
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled alcohol oxidation reactions by supported non-noble metal nanoparticles on chitin-derived N-doped carbons†
Daniele
Polidoro
 a,
Daniel
Ballesteros-Plata
b,
Alvise
Perosa
a,
Enrique
Rodríguez-Castellón
b,
Daily
Rodríguez-Padrón
*a and
Maurizio
Selva
*a
a,
Daniel
Ballesteros-Plata
b,
Alvise
Perosa
a,
Enrique
Rodríguez-Castellón
b,
Daily
Rodríguez-Padrón
*a and
Maurizio
Selva
*a
aDipartimento di Scienze Molecolari e Nanosistemi, Università Ca' Foscari di Venezia, 30123 Venezia, Italy. E-mail: daily.rodriguez@unive.it; selva@unive.it
bDepartment of Inorganic Chemistry, Facultad de Ciencias, Universidad de Málaga, Campus de Teatinos s/n, 29071 Málaga, Spain
First published on 20th February 2023
Abstract
A series of catalysts based on non-noble metal nanoparticles supported on chitin-derived N-doped carbons was prepared through a one-step protocol in the presence of EDTA as a ligand. Both the complexing properties of EDTA and the interaction of metal precursors with the nitrogen sites on the support surface allowed a good dispersion and a homogeneous distribution of the metal active sites. The synthesized materials were tested in the catalytic oxidation of alcohols to aldehydes and ketones. Particularly, the model reaction of oxidation of benzyl alcohol showed that iron- and even more molybdenum-based materials exhibited the best performance. At 130 °C and 20 bar, in the presence of air as an oxidant, the Mo–N/C-catalyzed reactions proceeded with excellent conversion and selectivity, both above 90%, towards the product of partial oxidation, benzaldehyde, and the catalytic performance was retained over 5 recycling runs without any loss of activity. The Mo-based system proved effective also for the conversion of some representative benzyl- and furyl-type alcohols bearing primary and secondary hydroxyl functions to the corresponding aldehydes/ketones, though the oxidation of aliphatic substrates was unsuccessful. All catalysts together with the Mo–N/C sample recovered after its use were fully characterized following a multi-technique approach involving XRD, N2-physisorption, XPS, and HRTEM-EDX analyses. Textural, morphological and chemical properties were thus compared and related to the observed trend of catalytic activity.
Introduction
The undoubted role of catalysis, involved in more than 90% of the chemical processes, has recently been highlighted and accentuated as a consequence of the current global warming trend.1 Indeed, the design of active, selective and stable catalysts, especially recyclable heterogeneous systems, is becoming increasingly significant to improve the energy efficiency, the environmental compatibility and the carbon footprint of any synthetic sequence.2–4 This scenario is well exemplified by a variety of transformations, among which oxidations have attracted extensive attention, and more particularly in this field, the selective conversion of alcohols to aldehydes and ketones is recognized for its importance.The oxidation of benzyl alcohol (BnOH) to benzaldehyde deserves a mention here not only for the fact that BnOH is a bio-based derived platform molecule and hence its upgrading represents a strategy of biomass valorisation, but also for the intrinsic value of the product benzaldehyde which is a versatile synthetic intermediate and a highly used flavouring agent.5–8
A variety of approaches with the use of different catalysts and oxidizing agents have been proposed for such a reaction. Peroxides, including hydrogen peroxide and t-butyl hydroperoxide, molecular oxygen or air, have been used as oxidizing agents, with emphasis on the convenience of air for safety and economic reasons.9 The analysis of available catalysts is considerably more articulated. Permanganate- and chromate-based systems, endangered noble-metals as Au, Pd, and Rh, and biocatalytic pathways have been often described.10–14 Notwithstanding these catalysts offer good-to-excellent synthetic solutions, major issues are associated with: i) the toxicity of the active elements (Mn and Cr) that poses negative consequences for health and the environment, ii) the difficult reaction scalability due to the limited availability and high price of noble metals, and iii) the storage, the operational stability, and the difficult reusability of enzymes. Other strategies for the selective oxidations of primary/benzyl alcohols have been designed with the use of co-catalysts based on aminoxyl radicals, such as 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO),15 transition metal-based catalysts as molybdenum supported on metal organic frameworks (MOFs),16,17 and bimetallic catalysts.18–20 In the latter respect, an interesting recent study has focused on the 3D-printing of bi-metallic (namely, Fe and Co) monoliths for the continuous-flow oxidation of benzyl alcohol.21,22
Alternative solutions are also emerging through the synthesis of carbocatalysts, especially nano-sized materials. For example, carbon nanotubes (CNTs) have been used for the activation of a common oxidant such as peroxymonosulfate (PMS) by which benzyl alcohol was converted into benzaldehyde with a selectivity and yield of 83% and 57%, respectively, at 50 °C.23 The carbocatalysis-assisted PMS activation proceeded via a radical process where surface carbonyl groups on CNTs promoted both O–O bond cleavage of PMS and a redox cycle to generate ˙OH and SO4˙− radicals.
Inspired by these models, we wish to propose herein a new class of oxidation catalysts based on non-noble metal nanoparticles supported on N-doped carbons (N/C). The literature describes the preparation of N/C materials starting from different N-containing polymers, either bio-based ones such as chitosan or of fossil origin such as polyvinylpyrrolidone.24,25 In this work instead, chitin, the second most abundant biopolymer on Earth, has been used directly as a precursor for the catalyst support. A similar approach has been recently followed by us also within a research program for the valorisation of high-chitin content bio-waste (shrimp shells), where the synthesis of catalysts for some CO2-fixation reactions was carried out.5 It should be noted here that albeit innovative applications of chitin are desirable to design sustainable protocols, they are often highly challenging mostly because of the lack of solubility of chitin in almost all liquid solvents except for task-specific ionic liquids.26
This study has been finalized to the achievement of low-loading nickel, iron, cobalt and molybdenum entities supported and dispersed on N/C networks, and to their further use for the catalysis of the oxidation of benzyl- and furyl-type alcohols, with emphasis on benzyl alcohol, to the corresponding aldehydes/ketones. It has been demonstrated that heteroatom-doping and especially N-doping of carbonaceous architectures has not only influenced the catalytic response, but the metal–N/C interactions in the investigated materials have favoured the incorporation of metallic precursors (such as cobalt, palladium and ruthenium, among others)27,28 and the formation of active sites during the catalyst synthesis. In the literature, similar metal–N/C (M–N/C) interactions have been already recognised for their role in the formation of interfacial sites active in several catalytic transformations.29
The performance of the M–N/C systems designed here was investigated by exploring the effects of different reaction parameters and the recycling/reusability opportunities in the presence of the most economic and safe oxidant such as air. The conversion of alcohols and selectivity towards the corresponding carbonyl derivatives were typically over 90%, and this outcome remained stable even after 5 cycles of catalyst reuse. The characterization of the catalytic samples was consistent with the formation of core–shell nanoparticles of metal/metal oxides with peculiar textural and physicochemical properties, and homogeneously supported on a N-doped carbonaceous support.
