
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutron scattering studies of the methanol-to-hydrocarbons reaction
Andrea
Zachariou
 a,
Alexander P.
Hawkins
b,
Paul
Collier
c,
Russell F.
Howe
d,
Stewart F.
Parker
ef and
David
Lennon
*f
a,
Alexander P.
Hawkins
b,
Paul
Collier
c,
Russell F.
Howe
d,
Stewart F.
Parker
ef and
David
Lennon
*f
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
bCentral Laser Facility, Science and Technology Facilities Council, Research Complex at Harwell, Rutherford Appleton Laboratory, Harwell, OX11 0QX, UK
cJohnson Matthey Technology Centre, Blounts Court, Sonning Common, Reading, RG4 9NH, UK
dDepartment of Chemistry, University of Aberdeen, Aberdeen, AB24 3UE, UK
eISIS Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Chilton, Oxon OX11 0QX, UK
fSchool of Chemistry, University of Glasgow, Joseph Black Building, Glasgow, G12 8QQ, UK. E-mail: David.Lennon@glasgow.ac.uk
First published on 7th March 2023
Abstract
The methanol-to-hydrocarbons reaction uses a zeolite catalyst (typically H-ZSM-5 or SAPO-34) to produce light olefins (mainly C2–C4) for chemical feedstocks and methylated benzenes for use as fuels. The reaction is a major industrial process world-wide, and consequently it has been extensively studied by a wide range of physical methods (e.g. reaction testing) and spectroscopic methods. One approach that has been used relatively little to-date is neutron scattering. Neutron methods can provide information about structure, diffusion at the microscopic scale and vibrational spectra that are not limited by selection rules, fluorescence or absorption. In this mini-review we give a short introduction to neutron scattering, then describe how neutron scattering provides insight into the Brønsted acid sites of the catalyst, then consider each of the three stages of the process; induction, steady-state and deactivation. We conclude with a perspective on how neutron scattering can further support studies of MTH chemistry.
Introduction
The MFI-type zeolite ZSM-5 was invented by the Mobil Oil Company in 19721,2 and has since been widely used in industry for reactions ranging from hydrocracking3 to toluene disproportionation4 (to generate benzene and xylenes) to methanol-to-hydrocarbons5 (MTH), (which includes methanol-to-olefins6 (MTO) and methanol-to-gasoline7 (MTG) chemistries).The MTG process was first commercialised by Mobil in New Zealand in 1986 using an HZSM-5 catalyst. Lurgi's methanol to olefins (MTO) process, also using HZSM-5, UOP-Statoil's and Dalian Institute of Chemical Physics (DICP) MTO process, both using a SAPO-34 catalyst8 and Topsoe's improved gasoline synthesis9 (TIGAS) using a proprietary zeolite catalyst followed. The availability of cheap methanol derived from natural gas was the initial driver for these technologies. More recently, methanol derived from coal has become the source of transport fuels or olefin feedstocks via MTG or MTO type processes.10
As a consequence of MTH's scientific novelty and commercial value, the process has been extensively studied and reviewed, for example.7,11–15 MTH typically exhibits three phases that merge into one another: an induction period, steady state operation and deactivation. For MTH using HZSM-5 at steady state operation, the process is generally accepted to occur by the dual cycle hydrocarbon pool mechanism,16,17 shown in Fig. 1. The induction period is the establishment of the hydrocarbon pool and deactivation is usually by coke formation.
 | ||
| Fig. 1 The dual cycle hydrocarbon pool mechanism operative for MTH using HZSM-5. The figure has been reproduced from ref. 122 with permission from Elsevier, copyright 2013. | ||
The MTH reaction has been investigated with a wide variety of methods, ranging from microreactor measurements to spectroscopic techniques, including, UV-vis,18–20 infrared,20–22 ESR23,24 and solid state (ss) NMR spectroscopies.14 All of the spectroscopic methods have strengths and limitations: assignment of spectra is difficult with UV-vis because the bands are broad and overlapping, infrared spectroscopy is largely restricted to the region >2000 cm−1 (because of absorption by the zeolite lattice), ESR requires paramagnetic species and product identification is difficult with ss-NMR. However, all of the methods either have been, or could be, used operando or in situ, which is a significant advantage.
Neutron scattering is very sensitive to hydrogen and has been used to investigate catalysts for many different processes that span commodity chemicals: methane reforming,25 Fischer–Tropsch synthesis,26 ammonia production,27,28 water gas shift,29 to catalysts for fine chemical synthesis30 to ethanol reforming.31 These systems, and several others, have been reviewed previously.32 All of the key species in MTH chemistry are hydrogenous: the Brønsted acid sites, methanol and the hydrocarbon pool contents, so our expectation was that neutron scattering methods would provide new insight into the MTH process.33
In the following sections we give a short introduction to neutron scattering, then describe how neutron scattering provides insight into the Brønsted acid sites of the catalyst, then consider each of the three stages of the process; induction, steady state and deactivation. We recognise that these are not discrete processes, there is considerable overlap between them. We will conclude with a perspective on how neutron scattering can further support studies of MTH chemistry. The particular advantages of neutron scattering for MTH chemistry will be highlighted throughout.
Neutron scattering
The theory34,35 and practice36–39 of neutron scattering have been comprehensively documented, so only a brief description will be given here. The utility of neutron scattering derives from the properties of the neutron, which is an uncharged, spin ½ particle with a mass of 1.009 amu, slightly greater than that of a proton (1.007 amu). Neutrons are scattered by atomic nuclei and, via the nuclear spin, by systems with unpaired electrons. The latter gives rise to the extensive applications of neutron scattering in magnetism.40 For catalysis, it is the atomic scattering that is crucial. The amount of scattering is determined by the cross section, in essence, the size of the target that the neutron “sees”. The cross section can be either coherent or incoherent. Coherent scattering describes the interference between the scattered waves caused by the scattering of a single neutron from all nuclei and provides information on long range order in the system: both structural (e.g. diffraction) and spectroscopic (e.g. phonon dispersion). Incoherent scattering measures the correlation between the positions of an atom at time zero and a later time. Each atom contributes independently to the total scattering, thus it acts as a local probe.Neutrons may be scattered elastically or inelastically, for the former there is no energy exchange between the neutron and the scattering nucleus, in the latter the energy exchange is non-zero (it may be positive – neutron energy gain, or negative – neutron energy loss). Elastic scattering gives information about location and inelastic about dynamics: neutrons tell you where atoms are and how they are moving. Note that for both elastic and inelastic scattering, the neutron changes direction on scattering, so in any scattering event there is neutron momentum transfer (Q, Å−1).
The cross section is both element and isotope dependent and this offers opportunities to manipulate the contrast in a system. However, the most significant effect is that the incoherent cross section for 1H is very large (80.26 barn, 1 barn = 10−28 m2) while the total cross section for most other elements (and isotopes, including deuterium) is ∼5 barn. The result is that for elastic scattering from hydrogenous materials it is generally advantageous to deuterate the sample, as the incoherent scattering results in a large background. For inelastic scattering, the large incoherent cross section of 1H means that the scattering is dominated by scattering from hydrogen.
As with any technique, there are advantages and disadvantages to neutron scattering. The advantages arise from the simplicity of the scattering process. This makes calculation of experimental observables tractable and there is a very strong synergy between neutron scattering and computational methods. As neutrons are scattered by nuclei, to a neutron, much of matter is empty space, consequently neutrons are highly penetrating (1 cm of steel is essentially transparent) which simplifies the design of experimental equipment: special materials for windows are not necessary. The major disadvantage of neutron scattering is that to produce sufficient neutrons to do useful science a central facility41 is needed and these are relatively scarce. Neutrons can be produced by nuclear fission of 235U in a reactor or by spallation, where a high power proton beam from an accelerator impacts on a heavy metal target. The leading research reactor is the Institute Laue Langevin42 (ILL, Grenoble, France). Spallation sources are fewer but are the next generation in neutron scattering. The major ones are: the ISIS Neutron and Muon Facility43 (ISIS, Didcot, UK), the Spallation Neutron Source44 (SNS, Oak Ridge, USA) and the Japan Proton Accelerator Research Complex45 (J-PARC, Tokai, Japan).
The sample size required for neutron scattering measurements of zeolites is significantly larger than for most laboratory methods. This arises from the relatively insensitive nature of the technique (to partly compensate for this, neutron beams are usually around 10–20 cm2 in area). Typically, for elastic scattering a few grams of sample are needed, for inelastic scattering, 5–10 g is typical. For the latter, the larger sample size arises from the need to have 0.25–0.5 g of the (hydrogenous) material of interest in the beam, whether this is a probe molecule (e.g. pyridine), a reference sample (e.g. a pure compound adsorbed in the zeolite) or the hydrocarbon pool itself. In order, to generate the large samples needed, dedicated preparation rigs have been developed.46,47
A current trend in catalysis is that operando measurements (simultaneous measurement of a sample property by spectroscopy and product composition by on-line mass spectroscopy and/or chromatography) are increasingly seen as the optimal approach. There are a few examples of operando neutron powder diffraction of catalysts32 (none of MTH), but, in general, neutron scattering methods do not lend themselves to this approach as the time resolution is relatively poor – usually minutes to hours, (a consequence of the low brightness of neutron sources) or measurements have to be made at low temperature (∼20 K).
In most cases a react-and-quench method is used. To validate that the samples prepared are relevant, laboratory microreactor studies are essential. These have two purposes: firstly to establish that the chemistry is representative of what is generally seen in the literature. Secondly, to provide a baseline against which the much larger samples prepared for neutron studies using the specialist apparatus46,47 can be compared.
