
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solar-driven CO2 reduction catalysed by hybrid supramolecular photocathodes and enhanced by ionic liquids†
Roger
Miró
 a,
Hilmar
Guzmán
b,
Cyril
Godard
*c,
Aitor
Gual
*a,
Federica
Zammillo
b,
Thomas J. S.
Schubert
d,
Boyan
Iliev
d,
Angelica
Chiodoni
e,
Simelys
Hernández
*b and
Miriam Díaz
de los Bernardos
*a
a,
Hilmar
Guzmán
b,
Cyril
Godard
*c,
Aitor
Gual
*a,
Federica
Zammillo
b,
Thomas J. S.
Schubert
d,
Boyan
Iliev
d,
Angelica
Chiodoni
e,
Simelys
Hernández
*b and
Miriam Díaz
de los Bernardos
*a
aEurecat, Technology Centre of Catalonia, Unit of Chemical Technologies, Tarragona, 43007, Spain. E-mail: miriam.diaz@eurecat.org
bCREST Group, Department of Applied Science and Technology (DISAT), Politecnico di Torino, Turin, Italy
cDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Tarragona, 43007, Spain
dIolitec Ionic Liquids Technology GmbH, Heilbronn, 74076, Germany
eCenter for Sustainable Future Technologies (IIT@Polito), Italian Institute of Technology, Turin, Italy
First published on 15th February 2023
Abstract
Photoelectrochemical carbon dioxide reduction (CO2) at ambient temperature and pressure was performed using molecular chromophores and catalyst assemblies on CuGaO2-based electrodes in an ionic liquid (IL) organic solution, acting as a CO2 absorbent and electrolyte. A simple and versatile methodology based on the silanization of the CuGaO2 electrode followed by electropolymerization provided a series of molecular and supramolecular hybrid photocathodes for solar driven CO2 reduction. Focusing on the cathodic half reactions, the most promising conditions for the formation of CO2 reduction products were determined. The results revealed a beneficial effect of the ionic liquid on the conversion of CO2 to formic acid and suppression of the production of hydrogen. The potentiality of anchoring supramolecular complexes on semiconductor photoelectrocatalysts was demonstrated to boost both carrier transport and catalytic activity with a FEred of up to 81% compared with the obtained FEred of 52% using bare CuGaO2 with formate as the major product.
Introduction
Over the last decade, the increase in CO2 concentration in the atmosphere, which could eventually cause irreversible effects on nature, has triggered much attention in chemical research on carbon capture and utilization (CCU) technologies.1,2 For CO2capture, the main challenge is to find an ideal absorbent that exhibits high selectivity and high capacity for CO2. In this context, ionic liquids (ILs) have demonstrated excellent capture capacity for CO2;3 in addition, they have also been widely adopted as electrolytes in the electrochemical conversion of CO2 since they exhibit a high CO2 solubility, low vapor pressure, high ionic conductivity and wide electrochemical windows.4 Furthermore, it was reported that ILs can interact with CO2 to decrease the overpotential and enhance the product selectivity.5 CO2 electroreduction studies also demonstrated that appropriate combinations of ILs and catalysts can enhance the generation of CO with faradaic efficiencies of up to 96%.6–8 Among the IL structures, 1-butyl-3-methylimidazolium triflate (BMIM·TfO) is a promising structure due to its high CO2 solubility (0.6–0.8 mol CO2 per molIL).9 Therefore, although it has not been often used as an electrolyte for the CO2 reduction reaction (CO2RR), herein it will be exploited instead of the most commonly employed BMIM·BF4, to avoid the issues related to the latter like the hydrolysis of the [BF4]− and the possible formation of HF under electrochemical CO2RR conditions.10Regarding CO2conversion, the main challenge is to efficiently activate and reduce CO2 to reach the zero-carbon footprint goal. In this respect, visible-light-driven CO2 reduction in combination with water oxidation is a promising solution as it involves the use of abundant water and inexhaustible solar energy, and constitutes one of the most representative models of artificial photosynthesis.11 Molecular metal complexes and semiconductors are promising candidate photocatalysts, which can reduce CO2 to CO, formic acid, formaldehyde or other hydrocarbons.12 Although both molecular metal complexes and semiconductors have strengths and weaknesses, their main limitations (i.e. low oxidation ability, low selectivity for reduction reactions and stability issues) can be overcome via the construction of suitable molecule/semiconductor hybrid materials. In this approach, the efficient electron transfer from a semiconductor to an immobilized molecular catalyst and the suppression of back electron transfer are crucial for reducing CO2 on the molecule/semiconductor hybrid material.13 To this end, both the conduction band potential of the semiconductor and the reduction potential of the molecular unit should be carefully designed.
Supramolecular hybrid photocatalysts based on ruthenium (Ru(II))–rhenium (Re(I)) complexes were reported to efficiently catalyze CO2 photoreduction under visible light with faradaic efficiencies of up to 87% with CO and HCOOH as the major products.12,13 Osamu Ishitani and co-workers developed a hybrid photocathode based on CuGaO2 and a supramolecular Ru–Re complex immobilized using phosphonic acid as an anchoring group.14 This system allowed them to obtain faradaic efficiencies of up to 81% with a CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio of 1.5:1 and to exceed the performance observed with a NiO electrode; however, the instability of the phosphonic acid anchoring group was the main drawback. Later, Meyer and co-workers adopted a new immobilization method based on the silanization of NiO, followed by electropolymerization of a molecular Ru complex and a molecular Re complex, thereby improving the overall energy conversion efficiency and the system stability during long-term operation, for up to 10 hours.15 Recently, Osamu Ishitani and co-workers performed a new immobilization process by the electropolymerization of a supramolecular Ru–Ru catalyst, obtaining a robust and selective system to CO and HCOOH for CO2 reduction coupled with water oxidation, which remained stable for 100 h with a total faradaic efficiency of 65%.16
:H2 ratio of 1.5:1 and to exceed the performance observed with a NiO electrode; however, the instability of the phosphonic acid anchoring group was the main drawback. Later, Meyer and co-workers adopted a new immobilization method based on the silanization of NiO, followed by electropolymerization of a molecular Ru complex and a molecular Re complex, thereby improving the overall energy conversion efficiency and the system stability during long-term operation, for up to 10 hours.15 Recently, Osamu Ishitani and co-workers performed a new immobilization process by the electropolymerization of a supramolecular Ru–Ru catalyst, obtaining a robust and selective system to CO and HCOOH for CO2 reduction coupled with water oxidation, which remained stable for 100 h with a total faradaic efficiency of 65%.16
Inspired by the progress in capture and conversion of CO2 demonstrated using ILs and given the results achieved with the different semiconductors and immobilization procedures, we report here the preparation of various hybrid assemblies based on CuGaO2 and a series of new molecular and supramolecular Ru and Re complexes. The photoelectrocatalytic CO2 reduction was carried out using BMIM·TfO as a catholyte and a i) Ru(II) photosensitizer complex (RuVLA) co-immobilized with a Re(I) catalyst complex (ReCAT), ii) supramolecular RuRe1 complex and iii) binuclear RuRe2 complex to compare the electron transfer efficiency using the different supramolecular complexes. Higher performance was achieved with the supramolecular and binuclear systems than with the co-immobilized complexes and the results revealed the beneficial role of the ionic liquid in promoting the conversion of CO2 into formic acid and C2+ alcohols, while improving the stability of the system and suppressing the production of H2.
Results and discussion
Synthesis and characterization of CuGaO2 and CuGaO2-VTES
X-ray diffraction analysis confirmed the successful synthesis of the CuGaO2 structure without other obvious impurity phases. The diffraction pattern (see ESI† Fig. S1) was in agreement with the typical delafossite structure with a hexagonal unit cell.17 The calculated unit cell parameters were 2.974 and 17.143 Å for a and c, respectively, with a crystallite size of 58.0 ± 1.1 nm calculated by the Scherrer formula. These values agree well with the JCPDS data (a = 2.977, c = 17.171 Å, JCPDS database 77-2495) of CuGaO2.17 The FESEM image (Fig. 1) of the obtained solid displays the surface morphology of the CuGaO2 particles, which exhibit a few micrometers in size with parallel lines on the surface indicating their laminar structure. Moreover, the energy dispersive X-ray analysis of the sample (see ESI† Fig. S2) confirmed the average Cu:Ga:O ratio of 1:1:2.
 | ||
| Fig. 1 FESEM images of the synthesized CuGaO2. | ||
The functionalized CuGaO2 (CuGaO2-VTES) catalyst was analyzed with FESEM-EDX (see Fig. S3 and S4†), which indicated that the morphology remains unchanged after functionalization while EDX analysis confirmed the presence of Si with an approximate Cu:Si ratio of 16:1.
Synthesis and characterization of Ru and Re molecular complexes
Four new Ru and Re molecular complexes containing a bpy-V ligand were successfully synthesized in high yield and characterized by NMR, MS, IR and UV-vis spectroscopy (Fig. 2).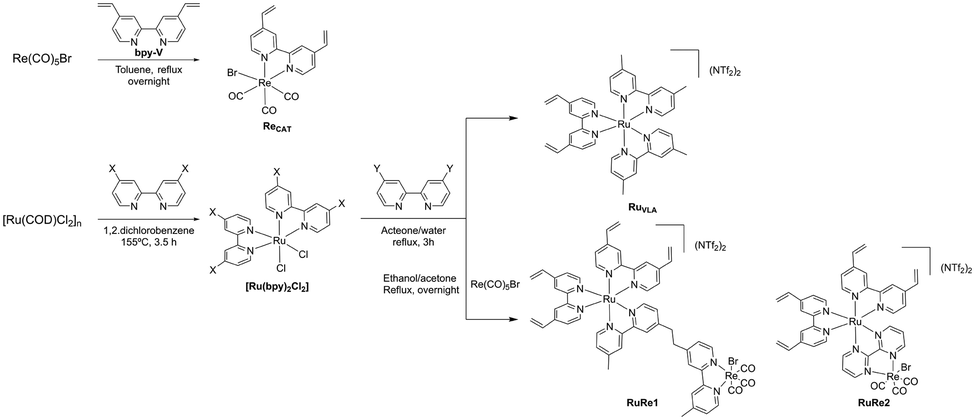 | ||
| Fig. 2 Schematic representation of the synthesis of the Ru and Re complexes used in this study. | ||
The synthesis of the ReCAT complex was performed using Re(CO)5Br as a precursor. This Re precursor and one equivalent of bpy-V ligand were dissolved in toluene and heated under reflux overnight to substitute two CO ligands, forming the ReCAT complex in high isolated yield (84%).
For the synthesis of RuVLA, the [Ru(COD)Cl2] precursor was first prepared starting from RuCl3·3H2O; then, the COD ligand was substituted by two 4,4′-dimethyl-2,2′-bipyridine (bpyMe2) ligands in 1,2-dichlorobenzene at 155 °C, obtaining the [Ru(bpyMe2)Cl2] complex. Finally, the remaining chloride ligands were substituted by one bpy-V ligand in an acetone/water solution obtaining the final desired complex in 74% isolated yield.
The complexes RuRe1 and RuRe2 were synthesized following the same synthetic strategy as the one adopted for RuVLA. The distance effect between Ru and Re was studied using these two complexes containing different bridge ligands between the Ru and Re. First, the [Ru(COD)Cl2] precursor was prepared, followed by the replacement of the COD ligand with two bpy-V ligands, obtaining the [Ru(bpy-V)Cl2] complex. Then, the chloride ligands were replaced by a 1,2-bis(4′-methyl-[2,2′-bipyridin]-4-yl)ethane or 2,2′-bipyrimidine ligand to obtain the corresponding Ru complexes. Finally, a reaction with one equivalent of Re(CO)5Br complex under reflux yielded the final RuRe1 and RuRe2 complexes in good isolated yields (64% and 67%, respectively). The coordination of the Re complex was confirmed by IR spectroscopy via the detection of the characteristic vibration bands of the CO ligands. The 1H and 13C NMR and HRMS analyses confirmed the structure of these complexes (see the ESI† for the detailed synthetic description and characterization of all the complexes).
Preparation of the RuRe@CuGaO2 photocathodes
Three photoelectrodes were prepared by electropolymerization of the synthesized homogeneous complexes (Fig. 2). The efficiency of the electropolymerization procedure of CuGaO2-VTES with the molecular complexes was corroborated by the continuous enhancement of the current densities after each scan (see ESI† S19). This observation suggested the in situ formation of polymeric layers onto the surface of the electrode. Furthermore, the presence of multiple vinyl groups in the diverse Ru and Re complexes led to a final layer-by-layer assembly of polymers, which translates into a different combination of interconnected complexes with the achievement of a high molecular loading on the surface of the electrodes.15,18The immobilization of Ru and Re species onto CuGaO2-VTES was supported by FESEM-EDX analysis. The obtained semi-quantitative results, the average of three different electrode zones, suggested the anchoring of equimolar amounts of Ru and Re species for both supramolecular Ru–Re complexes, whereas 2 moles of Re per mole of Ru were measured for the mixture of homogeneous complexes (see Table 1).
An estimation of the relative amount of Ru and Re immobilized with respect to the Cu deposited onto the support was performed to determine the efficiency of the anchoring approach. The approach used in the present work allowed a higher immobilization of the RuRe1 complex than that of the RuRe2 complex. Moreover, through the FESEM images, agglomerations were evidenced in all the electrodes containing the RuRe1 molecular complex (see Fig. 3).
 | ||
| Fig. 3 FESEM images and EDX analysis of the RuRe1@CuGaO2 electrode. a) EDX analysis of a region in the agglomeration section. b) EDX analysis of a region without agglomeration. c) Line scanning EDX analysis. | ||
The region containing the agglomeration (Fig. 3a) was analyzed by EDX and revealed a higher content of Ru and Re (Fig. 3b). These EDX results confirm that the agglomeration consists in RuRe1 supramolecular complexes interconnected between them. The line scanning EDX (Fig. 3c) performed in both regions, the agglomerated and non-agglomerated one, also confirms the presence of a higher content of Ru and Re in the agglomerated region compared to the non-agglomerated ones. Since the RuRe1 complex contains four anchoring groups, the coupling between molecular complexes could explain the agglomeration with high Ru and Re contents.
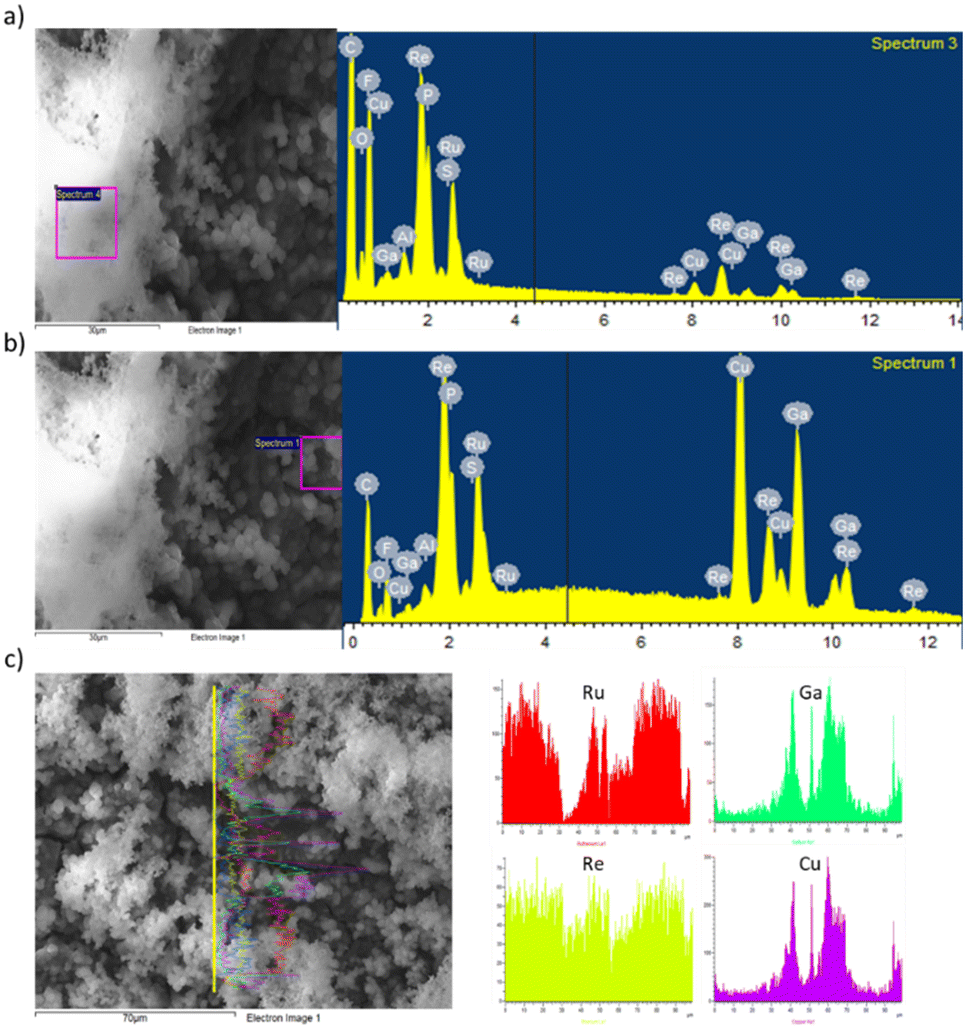
UV-visible absorption spectra of all the prepared electrodes were measured in diffuse reflectance mode (Fig. 4). The UV-vis spectrum of the bare CuGaO2 indicates no absorption in the visible range.17,19 However, a change was observed with the introduction of the Ru and Re complexes. Indeed, the electrode containing the molecular RuVLA and ReCAT complexes immobilized separately exhibited a small absorption band which starts at λ = 550 nm. Meanwhile, the electrode containing RuRe1 showed a large absorption band starting at λ = 600 nm and that with RuRe2 showed an absorption band throughout the visible light spectrum. These results are in concordance with previous reports where a characteristic peak appeared in the UV spectrum at around 464 nm, attributed to the singlet metal-to-ligand-charge-transfer absorption band of the Ru complex.18
 | ||
| Fig. 4 Diffuse-reflectance UV-visible absorption spectra of the prepared electrodes. | ||
Photoelectrochemical characterization of CuGaO2
There is a literature discussion about the electro- and photoelectrochemical activity of CuGaO2 in the CO2 reduction reaction.14,19 The photoelectrochemical and electrochemical properties of the bare CuGaO2 were first studied under dark conditions and under simulated sunlight irradiation. The current-potential curves (Fig. 5a) show a difference in the current between the dark and light conditions indicating that CuGaO2 is photocatalytically active under sunlight irradiation in a 0.3 M BMIM·TfO acetonitrile solution saturated with CO2. Indeed, from −0.3 to −1.2 V vs. Ag/AgCl, a difference in the current density values of about 19 μA cm−2 between darkness and light irradiation can be observed. | ||
| Fig. 5 a) Current–potential curves of the synthesized CuGaO2 with and without simulated sunlight irradiation in CO2 saturated 0.3 M BMIM·TfO acetonitrile solution. b) Dark/light chronoamperometry of CuGaO2 performed at −2.0 V vs. Ag/AgCl under the same conditions as those in a). | ||
Chopped dark/light chronoamperometry performed at −2.0 V (Fig. 5b) confirms the CuGaO2 photoactivity, showing an average photocurrent contribution of −0.195 mA cm−2.
