
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntimate ruthenium–platinum nanoalloys supported on carbon catalyze the hydrogenation and one-pot hydrogenation-coupling reaction of oxidized amino derivatives†
Miguel A.
Rivero-Crespo‡
 a,
Paula
Rubio-Marqués§
a,
Juan Carlos
Hernández-Garrido
b,
Marta
Mon
a,
Judit
Oliver-Meseguer
a and
Antonio
Leyva-Pérez
*a
a,
Paula
Rubio-Marqués§
a,
Juan Carlos
Hernández-Garrido
b,
Marta
Mon
a,
Judit
Oliver-Meseguer
a and
Antonio
Leyva-Pérez
*a
aInstituto de Tecnología Química (UPV-CSIC) Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: anleyva@itq.upv.es
bDepartamento de Ciencia de los Materiales e Ingeniería Metalúrgica y Química Inorgánica, Facultad de Ciencias, Universidad de Cádiz, Campus Universitario Puerto Real, 11510 Puerto Real, Cádiz, Spain
First published on 15th March 2023
Abstract
The use of bimetallic nanoparticles as catalysts in complex organic synthesis is rare, despite the widespread use of these materials in thermal C1 chemistry and electrocatalysis. Here we show that intimate RuPt nanoalloys, supported on charcoal, catalyze the synthesis of secondary and tertiary amines from a variety of amino derivatives, such as quinolines, isoquinolines, nitrobenzenes, oximes and nitriles, under mild reaction conditions. The organic reactions include not only direct hydrogenation reactions but also hydrogen-borrowing couplings in cascade, to access the desired amines in one-pot from readily available materials. The recyclable bimetallic solid catalyst is at least one order of magnitude more active than the monometallic counterparts in some cases, which illustrates the advantages of Ru–Pt co-operation during hydrogenation and hydrogen-borrowing reactions.
Introduction
Alloyed bimetallic nanoparticles (nanoalloys) are now accessible to researchers and their use in a great variety of fields has rapidly increased during the last years.1 A representative bimetallic combination is ruthenium–platinum (RuPt), well-known during years in nanoalloyed form, and with extensive use in thermo- and electro-catalysis, for instance for carbon monoxide oxidation,2 hydrogen and oxygen evolution reactions (HER and OER),3 methanol oxidation and fuel cell applications,4 and hydrodeoxygenation reactions,4g among other reactions.5 Related to this, RuNi nanoalloys have also been employed as catalysts.6 In striking contrast, little use of RuPt nanoalloys has been done in more complex organic synthesis. To our knowledge, the selective hydrogenation of benzene to 1,4-cyclohexadiene7a and the reduction of phthalates7b have been some of the few reported examples with relatively complex organic reactions catalysed by RuPt nanoalloys.Reactions involving the selective transferring of H atoms constitute an atom-efficient, economic and environmentally-friendly way to prepare new chemicals in organic synthesis.8Fig. 1 shows two examples with broad industrial application,9 the classical Sabatier hydrogenation where H2 is activated, dissociated and transferred on a metal surface catalyst; and the hydrogen-borrowing coupling reaction, where the metal catalyst first receives dissociated H atoms from the substrate and then gives them back, after the coupling of two substrate molecules.10
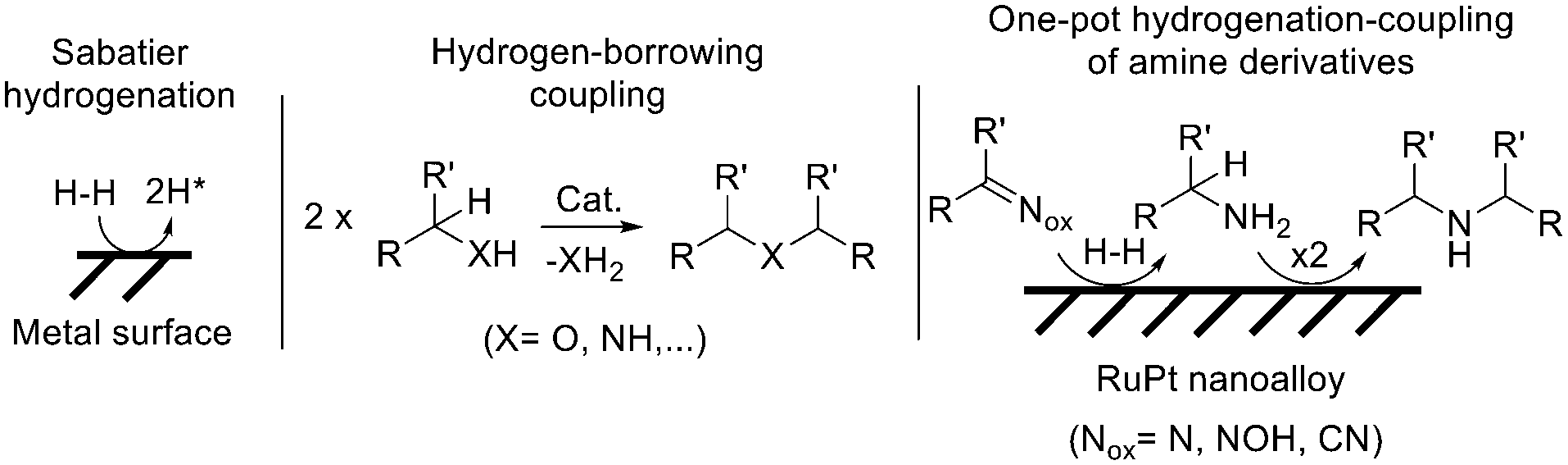 | ||
| Fig. 1 Management of H atoms by metal catalysts during classical hydrogenation and hydrogen-borrowing reactions, and the combination of both. The three reactions are studied in this work for different amines, with RuPt nanoalloyed catalysts. | ||
While the Sabatier reaction is commonly catalysed by Pt metal surfaces, the hydrogen-borrowing reaction is usually catalysed by Ru organometallic complexes; thus, the combination of both processes in one-pot with a single catalyst is not trivial.11,12 If achieved, the potential use of oxidised amino derivatives as starting materials for the synthesis of alkyl amines, after incorporation of an unlimited amount of external H atoms, will open new synthetic opportunities.
It is well-known that Pt(0) nanoparticles rank among the most active catalysts for the dissociation and transfer of H2 to unsaturated bonds such nitro and carbonyl derivatives,13 while low valence Ru complexes and nanoparticles are considered the most efficient catalysts for the hydrogenation of aromatics14,15 and the deaminative coupling of amines.16 Thus, it was envisioned here that a nanoparticle containing both Pt and Ru, intimately segregated, could catalyse in much better way than Pt and Ru, by separate, either the hydrogenation of amino-containing aromatic rings, the hydrogen-borrowing reaction of amines, or concomitantly both, enabling the one-pot hydrogenation–hydrogen-borrowing reaction of oxidized amine derivatives.17 It is shown herein that, indeed, Ru–Pt nanoalloys catalyse the synthesis of secondary and tertiary alkyl amines after hydrogenation of quinolines, isoquinolines, nitrobenzenes, nitriles or imines, and also of aromatic rings, enabling amine or oxime hydrogen-borrowing couplings during the reaction.
Results and discussion
Synthesis and characterisation of the catalysts.
Ru–Pt nanoalloys with molar ratios 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1 and 1:3, on activated charcoal (RuPt–C), were prepared by impregnation of the support with an aqueous solution of the corresponding amounts of H2PtCl4 and RuCl3. The total metal contain was typically kept in 5 wt%. The material was then dried at 100 °C and reduced under a H2/N2 flow (1:10) at 360 °C.18 Inductively coupled plasma-atomic emission spectroscopy (ICP-AES), after treatment of the solids with strong acid aqueous solutions and filtration, confirmed that the total metal amount was incorporated and persisted onto the solid. For the sake of comparison, monometallic Ru–C and Pt–C samples were prepared by the same methodology. Fig. 2 shows high resolution transmission electron microscopy (HR-TEM) images of different samples of RuPt–C, with well-dispersed, homogeneous nanocrystallites present in the catalysts, and with an average size around 5 nm. Table 1 shows the particle diameter size values for the different samples of RuPt–C, as well as of the monometallic Ru–C and Pt–C materials, the latter presenting average sizes of d = 9.6 and 2.3 nm, respectively (Fig. S1†). Powder X-ray diffraction (XRD) analyses confirmed the relative average size of the nanoparticles, after applying the Scherrer equation (Fig. S2†).
:1, 1:1 and 1:3, on activated charcoal (RuPt–C), were prepared by impregnation of the support with an aqueous solution of the corresponding amounts of H2PtCl4 and RuCl3. The total metal contain was typically kept in 5 wt%. The material was then dried at 100 °C and reduced under a H2/N2 flow (1:10) at 360 °C.18 Inductively coupled plasma-atomic emission spectroscopy (ICP-AES), after treatment of the solids with strong acid aqueous solutions and filtration, confirmed that the total metal amount was incorporated and persisted onto the solid. For the sake of comparison, monometallic Ru–C and Pt–C samples were prepared by the same methodology. Fig. 2 shows high resolution transmission electron microscopy (HR-TEM) images of different samples of RuPt–C, with well-dispersed, homogeneous nanocrystallites present in the catalysts, and with an average size around 5 nm. Table 1 shows the particle diameter size values for the different samples of RuPt–C, as well as of the monometallic Ru–C and Pt–C materials, the latter presenting average sizes of d = 9.6 and 2.3 nm, respectively (Fig. S1†). Powder X-ray diffraction (XRD) analyses confirmed the relative average size of the nanoparticles, after applying the Scherrer equation (Fig. S2†).
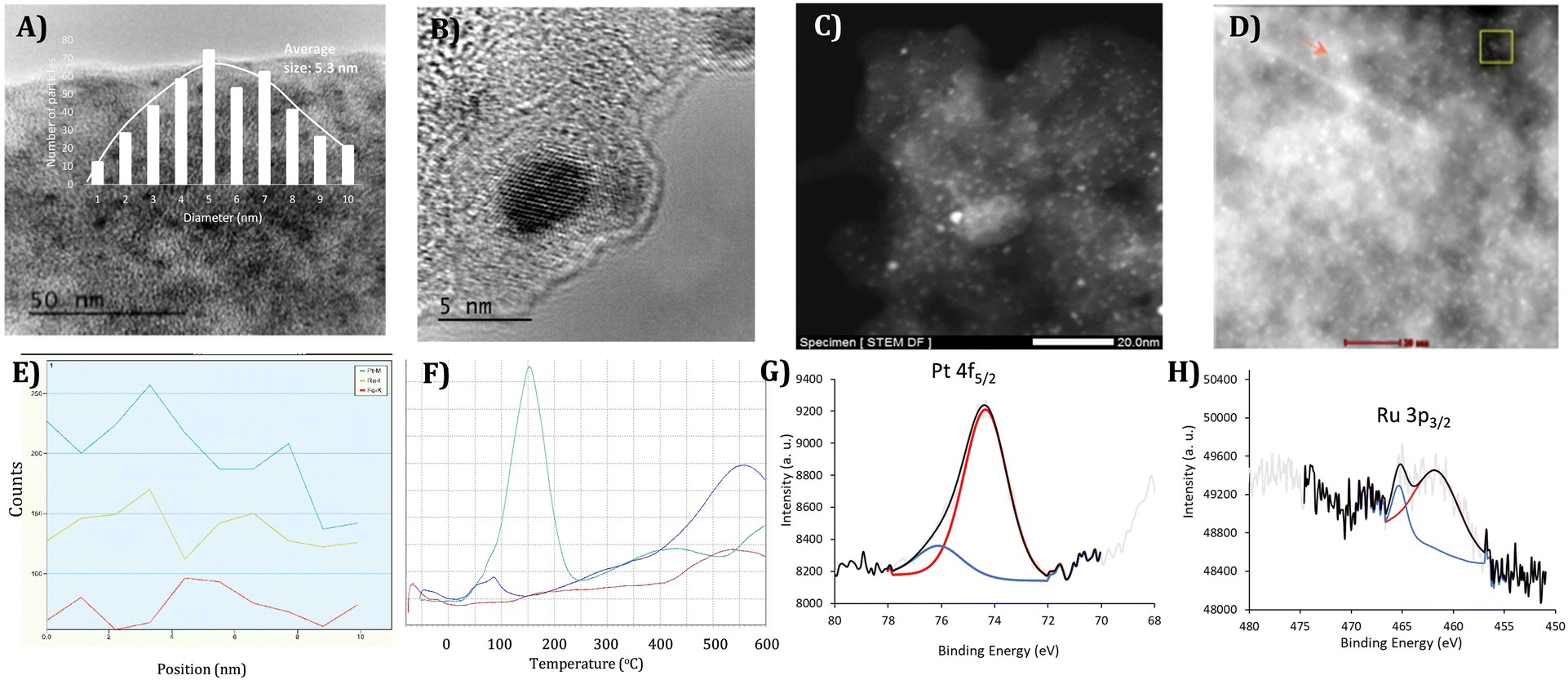 | ||
| Fig. 2 A) Representative HR-TEM image of 100 nm2 for RuPt–C (1:1), the inset shows the histogram taken from at least 5 different images and the calculated particle average size. B) Amplification of a RuPt nanocrystallite (1:1). C and D) Two representative STEM DF images of RuPt–C samples (1:1), the orange arrow points one typical nanoparticle. E) EDX sweeping spectra of the squared area in D), the signal for residual Fe on the support serves as a blank and follows a completely different pattern than the Ru and Pt signals. F) TPR spectra of Ru–C (blue line), Pt–C (red line) and a representative sample of RuPt–C (grey line, 1:1), after previous oxidation of the samples. G) Pt4f XPS of RuPt–C (1:1). H) Ru3p XPS of RuPt–C (1:1). | ||
| Entry | Catalyst | Particle diameter (d, nm) | Atoms on surface (%) |
|---|---|---|---|
| 1 | Ru–C | 9.6 | 7 |
| 2 | Pt–C | 2.3 | 19 |
| 3 | RuPt–C (2:1) |
6.5 | 11 |
| 4 | RuPt–C (1:1) |
5.3 | 13 |
| 5 | RuPt–C (1:3) |
4.0 | 13 |
Scanning transmission electron microscopy with annular dark field detector (STEM-DF) confirmed the metallic nature of the crystallites, and energy-dispersive X-ray spectroscopy (EDX) analysis, in Fig. 2D and E, shows that Ru and Pt are the only constituents of the nanocrystallites, and that both metals are intimately mixed at the sub-nanometer regime, since the signals of Ru and Pt are spatially coupled (see also Fig. S3†).
Scanning of some discrete particles in FESEM supports the presence of Ru and Pt in equal amounts in the 1:1 nanoalloy (Fig. S4†). However, in order to have a more precision measurement of an individual nanoalloy crystal, STEM-DF with a high-angle annular detector was employed (Fig. S5†). Spectra were collected by sweeping during just 625 ms, to avoid damaging of the nanoparticle, which was enough to observe the interaction of the nanoparticle with the electron beam despite the very low current (barely 100 pA) employed. As it can be seen, the quantification, with the atomic error measured, gives a molar Ru:Pt ratio of approximately 1:1. Besides, only intimate but not monometallic, shell–core or partially segregated particles could be observed. In view of the extremely good homogeneity of the nanoparticles on the material (Fig. S6†), these results strongly support the formation of intimate RuPt nanoalloys on the charcoal surface.
In order to confirm the formation of intimate RuPt nanoalloys, temperature-programmed reduction (TPR) experiments between −100 and 600 °C were performed, for Ru–C, Pt–C and RuPt–C samples, after previous oxidation under air. Fig. 2F shows that Ru needs a temperature of ≈500 °C to be reduced, Pt is extremely easy to be reduced, at ≈−50 °C, and the RuPt sample (1:1) shows an intermediate reduction temperature of 150 °C, with a signal in the RuO2 reduction temperature range (450 °C) that accounts for ∼15% of the total integrated area.18 The 2:1 and 1:3 RuPt–C samples consistently gave the intermediate temperature reduction peak as the major signal, at around 150 °C (Fig. S7†). The RuO2 signal decreases as the RuPt nanoalloy enriches in Pt, to give <10% of integrated area for pure Ru phase in the 1:3 RuPt–C sample. These results confirm that the three RuPt–C materials are mainly formed by intimate alloyed nanoparticles, which no more presents the chemophysical properties of the individual Ru or Pt aggregates but intermediate properties.
Fig. 2G and H shows the X-ray photoelectron spectroscopy (XPS) measurements of RuPt–C (1:1), with main peaks at binding energies of 74.4 and 461.6 eV for Pt 4f5/2 and Ru 3p3/2, respectively, which agrees well with both metals in zero valence state. Small contributions (around 15% for both) of 76.1 eV and 465.3 eV can be assigned to Pt2+ and Ru4+, respectively, associated with surface species.19
Catalytic results
We first focused on the selective hydrogenation of quinolines, a challenging reaction in organic chemistry since the basic N atom of the aromatic ring easily poisons and deactivates metallic catalysts.20Table 2 shows the regioselective hydrogenation of quinoline 1 to 1,2,3,4-tetrahydroquinoline 2 in the presence of Ru–C, Pt–C or RuPt–C catalysts. While both monometallic catalysts give <50% of 2 (entries 1–4), the three different RuPt nanoalloys consistently give good yields of 2 (44–83%, entries 6–13), regardless of the order of impregnation of the metal on the support (compare entries 10 and 12–13). The 1:3 RuPt nanoalloy proved catalytically superior to the 1:1 and 2:1 counterparts, and the total amount of metals can be decreased to 1 mol% without depletion in the final yield of 2 (entry 7). Even the amount of metals impregnated on the solid can be decreased ten times (to 0.5 wt%) without hampering the final yield, provided that a total catalytic metal amount of at least 1 mol% is employed (entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Metal | Mol%a | Conversionb (%) | Yieldb (%) |
| a Total amount of metals. b GC yield, using n-dodecane as an internal standard. c 10 times less metal wt% impregnated on the solid. d Pt impregnation first. e Ru impregnation first. | ||||
| 1 | Ru | 1 | 41 | 25 |
| 2 | 10 | 100 | 48 | |
| 3 | Pt | 1 | 9 | 0 |
| 4 | 10 | 74 | 35 | |
| 5 | RuPt (1:1) |
1 | 54 | 41 |
| 6 | 10 | 100 | 68 | |
| 7 | RuPt (1:3) |
1 | 94 | 83 |
| 8 | 10 | 100 | 80 | |
| 9 | RuPt (2:1) |
1 | 59 | 44 |
| 10 | 10 | 100 | 64 | |
| 11 | RuPt (1:1)c |
1 | 100 | 68 |
| 12 | RuPt (1:1)d |
10 | 100 | 73 |
| 13 | RuPt (1:1)e |
10 | 100 | 80 |

Fig. 3 shows that the different RuPt–C catalysts allow the selective hydrogenation not only of different quinolines (products 2, 4–18) but also of isoquinolines (product 3), benzofuranes (product 19) and benzoic methyl ester (product 20),21 in good yields and with good tolerance for halide (products 5, 6 and 10), alcohol (product 7), ether (product 9), cyano (product 12), carboxylic acid (product 13), amide (product 14) and ester substitutions (product 20).