There are a wide variety of neutron techniques that could be applied to study the MTH reaction. These are listed in Table 1, together with their strengths and limitations and whether they have been used to study MTH chemistry.
| Method | Strengths | Limitations | Used for MTH? |
|---|---|---|---|
| a NPD = neutron powder diffraction. b QENS = quasielastic neutron scattering. c INS = inelastic neutron scattering (neutron vibrational spectroscopy). d DINS = deep inelastic neutron scattering. | |||
| Neutron scattering (generally) | Sensitive to light atoms (H,C,O) | Limited availability – requires use of a central facility | Yes |
| Highly penetrating (simplifies cell design) | Overall sensitivity is low, large (2–20 g) samples are needed | ||
| Observables are readily calculated | Relatively slow; measurements generally require minutes to hours | ||
| No radiation damage | |||
| Elastic scattering | |||
| NPDa | Able to detect light atoms (especially H or D) in the presence of heavy atoms | Yes | |
| Neighbouring elements in the periodic table may have different scattering strengths | |||
| Can be used operando | |||
| No form factor (so wide Q range) | |||
| Total scattering | Can simultaneously provide information on microscopic, mesoscopic and macroscopic length scales | Complex analysis – modelling essential | No |
| Can study disordered/amorphous materials | |||
| H/D isotopic substitution is very informative | |||
| Sensitive to the local structure | |||
| Operando conceivable, but not demonstrated | |||
| Imaging | Operando possible | Time and spatial resolution inversely correlated | No |
| Inelastic scattering | |||
| QENSb | Provides direct measurement of diffusion (rotational and/or translational) on the ps – ∼1 μs timescale | Different instruments needed to cover the complete time range | Yes |
| Timescales very well-matched to that of molecular dynamics calculations | Restricted to a single mobile species (very difficult/impossible to distinguish multiple species) | ||
| Operando under development | |||
| INSc | Vibrational spectrum | Most measurements have to be made at 20 K or less | Yes |
| Entire 0–4000 cm−1 range accessible | Hydrogenous adsorbates only | ||
| No selection rules | Operando only rarely possible | ||
| Intensities straightforward to calculate – strong synergy with ab initio methods | |||
| DINSd | Quantitative elemental analysis | Analysis complex | No |
| In situ temperature determination possible | Operando possible, but the time resolution is very poor – generally requires hours per measurement | ||
| Can be carried out at temperatures relevant for catalysis | |||
Catalyst characterisation
Key to understanding the properties of a zeolite is a knowledge of the number and location of the Brønsted acid sites. The number of acid sites depends on the Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al ratio, as each site consists of an Si–O(H)–Al bridge. There are a number of techniques that can be used to determine the number of acid sites, but 29Si ss-NMR and ammonia temperature programmed desorption (NH3-TPD) are the usual methods. Infrared spectroscopy can also be used, but it is complicated by uncertain knowledge of the extinction coefficients of the various species present.
:Al ratio, as each site consists of an Si–O(H)–Al bridge. There are a number of techniques that can be used to determine the number of acid sites, but 29Si ss-NMR and ammonia temperature programmed desorption (NH3-TPD) are the usual methods. Infrared spectroscopy can also be used, but it is complicated by uncertain knowledge of the extinction coefficients of the various species present.
Fig. 2 shows the infrared (DRIFTS) and inelastic neutron scattering (INS) spectra of calcined and dried HZSM-5.48 The diffuse reflectance infrared spectrum, Fig. 2a, below 2000 cm−1 is dominated by the Si–O and Al–O framework modes and their overtones and are distorted by dispersion effects from specular reflectance. These are not associated directly with the active sites and the spectrum provides little useful information in this region (we note that transmission infrared spectroscopy through pressed disks of zeolite is not subject to dispersion effects and gives useful spectra down to ∼1300 cm−1). In contrast, the O–H stretch modes located above 3500 cm−1 are clearly seen. The strongest of these is at 3592 cm−1 and is assigned to Brønsted acid groups within the zeolite. The weaker, separate peak at 3735 cm−1 results from the silanol Si–O–H groups that terminate the zeolite framework at its exterior surfaces and at internal defects. The peak between these two modes at 3648 cm−1 is associated with the presence of extra-framework aluminium species (EFAl).
 | ||
| Fig. 2 Infrared at 423 K (a) and INS at ∼20 K (b) and (c) spectra of calcined, dried HZSM-5. (b) Recorded with MAPS using incident energies of 2017 cm−1 (left) and 5244 cm−1 (right). Insets show ×10 expansions of selected regions to make the O–H related modes more apparent. (c) Recorded with TOSCA. The inset shows ×4 ordinate expansion of the internal mode region. (d) The O–H stretch region as recorded by MAPS, after baseline correction and curve resolution into the Brønsted acid sites (orange), EFAl (olive) and silanols (red). The data shown in (d) are from a different measurement than that shown in the right of (b). The MAPS spectra in (b) and (d) are integrated over the momentum transfer range 0 ≤ Q ≤ 10 Å−1. Reproduced from ref. 48 under a Creative Commons Attribution 4.0 Unported Licence (CC-BY). | ||
The corresponding INS spectra are shown in Fig. 2b and c for data recorded on two different spectrometers, MAPS and TOSCA respectively. The two instruments are complementary, for reasons explained elsewhere,49 MAPS enables the C–H and O–H stretch region above 2500 cm−1 to be observed, while TOSCA is optimal for the region below 2000 cm−1. The low intensity of the zeolite spectra owing to the small scattering cross sections of the framework atoms are immediately apparent and enables the entire 0–4000 cm−1 range to be accessed. This is one of the major advantages of INS for catalyst studies.
While the scattering cross sections of Al, Si and O are only ∼10% that of 1H (∼80 barn), there are more than ten times as many of them as there are hydrogen atoms. Comparison with the spectra of a variety of silicas,50,51 shows that the weak modes at 445, 535, 800, 1115 and 1205 cm−1 are Si–O modes. The O–H stretching modes are observed by MAPS.
The intensity of a mode in the INS depends on the momentum transfer (Q, Å−1) and the amplitude of vibration of the atoms in the mode and their scattering cross section.39 In the harmonic approximation, the amplitude of vibration depends only on the reduced mass of the atoms and the energy transfer. This means that the intensity per oscillator, is independent of the material, it only depends on the type of oscillator: sp3 C–H stretch, sp2 C–H stretch, O–H stretch. In particular, it does not depend on the electronic structure of the molecule, as is the case for infrared and Raman intensities. By calibration with a suitable reference material, it is possible to quantify the number of oscillators in the beam.52 In the present case brucite, Mg(OH)2, was used to create a calibration curve, which was linear as expected. The MAPS INS spectrum in the O–H stretch region was resolved into the three hydroxyl components identified in the infrared spectrum, as shown in Fig. 2d, and the amounts of each quantified48 (we note that the transition energies for the O–H stretch modes in Fig. 2d differ from those observed by infrared spectroscopy shown in Fig. 2a. The reasons for the difference are currently under investigation). Table 2 shows the results and equating the Brønsted acid site concentration to aluminium in the zeolite framework gives a framework Si:Al ratio of 28.
| OH site | OH concentration mg gZSM-5−1 | OH/unit cell |
|---|---|---|
| Brønsted | 9.77 | 3.26 |
| Extra framework Al | 3.32 | 1.11 |
| Silanol | 1.98 | 0.66 |
The Si:Al ratio was also determined by X-ray fluorescence (XRF) and gave a value of Si:Al ∼ 16. The difference between the XRF and INS values is attributed to the presence of EFAl and indicates that approximately half of the Al is present as EFAl. 27Al ss-NMR shows that 30% of the Al is present as EFAl,48 in approximate agreement with our estimate. We note that 27Al ss-NMR is not strictly quantitative, as quadrupolar broadening caused by the 5/2 spin can render the Al NMR invisible. The nature of the EFAl is still debated but several of the postulated EFAl species have one or more hydroxyl groups attached.53Table 2 shows that only ∼⅓ of the EFAl Al ions have hydroxyls attached.
INS spectroscopy has long been used to observe the hydroxyl bending modes in zeolites.54–57 In a recent development, INS spectroscopy was combined with DFT calculations to determine the location of the Brønsted acid sites in LTA zeolites.58,59 For a given zeolite structure there are a limited number of possible locations for the aluminium and hence the Brønsted acid sites. By calculating the INS spectra of all of the possible arrangements in LTA-5 (a specially synthesised high-silica form of LTA with Si:Al = 5, the usual form of LTA has Si:Al = 1) and comparing them to the experimental INS spectrum, Lemishko and co-workers58,59 showed that it was possible to deduce the most likely locations. Fig. 3 shows the comparison of observed and calculated spectra, structure “d5” is considered to be the best match based on a computational procedure to assess the similarity between two spectra. The structure is shown on the right of Fig. 3 and “shows Al atoms to be homogeneously distributed in the framework and protons distributed relatively far from each other”.59
 | ||
| Fig. 3 Left: Comparison between the experimental INS spectrum of LTA-5 (a specially synthesised high-silica form of LTA with Si:Al = 5) and the hydrogen partial density of states, gH(ω), of the fundamental vibrations calculated by periodic DFT for the models of the possible Brønsted acid site locations. Right: “d5” the structure that produces the spectrum that best matches the INS results. Note that the hydroxyls are distributed relatively far from each other. The figure has been reproduced from ref. 59 with permission from Elsevier, copyright 2013. | ||
Powder X-ray diffraction (XRD) has long been used to characterise zeolites, however, it is incapable of locating the hydrogens of the acid sites owing to their negligible electron density and the very similar scattering power of Al and Si. Neutron powder diffraction (NPD) has successfully located the Brønsted acid sites in certain zeolites, however, these were specifically synthesised for the measurements and contained little or no EFAl.60–62
In an attempt to directly locate the acid sites, we measured NPD data from our material.48 HZSM-5 exhibits a “low temperature” monoclinic form, space group P21/n, and a “high temperature” orthorhombic form, space group Pnma. The transition temperature depends on several factors including the Si:Al ratio, the state of hydration and the presence (or not) of molecules in the channels. Powder XRD data of our material can be assigned as the orthorhombic structure. Structural models were refined from the NPD data in both the monoclinic and orthorhombic space groups. Both resulted in good agreement, with the monoclinic fit being marginally better. The monoclinic structure is very close to the orthorhombic one: the β angle deviates from 90° by less than 0.1° and this is unobservable by conventional X-ray diffraction instrumentation. NPD is more sensitive to the oxygen positions than powder XRD and this probably accounts for why NPD finds the monoclinic structure and powder XRD finds the orthorhombic structure.
