
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect analysis at temporal and molecular level of deactivating coke species formed on zeolite catalysts with diverse pore topologies†
Idoia
Hita
 a,
Hend Omar
Mohamed
a,
Yerrayya
Attada
a,
Naydu
Zambrano
a,
Wen
Zhang
b,
Adrian
Ramírez
c and
Pedro
Castaño
*ad
a,
Hend Omar
Mohamed
a,
Yerrayya
Attada
a,
Naydu
Zambrano
a,
Wen
Zhang
b,
Adrian
Ramírez
c and
Pedro
Castaño
*ad
aMultiscale Reaction Engineering, KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: pedro.castano@kaust.edu.sa
bAnalytical Chemistry Core Labs, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
cKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
dChemical Engineering Program, Physical Science and Engineering (PSE) Division, King Abdullah University of Science and Technology, Saudi Arabia
First published on 11th January 2023
Abstract
The mechanistic understanding of coke formation on zeolites is elusive, given the limitations for the extraction and analysis of coke species. Here, we analyze the evolution of deactivating coke species over time on the surface of Ni/ZSM-5, β and Y zeolites during ethylene oligomerization, which is a representative coke-forming reaction. We present a method that directly analyzes the coke species using high-resolution mass spectrometry on the used catalysts and their isolated soluble and insoluble coke fractions (indirect analysis). Along with other techniques, we report a full-picture temporal evolution of coke and its fractions at the molecular level to establish a correlation between the dominant reaction mechanisms, the location of coke within the catalyst structure, and the observed performance of the catalysts. Ab initio calculations are performed to understand the preferable location of coke. We delve deep into the peculiar deactivation mechanism of ZSM-5 zeolite (MFI structure), which shows two less intuitive parallel deactivation pathways.
1. Introduction
Coke fouling is a recurrent and long-time companion of catalytic reactions, often considered an “inevitable disease” in the field.1 It affects catalytic processes in many ways, such as lowering the stability, “capturing” carbon that eventually becomes CO2 in an oxidative regeneration step, or forcing catalyst and reactor modifications, among many others. The cost of coke fouling is tremendous, not only economically but also in terms of the carbon footprint of CO2 production. For instance, the estimated yearly CO2 emissions from fluid catalytic cracking (FCC) units are 200–240 million tons (assuming a total processing capacity of over 20 million barrels per day).2Ethylene (C2H4) oligomerization is a representative reaction suffering from coke deactivation and a way to obtain larger olefins (CnH2n, n ≥4), which have a limited yet interesting market in the manufacturing of detergents, lubricants, surfactants, coatings, synthetic fibers, resins, packaging, and fuels and for the termination of certain polymerizations (linear α-olefins). Moreover, ethylene oligomerization is a key intermediate step in the zeolite-catalyzed conversion of renewable feedstock like methanol, dimethyl ether (DME), or biomass pyrolysis oil.3–8 Ethylene has a much broader annual market of more than USD 200 billion and is expected to grow up to USD 270 billion by 2023.9 To ensure a future supply, other more environmentally sustainable sources of ethylene are being explored.10–13 The oligomerization of ethylene is thus an interesting avenue that has called for deep research in both the homogeneous and the heterogeneous catalytic fields.14 In the latter, the most promising results have been obtained with Ni2+ supported on zeolites,15 which are prone to catalyst deactivation16 and behave as bifunctional catalysts,17 having parallel reactions in metallic and acidic sites. In addition, olefins are key intermediates and model reactants in coke formation mechanisms.18,19
The molecular-level elucidation of coke species has been a prominent analytical objective given the necessity of understanding coke fouling mechanisms at a deeper level.19,20 The lack of precise analyses of coke composition has grown at the same rate we have developed a picture of coke as a highly aromatic structure given its very low H/C ratios.21In situ techniques, from spectroscopy22,23 to microscopy–tomography24,25 techniques, have contributed substantially to the understanding of the coke formation mechanisms. To date, in situ techniques cannot resolve the molecular-level characterization, and ex situ techniques, such as disaggregation and extraction26 or extraction alone,27,28 are widely used instead. These ex situ methods enable liquid separation from the solid phase, classifying coke into soluble and insoluble fractions. Soluble coke structures with up to 16 carbon atoms can be easily analyzed by conventional techniques such as gas chromatography. However, the composition of the heaviest pool of soluble fraction molecules, together with all insoluble coke, remains largely unknown. Efforts using (matrix-assisted) laser desorption ionization time-of-flight mass spectrometry ((MA)LDI-TOF MS) have noticeably advanced the molecular-level characterization of heavy coke species.29–34 However, this technique does not have enough mass accuracy to resolve the individual chemical formula of each species.
The main deficiencies of the methods developed before arise since they use (1) multi-step treatments to dissolve the catalysts and extract small quantities of coke species, (2) analytical techniques that can only detect the lighter fraction of coke, such as gas chromatography, or (3) analytical techniques that do not have enough resolution to distinguish the nature of these species such as (MA)LDI-TOF MS.
Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR MS) has an unparalleled resolution compared with any other technique of this kind, to the point of resolving the atomic composition of every single molecule of the mixture (that can be ionized). In fact, FT-ICR MS is transforming the way we understand coke growth in the methanol-to-olefin (MTO) reaction in terms of (i) the unusual mechanism of internal coke growth in SAPO-34 catalysts,35 (ii) the temporal analysis of the same internal coke,36 and (iii) the growth mechanisms on the catalysts when water is present in the reaction medium.37 All these previous studies involve the analysis of the insoluble or soluble species (solving limitations 2 and 3) after catalyst dissolution–extraction, but neither total nor direct coke analysis has been successfully conducted on untreated spent catalysts (to solve limitation 1). Together with the temporal evolution, the whole picture of the coke formation mechanisms remains incomplete.
Our work provides a comprehensive molecular-level and temporal analysis of coke formed on Ni supported on different pore-network zeolite catalysts. We analyze and compare the composition of the “whole” coke obtained through its direct analysis as well as its soluble/insoluble counterparts previously isolated through a dissolution–extraction procedure. Comparing the compositions of the reaction medium and coke as well as the properties of the catalysts and supported by adsorption calculations, we propose the formation and growth mechanisms of coke in catalysts with different zeolite pore topologies.
2. Experimental
2.1. Catalyst synthesis and characterization techniques
Four Ni-containing catalysts were prepared using four different zeolites as supports (all provided by Zeolyst): H-Y (CBV712, Si/Al = 6), H-ZSM-5 with Si/Al = 40 (CBV8014), H-ZSM-5 with Si/Al = 15 (CBV3024E), and H-β (CP814E*, Si/Al = 12.5). All the zeolite supports were calcined at 550 °C for 5 h in static air using a ramp of 5 °C min−1 to attain their protonic form. Each zeolite was impregnated dropwise using a vortex mixer with ∼0.5 mL g−1 of a 1 M solution of Ni(NO3)2·6H2O (Sigma Aldrich) as a salt precursor, targeting a nominal Ni content of 3 wt%. After impregnation, the catalysts were dried overnight at 100 °C and subsequently calcined at 550 °C in static air under atmospheric conditions for 5 h using a temperature ramp of 5 °C min−1. Lastly, they were pelletized, crushed, and sieved to a particle size of 0.15–0.30 mm. The catalysts were designated as NiY, NiZ40, NiZ15 (subindices indicate the Si/Al ratio), and Niβ.The metal content in the catalysts was measured through inductively coupled plasma-optical emission spectrometry (ICP-OES) using an Agilent 5100 unit. Around 20 mg of sample were digested using an Ultra WAVE instrument (Milestone) in an acid solution of HF (1 mL), HCl (1 mL), and HNO3 (5 wt%, 3 mL) at 250 °C and 130 bar for 40 min. Digested samples were diluted to 50 ml with HNO3 (1% in water) and analyzed.
The fresh catalysts' specific surface area, micropore volume, and pore size distribution were characterized by N2 adsorption–desorption at −196 °C in a Micromeritics ASAP 2420 instrument. Prior to N2 adsorption, the samples were outgassed at 350 °C for 10 h under vacuum. The specific surface area (SBET) and pore size distribution were calculated using the Brunauer–Emmett–Teller method, while the micropore volume (Vmicro) was obtained using the t-plot method. The average pore size (dpore) was estimated using the Barrett–Joyner–Halenda (BJH) method.
