
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of statistical learning and mechanistic modelling towards mapping the substrate electronic space in a Cu-catalyzed Suzuki–Miyaura coupling†
Francisco A.
Gómez-Mudarra
 ab,
Gabriel
Aullón
ab and
Jesús
Jover
*ab
ab,
Gabriel
Aullón
ab and
Jesús
Jover
*ab
aSecció de Química Inorgànica, Departament de Química Inorgànica i Orgànica, Universitat de Barcelona, Martí i Franquès 1-11, 08028, Barcelona, Spain. E-mail: jjovermo@ub.edu
bInstitut de Química Teòrica i Computacional (IQTC-UB), Universitat de Barcelona, Martí i Franquès 1-11, 08028, Barcelona, Spain
First published on 24th January 2023
Abstract
Cu-catalyzed Suzuki–Miyaura coupling between highly fluorinated aryl boronate esters and aryl iodides has been studied with DFT calculations. The reaction mechanism proposed suggests that the oxidative addition of the aryl iodide onto the copper catalyst is the rate-determining step. Several alternative reactions, in which the initial substrates have been replaced by para- and meta-substituted reactants, have been computed to build a reaction barrier database that accounts for the electronic properties of a wide range of functional groups. Statistical learning techniques, based on multi linear regression (MLR) modeling, allow interpreting the electronic effects of both substrates onto the overall reaction barrier, indicating that both electron poor aryl boronate esters and aryl iodides produce lower reaction barriers. The reaction barrier database and the MLR models can be combined to generate an electronic substrate map that easily enables estimating the reaction outcome for analogous Suzuki–Miyaura reactions between aryl boronate esters and iodobenzenes.
Introduction
The use of homogeneous catalysts based on first-row (or base) transition metals (Mn, Fe, Co, Ni, and Cu) as a replacement for their precious metal analogs (Ru, Rh, Pd, Ir, Pt, Au) in cross coupling reactions has experienced a large increase in recent years.1–3 This replacement obeys both environmental and economic factors; first-row transition metals are usually less toxic,4 cheaper, and more abundant5 than their precious second- and third-row transition metal counterparts, and if handled properly, they can provide equally active catalytic platforms. However, there are some important challenges to be addressed for base metals to be implemented as general catalysts, and these issues arise from their inherent reactivity, which can lead to problems with 1) the stability of the active species in presence of air, 2) the propensity to generate radical species through single-electron transfer processes (seriously affecting the selectivity of the reaction), and 3) the characterization of high-spin paramagnetic species, which requires less traditional techniques. The study of base metal-catalyzed cross-coupling reactions is experiencing a fast development and many classical reactions, usually carried out with precious metals, already showcase entire functional versions that employ base metal homogeneous species as catalysts. For instance, it is now possible to find classical cross-coupling reactions promoted by homogeneous catalysts based on manganese,6–9 iron,10–14 cobalt,15–19 and nickel.20–24 On the other hand, the study of copper-catalyzed homogeneous cross-coupling reactions is one of the most active research areas related to base metal catalysis.25–27 Currently, it is possible to find a recent example for almost any of the classical coupling reactions such as Heck,28 Sonogashira,29 Stille,30 Hiyama,31 and Buchwald–Hartwig.32 The Suzuki–Miyaura reaction, one of the most representative metal-catalyzed cross-couplings, has several versions in which the typical palladium catalysts have been replaced by analogous copper complexes.33–39 Recently, a Cu-catalyzed Suzuki–Miyaura coupling of highly fluorinated aryl boronate esters with aryl iodides has been reported.40 This procedure allows preparing polyfluorobiphenyl products that present a wide variety of potential applications in the fields of medicinal chemistry,41,42 electron-transport materials,43,44 and supramolecular chemistry.45,46 The general experimental setup for this reaction showcases the coupling between 2,3,4,5,6-pentafluorophenylboronic acid pinacol ester (C6F5Bpin) and iodobenzene in the presence of the 1,10-phenanthroline (phen) ligand, the CsF base, and copper(I) iodide, which probably takes the shape of [CuI(phen)] as the initial catalyst (Scheme 1). One of the main interests of our group consists of computationally studying metal-catalyzed fluorination processes to find plausible reaction mechanisms that could allow to better understand the underlying chemistry in these systems.47–52 Herein, we will study the reaction mechanism associated with the copper-catalyzed Suzuki–Miyaura shown in Scheme 1 and, afterward, we will explore the impact of electronic effects of both substrates, i.e., the aryl boronate ester and the aryl iodide, onto the activation barrier of the reaction. This procedure will be carried out by recomputing the catalytic cycle and hence creating a large activation barrier database for a varied range of para- and meta-substituted boronates and iodobenzenes. Finally, we will employ the reaction barrier database to explore the electronic substrate space with statistical learning techniques and to construct a substrate map, which will be able to relate the performance of each catalytic system with the electronic properties of both substrates. | ||
| Scheme 1 Cu-catalyzed Suzuki–Miyaura coupling between C6F5Bpin and iodobenzene, as described in ref. 40. | ||
Computational details
All the structures have been fully optimized in N,N-dimethylformamide (DMF) using the Gaussian1653 suite of programs with the B3LYP54–57 functional. In the geometry optimization process, the 6-31G* basis set58–60 was used to describe all atoms except for Cu and I, for which the Stuttgart double zeta basis set (SDD),61–64 along with the associated ECP to describe the core electrons, was employed. Ultrafine integration grids have been used to ensure satisfactory convergence. This is necessary because some of the species under study present a number of low frequency vibrational modes (<100 cm−1) that contribute significantly to the entropy and must be computed properly. In all cases, the solvation energies in N,N-dimethylformamide were computed with the (IEF-PCM)65,66 continuum dielectric solvation model using the SMD67 radii and non-electrostatic terms. Dispersion effects have been included in the optimization process using the D3 method of Grimme.68 For some species, namely, I6 and RETS (vide infra), additional features have to be included to produce the final geometries; in both cases, the maximum displacement step was set to 0.05 Bohr, and vibrational frequencies were computed during each optimization step. In all cases, frequency calculations are carried out to ensure the nature of stationary points and transition states. This computational scheme is named BS1.Additional single point calculations on the optimized geometries have been employed to obtain improved Gibbs energy values with larger basis sets (scheme BS2). In these calculations, the B3LYP functional was kept and the 6-311+G** all-electron basis set60,69,70 was employed for all the atoms except Cu and I, which were described with the aug-cc-pVTZ-PP basis set.71,72 These single point calculations also include the solvation and dispersion effects. These computational settings have been used previously for studying similar copper-catalyzed reactions.49,50,52
To improve the speed of calculations, the pinacolborane group (Bpin) was simplified to the corresponding [1,3,2]dioxaborolane analog. This approximation was validated by computing the initial reaction steps between PhI and C6F5Bpin with the full and simplified systems, which produced very similar relative Gibbs energies for the formation of intermediate I3 (0.6 and 0.1 kcal mol−1), and for the transmetalation transition state TMTS (8.1 and 7.4 kcal mol−1). Therefore, all the references to the Bpin group throughout the manuscript refer to the [1,3,2]dioxaborolane moiety.
The NBO charges have been computed employing the NBO7 program73 using the BS2 computational scheme.
The computed Gibbs energies were corrected to use a standard state corresponding to species in solution with a standard concentration of 1 M. The final Gibbs energies at a given temperature, based on the rigid rotor/harmonic oscillator approach to statistical mechanics, were obtained using the following formula

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(C°/C1atm), where C° is the standard reference state concentration (1 M), and C1atm is the concentration of an ideal gas under the standard P = 1 atm conditions at a given temperature. Therefore, for an ideal gas at 1 atm and at 130 °C, C1atm = 1/Vm = P/RT = 0.030 M; numerically, this correction term equals 2.80 kcal mol−1 per molecule. The values of EBS2, Hcorr,BS1, TSBS1, and G° at 130 °C can be found in Tables S1–S3.†
ln(C°/C1atm), where C° is the standard reference state concentration (1 M), and C1atm is the concentration of an ideal gas under the standard P = 1 atm conditions at a given temperature. Therefore, for an ideal gas at 1 atm and at 130 °C, C1atm = 1/Vm = P/RT = 0.030 M; numerically, this correction term equals 2.80 kcal mol−1 per molecule. The values of EBS2, Hcorr,BS1, TSBS1, and G° at 130 °C can be found in Tables S1–S3.†
All the statistical analysis procedures—parameter correlations, construction of simple and multilinear regressions, external validation, and X-/Y-scrambling processes—have been performed with Excel.
Results and discussion
Determination of the reaction mechanism
The first stage of this study consists of exploring the reaction shown in Scheme 1 to propose a plausible reaction sequence for the coupling of C6F5Bpin and iodobenzene (Scheme 2, the 3D representation of the species involved can be found in the ESI,† Fig. S1). In the process of finding a suitable mechanism, different reaction pathways have been considered and computed; finally, the pathway displaying the lowest energy requirements has been selected. | ||
Scheme 2 Plausible reaction mechanism for the Cu-catalyzed Suzuki–Miyaura coupling between C6F5Bpin and PhI (top) and its corresponding Gibbs energy profile (bottom); in all cases, the bidentate phen ligand is represented as  . . | ||
Recently, a catalytic system, reported by Lledós and Casares,74 was reported to produce a similar pathway for the reaction between [Cu(bipy)(C6F5)] and aryl iodides, which generates biaryl products. Following this pathway, the reaction starts with the replacement of iodide by fluoride on the initial [CuI(phen)] catalyst (I1) to produce intermediate I2. This reaction stage is exergonic by 4.6 kcal mol−1 and has been previously reported to proceed smoothly in similar reaction conditions;75 hence, the transition state governing this transformation has not been sought. Then, C6F5Bpin comes in and attaches onto the fluoride ligand through the boron atom, delivering intermediate I3, which is 4.7 kcal mol−1 higher in energy than the previous species. Since fluoride is not sterically hindered, this addition is considered to be barrierless. The C6F5 group is transferred from boron to copper through the corresponding transmetalation transition state (TMTS), while fluoride detaches from the metal to form FBpin, which is released into the reaction mixture. This step requires further 7.4 kcal mol−1, indicating that the overall barrier up to this point is 12.0 kcal mol−1. After that, the organometallic complex [Cu(C6F5)(phen)] (I4) is obtained, and the relative Gibbs energy goes down by 23 kcal mol−1. Iodobenzene then reacts with I4 to form an encounter complex (I5), in which the incoming aryl ring exhibits a π-stacking interaction with the C6F5 group; this addition is almost thermoneutral. At this point, the oxidative addition takes place and the C–I bond is cleaved onto the copper; the most favorable transition state (OATS) adopts a square pyramid geometry in which both aryl rings are placed cis to each other, while one of the nitrogen donor atoms of phen occupies the axial coordination site. The oxidative addition step has an energy requirement of 24.2 kcal mol−1. The trans oxidative addition transition state (trans-OATS) was also computed and found to require a slightly higher energy investment (25.4 kcal mol−1). Alternative transition states with different ligand arrangements and trigonal bipyramidal geometries have been computed and found to produce the same cis and trans transition states described above.
Dinuclear oxidative addition transition states were not taken into consideration because of the low concentration of the initial catalytic species.76 After the oxidative addition, intermediate I6 should be formed; however, all the attempts to optimize this formally Cu(III) species with the regular computational settings, including different trigonal bipyramidal and square pyramidal complexes, have led to the formation of the final product (PhC6F5), indicating that the reductive elimination process seems to be barrierless. This behavior has been reported before by Bolm in a computational study of a copper-catalyzed Sonogashira coupling.77 The stabilization of this structure as a local minimum was attempted by adding additional keywords during the geometry optimization procedure (vide supra) but they have not been able to produce the desired intermediate. Then, the angle between both aryl substituents on I6 was frozen at different values and the complexes were optimized. These calculations produce the expected copper(III) intermediate as a local minimum (all vibrational frequencies positive) with a CPh–Cu–CC6F5 angle of 93°; unfortunately, removing the angle constraint in a further reoptimization produces the reductive elimination of PhC6F5. Therefore, we decided to study the potential energy surface in the forward direction from I6 by performing a relaxed potential energy surface scan of the CPh–Cu–CC6F5 angle—starting at 93° with 5° decrements until the angle takes a value of 48°. The relaxed scan indicates that the energy smoothly decreases as the angle takes lower values, with no clear sign of an energy requirement to overcome the reductive elimination barrier, supporting the idea that intermediate I6 is not stable and, whenever formed, automatically evolves into the final products (Fig. S2†). Nevertheless, intermediate I6 could be considered a transient species in the reaction pathway and has been kept as such and further analyzed. This intermediate adopts a square pyramid geometry with one of the N atoms of the phen ligand taking the apical coordination position while both aryl rings are placed cis to each other in the equatorial plane. The relative Gibbs energy of I6 is −3.4 kcal mol−1, well below to that found for the trans-I6 isomer (2.8 kcal mol−1), in which the phenyl and the pentafluorophenyl rings are placed trans to each other in the equatorial plane of the complex. Intermediate I6, which should formally be a copper(III) species, shows a certain ligand field inversion since its occupied frontier orbitals (HOMO, HOMO−1, etc., computed at the BS2 level) have mainly ligand character instead of showcasing the expected doubly occupied non-bonding d-orbitals of the copper center, which appear at lower energies (see ESI† and Fig. S3).78 This particular electronic structure is probably responsible for the unstable nature of intermediate I6, in which a certain oxidation state cannot be assigned to the copper center. Even so, NBO calculations state that the charge of copper increases from 0.68 to 1.07 when I4 is transformed into I6, indicating the oxidation of the metal during the oxidative addition stage. Other alternative pathways were computed to ascertain that the reaction proceeds through the sequence shown in Scheme 2. First, the outer-sphere electron transfer between I4 and PhI to produce the corresponding [Cu(C6F5)(phen)]+ and PhI− species has been computed to require an energy investment of more than 30 kcal mol−1, which rules out the possibility of the reaction proceeding through this pathway. In addition, the reaction sequence from I4 + PhI to I1 + PhC6F5 does not correspond to a σ-complex-assisted metathesis. This can be observed from the IRC analysis79,80 of the oxidative addition transition state, which produces the expected species close to the I6 intermediate (Fig. S4†).
As stated above, I6 is not stable and directly produces the final PhC6F5 product and the initial catalyst I1. However, during the reviewing process of this manuscript, in particular from the relaxed potential energy surface scan from I6, we were able to identify a plausible transition state for the reductive elimination stage (RETS). Although this transition state is not a “traditional” saddle point, we decided to keep it in the overall catalytic cycle for completeness. As expected from the relaxed potential energy surface scan described above, this transition state should be found at a Gibbs energy value slightly below that of I6 (−4.3 kcal mol−1). After RETS, the newly formed [CuI(phen)] (I1) and PhC6F5 fragments do not strongly interact; thus, we consider that no significant intermediate is formed after this transition state. At this point, the reaction goes back to the starting point and a large amount of energy is released (ca. 55 kcal mol−1) due to the C–C bond formation of PhC6F5. The overall energy barrier (ΔG‡DFT from now on), computed as the Gibbs energy difference between I5 and OATS, corresponds to the oxidative addition step and spans over 24.2 kcal mol−1.