Experimental part
Material and equipment
Benzyl alcohol, furfuryl alcohol, 1,5-bis(hydroxymethyl)furan, 1-phenylethanol, vanillyl alcohol, 1-pentanol, cyclopentanol, acetonitrile, 2-propanol, chitin, microcrystalline cellulose, EDTA, CoCl2·6H2O, Fe(NO3)3·9H2O, and Ni(Ac)2·4H2O, (NH4)MoO4 were commercially available compounds sourced from Sigma-Aldrich. If not otherwise specified, reagents and solvents were employed without further purification. Air gas was purchased from SIAD, Italy. GC-MS (EI, 70 eV) analyses were performed on an HP5-MS capillary column (L = 30 m, ∅ = 0.32 mm, film = 0.25 mm). GC (flame ionization detector; FID) analyses were performed with an Elite-624 capillary column (L = 30 m, Ø = 0.32 mm, film = 1.8 mm). 1H, 13C NMR spectra were recorded using a Bruker Advance III HD 400 WB equipped with a 4 mm CP/MAS probe, at 400 and 101 MHz, respectively. Chemical shifts were reported downfield from tetramethylsilane (TMS) and MeOD, DMSO-d6 and CDCl3 were used as solvents. All reactions were performed in duplicate to verify reproducibility.Synthesis of supported non-noble nanoparticles on chitin derived N-doped carbonaceous materials
In a typical synthesis, the salt chosen as a metal precursor (1 mmol) was dissolved in 2-propanol (60 mL), together with EDTA (1 g). Subsequently, chitin (5 g) was added to the mixture, which was kept under stirring for 9 h at 80 °C under reflux. The suspension was filtered and the so-obtained solid was dried at 100 °C overnight and finally, heated at 500 °C (heating rate was 5 °C min−1) under a N2 flow (10 mL min−1) for 1 h. The catalytic samples achieved by this procedure were labelled as Ni–N/C, Fe–N/C, Co–N/C and Mo–N/C for the nickel, iron, cobalt and molybdenum catalysts, respectively. Additionally, two molybdenum-based samples were prepared under identical conditions, but i) in the absence of EDTA and ii) replacing chitin with cellulose. These two solids were labelled Mo–N/C_b and Mo–C, respectively. The resulting materials were ground to powder (particle size <200 μm) and stored in an oven (60 °C, 15 mbar) until further use. The yield of the obtained materials was ca. 25 ± 5%, based on the total weight of chitin and the metal precursor used.Material characterization
The analysis of the chemical composition on the surface of the solids was carried out by XPS (X-ray Photoelectron Spectroscopy) with a Physical Electronics VersaProbe II Scanning XPS Microprobe equipped with a monochromatic X-ray Al Kα radiation source at a vacuum of 10−7 Pa. The binding energies were referenced to the C 1s peak from adventitious carbon at 284.8 eV. High-resolution spectra were recorded using a concentric hemispherical analyzer at a 29.35 eV constant energy pass, using a 200 μm diameter analysis area, and the pressure in the analysis chamber was kept below 5 × 10−6 Pa. PHI ACCESS ESCA-F V6 software was used for data acquisition and analysis. A Shirley-type background was subtracted from the signals. The recorded spectra were always analyzed with Gauss–Lorentz curves in order to determine more accurately the binding energy of the atomic levels of the different elements.Laboratory X-ray powder diffraction (XRPD) patterns were collected on a PANanalytical X'Pert Pro automated diffractometer in the central research facilities (SCAI) at the University of Málaga. Powder patterns were recorded in Bragg–Brentano reflection configuration by using a Ge(111) primary monochromator (Cu Kalfa1) and an X'Celerator detector with a step size of 0.0167° (2θ). The powder patterns were recorded between 4 and 70° in 2θ with an equivalent counting time of ∼60 s per step.
N2 physisorption measurements were conducted on a Micromeritics TriStar 3000 instrument. The samples were outgassed at 120 °C for 2 h. Then, adsorption–desorption isotherms were recorded at −196 °C. The specific surface areas were calculated by the BET method; the pore volumes were calculated from adsorption isotherms and the pore size distributions were estimated using the Barrett–Joyner–Halenda (BJH) algorithm available as a built-in software from Micromeritics.
High Resolution Transmission Electron Microscopy (HRTEM) was carried out with a TALOS F200x instrument also operating in STEM mode (Scanning Transmission Electron Microscopy), equipped with a HAADF detector, at 200 kV and 200 nA. The microanalysis was carried out with an EDX Super-X system provided with 4 X-ray detectors and an X-FEG beam. Metal leaching was determined by ICP-MS using an Elan DRC-e (PerkinElmer SCIEX) spectrometer.
Typical oxidation reaction procedure and product analysis
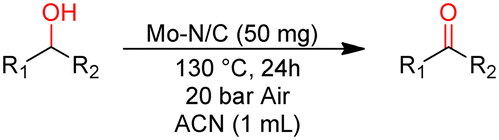
Experiments were performed in a 25 mL tubular reactor of borosilicate glass (Pyrex) loaded with benzyl alcohol (1 mmol, 108 mg), catalyst (50 mg) and acetonitrile (1 mL). The reactor was then placed in a jacketed stainless-steel autoclave equipped with a manometer and two needle valves. The autoclave was pressurized with the desired air pressure and heated by circulating oil at the desired temperature. The mixture was kept under magnetic stirring at a rate of 900 rpm. At the end of the tests, the autoclave was cooled to room temperature and gently purged. Upon the completion of the reaction, an aliquot of the reaction mixture was withdrawn by a syringe and analysed by GC-FID to determine the reaction conversion and selectivity and rotary evaporated (60 °C, 15 mbar). The obtained dried sample was analysed by GC-MS and NMR in CDCl3 or DMSO-d6 solvent. All characterization data are reported in the ESI† section (Fig. S3–S20).Results and discussion
Synthesis and characterization of the catalysts
 | ||
| Fig. 1 Schematic representation of the synthetic procedure of the supported metal nanoparticles on N-doped carbons. | ||
The four materials obtained as described in Fig. 1 were fully characterized through a multi-technique approach, including XRD, N2-physisorption, XPS and HRTEM-mapping analyses.
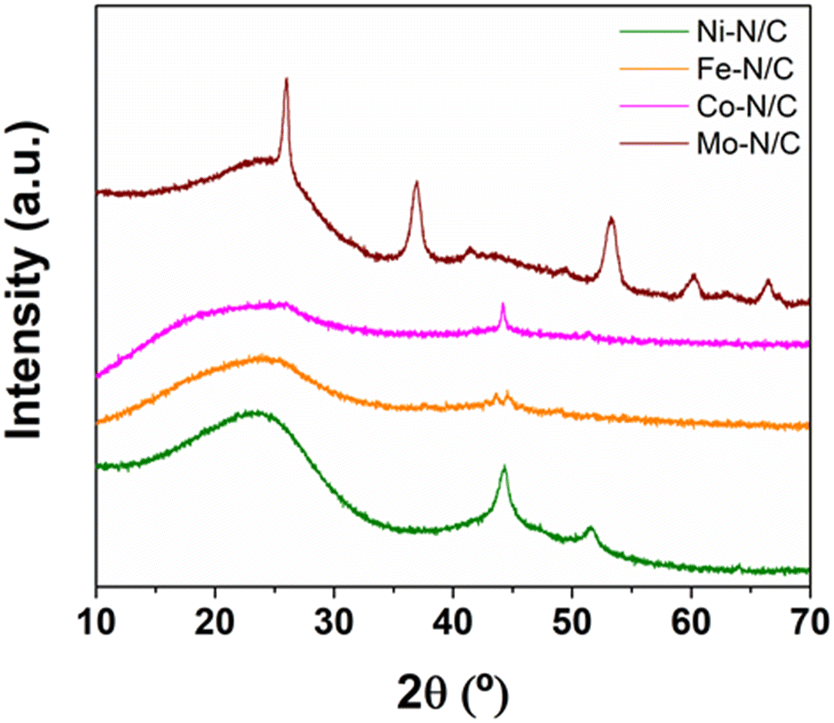 | ||
| Fig. 2 XRD patterns of the metal-supported N-doped carbonaceous materials. | ||
The textural properties of the samples were investigated by N2-physisorption analysis. The four samples displayed a similar behavior, with a type IV isotherm and type II adsorption hysteresis, corresponding to the formation of mesoporous materials and the presence of disordered networks (Fig. 3). The textural properties of the prepared catalysts, including surface area, pore diameter and pore volume are described in Table 1. The relatively slight differences observed within the investigated materials were most probably determined by the nature of the metal precursors employed for the synthesis.
 | ||
| Fig. 3 N2 physisorption isotherms at −196 °C of the prepared supported metal nanoparticles on N-doped carbon. A: Ni–N/C; B: Fe–N/C; C: Co–N/C and D: Mo–N/C. | ||
| Material | S BET [m2 g−1] | D BJH (nm) | V BJH [cm3 g−1] |
|---|---|---|---|
| a S BET: specific surface area was calculated by the Brunauer–Emmett–Teller (BET) equation. b D BJH: mean pore size diameter was calculated by the Barret–Joyner–Halenda (BJH) equation. c V BJH: pore volumes were calculated by the Barret–Joyner–Halenda (BJH) equation. | |||
| Ni–N/C | 360 | 2.7 | 0.25 |
| Fe–N/C | 234 | 2.5 | 0.15 |
| Co–N/C | 298 | 2.3 | 0.17 |
| Mo–N/C | 285 | 2.9 | 0.20 |
The chemical properties and composition of the synthesized materials were explored by XPS analysis. Fig. 4 depicts the XPS spectra of the metal components, namely Ni, Fe, Co and Mo in the four materials, respectively. The results confirmed the incorporation of the metal entities on the surface of the N-doped carbonaceous supports.
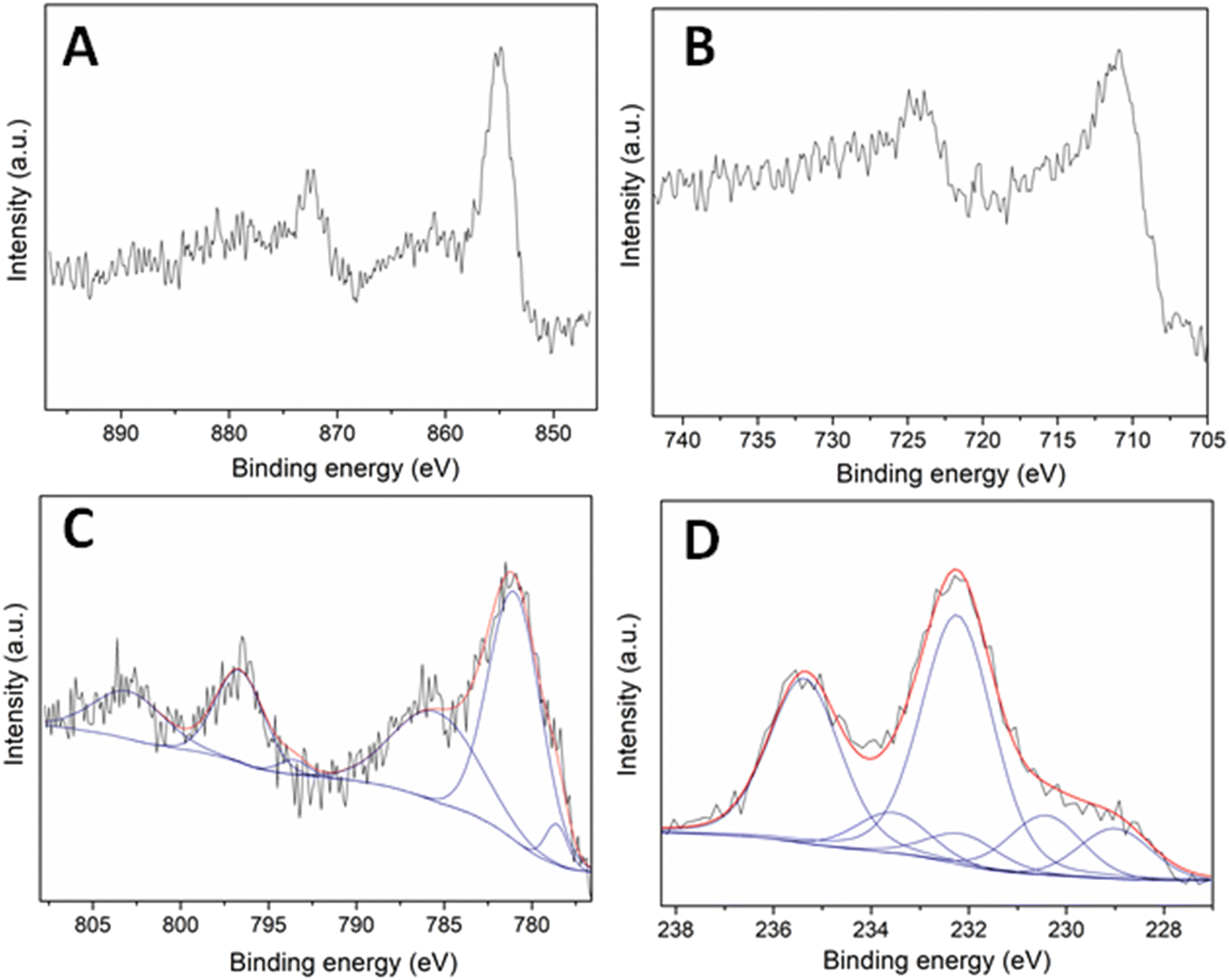 | ||
| Fig. 4 XPS spectra of the prepared samples in A) the Ni 2p XPS region for the Ni–N/C; B) the Fe 2p XPS region for the Fe–N/C; C) the Co 2p XPS region for the Co–N/C and D) the Mo 3d XPS region for the Mo–N/C. | ||
The Ni 2p core level spectrum of the Ni–N/C sample indicated two main bands around 855.0 and 872.8 eV, consistent with the doublet Ni 2p3/2–Ni 2p1/2, respectively (Fig. 4A). The distance between the two signals was 17.8 eV: this confirmed the presence of Ni(0) entities, in agreement with XRD data. However, the Ni 2p3/2 signal at a binding energy of 855.0 eV was indicative of the concomitant formation of Ni(II). XPS and XRD results therefore suggested the occurrence of a core–shell architecture where a core of metallic nickel was enveloped by nickel oxide.
The Fe 2p core level spectrum of the Fe–N/C sample exhibited two main signals with maxima at ca. 710.9 and 724.4 eV corresponding to the doublet Fe 2p3/2–Fe 2p1/2 (Fig. 4B). Such binding energies were consistent with the presence of iron oxide species, differently from the conclusions raised from XRD data. Both analyses together again suggested the generation of core–shell structures with a metallic center and a metal oxide shell.
The Co 2p core level spectrum of the Co–N/C material displayed the presence of six contributions located at 778.5, 781.2, 785.9, 793.7, 796.5 and 803.2 eV (Fig. 4C). The peaks located at 778.5 and 793.7 eV were assigned to the doublet Co 2p3/2 and Co 2p1/2 of metallic cobalt. The low-intensity observed for such signals was attributed to a lower content of Co(0) on the sample surface. On the other hand, the contributions at 781.2, 785.9, 796.5 and 803.2 eV were associated with the doublet Co 2p3/2, Co 2p1/2 and the corresponding satellite signals of cobalt oxide species. The higher intensity of such signals compared to those of metallic cobalt suggested that core–shell architectures were plausible also in this case.
The Mo 3d XPS region of the Mo–N/C sample is shown in Fig. 4D. Several signals at 229.0, 230.4, 232.3, 232.4, 233.5 and 235.3 eV were observed. The bands located at binding energies of 229.0 and 232.4 eV have been previously assigned in the literature to Moδ+ (0 < δ < 4) entities in molybdenum nitride, indicating the formation of Mo–N bonds.36 The peaks at 230.4 and 233.5 eV were instead attributed to Mo 3d5/2 and Mo 3d3/2 contributions of Mo(IV) entities in MoO2, while the signals at 232.3 and 235.3 eV were ascribed to Mo 3d5/2 and Mo 3d3/2 contributions of Mo(VI) entities in MoO3. Albeit these results generally corroborated the conclusions from XRD analyses, a difference emerged between the two techniques: the XPS spectrum revealed a content of MoO3 on the material surface higher than that detected by XRD, while XRD provided a content of MoO2 in the bulk sample greater than that noticed by XPS. Also for Mo–N/C, the characterization analyses agreed with the formation of core–shell architectures where a core mainly comprised of MoO2 was surrounded by MoO3, with a minor presence of Mo–N groups.
XPS analysis of C 1s, O 1s and N 1s regions of the four materials are included in Fig. S1 (ESI†). Similar results were obtained in all cases. The high-resolution C 1s core level spectrum was deconvoluted into four contributions located around 284.7, 286.3, 287.8 and 289.3 eV attributed to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (graphitic, aromatic carbon) and C–OH, C–N/C–O, and CO carbonate bonds, respectively. Moreover, the N 1s core level spectrum displayed the presence of two main contributions at ca. 398.6 and 400.8 eV due to pyridinic and pyrrolic groups, respectively. Finally, the O 1s XPS region of the prepared materials showed two contributions at 531.4 and 533.1 eV related to the lattice oxygen of metal oxides and to adsorbed/bonded water, respectively.
C (graphitic, aromatic carbon) and C–OH, C–N/C–O, and CO carbonate bonds, respectively. Moreover, the N 1s core level spectrum displayed the presence of two main contributions at ca. 398.6 and 400.8 eV due to pyridinic and pyrrolic groups, respectively. Finally, the O 1s XPS region of the prepared materials showed two contributions at 531.4 and 533.1 eV related to the lattice oxygen of metal oxides and to adsorbed/bonded water, respectively.
HRTEM images are displayed in Fig. 5. The presence of a laminar structure of the carbonaceous support was noticed, where various N-doped carbon layers were found superimposed to each other. This evidence was especially clear in the case of the Mo–N/C sample. HRTEM micrographs proved the formation of homogeneously distributed nanoparticles in each case, where the size and dispersion were also influenced by the nature of the used metal precursors. The Ni–N/C, Fe–N/C, Co–N/C and Mo–N/C samples exhibited the formation of nanoparticles with mean diameters of (8 ± 2) nm, (28 ± 2) nm, (30 ± 2) nm and (31 ± 2) nm, respectively. HRTEM micrographs and mapping analysis confirmed the formation of highly dispersed and homogeneously distributed metallic entities in the four prepared samples. The formation of metal agglomerates occurred only in the case of the Mo–N/C material (Fig. 6).
 | ||
| Fig. 5 HRTEM micrographs of the prepared catalytic materials. | ||
 | ||
| Fig. 6 HRTEM-EDX micrographics of the prepared materials. | ||
Catalytic activity
Benzyl alcohol was chosen as a model substrate to start investigating the oxidation performance of the above-described metal nanoparticles supported on N-doped carbon materials. Air was selected as the oxidant for both safety and economic reasons. Batch experiments were carried out in a stainless-steel autoclave in which a mixture of benzyl alcohol (1 mmol, 108 mg) and the selected catalyst (50 mg) was set to react at different temperatures (100 to 150 °C) under 40 bar of air for 24 h. NMR and GC/MS analyses of the reaction mixture confirmed the formation of the products 1a (benzaldehyde) and 1b (benzoic acid) (Scheme 1). All characterization data are reported in the ESI† section (Fig. S3–S20). Additional (blank) experiments were also performed without any catalyst and by using N-doped carbon obtained directly from chitin: in these cases, however, the conversion of BnOH was very low, between 2 and 7%, even operating at 150 °C (24 h, 40 bar). The selectivity for 1a was remarkable (>99%). | ||
| Scheme 1 Schematic representation of the benzyl alcohol oxidation reaction. | ||
Fig. 7 shows the results of the conversion of benzyl alcohol and the selectivity towards 1a and 1b achieved with the supported metal-based nanoparticles as catalysts.
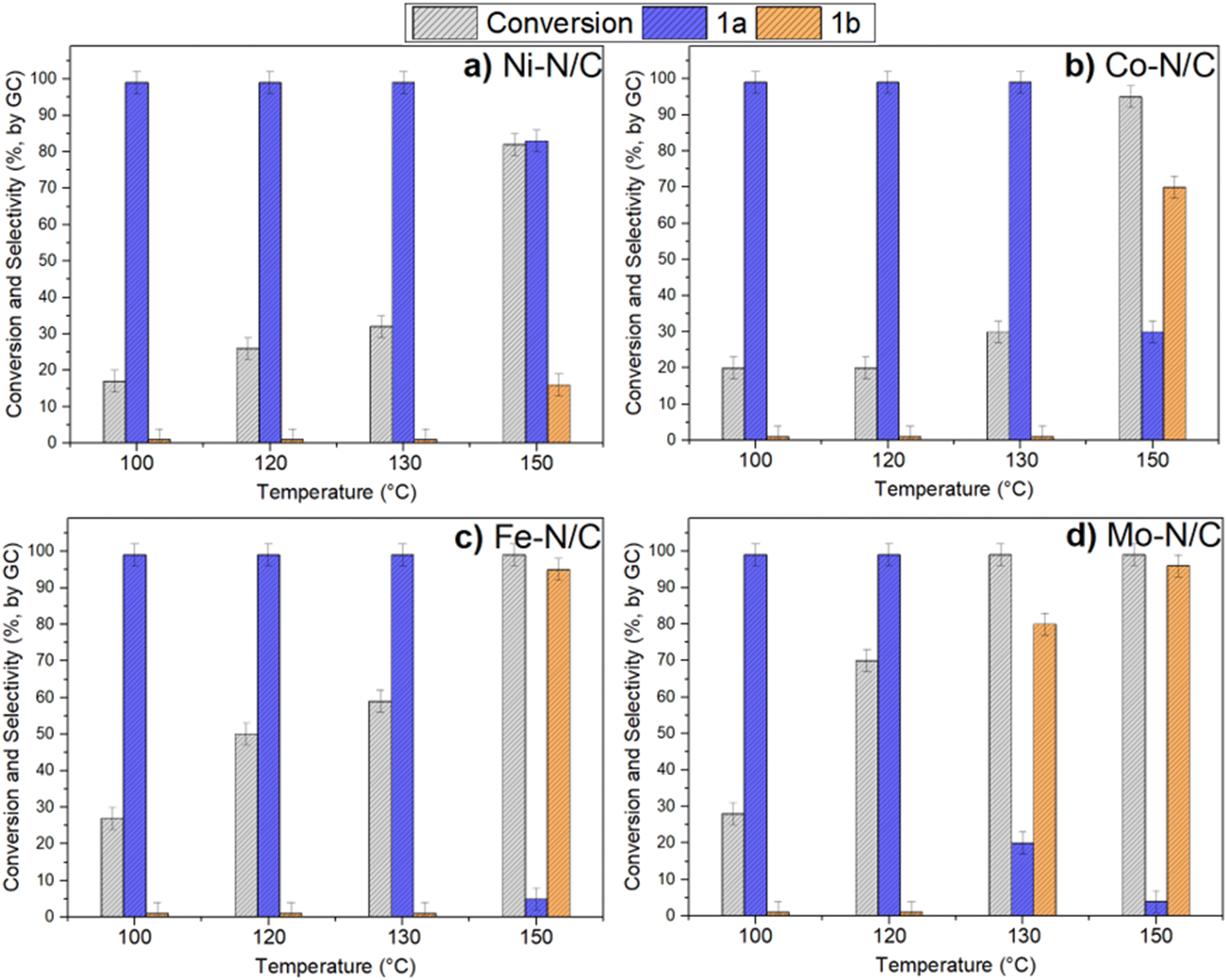 | ||
| Fig. 7 Catalyst screening in the oxidation reaction of benzyl alcohol at different temperatures. Reaction conditions: benzyl alcohol (1 mmol, 108 mg), catalyst (50 mg), acetonitrile (1 mL) at 130 °C under 40 bars of air for 24 h. Conversion and selectivity were determined by GC. | ||
In the presence of Ni–N/C, the conversion increased from ca. 17% to 32% by increasing the reaction temperature from 100 °C to 130 °C, while the selectivity towards 1a was steady and greater than 99% (Fig. 7a). On the other hand, an enhancement of T up to 150 °C made the conversion further increase to 82%, but this brought about a change in the product distribution due to the concurrent formation of benzaldehyde (83%) and benzoic acid (16%).
A different behavior was noticed when using Co–N/C (Fig. 7b). From 100 °C to 130 °C, the conversion gradually increased from 20% to 30%, respectively, with the exclusive formation of 1a; at 150 °C, instead, the reaction was almost quantitative (95%), but the predominant product became benzoic acid (1b, 70%). No other derivatives than benzaldehyde (30%) were observed.
Fe–N/C proved an even more active catalyst compared to Ni- and Co-based systems. At 130 °C, it allowed the same excellent selectivity toward 1a (>99%), but a greater conversion up to 57%. A drastic variation in the product distribution was instead observed at 150 °C: indeed, benzyl alcohol was almost quantitatively oxidized into benzoic acid (1b, 95%) (Fig. 7c). It should be noted here that iron/iron oxide-based systems are receiving increasing attention from the scientific community because their catalytic performance is often coupled to other desirable properties including low toxicity, good dispersibility and easy separation. The latter is favored by the magnetic behavior of iron (alike Ni and Co) that makes magnetic recovery from bulk reaction solutions a highly efficient technique.6,37–41
The last tested sample, Mo–N/C, was confirmed as the most active and promising system among those investigated here (Fig. 7d). The performance of Mo–N/C as an oxidation catalyst emerged by comparing the reaction T and the product distribution: a good conversion (70%) and 1a selectivity (100%) were both reached at 120 °C, while at 130 °C, a quantitative reaction took place with the prevalent formation of the fully oxidized acid derivative 1b (80%). Finally, the increase of T up to 150 °C resulted in the complete oxidation of benzyl alcohol to benzoic acid (1b, 96%).