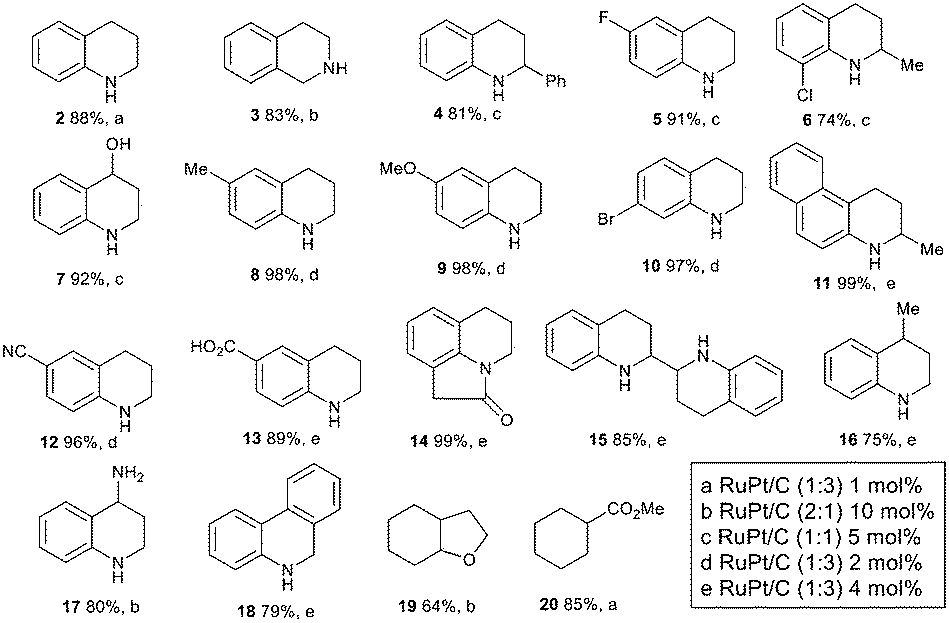 | ||
| Fig. 3 Hydrogenation of quinolines, isoquinolines, benzofuranes and arenes using different RuPt nanoparticles on carbon. Reaction conditions: THF (0.25 M), H2 (10 atm), RuPt/C, 60 °C, 18 h. GC or NMR yield. | ||
It must be noticed that the partially hydrogenated bicyclic aromatic products are stable under hydrogenating reaction conditions, and resist a further hydrogenation of the second aromatic ring. However, if the RuPt–C catalyst is active enough, it may be expected that the hydrogenation of anilines will occur under similar reaction conditions. Table 3 shows the results for the hydrogenation of nitrobenzene 21 at 60 °C in the presence of Ru–C, Pt–C or RuPt–C catalysts. Here, the nitro group should be easily hydrogenated to give aniline in one-pot. Indeed, 1 mol% of the Ru and Pt catalysts give aniline 22 as the major product (entries 1 and 2), however, the different RuPt nanoalloy compositions give the corresponding hydrogenation-coupling product, i.e. dicyclohexylamine 24, in significant amounts, to finally achieve a 94% yield of 24 when 10 mol% of RuPt (2:1) is used (entry 8). In contrast to the hydrogenation of quinolines and derivatives, the hydrogenation-coupling reaction proceeds better with Ru-rich rather than Pt-rich nanoalloys (compare entries 5, 8 and 9). These results reflect the importance of studying different metal compositions of the supported nanoalloy in catalytic studies.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Metal | Mol%a | 22 (%) | 23 (%) | 24 (%) | Selectivity to 24 (%) |
| a Total amount of metals. b GC yield, using n-dodecane as an external standard. c 10 times less metal wt% impregnated on the solid. d Pt impregnation first. e Ru impregnation first. | ||||||
| 1 | Ru | 1 | 99 | — | — | — |
| 2 | Pt | 1 | 92 | 8 | — | — |
| 3 | RuPt (1:1) |
1 | 79 | — | 6 | 79 |
| 4 | 5 | 28 | 2 | 19 | 28 | |
| 5 | 10 | 5 | 2 | 17 | 17 | |
| 6 | RuPt (2:1) |
1 | 41 | 26 | 33 | 56 |
| 7 | 5 | 67 | 8 | 25 | 76 | |
| 8 | 10 | — | 6 | 94 | 94 | |
| 9 | RuPt (1:3) |
10 | — | 12 | 88 | 88 |
| 10 | RuPt (1:1)c |
1 | 57 | 6 | 12 | 12 |
| 11 | RuPt (1:1)d |
10 | 45 | 4 | 11 | 11 |
| 12 | RuPt (1:1)e |
10 | 0 | 4 | 36 | 36 |
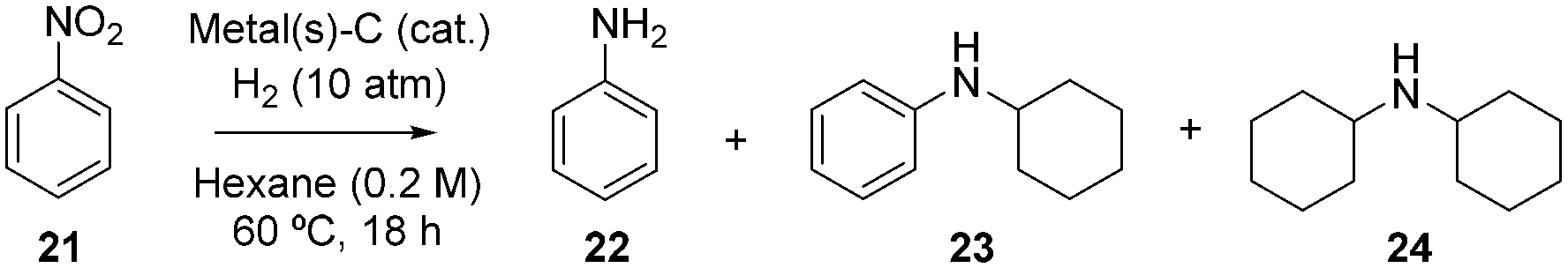
The order of impregnation of the metal in the support is not catalytically relevant (compare entries 5 and 11–12) and the amount of impregnated metal in the solid has also low impact in the results (compare entries 3 and 10). Other reaction conditions tested, which include different reaction temperatures (25 to 100 °C), solvent (dichloromethane, 1,4-dioxane, dimethylsulfoxide and water), substrate concentration (neat) and support (Al2O3), did not improve the results.
The superiority of RuPt nanoalloys vs. the separate metals to catalyse the hydrogenation of aniline is in line with previous literature.6,22–24 However, the ability of RuPt–C to catalyse the amine coupling is unique, and nicely complements the direct hydrogenation. With these results in hand, the coupling of other oxidized amine derivatives, in this case oximes, was attempted. Oximes have been rarely used as coupling partners in hydrogen borrowing reactions,25 despite they are intermediates during the hydrogenation of nitrocompounds.26 Thus, the possibility that oximes could also participate in the one-pot hydrogenation-coupling process, with the RuPt–C catalysts, was tested.
Table 4 shows that cyclohexanone oxime 25 reacts in the presence of 5 mol% of different RuPt–C samples to give dicyclohexanamine 24 in 89–94% yields, while Ru–C and Pt–C gave 37 and 67% yields, respectively. Kinetic experiments showed that the initial turnover frequency (TOF0) for the RuPt–C catalysts approaches 10000 h−1, one order of magnitude higher than for monometallic catalysts (≈500 and 2300 h−1 for Ru and Pt, respectively). Although it is true that the concept of TOF is questionable for nanoparticles since it considers that all the surface atoms are active during catalysis, the TOF is at least a quantitative approach to compare between different nanoparticle sizes.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yielda (%) | TOF0b (h−1) |
| a GC yield, using n-dodecane as an external standard. b Calculated as initial rate of the reaction per superficial atoms, considering perfect nanocubes (see next section). | |||
| 1 | Ru–C | 37 | 535 |
| 2 | Pt–C | 67 | 2353 |
| 3 | RuPt–C (1:1) |
92 | 4285 |
| 4 | RuPt–C (2:1) |
94 | 8300 |
| 5 | RuPt–C (1:3) |
89 | 14250 |

Fig. 4 shows the one-pot synthesis and then reductive coupling of oximes, from aldehydes and hydroxylamine hydrochloride, to yield the symmetric secondary amines 27a–b as coupling products in excellent yields. Since the participation of nitrobenzenes and oximes in amine coupling under hydrogenating conditions opens the door to the use of other amine precursors, nitriles were also tested,27 and Fig. 4 also shows that they can be coupled to oximes to give amines 28a–d and 29a–d in reasonable yields.
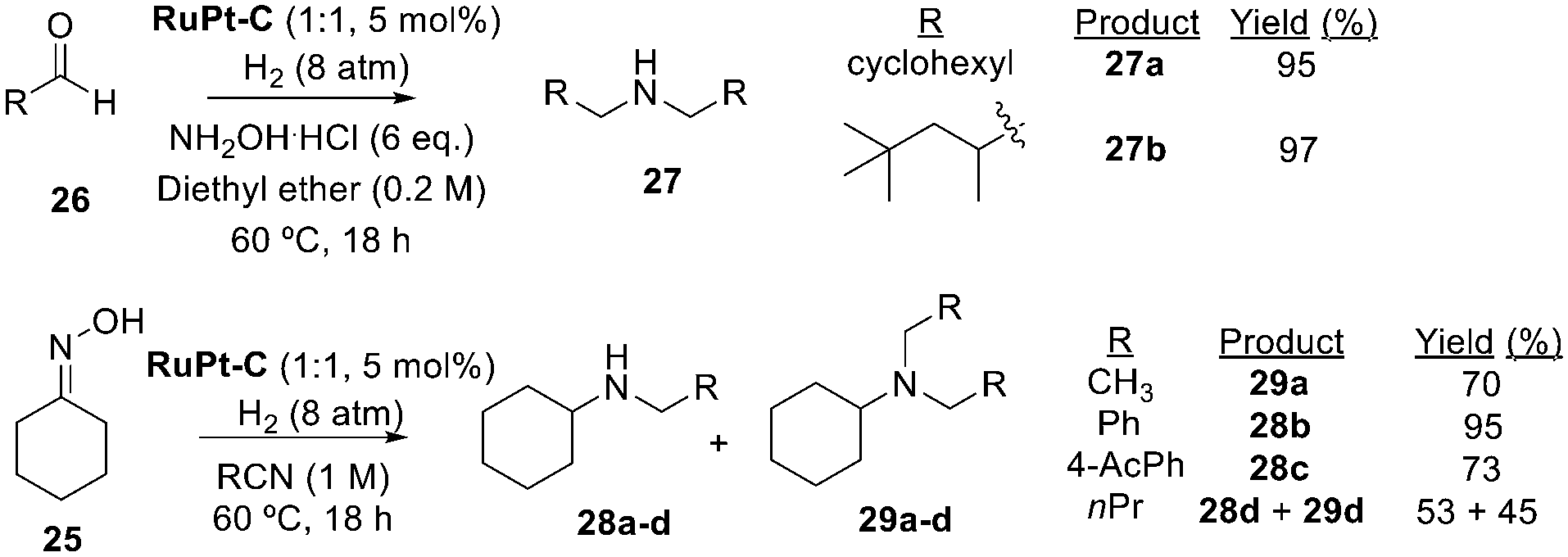 | ||
| Fig. 4 One-pot synthesis of secondary and tertiary amines catalysed by RuPt–C (1:1) from aldehydes 26 and hydroxylamine chloride (top), and from oxime 25 and nitriles (bottom). | ||
The RuPt–C (1:1) catalyst could be reused up to 4 times for the coupling of cyclohexanone oxime 25, with a decrease in the final yield from 92 to 77% yield. A hot filtration test shows that catalytically active species are not present in solution (Fig. S8†), which confirms the heterogenous nature of the catalysis but does not explain the loss of catalytic activity throughout the reuses. A possible cause for catalyst deactivation is aggregation of the nanoalloy particles during reaction. However, XRD analysis of the spent catalyst did not show any aggregation of the RuPt NPs according to the Scherrer equation (Fig. S9†). Another possible cause for catalyst deactivation is poisoning of the surface with heavily adsorbed organic products. To study this, an elemental analysis of the thoroughly washed used catalyst was performed, and it was found that the spent solid catalyst contained an additional 2.2% of nitrogen after reaction (Table S1†). Thus, the loss of catalytic activity can be ascribed to adsorbed nitrogen species derived from the reactant which, however, does not severely hamper the recyclability of the solid catalyst. It has to be noticed that while the RuPt nanoparticles are stable according to the Scherrer equation, the Ru nanoparticles agglomerate, at least to 2.5 times bigger average diameter sizes (Fig. S9†), and the latter also adsorb organic compounds (Table S1†).
Catalytic reaction mechanism
The catalytic activity of the supported nanoparticles can be referred to the surface atoms, in contact with the reagents. Thus, the % of surface atoms for each catalyst was calculated (Fig. S10†). For that, the following formula was employed: d = 1.105 × 0.29 × Ntotal1/3, where the diameter of the nanoparticle (d, obtanied from HR-TEM experiments) was used to calculate the number of total atoms (Ntotal) in the (Ru)Pt nanoparticle. The approach of considering nanocubes instead of circular particles facilitates the calculation and does not give much error for small nanoparticles, as it is our case here. With values taken from typical crystallographic distances for Ru and Pt, the % of atoms in surface for each catalyst could be calculated. The results are shown in Table 1, above. With these data in hand, the turnover frequencies for the individual reactions and catalysts can now be calculated, since initial rates for each solid catalyst are addressed from the initial points in the corresponding kinetic curve, where the particle size should still be maintained, at least in a first approximation. To check this, the RuPt–C (1:1) solid was treated with an atmosphere of H2 in the pre-chamber of the XPS instrument, at 60 °C for 1 h (Fig. S11†). The main peaks observed for Ru in the RuPt–C (1:1) sample before treatment appear at 460.0 eV (8%) and 461.6 eV (75%) for Ru, that corresponds to Ru0 and Ruδ+, respectively, and at 74.3 eV for Pt, which corresponds to Pt0 (85%). Despite the amplitude of the signals, it can be considered that the Ru 3p and Pt 4f peaks are mainly unmodified after H2 treatment, which confirms that the RuPt–C (1:1) catalyst keeps the observed structure under the catalytic hydrogenation reactions.
Then, we studied the hydrogenation reaction with a variety of aromatic molecules. First, three aromatic rings with very different electron density (anisole, tert-butylbenzene and trifluoromethylbenzene; Table S2 and Fig. S12†) were tested.28 It was found that the richer Pt nanoalloys, i.e. RuPt (1:3), were the more active catalysts, despite Pt–C was merely inactive. Notice that not only Ru–C and Pt–C by separate, were low active for the reaction, but also a physical mixture of Ru–C and Pt–C was also inactive. The low catalytic activity observed for the Ru/C catalyst can be explained by a partial oxidation on surface, which is easy considering the slow size of the crystallites and is in accordance with the XPS measurements. Without Pt in the nanoparticle, H2 splitting should be more difficult. Regarding substrate substitution, the different RuPt–C catalysts roughly obey the Taft rule, i.e. aromatic rings with bulky substituted groups reacts sluggishly due to steric constraints on the extended metal surface, as assessed here with tert-butylbenzene (see Table S2†). This result further proves the heterogeneous nature of the catalyst, in accordance with the classical Horiuti–Polanyi mechanism for the hydrogenation of aromatics on metal surfaces.29 A second study on the aromatic hydrogenation reaction was performed with acetophenone as a substrate (Tables S3–S5 and Fig. S13†), where it was found again that the RuPt nanoalloy (1:3) was much more catalytically active than the Ru-richer Pt nanoalloys and also than the monometallic counterparts, achieving complete hydrogenation of the substrate. A third study was performed with para-vinyl benzoic acid, a substrate containing alkene, aromatic and carboxylic acid functionalities (Table S6†). The results showed that the alkene functionality is much more rapidly hydrogenated than the arene ring on the RuPt–C (1:1) nanoalloy catalyst, while the carboxylic acid remains unreacted. These results prove that the catalytic activation of the organic functional group controls the reaction rate, and that H2 dissociation easily occurs on the RuPt–C nanoalloy. Overall, the results on the hydrogenation of different arene rings support the key role of nanoalloyed Pt as the more active metal for the catalysis.