Exploration of substrate electronic effects
The computed Gibbs energy profile, in which the oxidative addition is the rate-limiting stage, indicates that an electronic effect should be observed on the overall ΔG‡DFT value when iodobenzene is replaced by substrates bearing different substituents in the para position of the ring (Scheme 3). This possibility has been explored by computing the complete reaction mechanism for six additional para-substituted iodobenzenes (p-R1-C6H4I, where R1 = NO2, CF3, F, Me, OMe, and NMe2) that cover almost all the range of electronic donation properties, with NO2 as the most electron-withdrawing and NMe2 as the most electron-donating substituents. Experimentally, the electronic effect for some of these substituents could not be observed clearly (reactions were reported for different p-R1-C6H4I substrates with R1 = F, CF3, Me, and OMe substituents), probably because the reaction times were too long (18 hours) and all the aryl iodides produced almost quantitative isolated yields.40 However, there are experimental data supporting the electronic effect of the iodobenzene substrate onto the overall performance of the reaction; competition experiments show that the reaction of a 1:1 mixture of p-Me–C6H4I and p-CF3–C6H4I with C6F5Bpin produces a 1:3 product ratio, indicating that the latter substrate, which has an electron withdrawing substituent, generates a more efficient catalytic system. In fact, the authors state that “aryl halides bearing electron-withdrawing groups are more reactive and give better yields compared to aryl halides bearing electron-donating groups”, confirming the electronic effect of the iodobenzene substrate onto the reaction barrier.
 | ||
| Scheme 3 Cu-catalyzed Suzuki–Miyaura coupling between C6F5Bpin and different para-substituted iodobenzenes. | ||
Computationally, the electronic effect of the para substituent can be clearly ascertained by the ΔG‡DFT values, which range between 22.2 and 27.4 kcal mol−1 (Fig. 1), all of them low enough to produce operative reactions under the experimental conditions. In all cases, ΔG‡DFT is computed as the energy difference between OATS and (I4 + p-R1-C6H4I), indicating that these values also include the formation of intermediate I5, which is always slightly higher in energy than the latter. As should be expected in an oxidative rate-determining step, the electron-withdrawing groups produce lower barriers than their electron-rich counterparts. As before, intermediate I6 could not be computed for these reactions because it automatically reductively eliminates to deliver the final product and the initial catalyst (I1); therefore, intermediate I6 and RETS have not been further computed for any of these systems. At this point, we tried to correlate these computed ΔG‡DFT values with different electronic features of the para substituents of the aryl iodides. At first, and since the iodobenzene is accepting the electrons during the oxidative addition stage, we used the electrophilic conceptual density functional theory indices, namely, electrophilicity (ω)81 and electroaccepting power (ω+)82 of the p-R1-C6H4I substrates. However, none of these parameters showed a good correlation with ΔG‡DFT. Then, we turned upon the classical Hammett para substituent constants (σp).83 In this case, the correlation between σp and ΔG‡DFT is better (R2 = 0.905, rmse = 0.53 kcal mol−1, MAE = 0.43 kcal mol−1) but the limited availability of σp values for more complex systems, e.g., the C6F5 ring or doubly substituted aryl rings, prompted us to search for a more general parameter to correlate with the DFT computed energy barriers.
 | ||
| Fig. 1 Computed ΔG‡DFT for reactions between different p-R1-C6H4I species and C6F5Bpin. | ||
Thus, we next explored the correlation of the ΔG‡DFT values with the electric properties of the carbon bound to iodide (Cipso) in p-R1-C6H4I, which is the main reactive point of these substrates toward the oxidative addition. The Cipso electrostatic potential84 shows just a moderate correlation with ΔG‡DFT (R2 = 0.876, rmse = 0.60 kcal mol−1, MAE = 0.54 kcal mol−1). In addition, the Mulliken,85 NBO,73 and CM5 (ref. 86) charges were computed and correlated with the reaction barriers. The Mulliken and NBO charges did not show any relevant correlation; on the other hand, the CM5 charge of Cipso (qC from now on, Table S5†) showed a very good negative correlation with ΔG‡DFT (R2 = 0.946, rmse = 0.40 kcal mol−1, MAE = 0.34 kcal mol−1, Fig. 2), indicating that the substrates displaying more positive qC values, i.e., those bearing electron-withdrawing para-substituents, produce lower reaction barriers. This behavior allows the interpretation of the substrate effect on the reaction barrier; when an electron-withdrawing group is placed at the para position of the iodobenzene, the electron density of the ring is depleted toward that group. In this situation, the ipso carbon has a more positive charge that consequently produces an increased electrophilic character and a higher tendency for getting electrons from copper, thus lowering the oxidative addition transition state. The opposite behavior should be expected when an electron-donating group is placed in the para position of iodobenzene.
 | ||
| Fig. 2 ΔG‡LRvs. ΔG‡DFT for reactions between p-R1-C6H4I and C6F5Bpin, and linear correlation between ΔG‡LRand the CM5 charge of the ipso carbon atom of p-R1-C6H4I species (qC). | ||
Next, we sought whether the electronic effects of the aryl boronate ester could also play a role on the reaction outcome; this was done by replacing the C6F5 ring of the original substrate by para-substituted phenyl rings p-R2-C6H4Bpin (R2 = NO2, CF3, F, H, Me, OMe and NMe2) that cover all the electron donating range, and then recomputing the complete reaction mechanism with iodobenzene as the counterpart (Scheme 4).
 | ||
| Scheme 4 Cu-catalyzed Suzuki–Miyaura coupling between iodobenzene and different para-substituted aryl ester boronates. | ||
Experimentally, the substrate effect of the boronate onto the reaction barrier can be observed in the reaction between bromobenzene and C6F5Bpin, m,m′-(CF3)2C6H3Bpin and m,m′-Me2–C6H3Bpin, which produce yields of 87%, 35%, and traces, respectively. These experiments again suggest that electron-poor aryl boronate esters produce more efficient reactions than their electron-rich counterparts.40 Computationally, a clear electronic dependence on the identity of the R2 substituent was observed for ΔG‡DFT, with electron-withdrawing groups producing lower barriers that increase as the electron donation ability of R2 increases (Fig. 3). The computed ΔG‡DFT values for this series of reactions show a larger span than those from the aryl iodide substrates—from 24.2 kcal mol−1 for C6F5Bpin to 37.0 kcal mol−1 for p-NMe2–C6H4Bpin. This electronic effect arises from how the Gibbs energy surface changes when the p-substituted phenyl ring replaces C6F5 on the aryl boronate ester. As stated above, ΔG‡DFT for the C6F5Bpin + PhI couple is computed as the energy difference between I5 and OATS (for the other aryl iodides is the energy difference between I4 + p-R1-C6H4I and OATS). However, as soon as C6F5Bpin is replaced by para-R2-C6H4Bpin, the Gibbs energy of all the species following the coordination of the aryl boronate ester (i.e., the formation of I3) is much higher and the overall reaction barrier is found between OATS and I2. This means that ΔG‡DFT must be computed as the Gibbs energy difference between (OATS + FBpin) − (I2 + p-R1-C6H4I + p-R2-C6H4Bpin), as shown in Scheme 5. In this way, it seems obvious why the different p-R2-C6H4Bpin substrates produce varying barriers and how the electronic effect of R2 is projected onto ΔG‡DFT.
 | ||
| Fig. 3 Computed ΔG‡DFT for reactions between PhI and different p-R2-C6H4Bpin species. | ||
 | ||
| Scheme 5 Gibbs energy profiles for reactions between PhI and different p-R2-C6H4Bpin species. | ||
With the computed barriers of the eight p-R2-C6H4Bpin substrates at hand, we tried to find a quantitative correlation with the electronic features of the para-R2 substituents. As before, the Hammett para substituent constants (σp)83 provide a quite good correlation (R2 = 0.985, rmse = 0.33 kcal mol−1, MAE = 0.28 kcal mol−1) but the C6F5 ring must be excluded of this procedure since its σp value has not been experimentally determined. Again, we explored the atomic electrostatic potential84 and the Mulliken,85 NBO,73 and CM5 (ref. 86) charges of the boron atom in the p-R2-C6H4Bpin substrates. In this case, selecting the boron atom is favorable for various reasons: 1) boron is the anchoring point of the aryl boronate ester onto I2 prior to transmetalation, 2) boron has a direct effect onto the para-R2 group transfer in TMTS, and 3) the stability of the species formed after TMTS shows a trend with respect to the p-R2-C6H4Bpin to FBpin transformation. As above, the Mulliken and NBO charges show very poor correlation with ΔG‡DFT. On the other hand, the electrostatic potential of the boron atom shows a very good correlation with the computed Gibbs energy barriers: R2 = 0.984, rmse = 0.47 kcal mol−1, and MAE = 0.43 kcal mol−1. As in the case of aryl iodides, the CM5 charge of the boron atom in p-R2-C6H4Bpin (qB from here on, Table S4†) shows also a very good linear relationship with ΔG‡DFT (R2 = 0.983, rmse = 0.49 kcal mol−1, MAE = 0.40 kcal mol−1) and was selected as the descriptor for quantitatively reproducing the overall reaction barrier with a simple linear regression for the studied set of p-R2-C6H4Bpin substrates reacting with iodobenzene (Fig. 4). In principle, both the boron electrostatic potential and the qB parameters could be employed, but the usage of an atomic charge seems to be more chemically intuitive.