Acidic properties were measured through temperature-programmed desorption of ammonia (NH3-TPD) in an Altamira AMI-200 instrument. After pretreatment under Ar (25 mL min−1) at 550 °C and 30 min, the samples (ca. 50 mg) were saturated for 1 h at 120 °C under 15 mL min−1 of a 3% NH3/He stream diluted in 25 mL min−1 Ar. Subsequently, a temperature-programmed desorption (TPD) analysis was performed up to 600 °C at a warm-up rate of 5 °C min−1 under a He stream (10 mL min−1). The NH3 signal was monitored using a thermal conductivity detector (TCD), and the obtained results were analyzed using AMI-Analysis v2.21 software. The amount of Brønsted and Lewis acid sites (BASs and LASs, respectively) on the catalysts were quantified by FTIR spectroscopy with pyridine adsorption on a Nicolet 6700 spectrophotometer operating in transmission mode. Prior to pyridine adsorption, the samples were degassed at 450 °C applying a temperature ramp of 10 °C min−1 and 10−4 mbar pressure for 16 h. Pyridine adsorption was performed at room temperature. After removing the physisorbed pyridine at 150 °C, the IR spectra were recorded.
Sample crystallinities were studied by X-ray diffraction (XRD) in a Bruker D8 Advance diffractometer operating at 40 kV and 40 mA with Cu Kα radiation of 0.154 nm. The diffraction pattern was recorded over a 2θ range of 5–90° at a step size of 0.02°.
The morphologies of the fresh catalysts were examined using SEM in a Nova Nano SEM 240 unit. In addition, TEM and high-resolution transmission electron microscopy (HR-TEM) were performed on an FEI-Titan ST electron microscope operated at 300 kV. Prior to analysis, the fresh catalyst samples were dispersed in ethanol using an ultrasonic bath, and the obtained suspension was placed onto a carbon-coated copper grid.
2.2. Ethylene oligomerization and product analysis
Ethylene oligomerization runs were performed in a high-throughput four-channel Flowrence reactor from Avantium. The channels consisted of 300 mm stainless steel tubes (inner diameter = 2 mm) located within a furnace. On each of the reactors, 120 mg of catalyst was loaded. A 200 μL silicon carbide (SiC) layer and quartz wool were placed below the catalyst bed to ensure optimal isothermal operation, prevent catalyst dragging and facilitate recovery of the used catalyst after reaction completion. Before the reaction, the catalysts were pretreated in situ under a helium flow (5 mL min−1) at 550 °C for 2 h. Afterward, the reaction temperature was set at 300 °C, and the reactors were pressurized until they reached the reaction conditions at 35 bar. Once reached, the feed mixture was switched on, which consisted of ethylene and helium in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. Nitrogen (20 mL min−1) was introduced at the reactor outlet as a diluent for the GC analysis. One of the four channels was used as a blank reference, and He was used for internal standard calculation. The evolved reaction gases were analyzed online using an Agilent 7890B gas chromatograph (GC) provided with two sample loops. One of the sample loops directed the sample to a TCD channel where He, H2, and CH4 were separated. The second sample loop directed the sample to an Innowax pre-column (length = 5 m, outer diameter = 0.20 mm, 0.4 μm film) and subsequently to a Gaspro column (length = 30 m, outer diameter = 0.32 mm). After 30 s, the gases were sent to an Innowax column (length = 45 m, outer diameter = 0.20 mm, 0.4 μm film) followed by a flame ionization detector (FID). In the latter column, one-to-eight-carbon paraffins and olefins were separated as well as larger (>9 carbons) paraffins and olefins, the benzene/toluene/xylene (BTX) fraction, and aromatics with ≥9 carbons.
:2 ratio. Nitrogen (20 mL min−1) was introduced at the reactor outlet as a diluent for the GC analysis. One of the four channels was used as a blank reference, and He was used for internal standard calculation. The evolved reaction gases were analyzed online using an Agilent 7890B gas chromatograph (GC) provided with two sample loops. One of the sample loops directed the sample to a TCD channel where He, H2, and CH4 were separated. The second sample loop directed the sample to an Innowax pre-column (length = 5 m, outer diameter = 0.20 mm, 0.4 μm film) and subsequently to a Gaspro column (length = 30 m, outer diameter = 0.32 mm). After 30 s, the gases were sent to an Innowax column (length = 45 m, outer diameter = 0.20 mm, 0.4 μm film) followed by a flame ionization detector (FID). In the latter column, one-to-eight-carbon paraffins and olefins were separated as well as larger (>9 carbons) paraffins and olefins, the benzene/toluene/xylene (BTX) fraction, and aromatics with ≥9 carbons.
Ethylene conversion Xethylene is defined by eqn (1) as follows:
 | (1) |
The selectivity Si of products with one to nine carbons (C1 to C9) is defined as:
 | (2) |
The selectivity towards coke Scoke is defined as:
 | (3) |
![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) is the average conversion for a certain time-on-stream range which is calculated by integrating the conversion profile over time.
is the average conversion for a certain time-on-stream range which is calculated by integrating the conversion profile over time.
2.3. Analysis of the total coke species, soluble and insoluble cokes
A complete overview of the protocols and analyses performed on the reaction products and different coke fractions is provided in Fig. 1. First, the deposited coke was quantified as recovered from the oligomerization reactor by combined thermogravimetry and temperature-programmed oxidation (TG-TPO) in a TGA/DSC 1 STAR System by Mettler Toledo. Approximately 5 mg of deactivated catalyst sample were analyzed per experiment. A primary sweeping of the adsorbed organic species was performed under a He atmosphere (50 mL min−1), ramping up to 300 °C at 10 °C min−1 and maintaining an isotherm for 30 min. Subsequently, the furnace temperature was cooled to 100 °C and the gas atmosphere was switched to air (50 mL min−1). To avoid possible weight effects from Ni oxidation on the TG quantification, the CO2 profiles registered in the MS from the combustion profiles were integrated. After the sample was stabilized for 10 min, a combustion ramp of 5 °C min−1 was applied up to 800 °C. The signal for the evolved CO2 gas from the combustion was registered in an OmniStar™ mass spectrometer by Pfeiffer Vacuum. The Gaussian deconvolution of the CO2 profiles obtained from coke combustion was conducted using OriginPro 9.6 software. | ||
| Fig. 1 Description of the workflow applied to reaction products with an emphasis on the coke characterization techniques. | ||
Molecular-level characterization of the coke deposited in the deactivated catalysts was conducted through Fourier-transform ion cyclotron resonance mass spectrometry using laser desorption ionization (LDI FT-ICR MS) in a Bruker SolariX XR 9.4 Tesla instrument, and the FT-ICR MS spectra were treated using Bruker Data Analysis V4.5 software. Chemical formula assignments were performed with Composer software (Sierra Analytics, Modesto, CA, USA). From the predicted molecular formula (CcHhOoNnSs), the DBE number of a given molecule was calculated using eqn (4).