Photoelectrochemical reduction of CO2 using the CuGaO2 photocathode
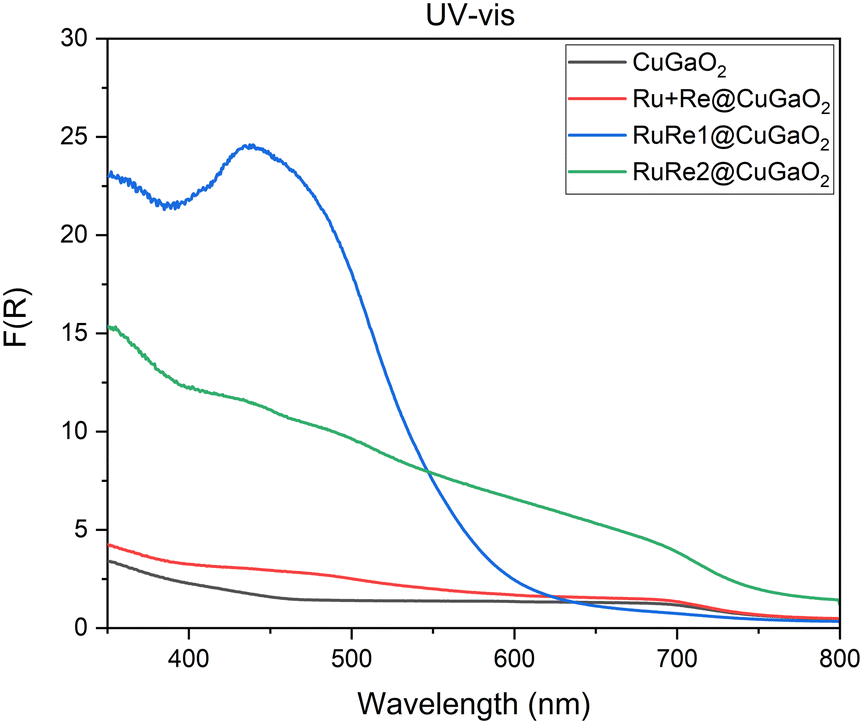
The effect of light and current intensity on the CO2 reduction performance was investigated on CuGaO2 by performing a current screening under both light and dark conditions and analyzing the gas and liquid products in each experiment. Short chronopotentiometry experiments of 30 min were performed: based on cyclic voltammetry (see ESI† Fig. S27), different current intensities were applied, specifically −0.2, −1.5, −3, −6, −10 and −20 mA cm−2, with the aim to observe the CO2 reduction product behavior in each experiment.As can be observed in Fig. 6, a significant difference was highlighted between dark and light conditions. Under dark conditions, a major selectivity towards CO2 reduction products and a faradaic efficiency to H2 lower than 10% were obtained. In contrast, under simulated sunlight irradiation conditions, a substantial increase in hydrogen production was observed. In both cases, the total faradaic efficiency decreased when higher current densities were applied. This behavior was attributed to the instability of the catalyst, i.e., self-reduction of CuGaO2.20
 | ||
| Fig. 6 Faradaic efficiencies of gas and liquid products produced in 30 min chronopotentiometry measurements at different current intensities on CuGaO2 electrodes in a CO2 saturated 0.3 M BMIM·TfO acetonitrile solution. a) Experiment without simulated sunlight irradiation. b) Experiment under simulated sunlight irradiation. | ||
Under both dark and light conditions, formate was the major CO2 reduction product, followed by CO. Small quantities of propanol, ethanol, and methanol were also detected. The production of CO and H2 was monitored during the experiment and was stable during the time of chronopotentiometry.
Fig. 7 shows the comparison of the chronopotentiometry measurements between dark and light conditions. The chronopotentiometry measurements performed at the same current intensity under visible light irradiation (dashed line) and in the dark (solid line) revealed a difference in the corresponding potential values. In general, lower potential values (in absolute values) were measured under light conditions than in the dark. However, the opposite behavior was observed when the chronopotentiometry was performed at −10 and −20 mA cm−2.
 | ||
| Fig. 7 Chronopotentiometry curves applying different current intensities under light irradiation (dashed line) and in the dark (solid line) on CuGaO2 electrodes in a CO2 saturated 0.3 M BMIM·TfO acetonitrile solution. The line colors are the same for the chronopotentiometry measurements performed at the same current intensity. | ||
Chronopotentiometry (CP) experiments for 120 minutes were performed in the dark and under visible light irradiation by applying −1.5 mA cm−2 to evaluate the stability of the CO2RR over time with the CuGaO2 bare material. At short reaction times, CO was the main product. However, at longer reaction times, the CO production decreased under both dark and light conditions (Fig. 8). When the experiment was performed in the dark, a constant decrease of CO selectivity was observed along with a constant increase in H2 production. Under light irradiation, the CO production decreased more rapidly and after ca. 40 min, H2 became the main product. The results obtained show that the total faradaic efficiencies are higher in the dark (FEtotal = 71% in the dark vs. 52% under light), confirming the quick deactivation of the bare CuGaO2 under light irradiation. These results were in agreement with previously reported data.19
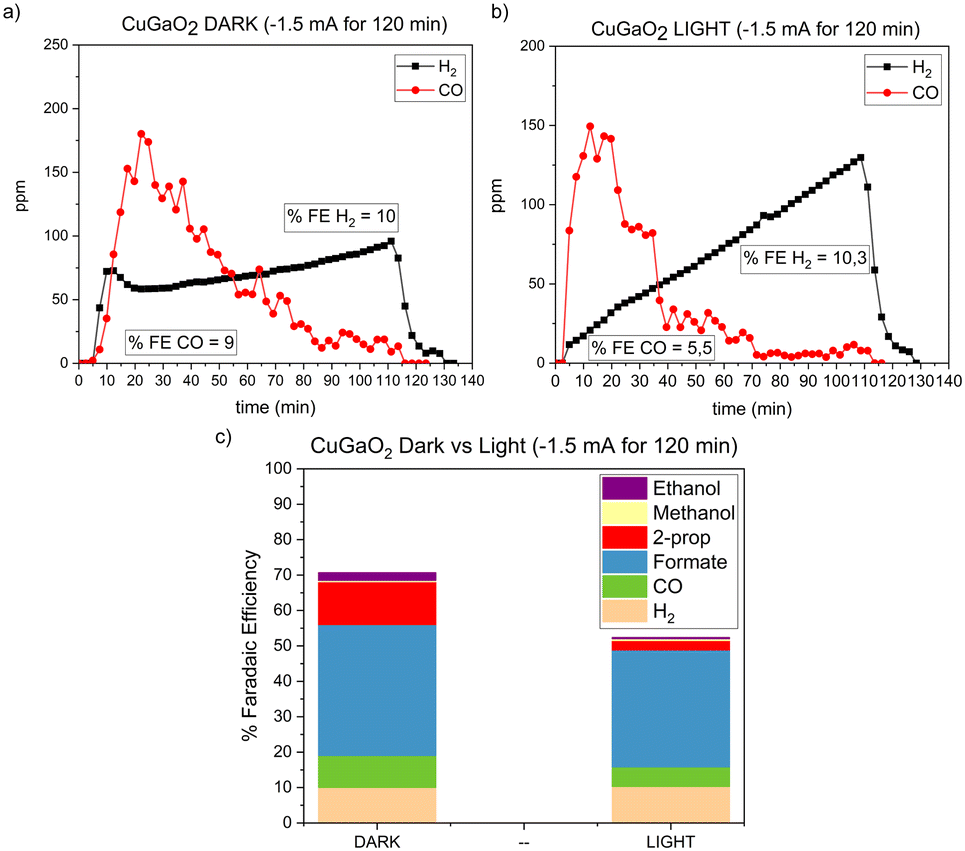 | ||
| Fig. 8 Chronopotentiometry performed at −1.5 mA cm−2 for 120 min in 0.3 M BMIM·TfO acetonitrile solution saturated with CO2. a) CO and H2 production under dark conditions. b) CO and H2 production under light conditions. c) Faradaic efficiencies under dark and light conditions. | ||
Photoelectrochemical properties of Ru + Re@CuGaO2
The photoelectrochemical properties of the prepared CuGaO2 electrode bearing a mixture of RuVLA and ReCAT (Ru + Re@CuGaO2) were studied and compared with those of the bare CuGaO2 electrode. The experiments were performed using simulated sunlight irradiation in an acetonitrile solution containing 0.3 M of BMIM·TfO and saturated with CO2. The current–potential curves (Fig. 9a) show a difference in the current response between dark and light conditions. Interestingly, Ru + Re@CuGaO2 exhibited an approximate current difference of 32 μA cm−2, which constituted a higher photo-response than using the bare CuGaO2 (19 μA cm−2) and indicated that the higher photocurrent was induced by the injection of electrons from CuGaO2 to the Ru photosensitizer. | ||
| Fig. 9 a) Current–potential curves of the synthesized Ru + Re@CuGaO2 with and without light irradiation. b) Dark/light chronoamperometry of the Ru + Re@CuGaO2 electrode performed at −2.0 V vs. Ag/AgCl in a CO2 saturated 0.3 M BMIM·TfO acetonitrile solution. | ||
Dark/light chronoamperometry performed at −2.0 V (Fig. 9b) confirmed the activity under light irradiation of the Ru + Re@CuGaO2 photocathode, showing an average photocurrent contribution of −0.204 mA cm−2 in a CO2 saturated 0.3 M BMIM·TfO acetonitrile solution. A comparison between Fig. 5b and 9b evidence that the two photoelectrodes gave a similar average photocurrent contribution at this fixed potential value. Nonetheless, Ru + Re@CuGaO2 was revealed to be more stable over time.
Photoelectrochemical reduction of CO2 using the Ru + Re@CuGaO2 photocathode
The selectivity of the Ru + Re@CuGaO2 electrode for the CO2RR was evaluated under simulated sunlight irradiation. As for the bare CuGaO2, short chronopotentiometry experiments of 30 min were performed by applying different constant current densities: −0.2, −1.5, −3, −6, −10 and −20 mA cm−2, respectively. The results are displayed in Fig. 10. | ||
| Fig. 10 Faradaic efficiencies of gas and liquid products produced in 30 min chronopotentiometry at different current intensities in a 0.3 M BMIM·TfO acetonitrile solution saturated with CO2 under simulated sunlight irradiation with the Ru + Re@CuGaO2 electrode. | ||
In general, the formation of CO2 reduction products was enhanced by the immobilization of Ru and Re molecular complexes onto the CuGaO2 semiconductor. More specifically, at current density values lower than −10 mA cm−2, the H2 production decreased along with an increase in the selectivity to CO2 reduction products. It should be noted that with the Ru + Re@CuGaO2 electrode, a total faradaic efficiency of 99% was reached at −1.5 mA cm−2, with a FEH2 of only 7%. A detrimental effect was evidenced at more negative currents with a clear decrease in the total FE, which could be due to the catalyst modification under these conditions. In view of these results, a current value of −1.5 mA cm−2 was selected for conducting the subsequent analyses. A blank control experiment was carried out using the Ru + Re@CuGaO2 electrode in a N2 saturated 0.3 M BMIM·TfO acetonitrile solution. After applying −1.5 mA cm−2 for 30 min, any CO2 reduction product was detected, confirming that the products originate from CO2.
Next, the effect of the reaction medium was evaluated. The reactions were carried out in the following CO2 saturated solutions: (i) 0.3 M BMIM·TfO in acetonitrile, chosen in view of the good results obtained using this IL in some recent studies,6–8 (ii) 0.3 M Et4N·PF6 in acetonitrile, to compare the use of an IL to that of a simple quaternary ammonium salt,21,22 and (iii) an aqueous solution of 0.1 M KHCO3, which was selected as a typical aqueous electrolyte.23
In aqueous medium, a constant H2 production was obtained with a total FE of 80% including a FE to formate (the main CO2RR product) of 14% (Fig. 11). As could be expected, in organic media (acetonitrile solutions of BMIM·TfO and Et4N·PF6), a higher selectivity to CO2 reduction products was obtained with formate as the major product. Interestingly, C2+ products like ethanol and propanol were also promoted in the organic solvents, and in particular with the IL electrolyte. This suggests the role of the imidazolium and ammonium-based electrolytes as co-catalysts that may also influence the CO2RR mechanism. Indeed, C2+ product formation requires the co-presence of adsorbed CO (CO*) and CHx ( ) intermediates, which react by CO*–CO*dimerization or CO*–
) intermediates, which react by CO*–CO*dimerization or CO*– coupling at the catalyst surface.24
coupling at the catalyst surface.24
 | ||
| Fig. 11 Faradaic efficiencies of gas and liquid products produced in 120 min chronopotentiometry at −1.5 mA cm−2, under continuous simulated sunlight irradiation, using different electrolytes, 0.3 M Et4N·PF6 in acetonitrile, 0.1 M KHCO3 in H2O and 0.3 M BMIM·TfO in acetonitrile, all saturated with CO2. | ||
Comparing the chronoamperometry curves, the experiment performed using acetonitrile 0.3 M BMIM·TfO as an electrolyte revealed a stable voltage of −1.9 V vs. Ag/AgCl (Fig. 12a), whereas using 0.3 M Et4N·PF6 in acetonitrile resulted in a variation of the voltage along the potential window from −1.8 V to −1.6 V. Using the aqueous solution of 0.1 M KHCO3, a stable voltage at −1.6 V vs. Ag/AgCl was also observed. It should be highlighted that the potential associated with the experiments seems to correlate with the product selectivity, since high hydrogen faradaic efficiencies and low formate faradaic efficiencies were achieved in the aqueous solution of 0.1 M KHCO3 at −1.6 V, whereas the opposite trend was observed using 0.3 M BMIM·TfO in acetonitrile as the electrolyte at −1.9 V. In the case of 0.3 M Et4N·PF6 in acetonitrile as the electrolyte, the selectivity varied with the potential along the experiment. In fact, considering the H2 and CO production during the test when using Et4N·PF6 (Fig. 12b), the CO production decreased severely after 40 min with a concomitant increase in hydrogen production.
 | ||
| Fig. 12 a) 120 min chronopotentiometry using the Ru + Re@CuGaO2 catalyst applying −1.5 mA cm−2 under simulated sunlight irradiation in a CO2 saturated solution using different reaction media. b) CO and H2 production during the 120 min chronopotentiometry using the Ru + Re@CuGaO2 catalyst at −1.5 mA cm−2 under the same conditions as those in a) using 0.3 M Et4N·PF6 in acetonitrile as an electrolyte. | ||
Based on these results, the beneficial effect of BMIM·TfO as the electrolyte was attributed to the high CO2 solubility9 and its potential role in lowering the activation energy of CO2 reduction.25 Then, 0.3 M BMIM·TfO in acetonitrile was selected as the electrolyte to study the immobilization of the different molecular complexes synthesized onto the CuGaO2 semiconductor.