The analysis did indicate the presence of atoms at a reasonable distance from the framework (∼2.5 Å). These can be plausibly assigned to the EFAl, although the assignment is not definitive and further work is required. The complexity of the material (i.e. multiple possible sites for Al (and hence H/D), the presence of significant amounts of extra framework Al (that contributes background), the low concentration of framework Al (and hence H/D)) experienced in the structural analysis of our ZSM-5, serves to emphasise the difficulty of studying real industrial materials.
The three stages of the catalyst's life
Induction
The induction period seen in MTH chemistry is the time taken to establish the hydrocarbon pool, Fig. 1. The formation of the first C–C bond has been the subject of intense discussion7 and is still actively debated.20,63 By having the entire spectrum available it was hoped that INS would provide some insight into this problem. Unfortunately this turned out not to be the case, the spectra were weak and complex with few distinguishing features.64 The weakness of the spectra derives from there being relatively few species present and the complexity indicates that no single molecule dominates the early stages of the hydrocarbon pool formation and that multiple species are involved.However, a crucial step in the induction period, and indeed throughout the catalyst's lifetime, is transport of methanol into the zeolite and removal of the products from it. The transport depends upon diffusion within the zeolite and this is a property that quasielastic neutron scattering (QENS) is ideally suited to investigate.
The fundamental principles of the QENS technique are described elsewhere.38 In brief, a QENS experiment measures the broadening of the elastic line as a function of the scattering angle, which is directly related to the momentum transfer, Q.38 The variation in width of the elastic line with Q is characteristic of the type of motion: whether it is translational or rotational diffusion, and also how it is executed; continuous or jump diffusion or rotation in a confined volume. The time scales that QENS accesses, picoseconds to (almost) microseconds, match that of classical molecular dynamics (MD) and MD simulations are extensively used to analyse QENS data.65
The diffusion of methanol in ZSM-5 has been extensively investigated by QENS.66–71 This has proven controversial. O'Malley and co-workers67 found that methanol was immobile at room temperature on the time scale of the spectrometer, see Fig. 4, whereas methanol in HY was mobile. An INS study of the same samples used for the QENS found no evidence for the OH functionality in methanol and it was concluded that the methanol had methoxylated the Brønsted acid sites. This had occurred at room temperature, which was unprecedented, methoxylation usually occurs around 523 K,72 at room temperature hydrogen bonded methanol is seen by infrared spectroscopy.73 This work sparked an intense experimental and computational program.74–78 This showed that most of the methanol was present in the hydrogen bonded form but a minority did result in methoxylation. The absence of the OH functionality in the INS spectrum was shown to be a consequence of the very strong hydrogen bonding that broadened the bands to such an extent that they disappeared into the background.79 It was also found that methoxylation was dependent on the number of methanol molecules present in the zeolite pore. The immobility of methanol in HZSM-5 is attributed to the strong hydrogen bonding that makes the diffusion an order of magnitude slower80 than in other zeolites e.g. HY81 and so falls outside the time window of the QENS spectrometer used. Methane has been proposed82 as a surrogate for methanol as it avoids the problems caused by hydrogen bonding. Unfortunately, it has a very limited useful temperature range and a heavier alkane would probably be more informative.
 | ||
| Fig. 4 QENS spectra obtained of methanol loaded in zeolites H-ZSM-5 and HY at Q = 0.9 Å−1 at 298 K, the dotted red line represents the resolution data taken at 5 K. The close fit to the resolution function in H-ZSM-5 shows that the methanol is immobile on the timescale of the spectrometer, unlike the significant broadening in HY showing diffusing methanol (1 meV = 8.067 cm−1). Reproduced from ref. 67 under a Creative Commons Attribution 3.0 Unported Licence (CC-BY). | ||
Generally, with methanol, only rotational diffusion or translational diffusion within a pore is seen, long range translational diffusion is not seen. In most cases, there is a substantial fraction (up to 70%) that is immobile on the picosecond timescale. A QENS study68 that compared a fresh catalyst with samples that had been used for MTH for three days at 623 and 673 K, found that methanol was immobile in the fresh and 623 K reacted samples but that ∼40% was mobile in the 673 K reacted catalyst. This was ascribed to the development of mesoporosity in the material caused by removal of framework Al. BET measurements confirmed the presence of induced mesoporosity and 27Al ss-NMR showed the removal of Al. The development of mesoporosity is significant because this is likely to be what happens to the catalyst in industrial operation.
Steady state
The MTH reaction at steady state has been extensively investigated by INS spectroscopy.64,83,84 The reaction is especially suited to INS because the zeolite is transparent to neutrons (so the diagnostic “fingerprint” region, 400–1600 cm−1, is visible), the species of interest are hydrogenous and the darkening of the zeolite that occurs as the reaction progresses, does not hinder the spectroscopy.Fig. 5 compares the INS spectra of ZSM-5 after MTH reaction at 573 K for 60 h, 623 K for 110 h and 673 K for 44 h.64 The 673 K reaction was run with a reduced contact time that favours alkene formation over aromatic synthesis85 and so slows deactivation. It can be seen that all three temperatures result in very similar spectra. This is the “vibrational fingerprint of the hydrocarbon pool”.
 | ||
| Fig. 5 INS spectra of ZSM-5 after MTH reaction at: (i) 573 K for 60 h; (ii) 623 K for 110 h; (iii) 673 K for 44 h. The spectra are normalised to the sample mass. (a) Spectra collected using the TOSCA spectrometer and (b) spectra collected with the MAPS spectrometer, integrated over the range 0 ≤ Q ≤ 10 Å−1. Adapted from ref. 64 under a Creative Commons Attribution Licence (CC-BY). | ||
It is apparent from Fig. 5 that no single molecular entity is present; rather the spectra represent a mixture of hydrocarbon species. The question arises as to what these might be. The finite size of the channels in the zeolite limit the possible species to methylated benzenes, naphthalenes and anthracene.86–89 The INS spectra of benzene and all its methylated derivatives, C6H6−x(CH3)x (x = 0–6), as well as cyclic and polycyclic alkenes and aromatic molecules were recorded as candidate species.90 Synthetic spectra were generated by adding spectra of the model compounds in an equimolar ratio; similar hydrocarbons were added together in order to highlight common spectral features. This is only possible because of the quantitative nature of the INS technique. The results of this process are shown in Fig. 6, where the simulated spectra are compared to the spectrum for the long-run steady-state sample, 623–110 h.
 | ||
| Fig. 6 Experimental spectrum of the catalyst reacted for 110 h at 623 K (i) compared with the synthetic spectra generated by equimolar additions of spectra of the model compounds. The colour coding highlights regions that are characteristic of the different hydrocarbons. (a) Comparison with methylated benzenes: (ii) methylated benzenes with four or more methyl groups; (iii) methylated benzenes with up to three methyl substitutions. (b) Comparison with polycyclic aromatic molecules: (iv) all polycyclics and (v) naphthalenes. Sections (c) and (d) illustrate the principal contributors to the spectra: (c) methylated benzenes with ≥4 methyl groups; (d) naphthalenes; (e) anthracenes and pyrene. Adapted from ref. 64 under a Creative Commons Attribution Licence (CC-BY). | ||
A doublet of peaks at 1354 cm−1 and 1458 cm−1 are evident in all plots in Fig. 5a, these are assigned to symmetric and asymmetric CH3 bending modes of highly methylated benzenes (≥4 methyl groups, those with three or less methyl groups do not show these bands, Fig. 6aiii). The same doublet is also seen in methylated polycyclic aromatics (Fig. 6b), hence part of the intensity could be the result of those species as well. Peaks near 1200 cm−1 are observed in the summed spectra of polycyclic molecules (Fig. 6b), but these features are absent in the spectra of methylated benzenes (Fig. 6a). So the strong peak at 1200 cm−1 with a high energy shoulder at 1246 cm−1 is assigned to C–H bending modes of polycyclic aromatic molecules, such as naphthalene. Comparison of the ratio of the 1200 cm−1 peak with other peaks in the reference compounds shows that for all the polycyclic hydrocarbons it has approximately the same intensity as the peak found at 910 cm−1, a CH wag. However, in the steady-state MTH spectrum (Fig. 5a) the relative intensity of the two peaks (1200 cm−1 and 910 cm−1) is different, with the 1200 cm−1 peak being significantly more intense. This suggests that there is another species that is also contributing to the intensity of the 1200 cm−1peak.