 | (4) |
2.4. Density functional theory (DFT) considerations
Dispersion-corrected adsorption energy calculations of aromatic compounds on H-ZSM-5 zeolite and Ni/ZSM-5 catalysts were performed, applying the first principle density functional theory (DFT) using the Vienna Ab initio Simulation Package (VASP).38–40 The electron exchange and correlation interactions were modeled using the generalized gradient approximation (GGA)41 with the Perdew–Burke–Ernzerhof (PBE) functional.42 The electron–ion interactions were defined using the projector-augmented wave (PAW) method. The weak binding systems were considered using the vdW D3 correction proposed by Grimme.43 A plane-wave basis set was used to describe the valence electrons with an energy cut-off of 400 eV. The Brillouin zone, sampled at the Monkhorst–Pack k-point grid,44 was used to sample the 36T model H-ZSM-5 and NiZ40. The equilibrium geometries were reached for all total energy optimization once the atomic forces on every atom were smaller than 0.05 eV Å−1 with a total energy convergence criterion of 1.0 × 10−6 eV. A cubic box of 15 Å × 15 Å × 15 Å was used for the gas-phase molecule.The adsorption energy Eads of aromatic compounds was calculated as
| Eads = EB+Ni/ZSM-5 − EB − ENi/ZSM-5 | (5) |
3. Results
3.1. Catalyst properties and impact on the reaction pathways
We prepared four oligomerization catalysts by impregnating a nominal amount of 3 wt% (actual contents of 2.69–2.98 wt%, see Table 1) Ni into four zeolites with different topologies: Y (framework code: FAU), ZSM-5 (MFI, with two different Si/Al ratios), and β (BEA) (see Fig. 2).45 The MFI framework has 10-membered ring (10-MR) pores of 0.51–0.56 nm interconnected in two directions (90°) of the space, with one straight and the other zigzag. The BEA framework has 12-MR pores of 0.56–0.77 nm with straight channels, creating intersections. Lastly, the FAU framework has 12-MR pores of 0.74 nm between sodalite cages (typically named supercages) of 1.2 nm, creating zigzag channels and four apertures per cage.| NiY | NiZ40 | NiZ15 | Niβ | |
|---|---|---|---|---|
| Ni content (%) | 2.69 | 2.80 | 2.86 | 2.98 |
| S BET (m2 g−1) | 606 | 346 | 303 | 465 |
| V pore (cm3 g−1) | 0.424 | 0.195 | 0.185 | 0.819 |
| V micro (cm3 g−1) | 0.227 | 0.112 | 0.120 | 0.143 |
| % micro | 53.5 | 57.4 | 64.9 | 17.5 |
| d pore (Å) | 64 | 43 | 58 | 177 |
| Total acidity (μmolNH3 g−1) | 483 | 235 | 671 | 362 |
| BAS (μmol g−1) | 131 | 41 | 105 | 19 |
| LAS (μmol g−1) | 82 | 546 | 380 | 1088 |
| BAS/LAS | 1.59 | 0.08 | 0.28 | 0.02 |
 | ||
| Fig. 2 TEM images of the four prepared Ni/zeolite catalysts and structures of the zeolites used for the (a) NiZ15, (b) NiZ40, (c) NiY, and (d) Niβ catalysts. | ||
The specific surface areas in Table 1 (Fig. S2†) range from 606 to 303 m2 g−1, following the trend NiY > Niβ > NiZ40 ≈ NiZ15. The NiY catalyst having the highest surface area is also in line with it having the highest micropore volume (0.227 cm3 g−1), which is 34–51% higher than the other catalysts. The Niβ catalyst shows the highest average pore diameter (177 Å), which is also in agreement with its structure having the lowest proportion of micropores. The acid site characterization (Fig. S3 and S4†) shows that the most acidic catalysts are NiZ15 (total acidity: 671 μmol g−1; Brønsted acid sites (BASs): 105 μmol g−1) and NiY (total: 483 μmol g−1; BASs: 131 μmol g−1). Conversely, the least acidic catalysts correspond to those having relatively higher proportions of Lewis acid sites (LASs) than BASs: NiZ40 (total: 235 μmol g−1; BASs: 41 μmol g−1) and Niβ (total: 362 μmol g−1; BASs: 19 μmol g−1) catalysts.
The X-ray diffraction (XRD) patterns for the fresh catalysts (Fig. S5†) show the characteristic sharp diffraction peaks of MFI, BEA, and FAU structures. All samples exhibit peaks at 2θ = 37.3° and 43.3°, which are assigned to the (111) and (200) NiO crystal planes, respectively.46 The morphology of the prepared catalysts was further investigated using scanning electron microscopy (SEM) as shown in Fig. S6.† The NiO particle size distributions observed through high-resolution transmission electron microscopy (HR-TEM) of the different catalyst surfaces (Fig. S7†) are of 20 ± 18 nm. As demonstrated by the TEM images of Fig. 2, all catalysts show a relatively uniform Ni particle distribution. Given the differences in micropore size, connectivity, acidity, and acid site nature, we expect that the differences among the catalysts arise from the zeolite properties themselves.
The temporal evolutions of the reaction products using the different Ni-supported catalysts are displayed in Fig. 3. We observe a drop in the ethylene conversion from the beginning of the reaction for all catalysts. The NiY catalyst (Fig. 3a) provides a high initial ethylene conversion of almost 100% but deactivates down to 43% after 10 h. The NiZ40 and NiZ15 catalysts (Fig. 3b and c) both have the lowest initial conversion and progressively deactivate throughout the reaction, reaching conversions of 20% and 27%, respectively, after 10 h on stream. Conversely, the Niβ catalyst (Fig. 3d) maintains a conversion of >90% over 10 h on stream. Such a greater level of stability is due to the larger pore size and lower proportion of micropores (Table 1) that constitute mesoporous materials, which facilitate the diffusion of large oligomers towards the outside of the catalyst particle and partially prevent pore blockage. The rest of the catalysts have a higher proportion of micropores (54–65%, Table 1), and this characteristic hinders oligomer diffusion in the HY zeolite supercages (see Fig. 2c).
 | ||
| Fig. 3 Effect of catalyst type on the evolution over time on stream (TOS) of the ethylene conversion and product yields using the (a) NiY, (b) NiZ40, (c) NiZ15 and (d) Niβ catalysts. | ||
Ethylene oligomerization will preferentially occur through the Cossee–Arlman mechanism and over the Ni2+/acid sites.47–49 This means that the heavy C10+ oligomers and paraffins are predominant at the beginning of the reaction (instead of cyclic species considered in the metalacyclic mechanism), especially when the conversion is high and the extent of coke deposition is still limited. Under these initial conditions, Ni2+ sites in the vicinity of acid sites are responsible for the initial ethylene dimerization and subsequent olefin oligomerization. The products of this initial oligomerization can diffuse and react further on the BASs through cracking and hydride transfer reactions, following a reaction scheme like the one in Fig. S8.† This means that heavy oligomers are the ones that suffer hydride transfers that lead to high-molecular-weight dehydrogenated molecules, which remain trapped in the zeolite pores and deposit on the catalyst surface. After 2 h on stream, the olefin concentration increases, particularly for the NiY catalyst (Fig. 3a), which provides up to 78.4% total olefin and 65.7% butene selectivity after 10 h on stream.
The NiZ15 and NiZ40 catalysts (Fig. 3b and c) provide relatively similar product distributions even though the former has almost three times as many acid sites as the latter (Table 1). Thus, under these conditions, our results demonstrate that the metallic Ni2+ ions are the controlling active sites on the catalysts. The oligomer ratios do not follow the statistical Schulz–Flory distribution, and all the catalysts show higher selectivity towards dimerization and tetramerization products, particularly for NiY (Fig. 3a) and Niβ (Fig. 3d) catalysts. These results confirm that although Ni2+ constitutes the main active oligomerization sites, a certain degree of cooperation with the acid sites occurs. The extent of this cooperation depends not only on the individual number of sites but also on the degree of interaction between them.50
3.2. Effect of pore topology on coke deposition and nature
The retained coke species on the catalysts were quantified through thermogravimetric techniques. Fig. S9† shows a typical CO2 or combustion profile of coke, evidencing the coexistence of two types of coke51 deconvoluted by two Gaussian curves (Fig. S10†): (I) a lighter, more aliphatic or more accessible coke at 384–414 °C and (II) a heavier, more aromatic coke likely trapped in the micropores at 498 °C. Fig. 4 shows the temporal evolution of the total coke contents deposited on the catalysts, which confirms the two coke formation/growth stages originally reported by Guisnet.52 These steps consist of active site poisoning caused by reaction products/intermediates up to 2 h on stream, followed by a pore blockage.53 The first step can be regarded as chemisorption, given the Langmuir adsorption behavior up to the formation of the monolayer.52 At this stage, the formed oligomers sequentially make the ring and aromatize on the active site of the catalyst.54 This stage of site poisoning coincides with a faster conversion decay over the initial 4 h on stream (Fig. 3) and affects the most active sites of the catalyst. However, as these sites are blocked, some undesired reactions, such as hydride transfer, decelerate even more than the oligomerization (Fig. S7†), causing an increase in the selectivity of desired oligomers (OL4, OL6, OL8).55,56 In contrast, the second stage of coke formation takes place when the coking degree is high (total coke content >100 mgcoke gcatalyst−1), forming a multilayered structure that plugs the pores, such as that observed for the Niβ catalyst at 10 h (Fig. 4d).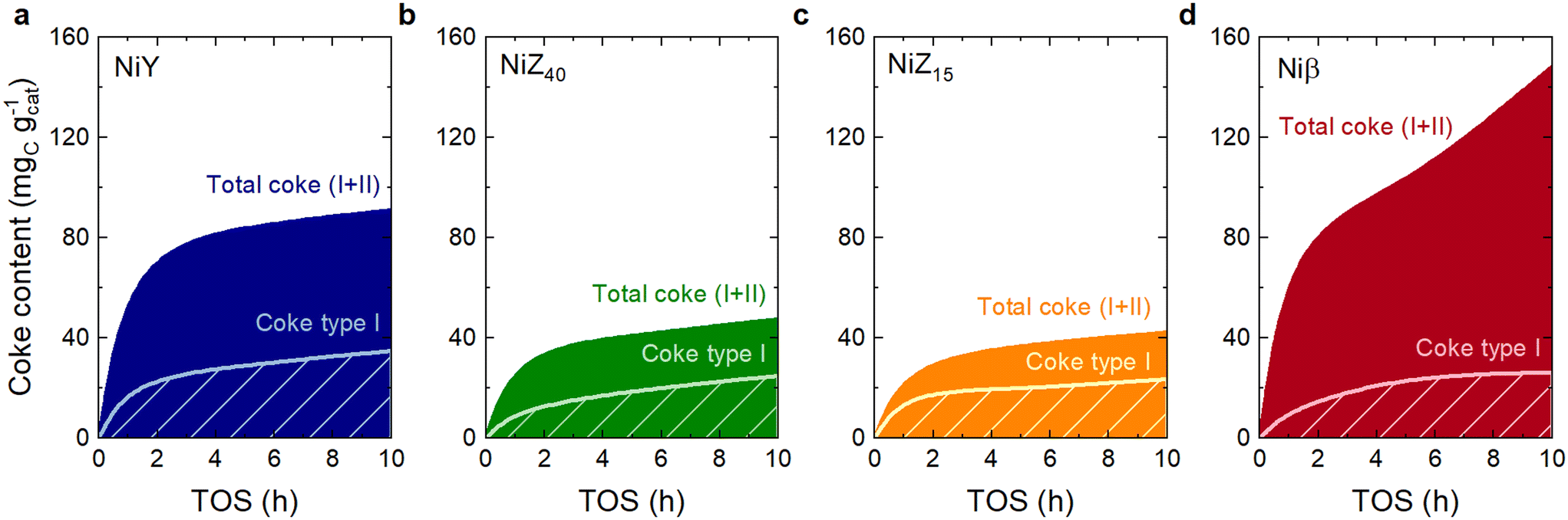 | ||
| Fig. 4 Dynamics of the total amount of the formed coke fractions over the (a) NiY, (b) NiZ40, (c) NiZ15 and (d) Niβ catalysts. The individual fractions of coke I and II have been calculated via Gaussian deconvolution of the CO2 profiles in Fig. S10.† | ||
There is a correlation between the coke content deposited during the active site poisoning and the micropore volume (Table 1), indicating that coke formation in this stage happens predominantly, but not exclusively, within the micropores of the catalysts. This is an interesting factor because it proves that BASs are responsible for the formation and development of the precursors of coke. Thus, the shapes of the micropores in the NiY (forming supercages) and Niβ catalysts (with a significantly wider diameter) directly impact the amounts of coke deposited during the poisoning stage. Conversely, due to their pore topology, the ZSM-5 zeolite catalysts (Fig. 4b and c) form the lowest amounts of coke. A combination of high pore connectivity, absence of cages, and smaller pore size limits the formation of bulky coke precursors. It enables their diffusion towards the outsides of the catalyst particles.57–59 These catalytic features lead to not only lower ethylene conversions (Fig. 3a–d) but also a reaction medium consisting of lighter compounds, hence with a lower concentration of coke precursors and a lower chance of occurrence of hydride transfer reactions that promote coke formation.
Coke formation is an auto-inhibited process since as the coke grows, the rate of coke formation decreases both at the site-poisoning and at the pore-blockade stages: fewer sites and pores are available.60 Due to this, the coke formation selectivity (Fig. S11†) is higher right at the beginning of the reaction. It decreases over time on stream until the steady state is attained after 5 h on stream when the selectivity towards coke reaches an approximately constant value which is also analogous for all catalysts. Despite an overall lower coke formation, the NiZ40 catalyst presents the highest total coke selectivity at 1 h time on stream, as a consequence of a more pronounced formation of coke II species, promoted by the diffusion mentioned above of coke precursors towards the outside of the catalyst particle. This effect causes larger (more aromatic) coke species to grow without steric impediments in pores of larger size (coke I) that, at the same time, burn at lower temperatures due to the presence of Ni on the catalytic surface (Fig. 2).
Fig. 5 displays the variation of the coke I:II peak intensity ratio with the total coke content. The trends reaffirm that for the NiY, NiZ40, and NiZ15 catalysts, coke first forms in the micropores of the catalysts (coke II) to an extent which is determined by the proportion and size of the micropores. It then expands outside the micropores and towards larger or wider pores (coke I). We also observe that compared to NiY, the formation of coke type I occurs at a faster (almost identical) rate over NiZ catalysts. Conversely, the proportion of coke II continues to increase over time for the Niβ catalyst due to a combination of wider micropores and a higher proportion of mesopores (Table 1), which enables coke II species to keep growing steadily at a more rapid rate than coke I. Even though the Niβ catalyst has the highest coke content (Fig. 4d), it also provides the highest ethylene conversion and a greater proportion of heavy C10+ compounds and paraffins (Fig. 3d). That is, the mesopores of this catalyst can retain coke species without affecting the diffusion of ethylene towards the active Ni2+ and acid sites, which remain available in a higher proportion in contrast to the rest of the catalysts.
 | ||
| Fig. 5 Variation of the ratio between the intensities of the peaks corresponding to coke types I and II (Fig. S10†) versus the total coke content for all catalysts. | ||
3.3. Direct molecular-level characterization of the total coke, and formation mechanism
The molecular-level and temporal mass intensities of the coke species formed on the catalysts at 1, 5, and 10 h on stream are displayed in Fig. 6. While (MA)LDI-TOF has already been reported for the analysis of coke species,30,61 to the best of our knowledge we are the first to combine FT-ICR MS with LDI for the direct ionization of the coke species without any prior used catalyst treatment. In all cases, we could detect coke species within the m/z range from 300 Da to 800 Da. The 12-MR zeolites (NiY and Niβ catalysts) exhibit a different behavior from the 10-MR zeolites (NiZ40 and NiZ15 catalysts). On the one hand, the spectra for the species retained on the 12-MR zeolites (Fig. 6a–c and j–l) show two distinct Gaussian-type distributions at 370–430 Da (M1 and M6 for NiY and Niβ, respectively) and a secondary signal at 450–600 Da (M2a–M2b–M2c and M7a–M7b–M7c, respectively). While the M1 and M6 distributions remain mostly unchanged over time on stream, the signals in the 450–600 Da range for these catalysts steadily widen and shift towards heavier average m/z values. On the other hand, over the 10-MR zeolites (NiZ40 and NiZ15, Fig. 6d–i) we detect the formation of a relatively broad and heterogeneous distribution of species across 420–500 Da (M3) after 1 h of reaction (overall bulkier than those observed at the same TOS using 12-MR zeolites, see Fig. 6a and j), which splits with time into two fractions: M4 (330–350 Da, lighter in nature than M3) and M5 (350–800 Da). | ||
| Fig. 6 LDI FT-ICR MS spectra for the deactivated (a–c) NiY, (d–f) NiZ40, (g–i) NiZ15, and (j–l) Niβ catalysts at 1, 5, and 10 h on stream. | ||
The periodicity of the signals in the 300–450 Da range (Fig. S12†) show distinctive –CH2– patterns for all catalysts, which indicates that the lightest coke fractions (M1, M4 and M6 in Fig. 6) are aromatic structures (islands) with different alkylation degrees indicative of internal coke. The heaviest fractions at >450 Da (M2a–M2b–M2c, M5, and M7a–M7b–M7c) show much more heterogeneous spectra with no regular patterns detected, especially for the 10-MR zeolites (NiZ40 and NiZ15 catalysts), indicating that these fractions have grown without steric impediments and in a comparatively “disorganized” fashion. From these results, we can infer that all coke species continue aging over time mainly through successive alkylation and aromatization reactions.62 However, external species that are more prominent on the surface of 10-MR zeolites are subject to these reactions for longer, thus developing their composition more.
Given the aromatic nature of coke precursors, we also calculated the adsorption energies for different aromatics (with up to four aromatic rings and considering different ring configurations) on different catalyst sites using DFT calculations, as shown in Fig. 7. We select the ZSM-5 zeolite as a reference zeolite to perform the calculations. The siting of the Ni2+ site within the various zeolite T-sites available is a very important subject, as it can influence the calculations significantly. We have considered a specific T-site for the insertion of the Ni2+ site based on previous studies.63–65 We have also considered a cluster model representative of the current system in accordance with previous studies.66,67
 | ||
| Fig. 7 Adsorption energies of various aromatic species containing up to 4 aromatic rings on various configurations over different active sites. | ||
The adsorption energies point out that the exothermicity of the adsorption of the aromatic molecules primarily depends on the type of site upon which it is adsorbing, and such molecules adsorb more strongly over Ni2+/acid than over zeolite acid sites. Secondarily, aromatic structures with a higher number of rings (>2) in their structure also tend to adsorb more stably than mono- or di-aromatics. Likewise, the adsorption of “linear” 3- and 4-ring aromatics (3L, anthracene; 4L, tetracene) is consistently more exothermic than that of their non-linear counterparts (phenanthrene and chrysene, respectively). Similar observations have been reported by Dang et al.67 over an γ-Al2O3 (110) surface. These results indicate that a driving force exists for the coke precursors (aromatics such as the ones modeled) to diffuse to the Ni2+/acid sites and grow through the addition of aromatic rings in a linear fashion. Due to the steric hindrance of the nanometer-sized micropore confinements and the FT-ICR MS patterns (Fig. S12†), we can fathom the emergence of linear aromatic islands with varying degrees of alkylated bridges and terminations inside the catalyst micropores.
From the predicted molecular formulas, we were able to compile the isoabundance plots in Fig. S13–S16,† which show the distributions of the detected hydrocarbon coke species, providing information on their carbon number and also H/C ratio (which can be inferred from their DBE number). From these graphs and the results derived from Fig. 4 and 5, we can elucidate the potential coke structures and schematize the reactions occurring in the coke, as displayed in Fig. 8. Over 12-MR zeolites (NiY and Niβ catalysts, Fig. 8a and c, respectively), early coke species (M1 and M6 in Fig. 6) start forming within the catalyst micropores. These species progressively evolve towards heavier and more unsaturated molecules, which grow inside the interstices of the pores. This mechanism is similar to the one observed for an 8-MR zeolite (SAPO-34) during the methanol-to-olefin reaction.35 The FT-ICR can resolve the mass and predict a molecular formula for a given compound; it cannot discern the exact arrangement of that molecule. This means that the full-potential application of high-resolution MS for the direct analysis of coke can only be attained when rationalizing the obtained results and considering the catalyst features (Table 1) and the composition of the rest of the reaction medium (Fig. 3). In the case of 12-MR zeolites, tentative M1 and M6 species can have an identical formula (C30H36) and DBE. Still, due to the higher availability of active sites during the reaction (aided by wider pores, see Table 1) and its higher selectivity toward heavy compounds (Fig. 3d), the coke species formed after 1 h of reaction over the Niβ catalyst (M6) will likely present a more aromatic nature than those formed over the NiY catalyst (M1). The patterns in Fig. S12† and the mechanisms shown in Fig. 8 indicate that the growth of these initial species into M2a–c and M7a–c lumps occur by adding alkyl substituents and/or aromatic rings which are preferentially arranged linearly (Fig. 7). In the case of the Niβ catalyst (Fig. 8c), the composition of the reaction medium will cause coke species to aromatize at a faster rate than NiY species (Fig. 8a), enabling the formation of very bulky aromatics and an overall high amount of total coke (Fig. 3d) during the pore blockage phase. This higher unsaturation of the bigger coke species (C >50) formed over the Niβ catalyst is also evidenced by their DBE numbers in Fig. S16† in contrast to Fig. S13† for NiY species.
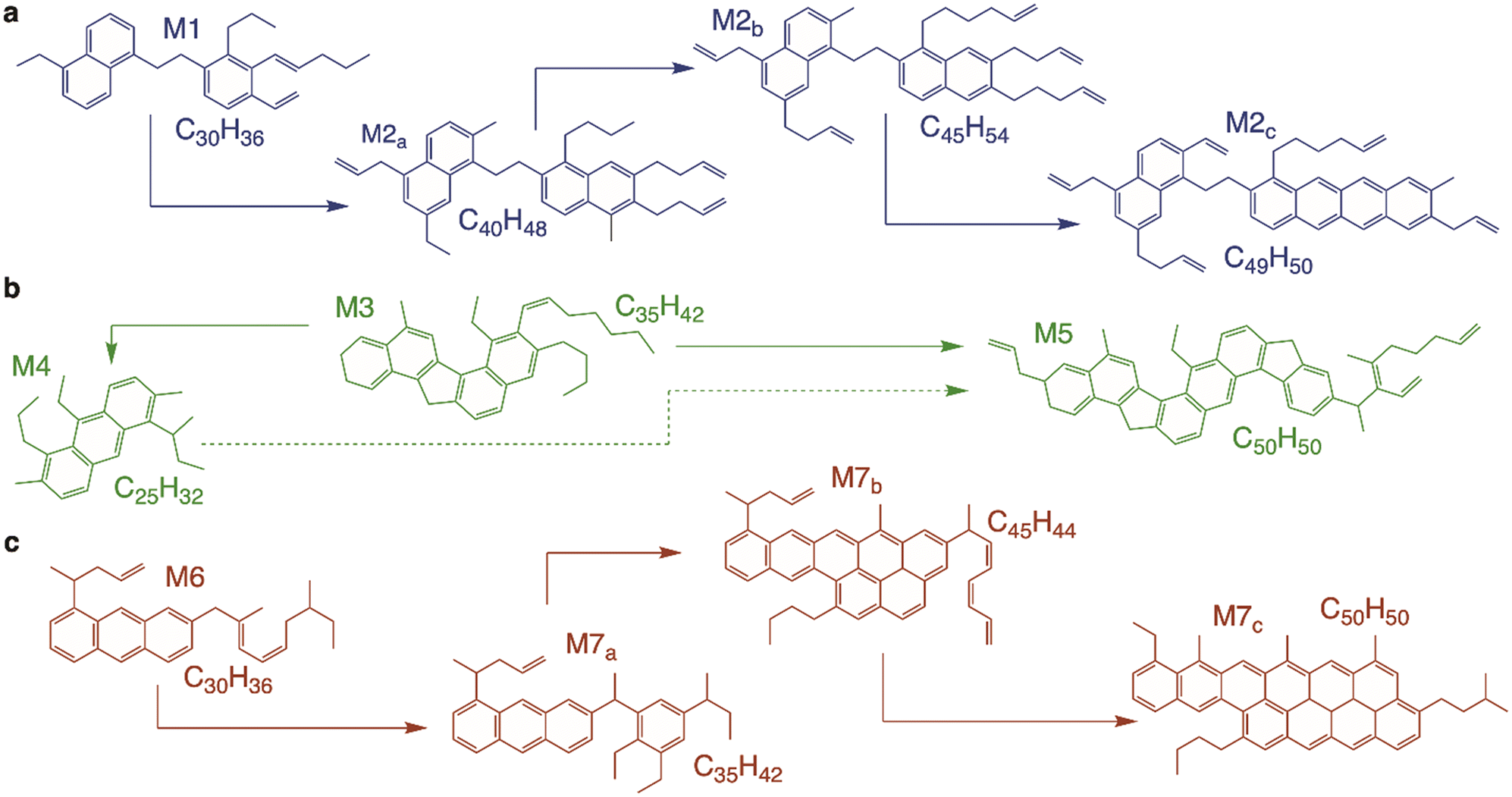 | ||
| Fig. 8 Tentative molecular depictions of the hydrocarbon coke species formed on the (a) NiY, (b) NiZ and (c) Niβ catalysts based on the predicted molecular formulae obtained through FT-ICR/MS analysis. | ||
On the contrary, the growth mechanism of the 10-MR zeolites (Fig. 8b) is unusual: first, the initial pool of coke species (M4 in Fig. 6) is formed during the site poisoning stage on the acid sites of the catalyst and inside the micropores. Then, these species “unfold” into two main fractions: a lighter M3 fraction and a secondary heavier M5 fraction whose FT-ICR MS distribution decreases and broadens over time. Thus, the interpretation of this effect is the interconversion of M3 coke into M4 inside the pores of the zeolite, as shown in Fig. 8b. On the other hand, the distribution of M5 species keeps widening, showing a very heterogeneous distribution of species, indicating that these species are partially located in the exterior of the catalysts. While quantification of species cannot be performed directly from FT-ICR MS spectra, we can combine these results with those in Fig. 6 and confirm that M4 species (attributable to coke I, more external) are the predominant ones after 10 h of reaction over both 10-MR zeolites, and even after 5 h in the case of the NiZ15 catalyst (Fig. 6h). Conversely, using 12-MR zeolite coke type II was predominant in all cases (Fig. S10†), despite decreasing in proportion over time on stream, because of the presence of supercages (NiY) and much wider pores (Niβ) on the catalysts as shown in Fig. 2.
