Ma, Q. Song and X.-F. Wu, Sci. China: Chem., 2021, 64, 1630–1657 CrossRef CAS.
-
M. Braun, J. Jaunzems and M. Kusakabe, WO2012025469A1, 2012.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experiments, experimental procedures, catalysts' physicochemical information, and spectral information of products. See DOI: https://doi.org/10.1039/d3cy00182b |
| ‡ The substance amount of substrate-based space velocity (SVmol) is defined as the value of the feed amount of the substrate supplied to the amount of catalytically active site in the reactor per unit period. |
| § From the viewpoint of an actual experimental situation, the minimum substrate feed rate should be set to more than 0.1 mL min−1 to maintain stable flow while preventing clogging. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab