
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the mechanism and origins of selectivity in palladium-catalysed carbene insertion cross-coupling reactions†
Gavin
Lennon
 ,
Christina
O'Boyle
,
Andrew I.
Carrick
and
Paul
Dingwall
*
,
Christina
O'Boyle
,
Andrew I.
Carrick
and
Paul
Dingwall
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, Antrim BT7 1NN, UK. E-mail: p.dingwall@qub.ac.uk
First published on 1st November 2022
Abstract
The mechanism of palladium-catalysed carbene insertion cross-coupling reactions was studied by variable time normalisation analysis (VTNA), NMR spectroscopy, tandem eletrospray ionisation-mass spectrometry (ESI-MS), and density functional theory calculations. VTNA revealed a zero-order dependence on benzyl bromide and base, a first-order dependence on diazo substrate and Pd, and a negative first-order dependence on the PPh3 ligand. These results suggest rate determining carbene formation and the existence of an off-cycle bisphosphine turnover determining intermediate prior to this step. A time-adjusted same excess protocol showed that, while the catalytic cycle was stable, an induction period was present when the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPh3 ratio was greater than 1:3. NMR spectroscopic study revealed a large degree of phosphorous speciation, with control experiments allowing each species present to be identified. The turnover determining intermediate implicated during kinetic studies was identified, along with a second, η3-benzyl, turnover determining intermediate placed off the catalytic cycle after carbene insertion but prior to β-hydride elimination. ESI-MS of the reaction mixture was conducted, providing additional evidence for the existence of both turnover determining intermediates. Theoretical investigations suggest carbene insertion as the origin of selectivity, rather than β-hydride elimination as is generally assumed. Selectivity during carbene insertion was found to be governed by frontier molecular orbital interactions which heavily favour (E)-selectivity. Energy span and degree of TOF control analysis strongly support experimental observations and mechanistic rationale, validating theoretical work. Beyond defining aspects of the mechanism of palladium carbene insertion reactions, this study reveals that, when a pendant migrating group is present, carbene insertion is the selectivity determining step. These results can be used to rationalise unexpectedly high selectivities reported for similar reactions in the literature, suggesting this to be a general model for selectivity in palladium-catalysed carbene insertion reactions.
:PPh3 ratio was greater than 1:3. NMR spectroscopic study revealed a large degree of phosphorous speciation, with control experiments allowing each species present to be identified. The turnover determining intermediate implicated during kinetic studies was identified, along with a second, η3-benzyl, turnover determining intermediate placed off the catalytic cycle after carbene insertion but prior to β-hydride elimination. ESI-MS of the reaction mixture was conducted, providing additional evidence for the existence of both turnover determining intermediates. Theoretical investigations suggest carbene insertion as the origin of selectivity, rather than β-hydride elimination as is generally assumed. Selectivity during carbene insertion was found to be governed by frontier molecular orbital interactions which heavily favour (E)-selectivity. Energy span and degree of TOF control analysis strongly support experimental observations and mechanistic rationale, validating theoretical work. Beyond defining aspects of the mechanism of palladium carbene insertion reactions, this study reveals that, when a pendant migrating group is present, carbene insertion is the selectivity determining step. These results can be used to rationalise unexpectedly high selectivities reported for similar reactions in the literature, suggesting this to be a general model for selectivity in palladium-catalysed carbene insertion reactions.
Introduction
It is only relatively recently that carbene precursors have emerged as a new type of cross-coupling partner in palladium catalysis.1 The power and utility of this transformation has proven to be impressive, with the extensive synthetic scope the subject of several detailed and excellent reviews.2–7 The mechanism is widely assumed to proceed via the catalytic cycle outlined in Scheme 1.1,5,8 Oxidative addition of Pd(0), I, to a standard electrophilic partner, such as an aryl or benzyl halide, forms an oxidative addition complex, II. Formation of the central palladium carbene, III, then follows. This is typically, although not always,9 through reaction with a diazo compound. Stabilised or semi-stabilised diazo substrates can be utilised directly, while N-tosylhydrazones precursors can generate the diazo compound in situ, negating associated safety issues.10–12 Carbene insertion then occurs, forming an alkyl palladium, IV, followed by product forming β-hydride elimination and regeneration of the palladium catalyst. | ||
| Scheme 1 Proposed catalytic cycle for palladium-catalysed carbene insertion cross-coupling reactions and summary of previous experimental works in the literature. | ||
To date, few mechanistic studies have been reported. Experimentally, it has been shown that oxidative addition is likely a key elementary step and that β-hydride elimination is likely occurring but is not rate-limiting (Scheme 1).13,14 With N-tosylhydrazones as a diazo substrate precursor, pressurized sample infusion-electrospray ionization-mass spectrometry determined that rate-limiting oxidative addition was exclusive to aryl chlorides, while tentative evidence implied in situ formation of the diazo substrate was turnover determining for aryl bromides and iodides.15 Newman and co-workers recently used this mechanistic cycle as the basis for their strategy to overcome scope limitations, with slow addition of diazo substrate controlling catalyst speciation and increasing reaction yields. A theoretical study supported oxidative addition of the aryl halide as rate limiting.16
While individual steps have been implicated on the catalytic cycle, there has been no investigation surrounding selectivity, particularly in the formation of tri-substituted alkenes. Selectivity has been widely assumed to originate during β-hydride elimination,17,18 despite the major product arising from the more sterically encumbered transition state. Neither does this assumption account for the different levels of selectivity when forming the same product from either a stabilised diazo compound (1) and a benzyl halide (2),13 or a semi-stabilised diazo compound (3) and an aryl halide (4).14 Here the movement of a single carbon atom between starting materials results in a near perfect 20:1 (E)-selectivity reducing to 5:1, despite supposedly identical β-hydride elimination transition states (Scheme 2).
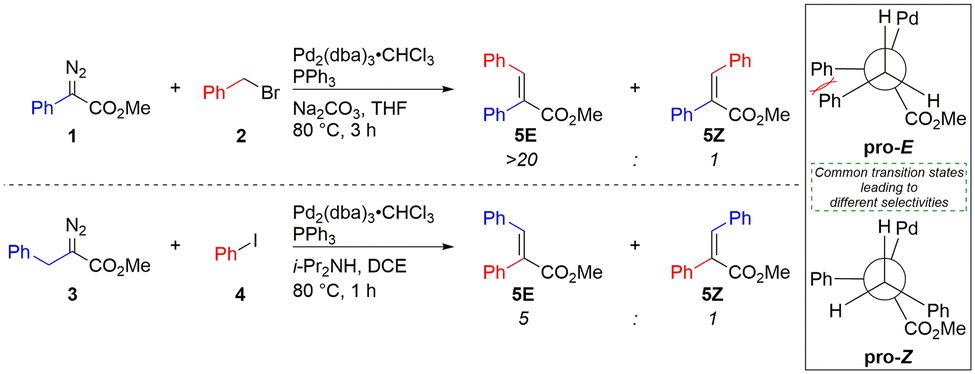 | ||
| Scheme 2 Forming the same products using either a stabilised diazo compound (1) and benzyl halide (2),13 or a semi-stabilised diazo compound (3) and an aryl halide (4),14 results in different selectivities, despite apparently identical selectivity determining β-hydride elimination steps in which the major product also results from the more sterically encumbered transition state. | ||
Here, we report the in-depth mechanistic study of palladium-catalysed carbene coupling for the formation of 1,1,2-substituted alkenes using kinetic studies, NMR spectroscopic investigation of off-cycle and parasitic species, ESI-MS investigation of palladium containing intermediates, and theoretical insights to determine the origin of high (E)-selectivity.
Results and discussion
Kinetic study
We chose, as a model reaction, work by Yu and co-workers.13 Minor modifications were made to the original conditions to remove side reactions and ensure a homogeneous reaction mixture (Scheme 3).19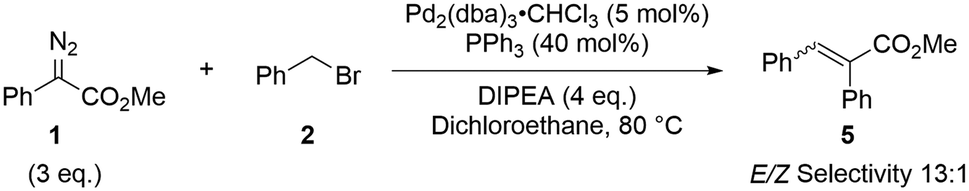 | ||
| Scheme 3 Optimised reaction conditions used in this study.13 | ||
We began with a study of the reaction kinetics, a “time-adjusted same excess” protocol of reaction progress kinetic analysis (RPKA) allowed us to first investigate the stability of the catalytic cycle (Fig. 1).20,21
 | ||
| Fig. 1 “Time-adjusted” same excess study conditions [DIPEA] 0.4 M, [Pd] 0.01 M, [PPh3] 0.04 M, dichloroethane (3 mL), 80 °C. Standard Conditions: [2] 0.1 M, [1] 0.3 M; same excess: [2] 0.05 M, [1] 0.25 M. The “time-adjusted” profile has the same [2] and [1] as the standard conditions profile at the point marked by the arrows. | ||
Overlay, and so confirmation of stability of the catalytic cycle, is observed towards the end of the reaction, while an induction period is observed during the first half hour. The reaction clearly takes time to achieve steady state but does not suffer from inhibition or deactivation. Pd precursors are well known to play an important role in speciation, particularly during activation. However, this was ruled out in our case as identical induction periods were observed for both Pd2(dba)3 and Pd(PPh)4 and additional dba had no effect on either the induction period or the rest of the reaction.19 Instead, the induction period was found to decrease on decreasing ratio of Pd:PPh3, disappearing completely at a 1:3 ratio, with a clear negative order in PPh3 observed (Fig. 2). While the reaction is fastest at a Pd:PPh3 ratio of 1:2, the final yield is reduced and virtually no activity is observed at a ratio of 1:1. This suggests a scenario where a minimum Pd:PPh3 ratio of 1:2 is required to guarantee stability of the catalytic cycle. The lower final yield at this ratio hints at the removal of PPh3 from the system resulting in early catalyst death.
 | ||
| Fig. 2 Time-course profiles of differing Pd:PPh3 loadings demonstrating the disappearance of the induction period at low Pd:PPh3 ratios. [Pd] 0.01 M, [2] 0.1 M, [1] 0.3 M, [DIPEA] 0.4 M, dichloroethane (3 mL), 80 °C. | ||
An induction period presumably arises due to the ability of the system to generate catalytically active low PPh3 ligated Pd species, likely mono-ligated,22 where slow activation is the result of slow PPh3 dissociation from Pd(PPh3)≥2 complexes.23 That the reaction fails below a Pd:PPh3 ratio of 1:2 is indicative of a bisphosphine intermediate probably playing an important role as a reservoir species in stabilising the catalytic cycle, helping maintain an effective steady-state concentration of active catalyst.21,24
We chose variable time normalisation analysis (VTNA) to probe the concentration dependencies of the catalyst and substrates.25,26 First-order behaviour in Pd was observed when comparing experiments below a Pd:PPh3 ratio of 1:3 in which the induction period was minimised (Fig. 3). At higher ratios, where the induction period was still present, unrealistic orders of greater than 1 were observed due to difficulties in achieving visual overlay.19
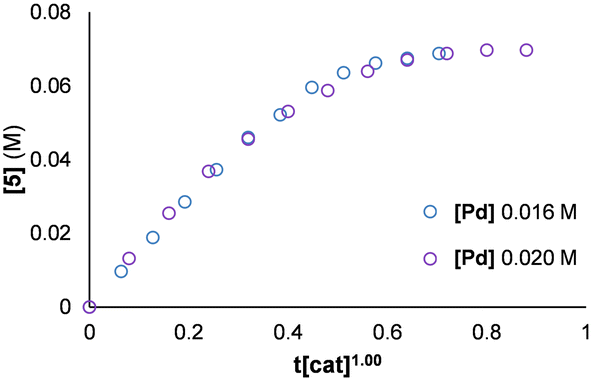 | ||
| Fig. 3 Graphical rate analysis determining order in Pd. [2] 0.1 M, [1] 0.3 M, [DIPEA] 0.4 M, [PPh3] 0.04 M, dichloroethane (3 mL), 80 °C. | ||
The system demonstrated zero-order kinetics with respect to benzyl bromide (2) and DIPEA. Comparatively, first-order kinetics in diazo substrate (1) and a negative order dependency in PPh3 were observed (Fig. 4).
 | ||
| Fig. 4 Different excess studies indicating the order in reaction with respect to each reagent. [2] 0.1 M, [1] 0.3 M, [DIPEA] 0.4 M, [Pd] 0.01 M, [PPh3] 0.04 M, dichloroethane (3 mL), 80 °C. | ||
Zero-order behaviour in DIPEA suggests that the turnover determining states occur prior to catalyst regeneration. Negative order behaviour in PPh3 is indicative of off-cycle binding interactions, and reservoir species, in which PPh3 effectively decreases the active Pd content on the catalytic cycle. First-order in diazo substrate (1) and zero-order in benzyl bromide (2) are consistent with turnover determining formation of the palladium carbene taking place after facile oxidative addition. Further investigation of the kinetics at lower Pd concentrations found a change to positive-order behaviour in benzyl bromide (2) but identical behaviours for all other components, suggesting oxidative addition becomes more challenging at lower Pd loading.19 Taken together, these experimental and kinetic results point towards an off-cycle catalytic resting state after oxidative addition but prior to carbene formation. If this is so, such a resting state might be observed spectroscopically as a turnover determining intermediate of the reaction.
Spectroscopic study
We turned to 31P NMR spectroscopy to attempt to identify intermediate species consistent with the above kinetic results. In situ reaction monitoring revealed a much larger degree of phosphorous speciation than we first anticipated (Fig. 5). | ||
| Fig. 5 31P{1H} NMR (243 MHz) at 80 °C in d8-toluene of a) in situ model reaction; b) Pd(PPh3)2(Br)(Bn) 6, [6] = 0.01 M; c) in situ mixture of 2 and PPh3, [2] = 0.30 M, [PPh3] = 0.04 M forming phosphazine 7; d) in situ mixture of 8 and Pd2(dba)3·CHCl3/PPh3, [8] = 0.04 M, [Pd] = 0.01 M, [PPh3] = 0.04 M; e) in situ mixture of Pd2(dba)3·CHCl3/PPh3 forming Pd(PPh3)n, [Pd] = 0.01, [PPh3] = 0.04 M, that a sharp line exists for this species is likely due to fast exchange at the high temperature at which the NMR was acquired. Assignments relative to Bu3P(O) as internal standard. Reaction conditions [2] 0.1 M, [1] 0.3 M, [DIPEA] 0.4 M, [Pd] 0.01 M, [PPh3] 0.04 M, d8-toluene (0.6 mL), 80 °C. | ||
The signal at δ 22.0 ppm can be assigned to the oxidative addition product 6. This was prepared independently and, at synthetically relevant 80 °C, was found to coalesce into a singlet.27 This species is the most likely candidate suggested as an off-cycle resting state by kinetic analysis. Analysing interactions between PPh3 and each reaction component, alone and in combination, allowed us to identify all but one of the remaining peaks. Three signals, at δ 18.8, δ 17.2 and δ 16.3 ppm are the result of side reactions between the diazo substrate (1) and PPh3. Addition of PPh3 to the diazo substrate results in reversible formation of phosphazine 7, assigned unambiguously via X-ray crystallography.19 On dissolution, free PPh3 and diazo substrate (1) are observed by 1H and 31P NMR along with the two peaks at δ 17.2 and δ 16.3 ppm which are presumably interconverting (E)- and (Z)-isomers of 7. The signal at δ 18.8 ppm is the phosphorane 8, which was prepared independently and again characterised unambiguously via X-ray crystallography.19 PPh3 also undergoes a side reaction with the benzyl bromide (2) substrate to form benzyltriphenylphosphonium salt (9). This species is insoluble in d8-toluene, and so is undetectable by NMR, but is visible as a white solid in the NMR tube. This compound is partially soluble in dichloroethane, used during the kinetic study, and its formation was found to be reversible under these reaction conditions.19
Activation of Pd2(dba)3 and excess PPh3 in d8-toluene at 80 °C leads to two peaks at δ ca. 23 and δ 23.6 ppm, presumably Pd(PPh3)≥3 species.28–32 Identical peaks are observed upon heating of Pd(PPh3)4 to 80 °C, confirming that at reaction temperatures, dba is effectively de-ligated.
All intermediates observed by 31P NMR decrease in concentration over the course of the reaction, except phosphorane 8, which increases.19 As the same excess experiment showed that the reaction reaches steady state, any observed species implicated with the catalytic cycle must be off-cycle intermediates. Otherwise, the fundamental assumption of the steady state approximation would be violated. Formation of the palladium carbene must take place on a monophosphine intermediate. To take place on the bisphosphine would place the oxidative addition intermediate 6 on, rather than off, the catalytic cycle. By the same logic, oxidative addition must also take place via a mono-, rather than bis-, phosphine intermediate. These observations rationalise the negative order observed for PPh3 as well as the requirement for a Pd:PPh3 ratio of at least 1:2 for an optimally productive catalytic cycle. For the other observed species, none are implicated in the reaction mechanism; the reversibility of their formation allows them to function as a source of substrates (i.e. benzyl bromide (2) and PPh3 in the case of the benzyltriphenylphophonium salt (9), or PPh3 and diazo substrate (1) for phosphazine (7) but none, when tested in isolation, resulted in product formation under reaction conditions.19
A final unknown signal at δ 24.6 ppm could not be assigned via control experiments, and 2D NMR techniques proved inconclusive.19 We wondered if this signal could possibly be a second intermediate linked to the catalytic cycle. To test this hypothesis, we performed the reaction with two different starting materials, an electron poor diazo compound (10) and electron poor benzyl bromide derivative (11) (Fig. 6).
 | ||
| Fig. 6 31P{1H} NMR (243 MHz) at 80 °C in d8-toluene. Comparison of 31P spectra of differing starting materials. a) 10 and Pd2(dba)3·CHCl3/PPh3; b) in situ reaction of 2 and 10 substrates; c) in situ reaction of 11 and 1 substrates; d) in situ model reaction. Reaction conditions [2]/[11] 0.1 M, [1]/[ 10] 0.3 M, [DIPEA] 0.4 M, [Pd] 0.01 M, [PPh3] 0.04 M, d8-toluene (0.6 mL), 80 °C. | ||
If the species at δ 24.6 ppm were to involve either substrate, then a reaction substituted with an electron poor derivative would result in a downfield chemical shift of the 31P signal, provided a trans relationship between the PPh3 and the ligand containing the substrate existed. To our delight, such a downfield shift was observed for both substituted starting materials, indicating this 31P signal is likely an intermediate linked to the catalytic cycle. The signal is a sharp singlet and is unlikely to be a bisphosphine, as the required cis-orientation of two PPh3 groups around the four-coordinate Pd would result in a doublet of doublets. As changing both substrates resulted in a downfield shift, both must be present in a single ligand trans to PPh3. This suggests carbene insertion has already occurred, but product forming β-hydride elimination has not. In order to fit with the kinetic data, other species on which a rate dependence could be observed are unlikely to be involved. Finally, as the signal intensity was observed to decrease over the course of the reaction it is also likely an off-cycle species.
The on-cycle alkyl palladium intermediate from which β-hydride elimination must occur is also an η1-benzyl complex. η1-benzyl complexes are well known to exist in equilibria with their η3-benzyl isomers through a variety of mechanisms.33,34 η3-benzyl or allyl complexes exist as catalyst precursors and are commonly proposed as on-cycle organometallic intermediates.35–38 The η3-isomer of this on-cycle η1-intemediate would represent an off-cycle species reversibly connected to the catalytic cycle and is the most likely candidate for this unknown 31P signal. Such intermediates have been identified computationally as a turnover determining intermediate in the study of similar systems involving aryl insertion into palladium carbenes.9,16
Curiously, the signal previously determined to be Pd(PPh3)n at δ 23.6 ppm also experienced a downfield shift when substituting an electron poor diazo compound (10). Activation of Pd2(dba)3 and PPh3 in d8-toluene at 80 °C in the presence of 1 results in no change to the 31P chemical shifts. However, in the presence of electron poor 10 the signal is observed to shift downfield to δ 24.1 ppm, suggesting that electron deficient diazo compounds are capable of complexing to Pd(PPh3)n species. Such siphoning of Pd to an off-cycle species would likely reduce the amount of active catalyst present on the catalytic cycle and would explain slower reactions, and poorer yields, observed when using electron poor diazo-compounds in palladium carbene insertion reactions.13,39–42 Indeed, in situ1H NMR time course data showed the reaction with electron poor 10 to be significantly slower than with 1. Of note, the signal for the oxidative addition product resting state (6) at δ 22.0 does not shift on substitution to an electron poor benzyl bromide derivative (11), confirming the cis relationship between this ligand and either PPh3 ligand.
Mass spectrometry
To corroborate our interpretation of the NMR data, the reaction was investigated by ESI-MS. To probe for Pd containing species an aliquot of the crude reaction mixture was analysed by positive ion-mode ESI LC/HRMS.Two Pd containing species were observed (Fig. 7).19 Under positive ion spray conditions, no bromine ligands on Pd were observed, with the resulting vacant site ligated by advantageous PPh3 which is present in 4-fold excess to Pd under reaction conditions.
 | ||
| Fig. 7 Ions observed by HRMS that support the existence of two off-cycle resting states 14 and 15. Compounds 14 and 15 as drawn are likely structures only. | ||
As expected, the major component is an ion of mass corresponding to the bis-ligated off-cycle oxidative addition resting state (14) with small quantities of another ion (15). Several species of the mass of 15 exist on the proposed catalytic cycle. Once the palladium carbene has formed, there is no change in mass until the product is released from Pd. Although these ESI-MS data cannot explicitly determine either ion's structure, taken in tandem with the 31P NMR studies they support the existence of an off-cycle η3-benzyl intermediate (15) as a second catalytic resting state.
Origin of selectivity
Having experimentally probed the mechanism of the reaction and uncovered likely turnover determining states, we were keen to further investigate the off-cycle η3-benzyl resting state as well as pinpoint the origin of high (E)-selectivity observed in this reaction. Our first step was to check the stability of the product to equilibration under reaction conditions, either via re-entry to the catalytic cycle through a reversible product formation step or by some other pathways involving Pd catalysis. No decrease in (Z)-product (5) was observed when an aliquot was added at the start of the reaction, ruling out either of these possibilities.19 To further explore mechanistic details, quantum chemical calculations (B3PW91-D3-PCMToluene/6-311+G(d,p)/SDD//B3PW91-D3/6-31G(d,p)/SDD) were carried out (Fig. 8). Dispersion corrections were included at both the optimisation and single point energy calculations as, in previous work, we have found this essential to replicate the presence of multiple experimentally determined resting states as well as reaction selectivity.43,44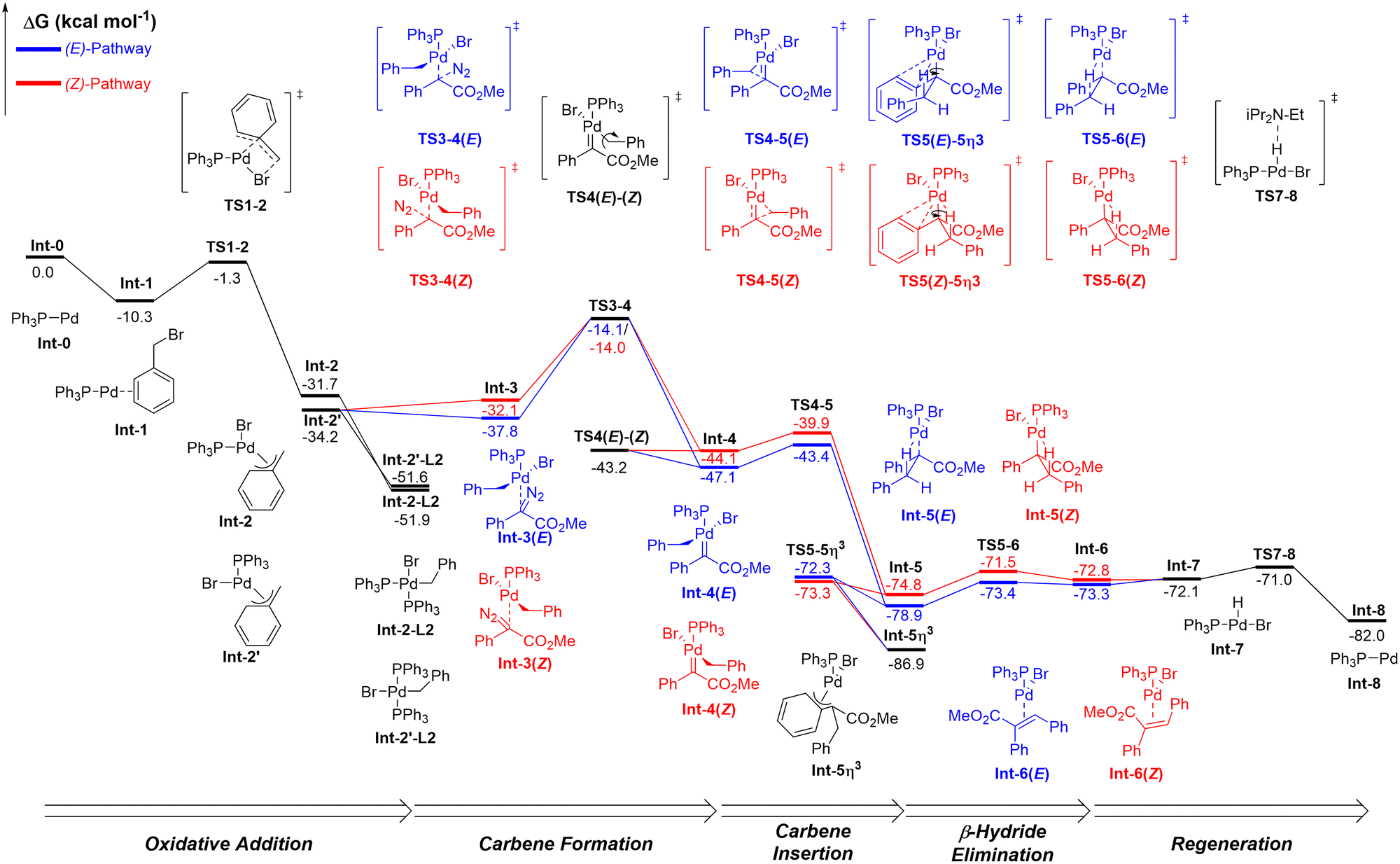 | ||
| Fig. 8 Free energy profiles for the major (E)-selective (blue) and minor (Z)-selective (red) reaction pathways. Theoretical reaction analysis performed at B3PW91-D3-PCMToluene/6-311+G(d,p)/SDD//B3PW91-D3/6-31G(d,p)/SDD energies are Gibbs free energies in kcal mol−1. | ||
The catalytic cycle initiates with oxidative addition of the electrophile to mono-ligated Pd,45 Int-0, affording two η3-benzyl species', Int-2 and Int-2′, which are in equilibrium with bisphosphine off-cycle intermediates Int-2-L2 and Int-2′-L2, the first experimentally determined oxidative addition turnover determining intermediate. Cis–trans isomerisation of square-planar Pd oxidative addition complexes can occur by a number of pathways, typically these processes are faster than other steps in a cross-coupling catalytic cycle and do not limit these reactions, here we show only the thermodynamic consequences.46
The availability of different coordination sites on Pd isomers Int-2 and Int-2′ means the resulting palladium carbene can potentially form either trans to a bromine or PPh3 ligand. Formation of the carbene trans to PPh3viaTS3–4 is 1.4 kcal mol−1 more energetically favourable than formation trans to the bromine, making this the dominant pathway. The resulting conformation around Pd that this leads to later in the cycle is in good agreement with that determined via NMR, giving confidence in this finding. Only the pathway originating from isomer Int-2′ will be discussed from here on. Full details of the pathway initiated from Int-2, trans to bromine, are available in the SI.19
The transition state for carbene formation, TS3–4, represents the first possible point on the catalytic cycle at which bifurcation into pro-(E) and pro-(Z) pathways can occur due to approach of differing pro-chiral faces of the diazo compound. The orientation of the benzyl group relative to the two groups on the carbene allows assignment of pro-(E) and pro-(Z) pathways during TS3–4 and for the resulting carbene Int-4. However, selectivity was found to be negligible at this stage, with a 0.1 kcal mol−1 difference between transition states for each pathway.
At carbene Int-4, a simple rotation around the Pd–CBenzyl bond, TS4(E)–(Z), allows the pathways to interconvert (Fig. 9). Although this is blocked in one direction by the bulky PPh3 ligand, the carbene itself is planar, meaning the barrier to rotation in this direction is low. From Int-4(E) the transition states for rotation, TS4(E)–(Z), and insertion, TS4–5(E), are essentially isoenergetic, resulting in 1:1 selectivity towards Pd–alkyl Int-5(E) and pro-(Z) carbene Int-4(Z). However, from Int-4(Z), a large 3.3 kcal mol−1 energy difference favours rotation to Int-4(E) over insertion to Int-5(Z). Any Int-4(Z) produced from TS3–4 or TS4(E)–(Z) will therefore rotate to Int-4(E) and insert, following the pro-(E) pathway, making the pro-(Z) pathway essentially unproductive.
 | ||
| Fig. 9 Relative energy profiles for the pro-(E) (blue) and pro-(Z) (red) product forming pathways originating from the bifurcation of the catalytic cycle at the carbene insertion step. Theoretical reaction analysis performed at B3PW91-D3-PCMToluene/6-311+G(d,p)/SDD//B3PW91-D3/6-31G(d,p)/SDD energies are Gibbs free energies in kcal mol−1. | ||
Considering carbene insertion as the first selectivity-determining step, a 3.5 kcal mol−1 preference exists for pro-(E) insertion over pro-(Z). With the benzyl ring syn to the carbene phenyl it is easy to imagine favourable π–π stacking non-covalent interaction between the two rings during this transition state. However, removal of dispersion corrections during single point energy calculations still leaves a large 2.6 kcal mol−1 preference for pro-(E) insertion. Examining the molecular orbitals of the transition state supplies another reasoning. The HOMO sits predominantly on the migrating benzyl ring while the LUMO sits predominantly on the carbene phenyl ring (Fig. 10). This highly favourable overlap is responsible for the drastic lowering of the barrier to pro-(E) insertion. That the barrier to pro-(Z) insertion is significantly higher is a combination of the lack of HOMO–LUMO overlap and steric effects of the other group on the carbene. It is the combination of these effects during carbene insertion that we suggest is the origin of the unexpectedly high (E)-selectivity in this reaction.
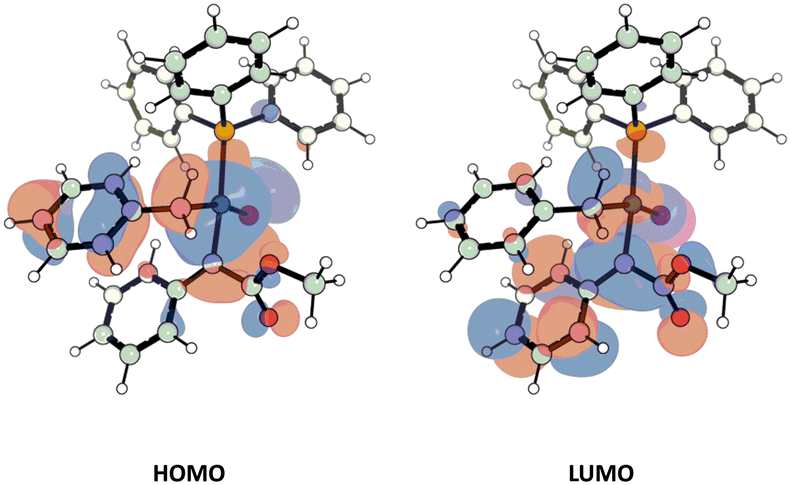 | ||
| Fig. 10 Molecular orbitals of Pd-carbene TS4–5(E) where the HOMO resides on the migrating benzyl group and LUMO on the carbene phenyl ring. | ||
Carbene insertion and subsequent formation of Pd–alkyl Int-5 is extremely exergonic and therefore irreversible. Here, a second opportunity for interconversion between the pro-(E) and pro-(Z) pathways arises. In theory, direct rotation around the pro-alkene C–C bond is possible, although we find this transition state energetically unfavourable. Instead, rotation around the Pd–carbon bond, viaTS5-5η3, results in the formation of off-cycle intermediate Int-5η3 representing the lowest point on the potential energy surface. We suggest Int-5η3 is the off-cycle intermediate observed by NMR and implicated via ESI-MS studies due to its high thermodynamic stability relative to other intermediates of identical mass. This species is in clear equilibria with its η1-benzyl complex, the on-cycle Pd–alkyl Int-5, where the additional site on Pd is filled with an agostic Pd–H interaction.
The barrier for η1–η3 interconversion viaTS5-5η3 is very similar to the barrier for β-hydride elimination, TS5–6, in either pathway (Fig. 9). As with interconversion between Int-4(E) and Int-4(Z) earlier in the catalytic cycle, the interconversion will not be a rapid equilibration or a Curtin–Hammett like scenario that would negate the selectivity set in previous steps of the cycle.47 Instead, with pathways so close in energy, interconversion would become possible to the degree that β-hydride elimination, TS5–6, either reinforces or erodes selectivity set during carbene insertion, TS4–5. TS5–6(E) is 1.9 kcal mol−1 lower in energy than TS5–6(Z), making β-hydride elimination also (E)-selective, though to a lesser extent than carbene insertion. This will result in only a minor erosion of the near perfect (E)-selectivity set earlier in the catalytic cycle.19
(E)-Selectivity during β-hydride elimination is not immediately anticipated due to the expected steric clash between adjacent phenyl groups. However, stabilising non-covalent interactions, particularly between the PPh3 ligand and phenyl aryl ring appear to be the cause of the added stability of the pro-(E) pathway, with removal of dispersion corrections during single point calculations resulting in isoenergetic transition states.19
That selectivity is set during carbene insertion explains the experimentally observed differences when forming the same product from either a benzyl halide 2 and stabilised diazo compound 1 or an aryl halide 4 and semi-stabilised diazo compound 3 (Scheme 2). Coupling 4 with 3 would result in the insertion of a phenyl group into a palladium carbene, followed by β-hydride elimination. With a phenyl group there can be no possible bifurcation during carbene insertion, TS4–5. This choice of substrates would then enter our model at intermediate Int-5η3 (Fig. 9). Newman and co-workers have computationally studied a similar insertion of a phenyl group to a palladium carbene leading to disubstituted alkenes,16 finding carbene insertion leading directly to a Pd η3-benzyl intermediate similar to Int-5η3. With an aryl halide substrate, β-hydride elimination now represents the only step at which selectivity can be set. Our calculations show significantly lower (E)-selectivity possible during β-hydride elimination (1.9 kcal mol−1) vs. carbene insertion (3.5 kcal mol−1) qualitatively matching experimental results.
Other palladium carbene insertion reactions display high levels of stereoselectivity, often >20:1. Examples from the literature include benzyl insertions to construct trifluoromethylated alkenes,40 alkenylphosphonates,41 substituted furans,9 as well as allyl insertions in the formation of various dienes (Scheme 4).39,40,48 These results suggest that carbene insertion as the origin of selectivity may be a general model for palladium carbene insertions when a pendant group allows bifurcation during carbene insertion.
 | ||
| Scheme 4 Literature examples of palladium-catalysed carbene insertion reactions displaying high levels of (E)-selectivity which can be mechanistically explained through carbene insertion as a selectivity determining step.9,39–41,48 | ||
A degree of TOF control analysis of the energy span model of Kozuch and Shaik49,50 suggests that two intermediates Int-5η3 and Int-2′-L2 and two transition states, TS1–2 and TS3–4, share the roles of turnover determining intermediates and turnover determining transition states. Maximising the energy span results in similar barriers of 38.8 kcal mol−1 from Int-5η3 to TS1–2 and 37.8 kcal mol−1 from Int-2′-L2 to TS3–4. Our DFT calculations have correctly predicted the existence of two experimentally observed turnover determining intermediates. The experimental observations of negative order in PPh3 and first order in diazo compound (1) are correctly reproduced. During the reaction induction period, as well as at lower Pd loadings, we find the reaction displays positive order kinetics in benzyl bromide (2).19 Computational studies, and our application of the energy span model, are energy-based representations which do not take into account the effects of reactant, product, or catalyst concentrations which clearly have a large experimental effect at the chosen conditions in this reaction. Qualitatively, experimentally observed kinetic orders, as well as the existence of multiple observed turnover determining intermediates, are reproduced by our DFT calculations, increasing the confidence in our model for stereoselectivity.
Conclusions
In summary, combined experimental and computational studies have been undertaken on a model palladium-catalysed carbene insertion reaction to better understand both the reaction mechanism and the origin of selectivity in the formation of tri-substituted alkenes. Kinetic profiling, NMR spectroscopic study of catalytically competent intermediates and ESI-MS studies concluded carbene formation as the turnover determining transition step and the presence of two, off-cycle turnover determining intermediates. The presence of an induction period was found to depend on the ratio of Pd:PPh3, the removal of which was essential to determine the order in catalyst. Theoretical studies provided corroboration for the experimental findings as well as insights into the origin of selectivity for these reactions, proposed to occur during carbene insertion rather than the previously assumed β-hydride elimination. An energy span and degree of TOF control analysis paralleled experimental results, suggesting two turnover determining transition states and two turnover determining intermediates. The mechanism of palladium carbene insertion has now been investigated to a high level of detail and a novel origin of selectivity proposed. Near perfect selectivity for a single stereoisomer is set during carbene insertion if bifurcation can occur at this step. This is aided by frontier molecular overlap between the migrating group and carbene substituent which favours one pathway over the other. This is proposed as a general model for selectivity in palladium-catalysed carbene insertion reactions with a pendant migratory group and can mechanistically account for previously unexplained selectivities reported in the literature. These conclusions offer crucial insights into the fundamental nature of an extensive, yet ever evolving, class of catalytic transformations and will aid the rational design of more efficient and selective synthetic strategies.
Data availability
Data for this paper, including experimental kinetic, NMR, and mass spec, as well as theoretical geometry optimisation, single point, and IRC calculations, are available from https://pure.qub.ac.uk with DOI: https://doi.org/10.17034/7d12b2fe-39bb-48e4-8070-330ceabdcf0b.Author contributions
PD conceived and supervised the study. Experiments were performed and analysed by GL with support from CO'B and AIC. DFT calculations were performed by PD. The manuscript was written by GL and PD.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research was funded by the Engineering and Physical Sciences Research Council (GL – EP/N509541/1) and a Queen's University Belfast Startup Fund (PD). Low resolution LCMS facilities were funded through an EPSRC block equipment grant for early career researchers (EP/S018077/1). We thank Dr Peter C. Knipe (QUB) for assistance with the determination of X-ray structures. We also thank Mr Conor McGrann (QUB) and Mr Richard Murphy (QUB) for their assistance with conducting high resolution mass spectrometry and NMR experiments.Notes and references
- K. L. Greenman, D. S. Carter and D. L. Van Vranken, Tetrahedron, 2001, 57, 5219 CrossRef.
- Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 2011, 1015 CrossRef.
- Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef PubMed.
- Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef.
- Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef PubMed.
- M. Peña-López and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 46 CrossRef.
- Y. Xia and J. Wang, J. Am. Chem. Soc., 2020, 142, 10592 CrossRef CAS PubMed.
- K. L. Greenman and D. L. Van Vranken, Tetrahedron, 2005, 61, 6438 CrossRef CAS.
- Y. Xia, S. Qu, Q. Xiao, Z.-X. Wang, P. Qu, L. Chen, Z. Liu, L. Tian, Z. Huang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 13502 CrossRef CAS.
- T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed.
- S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67 CrossRef CAS.
- W.-Y. Yu, Y.-T. Tsoi, Z. Zhou and A. S. C. Chan, Org. Lett., 2009, 11, 469 CrossRef CAS.
- C. Peng, G. Yan, Y. Wang, Y. Jiang, Y. Zhang and J. Wang, Synthesis, 2010, 2010, 4154 CrossRef.
- G. T. Thomas, K. Ronda and J. S. McIndoe, Dalton Trans., 2021, 50, 15533 RSC.
- R. J. Sullivan, G. P. R. Freure and S. G. Newman, ACS Catal., 2019, 9, 5623 CrossRef CAS.
- J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS.
- K. Ishitobi, K. Muto and J. Yamaguchi, ACS Catal., 2019, 9, 11685 CrossRef CAS.
- See ESI.†.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302 CrossRef CAS.
- R. D. Baxter, D. Sale, K. M. Engle, J. Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 4600 CrossRef CAS PubMed.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366 CrossRef CAS.
- E. R. Strieter, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 13978 CrossRef CAS.
- A. C. Ferretti, C. Brennan and D. G. Blackmond, Inorg. Chim. Acta, 2011, 369, 292 CrossRef CAS.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028 CrossRef.
- J. Bures, Angew. Chem., Int. Ed., 2016, 55, 16084 CrossRef CAS.
- C. M. Crawforth, S. Burling, I. J. S. Fairlamb, A. R. Kapdi, R. J. K. Taylor and A. C. Whitwood, Tetrahedron, 2005, 61, 9736 CrossRef CAS.
- C. Amatore, A. Jutand and M. A. M'Barki, Organometallics, 1992, 11, 3009 CrossRef CAS.
- C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254 CrossRef CAS.
- I. J. S. Fairlamb, Org. Biomol. Chem., 2008, 6, 3645 RSC.
- S. Sawadjoon, A. Orthaber, P. J. R. Sjöberg, L. Eriksson and J. S. M. Samec, Organometallics, 2014, 33, 249 CrossRef CAS.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302 CrossRef CAS.
- Y. Becker and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 845 CrossRef CAS.
- L. E. Crascall and J. L. Spencer, J. Chem. Soc., Dalton Trans., 1992, 3445, 10.1039/dt9920003445.
- A. M. Johns, J. W. Tye and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 16010 CrossRef CAS.
- A. M. Johns, M. Utsunomiya, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 1828 CrossRef CAS.
- S. Zhang, Y. Yamamoto and M. Bao, Adv. Synth. Catal., 2021, 363, 587 CrossRef CAS.
- R. Kuwano, Y. Kondo and Y. Matsuyama, J. Am. Chem. Soc., 2003, 125, 12104 CrossRef CAS.
- K. Wang, S. Chen, H. Zhang, S. Xu, F. Ye, Y. Zhang and J. Wang, Org. Biomol. Chem., 2016, 14, 3809 RSC.
- X. Wang, Y. Xu, Y. Deng, Y. Zhou, J. Feng, G. Ji, Y. Zhang and J. Wang, Chem. – Eur. J., 2014, 20, 961 CrossRef CAS PubMed.
- Y. Zhou, F. Ye, X. Wang, S. Xu, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 6109 CrossRef CAS PubMed.
- X. Zhao, J. Jing, K. Lu, Y. Zhang and J. Wang, Chem. Commun., 2010, 46, 1724 RSC.
- S. Gallarati, P. Dingwall, J. A. Fuentes, M. Bühl and M. L. Clarke, Organometallics, 2020, 39, 4544 CrossRef CAS.
- P. Dingwall, J. A. Fuentes, L. Crawford, A. M. Z. Slawin, M. Buhl and M. L. Clarke, J. Am. Chem. Soc., 2017, 139, 15921 CrossRef CAS.
- J. Jover, N. Fey, M. Purdie, G. C. Lloyd-Jones and J. N. Harvey, J. Mol. Catal. A: Chem., 2010, 324, 39 CrossRef CAS.
- J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164 RSC.
- J. Bures, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 6741 CrossRef CAS PubMed.
- S. Chen and J. Wang, Chem. Commun., 2008, 4198, 10.1039/B806970K.
- E. Solel, N. Tarannam and S. Kozuch, Chem. Commun., 2019, 55, 5306 RSC.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01702d |
| This journal is © The Royal Society of Chemistry 2023 |