In the MTH spectrum (Fig. 6a(i)) a sharp band is located at 600 cm−1. This band is only present in the spectra of naphthalene model compounds and, consequently, it is assigned to the double aromatic ring distortion of naphthalenes. The broad peaks below 400 cm−1 are largely attributed to the methyl torsions of the methylated benzenes and the polycyclic species. These usually give very strong INS features, as there is a large amplitude of motion of the hydrogen atoms. We have shown elsewhere90 that both the degree of methyl substitution on the aromatic rings and the local environment strongly influence the methyl torsion transition energies, which explains why there are no distinct methyl torsion peaks present in the HCP spectrum.
In some variations of the MTH process utilising a fixed bed reactor, the feedstock is an equilibrated mixture of methanol, dimethylether (DME) and water.91 Recent work has suggested that the mechanistic steps of methanol and of DME, when used as feedstock, may not be the same.92,93 DME converts faster, and at lower temperatures, to olefins than methanol. Deactivation studies have also shown that the catalyst deactivates at a faster rate with DME. This has been attributed to the lower water concentration and the faster kinetics of the reaction when DME is used.92
If the mechanism has changed, then it would be reasonable to expect that the HCP has also changed and this should be reflected in the INS spectrum. A study94 using DME as the feedstock confirmed the previous work that the catalyst deactivated much faster but that the HCP “fingerprint” was essentially the same as that shown in Fig. 5a. INS spectra in the C–H stretch region show that the aromatic to aliphatic ratio in the coke deposits is considerably higher for DME in comparison with methanol conversion. Together with the reduced catalyst lifetime, this is attributed to the lower levels of water present in DME conversion because this reduces the regeneration of the acid sites needed for methylation of aromatic species in the zeolite. The issue of by-product water molecules being responsible for the regeneration of the Brønsted acid sites is thought to be a key factor in sustained catalytic performance.
As noted in the section on the induction period, the formation of the first C–C bond is still controversial. A recent proposal is that C–C bonds are created via methyl acetate (MeOAc) as the first intermediate.95,96 Formation of MeOAc from methanol or DME requires the feedstock to undergo a carbonylation reaction, this requires a source of CO, which may result from dehydrogenation of methanol. Methanol and DME carbonylation to form MeOAc has been reported mainly with a mordenite acid zeolite catalyst, although good selectivity towards MeOAc at relatively lower temperatures has also been observed with HZSM-5.
An INS study97 explored the reactivity of methanol–MeOAc mixtures over an HZSM-5 catalyst at temperatures representative of that employed in MTH chemistry, with a focus on the effects of MeOAc on the catalyst lifetime. Fig. 7 shows a comparison of the blank zeolite, pure MeOAc and the zeolite loaded with MeOAc at room temperature. This emphasises the negligible contribution of the zeolite to the spectrum. It also shows that MeOAc is adsorbed intact, as the bands occur at very similar transition energies. The exception is the methyl torsion at 235 cm−1, that has downshifted to 150 cm−1. Methyl torsions are sensitive90 to their local environment and this is a striking example.
 | ||
| Fig. 7 INS spectra of the blank zeolite (black), methyl acetate (orange) and ZSM-5 exposed to methyl acetate at ambient temperature (grey). Reproduced from ref. 97 under a Creative Commons Attribution 4.0 International License (CC-BY). | ||
Fig. 8 shows INS spectra of the catalyst as a function of the concentration of MeOAc in the feed stream at 623 K. The intensities of the bands due to hydrocarbon coke species increase with increasing MeOAc content in the feed, (the balance is methanol). This trend additionally reflects that observed for the retained coke content, which ranged from 2.53 wt% in the 0% MeOAc feed to 9.76 wt% in the 100% MeOAc feed. The spectra measured from catalysts exposed to 30% or more MeOAc resemble those shown in Fig. 5 for a pure methanol feed and also those seen for HZSM-5 zeolite catalysts reacted with DME at 623 K.9,44 Crucially, there is no correspondence with the spectrum of adsorbed MeOAc (Fig. 7).
 | ||
| Fig. 8 INS spectra of ZSM-5 samples reacted at 623 K for 6 h with a feed of 0% MeOAc, 10% MeOAc, 30% MeOAc, 60% MeOAc and 100% MeOAc (the balance is methanol). The spectra are offset vertically for clarity. Reproduced from ref. 97 under a Creative Commons Attribution 4.0 International License (CC-BY). | ||
The presence of high concentrations of MeOAc accelerates catalyst deactivation by enhancing the formation of methylated aromatic coke compounds that block active sites within the zeolite: to reach the same coke content as that of the 100% MeOAc sample required 100 hours on stream with a methanol feed, versus the 6 hours for the sample in Fig. 8.
The spectra also show that the nature of the HCP is largely independent of the feedstock: methanol, DME and MeOAc all result in essentially the same spectrum, showing that the HCP is comparable in all three cases. For methanol, temperature also appears to play only a minor role, its major effect is how quickly the HCP is established, the contents are the same.
Deactivation
MTH catalysts generally deactivate by coke formation.15,19 A deactivated catalyst is characterised by a coke content of 14–18 wt%.98 By carrying out the MTH reaction at 623 K for 44 hours, but with a longer contact time than for the sample shown in Fig. 5(iii), we obtained a sample with 20.2 wt% coke.64 The INS spectra are shown in Fig. 9. These are markedly different from the spectra at steady state, Fig. 5. The methyl doublet at 1354 cm−1 and 1458 cm−1 is still present but much weaker, showing that methylated aromatics are less prevalent than at steady state. The spectra are dominated by peaks around 880 and 1200 cm−1. These do not match the spectrum of graphite, Fig. 9a(iii),99 but are a good match to the spectrum of glassy carbon, Fig. 9a(ii).100 This consists of mostly sp2 carbon atoms, as with graphite but with much smaller sheets and edge termination by adatoms, so it is similar to (but distinct from) amorphous carbon. The nature of glassy carbon means that it must grow on the outer surfaces of the zeolite, presumably by condensation of polycyclic aromatic molecules. The growth results in pore blocking of the zeolite and consequent deactivation. The formation of the glassy carbon would also account for the depletion of the hydrocarbon pool, as this is the source of the polycyclic aromatic molecules and these cannot be replenished because of the pore blocking.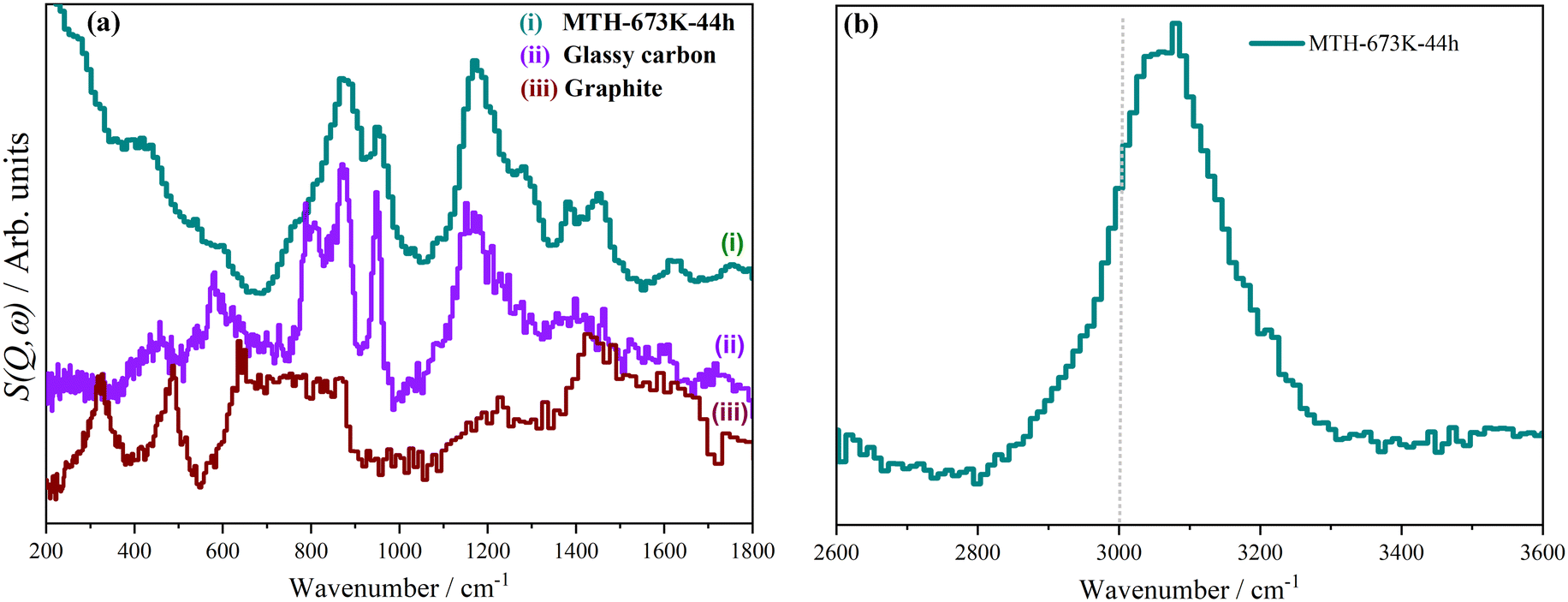 | ||
| Fig. 9 INS spectra of (i) ZSM-5 after MTH reaction at: 673 K for 44 h, reference INS spectra of: (ii) glassy carbon98 and (iii) graphite.97 (a) Spectra collected using the TOSCA spectrometer and (b) spectra collected with the MAPS spectrometer integrated over the range 0 ≤ Q ≤ 10 Å−1. Adapted from ref. 64 under a Creative Commons Attribution Licence (CC-BY). | ||
The C–H stretch region, Fig. 9b, shows that most of the hydrogen is bonded to sp2 hybridised carbon, as the majority of the peak lies above 3000 cm−1. This is consistent with the low energy spectrum which shows that the hydrogen is largely located at the edges of the graphene planes. INS spectroscopy is quantitative, thus the ratio of the areas of the sp3 C–H to the sp2 C–H directly measures the ratio present in the sample. Curve fitting Fig. 5b and 9b, gives sp3 C–H:sp2 C–H ratios of 1:1.36 (573 K-60 h), 1:1.37 (623 K-60 h), 1:3.54 (673 K-60 h, steady state) and >1:20 (673 K-44 h, deactivated). As the majority species in the HCP are highly methylated benzenes, it would be expected that the C–H stretch mode would be largely aliphatic sp3 C–H. The spectra show that this is not the case and that sp2 C–H dominates. The presence of the glassy carbon-type coke accounts for these ratios, since this contains almost entirely sp2 C–H. This also accounts for the mismatch in the ratio of the 800 and 1200 cm−1 peaks noted in the section on steady state. The data shows unambiguously that coke grows continuously throughout the reaction; it is not just a late stage product.
Conclusions and outlook
The aim of this mini-review is to demonstrate how the use of neutron-based techniques is useful for the study of MTH chemistry. It is to be emphasised that neutrons are “the technique of last resort” and behind each of the examples described or cited here, there is a raft of conventional characterisation methods including microreactor testing, surface area determination by nitrogen physisorption, NH3-TPD, TPO, ss-NMR and infrared spectroscopy amongst others. In particular, microreactor testing is crucial because to produce the large samples (typically ∼2–10 g) needed for neutron studies dedicated apparatus is required46,47 and it is essential to ensure that the large samples produced are representative of the chemistry. Comparison with microreactor data provides this assurance.As is apparent, most of the work to date has used QENS or INS. QENS provides direct access to diffusion constants on the microscopic scale and also informs as to the type of diffusion, whether it is localised (rotational) diffusion or long range (translational) diffusion. It has become clear that the strong hydrogen bonding of methanol to the zeolite results in translational diffusion that is an order of magnitude slower than expected. It is possible to access this time domain by using neutron spin echo (NSE).38 There are examples where this has been used for alkanes101–104 or aromatics105–109 in zeolites and we are currently exploring its use to study methanol in HZSM-5. QENS is also able to study translational and/or rotational diffusion in used catalysts, whether coked or dealuminated (or both) and it is clear that the possible motions are modified. This is obviously relevant to the industrial use of these materials.
INS spectroscopy has the huge advantage that it can access the entire 0–4000 cm−1 spectral range, including the diagnostic ‘fingerprint region’ (400–1800 cm−1). This attribute has allowed the hydrocarbon pool to be characterised without having to destroy the zeolite matrix (e.g. by dissolution in HF solution). The colour of the catalyst is also irrelevant to INS, so coked samples can be readily studied. This is a feature that has not been exploited until now for zeolite chemistry and has already provided insights into the nature of the coke. Deactivation is a major issue in heterogeneous catalysis in general and coking is a particular problem. INS has the ability to provide insights into the nature of the material and hence (hopefully) the mechanism of formation.
The use of INS to quantify the hydroxyls present is a unique attribute of the technique. The work that compared INS spectra and DFT calculations to locate the acid sites is a method that clearly has potential for other zeolites than LTA.58,59 The possibility to combine both approaches may enable the acid sites to be located and quantified in cases where diffraction methods fail.
Two neutron techniques that have been little used for zeolite investigations are neutron diffraction and neutron imaging. In principle, single crystal neutron diffraction can locate the Brønsted acid sites directly because it can observe the protons, unfortunately the difficulty of preparing suitable size zeolite single crystals means that this is still only an aspiration. Neutron powder diffraction should also be able to detect the protons (deuterons) of the acid sites and this has been done in a few cases.60–62 However, in real industrial materials this is complicated by the presence of multiple possible sites, the low concentration of Al (and hence H or D) and especially by EFAl. A more promising avenue is to use NPD to characterise adsorbates in zeolites. There are surprisingly few examples e.g.109–114 where this has been done and there are clearly opportunities here.
Neutron imaging115 provides information on the μm to cm length scale, thus it is highly complementary to the microscopic information provided by QENS. To date, there are very few imaging studies of catalysts116–120 (we exclude the extensive body of work on imaging of water flow in fuel cells121) and none that that have involved MTH. Neutron imaging has the advantage that it is usable at any temperature, so there are possibilities for operando studies.
Neutron beams are a scarce resource and are only available at central facilities, so obtaining access might be considered to be difficult. In practice, all the major facilities (ISIS, ILL, SNS, J-PARC) make the process of obtaining beam time as straightforward as possible and all operate a ‘free-at-the-point-of-use’ policy.
The limited capacity of neutron scattering means that the use of neutrons to study catalysts and catalytic processes will always be complementary to conventional methods of zeolite characterisation and catalysis. Nonetheless, the unique perspective provided by neutrons on structure, composition and diffusion means that the use of these methods to study zeolites and related materials will only grow.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work described here has involved many people and we would like to thank our collaborators for their contributions to this project: Nathan Barrow and Jonathan Bradley (Johnson Matthey), Santhosh Matam and Richard Catlow (Cardiff), Suwardiyanto (Jember, Indonesia), Alex O'Malley (Bath), Ali Hameed and James McGregor (Sheffield), Janet Skakle (Aberdeen), Chin Yong and Ilian Todorov (Scientific Computing Department, STFC), Ian Silverwood, Daniel Nye, Ronald Smith and Gavin Stenning (ISIS). Johnson Matthey Plc. is thanked for supplying the ZSM-5 zeolite and for financial support through the provision of industrial CASE studentships in partnership with the EPSRC (APH (EP/P510506/1), AZ (EP/N509176/1)). The resources and support provided by the UK Catalysis Hub via membership of the UK Catalysis Hub consortium and funded by EPSRC grants EP/R026815/1 and EP/R026939/1 and for hosting APH and AZ are gratefully acknowledged. This research has been performed with the use of facilities and equipment at the Research Complex at Harwell; the authors are grateful to the Research Complex for this access and support. The ISIS Neutron and Muon Facility is thanked for access to neutron beam facilities.References
- R. J. Argauer and G. R. Landolt, Crystalline zeolite ZSM-5 and method of preparing the same, US Pat., 3702866, 1972 Search PubMed.
- T. F. Degnan, G. K. Chitnis and P. H. Schipper, History of ZSM-5 fluid catalytic cracking additive development at Mobil, Microporous Mesoporous Mater., 2000, 35-36, 245–252 CrossRef CAS.
- M. A. den Hollander, M. Wissink, M. Makkee and J. A. Moulijn, Gasoline conversion: reactivity towards cracking with equilibrated FCC and ZSM-5 catalysts, Appl. Catal., A, 2002, 223, 85–102 CrossRef CAS.
- D. H. Olson and W. O. Haag, Structure-selectivity relationship in xylene isomerization and selective toluene disproportionation, in Catalytic Materials: Relationship Between Structure and Reactivity, ed. T. E. Whyte, R. A. Dalla Betta, E. G. Derouane and R. T. K. Baker, ACS Symposium Series, 1984, ch. 14, pp. 275–307 Search PubMed.
- S. S. Ali and H. A. Zaidi, Experimental and kinetic modeling studies of methanol transformation to hydrocarbons using zeolite-based catalysts: A review, Energy Fuels, 2020, 34, 13225–13246 CrossRef CAS.
- M. Yang, D. Fan, Y. Wei, P. Tian and Z. Liu, Recent progress in methanol-to-olefins (MTO) catalysts, Adv. Mater., 2019, 31, 1902181 CrossRef CAS PubMed.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, The formation and degradation of active species during methanol conversion over protonated zeotype catalysts, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- M. Ye, H. Li, Y. Zhao, T. Zhang and Z. Liu, MTO processes development: The key of mesoscale studies, in Advances in Chemical Engineering, Mesoscale Modeling in Chemical Engineering Part II, ed. G. B. Marin and J. Li, Academic Press, Boston, 2015, ch. 5, vol. 47, pp. 279–335 Search PubMed.
- J. Topp-Jørgensen, Topsøe integrated gasoline synthesis - the TIGAS process, in Studies in Surface Science and Catalysis, Methane Conversion, ed. D. M. Bibby, C. D. Chang, R. F. Howe and S. Yurchak, 1988, vol. 36, pp. 293–305 Search PubMed.
- J. Ding and W. Hua, Game changers of the C3 value chain: gas, coal, and biotechnologies, Chem. Eng. Technol., 2013, 36, 83–90 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Recent trends and fundamental insights in the methanol-to-hydrocarbons process, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- S. Ilias and A. Bhan, Mechanism of the catalytic conversion of methanol to hydrocarbons, ACS Catal., 2013, 3, 18–31 CrossRef CAS.
- A. Galadima and O. Muraza, From synthesis gas production to methanol synthesis and potential upgrade to gasoline range hydrocarbons: A review, J. Nat. Gas Sci. Eng., 2015, 25, 303–316 CrossRef CAS.
- C. Wang, J. Xu and F. Deng, Mechanism of methanol-to-hydrocarbon reaction over zeolites: A solid-state NMR perspective, ChemCatChem, 2020, 12, 965–980 CrossRef CAS.
- H. Schulz, About the mechanism of methanol conversion on zeolites, Catal. Lett., 2018, 148, 1263–1280 CrossRef CAS.
- I. M. Dahl and S. Kolboe, On the reaction mechanism for propene formation in the MTO reaction over SAPO-34, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- J. L. White, Methanol-to-hydrocarbon chemistry: The carbon pool (r)evolution, Catal. Sci. Technol., 2011, 1, 1630–1635 RSC.
- M. J. Wulfers and F. C. Jentoft, The role of cyclopentadienium ions in methanol-to-hydrocarbons chemistry, ACS Catal., 2014, 4, 3521–3532 CrossRef CAS.
- J. Goetze and B. M. Weckhuysen, Spatiotemporal coke formation over zeolite ZSM-5 during the methanol-to-olefins process as studied with operando UV-vis spectroscopy: a comparison between H-ZSM-5 and Mg-ZSM-5, Catal. Sci. Technol., 2018, 8, 1632–1644 RSC.
- J. Valecillos, H. Vicente, A. G. Gayubo, A. T. Aguayo and P. Castaño, Spectro-kinetics of the methanol to hydrocarbons reaction combining online product analysis with UV–vis and FTIR spectroscopies throughout the space time evolution, J. Catal., 2022, 408, 115–127 CrossRef CAS.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzy, Probing zeolites by infrared spectroscopy, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC.
- I. B. Minova, S. K. Matam, A. Greenaway, C. R. A. Catlow, M. D. Frogley, G. Cinque, P. A. Wright and R. F. Howe, Elementary steps in the formation of hydrocarbons from surface methoxy groups in HZSM-5 seen by synchrotron infrared microspectroscopy, ACS Catal., 2019, 9, 6564–6570 CrossRef CAS.
- F. F. Madeira, H. Vezin, N. S. Gnep, P. Magnoux, S. Maury and N. Cadran, Radical species detection and their nature evolution with catalyst deactivation in the ethanol-to-hydrocarbon reaction over HZSM-5 zeolite, ACS Catal., 2011, 1, 417–424 CrossRef CAS.
- S. Hamieh, C. Canaff, K. Ben Tayeb, M. Tarighi, S. Maury, H. Vezin, Y. Pouilloux and L. Pinard, Methanol and ethanol conversion into hydrocarbons over H-ZSM-5 catalyst, Eur. Phys. J.: Spec. Top., 2015, 224, 1817–1830 CAS.
- A. R. McFarlane, I. P. Silverwood, E. L. Norris, R. M. Ormerod, C. D. Frost, S. F. Parker and D. Lennon, The application of inelastic neutron scattering to investigate the steam reforming of methane over an alumina-supported nickel catalyst, Chem. Phys., 2013, 427, 54–60 CrossRef CAS.
- N. G. Hamilton, I. P. Silverwood, R. Warringham, J. Kapitán, L. Hecht, P. B. Webb, R. P. Tooze, S. F. Parker and D. Lennon, Vibrational analysis of an industrial Fe-based Fischer-Tropsch catalyst employing inelastic neutron scattering, Angew. Chem., Int. Ed., 2013, 52, 5608–5611 CrossRef CAS PubMed.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schlögl, The Haber–Bosch process revisited: On the real structure and stability of “ammonia iron” under working conditions, Angew. Chem., Int. Ed., 2013, 52, 12723–12726 CrossRef CAS PubMed.
- J. Kammert, J. Moon, Y. Cheng, L. Daemen, S. Irle, V. Fung, J. Liu, K. Page, X. Ma, V. Phaneuf, J. Tong, A. J. Ramirez-Cuesta and Z. Wu, Nature of reactive hydrogen for ammonia synthesis over a Ru/C12A7 electride catalyst, J. Am. Chem. Soc., 2020, 143, 7655–7667 CrossRef PubMed.
- F. Polo-Garzon, V. Fung, L. Nguyen, Y. Tang, F. Tao, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, G. S. Foo, M. Zhu, I. E. Wachs, D. Jiang and Z. Wu, Elucidation of the reaction mechanism for high-temperature water gas shift over an industrial-type copper−chromium−iron oxide catalyst, J. Am. Chem. Soc., 2019, 141, 7990–7999 CrossRef CAS PubMed.
- P. W. Albers, K. Möbus, C. D. Frost and S. F. Parker, Characterisation of beta palladium hydride formation in the Lindlar catalyst and in carbon supported palladium, J. Phys. Chem. C, 2011, 115, 24485–24493 CrossRef CAS.
- W. Fang, C. Pirez, S. Paul, M. Capron, H. Jobic, F. Dumeignil and L. Jalowiecki-Duhamel, Room temperature hydrogen production from ethanol over CeNixHzOy nano-oxyhydride catalysts, ChemCatChem, 2013, 5, 2207–2216 CrossRef CAS.
- P. W. Albers, D. Lennon and S. F. Parker, Catalysis, ed. F. Fernandez-Alonso and D. L. Price, Academic Press, 2017, ch. 5, vol. 49, pp. 279–348 Search PubMed.
- R. F. Howe, J. McGregor, S. F. Parker, P. Collier and D. Lennon, Application of inelastic neutron scattering to the methanol-to-gasoline reaction over a ZSM-5 catalyst, Catal. Lett., 2016, 46, 1242–1248 CrossRef PubMed.
- G. L. Squires, Introduction to the Theory of Thermal Neutron Scattering, Cambridge University Press, 2012 Search PubMed.
- A. T. Boothroyd, Principles of Neutron Scattering from Condensed Matter, Oxford University Press, 2020 Search PubMed.
- B. T. M. Willis and C. J. Carlile, Experimental Neutron Scattering, Oxford University Press, 2013 Search PubMed.
- E. H. Kisi and C. J. Howard, Applications of Neutron Powder Diffraction, Oxford Science Publications, 2012 Search PubMed.
- M. T. F. Telling, A Practical Guide to Quasi-elastic Neutron Scattering, Royal Society of Chemistry, 2020 Search PubMed.
- P. C. H. Mitchell, S. F. Parker, A. J. Ramirez-Cuesta and J. Tomkinson, Vibrational Spectroscopy with Neutrons, With Applications in Chemistry, Biology, Materials Science and Catalysis, World Scientific, Singapore, 2005 Search PubMed.
- A. Furrer, J. Mesot and T. Strässle, Neutron Scattering in Condensed Matter Physics, World Scientific, Singapore, 2009 Search PubMed.
- https://neutronsources.org/neutron-centres/, Accessed on 11/10/22.
- https://www.ill.eu/, Accessed on 11/10/22.
- https://www.isis.stfc.ac.uk/Pages/home.aspx, Accessed on 11/10/22.
- https://neutrons.ornl.gov/sns, Accessed on 11/10/22.
- https://j-parc.jp/en/jparc.html, Accessed on 11/10/22.
- R. Warringham, D. Bellaire, S. F. Parker, J. Taylor, C. M. Goodway, M. Kibble, S. R. Wakefield, M. Jura, M. P. Dudman, R. P. Tooze, P. B. Webb and D. Lennon, Sample environment issues relevant to the acquisition of inelastic neutron scattering measurements of heterogeneous catalyst samples, J. Phys.: Conf. Ser., 2014, 554, 012005 CrossRef.
- S. Tan, Y. Cheng, L. L. Daemen and D. A. Lutterman, Design of a facility for the in situ measurement of catalytic reaction by neutron scattering spectroscopy, Rev. Sci. Instrum., 2018, 89, 014101 CrossRef PubMed.
- A. Zachariou, A. P. Hawkins, R. F. Howe, J. Skakle, P. Collier, N. S. Barrow, D. Nye, R. Smith, G. Stenning, S. F. Parker and D. Lennon, Counting the acid sites in a commercial ZSM-5 zeolite catalyst, ACS Phys. Chem. Au, 2023, 3, 74–83 CrossRef CAS PubMed.
- S. F. Parker, D. Lennon and P. W. Albers, Vibrational spectroscopy with neutrons – a review of new directions, Appl. Spectrosc., 2011, 65, 1325–1341 CrossRef CAS.
- P. W. Albers, G. Michael, H. L. Rotgerink, M. Reisinger and S. F. Parker, Low frequency vibrational dynamics of amorphous and crystalline silica, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 1016–1020 CrossRef CAS.
- S. F. Parker, U. Klehm and P. W. Albers, Differences in the morphology and vibrational dynamics of crystalline, glassy and amorphous silica – commercial implications, Mater. Adv., 2020, 1, 749–759 RSC.
- I. P. Silverwood, N. G. Hamilton, C. J. Laycock, J. Z. Staniforth, R. M. Ormerod, C. D. Frost, S. F. Parker and D. Lennon, Quantification of surface species present on a nickel/alumina methane reforming catalyst, Phys. Chem. Chem. Phys., 2010, 12, 3102–3107 RSC.
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S.-B. Liu and A. Zheng, Origin and structural characteristics of tri-coordinated extra-framework aluminum species in dealuminated zeolites, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS PubMed.
- M. J. Wax, R. R. Cavanagh, J. J. Rush, G. D. Stucky, L. Abrams and D. R. Corbin, A neutron scattering study of zeolite Rho, J. Phys. Chem., 1986, 90, 532–534 CrossRef CAS.
- H. Jobic, Observation of the fundamental bending vibrations of hydroxyl groups in HNa-Y zeolite by neutron inelastic scattering, J. Catal., 1991, 131, 289–293 CrossRef CAS.
- W. P. J. H. Jacobs, H. Jobic, J. H. M. C. van Wolput and R. A. van Santen, Fourier transform infrared and inelastic neutron scattering study of HY zeolites, Zeolites, 1992, 12, 315–319 CrossRef CAS.
- W. P. J. H. Jacobs, J. H. M. C. van Wolput, R. A. van Santen and H. Jobic, A vibrational study of the OH and OD bending modes of the Brønsted acid sites in zeolites, Zeolites, 1994, 14, 117–125 CrossRef CAS.
- T. Lemishko, S. Valencia, F. Rey, M. Jiménez-Ruiz and G. Sastre, Inelastic neutron scattering study on the location of Brønsted acid sites in high silica LTA zeolite, J. Phys. Chem. C, 2016, 120, 24904–24909 CrossRef CAS.
- T. Lemishko, M. Jiménez-Ruiz, F. Rey, S. Valencia, T. Blasco, A. V. Moya and G. Sastre, Inelastic neutron scattering study of the aluminium and Brønsted site location of acid in aluminosilicate LTA zeolites, J. Phys. Chem. C, 2018, 122, 11450–114954 CrossRef CAS.
- J. Jirak, S. Vratislav, J. Zajicek and V. Bosacek, The localization of protons in decationated zeolites by neutron diffraction, J. Catal., 1977, 49, 112–114 CrossRef.
- A. Martucci, A. Alberti, G. Cruciani, P. Radaelli, P. Ciambelli and M. Rapacciulo, Location of Brønsted sites in D-ferrierite by neutron powder diffraction, Microporous Mesoporous Mater., 1999, 30, 95–101 CrossRef CAS.
- A. Martucci, G. Cruciani, A. Alberti, C. Ritter, P. Ciambelli and M. Rapacciuolo, Location of Brønsted sites in D-mordenites by neutron powder diffraction, Microporous Mesoporous Mater., 2000, 35−36, 405–412 CrossRef CAS.
- W. Zhang, Y. Zhi, J. Huang, X. Wu, S. Zeng, S. Xu, A. Zheng, Y. Wei and Z. Liu, Methanol to olefins reaction route based on methylcyclopentadienes as critical intermediates, ACS Catal., 2019, 9, 7373–7379 CrossRef CAS.
- A. Zachariou, A. P. Hawkins, Suwardiyanto, P. Collier, N. Barrow, R. F. Howe, S. F. Parker and D. Lennon, New spectroscopic insight into the deactivation of a ZSM-5 methanol-to-hydrocarbons catalyst, ChemCatChem, 2021, 13, 2625–2633 CrossRef CAS.
- J. Armstrong, A. J. O'Malley, M. R. Ryder and K. T. Butler, Understanding dynamic properties of materials using neutron spectroscopy and atomistic simulation, J. Phys. Commun., 2020, 4, 072001 CrossRef CAS.
- H. Jobic, A. Renouprez, M. Bée and C. Poinsignon, Quasi-elastic neutron scattering study of the molecular motions of methanol adsorbed on H-ZSM-5, J. Phys. Chem., 1986, 90, 1059–1065 CrossRef CAS.
- A. J. O'Malley, S. F. Parker, A. Chutia, I. P. Silverwood, V. García-Sakai and C. R. A. Catlow, Framework dependent room temperature methoxylation in zeolites: insight into the first step of the methanol-to-hydrocarbons process, Chem. Commun., 2016, 52, 2897–2900 RSC.
- S. K. Matam, A. J. O'Malley, C. R. A. Catlow, Suwardiyanto, P. Collier, A. Hawkins, A. Zachariou, D. Lennon, I. Silverwood, S. F. Parker and R. F. Howe, The effects of MTG catalysis on methanol mobility in ZSM-5, Catal. Sci. Technol., 2018, 8, 3304–3312 RSC.
- T. Omojola, I. P. Silverwood and A. J. O'Malley, Molecular behaviour of methanol and dimethyl ether in H-ZSM-5 catalysts as a function of Si/Al ratio: a quasielastic neutron scattering study, Catal. Sci. Technol., 2020, 10, 4305–4320 RSC.
- C. H. Botchway, R. Tia, E. Adei, A. J. O'Malley, N. Y. Dzade, C. Hernandez-Tamargo and N. H. de Leeuw, Influence of topology and Brønsted acid site presence on methanol diffusion in zeolites Beta and MFI, Catalysts, 2020, 10, 1342 CrossRef CAS.
- S. K. Matam, C. R. A. Catlow, I. P. Silverwood, S. F. Parker and A. J. O'Malley, Methanol dynamics in H-ZSM-5 with Si/Al ratio of 25: a quasi-elastic neutron scattering (QENS) study, Top. Catal., 2021, 64, 699–706 CrossRef CAS.
- T. R. Forester and R. F. Howe, In situ FTIR studies of methanol and dimethyl ether ZSM-5, J. Am. Chem. Soc., 1987, 109, 5076–5082 CrossRef CAS.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Spano and F. Geobaldo, IR spectroscopy of neutral and ionic hydrogen-bonded complexes formed upon interaction of CH3OH, C2H5OH, (CH3)2O, (C2H5)2O and C4H8O with H-Y, H-ZSM-5 and H-mordenite: Comparison with analogous adducts formed on the H-Nafion superacidic membrane, J. Chem. Soc., Faraday Trans., 1996, 92, 4863–4875 RSC.
- S. K. Matam, R. F. Howe, A. Thetford and C. R. A. Catlow, Room temperature methoxylation in zeolite H-ZSM-5: an operando DRIFTS/mass spectrometric study, Chem. Commun., 2018, 54, 12875–12878 RSC.
- S. A. F. Nastase, A. J. O'Malley, C. R. A. Catlow and A. J. Logsdail, Computational QM/MM investigation of the adsorption of MTH active species in H-Y and H-ZSM-5, Phys. Chem. Chem. Phys., 2019, 21, 2639–2650 RSC.
- S. K. Matam, S. A. F. Nastase, A. J. Logsdail and C. R. A. Catlow, Methanol loading dependent methoxylation in zeolite H-ZSM-5, Chem. Sci., 2020, 11, 6805–6814 RSC.
- S. A. F. Nastase, P. Cnudde, L. Vanduyfhuys, K. De Wispelaere, V. Van Speybroeck, C. R. A. Catlow and A. J. Logsdail, Mechanistic insight into the framework methylation of H-ZSM-5 for varying methanol loadings and Si/Al Ratios using first-principles molecular dynamics simulations, ACS Catal., 2020, 10, 8904–8915 CrossRef CAS PubMed.
- S. A. F. Nastase, A. J. Logsdail and C. R. A. Catlow, QM/MM study of the reactivity of zeolite bound methoxy and carbene groups, Phys. Chem. Chem. Phys., 2021, 23, 17634–17644 RSC.
- A. Zachariou, A. P. Hawkins, R. F. Howe, N. Barrow, J. Bradley, P. Collier, D. Lennon and S. F. Parker, A spectroscopic paradox: the interaction of methanol with ZSM-5 at room temperature, Top. Catal., 2021, 64, 672–684 CrossRef CAS.
- C.-L. M. Woodward, A. J. Porter, K. S. C. Morton and A. J. O'Malley, Methanol diffusion in H-ZSM-5 catalysts as a function of loading and Si/Al ratio: A classical molecular dynamics study, Catal. Commun., 2022, 164, 106415 CrossRef CAS.
- A. J. O'Malley, V. García-Sakai, I. P. Silverwood, N. Dimitratos, S. F. Parker and C. R. A. Catlow, Methanol diffusion in zeolite HY: A combined quasielastic neutron scattering and molecular dynamics simulation study, Phys. Chem. Chem. Phys., 2016, 18, 17294–17302 RSC.
- A. P. Hawkins, A. Zachariou, I. P. Silverwood, C. Yong, P. Collier, I. Todorov, R. F. Howe, S. F. Parker and D. Lennon, Combining quasielastic neutron scattering and molecular dynamics to study methane motions in ZSM-5, J. Chem. Phys., 2022, 157, 184702 CrossRef CAS PubMed.
- Suwardiyanto, R. F. Howe, E. K. Gibson, C. R. A. Catlow, A. Hameed, J. McGregor, P. Collier, S. F. Parker and D. Lennon, An assessment of hydrocarbon species in the methanol-to-hydrocarbon reaction over a ZSM-5 catalyst, Faraday Discuss., 2017, 197, 447–471 RSC.
- A. Zachariou, A. P. Hawkins, D. Lennon, S. F. Parker and R. F. Howe, Neutron spectroscopy studies of methanol to hydrocarbons catalysis over ZSM-5, Catal. Today, 2020, 368, 20–27 CrossRef.
- M. Zhang, S. Xu, Y. Wei, J. Li, J. Wang, W. Zhang, S. Gao and Z. Liu, Changing the balance of the MTO reaction dual-cycle mechanism: Reactions over ZSM-5 with varying contact times, Chin. J. Catal., 2016, 37, 1413–1422 CrossRef CAS.
- F. Bleken, W. Skistad, K. Barbera, M. Kustova, S. Bordiga, P. Beato, K. P. Lillerud, S. Svelle and U. Olsbye, Conversion of methanol over 10-ring zeolites with differing volumes at channel intersections: Comparison of TNU-9, IM-5, ZSM-11 and ZSM-5, Phys. Chem. Chem. Phys., 2011, 13, 2539–2549 RSC.
- J. Goetze, F. Meirer, I. Yarulina, J. Gascon, F. Kapteijn, J. Ruiz-Martínez and B. M. Weckhuysen, Insights into the activity and deactivation of the methanol-to-olefins process over different small-pore zeolites as studied with operando UV–vis spectroscopy, ACS Catal., 2017, 7, 4033–4046 CrossRef CAS PubMed.