 | ||
| Fig. 4 ΔG‡LRvs. ΔG‡DFT for reactions between PhI and p-R2-C6H4Bpin, and linear correlation between ΔG‡LR and the CM5 charge of the boron atom of p-R2-C6H4Bpin species (qB). | ||
The linear relationship described by qB is negative with respect to this parameter, indicating that the substrates with more positive qB values produce lower energy barriers. Since the aryl boronate ester is a Lewis acid, adding an electron withdrawing substituent to the para position of the ring produces a larger qB value and enhances the possibility of coordinating to the fluoride ligand in I2, hence increasing the overall reactivity of the system. The opposite behavior will be expected when an electron-donating group is placed in the para position of the aryl boronate ester.
Statistical learning for reaction barrier estimation
Up to this point, we have observed that the reaction barrier depends on the nature of both the aryl iodide and the aryl boronate ester substrates. In addition, we have stated that the activation barrier can be modeled from the CM5 charges of the key atoms of these substrates by linear regression methods. Therefore, we thought about building a quantitative structure–property relationship (QSPR) multilinear regression (MLR) mathematical model that allows estimating the overall barrier (ΔG‡MLR) for any pair of substrates directly from the computed qC and qB values. This kind of approach has been extensively employed by the group of Sigman in recent years.87–89 The QSPR model will take the form shown is eqn (1)| ΔG‡MLR = a + b1·qC + b2·qB | (1) |
 | ||
| Scheme 6 Cu-catalyzed Suzuki–Miyaura coupling between para-substituted iodobenzenes and para-substituted aryl boronate esters. | ||
We have computed the complete Gibbs energy profiles for 18 additional p-R1-C6H4I/p-R2-C6H4Bpin substrate pairs within three different series (see ESI†): 1) p-NO2–C6H4I with p-R2-C6H4Bpin, 2) p-NMe2–C6H4I with p-R2-C6H4Bpin, and 3) p-R1-C6H4I with p-NMe2–C6H4Bpin (where R1 and R2 = NO2, CF3, F, H, Me, OMe, and NMe2). In principle, these series of substrate pairs contain the maximum variability on the electron-donating properties for R1 and R2. The computed energy profiles show that, in all cases, the highest energy stationary point is the oxidative addition transition state (OATS), and ΔG‡DFT must be computed as the energy difference between (OATS + FBpin) and (I2 + p-R1-C6H4I + p-R2-C6H4Bpin), following the trend shown in Scheme 5. The only exceptions to this behavior are the p-R1-C6H4I/C6F5Bpin pairs, where the overall barrier is found between OATS and I4 + p-R1-C6H4I. At this point, and to provide the database with a higher variability, new para-substituents were included in both p-R1-C6H4I and p-R2-C6H4Bpin substrates: R1, R2 = CN, COMe, Cl, tBu, OH, and NH2. The electron-donating properties of these new substituents are in between those of the substituents employed before and, thus, these groups should produce activation barriers located in the range of 22–39 kcal mol−1, which correspond to the most electron withdrawing (p-NO2–C6H4I/C6F5Bpin) and most electron-donating (p-NMe2–C6H4I/p-NMe2–C6H4Bpin) pairs, respectively. Therefore, ΔG‡DFT was computed for several para-substituted substrate pairs (71) by computing only the species needed, i.e., p-R1-C6H4I, p-R2-C6H4Bpin and their corresponding OATS (I2 and FBpin are also required but have been computed previously). At this point, 103 ΔG‡DFT values have been computed for p-R1-C6H4I/p-R2-C6H4Bpin substrate pairs (Table S6†), and the MLR model can be constructed. Usually, when building QSPR models, the dataset is randomly split into 80% for model selection (training set or tset) and 20% to validate the final model (prediction set or pset). In this case, the training set used for building the MLR model has been constructed to comprise a balanced amount of p-R1 and p-R2 substituents, i.e., each substituent appears 4 times as R1 and R2, except for R1 = NMe2, H, and NO2, which are employed 5 times (Fig. S5†). Thereby, the tset used to build the MLR model includes 56 substrate pairs, which is ca. 50% of the dataset. All the other substrate pairs (47 points in the database) will be used as prediction set to validate the MLR as external predictors (Fig. S5†). The MLR model obtained, along with some of the relevant statistical parameters from both tset and pset, are shown in Table 1; the computed ΔG‡MLR values and the associated residual errors can be found in Table S7.† The cross-correlation coefficient between qC and qB for the reactions within the training set is as low as 0.002.
| MLR equation |
|
||||
| Standard MLR |
|
||||
| tset | pset | ||||
| R 2 | 0.965 | 0.971 | |||
| R loo 2 | 0.964 | — | |||
| rmse (kcal mol−1) | 0.65 | 0.64 | |||
| MAE (kcal mol−1) | 0.54 | 0.50 | |||
The statistical results of the MLR model are very good; for the training set, the correlation coefficient (R2) is 0.965 and the associated errors rmse and MAE are very low, 0.65 and 0.54 kcal mol−1, respectively. Similar values are obtained for the prediction set, showing the robustness and the high prediction capacity of the model. Fig. 5 shows a plot of ΔG‡MLRvs. ΔG‡DFT for all the substrate pairs studied (including training and prediction sets). As in the previous linear regressions, both qC and qB showcase negative signs, indicating that larger charge values would produce lower activation barriers for both substrates. Hence, the physicochemical interpretation for both parameters remains the same as above. Adding an electron withdrawing substituent to the para position of the iodobenzene depletes the electron density of the ipso carbon, causing an increase of qC and a higher tendency for the substrate to engage in the oxidative addition stage, which entails receiving electrons from the copper atom in I4. On the other hand, adding an electron withdrawing group to the para position of the aryl boronate ester produces a stronger Lewis acid, which is more easily activated by I2. In both cases, the presence of electron donating groups produce the opposite effect.
 | ||
| Fig. 5 ΔG‡MLRvs. ΔG‡DFT for the training and prediction sets of different substrate pairs. | ||
The distribution of the predicted residual errors for the 103 barriers is shown in Fig. S6.† As may be observed, 89% of these values display an error lower than 1 kcal mol−1, which is often considered as the chemical accuracy limit. The highest and lowest computed residual errors are 1.40 and −1.35 kcal mol−1, respectively, well below three times the rmse of the MLR. In addition, only five substrate pairs have residual errors larger than twice the rmse (Table S7†), which indicates the goodness of the MLR model obtained. According to the regression coefficients found for the standardized MLR (in which both descriptors have been autoscaled to produce variables with zero mean and unit variance) shown in Table 1, qB is ca. 2.75 times more important than qC in the estimation of the reaction barrier, suggesting that the nature of the aryl boronate ester plays a major role in the expected reactivity of the system. Y-Randomization experiments were carried out to ensure that the good performance of the MLR model was not due to chance effects; the average R2, rmse, and MAE values obtained for ten different scrambling procedures of the computed barriers are 0.022, 4.69 kcal mol−1, and 3.68 kcal−1, respectively, showing that chance correlation did not play any significant role in the resulting statistics. As an additional validation procedure, we have repeated the MLR modeling ten times by randomly varying the splitting of the whole dataset into training and prediction sets. The average statistical parameters obtained after this procedure are very similar to those reported above; tset: R2 = 0.971, rmse = 0.64 kcal mol−1, and MAE = 0.52 kcal mol−1; pset: R2 = 0.963, rmse = 0.65 kcal mol−1, and MAE = 0.43 kcal mol−1, demonstrating again the goodness of the proposed MLR model.
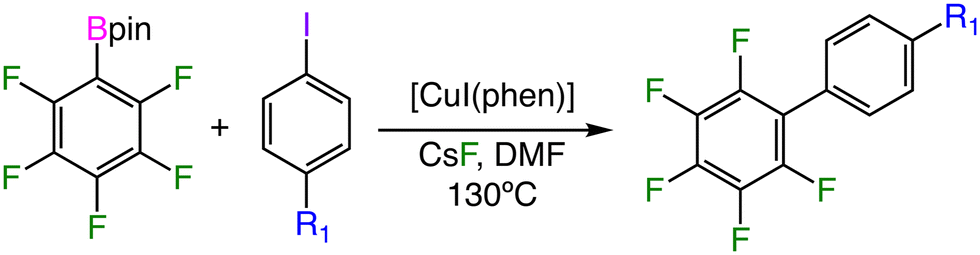
Since the electronic effect of the para-substituent on the computed barrier is transmitted through the qC and qB descriptors, it should be expected that the activation barrier of other substrates, for which the electronic properties are in between those employed to construct the MLR, could also be computed. At this point, we decided to use the MLR model to predict the activation barrier for substrate pairs bearing the substituents included in Fig. S5† in either para- or meta-positions. Thus, we built three different subsets of reactions between m-R1-C6H4I/p-R2-C6H4Bpin (m-R1/p-R2), p-R1-C6H4I/m-R2-C6H4Bpin (p-R1/m-R2), and m-R1-C6H4I/m-R2-C6H4Bpin (m-R1/m-R2) substrates, which contain 17, 16, and 17 substrate pairs respectively. The results obtained for these new external prediction sets are very good and the ΔG‡MLR derived values are quite close to those computed by DFT (Fig. 6 and Table S8†). As may be observed, the results for the m-R1/p-R2 and p-R1/m-R2 subsets are very close to those obtained for the original training and prediction sets both in terms of R2 coefficients and error values. The correlation between ΔG‡MLR and ΔG‡DFT shows that the m-R1/p-R2 subset produces both positive and negative errors (points both above and below the perfect fit), while the p-R1/m-R2 substrate pairs mainly produce ΔG‡MLR values lower than their DFT counterparts (points below the diagonal in Fig. 6). This could be attributed to an incipient appearance of steric interactions between the meta substituent of the substrates with other parts of the catalytic system, mainly between both aryl rings in the oxidative addition transition state.
 | ||
| Fig. 6 ΔG‡MLRvs. ΔG‡DFT for reaction between substrate pairs that include para- and meta-substituted substrate pairs, and statistical parameters for these reaction subsets (* in kcal mol−1). | ||
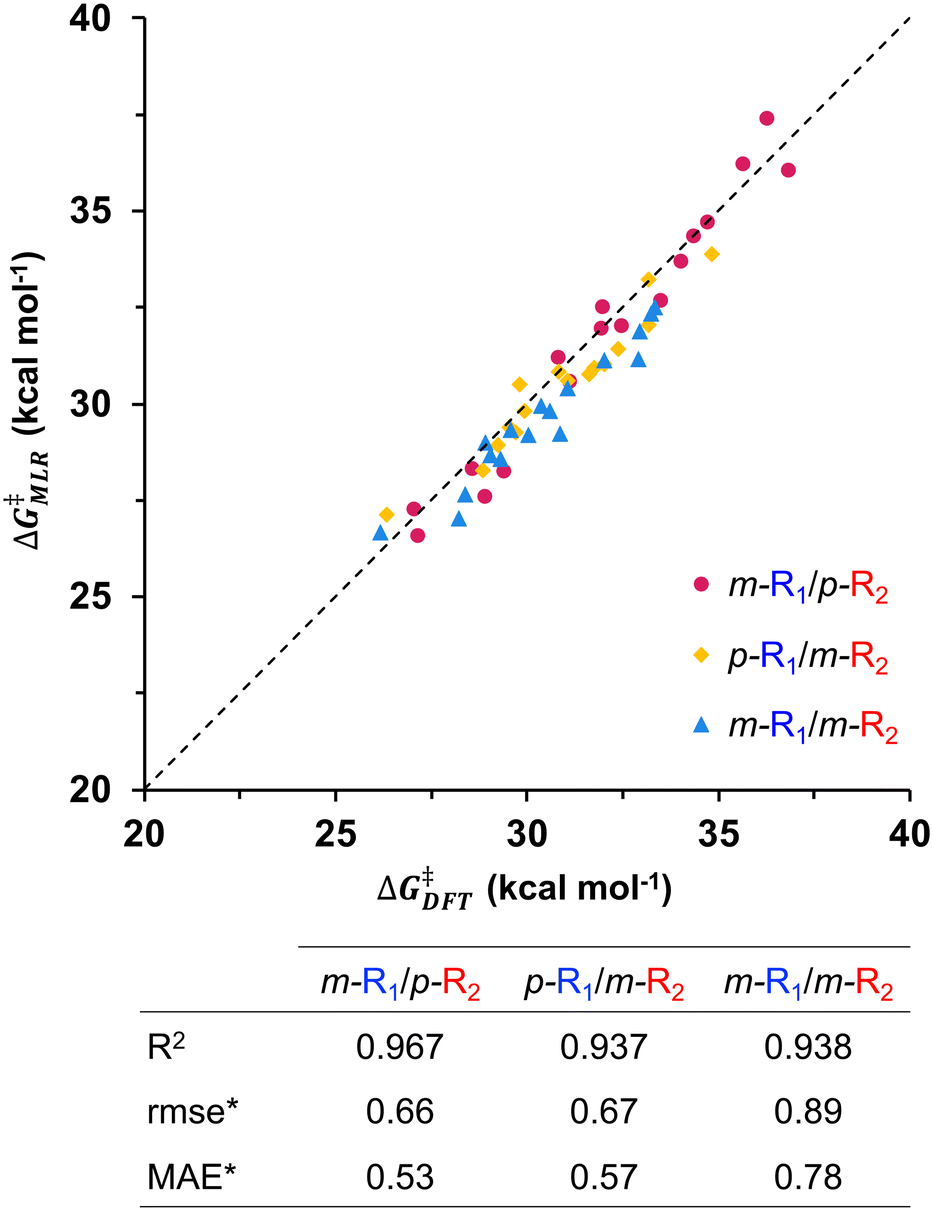
This same behavior is stressed for the m-R1/m-R2 substrate pairs, which display the largest errors, probably because the steric interactions are becoming increasingly more important. Nevertheless, the statistical parameters of this reaction subset remain at a very good level, demonstrating that the MLR developed with only para-substituted substrates can also capture the electronic effects of meta-substituents. The deviation between ΔG‡MLR and ΔG‡DFT can be further observed for reactions where one of the substrates is doubly substituted at the meta-positions, i.e., m,m′-(R1)2-C6H3I or m,m′-(R2)2-C6H3Bpin. We have computed 10 additional systems comprising five m,m′-(R1)2-C6H3I/p-R2-C6H4Bpin (m,m′-(R1)2/p-R2) and five p-R1-C6H4I/m,m′-(R2)2-C6H3Bpin (p-R1/m,m′-(R2)2) substrate pairs and compared their MLR and DFT barriers (Fig. 7). Although the agreement found between the barriers is still quite good, both sets show larger errors and a clear deviation from the perfect fit can be observed (Table S9†), most probably because the electronic effects are starting to be blurred by the incipient appearance of steric interactions of the meta-substituents within the catalytic species. Obviously, ortho-substituted substrates produce even larger barrier differences because the steric hindrance introduced by the substituent is more pronounced; for this reason, substrate pairs including this substitution pattern have not been included in this study.
 | ||
| Fig. 7 ΔG‡MLRvs. ΔG‡DFT for reaction between substrate pairs that include one para- and one doubly substituted meta-substrate pairs, and statistical parameters for these reaction subsets (* in kcal mol−1). | ||
Mapping the substrate electronic space of the reaction
Finally, the computed barrier dataset can be employed to map the electronic chemical space of the substrates for the studied Suzuki–Miyaura reaction. To do this, a new MLR, similar to eqn (2), has been constructed employing all the single para- and meta-substituted iodobenzenes and aryl boronate esters (153 substrate pairs, eqn (4)).| ΔG‡MAP = 166.36 − 68.94qC − 339.86qB | (4) |
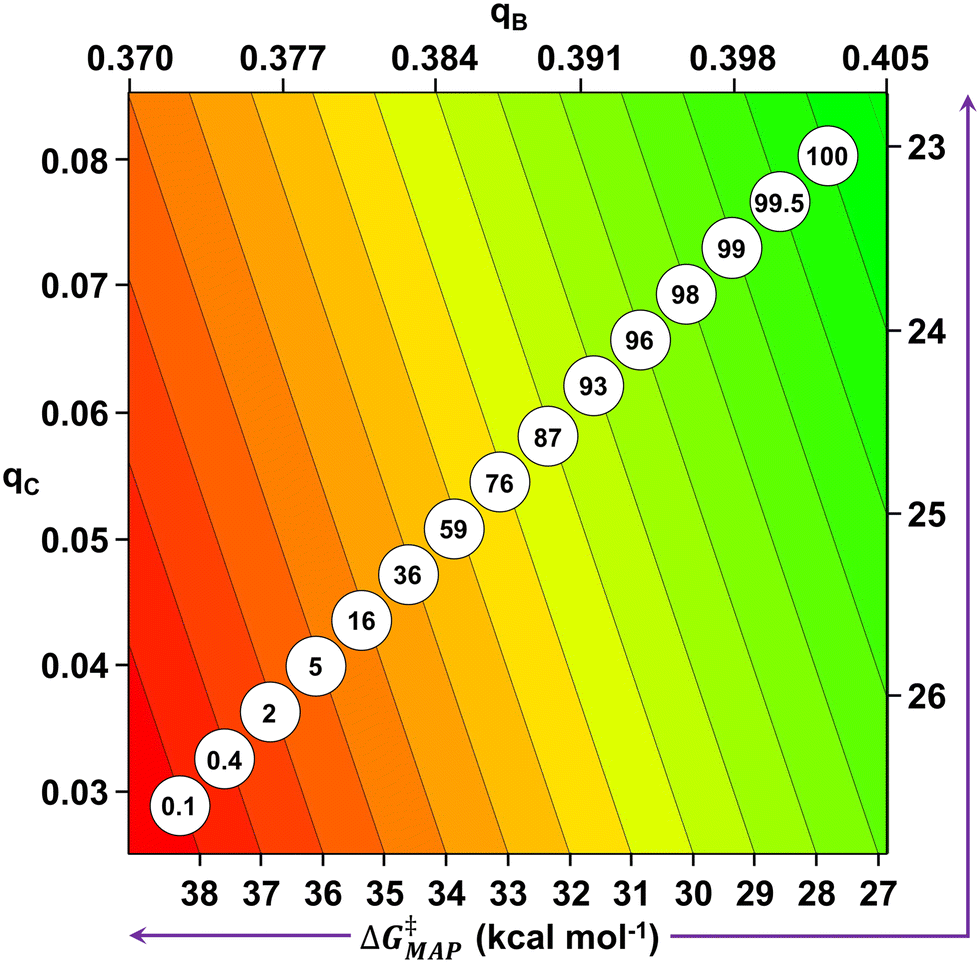 | ||
| Fig. 8 Map of electronic substrate space for the Suzuki–Miyaura reaction between iodobenzenes and aryl boronate esters as a function of qC and qB. The black diagonal lines correspond to the representation of eqn (4) for the ΔG‡MAP values shown in the bottom and right axes. The computed 18-hour yield for each ΔG‡MAP value appears circled onto the corresponding regression line. | ||
Conclusion
A plausible mechanism has been proposed for the copper-catalyzed Suzuki–Miyaura coupling between iodobenzenes (R1-C6H4I) and aryl boronate esters (R2-C6H4Bpin). The reaction seems to follow the typical transmetalation/oxidative addition/reductive elimination sequence for a copper-catalyzed process. The copper(III) intermediate, which should be formed after the oxidative addition, is unstable and automatically evolves into the final product through a low-energy reductive elimination barrier. This intermediate, with formal +3 oxidation state, shows a certain ligand field inversion that can be observed in its occupied frontier orbitals, which mainly display ligand contributions.The reaction mechanism has been recomputed for different substrate pairs; in all cases, the highest energy transition state corresponds to the oxidative addition of the iodobenzene onto the copper(I) organometallic intermediate [Cu(C6F5)(phen)].
A large database of reaction barriers for several substrate pairs (153 different reactions) has been constructed by computational means. These reactions feature a wide variety of para- and meta-substituted iodobenzenes and aryl boronate esters, which cover the full range of electronic properties of both the substrates.
The electronic effects of both substrates on the reaction barrier are captured by the computed CM5 charges of the ipso carbon of the iodobenzene (qC) and the boron atom of the aryl boronate ester (qB), respectively. These parameters can be related with the propensity of both substrates to engage in the catalytic cycle. qC modulates the ability of the aryl iodide to participate in the oxidative addition stage, with larger values producing lower activation barriers. qB indicates the tendency of the aryl boronate ester to behave as Lewis acid and shows that electron-withdrawing substituents generate more reactive substrates and consequently lower activation barriers.
The qC and qB parameters, combined with the reaction barrier database, can be employed to derive multilinear regression (MLR) models that allow computing and predicting the activation energy in reactions between monosubstituted para- and meta-substituted reactants. The MLR equations show very small errors, typically lower than 1 kcal mol−1, in all cases except for those in which one of the substrates has a double meta-substitution.
The MLR model comprising all the computed reactions allows building an electronic substrate map, based on qC and qB, which can be employed to easily allocate the activation barrier and the yield for any pair of substrates used in the studied Suzuki–Miyaura reaction.
Author contributions
FAGM was involved in the investigation and data curation of this manuscript. GA participated in the writing – review & editing procedure. JJ oversaw the conceptualization, formal analysis, visualization, and writing the original and revised version of this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from MICINN (PGC2018-093863-B-C21) and the Spanish Structures of Excellence María de Maeztu program (MDM-2017-0767) is gratefully acknowledged. FAGM thanks the MICINN and the FSE program for the predoctoral grant PRE2019-091164.Notes and references
- P. Chirik and R. Morris, Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS PubMed.
- M. Beller, Introduction: First Row Metals and Catalysis, Chem. Rev., 2019, 119, 2089–2089 CrossRef CAS PubMed.
- S. Shekhar, T. S. Ahmed, A. R. Ickes and M. C. Haibach, Recent Advances in Nonprecious Metal Catalysis, Org. Process Res. Dev., 2022, 26, 14–42 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Which Metals are Green for Catalysis? Comparison of the Toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au Salts, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed.
- Mineral commodity summaries 2022, Reston, VA, 2022.
- M. Rueping and W. Ieawsuwan, A Manganese-Catalyzed Cross-Coupling Reaction, Synlett, 2007, 2, 0247–0250 CrossRef.
- T. C. Atack and S. P. Cook, Manganese-Catalyzed Borylation of Unactivated Alkyl Chlorides, J. Am. Chem. Soc., 2016, 138, 6139–6142 CrossRef CAS PubMed.
- B. E. Petel, M. Purak and E. M. Matson, Manganese-Catalyzed Kumada Cross-Coupling Reactions of Aliphatic Grignard Reagents with N-Heterocyclic Chlorides, Synlett, 2018, 29, 1700–1706 CrossRef CAS.
- K. Das, K. Sarkar and B. Maji, Manganese-Catalyzed Anti-Markovnikov Hydroamination of Allyl Alcohols via Hydrogen-Borrowing Catalysis, ACS Catal., 2021, 11, 7060–7069 CrossRef CAS.
- T. L. Mako and J. A. Byers, Recent advances in iron-catalysed cross coupling reactions and their mechanistic underpinning, Inorg. Chem. Front., 2016, 3, 766–790 RSC.
- H. Xiong, Y. Li, B. Qian, R. Wei, E. V. Van der Eycken and H. Bao, Iron(II)-Catalyzed Heck-Type Coupling of Vinylarenes with Alkyl Iodides, Org. Lett., 2019, 21, 776–779 CrossRef CAS PubMed.
- M. P. Crockett, A. S. Wong, B. Li and J. A. Byers, Rational Design of an Iron-Based Catalyst for Suzuki–Miyaura Cross-Couplings Involving Heteroaromatic Boronic Esters and Tertiary Alkyl Electrophiles, Angew. Chem., Int. Ed., 2020, 59, 5392–5397 CrossRef CAS PubMed.
- J. Zhang, S. Wang, Y. Zhang and Z. Feng, Iron-Catalyzed Cross-Coupling Reactions for the Construction of Carbon-Heteroatom Bonds, Asian J. Org. Chem., 2020, 9, 1519–1531 CrossRef CAS.
- Q. Wang, Q. Sun, Y. Jiang, H. Zhang, L. Yu, C. Tian, G. Chen and M. J. Koh, Iron-catalysed reductive cross-coupling of glycosyl radicals for the stereoselective synthesis of C-glycosides, Nat. Synth., 2022, 1, 235–244 CrossRef.
- S. Asghar, S. B. Tailor, D. Elorriaga and R. B. Bedford, Cobalt-Catalyzed Suzuki Biaryl Coupling of Aryl Halides, Angew. Chem., Int. Ed., 2017, 56, 16367–16370 CrossRef CAS PubMed.
- F. Kreyenschmidt, S. E. Meurer and K. Koszinowski, Mechanisms of Cobalt/Phosphine-Catalyzed Cross-Coupling Reactions, Chem. – Eur. J., 2019, 25, 5912–5921 CrossRef CAS PubMed.
- M.-Z. Lu, X. Ding, C. Shao, Z. Hu, H. Luo, S. Zhi, H. Hu, Y. Kan and T.-P. Loh, Direct Hiyama Cross-Coupling of (Hetero)arylsilanes with C(sp2)–H Bonds Enabled by Cobalt Catalysis, Org. Lett., 2020, 22, 2663–2668 CrossRef CAS PubMed.
- J. R. Ludwig, E. M. Simmons, S. R. Wisniewski and P. J. Chirik, Cobalt-Catalyzed C(sp2)–C(sp3) Suzuki–Miyaura Cross Coupling, Org. Lett., 2021, 23, 625–630 CrossRef CAS PubMed.
- L. R. Mills, D. Gygi, J. R. Ludwig, E. M. Simmons, S. R. Wisniewski, J. Kim and P. J. Chirik, Cobalt-Catalyzed C(sp2)–C(sp3) Suzuki–Miyaura Cross-Coupling Enabled by Well-Defined Precatalysts with L,X-Type Ligands, ACS Catal., 2022, 12, 1905–1918 CrossRef CAS PubMed.
- L. Guo, W. Srimontree, C. Zhu, B. Maity, X. Liu, L. Cavallo and M. Rueping, Nickel-catalyzed Suzuki–Miyaura cross-couplings of aldehydes, Nat. Commun., 2019, 10, 1957 CrossRef PubMed.
- J. E. A. Russell, E. D. Entz, I. M. Joyce and S. R. Neufeldt, Nickel-Catalyzed Stille Cross Coupling of C–O Electrophiles, ACS Catal., 2019, 9, 3304–3310 CrossRef CAS PubMed.
- S. Bhakta and T. Ghosh, Emerging Nickel Catalysis in Heck Reactions: Recent Developments, Adv. Synth. Catal., 2020, 362, 5257–5274 CrossRef CAS.
- Y. Li, Y. Luo, L. Peng, Y. Li, B. Zhao, W. Wang, H. Pang, Y. Deng, R. Bai, Y. Lan and G. Yin, Reaction scope and mechanistic insights of nickel-catalyzed migratory Suzuki–Miyaura cross-coupling, Nat. Commun., 2020, 11, 417 CrossRef CAS PubMed.
- Q. Fan, H. Sun, S. Xie, Y. Dong, X. Li, O. Fuhr and D. Fenske, Nickel-Catalyzed Sonogashira Coupling Reactions of Nonactivated Alkyl Chlorides under Mild Conditions, Organometallics, 2021, 40, 2240–2245 CrossRef CAS.
- S. Thapa, B. Shrestha, S. K. Gurung and R. Giri, Copper-catalysed cross-coupling: an untapped potential, Org. Biomol. Chem., 2015, 13, 4816–4827 RSC.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Selected Copper-Based Reactions for C−N, C−O, C−S, and C−C Bond Formation, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- L.-J. Cheng and N. P. Mankad, C–C and C–X coupling reactions of unactivated alkyl electrophiles using copper catalysis, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC.
- C. Tang, R. Zhang, B. Zhu, J. Fu, Y. Deng, L. Tian, W. Guan and X. Bi, Directed Copper-Catalyzed Intermolecular Heck-Type Reaction of Unactivated Olefins and Alkyl Halides, J. Am. Chem. Soc., 2018, 140, 16929–16935 CrossRef CAS PubMed.
- X. Mo, H. Huang and G. Zhang, Tetrasubstituted Carbon Stereocenters via Copper-Catalyzed Asymmetric Sonogashira Coupling Reactions with Cyclic gem-Dihaloketones and Tertiary α-Carbonyl Bromides, ACS Catal., 2022, 12, 9944–9952 CrossRef CAS.
- V. Lee, Application of copper(i) salt and fluoride promoted Stille coupling reactions in the synthesis of bioactive molecules, Org. Biomol. Chem., 2019, 17, 9095–9123 RSC.
- A. Adenot, L. Anthore-Dalion, E. Nicolas, J.-C. Berthet, P. Thuéry and T. Cantat, A Copper(I)-Catalyzed Sulfonylative Hiyama Cross-Coupling, Chem. – Eur. J., 2021, 27, 18047–18053 CrossRef CAS PubMed.
- S. Roy, M. M. Dutta, M. J. Sarma and P. Phukan, Accelerating Effect of DMAP on CuI Catalyzed Buchwald-Hartwig C-N Coupling: Mechanistic Insight to the Reaction Pathway, ChemistrySelect, 2019, 4, 13094–13098 CrossRef CAS.
- Z.-Q. Zhang, C.-T. Yang, L.-J. Liang, B. Xiao, X. Lu, J.-H. Liu, Y.-Y. Sun, T. B. Marder and Y. Fu, Copper-Catalyzed/Promoted Cross-coupling of gem-Diborylalkanes with Nonactivated Primary Alkyl Halides: An Alternative Route to Alkylboronic Esters, Org. Lett., 2014, 16, 6342–6345 CrossRef CAS PubMed.
- Y.-Y. Sun, J. Yi, X. Lu, Z.-Q. Zhang, B. Xiao and Y. Fu, Cu-Catalyzed Suzuki–Miyaura reactions of primary and secondary benzyl halides with arylboronates, Chem. Commun., 2014, 50, 11060–11062 RSC.
- S. K. Gurung, S. Thapa, B. Shrestha and R. Giri, Copper-catalysed cross-couplings of arylboronate esters with aryl and heteroaryl iodides and bromides, Org. Chem. Front., 2015, 2, 649–653 RSC.
- P. Basnet, S. Thapa, D. A. Dickie and R. Giri, The copper-catalysed Suzuki–Miyaura coupling of alkylboron reagents: disproportionation of anionic (alkyl)(alkoxy)borates to anionic dialkylborates prior to transmetalation, Chem. Commun., 2016, 52, 11072–11075 RSC.
- T. Hergert, B. Varga, A. Thurner, F. Faigl and B. Mátravölgyi, Copper-facilitated Suzuki-Miyaura coupling for the preparation of 1,3-dioxolane-protected 5-arylthiophene-2-carboxaldehydes, Tetrahedron, 2018, 74, 2002–2008 CrossRef CAS.
- S.-P. Jiang, X.-Y. Dong, Q.-S. Gu, L. Ye, Z.-L. Li and X.-Y. Liu, Copper-Catalyzed Enantioconvergent Radical Suzuki–Miyaura C(sp3)–C(sp2) Cross-Coupling, J. Am. Chem. Soc., 2020, 142, 19652–19659 CrossRef CAS PubMed.
- Z. Zhang, B. Górski and D. Leonori, Merging Halogen-Atom Transfer (XAT) and Copper Catalysis for the Modular Suzuki–Miyaura-Type Cross-Coupling of Alkyl Iodides and Organoborons, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed.
- Y. P. Budiman, A. Friedrich, U. Radius and T. B. Marder, Copper-Catalysed Suzuki-Miyaura Cross-Coupling of Highly Fluorinated Aryl Boronate Esters with Aryl Iodides and Bromides and Fluoroarene−Arene π-Stacking Interactions in the Products, ChemCatChem, 2019, 11, 5387–5396 CrossRef CAS.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- C. Zhang, K. Yan, C. Fu, H. Peng, C. J. Hawker and A. K. Whittaker, Biological Utility of Fluorinated Compounds: from Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation, Chem. Rev., 2022, 122, 167–208 CrossRef CAS.
- Y. Sakamoto, T. Suzuki, A. Miura, H. Fujikawa, S. Tokito and Y. Taga, Synthesis, Characterization, and Electron-Transport Property of Perfluorinated Phenylene Dendrimers, J. Am. Chem. Soc., 2000, 122, 1832–1833 CrossRef CAS.
- M. Moral, A. Navarro, A. Garzón-Ruiz and E. M. García-Frutos, Tuning the Crystal Packing and Semiconductor Electronic Properties of 7,7′-Diazaisoindigo by Side-Chain Length and Halogenation, J. Phys. Chem. C, 2019, 123, 153–164 CrossRef CAS.
- H. Yi, M. Albrecht, A. Valkonen and K. Rissanen, Perfluoro-1,1′-biphenyl and perfluoronaphthalene and their derivatives as π-acceptors for anions, New J. Chem., 2015, 39, 746–749 RSC.
- I. A. Rather, S. A. Wagay and R. Ali, Emergence of anion-π interactions: The land of opportunity in supramolecular chemistry and beyond, Coord. Chem. Rev., 2020, 415, 213327 CrossRef CAS.
- J. Jover, Computational insights into nucleophilic copper-catalyzed trifluoromethylation of aryl halides, ACS Catal., 2014, 4, 4389–4397 CrossRef CAS.
- J. Jover, F. M. Miloserdov, J. Benet-Buchholz, V. V. Grushin and F. Maseras, On the feasibility of nickel-catalyzed trifluoromethylation of aryl halides, Organometallics, 2014, 33, 6531–6543 CrossRef CAS.
- J. Jover, Quantitative DFT modeling of product concentration in organometallic reactions: Cu-mediated pentafluoroethylation of benzoic acid chlorides as a case study, Phys. Chem. Chem. Phys., 2017, 19, 29344–29353 RSC.
- J. Jover, Zn(II) Byproduct Enhances the Cu-Catalyzed Cross-Coupling of Bromozinc Difluorophosphonate with Iodobenzoates: A DFT Study, Organometallics, 2018, 37, 327–336 CrossRef CAS.
- J. Jover, Nickel-catalyzed aryl trifluoromethyl sulfide synthesis: A DFT study, Catal. Sci. Technol., 2019, 9, 5962–5970 RSC.
- J. Echeverría and J. Jover, Eur. J. Org. Chem., 2022, e202101243 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular Orbital Methods. 12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. H. Dunning and P. J. Hay, in Methods of Electronic Structure Theory, ed. H. F. Schaefer, Springer US, Boston, MA, 1977, pp. 1–27 Search PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the first row transition elements, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Ab initio energy-adjusted pseudopotentials for elements of groups 13–17, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- J. M. L. Martin and A. Sundermann, Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe, J. Chem. Phys., 2001, 114, 3408–3420 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, Self-Consistent Molecular-Orbital Methods. 25. Supplementary Functions for Gaussian-Basis Sets, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- K. A. Peterson and C. Puzzarini, Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements, Theor. Chem. Acc., 2005, 114, 283–296 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- O. Lozano-Lavilla, P. Gómez-Orellana, A. Lledós and J. A. Casares, Transmetalation Reactions Triggered by Electron Transfer between Organocopper Complexes, Inorg. Chem., 2021, 60, 11633–11639 CrossRef CAS PubMed.
- M. Ohashi, N. Ishida, K. Ando, Y. Hashimoto, A. Shigaki, K. Kikushima and S. Ogoshi, CuI-Catalyzed pentafluoroethylation of Aryl Iodides in the Presence of Tetrafluoroethylene and Cesium Fluoride: Determining the Route to the Key pentafluoroethyl CuI Intermediate, Chem. – Eur. J., 2018, 24, 9794–9798 CrossRef CAS PubMed.
- G. Marcos-Ayuso, A. Lledós and J. A. Casares, Copper(i) activation of C–X bonds: bimolecular vs. unimolecular reaction mechanism, Chem. Commun., 2022, 58, 2718–2721 RSC.
- L.-H. Zou, A. J. Johansson, E. Zuidema and C. Bolm, Mechanistic Insights into Copper-Catalyzed Sonogashira–Hagihara-Type Cross-Coupling Reactions: Sub-Mol% Catalyst Loadings and Ligand Effects, Chem. – Eur. J., 2013, 19, 8144–8152 CrossRef CAS PubMed.
- I. M. DiMucci, J. T. Lukens, S. Chatterjee, K. M. Carsch, C. J. Titus, S. J. Lee, D. Nordlund, T. A. Betley, S. N. MacMillan and K. M. Lancaster, The Myth of d8 Copper(III), J. Am. Chem. Soc., 2019, 141, 18508–18520 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, Accurate reaction paths using a Hessian based predictor–corrector integrator, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, Using Hessian Updating To Increase the Efficiency of a Hessian Based Predictor-Corrector Reaction Path Following Method, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS PubMed.
- R. G. Parr, L. V. Szentpály and S. Liu, Electrophilicity Index, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- J. L. Gázquez, A. Cedillo and A. Vela, Electrodonating and Electroaccepting Powers, J. Phys. Chem. A, 2007, 111, 1966–1970 CrossRef PubMed.
- C. Hansch and A. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley & Sons, New York, 1979 Search PubMed.
- B. G. Johnson, P. M. W. Gill, J. A. Pople and D. J. Fox, Computing molecular electrostatic potentials with the PRISM algorithm, Chem. Phys. Lett., 1993, 206, 239–246 CrossRef CAS.
- R. S. Mulliken, Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef CAS.
- J.-Y. Guo, Y. Minko, C. B. Santiago and M. S. Sigman, Developing Comprehensive Computational Parameter Sets To Describe the Performance of Pyridine-Oxazoline and Related Ligands, ACS Catal., 2017, 7, 4144–4151 CrossRef CAS.
- J. P. Reid, R. S. J. Proctor, M. S. Sigman and R. J. Phipps, Predictive Multivariate Linear Regression Analysis Guides Successful Catalytic Enantioselective Minisci Reactions of Diazines, J. Am. Chem. Soc., 2019, 141, 19178–19185 CrossRef CAS PubMed.
- B. C. Haas, A. E. Goetz, A. Bahamonde, J. C. McWilliams and M. S. Sigman, Predicting relative efficiency of amide bond formation using multivariate linear regression, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2118451119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The computed barriers, energy terms to compute the Gibbs energy profiles and the MLR modeling can be found in the ESI file. All the computed structures (G16 input and output files) have been uploaded to the ioChem-BD database, and can be retrieved in the following link: https://doi.org/10.19061/iochem-bd-1-256. See DOI: https://doi.org/10.1039/d2cy02039d |
| This journal is © The Royal Society of Chemistry 2023 |