All in all, we demonstrate that the direct analysis of the total deposited coke over spent catalysts (without pretreatments) through high-resolution MS, and in combination with conventional coke quantification techniques for further rationalization of the obtained spectra, is a highly powerful tool for understanding coke formation mechanisms at the molecular level. We must also consider that through LDI FT-ICR MS, we might face certain limitations regarding the ionization of the coke species, which can hinder the detection of the “full picture” of coke species. These ionization limitations can be caused by either the nature of the coke species and/or their location within the catalyst structure (either more accessible or trapped within blocked pores). For a detailed understanding of the potential limits of the direct coke analysis, in the following section, we individually analyze the different coke fractions after HF zeolite disaggregation and subsequent isolation through organic extraction of the soluble and insoluble cokes. Given the peculiar deactivation mechanism on 10-MR zeolites, we decided to focus on these catalysts.
3.4. Molecular-level characterization of the different coke fractions
Following the protocol specified in Fig. 1, we isolated the soluble and insoluble coke fractions from a deactivated 10-MR zeolite (NiZ40 catalyst) at different times on stream (1, 5 and 10 h) and analyzed both separately. The GC analysis (Fig. S17†) reveals a decreasing proportion of soluble coke over time in the total formed coke using 10-MR zeolites, with weight fractions of 14%, 11%, and 10% at 1, 5, and 10 h on stream, respectively, concerning the total coke deposited on the NiZ40 catalyst.19 This evolution over time on stream (and upon a progressively greater overall coke formation, see Fig. 4) of the soluble coke exemplifies the transformation of a higher proportion of alkylated monoaromatics into structures with greater aromaticity and weight and a lower degree of alkylation.From the 1H NMR spectra (Fig. S18 and Table S1†) of the soluble cokes from the same used NiZ40 catalyst, we detected an increase in the aromatic hydrogens linked to aromatic carbons in the polyaromatic structures as well as alkyl and naphthenic hydrogens linked to aromatic systems, as displayed in Fig. S19.† Overall, the aromatic hydrogen content increased with time on stream as a result of the bulkier coke species growing outside the catalyst micropores, more accessible for alkylation and aromatization reactions, and also with a higher H/C ratio than the coke species retained within the micropores. From the profiles of the carbon and DBE distributions derived from the FT-ICR MS analysis of the different coke fractions (total, soluble, and insoluble) in Fig. 9, we can compare the compositional molecular-level dynamics of the total coke analyzed directly and its independent soluble and insoluble fractions. This is an important step forward in understanding not only the formation mechanism but also the spatial distribution of the different types of coke within the zeolite framework, which can only be attained through FT-ICR MS and by applying the disaggregation–extraction protocol (Fig. 1).
 | ||
| Fig. 9 Comparison of the carbon and DBE number distributions obtained using FT-ICR MS to analyze the (a and b) total coke, (c and d) soluble coke, and (e and f) insoluble coke fractions formed on the NiZ40 catalyst at 1, 5, and 10 h on stream. | ||
Through the direct LDI FT-ICR MS analysis of total coke, we can discern the split of the total coke on the used NiZ40 catalyst from a single peak toward a bimodal distribution in terms of carbon number (Fig. 9a) and unsaturation degree (Fig. 9b), which is in line with the behavior observed from the coke combustion profiles in Fig. S10.† After 1 h on stream, the soluble and insoluble coke carbon number distributions are relatively similar (Fig. 9b and c), while the unsaturation degree of insoluble coke (Fig. 9e and f) is higher in contrast. This indicates that secondary reactions (hydride transfer, aromatization) from soluble coke are occurring to a significant extent. However, at longer times on stream, soluble and insoluble coke species evolve independently. On the one hand, the carbon number distribution of soluble coke (Fig. 9c) barely changes over time on stream, becoming slightly more unsaturated (Fig. 9d) as the reaction progresses. On the other hand, insoluble coke (Fig. 9e and f) evolves more dramatically towards more aromatic and more unsaturated types of species, particularly after 5 h on stream. These observations evidence the limitations of the direct LDI ionization of total coke (Fig. 9a and b), and more specifically for discerning the dynamics of the insoluble coke fraction, which remains largely “invisible” unless isolated for its individual analysis. Contrary to the well-established premise of insoluble coke being deposited on the outermost layers of the catalyst due to the spatial limitations of micropores for accommodating large aromatic structures,37,68 we prove that a fraction of it can also remain trapped within narrower and/or blocked pores, hence not exposed to ionization in the FT-ICR MS analysis of total coke.
FT-ICR MS analysis of isolated coke fractions also provides valuable information for a more accurate interpretation of the dynamics observed via TG-TPO MS. While in Fig. 4 and S10† there is an apparent increase in the amount of coke I (lighter in nature) over time on stream, the FT-ICR MS results indicate that a fraction of what is quantified as coke I via TG-TPO is likely insoluble coke located in the outermost part of the catalyst cavities, therefore more accessible for oxidation/combustion. This overestimation of coke I through TG-TPO MS can be conditioned by the high proportions of insoluble coke (>86%) in contrast to soluble coke and becomes more pronounced as the content of insoluble coke increases.
4. Discussion
Ethylene oligomerization occurs through formation of long oligomers in the metallic and acid sites of the catalyst (preferentially in the former ones). From the coke formation point of view, cyclic species are not necessarily formed on the oligomerization sites. These long oligomers diffuse through the zeolite micropores to find acid sites and proceed through carbocation chemistry involving further oligomerization, β-scission, and bimolecular hydride transfer (Fig. S8†). As a product of the latter reaction, aromatics form, which can either diffuse or stay in the pores. The 12-MR zeolites (HY and Hβ) produce a greater amount of high-molecular-weight oligomers over the Ni2+ sites due to the lower diffusional barrier in these zeolites and the higher chance of bimolecular reactions.Once the carbocation chemistry proceeds over the acidic zeolite sites, the heavier molecules formed in the site poisoning stage by hydride transfer with aromatic islands (M1 and M6 species, Fig. 8) do not escape the framework; rather, they continue aging and growing in the pore blocking stage of coke formation (M2c and M7c species, Fig. 8). This effect simultaneously limits the number of coke precursors that react further over the Ni2+ from the acid sites, causing the former sites to keep their activity for longer. Conversely, the 10-MR zeolite (H-ZSM-5) exchanged with Ni2+ produces oligomers of lower molecular weight (due to higher diffusional barriers), and the molecules formed either remain trapped in the micropores or escape the zeolite framework due to the MFI zeolite framework's relatively high micropore connectivity. The latter species are strongly adsorbed on the NiO (out of the zeolite), where they keep growing less selectively into bulkier and more condensed coke species.
These mechanisms explain the nature of the coke species and their evolution depending on the pore topology of the catalysts. They also explain why 12-MR zeolites retain higher conversion due to maintained Ni2+ site accessibility. In the 10-MR zeolites, the “expulsion” of coke precursors out of the framework leads to stronger adsorption of these over the NiO and the simultaneous deactivation of Ni and acid zeolite sites. This is the reason behind the relatively similar performance and lifetime of 10-MR zeolites with substantially different numbers of acid sites. These coke mechanisms on ZSM-5 zeolites have also been reported using other techniques, such as operando X-ray diffraction.69
In terms of carbon number, this external coke is measured within the insoluble coke fraction and the total coke is measured by direct analysis. Interestingly, that does not happen in terms of the DBE, i.e., the external coke measured within the insoluble coke fraction (with DBE >ca. 35, Fig. 9f) is not detected within the total coke (Fig. 9a and b). This means that in our developed technique of direct analysis of coke, there is a fraction of coke that is external, with a very high carbon number (>50), and aromatic (DBE > 35), that cannot be analyzed due to its partial entrapment within the pores of the catalysts. However, our technique can detect aromatic structures growing inside the pores (soluble coke) without dissolving the catalyst structure.
The results presented here from combined direct coke and soluble–insoluble coke analysis using FT-ICR combined with high-resolution mass spectrometry can capture a nearly complete description of the coke dynamics at a molecular level. The direct analysis of coke fractions captures the dynamics of relatively light coke fractions trapped in the catalyst (soluble coke) and overlooks the one of a small fraction of insoluble, very aromatic coke. We also prove that the internal coke is the one that can keep a narrow distribution of composition over time, whereas the external coke widens due to disorganized growth. The term “external” should be pictured with species that grow outside the micropores of the catalyst but maintain a certain degree of entrapment within the catalyst, which is why these species cannot be fully analyzed with the direct method. All in all, this kind of analysis puts the controlling effect of coke in the overall zeolite catalysis in place and it can be expanded to other chemistries to develop more advanced models, reactors, and catalysts.
5. Conclusions
In this work, we have elucidated the nature, location, and evolution of the analytically elusive coke species formed on zeolite catalysts of different pore topologies (ZSM-5, β, and Y) during ethylene oligomerization. We demonstrate that through the application of laser desorption ionization Fourier-transform ion cyclotron resonance combined with high-resolution mass spectrometry (LDI FT-ICR MS), it is possible to perform the direct analysis of the coke deposited over the deactivated catalysts and resolve the coke species at the molecular level.We have also concluded that for rationalizing the obtained results, more conventional coke characterization techniques are also required (i.e., thermogravimetric temperature-programmed oxidation) as well as a complete description of the composition of the reaction medium to elucidate the coke precursors. Further adsorption calculations demonstrate where the coke-deactivating species are strongly adsorbed, which in our case is on Ni2+ compared to acid sites.
To explore the potential and limitations of the direct coke analysis, we have compared this novel approach with the more standard analysis of the individual coke fractions after extraction (soluble and insoluble coke). Our results demonstrate that, comparatively, the direct analysis of coke is efficient for detecting soluble coke species. Still, a fraction of insoluble coke remains “invisible” unless isolated through HF treatment and subsequent extraction. Furthermore, we have also provided evidence that contrary to the conventional assumptions, a fraction of the insoluble coke can also remain trapped within the zeolite pore structure. The methodologies developed in this work are paramount steps in explaining the coke formation and evolution/growth pathways.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was conducted thanks to the financial support of the King Abdullah University of Science and Technology (KAUST, BAS/1/1403). We acknowledge Mohammed Khalid for his assistance with ICP-OES analysis.References
- A. Primo and H. Garcia, Chem. Soc. Rev., 2014, 43, 7548–7561 RSC
.
-
R. Sadeghbeigi, Fluid Catalytic Cracking Handbook: An Expert Guide to the Practical Operation, Design, and Optimization of FCC Units, Butterworth-Heinemann, Waltham, MA, 2012 Search PubMed
- I. Hita, T. Cordero-Lanzac, F. J. García-Mateos, M. J. Azkoiti, J. Rodríguez-Mirasol, T. Cordero and J. Bilbao, Appl. Catal., B, 2019, 118112 CrossRef CAS
- Y. Xue, J. Li, P. Wang, X. Cui, H. Zheng, Y. Niu, M. Dong, Z. Qin, J. Wang and W. Fan, Appl. Catal., B, 2021, 280, 119391 CrossRef CAS
- A. G. Gayubo, A. Alonso, B. Valle, A. T. Aguayo and J. Bilbao, Appl. Catal., B, 2010, 97, 299–306 CrossRef CAS
- T. Shoinkhorova, A. Dikhtiarenko, A. Ramirez, A. Dutta Chowdhury, M. Caglayan, J. Vittenet, A. Bendjeriou-Sedjerari, O. S. Ali, I. Morales-Osorio, W. Xu and J. Gascon, ACS Appl. Mater. Interfaces, 2019, 11, 44133–44143 CrossRef CAS PubMed
- M. Dusselier, M. A. Deimund, J. E. Schmidt and M. E. Davis, ACS Catal., 2015, 5, 6078–6085 CrossRef CAS
- Y. Ni, Y. Liu, Z. Chen, M. Yang, H. Liu, Y. He, Y. Fu, W. Zhu and Z. Liu, ACS Catal., 2019, 9, 1026–1032 CrossRef CAS
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS
- K. Gong, Y. Wei, Y. Dai, T. Lin, F. Yu, Y. An, X. Wang, F. Sun, Z. Jiang and L. Zhong, Appl. Catal., B, 2022, 316, 121700 CrossRef CAS
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed
- Z. Yang, Z. Zhang, Y. Liu, X. Ding, J. Zhang, J. Xu and Y. Han, Appl. Catal., B, 2021, 285, 119815 CrossRef CAS
- P. Zhang, L. Ma, F. Meng, L. Wang, R. Zhang, G. Yang and Z. Li, Appl. Catal., B, 2022, 305, 121042 CrossRef CAS
- N. J. LiBretto, Y. Xu, A. Quigley, E. Edwards, R. Nargund, J. C. Vega-Vila, R. Caulkins, A. Saxena, R. Gounder, J. Greeley, G. Zhang and J. T. Miller, Nat. Commun., 2021, 12, 2322 CrossRef CAS PubMed
- H. Olivier-Bourbigou, P. A. R. Breuil, L. Magna, T. Michel, M. F. Espada Pastor and D. Delcroix, Chem. Rev., 2020, 120, 7919–7983 CrossRef CAS PubMed
- N. M. Eagan, M. D. Kumbhalkar, J. S. Buchanan, J. A. Dumesic and G. W. Huber, Nat. Rev. Chem., 2019, 3, 223–249 CrossRef CAS
- E. Koninckx, P. S. F. Mendes, J. W. Thybaut and L. J. Broadbelt, Appl. Catal., A, 2021, 624, 118296 CrossRef CAS
- P. Castaño, G. Elordi, M. Ibañez, M. Olazar and J. Bilbao, Catal. Sci. Technol., 2012, 2, 504–508 RSC
- M. Guisnet and P. Magnoux, Appl. Catal., A, 2001, 212, 83–96 CrossRef CAS
- W. Chen, G. Li, X. Yi, S. J. Day, K. A. Tarach, Z. Liu, S.-B. Liu, S. C. Edman Tsang, K. Góra-Marek and A. Zheng, J. Am. Chem. Soc., 2021, 143, 15440–15452 CrossRef CAS PubMed
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS
- A. Devaraj, M. Vijayakumar, J. Bao, M. F. Guo, M. A. Derewinski, Z. Xu, M. J. Gray, S. Prodinger and K. K. Ramasamy, Sci. Rep., 2016, 6, 37586–37597 CrossRef CAS PubMed
- B. Liu, D. R. Slocombe, J. Wang, A. Aldawsari, S. Gonzalez-Cortes, J. Arden, V. L. Kuznetsov, H. AlMegren, M. AlKinany, T. Xiao and P. P. Edwards, Nat. Commun., 2017, 8, 514 CrossRef CAS PubMed
- Q. Qian, J. Ruiz-Martínez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel and B. M. Weckhuysen, Catal. Today, 2014, 226, 14–24 CrossRef CAS
- D. S. Wragg, G. N. Kalantzopoulos, D. K. Pappas, I. Pinilla-Herrero, D. Rojo-Gama, E. Redekop, M. Di Michiel, P. Beato, L. F. Lundegaard and S. Svelle, J. Catal., 2021, 401, 1–6 CrossRef CAS
- F. A. Ali, A. Hauser, H. A. Abdullah and A. Al-Adwani, Energy Fuels, 2006, 20, 45–53 CrossRef CAS
- S. R. Bare, F. D. Vila, M. E. Charochak, S. Prabhakar, W. J. Bradley, C. Jaye, D. A. Fischer, S. T. Hayashi, S. A. Bradley and J. J. Rehr, ACS Catal., 2017, 7, 1452–1461 CrossRef CAS
- N. A. Alawani, H. Muller, S. K. Panda, A. A. Al-Hajji and O. R. Koseoglu, Energy Fuels, 2019, 34, 179–187 CrossRef
- K. Ben Tayeb, S. Hamieh, C. Canaff, H. Nguyen, H. Vezin and L. Pinard, Microporous Mesoporous Mater., 2019, 289, 109617 CrossRef CAS
- L. Pinard, S. Hamieh, C. Canaff, F. Ferreira Madeira, I. Batonneau-Gener, S. Maury, O. Delpoux, K. Ben Tayeb, Y. Pouilloux and H. Vezin, J. Catal., 2013, 299, 284–297 CrossRef CAS
- A. C. Rodriguez, M. E. Sad, H. Cruchade, L. Pinard and C. L. Padró, Microporous Mesoporous Mater., 2021, 320, 111066 CrossRef CAS
- A. Feller, J. O. Barth, A. Guzman, I. Zuazo and J. A. Lercher, J. Catal., 2003, 220, 192–206 CrossRef CAS
- T. Cordero-Lanzac, I. Hita, A. Veloso, J. M. Arandes, J. Rodríguez-Mirasol, J. Bilbao, T. Cordero and P. Castaño, Chem. Eng. J., 2017, 327, 454–464 CrossRef CAS
- B. Matas Güell, I. Babich, K. P. Nichols, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Appl. Catal., B, 2009, 90, 38–44 CrossRef
- N. Wang, Y. Zhi, Y. Wei, W. Zhang, Z. Liu, J. Huang, T. Sun, S. Xu, S. Lin, Y. He, A. Zheng and Z. Liu, Nat. Commun., 2020, 11, 1079 CrossRef CAS PubMed
- M. Gao, H. Li, W. Liu, Z. Xu, S. Peng, M. Yang, M. Ye and Z. Liu, Nat. Commun., 2020, 11, 3641 CrossRef CAS PubMed
- J. Valecillos, I. Hita, E. Sastre, A. T. Aguayo and P. Castaño, ChemCatChem, 2021, 13, 3140–3154 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4978 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- K. Hu, M. Wu, S. Hinokuma, T. Ohto, M. Wakisaka, J. Fujita and Y. Ito, J. Mater. Chem. A, 2019, 7, 2156–2164 RSC
-
International Zeolite Asociation (IZA), Database of zeolite structures, 2017 Search PubMed
- H. Sato, T. Minami, S. Takata and T. Yamada, Thin Solid Films, 1993, 236, 27–31 CrossRef CAS
- R. Y. Brogaard and U. Olsbye, ACS Catal., 2016, 6, 1205–1214 CrossRef CAS
- R. Joshi, A. Saxena and R. Gounder, Catal. Sci. Technol., 2020, 10, 7101–7123 RSC
- R. Joshi, G. Zhang, J. T. Miller and R. Gounder, ACS Catal., 2018, 8, 11407–11422 CrossRef CAS
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 CrossRef CAS PubMed
- A. Ochoa, B. Valle, D. E. Resasco, J. Bilbao, A. G. Gayubo and P. Castaño, ChemCatChem, 2018, 10, 2311–2321 CrossRef CAS
-
M. Guisnet, in Deactivation and Regeneration of Zeolite Catalysts, Imperial College Press, 2011, vol. 9, pp. 101–114 Search PubMed
- M. Guisnet, J. Mol. Catal. A: Chem., 2002, 182–183, 367–382 CrossRef CAS
- C. Wei, J. Li, K. Yang, Q. Yu, S. Zeng and Z. Liu, Chem Catal., 2021, 1, 1273–1290 CrossRef
- H. O. Mohamed, R. K. Parsapur, I. Hita, J. L. Cerrillo, A. Ramírez, K.-W. Huang, J. Gascon and P. Castaño, Appl. Catal., B, 2022, 316, 121582 CrossRef CAS
- D. Parmar, S. H. Cha, T. Salavati-fard, A. Agarwal, H. Chiang, S. M. Washburn, J. C. Palmer, L. C. Grabow and J. D. Rimer, J. Am. Chem. Soc., 2022, 144, 7861–7870 CrossRef CAS PubMed
- P. Castaño, G. Elordi, M. Olazar, A. T. Aguayo, B. Pawelec and J. Bilbao, Appl. Catal., B, 2011, 104, 91–100 CrossRef
- S. Lee and M. Choi, J. Catal., 2019, 375, 183–192 CrossRef CAS
- I. Hita, T. Cordero-Lanzac, G. Bonura, C. Cannilla, J. M. Arandes, F. Frusteri and J. Bilbao, J. Ind. Eng. Chem., 2019, 80, 392–400 CrossRef CAS
- S. Lin, Y. Zhi, W. Chen, H. Li, W. Zhang, C. Lou, X. Wu, S. Zeng, S. Xu, J. Xiao, A. Zheng, Y. Wei and Z. Liu, J. Am. Chem. Soc., 2021, 143, 12038–12052 CrossRef CAS PubMed
- H. S. Cerqueira, C. Sievers, G. Joly, P. Magnoux and J. A. Lercher, Ind. Eng. Chem. Res., 2005, 44, 2069–2077 CrossRef CAS
- M. Díaz, E. Epelde, J. Valecillos, S. Izaddoust, A. T. Aguayo and J. Bilbao, Appl. Catal., B, 2021, 291, 120076 CrossRef
- Z. Feng, X. Liu, Y. Wang and C. Meng, Molecules, 2021, 26, 2234 CrossRef CAS PubMed
- Y. V. Joshi and K. T. Thomson, J. Catal., 2005, 230, 440–463 CrossRef CAS
- T. C. Brüggemann, D. G. Vlachos and F. J. Keil, J. Catal., 2011, 283, 178–191 CrossRef
- E. A. Pidko, E. J. M. Hensen and R. A. van Santen, J. Phys. Chem. C, 2008, 112, 19604–19611 CrossRef CAS
- Y. Dang, Y. Liu, X. Feng, X. Chen and C. Yang, Appl. Surf. Sci., 2019, 486, 137–143 CrossRef CAS
- M. Guisnet, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2009, 305, 69–83 CrossRef CAS
- G. N. Kalantzopoulos, D. Rojo Gama, D. K. Pappas, I. Dovgaliuk, U. Olsbye, P. Beato, L. F. Lundegaard, D. S. Wragg and S. Svelle, Dalton Trans., 2022, 51, 16845–16851 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01850k |
| This journal is © The Royal Society of Chemistry 2023 |