Then, the hydrogen-borrowing coupling reaction of 22 was studied (Table S7†). It was found that Ru–C was very active in forming product 23, but not 24, if a stoichiometric amount of cyclohexylamine is added to the reaction mixture. The fact that product 23 (aromatic–aliphatic) is exclusively formed with the Ru–C catalyst, and not product 24 (aliphatic–aliphatic), and that cyclohexylamine is required for the coupling to proceed, strongly supports that Ru sites are mainly involved in the coupling but not in the hydrogenation reaction. In accordance, Pt–C is totally inactive for the coupling reaction under the same reaction conditions, while the RuPt–C (1:1) nanoalloy catalyst gives the fully hydrogenated product 24. These results corroborate that Ru metal atoms mainly constitute the active metal sites for the hydrogen-borrowing coupling reaction.
The above catalytic results and conclusions are in line with the expected electronic properties of the Pt and Ru atoms in the RuPt nanoalloy. Pt is more electronegative than Ru, and since both are in formal zero oxidation state, Pt removes some electron density from Ru to get itself slightly more electron rich, while Ru gets more electron poor within the intimate nanoalloy. The value of relative electron density transferred from Ru to Pt has been calculated to be ∼0.04 e−.30 Thus, in one hand, nanoalloyed Pt is more prone to dissociate H2 through transferring of electron density to the antibonding σ* orbital of H2, then facilitating hydrogenation reactions. And in the other hand, nanoalloyed Ru is slightly more cationic to coordinate and activate the C–N bonds of the amine, thus manifolding the hydrogen-borrowing coupling. XPS measurements of the different RuPt–C solids confirm the electronic transferring of Ru to Pt.31Fig. 5 shows that lower and higher binding energies values for Pt and Ru, respectively, can be found in the different RuPt nanoalloys, compared to the monometallic materials. For instance, the peaks corresponding to Pt 4f decrease in their binding energy (from 74.7 to 74.4 eV and from 77.3 to 76.0 eV, respectively) with the amount of Pt. Thus, one can say that electron poorer Ru atoms and electron richer Pt atoms are present in the nanoalloy.
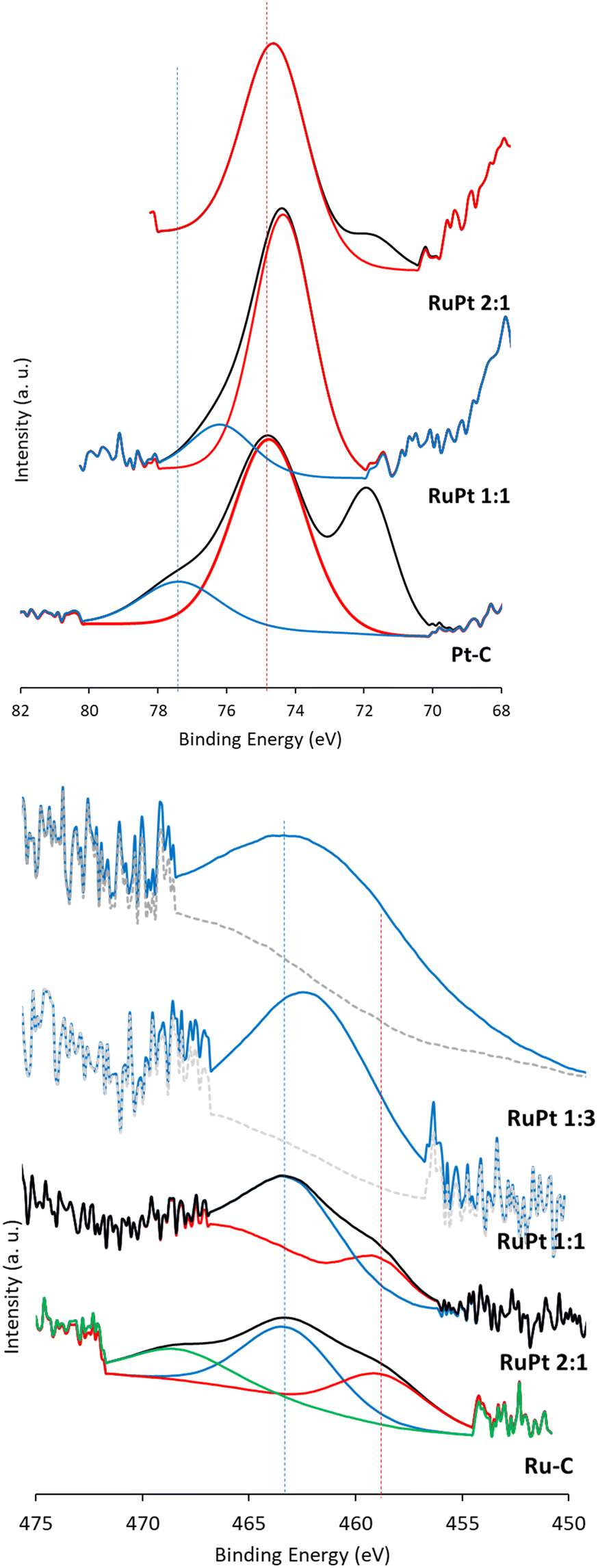 | ||
| Fig. 5 Deconvoluted Pt4f (top) and Ru3p (bottom) XPS measurements of the different solid catalysts, showing the electron transfer from Ru to Pt. For Pt, a shift to lower binding energies is found (more electron rich). For Ru, a shift to higher binding energies is found (more electron poor). Lines are a guide to the eye. Notice that the Pt 4f signal for the RuPt–C (1:3) alloy is not shown since residual pure Pt nanoparticles shift the signal to higher binding energies. Regarding the Ru signals, the alloy RuPt–C 1:1 does not show any metallic Ru signal, as it can be somewhat observed in the RuPt–C 2:1 alloy. | ||
Fig. 6 shows a plausible general mechanism for the hydrogenation and the one-pot hydrogenation and coupling reactions with intimate nanoalloyed RuPt–C catalysts.12 The oxidised alkyl or aromatic amine undergoes hydrogenation to primary alkyl amines over Pt sites on the catalyst. The hydrogenated molecule then participates in the hydrogen-borrowing event on the Ru sites of the catalyst, to give the corresponding secondary amine. Further hydrogenation can then occur, again on Pt sites. Depending on steric effects, the secondary amine can suffer, or not, a last dehydrogenation and coupling to form the tertiary amine. Alternatively, the secondary amine can attack a primary imine formed by dehydrogenation of the primary amine to form the tertiary amine. This mechanism is supported by the formation of the coupling product 24 when starting the reaction either from aniline 22 or cyclohexylamine, and also by the confirmation by gas-chromatography coupled to mass-spectrometry (GC-MS) that NH3 appears as a major by-product during reaction.
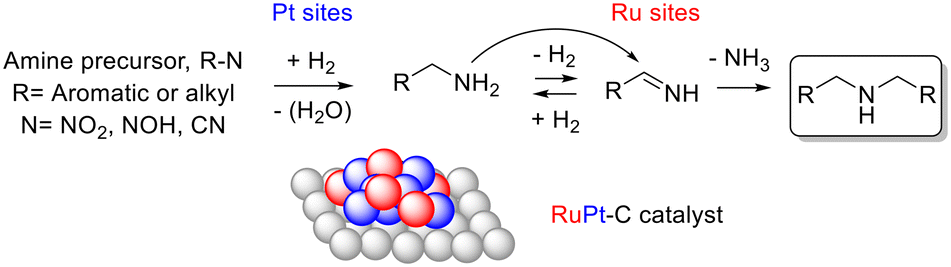 | ||
| Fig. 6 Proposed reaction mechanism for the hydrogenation and one-pot hydrogenation-coupling of nitrocompounds, oximes and/or nitriles, over RuPt–C catalysts and under a H2 atmosphere. | ||
Conclusions
RuPt nanoalloys supported on charcoal outperform the catalytic activity of monometallic Ru–C and Pt–C nanoparticles for the synthesis of secondary and tertiary amines from oxidized amine derivatives, under a hydrogen atmosphere. The solid catalyst is truly heterogeneous and recyclable. These results may stimulate new studies on nanoalloys as catalysts for complex organic synthesis, likely superior compared to monometallic counterparts, especially in tandem reactions.Author contributions
M. A. R.-C. performed and analysed the data of the catalytic tests; P. R.-M. synthesized and characterized the materials and performed some catalytic experiments; J. C. H.-G. carried out and interpreted the HAADF-STEM analyses of individual RuPt/C (1:1) nanoparticles; M. M. and J. O.-M. performed and interpreted some characterization to establish the catalyst structure–activity relationship; A. L.-P. conceived and coordinated the work, supervised the synthetic and catalytic part, and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the project PID2020–115100GB–I00 (funded by Spanish MCIINN, MCIN/AEI/10.13039/501100011033MICIIN) is acknowledged. Financial support by Severo Ochoa centre of excellence program (CEX2021–001230–S) is also gratefully acknowledged. The work has also been funded by Generalitat Valenciana, Grupos Emergentes (GV/2021/138). M. M. and J. O.-M. thanks MICIIN from a contract under the Juan de la Cierva program (FJC2019–040523–I and IJC2018–036514–I, respectively). M. A. R.-C. and P. R.-M. thank MEC for FPU contracts. We thank M. Cabrero-Antonino for performing some experiments.Notes and references
- (a) P. K. Gupta and L. Mishra, Nanoscale Adv., 2020, 2, 1774–1791 RSC; (b) M. M. Seitkalieva, D. E. Samoylenko, K. A. Lotsman, K. S. Rodygin and V. P. Ananikov, Coord. Chem. Rev., 2021, 445, 213982 CrossRef CAS.
- (a) S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333–338 CrossRef CAS PubMed; (b) M. J. S. Farias, W. Cheuquepan, A. A. Tanaka and J. M. Feliu, ACS Catal., 2017, 7, 3434–3445 CrossRef CAS; (c) P. Zhou, X. Hou, Y. Chao, W. Yang, W. Zhang, Z. Mu, J. Lai, F. Lv, K. Yang, Y. Liu, J. Li, J. Ma, J. Luo and S. Guo, Chem. Sci., 2019, 10, 5898–5905 RSC.
- (a) Z. Yang, D. Yang, Y. Wang, Y. Long, W. Huang and G. Fan, Nanoscale, 2021, 13, 10044–10050 RSC; (b) S. Zhu, X. Qin, F. Xiao, S. Yang, Y. Xu, Z. Tan, J. Li, J. Yan, Q. Chen, M. Chen and M. Shao, Nat. Catal., 2021, 4, 711–718 CrossRef CAS.
- (a) M. Bello, S. M. J. Zaidi, A. Al-Ahmed, S. Basu, D.-H. Park, K. S. Lakhi and A. Vinu, Microporous Mesoporous Mater., 2017, 252, 50–58 CrossRef CAS; (b) S. Xue, W. Deng, F. Yang, J. Yang, I. S. Amiinu, D. He, H. Tang and S. Mu, ACS Catal., 2018, 8, 7578–7584 CrossRef CAS; (c) J. Zhang, X. Qu, Y. Han, L. Shen, S. Yin, G. Li, Y. Jiang and S. Sun, Appl. Catal., B, 2020, 263, 118345 CrossRef CAS; (d) Q. Wang, S. Chen, J. Jiang, J. Liu, J. Deng, X. Ping and Z. Wei, Chem. Commun., 2020, 56, 2419–2422 RSC; (e) G. Montiel, E. Fuentes-Quezada, M. M. Bruno, H. R. Corti and F. A. Viva, RSC Adv., 2020, 10, 30631–30639 RSC; (f) Q. Wang, S. Chen, P. Li, S. Ibraheem, J. Li, J. Deng and Z. Wei, Appl. Catal., B, 2019, 252, 120–127 CrossRef CAS; (g) A. Torres, H. Shi, B. Subramaniam and R. V. Chaudhari, ACS Sustainable Chem. Eng., 2019, 7, 11323–11333 CrossRef CAS.
- For reviews see: (a) R. A. Adams and B. Captain, J. Organomet. Chem., 2004, 689, 4521–4529 CrossRef CAS; (b) E. Antolini, J. Solid State Electrochem., 2011, 15, 455–472 CrossRef CAS.
- (a) L. Zhu, S. Shan, V. Petkov, W. Hu, A. Kroner, J. Zheng, C. Yu, N. Zhang, Y. Li, R. Luque, C.-J. Zhong, H. Ye, Z. Yang and B. H. Chen, J. Mater. Chem. A, 2017, 5, 7869–7875 RSC; (b) L. Zhu, H. Zhang, W. Hu, J. Zheng, N. Zhang, C. Yu, H. Ye, Z. Yang and B. H. Chen, ChemCatChem, 2018, 10, 1998–2002 CrossRef CAS; (c) L. Zhu, H. Zhang, N. Ma, C. Yu, N. Ding, J.-L. Chen, C.-W. Pao, J.-F. Lee, Q. Xiao and B. H. Chen, J. Catal., 2019, 377, 299–308 CrossRef CAS.
- (a) A. Weilhard, G. Abarca, J. Viscardi, M. H. G. Prechtl, J. D. Scholten, F. Bernardi, D. L. Baptista and J. Dupont, ChemCatChem, 2017, 9, 204–211 CrossRef CAS; (b) Y. Xu, C. Wu, Y. Wang, Y. Zhang, H. Sun, H. Chen and Y. Zhao, Chin. Chem. Lett., 2021, 32, 516–520 CrossRef CAS.
- L. A. Oro, D. Carmona and J. M. Fraile, in Metal–catalysis in Industrial Organic Processes, The Royal Society of Chemistry, 2006, pp. 79–113 Search PubMed.
- For a review with relevant patents see: K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS and references therein.
- For reviews see: (a) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS; (b) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC; (c) G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed; (d) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- For reviews see: (a) P. Serna and A. Corma, ACS Catal., 2015, 5, 7114–7121 CrossRef CAS; (b) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed; (c) E. Podyacheva, O. I. Afanasyev, D. V. Vasilyev and D. Chusov, ACS Catal., 2022, 12, 7142–7198 CrossRef CAS.
- For some references see: (a) Y. Yamane, X. Liu, A. Hamasaki, T. Ishida, M. Haruta, T. Yokoyama and M. Tokunaga, Org. Lett., 2009, 11, 5162–5165 CrossRef CAS PubMed; (b) L. L. Santos, P. Serna and A. Corma, Chem. – Eur. J., 2009, 15, 8196–8203 CrossRef CAS PubMed; (c) A. Corma, T. Rodenas and M. J. Sabater, Chem. – Eur. J., 2010, 16, 254–260 CrossRef CAS PubMed; (d) P. Rubio-Marques, A. Leyva-Perez and A. Corma, Chem. Commun., 2013, 49, 8160–8162 RSC; (e) S. G. Oh, V. Mishra, J. K. Cho, B.-J. Kim, H. S. Kim, Y.-W. Suh, H. Lee, H. S. Park and Y. J. Kim, Catal. Commun., 2014, 43, 79–83 CrossRef CAS; (f) P. Rubio-Marques, J. C. Hernandez-Garrido, A. Leyva-Perez and A. Corma, Chem. Commun., 2014, 50, 1645–1647 RSC.
- For some references see: (a) H.-U. Blaser and M. Studer, Acc. Chem. Res., 2007, 40, 1348–1356 CrossRef CAS PubMed; (b) H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- For some reviews see: (a) A. Stanislaus and B. H. Cooper, Catal. Rev.: Sci. Eng., 1994, 36, 75–123 CrossRef CAS; (b) S.-C. Qi, X.-Y. Wei, Z.-M. Zong and Y.-K. Wang, RSC Adv., 2013, 3, 14219–14232 RSC; (c) P. Lara, K. Philippot and B. Chaudret, ChemCatChem, 2013, 5, 28–45 CrossRef CAS.
- For some representative references see: (a) T. Maegawa, A. Akashi, K. Yaguchi, Y. Iwasaki, M. Shigetsura, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2009, 15, 6953–6963 CrossRef CAS PubMed; (b) M. Zahmakiran, Y. Tonbul and S. Oezkar, Chem. Commun., 2010, 46, 4788–4790 RSC; (c) M. Fang and R. A. Sanchez-Delgado, J. Catal., 2014, 311, 357–368 CrossRef CAS.
- For some representative references see: (a) D. Hollmann, S. Bahn, A. Tillack and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 8291–8294 CrossRef CAS PubMed; (b) A. Tillack, D. Hollmann, K. Mevius, D. Michalik, S. Bahn and M. Beller, Eur. J. Org. Chem., 2008, 4745–4750 CrossRef CAS; (c) J. He, J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 9888–9891 CrossRef CAS PubMed; (d) D. Pingen, C. Muller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS PubMed; (e) M. Mon, R. Adam, J. Ferrando-Soria, A. Corma, D. Armentano, E. Pardo and A. Leyva-Pérez, ACS Catal., 2018, 8, 10401–10406 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- S.-Y. Huang, S.-M. Chang and C. Yeh, J. Phys. Chem. B, 2006, 110, 234–239 CrossRef CAS PubMed.
- E. A. Anumol, A. Halder, C. Nethravathi, B. Viswanath and N. Ravishankar, J. Mater. Chem., 2011, 21, 8721–8726 RSC.
- D. Ren, L. He, L. Yu, R.-S. Ding, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS PubMed.
- A. Karakulina, A. Gopakumar, I. Akcok, B. L. Roulier, T. LaGrange, S. A. Katsyuba, S. Das and P. J. Dyson, Angew. Chem., Int. Ed., 2016, 55, 292–296 CrossRef CAS PubMed.
- For a review see: J. M. Thomas, B. F. G. Johnson, R. Raja, G. Sankar and P. A. Midgley, Acc. Chem. Res., 2003, 36, 20–30 CrossRef CAS PubMed.
- For seminal works see: G. C. Bond and D. E. Webster, Proc. Chem. Soc., London, 1964, 398 CAS.
- For a represenative reference see: N. A. Dehm, X. Zhang and J. M. Buriak, Inorg. Chem., 2010, 49, 2706–2714 CrossRef CAS PubMed.
- (a) M. Searcey, S. S. Grewal, F. Madeo and P. G. Tsoungas, Tetrahedron Lett., 2003, 44, 6745–6747 CrossRef CAS; (b) E. Gebauer-Henke, W. Leitner, A. Prokofieva, H. Vogt and T. E. Mueller, Catal. Sci. Technol., 2012, 2, 2539–2548 RSC.
- A. Corma, P. Serna and H. Garcia, J. Am. Chem. Soc., 2007, 129, 6358–6359 CrossRef CAS PubMed.
- (a) H. Sajiki, T. Ikawa and K. Hirota, Org. Lett., 2004, 6, 4977–4980 CrossRef CAS PubMed; (b) R. Nacario, S. Kotakonda, D. M. D. Fouchard, L. M. V. Tillekeratne and R. A. Hudson, Org. Lett., 2005, 7, 471–474 CrossRef CAS PubMed.
- X. Cui, A.-E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Nat. Commun., 2016, 7, 11326 CrossRef PubMed.
- G. S. Fonseca, E. T. Silveira, M. A. Gelesky and J. Dupont, Adv. Synth. Catal., 2005, 347, 847–853 CrossRef CAS.
- M. Harada and H. Dexpert, J. Phys. Chem., 1996, 100, 565–572 CrossRef CAS.
- S. Alayoglu, P. Zavalij, B. Eichhorn, Q. Wang, A. I. Frenkel and P. Chupas, ACS Nano, 2009, 3, 3127–3137 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. S1–S13 and Tables S1–S7. See DOI: https://doi.org/10.1039/d2cy01846b |
| ‡ Current address: Swiss Federal Institute of Technology (ETH), Zurich, Switzerland. |
| § Current address: Pharmacoepidemiology and Pharmacovigilance Division, Medicines for Human Use Department, Spanish Agency for Medicines and Medical Devices (AEMPS), Madrid, Spain. |
| This journal is © The Royal Society of Chemistry 2023 |