Molybdenum oxides have been extensively employed in several fields including sensing and electrochemical applications though they have been used to a far lesser extent in catalysis.42 In general, the activity of heterogeneous catalysts can be influenced by several factors associated with the preparation procedure including, among others, the metal precursor, and the calcination temperature. The establishment of a relationship between the catalytic performance and the nature of its active sites is, therefore, anything but a trivial goal. Efforts in this direction have been accomplished by the known studies of Hugh, Stott, and Taylor that attempted a correlation between the surface chemical reactivity and the presence of unsaturated active sites at an atomic level, or the Brønsted–Evans–Polanyi (BEP) relation that offers an interpretation for volcano curves in heterogeneous catalysis based on the activation energy and the enthalpy change of elementary reactions. In some cases, a correlation between the d-band electronic distribution of transition metals and its affinity for adsorbates/reactants has been described.43–46 A similar tendency can be proposed also in this work by observing that the better catalytic performance (directly related to the affinity with the reactants) was achieved for metals with a less-occupied d-band. Indeed, in general terms, the following order of increasing catalytic activity can be suggested in the investigated reaction: Ni < Co < Fe < Mo. In addition, XPS and XRD characterization analyses have proved that the compresence of at least two different oxidized species such as MoO2 and MoO3 can play a role in the performance of the Mo–N/C system. Deeper investigations involving computational studies should be carried out to obtain more insights in this direction.
Whatever the reasons for the results of Fig. 7, given the behavior of Mo–N/C, this solid was selected to continue the study through a parametric analysis and substrate scope investigation. These aspects will be detailed in the following sections.
Influence of major reaction parameters and comparison to the literature
The effects of major reaction parameters, such as time (t) and air pressure (p), were investigated by performing two series of tests during which the oxidation of benzyl alcohol was carried out by changing: i) p from 10 to 50 bar, at 130 °C for 24 h; ii) t from 2 to 24 h, at 130 °C and 20 bar. The temperature was set to 130 °C according to the outcome of tests in Fig. 7. The results are shown in Fig. 8a and b. | ||
| Fig. 8 Influence of major reaction parameters on the catalytic performance of the Mo–N/C catalysts, considering a) air pressure and b) time. Reaction conditions: benzyl alcohol (1 mmol, 108 mg), Mo–N/C (50 mg), 130 °C, acetonitrile (1 mL). | ||
The pressure affected both the conversion and the product distribution. The increase of p from 10 to 20 bar and 30 bar favored the conversion from 82% to 95% and >99%, respectively (Fig. 8a). Thereafter, no further changes were observed at 40 and 50 bar. A dramatic influence was noticed on the reaction selectivity: in the range 10–20 bar, 1a was the predominant product (>91% selectivity), while from 30 to 50 bar, the full oxidation of BnOH to benzoic acid became the major process (1b: from 67 to 81%, respectively). Overall, the results were consistent with the availability of air (oxygen) dissolved in the reaction mixture: the greater the pressure, the greater the oxidant amount, the deeper the oxidation extent.47
Subsequent experiments were then carried out under 20 bar of air, by tuning the reaction time in an attempt to improve both the selectivity and the yield of the aldehyde derivative. This study, however, confirmed the result of Fig. 8a: the best conversion/selectivity compromise was achieved after 24 hours, resulting in a 95% conversion and a 91% selectivity towards 1a, respectively. Shorter reaction times, from 2 to 12 h, allowed the exclusive formation of benzaldehyde (>99%), but the conversion – though it gradually increased – did not exceed 63% (Fig. 8b).
The effect of temperature (T) was further evaluated by additional experiments carried out in the range of 100–150 °C, under 20 bar of air, for 24 h. However, the results reported in Fig. S2† proved that no significant improvement was achieved.
An in-depth analysis of the current literature was then performed to contextualize the achievements of Fig. 8b. Table 2 displays a comparison of various catalytic systems reported for the oxidative transformation of benzyl alcohol.
| Entry | Catalyst | Reaction conditions | Conversion (%) | Selectivity (%) | Ref. |
|---|---|---|---|---|---|
| a UHP: urea hydroperoxide. | |||||
| 1 | Au/HT | O2, 120 °C, 2 h | 72 | 99 | 48 |
| 2 | RuO2/TiO2 | Air, 240 °C | 92 | 98 | 49 |
| 3 | 1% PdFe/TiO2 | O2, 0.083 h, 50 °C | 3 | 100 | 11 |
| 4 | PdAu/HP-SAPO-5 | Air, 100 °C, 6 h | <20 | >99 | 12 |
| 5 | Pd–Cu/TiO2 | O2, 120 °C, 2 h | 35.9 | 98.7 | 20 |
| 6 | CuOx/SBA15 | H2O2, 80 °C/30 min | 73 | 54 | 50 |
| 7 | Nano-g-Fe2O3 | H2O2, 75 °C, 12 h | 32 | 97 | 51 |
| 8 | 0.5% SnOx@AP-ox | O2, 110 °C, 2 h | 5.8% | 85.3% | 13 |
| 9 | MnO2/GO | Air, 110 °C, 3 h | >99 | 96 | 52 |
| 10 | MoO2L·DMF complex | UHP,a 80 °C, 2 h | 99 | 92 | 16 |
| 11 | Mo–N/C | Air, 130 °C, 24 h | 95 | 91 | This work |
Several studies described catalysts based on Au, Ru, and Pd in the presence of both air and O2 as the oxidants (entries 1–4). In some cases, good conversion and selectivity were reached. One of the best results was obtained with RuO2/TiO2 by which BnOH was almost quantitatively converted to benzaldehyde (98%) using air though the reaction took place at a very high T of 240 °C (entry 2). Moreover, the use of noble metals should be limited for both costs and availability reasons (see Introduction).
Among non-precious metals including Cu, Fe, Sn and Mn, Mn-based systems offered the best options for the selective oxidation of BnOH (entries 6–10). In the form of both a heterogeneous catalyst as MnO2 supported on graphene oxide and a homogenous complex as a dioxomolybdenum Schiff base (MoO2L·DMF; L: from the condensation of 4-aminobenzohydrazide and 3-methoxy salicylaldehyde), these catalysts exhibit high performance with a conversion >99% and a selectivity of 92–96%, respectively, in the presence of air or urea hydroperoxide as an oxidant (entries 9–10). A similar behavior was achieved also with the Mo–N/C system proposed in this work (entry 11): albeit our reaction conditions were apparently more energy demanding (130 °C, 24 h) than those of entries 9–10 (80–110 °C, 2–3 h), Mo–N/C was still competitive in view of its cheap cost and more sustainable synthesis, especially if bio-waste derived chitin was used.
Additional experiments were conceived to shed light on the activity of the Mo–N/C sample. These were carried out by employing two other catalytic systems: i) a molybdenum-based catalyst prepared under the same conditions used for Mo–N/C, but in the absence of EDTA; ii) a second material synthesized in the absence of EDTA and by replacing chitin with cellulose as the carbon source. These samples were labelled as Mo–N/C_b and Mo–C, respectively. Such materials were tested for the oxidation of benzyl alcohol run at 130 °C, under 20 bar of air for 24 h. Compared to Mo–N/C, both samples allowed a lower conversion not exceeding 75–80%, but even more interesting, the selectivity turned out drastically different, since benzoic acid was the almost unique oxidation product (96%). This outcome strongly suggested that both the use of EDTA and the presence of nitrogen on the structure acted synergically to improve the catalytic activity and steer the selectivity towards the partial oxidation of benzyl alcohol, most likely thanks to a better dispersion of the metal active sites on the support surface, and to the occurrence of metal–nitrogen active sites as observed from the XPS results. It should be noted that the potential of such (Mo–N) species for the adsorption and activation of oxygen has been previously highlighted by other authors.53
Substrate scope
The Mo–N/C catalyst was tested also for the oxidation of both primary and secondary alcohols other than BnOH. To the scope, furfuryl alcohol (2), vanillyl alcohol (3), 1,5-bis(hydroxymethyl)furan (4), 1-phenylethanol (5), cyclopentanol (6), 2-pentanol (7), 1-hexanol (8), 2-amino-benzyl alcohol (9), 4-amino-benzyl alcohol (10) and 4-bromo-benzyl alcohol (11) were used. A mixture of the chosen alcohol (1 mmol) and Mo–N/C (50 mg) was set to react under the optimized conditions of Fig. 8, i.e. at 130 °C under 20 bar of air for 24 h. The results are summarized in Table 3, while GC-MS and 1H-13C NMR spectra are reported in the ESI† section (Fig. S3–S20). Conversion and selectivity were determined by GC (Table 3).|
|
|||
|---|---|---|---|
| Substrate | Conversion (%) | Main product (selectivity, %) | |
| Reaction conditions: alcohol (1 mmol), Mo–N/C (50 mg), ACN (1 mL), 130 °C, 20 bar air, 24 h. Conversion and selectivity were determined by GC. | |||
| 2 |
|
>99 |
 2a (>99)
2a (>99) |
| 3 |

|
>99 |
 3a (98)
3a (98) |
| 4 |

|
>99 |
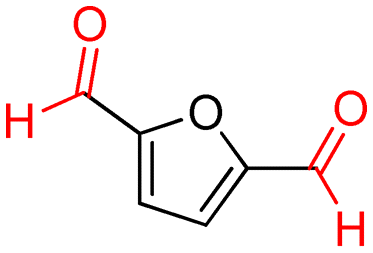 4a (98)
4a (98) |
| 5 |

|
>99 |
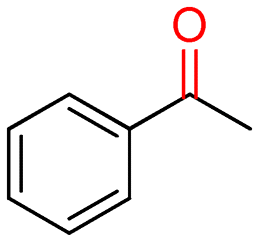 5a (30)
5a (30) |
| 6 |

|
7 |
 6a (>99)
6a (>99) |
| 7 |

|
2 |
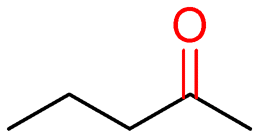 7a (>99)
7a (>99) |
| 8 |

|
3 |
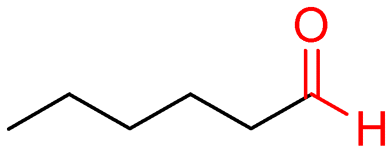 8a (>99)
8a (>99) |
| 9 |
|
>99 |
 9a (95)
9a (95) |
| 10 |
|
>99 |
 10a (>99)
10a (>99) |
| 11 |
|
79 |
 11a (>99)
11a (>99) |
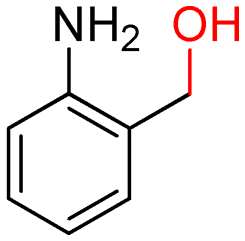
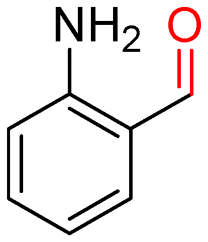
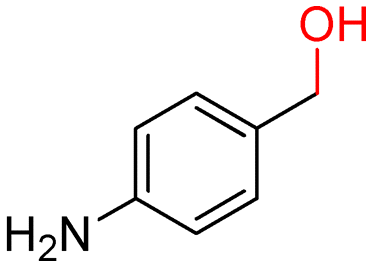
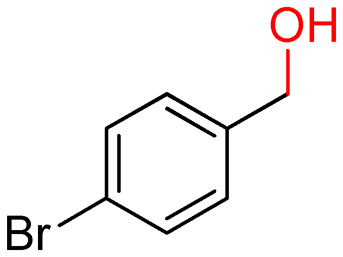
The batch oxidation protocol implemented for benzyl alcohol proved its general applicability for primary alcohols (2–3), and the primary diol 4. In these cases, the corresponding aldehyde products (2a–4a) from the partial hydroxyl oxidation were obtained with quantitative conversions and excellent selectivity (>98%). On the other hand, a remarkable different outcome was observed for secondary alcohols. 1-Phenylethanol (5) provided the desired product 5a with only 30% selectivity even if the conversion was quantitative [1-phenylethyl ether (40%) was found as the main side-product], while the oxidation of alcohols 6–7 proceeded with almost negligible conversion (2–7%, respectively). Overall, primary benzyl-/furyl-type alcohols were well oxidized using the Mo–N/C system, and even the hindered secondary benzyl alcohol 5 was converted to some extent to its ketone derivative 5a. However, the oxidation system was not successful for aliphatic secondary alcohols 6 and 7, neither for aliphatic primary ones such as 1-hexanol 8. Moreover, looking forward to investigating the catalytic activity of the designed material on different substituted benzyl alcohols, substrates 9, 10 and 11 were tested. In the case of substrates 9 and 10 the reaction proceeds with full conversion and excellent selectivity towards the desired aldehyde, being 95% and higher than 99% for products 9a and 10a, respectively. On the other hand, a 79% conversion of substrate 11 was reached with full selectivity towards the aldehyde. The lower conversion observed for 11 could be attributed to several factors, including the possible interactions of bromide with the metal nanoparticles on the N-doped carbonaceous surface, preventing the catalytic effect of the active centres for the desired reaction, besides the electron-withdrawing properties of the bromide group on the phenyl ring and its respective position to the CH2OH-group.54,55 A free-energy relationship is proposed, considering the substituted benzyl moieties already studied in this contribution. A non-linear relationship with a negative pseudo-slope was obtained (Fig. S23†). This indicates that 1) the substituent effect in the benzyl alcohol oxidation reaction, employing the Mo–N/C catalyst, is opposite to that of the Hammett standard reaction (ionization of substituted benzoic acids in water at 25 °C) and that 2) the non-linear relationship could be most likely attributed to the concomitant influence of two effects, namely the electron-withdrawing properties of Br together with the previously mentioned interactions of bromide with the metal nanoparticles.56–59
Beyond the scant reactivity expected for secondary versus primary hydroxyl groups, these results led us to hypothesize that stabilized aromatic species (e.g. radicals) of benzyl-/furyl-type alcohols adsorbed on the catalyst surface were crucial for the oxidation step. Even if the study of the reaction mechanism was beyond the scope of the paper, a further test was conducted by performing the oxidation of benzyl alcohol under optimized conditions employing the Mo–N/C system as the catalyst and additionally employing ascorbic acid as a radical scavenger. Interestingly, only a low conversion of ca. 20% was reached, therefore suggesting that most likely benzyl radicals were involved in the reaction pathway. Moreover, other radical scavengers, besides ascorbic acid, namely 1) phenol and 2) methanol were employed, performing the reaction under identical optimized conditions (130 °C, 20 bar of air), confirming the proposed reaction pathway. A quenching effect was as well observed by employing the aforementioned radical-scavengers. In particular, a 58% and 64% remaining conversion was observed by adding phenol and methanol, respectively, in comparison with 95% without the quenching effect. From such results it could be speculated that over ca. 40% of the total benzyl alcohol oxidation, employing Mo–N/C, could be most likely attributed to a radical-based pathway, in accordance with the results obtained by employing ascorbic acid as a radical scavenger and implying that benzyl radical species are probably involved in the reaction mechanism.
Catalyst reusability
The cost of the catalyst in a liquid-phase reaction may represent up to a third of the total cost of the process, implying that its loss by leaching or other reasons is critical, and its recovery and reuse are crucial.60,61 The stability and reusability of the Mo–N/C catalyst were therefore investigated by designing recycling experiments under the conditions shown in Fig. 8b [benzyl alcohol (1 mmol), Mo–N/C (50 mg), acetonitrile (1 mL), 130 °C, 20 bar, 24 h]. Once a first reaction was complete, the catalyst was filtered off, washed with methanol (30 mL) and dried overnight. The recovered catalyst was added with a fresh solution of benzyl alcohol (1 mmol) and acetonitrile (1 mL) and a new reaction was started. The recycling procedure was repeated six times, and the whole set of reactions was run twice to ensure reproducibility. The results are illustrated in Fig. 9. Both the conversion and the 1a selectivity were steady at 91–93% and 93–95%, respectively, during the six runs, thereby demonstrating that the overall performance of Mo–N/C was not altered over time by the reaction environment during the recycling tests and by the washing/restoring procedures. Furthermore, ICP-MS analysis of the reaction samples was carried out revealing certain leaching of 20 μg L−1, associated with Mo weakly linked to the N-doped carbonaceous support. In any case, it is worth mentioning that such leaching represents ca. 1% of the initial concentration of molybdenum in the sample. Notwithstanding the excellent outcome of this study, the catalyst recovered after its reuse was analyzed to investigate whether structural and chemical changes took place. This investigation is described in the next section. | ||
| Fig. 9 Catalyst reusability study. Reaction conditions for each run: benzyl alcohol (1 mmol, 108 mg), Mo–N/C (50 mg), acetonitrile (1 mL), at 130 °C under 20 bar of air for 24 hours. Conversion and selectivity were determined by GC. | ||
Post-recycling characterization
A post-recycling characterization of Mo–N/C was accomplished to obtain insights into the chemical, structural and morphological features of the sample after the catalytic reaction and its reuse, under the investigated conditions. N2 adsorption–desorption isotherms, displayed in Fig. 10A, showed that the material preserved a mesoporous network, as suggested from the type IV isotherm with type II adsorption hysteresis. However, a drastic change was observed in the adsorbed volume, with a surface area of 50 m2 g−1 only corresponding to a decrease of ca. 82% compared to the fresh sample. Also, the pore volume of 0.07 cm3 g−1 was almost 3 times lower than that of the fresh catalyst, while the mean pore diameter up to 5.6 nm was nearly doubled. Such results were consistent with the adsorption of organic moieties, which could lead to occlusion of smaller pores. | ||
| Fig. 10 Comparative post-characterization analyses of the Mo–N/C material. A) N2-physisorption isotherm of the sample before and after the catalytic tests. B) XRD patterns of the material before and after the catalytic tests. C) HRTEM-EDX images of the Mo–N/C_used sample. | ||
More pronounced differences were found in the XRD pattern of the reused material (Fig. 10B), which exhibited a degree of amorphization, together with the concomitant appearance of new signals. A marked signal around 13° was noticed with the decay of the signal around 51° that was characteristic of the fresh material. This evidence indicated the nearly complete oxidation of molybdenum species, shifting from a MoO2 and MoO3 mixture to a prevalent MoO3 phase.62
HRTEM-EDX results (Fig. 10C) and HRTEM micrographs (Fig. 11) of the reused Mo–N/C material did not exhibit significant modifications. The carbonaceous support retained a laminar structure, with the presence of supported nanoparticles with a mean diameter around (30 ± 2) nm. HRTEM-EDX analysis also showed a highly homogeneous distribution of all elements, and in particular, the active molybdenum sites maintained an excellent dispersion without agglomerated regions, indicating that no sintering occurred during the reaction.
 | ||
| Fig. 11 HRTEM micrographs of the Mo–N/C_used sample. | ||
The results of XPS analysis of the Mo–N/C_used material are reported in Fig. 12. Compared to the fresh sample, a drastic change was observed in the Mo 3d core level spectrum (Fig. 12A). The signals located at 232.7 and 235.9 eV were attributed to Mo(VI) species, thereby indicating that MoO3 was the predominant phase. In agreement with the XRD data, the active metal was completely oxidized during the investigated reaction. The C 1s, N 1s and O 1s spectra (Fig. 12B–D) did not display significant differences with respect to the same contributions in the XPS of the fresh material.
 | ||
| Fig. 12 A: Mo 3d, B: C 1s, C: N 1s and D: O 1s core level spectra of the Mo–N/C_used sample. | ||
This study highlighted that during its use, Mo–N/C underwent textural, morphological, and chemical modifications, but none of these changes apparently impacted on its catalytic performance. An unaltered property, however, was the size of the metal nanoparticles which was probably critical to preserve the efficiency of the system over time.
Conclusions
This work reports on a new family of supported non-noble metal nanoparticles that was prepared and tested for the catalytic oxidation of alcohols to aldehydes in the presence of air as an oxidant. Chitin was used as a renewable carbon source to provide a support for the catalyst, which was intrinsically doped with a high N content.A parametric analysis of the reaction using benzyl alcohol as a model substrate proved that an outstanding selectivity could be reached towards the product of the partial oxidation of the alcohol: in particular, using the molybdenum-based system (Mo–N/C), the best performing catalyst among those investigated, benzaldehyde was achieved with 91% selectivity at an almost quantitative conversion (95%). This result was comparable to some of the best achievements in the recent literature for the same reaction. Moreover, Mo–N/C was successfully recovered and recycled for up to 6 subsequent runs without any loss of activity. Different benzyl- and furyl-type primary alcohols provided conversion and aldehyde selectivity similar – if not better – than those obtained for benzyl alcohol, though the tested conditions were ineffective for secondary substrates and for aliphatic alcohols. The reasons for such a high activity/selectivity were investigated by characterizing the textural, morphological, and chemical properties of both a fresh and a used sample of Mo–N/C. This analysis was consistent with a significant alteration of the catalyst during its use: the surface area of the sample decreased by ca. 82%, and its composition changed from a MoO2 and MoO3 mixture (fresh) to a prevalent MoO3 phase (used), but the size of the metal nanoparticles [(30 ± 2) nm] did not vary. The latter (size) was probably responsible for the preservation of the efficiency of the system over time. XPS results also suggested that the presence of N as a dopant of the carbon-based support of the catalyst induced the formation of metal–nitrogen active sites which affected the reaction selectivity.
On a final note, multiple aspects contributed to the characteristics of sustainability of the protocol, among others the simple/economic synthesis of catalytic systems based on non-noble and not endangered metals, the use of a bio-based carbon support especially in view of using chitin from largely available bio-waste, the use of air as a safe oxidant, and the extension of the oxidation procedure for the upgrading of bio-based (benzyl-/furyl-) alcohols.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. P. and D. R. P. gratefully acknowledge Mr. Pippo Rizzo for his valuable support during the development of this work. D. R. P. acknowledges the project H2020-MSCA-COFUND-2019 “Global at Venice- Research and Training for Global Challenges”. D. B. P. and E. R. C. acknowledge the projects PID2021-126235OB-C32 of Ministerio de Ciencia e Innovación and UMA18-FEDERJA-126 of Junta de Andalucía and FEDER funds.References
- D. Rodríguez-Padrón, A. R. Puente-Santiago, A. M. Balu, M. J. Muñoz-Batista and R. Luque, ChemCatChem, 2019, 11, 18–38 CrossRef.
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- M. Elayaperumal, M. Duraisamy and H. Ganesh, Sci. Adv. Mater., 2018, 7, 183–206 Search PubMed.
- M. Xiong, Z. Gao and Y. Qin, ACS Catal., 2021, 11, 3159–3172 CrossRef CAS.
- D. Polidoro, A. Perosa, E. Rodríguez-Castellón, P. Canton, L. Castoldi, D. Rodríguez-Padrón and M. Selva, ACS Sustainable Chem. Eng., 2022, 10, 13835–13848 CrossRef CAS PubMed.
- D. Rodríguez-Padrón, A. M. Balu, A. A. Romero and R. Luque, Beilstein J. Org. Chem., 2017, 13, 1982–1993 CrossRef PubMed.
- S. Ostovar, H. Saravani and D. Rodríguez-Padrón, Renewable Energy, 2021, 178, 1070–1083 CrossRef.
- S. Ostovar, D. Rodríguez-Padrón, F. Saberi, A. M. Balu and R. Luque, Catalysts, 2019, 9, 348–360 CrossRef.
- N. Gunasekaran, Adv. Synth. Catal., 2015, 357, 1990–2010 CrossRef CAS.
- M. Nasrollahzadeh, M. Sajjadi, M. Shokouhimehr and R. S. Varma, Coord. Chem. Rev., 2019, 397, 54–75 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- P. Verma, M. E. Potter, A. E. Oakley, P. M. Mhembere and R. Raja, Nanomaterials, 2021, 11, 1–13 CrossRef PubMed.
- P. Wu, Z. He, Y. Liu, L. Song, C. Wang, E. Muhumuza, P. Bai, L. Zhao, S. Mintova and Z. Yan, ACS Appl. Mater. Interfaces, 2021, 13, 49780–49792 CrossRef CAS PubMed.
- J. Liu, S. Wu and Z. Li, Curr. Opin. Chem. Biol., 2018, 43, 77–86 CrossRef CAS PubMed.
- J. E. Nutting, K. Mao and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 10565–10570 CrossRef CAS PubMed.
- H. Kargar, M. Fallah-Mehrjardi, R. Behjatmanesh-Ardakani, K. S. Munawar, M. Ashfaq and M. N. Tahir, Transition Met. Chem., 2021, 46, 437–455 CrossRef CAS.
- Y. Chen, X. Zhang, X. Wang, R. J. Drout, M. R. Mian, R. Cao, K. Ma, Q. Xia, Z. Li and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 4302–4310 CrossRef CAS PubMed.
- X. Wang, H. Xie, J. G. Knapp, M. C. Wasson, Y. Wu, K. Ma, A. E. B. S. Stone, M. D. Krzyaniak, Y. Chen, X. Zhang, J. M. Notestein, M. R. Wasielewski and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 12092–12101 CrossRef CAS PubMed.
- W. Wang, R. Wang, X. Jiang, Z. H. He, K. Wang, Y. Yang and Z. T. Liu, Appl. Catal., A, 2022, 634, 118537 CrossRef CAS.
- H. M. Alshammari, Processes, 2021, 9, 1590–1601 CrossRef CAS.
- C. Jacquot, V. Middelkoop, A. Köckritz, A. Pohar, R. Bienert, S. Kellici, I. A. Bărăgău, B. Venezia, A. Gavriilidis, B. Likozar and A. M. Beale, Sustainable Mater. Technol., 2021, 30, e00329 CrossRef CAS.
- D. Rodriguez-Padron, A. Ahmad, P. Romero-Carrillo, R. Luque and R. Esposito, Trends Chem., 2022, 4, 739–753 CrossRef CAS.
- J. Li, M. Li, H. Sun, Z. Ao, S. Wang and S. Liu, ACS Catal., 2020, 10, 3516–3525 CrossRef CAS.
- A. Samikannu, L. J. Konwar, P. Mäki-Arvela and J. P. Mikkola, Appl. Catal., B, 2019, 241, 41–51 CrossRef CAS.
- H. Xu, S. Zhang, J. Geng, G. Wang and H. Zhang, Inorg. Chem. Front., 2021, 8, 2829–2834 RSC.
- J. L. Shamshina, Green Chem., 2019, 21, 3974–3993 RSC.
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Hävecker, A. Knop-Gericke and R. Schlögl, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS.
- K. Shi, M. Ren and I. Zhitomirsky, ACS Sustainable Chem. Eng., 2014, 2, 1289–1298 CrossRef CAS.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- B. Mi, X. Chen, C. Jiang, J. Wang, X. Chen, B. Zhang, X. Liu, Z. Liu and B. Fei, Catalysts, 2018, 8, 232–242 CrossRef.
- H. Ma, G. Ma, Y. Qi, Y. Wang, Q. Chen, K. R. Rout, T. Fuglerud and D. Chen, Angew. Chem., Int. Ed., 2020, 59, 22080–22085 CrossRef CAS PubMed.
- Y. Huang, H. Yan, C. Zhang, Y. Wang, Q. Wei and R. Zhang, Nanomaterials, 2021, 11, 2776–2790 CrossRef CAS PubMed.
- J. Li, P. Li, J. Li, Z. Tian and F. Yu, Catalysts, 2019, 9, 506–518 CrossRef CAS.
- R. K. Singhal, B. Gangadhar, H. Basu, V. Manisha, G. R. K. Naidu and A. V. R. Reddy, Am. J. Anal. Chem., 2012, 3, 76–82 CrossRef CAS.
- J. S. Kang, J. Kim, M. J. Lee, Y. J. Son, D. Y. Chung, S. Park, J. Jeong, J. M. Yoo, H. Shin, H. Choe, H. S. Park and Y. E. Sung, Adv. Sci., 2018, 5, 1–10 Search PubMed.
- E. C. S. Transactions and T. E. Society, ECS Trans., 2014, 58, 67–75 Search PubMed.
- D. Rodríguez-Padrón, A. D. Jodlowski, G. De Miguel, A. R. Puente-Santiago, A. M. Balu and R. Luque, Green Chem., 2018, 20, 225–229 RSC.
- D. Rodríguez-Padrón, A. R. Puente-Santiago, A. Caballero, A. Benítez, A. M. Balu, A. A. Romero and R. Luque, J. Mater. Chem. A, 2017, 5, 16404–16411 RSC.
- D. Rodriguez-Padrón, A. R. Puente-Santiago, A. M. Balu, A. A. Romero and R. Luque, Chem. Commun., 2017, 53, 7635–7637 RSC.
- D. Rodríguez-Padrón, A. R. Puente-Santiago, A. Caballero, A. M. Balu, A. A. Romero and R. Luque, Nanoscale, 2018, 10, 3961–3968 RSC.
- D. Rodríguez-Padrón, M. J. Muñoz-Batista, H. Li, K. Shih, A. M. Balu, A. Pineda and R. Luque, ACS Sustainable Chem. Eng., 2019, 7, 17030–17038 CrossRef.
- A. Akbari, M. Amini, A. Tarassoli, B. Eftekhari-Sis, N. Ghasemian and E. Jabbari, Nano-Struct. Nano-Objects, 2018, 14, 19–48 CrossRef CAS.
- D. Polidoro, C. Espro, N. Lazaro, O. Trentin, A. Perosa, S. M. Osman, D. Rodríguez-Padrón, R. Luque and M. Selva, Catal. Today, 2022 DOI:10.1016/j.cattod.2022.08.032.
- S. Mitchell, R. Qin, N. Zheng and J. Pérez-Ramírez, Nat. Nanotechnol., 2021, 16, 129–139 CrossRef CAS PubMed.
- I. Ro, J. Resasco and P. Christopher, ACS Catal., 2018, 8, 7368–7387 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, B. Hvolbæk, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC.
- S. Horstmann, A. Grybat and R. Kato, J. Chem. Thermodyn., 2004, 36, 1015–1018 CrossRef CAS.
- J. Chen, W. Fang, Q. Zhang, W. Deng and Y. Wang, Chem. – Asian J., 2014, 9, 2187–2196 CrossRef CAS PubMed.
- J. Tian, X. Hu, N. Wei, Y. Zhou, X. Xu, H. Cui and H. Liu, Sol. Energy Mater. Sol. Cells, 2016, 151, 7–13 CrossRef CAS.
- P. Cruz, Y. Pérez, I. Del Hierro and M. Fajardo, Microporous Mesoporous Mater., 2016, 220, 136–147 CrossRef CAS.
- F. Shi, M. K. Tse, M.-M. Pohl, A. Brückner, S. Zhang and M. Beller, Angew. Chem., 2007, 119, 9022–9024 CrossRef.
- Z. Hu, Y. Zhao, J. Liu, J. Wang, B. Zhang and X. Xiang, J. Colloid Interface Sci., 2016, 483, 26–33 CrossRef CAS PubMed.
- W. Wu, Q. Zhang, X. Wang, C. Han, X. Shao, Y. Wang, J. Liu, Z. Li, X. Lu and M. Wu, ACS Catal., 2017, 7, 7267–7273 CrossRef CAS.
- I. Krivtsov, M. Ilkaeva, E. I. García-López, G. Marcì, L. Palmisano, E. Bartashevich, E. Grigoreva, K. Matveeva, E. Díaz and S. Ordóñez, ChemCatChem, 2019, 11, 2713–2724 CrossRef CAS.
- I. Krivtsov, M. Ilkaeva, E. I. García-López, G. Marcì, L. Palmisano, E. Bartashevich, E. Grigoreva, K. Matveeva, E. Díaz and S. Ordóñez, ChemCatChem, 2019, 11, 2713–2724 CrossRef CAS.
- L. Calvanese, M. E. Cucciolito, A. D'Amora, G. D'Auria, A. Esposito, R. Esposito, L. Falcigno and F. Ruffo, Eur. J. Inorg. Chem., 2015, 24, 4068–4075 CrossRef.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- A. Chellamani and S. Harikengaram, J. Phys. Org. Chem., 2003, 16, 589–597 CrossRef CAS.
- L. Vugrin, M. Carta, F. Delogu and I. Halasz, Chem. Commun., 2023, 59, 1629–1632 RSC.
- I. Sádaba, M. L. Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- D. Polidoro, A. Perosa and M. Selva, ChemSusChem, 2022, 15, e202201059 CAS.
- L. O. Alemán-Vázquez, E. Torres-García, J. R. Villagómez-Ibarra and J. L. Cano-Domínguez, Catal. Lett., 2005, 100, 219–226 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00082f |
| This journal is © The Royal Society of Chemistry 2023 |