
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insight into low temperature SCR by ceria–manganese mixed oxides incorporated into zeolites†
Nicholas C.
Nelson
 a,
Tahrizi
Andana
a,
Kenneth G.
Rappé
*a and
Yong
Wang
ab
a,
Tahrizi
Andana
a,
Kenneth G.
Rappé
*a and
Yong
Wang
ab
aEnergy & Environment Directorate, Pacific Northwest National Laboratory, Richland, WA 99354, USA. E-mail: ken.rappe@pnnl.gov
bThe Gene and Linda Voiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, WA, USA
First published on 9th January 2023
Abstract
Recent efforts to increase the low-temperature activity of zeolite-based selective catalytic reduction (SCR) catalysts has led to the exploration of hybrid materials comprised of a metal oxide and zeolite phase. However, the role of each component in promoting low temperature activity and their interaction with each other is not well understood. Herein, we attempt to understand the low temperature promotion by synthesizing a series of ceria–manganese mixed oxides introduced to a H-SSZ-13 zeolite via incipient wetness impregnation of the oxide precursors. Our data suggests that the mixed oxide phase provides access to surface fast SCR reaction channels via SCR generation of adsorbed nitrogen dioxide and its derivatives. At low temperature (100–170 °C), where the SCR promotion is greatest, we show that it is unfavorable for the adsorbed nitrogen dioxide (derivatives) to react with ammonia to form ammonium nitrate. This implies that fast SCR pathways remain accessible on the oxide at low temperatures and are not blocked by ammonium nitrate deposits as they are on the pure zeolite component. We hypothesize that this a contributing factor for the observed low temperature SCR promotion. Our results may benefit the current understanding of hybrid SCO–SCR catalysts and lead to further technological development in this area.
Introduction
Ammonia selective catalytic reduction (NH3-SCR) over zeolite-based materials has seen expansive industrial adoption for the abatement of pollutants in diesel engines.1 A perennial challenge in these SCR devices is to maintain (near) stoichiometric conversion of criterion pollutants at ever decreasing temperatures. One strategy to meet this challenge leverages the faster SCR kinetics afforded by increasing the nitrogen dioxide (NO2) to nitric oxide (NO) ratio at the SCR inlet to the point of unity, NO2/NOx = 0.5.2 This strategy has been commercially implemented by introducing an oxidation catalyst (e.g., a DOC) upstream of the SCR device. The DOC increases the NO2/NOx ratio at the SCR inlet through NO oxidation; however, the benefits of this strategy on state-of-the-art zeolite-based systems are limited to temperatures above ca. 200 °C. This temperature limitation arises due to the sluggish NO oxidation kinetics of the DOC and reversible fouling of zeolite-based SCR catalysts caused by the association of adsorbed NO2 and NH3 to form ammonium nitrate (NH4NO3) deposits.2–4A nascent technology to alleviate the pitfalls associated with the DOC-SCR coupled system incorporates a selective catalytic oxidation (SCO) functionality directly onto the SCR catalyst.5 This SCR composite catalyst system mitigates the sluggish NO oxidation kinetics of the DOC by nullifying the energetically demanding desorption of NO2 and mitigates the effects of NO2 reduction by CO and hydrocarbons in the DOC, i.e. increases efficiency.4,5 These SCR composite catalysts are typically comprised of a Cu- or Fe-zeolite combined with a metal oxide phase that is introduced via a metal salt solution or mechanical mixing of the zeolite and preformed oxide.5 It is well established that the efficacy of these hybrid systems depend on the ability of the zeolite and oxide components to transfer reactive species to one another through surface diffusion as opposed to in the gas phase.6–9 However, the atomistic details have not yet been clarified owing to the complex nature of SCR chemistry and the equally complex nature of the composite catalysts.10
As alluded to above, the zeolite and oxide components are not additive in the sense that the oxide component catalyzes NO oxidation to provide the zeolite access to fast SCR channels.8,10 Nonetheless, the NO oxidation activity of the oxide typically runs parallel to the low temperature SCR promotion observed in the composite systems.6,10 This suggests that adsorbed NO2 and/or its derivatives (e.g. HNO2) are key intermediates that give rise to the low temperature promotion. Moreover, the oxidation of NO to NO2, or the formation of the active site that leads to NO oxidation, is widely regarded to be kinetically relevant under standard SCR conditions (NO2/NOx = 0) over commercial zeolite-based catalysts.11,12 Considering these observations, it may be prudent to view the SCR composite system through the lens of NO2-SCR (eqn (1)).
| 2NO2 + 2NH3 = N2 + NH4NO3 + H2O | (1) |
| 2NH3 + 2NO + NH4NO3 = 3N2 + 5H2O | (2) |
Our aim in this study was to identify potential contributing factors that lead to low temperature SCR promotion in SCR composite catalyst systems. To this end, we synthesized a series of hybrid materials by introducing ceria–manganese mixed oxides to H-SSZ-13 zeolite via incipient wetness impregnation of the oxide precursors. Ceria–manganese mixed oxides were chosen owing to their activity for NO oxidation.17 Our data suggests that the role of the ceria–manganese mixed oxide phase is to generate NO2 and its derivatives through NO oxidation. This provides the hybrid material access to surface fast SCR reaction channels that are not readily accessible during standard SCR over commercial Cu-based chabazite materials. However, the oxidation activity of the mixed oxide is not restricted to NO. The low oxidation selectivity results in parasitic NH3 oxidation and limits the SCR activity over the oxide component at and above intermediate temperatures (ca. >170 °C). An apparent role of the zeolite then is to provide a reservoir of accessible NH3 reductant to the oxide phase. In addition to this, the oxide component circumvents NH4NO3 fouling by disfavoring its formation. It is possible that this and the NO oxidation activity are key contributing factors for low temperature promotion over the oxide.
Experimental
Reagents
Cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O) was purchased from Alfa Aesar. Trimethyladamantylammonium hydroxide (TMAda-OH, ∼25 wt%) was purchased from Sachem Inc. Manganese(II) nitrate hydrate (Mn(NO3)2·xH2O), Aluminum(III) hydroxide (Al(OH)3), LUDOX AS-30, Davisil grade 645, pore size 150 Å, 60–100 mesh (SiO2), sodium hydroxide (NaOH),and ammonium nitrate (NH4NO3) were purchased from Sigma Aldrich. For Mn(NO3)2·xH2O, x was assumed equal to zero for calculation purposes.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaOH:SiO2:Al2O3:H2O = 10:10:98:4:2200) was obtained and transferred into a 125 ml Teflon-lined stainless-steel autoclave equipped with magnetic stirrer. The hydrothermal synthesis then proceeded with heating the autoclave in a sand bath at 160 °C for 96 h under continuous stirring (300 rpm). After the synthesis, the white solid was recovered via centrifugation and rinsed with deionized water. This process was repeated three times. The solid was then dried overnight under the flow of N2 at 70 °C and finally calcined in a muffle furnace at 550 °C for 4 h. The parent zeolite was then transformed into the NH4-form (NH4+-SSZ-13) via two-time ion-exchange with NH4NO3 aqueous solution (weight ratio of Na-SSZ-13:NH4NO3:H2O = 5:4:50) carried out at 80 °C for 2 h. The solid was recovered via centrifugation and rinsed with deionized water after each exchange. This recovery process was repeated three times. Finally, the material was calcined in air at 550 °C for 6 h with a 2 °C min−1 heating rate. The Si/Al ratio of 11.8 has been confirmed via ICP-AES.
:NaOH:SiO2:Al2O3:H2O = 10:10:98:4:2200) was obtained and transferred into a 125 ml Teflon-lined stainless-steel autoclave equipped with magnetic stirrer. The hydrothermal synthesis then proceeded with heating the autoclave in a sand bath at 160 °C for 96 h under continuous stirring (300 rpm). After the synthesis, the white solid was recovered via centrifugation and rinsed with deionized water. This process was repeated three times. The solid was then dried overnight under the flow of N2 at 70 °C and finally calcined in a muffle furnace at 550 °C for 4 h. The parent zeolite was then transformed into the NH4-form (NH4+-SSZ-13) via two-time ion-exchange with NH4NO3 aqueous solution (weight ratio of Na-SSZ-13:NH4NO3:H2O = 5:4:50) carried out at 80 °C for 2 h. The solid was recovered via centrifugation and rinsed with deionized water after each exchange. This recovery process was repeated three times. Finally, the material was calcined in air at 550 °C for 6 h with a 2 °C min−1 heating rate. The Si/Al ratio of 11.8 has been confirmed via ICP-AES.
Activity testing
All the catalytic tests were carried out in a fixed-bed reactor comprised of a vertically mounted quartz tube (12.7 mm OD) with a K-type thermocouple placed upstream of the catalytic bed, a PID-controlled tubular furnace, a MKS MultiGas™ 2030 FTIR Continuous Gas Analyzer, and a set of Brooks 5850E Series mass flow controller and Brooks 0254 Series control box. Typically, 25 mg of catalyst (60–80 mesh) and 150 mg of SiO2 (60–100 mesh) were homogeneously mixed and loaded into the reactor. The total reactant flow rate was set at 1.5 L min−1 (space velocity = 4.6 × 106 h−1 for bulk CexMn1−xOy and 1.7 × 106 h−1 for CexMn1−xOy/H-SSZ-13), and both NO oxidation and standard SCR tests were conducted with a heating rate of 10 °C min−1. For NO oxidation, the feed was comprised of 330 ppm NO and 15% O2. For standard SCR, the feed was comprised of 330 ppm NO, 330 ppm NH3, and 15% O2. The NO2 yield is defined below. All temperature programmed decomposition and reactions with NH4NO3 were carried out using the same setup. In a typical experiment, 18 μL of an NH4NO3 aqueous stock solution (0.492 mg μL−1) was impregnated onto ∼60 mg of catalyst. The material was dried in air at room temperature for >24 h prior to temperature programmed experiment.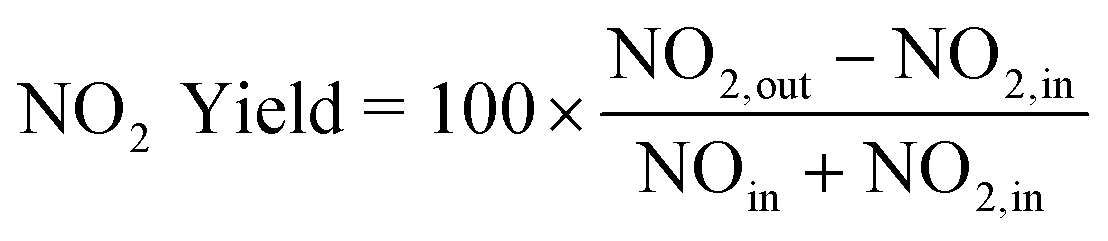
| NOx Conversion = 100* |

| N2 Yield = 100* |

Diffuse Reflectance Infrared Fourier Transform Spectroscopy
In situ DRIFTS analysis was performed on a Nicolet iS50R FTIR spectrometer equipped with an MCT/A detector and Harrick Praying Mantis™ high temperature reaction chamber (HVC-DRM-5) with ZnSe windows. Spectra were recorded with a mirror velocity of 1.8988 cm s−1, aperture setting of 32, spectral resolution of 4 cm−1 with zero-filling of two, and 16 scans per spectrum. Where applicable, the spectra were recorded with a time interval of 10 seconds and are reported in Kubelka-Munk (K-M) units. The gases were delivered to the reaction chamber using a gas manifold equipped with electronic mass flow controllers and two-position-four-port switching valves. Background spectra were collected under He flow at 150 °C. Prior to background acquisition the sample was pretreated to 500 °C under 2% O2/He and held for 15 min, followed by a 45 min purge under He flow at 500 °C. In a typical experiment, the gas feed composition was 330 ppm NO, 330 ppm NH3, and/or 2% O2 with a total flow rate of 100 mL min−1 (balanced by He) vented to atmosphere. Following each test, surface species were removed by repeating the pretreatment procedure.Characterization
Results and discussion
Our group recently studied the promotional role of a cerium-manganese mixed oxide phase incorporated into a Cu-SSZ-13 catalyst for low-temperature ammonia SCR activity.9 The promotional effect of the mixed oxide phase on SCR performance observed in that work depended on the proximity between the Cu zeolite and oxide phases. Incipient wetness impregnation of the mixed oxide onto the Cu-SSZ-13 and ball milling of the preformed mixed oxide and zeolite resulted in greater SCR promotion relative to simple physical mixing of the preformed mixed oxide and zeolite catalyst particles. Additionally, there were further differences in the impregnated and ball milled samples such as susceptibility of NH3 to non-selective (i.e., parasitic) oxidation. Here, we aim to better understand the promotional role of the oxide phase.We prepared a series of catalysts that are analogous to the CexMn1−xOy/Cu-SSZ-13 composite catalyst system. We omitted Cu from our catalyst system to enable a focused study on the role of the mixed oxide phase. This is justified on the basis that the activity of the CexMn1−xOy/Cu-SSZ-13 composite catalyst was the sum of the individual activity of the Cu-SSZ-13 and CexMn1−xOy/H-SSZ-13 phases in the low-to-intermediate temperature (<200 °C) regime; thus, the observed activity promotion observed in this regime resulted from the interaction of the mixed oxide and H-SSZ-13.9 Catalysts were prepared through incipient wetness impregnation of H-SSZ-13 with cerium and manganese nitrate salts with a nominal mixed oxide composition of CexMn1−xOy/H-SSZ-13 where x = 1, 0.7, 0.5.
The impetus for incorporating the mixed oxide phase was to provide an NO oxidation functionality to Cu-SSZ-13. Our hypothesis was that the NO oxidation functionality could enable fast SCR reaction pathways (NO2/NOx = 0.5) under a standard SCR gas feed (NO2/NOx = 0). To test this hypothesis, we measured the NO oxidation activity across the three composite catalysts. Fig. 1a shows that increasing the amount of Mn in the oxide phase of the composite catalyst resulted in an increase in the NO2 yield. The same trend was observed for the NOx conversion (Fig. 1c) and the N2 yield (Fig. S2†) during SCR over the composite catalyst.
 | ||
| Fig. 1 NO oxidation activity over CexMn1−xOy/H-SSZ-13 (a) without and (b) with pre-adsorbed NH3. Reaction conditions: [NO] = 325 ppm; [NO2] = 6 ppm; [O2] = 15%; [N2] = balance; flow rate = 1.5 SLM; GHSV = 1.7e + 6 h−1. c.) SCR activity over CexMn1−xOy/H-SSZ-13. Reaction conditions: same as (a and b) but with [NH3] = 330 ppm. | ||
A primary role of the zeolite phase in ammonia SCR is to provide a Bronsted acid site that stores NH3. Our previous work showed that NH3 storage at the Bronsted acid site was critical for the mixed oxide to promote low-temperature SCR in the composite catalyst.9 Considering this, we carried out NO oxidation activity experiments on the composite catalysts in the presence of pre-adsorbed NH3 to determine how zeolite-stored ammonia effects the NO2 yield. For this experiment, NO oxidation measurements were preceded by introducing 330 ppm NH3 to the NO oxidation feed (i.e., NO + O2 + NH3) at 100 °C for 30 min followed by purging for 10 min with the NO oxidation feed (i.e., NO + O2). The trend of NO oxidation activity amongst the composite catalysts remained unchanged as shown in Fig. 1b. However, there were distinct differences between the activity profiles in Fig. 1a and b. The most notable difference was curvature change of the NO oxidation light-off profiles following NH3 exposure. The curvature changes for CeO2, Ce0.7Mn0.3Oy, and Ce0.5Mn0.5Oy occurred around 270 °C, 200 °C, and 170 °C, respectively, and coincided with a slight increase in the NO concentration measured at the reactor outlet (Fig. S3†). In other words, the NO2 evolution that caused the change in curvature was not derived exclusively from oxidation of the NO inlet feed in that moment. This observation suggests that, in the presence of pre-adsorbed NH3, a metastable intermediate was formed under NO oxidation conditions and that at least one of the decomposition products was NO2. The decomposition temperature of this intermediate in our test profile depended on the catalyst composition as evidenced from Fig. 1b. This implies that the intermediate was either formed on or diffused to the oxide phase prior to decomposition. In the absence of zeolite, i.e., over the bulk mixed oxide, no curvature change was observed under an identical gas feed protocol (Fig. S4†). This demonstrates the requirement for stored ammonia in the zeolite to affect the curvature changes observed in Fig. 1. It is important to note that the rate of NO2 formation from the metastable intermediate exceeded the rate of NO2 formation from NO oxidation, which was the reason for the curvature change.
Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) was utilized to elucidate the species formed upon co-adsorption of NO, O2, and NH3. We began our study with the bulk mixed metal oxide to delineate species adsorbed on the mixed oxide phase and the zeolite phase. Adsorption of NO onto Ce0.7Mn0.3Ox at 150 °C (Fig. S5-a†) resulted in the formation of nitrite (1163 cm−1) and hyponitrite (1102 cm−1) species along with several lower intensity bands spanning from 1200 cm−1 to 1600 cm−1. The bands in the latter region are attributed to nitrate and its derivatives (e.g., N2O3).18,19 The latter assignment was supported by the time-dependent evolution of these bands upon co-adsorption of NO and O2 (Fig. S5-b†).
After the co-adsorption of NO and O2, we purged the DRIFTS cell with He and then admitted a feed of 330 ppm NH3 in He. The dynamic evolution of NO2-derived species during the experiment is shown in Fig. 2a. The detailed changes that occurred are beyond the scope of this manuscript, but general relevant observations are addressed. From the DRIFTS experiment, it can be concluded that adsorbed NO2 and/or its derivatives are perturbed upon exposure to NH3. This was especially noticeable within the 1600–1400 cm−1 region which is commonly associated with monodentate and bidentate nitrates.18 The perturbation can be interpreted as an interaction or association between NH3 and NO2-derived species. The nature of the perturbation was likely chemical, as opposed to physical, owing to the negative order of NH3 on NO2 yield during NO oxidation (Fig. 2b). The inhibition of gaseous NH3 on NO2 yield was caused by the relatively favorable SCR kinetics (Fig. S2†).
 | ||
| Fig. 2 a.) DRIFTS spectra recorded over bulk Ce0.7Mn0.3Oy under NH3 feed following pre-exposure to NO + O2 gas feed. A He purge was interjected between the NH3 and NO + O2 gas feed. b.) NO oxidation activity as a function of NH3 concentration and temperature over bulk Ce0.7Mn0.3Oy. Reaction conditions: [NO] = 325 ppm; [NO2] = 6 ppm; [O2] = 15%; [N2] = balance; flow rate = 1.5 SLM; GHSV = 4.6e + 6 h−1. | ||
The interaction between NH3 and NO2 co-adsorbates discussed above points toward a nitroamine (NHxNOy) intermediate that may have given rise to the low-temperature NO2 evolution shown in Fig. 1b. Although the exact form of this nitroamine complex remains unknown, we reasoned that its reactivity on the different oxide catalysts should scale proportionally with the reactivity of ammonia nitrate, NH4NO3. To assess this, we used NH4NO3 as a surrogate to the true nitroamine complex to gain a better understanding of how nitroamine complexes react over the mixed oxide component. The reactivity and product selectivity of NH4NO3 decomposition over the zeolite (in the absence of oxide) and the bulk oxide (in the absence of zeolite phase) were tested through impregnation of aqueous NH4NO3 followed by a temperature ramp under N2 (Fig. S6†). The plot in Fig. 3a shows that the main NH4NO3 decomposition product over the zeolite was N2O. The formation of N2O was a direct or primary decomposition product (eqn (3)) and is the expected product from the thermal decomposition of NH4NO3.
| NH4NO3 = N2O + 2H2O | (3) |
| NO + NH4NO3 = N2 + NO2 + 2H2O | (4) |
 | ||
| Fig. 3 Observed product distribution over various catalysts upon impregnation with aqueous NH4NO3 followed by heating under (a) N2 and (b) NO. The right y-axis in (b) represents the ratio of NO consumed to the amount of NO2 produced. Conditions: [NH4NO3] = 15 wt%; [NO] = 325 ppm; [N2] = balance; b = 10 °C min−1; F = 1 SLM. | ||
Performing analogous TPSR experiments over the composite catalysts, i.e., CexMn1−xOy/H-SSZ-13, resulted in similar desorption profiles (Fig. S9†). In contrast to the bulk oxide materials, but in agreement with H-SSZ-13, the amount of NO consumed over the composite materials were in fair agreement with the amount of NO2 evolved. The one-to-one stoichiometry suggests that most of the NH4NO3 resided on the zeolite after impregnation, as opposed to the oxide, and thereby minimized the autocatalytic decomposition of NH4NO3 during the TPSR. The diffusion of NH4NO3 from the zeolite to the oxide prior to its decomposition to NO2 is implicit in the previous statement since the decomposition profile was oxide dependent (Fig. S9†). These data indicate that the oxide component, and in particular the Mn-containing oxide component, had a higher catalytic activity for the decomposition of NH4NO3 to NO2 at intermediate temperatures (100–200 °C) relative to the zeolite. This temperature range coincides with the temperature range for SCR promotion by the mixed oxide in the composite system,9 which suggests there may be relationship between NH4NO3 decomposition and SCR promotion.
The TPSR experiments indicate that the curvature change documented in Fig. 1b was caused by NH4NO3 decomposition viaeqn (4). To explain, exposure of the CexMn1−xOy/H-SSZ-13 catalysts to NH3 prior to NO preferentially populated the zeolite with adsorbed ammonia. This is supported by the absent curvature change using the same protocol over the bulk Ce0.7Mn0.3Oy oxide (Fig. S4†) and the much higher uptake of NH3 on the zeolite compared to the bulk oxide (Fig. 4). As the temperature increased in Fig. 1b, NO oxidation to NO2 (and its derivatives) occurred over the oxide and reacted with NH3 from the zeolite to form NH4NO3, which subsequently reacted viaeqn (4). However, given the non-steady-state nature of the experiment in Fig. 1b, the interplay between these two catalyst phases and the formation of NH4NO3 is less clear under actual SCR conditions. Nonetheless, these observations, and the prior observation that NH3 storage (i.e., Brønsted acidity) was required for the low-temperature SCR promotion by the mixed oxide,9 provide a basis for understanding the synergy between the mixed oxide and zeolite components. In the absence of NH3 storage sites, the oxide phase (e.g., Ce1−xMnxOy) will catalyze the oxidation of NH3 to NOx at intermediate temperature, thereby restricting SCR performance (Fig. S7†). Thus, one apparent role of the zeolite phase in the CexMn1−xOy/H-SSZ-13 composite system is to provide an adsorption site for NH3 proximal to the oxide phase.
 | ||
| Fig. 4 NH3 temperature programmed desorption (TPD) over bulk Ce0.7Mn0.3Oy and Ce0.7Mn0.3Oy/SSZ-13. Both catalysts were heated to 500 °C in 15% O2 for 0.5 h, followed by cooling under He, admittance of 350 ppm NH3 for 0.5 h at 50 °C, and heating at 10 °C min−1 under 1 SLM N2. | ||
The mixed oxide component catalyzes NO oxidation to NO2 (and derivatives) which, regardless of the underlying mechanism, allows access to fast SCR pathways under standard SCR gas feed. Table 1 summarizes possible SCR pathways that have been suggested by others.15,16,20,21 Please note that these are known net gas phase reactions and not elementary steps occurring on a surface. The reaction network N(1) and N(2) represent standard SCR pathways; they differ in the way that nitrous acid (HNO2) is formed which is regarded as the key intermediate leading to N2 formation. For N(1) nitrous acid is formed through NO2 disproportionation (R2) and nitric acid reduction by NO (R3). The latter reaction has been shown to occur under mild conditions;20,22 indeed, the curvature change in Fig. 1b was caused by the higher rates of the reactions Rn that comprise NH4NO3 decomposition by NO, N(3), relative to NO oxidation, R1. The former NO2 disproportionation (R2) reaction has also been shown to occur under mild conditions as it is a key step in NO2 SCR, which is favored at low temperatures relative to standard and fast SCR.13,23 This suggests that R1 is a rate-controlling step for N(1) and implies that the rate of N(1) will tend to increase over materials with higher NO oxidation activity. This agrees with the general correlation between NO oxidation and low-temperature SCR activity observed here and elsewhere.6,10
| Index | Net reaction | Reaction network | |||
|---|---|---|---|---|---|
| N(1) | N(2) | N(3) | N(4) | ||
| R1 | 2NO + O2 = 2NO2 | 1 | 1 | 0 | 0 |
| R2 | 2NO2 + H2O = HNO2 + HNO3 | 2 | 0 | 0 | 1 |
| R3 | NO + HNO3 = NO2 + HNO2 | 2 | 0 | 1 | 0 |
| R4 | NH3 + HNO2 = N2 + 2H2O | 4 | 4 | 1 | 1 |
| R5 | NO + NO2 = N2O3 | 0 | 2 | 0 | 0 |
| R6 | N2O3 + H2O = 2HNO2 | 0 | 2 | 0 | 0 |
| R7 | NH4NO3 = NH3 + HNO3 | 0 | 0 | 1 | -1 |
| N(1) | 4NO + O2 + 4NH3 = 4N2 + 6H2O | ||||
| N(2) | 4NO + O2 + 4NH3 = 4N2 + 6H2O | ||||
| N(3) | NO + NH4NO3 = N2 +NO2 +H2O | ||||
| N(4) | 2NH3 + 2NO2 = N2 + NH4NO3 + H2O | ||||
For reaction network N(2), nitrous acid is formed through the hydrolysis of dinitrogen trioxide (R6). This reaction occurs readily at room temperature24 and suggests that the rate limiting step for N(2) is either R1 or R5. Regardless of whether R1 or R5 is rate determining, the rate of N(2) will increase over materials with higher NO oxidation activity since the rate of R5 is proportional to the concentration of NO2. From the data obtained here it remains unclear whether N(1) or N(2) was kinetically dominate during standard SCR over the composite catalyst,9 yet the kinetic relevance of NO oxidation (R1) is manifested in both reaction networks. We hypothesize that the oxide phase in hybrid SCO–SCR systems catalyze the reactions that lead to nitrous acid formation and that these key intermediates react with NH3 from and/or on the zeolite phase (R4). In the absence of zeolite, R4 is limited by the oxidation of NH3 to NHxOy that occurred readily over Ce0.7Mn0.3Ox (Fig. S7†).
Reaction network N(3) represents the NH4NO3 decomposition pathway that occurred over the CexMn1−xOy oxide phase in the presence and absence of gaseous NO (Fig. 3). The dissociation of NH4NO3 (R7) is a prerequisite for N(3) to occur while the remaining reactions comprising N(3) (R3 and R4) occur below 100 °C. Thus, the dissociation reaction occurred readily over the ceria–manganese oxide phase relative to the zeolite phase, especially in the low temperature range (100–170 °C) (Fig. S6 and S7†). This may be a key contributing factor for the low temperature promotion observed in hybrid SCO–SCR systems. To be clear, fast and NO2 SCR reactions that yield only N2 and H2O are limited below ca. 170 °C on zeolite materials due to NH4NO3 precipitation, N(4). The ability of ceria–manganese oxide to disfavor NH4NO3 formation (R−7) prevents NH3 competing with NO for free nitrate (R3), and thereby, surface fast SCR reactions N(1) and N(2) can proceed at low temperature by circumventing reaction N(4).
Conclusions
It has been well-established that introducing a ceria–manganese mixed oxide to a zeolite provides access to low energy SCR pathways. We have identified a few possible contributing factors that may allow us to better understand the low energy pathways and rationalize the synergistic interaction between the mixed oxide and zeolite phase. Our data suggests that the oxide provides access to surface fast SCR channels through NO oxidation to adsorbed nitrogen dioxide and its derivatives. In the presence of nitrogen dioxide and ammonia, zeolites reversible deactivate from ammonium nitrate precipitation. However, ceria–manganese mixed oxides disfavor ammonium nitrate formation, and therefore, fast SCR reaction channels ostensibly remain accessible. We hypothesize that one primary role of the zeolite is to provide an ammonia reservoir for nitrogen dioxide (derivatives) to react with, since ammonia tends to oxidize on the surface of the mixed oxide. The unselective oxidation of these hybrid SCO–SCR materials remains a primary technological hurdle to their industrial deployment for diesel application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the U.S. Department of Energy (DOE), Energy Efficiency and Renewable Energy (EERE), Vehicle Technologies Office for the financial support of this work. PNNL is operated for the DOE by Battelle under contract number DE-AC05-76RL01830.Notes and references
- K. Kamasamudram, N. W. Currier, X. Chen and A. Yezerets, Catal. Today, 2010, 151, 212–222 CrossRef CAS.
- M. Koebel, M. Elsener and G. Madia, Ind. Eng. Chem. Res., 2001, 40, 52–59 CrossRef CAS.
- F. Gao, Y. Wang, M. Kollár, N. M. Washton, J. Szanyi and C. H. F. Peden, Catal. Today, 2015, 258, 347–358 CrossRef CAS; Y. Cui and F. Gao, Emiss. Control Sci. Technol., 2019, 5, 124–132 CrossRef.
- A. Russell and W. S. Epling, Catal. Rev.: Sci. Eng., 2011, 53, 337–423 CrossRef CAS.
- T. Andana, K. G. Rappé, F. Gao, J. Szanyi, X. Pereira-Hernandez and Y. Wang, Appl. Catal., B, 2021, 291, 120054 CrossRef CAS.
- M. Salazar, R. Becker and W. Grünert, Appl. Catal., B, 2015, 165, 316–327 CrossRef CAS.
- A. I. Mytareva, A. Y. Stakheev, G. N. Baeva, D. A. Bokarev, A. L. Kustov and J. R. Thøgersen, Top. Catal., 2016, 59, 919–924 CrossRef CAS.
- M. Salazar, S. Hoffmann, O. P. Tkachenko, R. Becker and W. Grünert, Appl. Catal., B, 2016, 182, 213–219 CrossRef CAS; T. Liese, E. Löffler and W. Grünert, J. Catal., 2001, 197, 123–130 CrossRef.
- T. Andana, K. G. Rappé, N. C. Nelson, F. Gao and Y. Wang, Appl. Catal., B, 2022, 316, 121522 CrossRef CAS.
- A. Y. Stakheev, A. I. Mytareva, D. A. Bokarev, G. N. Baeva, D. S. Krivoruchenko, A. L. Kustov, M. Grill and J. R. Thøgersen, Catal. Today, 2015, 258, 183–189 CrossRef CAS.
- A. A. Verma, S. A. Bates, T. Anggara, C. Paolucci, A. A. Parekh, K. Kamasamudram, A. Yezerets, J. T. Miller, W. N. Delgass, W. F. Schneider and F. H. Ribeiro, J. Catal., 2014, 312, 179–190 CrossRef CAS; T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef; F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef PubMed.
- C. Paolucci, A. A. Verma, S. A. Bates, V. F. Kispersky, J. T. Miller, R. Gounder, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11828–11833 CrossRef CAS PubMed; C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef PubMed.
- M. Iwasaki and H. Shinjoh, Appl. Catal., A, 2010, 390, 71–77 CrossRef CAS.
- H. Kubota, C. Liu, T. Toyao, Z. Maeno, M. Ogura, N. Nakazawa, S. Inagaki, Y. Kubota and K.-i. Shimizu, ACS Catal., 2020, 10, 2334–2344 CrossRef CAS; C. Liu, G. Malta, H. Kubota, K. Kon, T. Toyao, Z. Maeno and K.-I. Shimizu, J. Phys. Chem. C, 2021, 125, 13889–13899 CrossRef.
- P. Forzatti, I. Nova and E. Tronconi, Angew. Chem., Int. Ed., 2009, 48, 8366–8368 CrossRef CAS PubMed.
- F. Marchitti, E. B. Hemings, I. Nova, P. Forzatti and E. Tronconi, Emiss. Control Sci. Technol., 2016, 2, 1–9 CrossRef CAS.
- G. Qi and W. Li, Catal. Today, 2015, 258, 205–213 CrossRef CAS.
- M. Y. Mihaylov, E. Z. Ivanova, G. N. Vayssilov and K. I. Hadjiivanov, Catal. Today, 2020, 357, 613–620 CrossRef CAS.
- M. Y. Mihaylov, E. Z. Ivanova, H. A. Aleksandrov, P. S. Petkov, G. N. Vayssilov and K. I. Hadjiivanov, Mol. Catal., 2018, 451, 114–124 CrossRef CAS.
- A. Grossale, I. Nova and E. Tronconi, J. Catal., 2009, 265, 141–147 CrossRef CAS.
- Q. Sun, Z.-X. Gao, H.-Y. Chen and W. M. H. Sachtler, J. Catal., 2001, 201, 89–99 CrossRef CAS; A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, J. Catal., 2008, 256, 312–322 CrossRef.
- A. Savara, M.-J. Li, W. M. H. Sachtler and E. Weitz, Appl. Catal., B, 2008, 81, 251–257 CrossRef CAS; Y. H. Yeom, J. Henao, M. J. Li, W. M. H. Sachtler and E. Weitz, J. Catal., 2005, 231, 181–193 CrossRef.
- A. Grossale, I. Nova and E. Tronconi, Catal. Lett., 2009, 130, 525–531 CrossRef CAS; I. Nova, C. Ciardelli, E. Tronconi, D. Chatterjee and B. Bandl-Konrad, Catal. Today, 2006, 114, 3–12 CrossRef.
- G. Y. Markovits, S. E. Schwartz and L. Newman, Inorg. Chem., 1981, 20, 445–450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalytic activity, infrared spectra, temperature programmed experiments. See DOI: https://doi.org/10.1039/d2cy01921c |
| This journal is © The Royal Society of Chemistry 2023 |