- M. Signorile, D. Rojo-Gama, F. Bonino, P. Beato, S. Svelle and S. Bordiga, Topology-dependent hydrocarbon transformations in the methanol-to-hydrocarbons reaction studied by operando UV-Raman spectroscopy, Phys. Chem. Chem. Phys., 2018, 20, 26580–26590 RSC.
- S. F. Parker and A. J. Kombanal, How many molecules can fit in a zeolite pore? Implications for the hydrocarbon pool mechanism of the methanol-to-hydrocarbons process, Catalysts, 2021, 11, 1204 CrossRef CAS.
- A. Zachariou, A. P. Hawkins, P. Collier, R. F. Howe, D. Lennon and S. F. Parker, The methyl torsion in unsaturated compounds, ACS Omega, 2020, 5, 2755–2765 CrossRef CAS PubMed.
- F. J. Keil, Methanol-to-hydrocarbons: process technology, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- P. Pérez-Uriarte, M. Gamero, A. Ateka, M. Díaz, A. T. Aguayo and J. Bilbao, Effect of the Acidity of HZSM-5 zeolite and the binder in the DME transformation to olefins, Ind. Eng. Chem. Res., 2016, 55, 1513–1521 CrossRef.
- J. S. Martinez-Espin, M. Mortén, T. V. W. Janssens, S. Svelle, P. Beato and U. Olsbye, New insights into catalyst deactivation and product distribution of zeolites in the methanol-to-hydrocarbons (MTH) reaction with methanol and dimethyl ether feeds, Catal. Sci. Technol., 2017, 7, 2700–2716 RSC.
- A. Zachariou, A. Hawkins, D. Lennon, S. F. Parker, Suwardiyanto, S. K. Matam, C. R. A. Catlow, P. Collier, A. Hameed, J. McGregor and R. F. Howe, Investigation of ZSM-5 catalysts for dimethylether conversion using inelastic neutron scattering, Appl. Catal., A, 2019, 569, 1–7 CrossRef CAS.
- Y. Liu, S. Müller, D. Berger, Y. Jelic, K. Reuter, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Formation mechanism of the first carbon–carbon bond and the first olefin in the methanol conversion into hydrocarbons, Angew. Chem., Int. Ed., 2016, 55, 5723–5726 CrossRef CAS PubMed.
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Initial carbon–carbon bond formation during the early stages of the methanol-to-olefin process proven by zeolite-trapped acetate and methyl acetate, Angew. Chem., Int. Ed., 2016, 55, 15840–15845 CrossRef CAS PubMed.
- A. Zachariou, A. P. Hawkins, P. Collier, R. F. Howe, S. F. Parker and D. Lennon, The effect of co-feeding methyl acetate on the H-ZSM5 catalysed methanol-to-hydrocarbons reaction, Top. Catal., 2020, 63, 370–377 CrossRef CAS.
- D. M. Bibby, N. B. Milestone, J. E. Patterson and L. P. Aldridge, Coke formation in zeolite ZSM-5, J. Catal., 1986, 97, 493–502 CrossRef CAS.
- J. K. Walters, R. J. Newport, S. F. Parker and W. S. Howells, A spectroscopic study of the structure of amorphous hydrogenated carbon, J. Phys.: Condens. Matter, 1995, 7, 10059–10073 CrossRef.
- S. F. Parker, S. Imberti, S. K. Callear and P. W. Albers, Structural and spectroscopic studies of a commercial glassy carbon, Chem. Phys., 2013, 427, 44–48 CrossRef CAS.
- H. Jobic, H. Paoli, A. Méthivier, G. Ehlers, J. Kärger and C. Krause, Diffusion of n-hexane in 5A zeolite studied by the neutron spin-echo and pulsed-field gradient NMR techniques, Microporous Mesoporous Mater., 2003, 59, 113–121 CrossRef CAS.
- H. Jobic, J. Kärger, C. Krause, S. Brandani, A. Gunadi, A. Methivier, G. Ehlers, B. Farago, W. Haeussler and D. M. Ruthven, Diffusivities of n-alkanes in 5A zeolite measured by neutron spin echo, pulsed-field gradient NMR, and zero length column techniques, Adsorption, 2005, 11(Suppl 1), 403–407 CrossRef.
- H. Jobic and B. Farago, Unidimensional diffusion of long n-alkanes in nanoporous channels, J. Chem. Phys., 2008, 129, 171102 CrossRef PubMed.
- A. J. O'Malley, C. R. A. Catlow, M. Monkenbusch and H. Jobic, Diffusion of isobutane in silicalite: A neutron spin-echo and molecular dynamics simulation study, J. Phys. Chem. C, 2015, 119, 26999–27006 CrossRef.
- H. Jobic, M. Bée and S. Pouget, Diffusion of benzene in ZSM-5 measured by the neutron spin−echo technique, J. Phys. Chem. B, 2000, 104, 7130–7133 CrossRef CAS.
- H. Jobic, A. Méthivier and G. Ehlers, Different diffusivities of xylene isomers in BaX zeolite measured by the neutron spin echo technique, Microporous Mesoporous Mater., 2002, 56, 27–32 CrossRef CAS.
- H. Jobic, H. Ramanan, S. M. Auerbach, M. Tsapatsis and P. Fouquet, Probing cooperative jump-diffusion in zeolites: Neutron spin-echo measurements and molecular dynamics simulations of benzene in NaX, Microporous Mesoporous Mater., 2006, 90, 307–313 CrossRef CAS.
- D. Bhange, C. Dejoie, F. Porcher, N. Malikova, P. Martinetto, E. Dooryhée and M. Anne, Dynamic study of N'N-dimethylparanitroaniline encapsulated in silicalite-1 matrix using neutron spin-echo spectroscopy, Eur. Phys. J.: Spec. Top., 2010, 189, 279–284 CAS.
- A. N. Fitch, H. Jobic and A. Renouprez, Localization of benzene in sodium-Y zeolite by powder neutron diffraction, J. Phys. Chem., 1986, 90, 1311–1318 CrossRef CAS.
- M. Sacerdote-Peronnet and B. F. Mentzen, Location of perdeuterated benzene sorbed at low pore-filling in a H·MFI material: A neutron powder diffraction study, Mater. Res. Bull., 1993, 28, 767–774 CrossRef CAS.
- G. Vitale, L. M. Bull, B. M. Powell and A. K. Cheetham, A neutron diffraction study of the acid form of zeolite Y and its complex with benzene, J. Chem. Soc., Chem. Commun., 1995, 2253–2254 RSC.
- B. F. Mentzen, Crystallographic determination of the positions of the monovalent H, Li, Na, K, Rb, and Tl cations in fully dehydrated MFI type zeolites, J. Phys. Chem. C, 2007, 111, 18932–18941 CrossRef CAS.
- C. A. Fyfea, J. S. Joseph, L. Lachlan, M. D. Cranswick and I. Swainson, Powder neutron diffraction determination of the structure of the o-xylene/zeolite ZSM-5 complex, Powder neutron diffraction determination of the structure of the o-xylene/zeolite ZSM-5 complex, Microporous Mesoporous Mater., 2008, 112, 299–307 CrossRef.
- G. Beltrami, A. Martucci, L. Pasti, T. Chenet, M. Ardit, L. Gigli, M. Cescon and E. Suard, L−Lysine amino acid adsorption on zeolite L: a combined synchrotron, x-ray and neutron diffraction study, ChemistryOpen, 2020, 9, 978–982 CrossRef CAS PubMed.
- N. Kardjilov, I. Manke, R. Woracek, A. Hilger and J. Banhart, Advances in neutron imaging, Mater. Today, 2018, 21, 652–672 CrossRef CAS.
- A. Borgschulte, R. Delmelle, R. B. Duarte, A. Heel, P. Boillatc and E. Lehmann, Water distribution in a sorption enhanced methanation reactor by time resolved neutron imaging, Phys. Chem. Chem. Phys., 2016, 18, 17217–17223 RSC.
- J. Terreni, M. Trottmann, R. Delmelle, A. Heel, P. Trtik, E. H. Lehmann and A. Borgschulte, Observing chemical reactions by time-resolved high-resolution neutron imaging, J. Phys. Chem. C, 2018, 122, 23574–23581 CrossRef CAS.
- G. Romanelli, T. Minniti, G. Skoro, M. Krzystyniak, J. Taylor, D. Fornalski and F. Fernandez-Alonso, Visualization of the catalyzed nuclear-spin conversion of molecular hydrogen using energy-selective neutron imaging, J. Phys. Chem. C, 2019, 123, 11745–11751 CrossRef CAS.
- J. Terreni, E. Billeter, O. Sambalova, X. Liu, M. Trottmann, A. Sterzi, H. Geerlings, P. Trtik, A. Kaestner and A. Borgschulte, Hydrogen in methanol catalysts by neutron imaging, Phys. Chem. Chem. Phys., 2020, 22, 22979–22988 RSC.
- M. Nikolic, F. Longo, E. Billeter, A. Cesarini, P. Trtik and A. Borgschulte, Combinatorial neutron imaging methods for hydrogenation catalysts, Phys. Chem. Chem. Phys., 2022, 24, 27394–27405 RSC.
- P. Boillat, E. H. Lehmann, P. Trtik and M. Cochet, Neutron imaging of fuel cells – Recent trends and future prospects, Curr. Opin. Electrochem., 2017, 5, 3–10 CrossRef CAS.
- M. W. Erichsen, S. Svelle and U. Olsbye, The influence of catalyst acid strength on the methanol to hydrocarbons (MTH) reaction, Catal. Today, 2013, 215, 216–223 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |