
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZeolites catalyze the halogen exchange reaction of alkyl halides†
Paloma
Mingueza-Verdejo
a,
Juan Carlos
Hernández-Garrido
 b,
Alejandro
Vidal-Moya
a,
Judit
Oliver-Meseguer
*a and
Antonio
Leyva-Pérez
*a
b,
Alejandro
Vidal-Moya
a,
Judit
Oliver-Meseguer
*a and
Antonio
Leyva-Pérez
*a
aInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, 46022, València, Spain. E-mail: joliverm@itq.upv.es; anleyva@itq.upv.es
bDepartamento de Ciencia de los Materiales e Ingeniería Metalúrgica y Química Inorgánica, Facultad de Ciencias, Universidad de Cádiz, Campus Universitario Puerto Real, 11510 Puerto Real, Cádiz, Spain
First published on 7th February 2023
Abstract
Simple aluminosilicates, including NaX and NaY zeolites, catalyze the selective halogen exchange reaction between two different alkyl organohalides, either in batch or in flow, without any additive or solvent, by simply heating at 130 °C and with <5 wt% of solid catalyst. The reaction protocol tolerates different functional groups and gives two new organohalides in good yields. Mechanistic studies unveil the key role of the zeolite oxygen atoms and countercations, to trigger the heterolytic scission of the R–X bond and generate intermediate alkoxy and halide species, respectively, which recombine within the zeolite framework. These results open a new way to synthesize organohalides with simple solid catalysts and expand the use of aluminosilicates in organic synthesis.
1 Introduction
Organic halides (R–X, R = alkyl or aryl; X = Cl, Br, I) are fundamental molecules in biology, industry and synthetic chemistry.1Fig. 1A shows that the synthesis of organohalides traditionally requires the use of dangerous and highly toxic halogenating agents such as X2 or HX, or expensive derivatives such as N-halosuccinimides (NXS). Thus, it is not surprising that the search for modern catalytic methods to prepare new R–X bonds is a topic of intense research.2Fig. 1B shows that the halogen exchange (halex) reaction (also known as the halogen switch reaction) is an attractive approach to prepare new organohalides since some halides are cheap and available in huge amounts, thus, the synthesis of an extensive gamut of organohalides from a small number of them is, in principle, feasible. It is worth commenting here that the halex reaction is indeed a halogen metathesis reaction or a Finkelstein reaction; however, these two terminologies will not be employed here since they also describe the reaction of an organohalide with an inorganic salt, i.e. AgX or NaI, respectively, and not with another organohalide reactant.3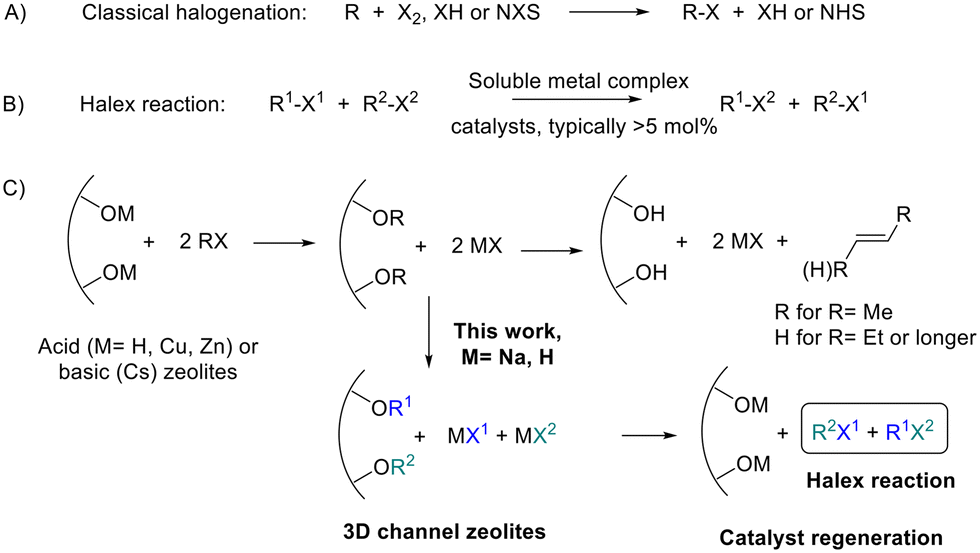 | ||
| Fig. 1 A) Classical halogenation reactions. B) Halogen exchange (halex) reaction. C) Acid or basic zeolite-mediated organohalide dehydro- and dehalogenation reactions, and the synthetic approach proposed here, the halex reaction catalyzed by Na(H)-zeolites. NXS: N-halosuccinimide. | ||
The halex reaction of aromatic halides is well-known to occur with first-row metal catalysts such as copper or nickel;4 however, alkyl halides are more difficult to activate. Different soluble catalysts have been described as representative examples of the latter, including 10 mol% of RhCl(PPh3)3,5 3 mol% of titanocene dihalides combined with trialkyl aluminum,6 and 10 mol% of copper iodide with trans-N,N′-dimethylcyclohexane-1,2-diamine ligands.7 These reaction protocols with unrecoverable metal catalysts are far from the principles of modern sustainable chemistry and, for instance, an efficient solid-catalyst for the halex reaction is difficult to find in the open literature. This lack of recoverable solid catalysts for the halex reaction is perhaps surprising when one considers that different solids are well-documented to activate the R–X bond,8 thus it seems that, under the right experimental conditions, a solid should become catalytically efficient for the halex reaction.
Zeolites are structured microporous aluminosilicates with an electronically defective network, compensated by extra-framework cations bound to the walls oxygen atoms (acid–base pair).9Fig. 1C shows that these acid–base pairs have been reported to break the R–X bond in organohalides and generate the corresponding alkene and HX as reaction products, since the dehydrohalogenation and dehalogenation reactions are favored over the halex reaction in acid or basic zeolites.8a–c,10 Zeolites have been described to activate the R–X bond in stoichiometric amounts, but not catalytically. Fig. 1C shows that, therefore, if the organohalide activation can be stopped at the intermediate alkoxy groups and halides,11 the reaction could then be diverted towards the desired halex reaction with an appropriate zeolite catalyst.
Na faujasites are the primary form of natural and synthetic zeolites, and the more common zeolites in the market, with NaX (Si/Al ratio = 1.2) and NaY (Si/Al = 2.6) as the most prominent examples. The Na zeolite is almost neutral, with less catalytic use than H and Cs faujasites, which are widely used as industrial catalysts. However, all these zeolites have common structural parameters, with tri-dimensional channels of ∼7 Å in diameter which intersect in supercages of ∼12 Å, surrounded and connected to small sodalite cages of ∼4 Å. The Na+ cations present in the sodalite cages of the faujasite have indeed been reported to assist the breaking of the R–X bond,12 after hosting the released halides13 and forming the corresponding alkoxy species.14 Therefore, it seems that simple Na faujasites may be able to catalyze the halex reaction, since they possess channels big enough to diffuse the organohalide molecules to the Na sites nearby the sodalite cages, where the reaction may start. Notice that the sodalite cages cannot catalyze the halex reaction in their own since they are too small to host any organohalide compound or generate alkoxy species, and that only the combination of Na and O atoms in the supercage–sodalite connection of the faujasite, with the stabilizing effect of the sodalite after hosting the released halide, may give an opportunity for the halex reaction to occur. As can be seen in Fig. 1C, the zeolite catalyst might be regenerated after each catalytic cycle provided that dehydro- or dehalogenation reactions are not started, which is intended to be avoided by the neutral nature of the Na zeolite.
2 Results and discussion
2.1 Batch reactions
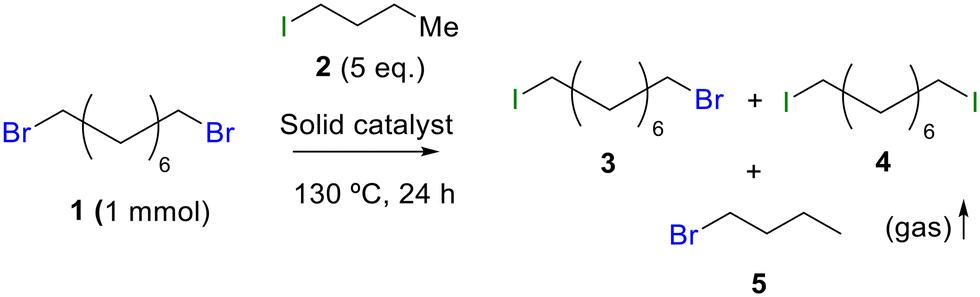
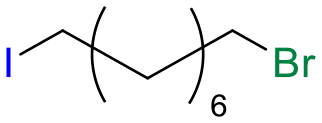
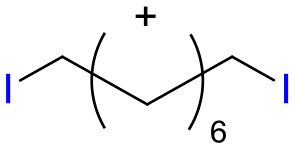
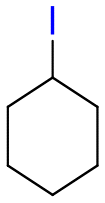
Table 1 shows the results for the halex reaction between dibromoderivative 1 and iododerivative 2 catalysed by 3.5 wt% of different aluminosilicates (10 mg of solid with respect to 1 mmol of dibromide 1, see the ESI† for the preparation of the different aluminosilicate materials tested).‡ A dihalogenated reactant was chosen in order to also follow the formation of mono-exchanged product 3. It can be seen that the reaction does not occur in the absence of a catalyst (entry 1) and that high-surface amorphous silica barely catalyzes the halex reaction (entry 2). However, simple silico-alumina catalyzes the reaction in good yield, but with poor selectivity to 4, even when we exchange H with Na or K in the solid (entries 3–5), with a conversion decrease for sodium cations. In striking contrast, faujasite zeolites Y and X are very good catalysts for the reaction, and the catalytic activity depends on the counterbalancing cation of the zeolite.15 Acid (entries 6 and 7) or basic zeolites (entry 12) give poor to moderate conversions, with most of the product being mono-exchanged product 3. In contrast, nearly neutral Na and K zeolites (entries 8–11) give excellent conversions and good yields of 4 at 130 °C after 24 h reaction time, particularly the NaY zeolite (see Table S1 in the ESI† for further optimization of the reaction conditions). When we used the beta zeolite (entry 13) and the corresponding Na and K ion exchanged zeolites (entries 14 and 15), we observed that the acid one (entry 13) is more active, with similar results to NaY, and the conversion and selectivity decrease when the cation basicity increases. The last zeolite tested was the protonic LTH, with moderate yields (entry 16). This zeolite has a one-dimensional (1D) channel with 12-ring undulating channels.16 Product 4 was easily isolated in >90% yield after filtration of the solid and evaporation of the volatiles. The halide compound 2 in excess can thus be recovered by distillation. Aromatic halides do not react under the present reaction conditions (Table S3†).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | Select. 3 (%) | Select. 4 (%) |
| a 13 wt% Al2O3. | ||||
| 1 | None | 0 | — | — |
| 2 | SiO2 | 28 | 92 | 8 |
| 3a | SiO2–Al2O3 | 93 | 47 | 53 |
| 4 | Na–SiO2–Al2O3 | 50 | 85 | 15 |
| 5 | K–SiO2–Al2O3 | 96 | 37 | 63 |
| 6 | HY zeolite | 14 | 94 | 6 |
| 7 | LiY zeolite | 35 | 89 | 11 |
| 8 | KY zeolite | 86 | 61 | 39 |
| 9 | NaY zeolite | 99 | 15 | 85 |
| 10 | NaX zeolite | 93 | 42 | 58 |
| 11 | KX zeolite | 98 | 23 | 77 |
| 12 | CsX zeolite | 57 | 77 | 23 |
| 13 | H-beta | >99 | 15 | 85 |
| 14 | Na-beta | 98 | 30 | 70 |
| 15 | K-beta | 93 | 44 | 56 |
| 16 | H-LTH | 68 | 79 | 21 |
The halex reaction catalyzed by aluminosilicates can be run in the presence of an oxygen atmosphere or not (Fig. S1†), bench zeolites perform similarly to dehydrated zeolites (Fig. S2†), and a hot-filtration test shows that the reaction is nearly stopped after filtration of the zeolite catalyst (Fig. S3†). These results illustrate the practicability of the aluminosilicate-catalyzed reaction protocol and the possibility of reusing the solid catalyst after reaction. In accordance, Table 2 shows that the NaY zeolite can be reused up to five times with just a little depletion in the final conversion, recovering the solid catalyst by simple gravity filtration, washing with hexane after each use and drying at 80 °C under vacuum. The ICP analysis of the liquid phase after use shows that only 0.66 ppm of Na was found in the reaction solution (0.03% of initial Na in the zeolite), it was not possible to perform ICP analysis on the solid since it could not be disaggregated under standard acid treatments. These results showcase the stability of the zeolite catalyst throughout the reuses.
| Use | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Conversion (%) | 98 | 97 | 97 | 94 | 82 |
| Selectivity to 3 (%) | 57 | 62 | 57 | 60 | 55 |
| Selectivity to 4 (%) | 43 | 38 | 43 | 40 | 45 |
2.2 In-flow reactions
In-flow processes are preferred in both academic laboratories and industry, particularly with solid catalysts, due to the higher throughput and easier monitoring of a liquid or gas stream passed through the insoluble solid catalyst, rather than separation–washing operation.17 In-flow halex reactions in the gas phase were early attempted in the literature, but with soluble catalysts and limited scope, since most of the organohalides are liquid in a wide range of temperatures.17e,fThus, aluminosilicates open a new opportunity to retake the in-flow halex processes. Fig. 2 shows the results for the in-flow reaction between 1 and 2 catalyzed by the NaY zeolite, using a fixed-bed tubular reactor at 0.1 mL min−1 flow rate and 130 °C reaction temperature.§ The zeolite catalyst was pelletized between 0.4 and 0.8 μm in order to have a smooth stream pass. The flow rate was adjusted to have 90% instead of 100% conversion of 1, in order to assess not only a decrease but also an increase of the reaction rate. The results show that the starting 90% conversion is kept within a ±10% margin during at least 6 h reaction time, after ∼1 h stabilization time of the zeolite within the reactor. The lower activity observed during the first 20 minutes of reaction is indeed due to the conditioning time of the catalyst, until the tubular reactor is filled with the reactant solution and we do not have any preferential ways (all the gaps are filled). The space–time yields (STYs) calculated for batch and flow reactors (0.21 and 1.16 mol L−1 h−1, respectively, see the ESI† for calculations) show that the efficiency of the catalytic system is 5 times higher for the in-flow reactor. The higher STY in flow can be explained by the better adjustment of the reaction conditions to maximize catalytic efficiency. The kinetics must be the same after excluding mass and heat transfer limitations. Iodine (I2) was not observed either visually or by UV-vis in the liquid.
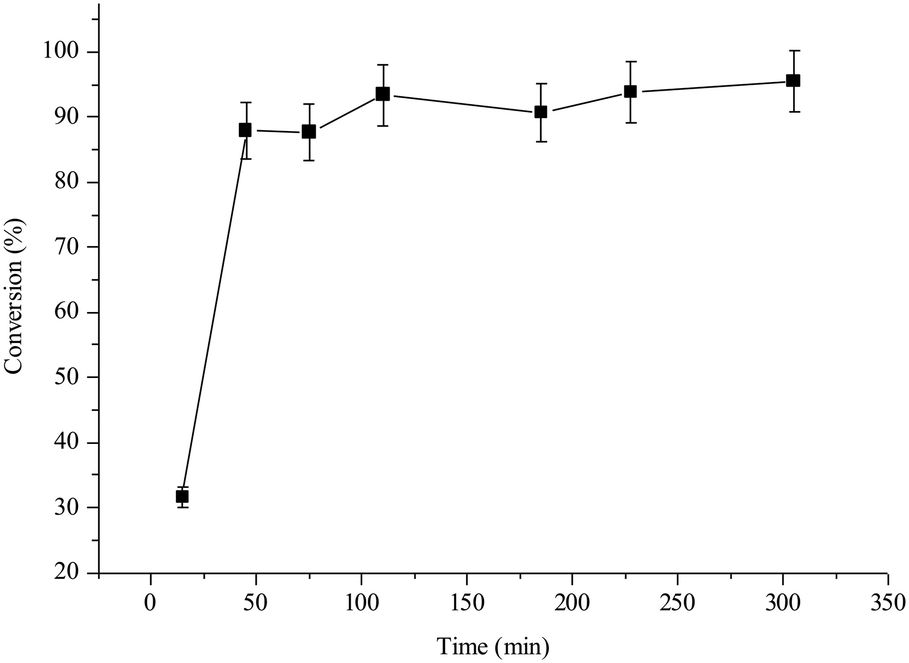 | ||
| Fig. 2 In-flow halex reaction of 1 and 2 with 10 mg of zeolite NaY (pelletized between 0.4 and 0.8 μm) in a fixed-bed tubular reactor, after passing a reactant feed flow rate of 0.1 mL min−1 at 130 °C. Error bars account for 5% uncertainty. | ||
2.3 Substrate scope
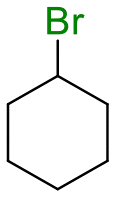
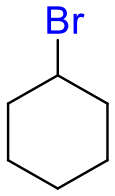
The group tolerance of the reaction was firstly studied by performing a robustness screening test in the presence of different molecules (Table S2†),18 and the results showed that the halex reaction catalyzed by the Na zeolite proceeds well with a variety of functional groups. Thus, the halex reaction was carried out with organohalides bearing different functionalities, and the results in Table 3 show that moderate to good yields of the corresponding products can be obtained. In particular, lactones (entry 5), carboxylic acids (entries 6 and 7), alkynes (entry 8), acetyl chlorides (entry 11) and benzyl groups (entry 12) are tolerated. Besides, not only linear primary but also secondary and tertiary alkyl halides engage well in the reaction. However, for cyclic alkyl halides, the conversions obtained are very low (entries 2, 4 and 5).|
|
|||||
|---|---|---|---|---|---|
| Run | Reagents | Time (h) | Product | Yield (%) | |
| 1 | 1 | 2 (5 equiv.) | 24 |
 3
3
|
93 |
 4
4
|
|||||
 5
5
|
|||||
| 2 |
 6
6
|
 7 (10 equiv.)
7 (10 equiv.) |
24 |
 8
8
|
27 |
| 3 |
 8
8
|
 9 (10 equiv.)
9 (10 equiv.) |
72 |
 6
6
|
51 |
| 4 |
 6
6
|
 10 (10 equiv.)
10 (10 equiv.) |
72 |
 11
11
|
16 |
| 5 |
 12
12
|
2 (10 equiv.) | 48 |
 13
13
|
15 |
| 6 |
 14
14
|
2 (10 equiv.) | 48 |
 15
15
|
80 |
| 7 |
 16
16
|
1 (10 equiv.) | 24 |
 17
17
|
>99 |
| 8 |
 18
18
|
1 (10 equiv.) | 72 |
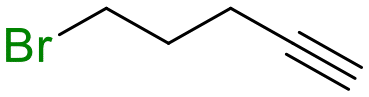 19
19
|
47 |
| 9 |
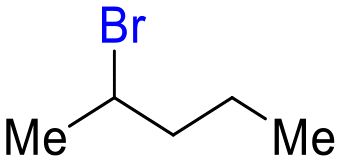 20
20
|
6 (10 equiv.) | 72 |
 21
21
|
69 |
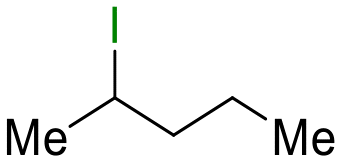
| 10 |
 22
22
|
2 (10 equiv.) | 24 |
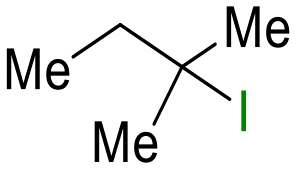 23
23
|
78 |
| 11 |
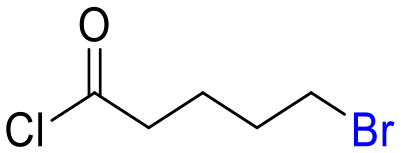 24
24
|
2 (10 equiv.) | 48 |
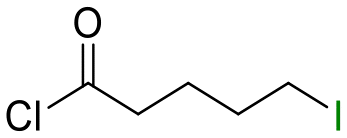 25
25
|
80 |
| 12 |
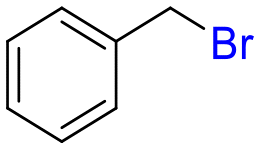 26
26
|
2 (10 equiv.) | 24 |
 27
27
|
>99 |

2.4 Nature of the catalytic sites
The nature of the zeolite catalytic sites was then studied. Fig. 3A shows that Na mordenite, a 1D Na zeolite with large dimensions such as Na faujasites but devoid of any sodalite cage, was nearly completely inactive for the reaction. It is true that Na mordenite has slightly small channels compared to NaY (5–6 vs. 7 Å, respectively) and that the framework is not exactly the same; however, the mordenite dimensions, with 1D channels, are enough to diffuse the reactants and products of the halex reaction tested, as the 3-D channels of NaY zeolite do. Besides, if NaI is impregnated in faujasite LiY (low active for the reaction, see entry 5 in Table 1), the halex reaction does not improve significantly (see also Fig. S4†). A control experiment with NaI as a reactant, in stoichiometric amounts and under optimized reaction conditions, confirms the inactivity of the free salt, obtaining a conversion below 10% (Fig. S5†).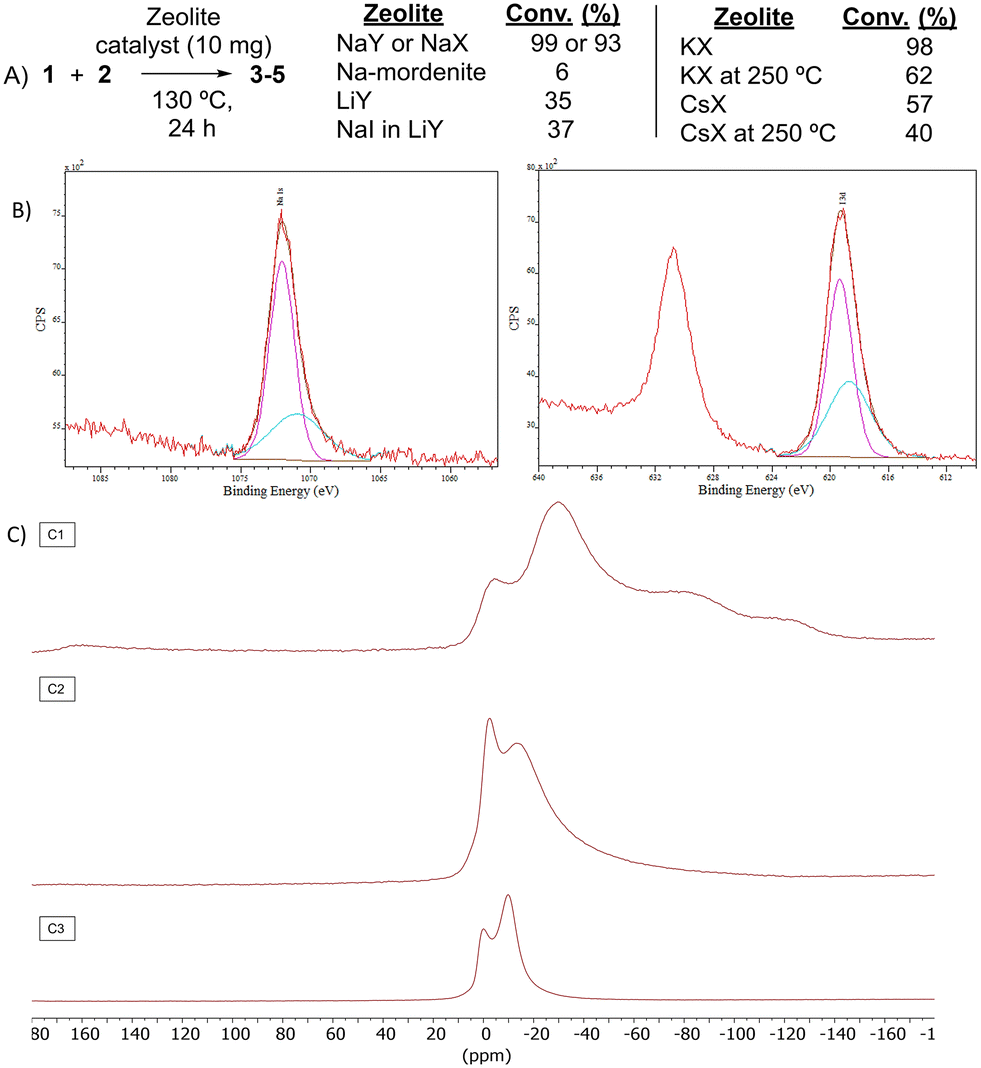 | ||
| Fig. 3 Study of the zeolite catalytic sites in the halex reaction. A) Catalytic results with different zeolites. B) Deconvoluted Na 1s (left) and I 3d (right) X-ray photoelectron spectrometry (XPS) of the NaX zeolite after reaction with iodobutane 2 at 130 °C. The blue area shows the deconvoluted part of the spectra. C) 23Na NMR spectra of the dehydrated NaX zeolite at 300 °C (C1), after adsorbing BuI (C2) and applying reaction conditions (C3). | ||
These results support the role of the Na+ cations in the faujasite sodalite cages during the halex reaction. Indeed, the Na+ cations in sodalite cages are the most difficult to exchange by other cations, thus, the catalytic activity of Li+, K+ and Cs+ exchanged zeolites could came from the residual Na+ cations in sodalite cages. This hypothesis is supported by the fact that Li+ exchanged faujasite still shows some catalytic activity despite being an acid and not a basic or neutral material (see Table 1). Indeed, Table 4 shows that NaY has twice the initial rate of NaX despite having half of the total Na+ cations. This enhanced catalytic activity for NaY with respect to NaX can be explained by the relatively higher wt% amount of Na+ sodalite cations with respect to the total Na+ cations in the zeolite, since most of the Na+ cations of NaX are localized in the supercage.19 Since Na-mordenite has a total Na value of 8 and Na–X has a total Na value of 88, we repeated the reaction with Na-mordenite but adding ten times more of zeolite; however, the result was still very poor (17% conversion).
To further confirm the catalytic role of the cations associated with sodalite cages in faujasites NaY and NaX, zeolites KX and CsX were treated at 250 °C under vacuum in order to force the exchange between sodalite Na+ and bulkier K+ and Cs+ cations.20
This more aggressive exchange procedure allows the bigger K+ and Cs+ cations to come into the sodalite cages and move some Na+ cations to the supercages and channels. Fig. 3A shows that these zeolites exchanged with K+ and Cs+ lost significant catalytic activity for the halex reaction, which strongly supports the key role of Na+ cations associated with sodalite cages during the catalytic halex reaction. Powder X-ray diffraction (PXRD) measurements confirm the integrity of the zeolites after the exchange procedures.
Fig. 3B shows the X-ray photoelectron spectrometry (XPS) analysis of the NaX zeolite after reaction with iodobutane 2 at 130 °C. The splitting of the Na 1s and 2s (Fig. S6†) bands in two peaks after reaction can be clearly seen, in an ∼3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, which correlates well with the corresponding splitting of the I 3d signal. A 2:3 ratio is found in NaY, in accordance with the double amount of Na+ cations associated with the sodalite cages (Fig. S7,† note the half relative intensities compared to NaX since half of the total Na+ cations are present in NaY). Thus, the relative signal areas are coherent with the formation of NaI adducts in the sodalite cages (smaller peaks) and also in the rest of the zeolite sites (higher peaks), for both NaX and NaY. Thermogravimetry (TG) analysis of the Na faujasites reacted with organohalide 2 shows the formation of two different types of NaI adducts along the whole zeolite structure, in an ∼3:1 ratio for NaX (Fig. S8†) and an ∼1.5:1 ratio for NaY (Fig. S9†), with the more tightly bound I atoms (desorbing at higher temperature) in a lower ratio. TG releases volatile products, which indicates that the butyl moiety in 2 is not retained in the zeolite after treatment at 130 °C (see below). The formation of I2 was not observed.21 These results confirm that inorganic iodides are the only form of I in the treated zeolites.
:1 ratio, which correlates well with the corresponding splitting of the I 3d signal. A 2:3 ratio is found in NaY, in accordance with the double amount of Na+ cations associated with the sodalite cages (Fig. S7,† note the half relative intensities compared to NaX since half of the total Na+ cations are present in NaY). Thus, the relative signal areas are coherent with the formation of NaI adducts in the sodalite cages (smaller peaks) and also in the rest of the zeolite sites (higher peaks), for both NaX and NaY. Thermogravimetry (TG) analysis of the Na faujasites reacted with organohalide 2 shows the formation of two different types of NaI adducts along the whole zeolite structure, in an ∼3:1 ratio for NaX (Fig. S8†) and an ∼1.5:1 ratio for NaY (Fig. S9†), with the more tightly bound I atoms (desorbing at higher temperature) in a lower ratio. TG releases volatile products, which indicates that the butyl moiety in 2 is not retained in the zeolite after treatment at 130 °C (see below). The formation of I2 was not observed.21 These results confirm that inorganic iodides are the only form of I in the treated zeolites.
The PXRD measurements of the Na faujasites reacted with different amounts of organohalide 2 (Fig. S10†) show that the intensity of the reflection associated with the extra-sodalite Na+ cations in the entrance of the supercages, with the smaller angle (2θ ∼ 6°), is nearly unaltered at lower amounts of 2 but decreases as the amount of iodide increases, which strongly supports that the sodalite Na+ cations are the first to be iodinated and, after that, the rest of the Na+ cations in the zeolite. The same decrease in the small angle reflection was found for NaX (Fig. S11†). Although low angle diffraction peaks in zeolites are easily influenced by several factors, these results are in good agreement with the higher iodide stabilization13 and slightly more basicity of Na sites in sodalite cages,19 which may trigger the R–I bond breaking.
Fig. 3C also shows the 23Na solid-state magic angle spinning nuclear magnetic resonance (23Na SS-MAS NMR) measurements of zeolite NaX before and after adsorbing and reacting BuI 2. The main signal of the bare zeolite, at ∼−7 ppm, can be assigned to sodalite Na+ cations,22 and it can be seen that the main Na signal after inclusion of 2 in the zeolite appears at −1.5 ppm, which is compatible with the formation of Na+–I− pairs.22 Indeed, this signal is still clearly visible after reaction, which strongly supports the formation of Na+–I− pairs with the Na+ cation in sodalite cages, after the release of the carbonaceous moiety of 2. The corresponding 13C SS-MAS NMR of adsorbed BuI 2 in NaX after reaction shows the disappearance of the butyl fragment from the zeolite cages (Fig. S12†).
At this point, another commercially available large pore 3D zeolite such as H-beta zeolite (Si/Al ∼ 17), with similar channel dimensions to zeolite Y but without sodalite cages, was also tested in the reaction. The result obtained (see entry 13 in Table 1 above) showed a similar catalytic activity to NaY (99% conversion, 85% selectivity towards 4), which indicates that different mechanisms can be operative depending on the zeolite used and the counteraction present. Na-beta zeolite was also prepared and tested showing a very similar conversion but less selectivity towards dihalogenated compound 4 (see entry 14 in Table 1 above) and, following a trend, K-beta zeolite performed the reaction with a bit lower conversion and even lower selectivity (entry 15). These results may reflect a shift towards more acid zeolites in beta with respect to Y, to achieve the optimum electronic density for the catalytic reaction.
In order to visually confirm the presence of I− within the zeolite, aberration corrected-scanning transmission electron microscopy (AC-STEM) measurements of the NaX zeolite reacted with 2 were performed (Fig. S13†). Fig. 4 shows the elemental mapping from chemical analysis by X-ray energy dispersive spectroscopy (EDS), where iodide can be seen to be homogeneously distributed along the zeolite structure (Fig. S14†). By means of AC-HAADF-STEM images coupled with the corresponding integrated differential phase contrast (iDPC-STEM) images, shown in Fig. 4, it is confirmed that iodine is atomically distributed within the zeolite without any evidence of agglomeration. It is worth noting here that the contrast in the iDPC images is roughly proportional to the atomic number Z instead of its square (as it is in HAADF-STEM mode), which drastically improves the capability in the light-element imaging visualization. By comparing both types of images, we can confirm the good atomic dispersion of iodine in the zeolite structure. Thus, according to the sensitivity of the HAADF technique to form images based on the −Z contrast, the homogeneity in contrasts observed, and the high atomic number of iodine compared to the other elements of the zeolite, these images together confirm the homogeneous dispersion of iodides in the zeolite NaX structure.
 | ||
| Fig. 4 Top and middle: Elemental analysis by X-ray energy dispersive spectroscopy (EDS) in aberration-corrected high-angle-annular-dark-field scanning transmission electron microscopy (AC-HAADF-STEM) of a NaX sample after reacting with BuI 2 at 130 °C. Bottom: AC-HAADF-STEM image and the corresponding integrated differential phase contrast (iDPC) image of the zeolite channels. | ||
With all the above results in hand, it can be concluded that the halex reaction catalyzed by faujasite zeolites occurs after formation of Na+–I− pairs, probably with the Na+ cations in sodalite cages. The reaction cannot take place inside the sodalite cage but only the iodine atom can (potentially) come inside this very small cage, and that the reaction must occur in the surroundings of the sodalite cage, which include the supercage and the 12MR ring openings. The catalytic sites for the alumina samples and beta zeolites remain unknown at this point, and they will be studied in due course in order to design, for instance, one-pot halex/acid-catalyzed organic reactions.
2.5 Reaction mechanism in catalytic Na-faujasites
The fate of the butyl moiety in 2 after reaction with NaX, dehydrated under vacuum at 300 °C to remove most of the water adsorbed, was also studied by gas chromatography coupled to mass spectrometry (GC-MS). Fig. 5A shows that the only product observed in the reaction atmosphere is 1-butene, corresponding to the dehydrohalogenation reaction, and any butane or carbon–carbon coupling products are not observed. This result implies that the Na zeolite should be protonated after reaction, with the H atoms released by the butyl group, and Fig. 5B shows the Fourier-transform infrared (FT-IR) spectrum of the Na zeolite after reaction, where the formation of additional silanol groups can be easily assessed by the appearance of a new broad band at ∼3400 cm−1. These results support heterolytic scission of the R–I bond, with the butyl group remaining as a zeolite-stabilized carbocation (zeolite-O−+Bu)23 to rapidly generate the expected alkene product.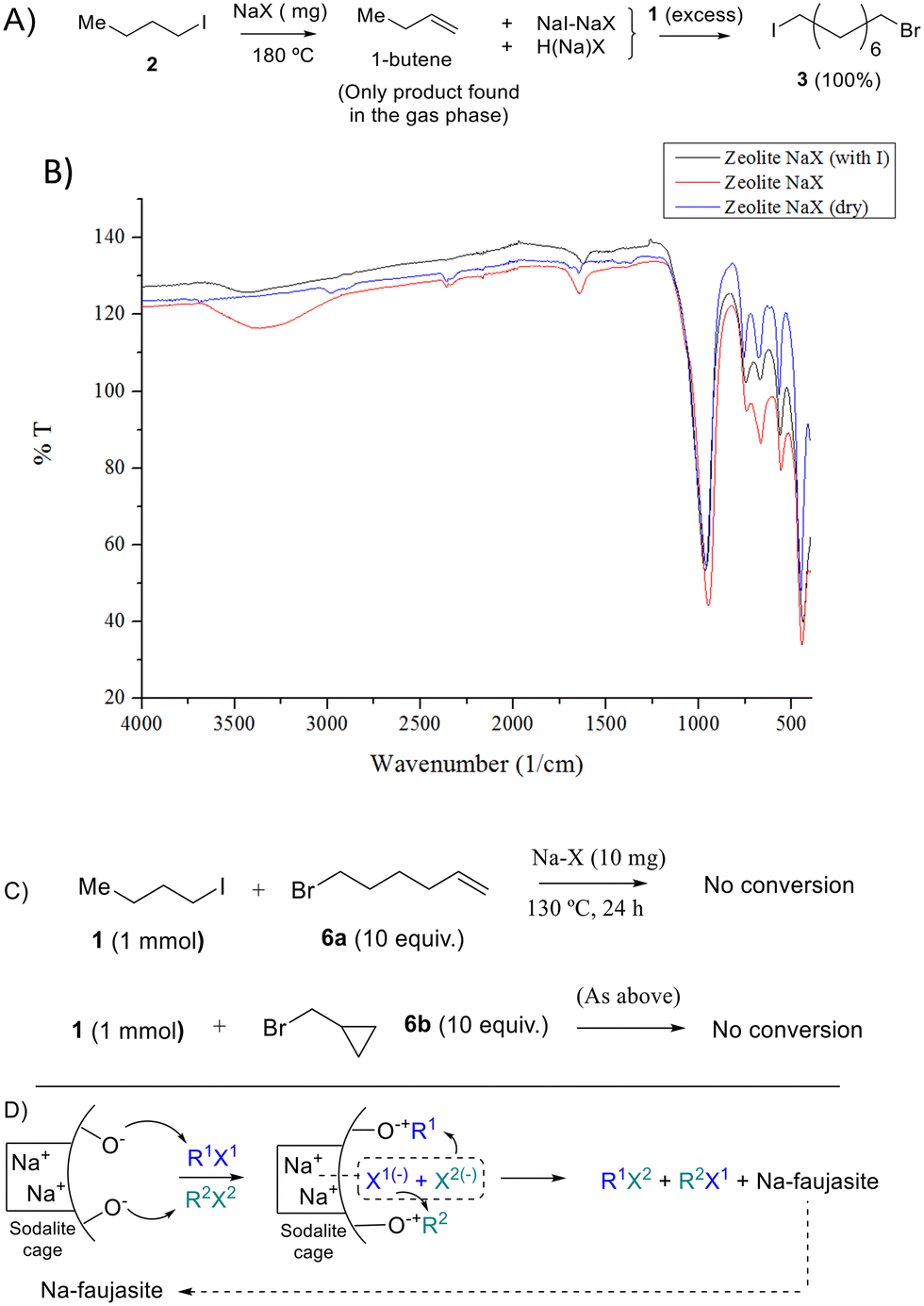 | ||
| Fig. 5 Evidence and proposed reaction mechanism for the Na faujasite-catalyzed halex reaction. A) Assessment of the products in the reaction of 2 with NaX, previously dehydrated under vacuum at 300 °C. B) Corresponding Fourier-transform infrared (FT-IR) spectrum of NaX after reaction. C) Radical clock experiments. D) Proposed reaction mechanism. | ||
Fig. 5A also shows that the intermediate NaX zeolite after reaction with 2, containing the NaI adducts, reacts quantitatively with dibromide 1 to give the halex product 3, which strongly supports that the first step of the halex reaction is the observed breaking of the R–X bond, i.e. in 2, and that the so-formed NaX adducts and zeolite-OR species are truly intermediates of the halex reaction. The intermediate NaI–NaX solid is stable to hexane washing and vacuum dryness, which illustrates the robustness of the NaI zeolite adduct. Of course, the formation of alkenes, HX and silanol groups does not occur when a second organohalide is present in the reaction to exchange with the corresponding halide, since the halide exchange process is favoured. Thus, at this point, we studied the nature of the R–X bond breaking within the zeolite. The lack of I2 formation during reaction indicates that halogen radicals are not formed;21 however, since radical cations are easily stabilized within zeolites as carbocations,24 radical clock molecules were used to completely discard a radical mechanism. Fig. 5C shows that probe molecules 6a and b do not react under optimized reaction conditions, nor the double bond in 6a is halogenated, which discards the formation of halogen radicals.25 Competitive experiments with primary, secondary and tertiary alkyl halides show that the order of reactivity is basically that expected for a nucleophilic substitution (iodide > bromide ≫ chloride compounds, primary > secondary > tertiary compounds) but not exactly the same (Fig. S15,† primary and secondary iodide and bromide > chloride and tertiary compounds), which supports the heterolytic scission of the R–X bond by the nucleophilic oxygen atoms on the internal zeolite surface. Calculation of the reaction enthalpy (ΔH≠) and entropy (ΔS≠) values in the transition state of the NaY-catalyzed halex reaction, using initial rates at different temperatures and the approximation of the Eyring–Polanyi equation (Fig. S16†), gives ΔH≠ = 79.8 kJ mol−1 and ΔS≠ = −20.7 J K−1, respectively. The entropic contribution in the transition state to the reaction feasibility confirms the positive effect of the molecular confinement provided by the zeolite, which significantly decreases the energy required to overcome the transition state of the reaction.26 Not only the zeolite structure but also its ionic strength can play a role during reaction,27 since the ionic pair O−+Na is able to manage the different chemical events of the halex reaction,15a,28 regardless of the presence of water or any solvent.29
Fig. 5D shows the proposed mechanism for the Na faujasite-catalyzed reaction, where two different R1X1 and R2X2 bonds are heterolytically broken and the corresponding carbocation and halide fragments are stabilized by the ionic environment of the faujasite, mainly by the sodalite cages, then rearranging to the new organohalide products R1X2 and R2X1. The order of activity is typical for a bimolecular nuclear substitution (SN2) but the heterolytic R–X bond cleavage induced by the zeolite could stabilize the carbocation (typical for a monomolecular nuclear substitution, SN1, mechanism). Moreover, the cyclic compounds do not work well, which favours the SN1 mechanism. It seems that a balance electronics/sterics is in play here, which makes sense considering the confined nature of the catalytic site. As an initial conclusion, we can suggest that the confined nature of the zeolites' catalytic sites inclines to the mechanism in favour of the SN1 mechanism (electronics) but apparently it would seem that an SN2 mechanism is in play because tertiary substrates are very impeded to react.
Conclusions
Commercially available, simple aluminosilicates such as Na faujasite (NaX and NaY) and H-beta zeolites catalyze the halogen exchange reaction between two different R–X molecules if R = alkyl, but not if R = aryl. The reaction protocol may be amenable for high-scale implementation since it does not require additives or solvents, employs just 3.5 wt% of solid catalyst either in batch or in flow, and proceeds to give two new organohalogenated products in good yields with different boiling points, thus distillable. The NaY zeolite catalyst can be reused up to five times in batch or used in flow for 6 h without apparent degradation, to give >800 product-to-catalyst (wt%) productivity. Mechanistic studies unveil the key role of the zeolite oxygen atoms and sodium sodalite cages in Na faujasites, to trigger the heterolytic scission of the R–X bond and generate intermediate alkoxy and halide species, respectively, within the zeolite framework. The mechanism for alumina and beta zeolite catalysts remains unknown and should be studied. Non-structured aluminosilicates are also active. These results show a route to exchange organic halides without the intervention of radicals30 and open new avenues for the use of (sodium) zeolites as catalysts for organic reactions.Author contributions
P. M.-V. carried out and interpreted the experimental part. J. C. H.-G. performed and interpreted the AC-STEM characterization. A. V.-M. carried out and participated in the interpretation of the SS-MAS NMR analyses. J. O.-M. supervised the work and wrote the manuscript. A. L.-P. conceived the idea, supervised the whole project and wrote the manuscript. All authors have contributed and agreed with the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of the Spanish projects PID2020-115100GB-I00 and PID2019-107578GA-I00, the Juan de la Cierva Program (contract number IJC2018-036514-I) and the Severo Ochoa Centre of Excellence Program CEX2021-001230-S (funded by MCIINN except for the former, funded by MCIN/AEI/10.13039/501100011033MICIIN) is acknowledged. Financial support of the Generalitat Valenciana, Grupos Emergentes Program (GV/2021/138) is also acknowledged. P.M. thanks ITQ for a contract. The AC-STEM data were obtained at the DME–UCA node of the Spanish Unique Scientific and Technological Infrastructure (ICTS) of Electron Microscopy of Materials ELECMIM.Notes and references
- G. W. Gribble, ARKIVOC, 2018, i, 372–410 Search PubMed.
- (a) Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS PubMed; (b) X. Dong, J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507–514 CrossRef CAS PubMed.
- (a) B. T. Worrell, J. E. Hein and V. V. Fokin, Angew. Chem., Int. Ed., 2012, 51, 11791–11794 CrossRef CAS PubMed; (b) A. Nitelet and G. Evano, Org. Lett., 2016, 18, 1904–1907 CrossRef CAS PubMed; (c) S. H. Yang, C. S. Li and C. H. Cheng, J. Org. Chem., 1987, 52, 691–694 CrossRef CAS.
- For a review, see: (a) T. D. Sheppard, Org. Biomol. Chem., 2009, 7, 1043–1052 RSC; (b) For an example of a first-row metal-mediated halex reaction, see: A. A. Cant, S. Champion, R. Bhalla, S. L. Pimlott and A. Sutherland, Angew. Chem., Int. Ed., 2013, 52, 7829–7832 CrossRef CAS PubMed.
- J. Wang, X. Tong, X. Xie and Z. Zhang, Org. Lett., 2010, 12, 5370–5373 CrossRef CAS PubMed.
- Y. Mizukami, Z. Song and T. Takahashi, Org. Lett., 2015, 17, 5942–5945 CrossRef CAS PubMed.
- A. Nitelet and G. Evano, Org. Lett., 2016, 18, 1904–1907 CrossRef CAS PubMed.
- (a) D. K. Murray, J. W. Chang and J. F. Haw, J. Am. Chem. Soc., 1993, 115, 4732–4741 CrossRef CAS; (b) D. K. Murray, T. Howard, P. W. Goguen, T. R. Krawietz and J. F. Haw, J. Am. Chem. Soc., 1994, 116, 6354–6360 CrossRef CAS; (c) S. Imhaoulène, L. Vivier, M. Guisnet, G. Pérot and M. Gubelmann, Tetrahedron, 1994, 50, 12913–12922 CrossRef; (d) A. Leyva-Perez, D. Combita-Merchan, J. R. Cabrero-Antonino, S. I. Al-Resayes and A. Corma, ACS Catal., 2013, 3, 250–258 CrossRef CAS; (e) M. Kolmer, A.-K. Steiner, I. Izydorczyk, W. Ko, M. Engelund, M. Szymonski, A.-P. Li and K. Amsharov, Science, 2020, 369, 571–575 CrossRef CAS PubMed.
- (a) A. Corma, J. Catal., 2003, 216, 298–312 CrossRef CAS; (b) Á. Berenguer Murcia, ISRN Nanotechnol., 2013, 2013, 257047 Search PubMed.
- (a) C. W. Kanyi, D. C. Doetschman, S.-W. Yang, J. Schulte and B. R. Jones, Microporous Mesoporous Mater., 2008, 108, 103–111 CrossRef CAS; (b) T. Xu, Q. Zhang, H. Song and Y. Wang, J. Catal., 2012, 295, 232–241 CrossRef CAS.
- (a) M. Franco, R. Nilton, G. B. Ferreira, A. C. O. Guerra, W. B. Kover, C. C. Turci and C. J. A. Mota, J. Am. Chem. Soc., 2008, 130, 1592–1600 CrossRef CAS PubMed; (b) L. Li, X.-S. Zhou, G.-D. Li, X.-L. Pan and J.-S. Chen, Angew. Chem., Int. Ed., 2009, 48, 6678–6682 CrossRef CAS PubMed.
- C. W. Kanyi, D. C. Doetschman, J. T. Schulte, K. Yan, R. E. Wilson, B. R. Jones, C. O. Kowenje and S.-W. Yang, Microporous Mesoporous Mater., 2006, 92, 292–299 CrossRef CAS.
- (a) A. Stein, G. A. Ozin and G. D. Stucky, J. Am. Chem. Soc., 1992, 114, 8119–8129 CrossRef CAS; (b) H. An, S. Kweon, S. Park, J. Lee, H.-K. Min and M. B. Park, Nanomaterials, 2020, 10, 2157 CrossRef CAS PubMed.
- W. Wang and M. Hunger, Acc. Chem. Res., 2008, 41, 895–904 CrossRef CAS PubMed.
- (a) P. Rubio-Marqués, M. A. Rivero-Crespo, A. Leyva-Pérez and A. Corma, J. Am. Chem. Soc., 2015, 137, 11832–11837 CrossRef PubMed; (b) F. Garnes-Portolés, R. Greco, J. Oliver-Meseguer, J. Castellanos-Soriano, M. Consuelo Jiménez, M. López-Haro, J. C. Hernández-Garrido, M. Boronat, R. Pérez-Ruiz and A. Leyva-Pérez, Nat. Catal., 2021, 4, 293–303 CrossRef.
- T. Ohsuna, B. Slater, F. Gao, J. Yu, Y. Sakamoto, G. Zhu, O. Terasaki, D. E. W. Vaughan, S. Qiu and C. R. A. Catlow, Chem. – Eur. J., 2004, 10, 5031–5040 CrossRef CAS PubMed.
- (a) S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS PubMed; (b) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC; (c) M. Á. Rivero-Crespo, M. Tejeda-Serrano, H. Pérez-Sánchez, J. P. Cerón-Carrasco and A. Leyva-Pérez, Angew. Chem., Int. Ed., 2020, 59, 3846–3849 CrossRef CAS PubMed; (d) G. Gambacorta, J. S. Sharley and I. R. Baxendale, Beilstein J. Org. Chem., 2021, 17, 1181–1312 CrossRef CAS PubMed; (e) E. Angeletti, P. Tundo, P. Venturello and F. Trotta, Br. Polym. J., 1984, 16, 219–221 CrossRef CAS; (f) P. Tundo, F. Trotta and G. Moraglio, J. Chem. Soc., Perkin Trans. 2, 1988, 1709–1712 RSC.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
- L. Zhang, T. N. Pham, J. Faria and D. E. Resasco, Appl. Catal., A, 2015, 504, 119–129 CrossRef CAS.
- P. Concepción-Heydorn, C. Jia, D. Herein, N. Pfänder, H. G. Karge and F. C. Jentoft, J. Mol. Catal. A: Chem., 2000, 162, 227–246 CrossRef.
- S. Y. Choi, Y. S. Park, S. B. Hong and K. B. Yoon, J. Am. Chem. Soc., 1996, 118, 9377–9386 CrossRef CAS.
- (a) A. Seidel, B. Schimiczek, U. Tracht and B. Boddenberg, Solid State Nucl. Magn. Reson., 1997, 9, 129–141 CrossRef CAS PubMed; (b) E. G. Bloor and R. G. Kidd, Can. J. Chem., 1968, 46, 3425–3430 CrossRef CAS.
- J. R. Cabrero-Antonino, A. Leyva-Perez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 5658–5661 CrossRef CAS PubMed.
- H. García and H. D. Roth, Chem. Rev., 2002, 102, 3947–4008 CrossRef PubMed.
- K. Smith, G. A. El-Hiti, M. E. W. Hammond, D. Bahzad, Z. Li and C. Siquet, J. Chem. Soc., Perkin Trans. 1, 2000, 2745–2752 RSC.
- E. M. Gallego, C. Paris, Á. Cantín, M. Moliner and A. Corma, Chem. Sci., 2019, 10, 8009–8015 RSC.
- (a) N. P. Schepp, W. Monk and F. L. Cozens, J. Am. Chem. Soc., 2004, 126, 1012–1013 CrossRef CAS PubMed; (b) N. Pfriem, P. H. Hintermeier, S. Eckstein, S. Kim, Q. Liu, H. Shi, L. Milakovic, Y. Liu, G. L. Haller, E. Baráth, Y. Liu and J. A. Lercher, Science, 2021, 372, 952–957 CrossRef CAS PubMed; (c) R. Gounder and E. Iglesia, Acc. Chem. Res., 2012, 45, 229–238 CrossRef CAS PubMed.
- H. Lee, Y. N. Choi, D.-W. Lim, M. M. Rahman, Y.-I. Kim, I. H. Cho, H. W. Kang, J.-H. Seo, C. Jeon and K. B. Yoon, Angew. Chem., Int. Ed., 2015, 54, 13080–13084 CrossRef CAS PubMed.
- Y. Liu, E. Baráth, H. Shi, J. Hu, D. M. Camaioni and J. A. Lercher, Nat. Catal., 2018, 1, 141–147 CrossRef CAS.
- (a) R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Eur. J. Org. Chem., 2018, 2769–2806 Search PubMed; (b) M. Tejeda-Serrano, V. Lloret, B. G. Márkus, F. Simon, F. Hauke, A. Hirsch, A. Doménech-Carbó, G. Abellán and A. Leyva-Pérez, ChemCatChem, 2020, 12, 2226–2232 CrossRef CAS PubMed; (c) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed; (d) Z. Zhang, B. Górski and D. Leonori, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental data, Tables S1–S3, Fig. S1–S16, additional references and compound characterization. See DOI: https://doi.org/10.1039/d2cy01933g |
| ‡ Representative reaction procedure for the Na zeolite-catalyzed reaction in batch. Reagents 1 (1 mmol) and 2 (5–10 equiv.) were introduced in a 2 ml vial with a magnetic stirrer and the solid catalyst (10 mg). The vial was sealed with a septum, placed in an oil-bath at 130 °C and allowed to react for 24 h. Aliquots of 25 μL were periodically taken and analysed by GC after diluting the reaction mixture in a vial with 1 mL of DCM containing N-dodecane (0.05 mmol) as an external standard. |
| § Representative reaction procedure for the Na zeolite-catalyzed reaction in flow. 1,8-Dibromooctane 1 (16.2 mmol) and 1-iodobutane 2 (5 equiv.) were placed in a 50.0 mL syringe. The mixture was pumped at atmospheric pressure at a flow rate of 0.1 mL min−1 on top of a stainless-steel tube with a 10 mm internal diameter filled with zeolite NaY previously pelletized (and sieved to a particle size of 0.4–0.8 mm) and glass wool to favour the uniform distribution of the flow through the catalyst bed and to avoid preferential pathways. The reaction took place at 130 °C and the samples were collected by gravity. The samples were analysed by GC using dodecane as an external standard. |
| This journal is © The Royal Society of Chemistry 2023 |