Ru and Re complex effect in the photoelectrochemical reduction of CO2 in ionic liquid
The current–potential curves under dark and light irradiation conditions and the chopped chronoamperometry curves for the RuRe1@CuGaO2 and RuRe2@CuGaO2 materials are displayed in Fig. 13.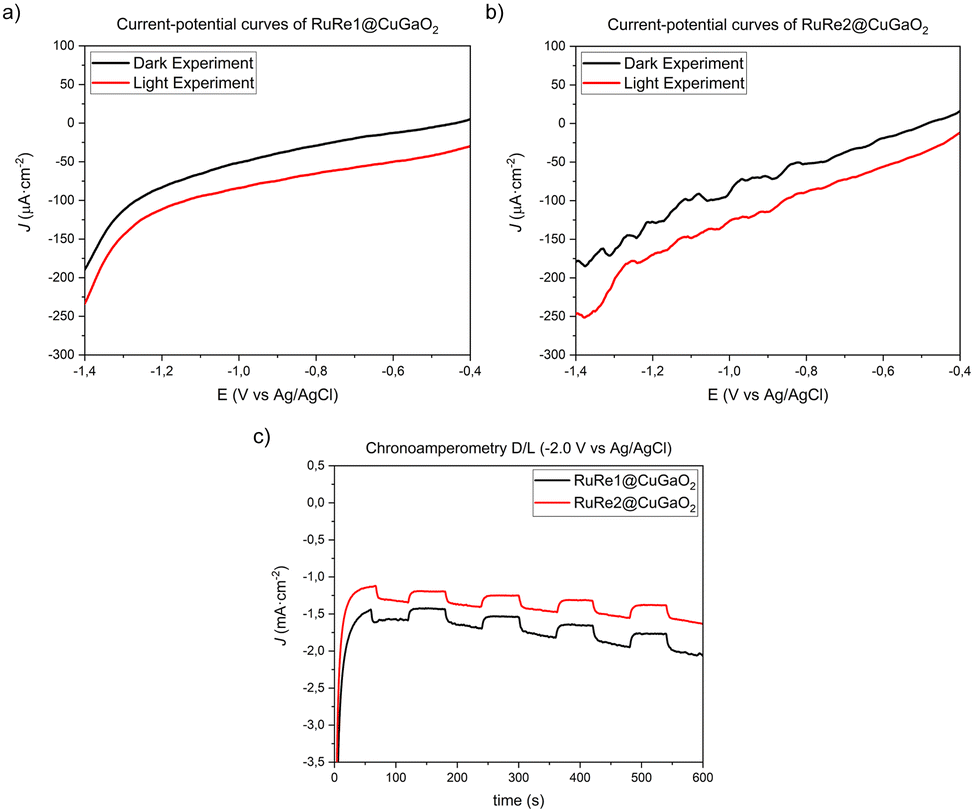 | ||
| Fig. 13 Current–potential curves with and without light irradiation in CO2 saturated 0.3 M BMIM·TfO acetonitrile solution of the a) RuRe1@CuGaO2 electrode and b) RuRe2@CuGaO2 electrode. c) Dark/light chronoamperometry of RuRe1@CuGaO2 (red line) and RuRe2@CuGaO2 (blue line) performed at −2.0 V vs. Ag/AgCl in 0.3 M BMIM·TfO acetonitrile solution saturated with CO2. | ||
In both cases, a difference in the current response between dark and light conditions was observed (Fig. 13). Slight differences in the measured photocurrent were observed for the electrodes containing the supramolecular complexes: RuRe1@CuGaO2 provided an average photocurrent of 36 μA cm−2 and RuRe2@CuGaO2 showed a value of 37 μA cm−2, from −0.4 to −1.2 V vs. Ag/AgCl. However, both electrodes exhibited a higher photoactivity than Ru + Re@CuGaO2 (with an average photocurrent of 32 μA cm−2 from −0.4 to −1.2 V vs. Ag/AgCl).
Nevertheless, when dark/light chronoamperometry at −2.0 V was performed (Fig. 13c), the electrodes containing the supramolecular complexes showed a lower average photocurrent contribution, −0.156 (RuRe1@CuGaO2) and −0.150 mA cm−2 (RuRe2@CuGaO2), than the Ru + Re@CuGaO2 electrode, −0.204 mA cm−2.
Subsequently, the effect of the different Ru/Re complexes on the CO2 photoelectroreduction selectivity were evaluated during 120 min chronopotentiometry by applying −1.5 mA cm−2 under simulated sunlight conditions. In Fig. 14a, the faradaic efficiencies of the gaseous and liquid products for each catalyst at this current density value are displayed. Using the bare CuGaO2 electrode, a low total faradaic efficiency was obtained (52%). With all the prepared hybrid materials, an increase in the faradaic efficiency was observed up to 81%, which evidenced the protective and stabilizing role of the Ru and Re complexes that hinder the CuGaO2 modification. Among them, similar faradaic efficiencies for H2 and CO2RR products were obtained; however, with the RuRe1 and RuRe2 complexes, lower external potentials were necessary at the same current intensity, which evidenced a higher generated photovoltage and a more efficient electron transfer when using the supramolecular systems rather than immobilizing separately the RuVLA and ReCAT complexes (Fig. 14b), which is in agreement with their superior UV-vis light absorption. Using the hybrid materials, a higher faradaic efficiency to formic acid was obtained compared with that of the bare CuGaO2, ca. 45% vs. 30%, respectively. Interestingly, the ethanol production and propanol production were enhanced with the hybrid materials and in particular with the binuclear RuRe2 complex. As previously mentioned, such results highlight the role of the Ru and Re complexes in co-catalyzing the multi-electron processes subsequent to the formate and CO formation at the catalyst surface.
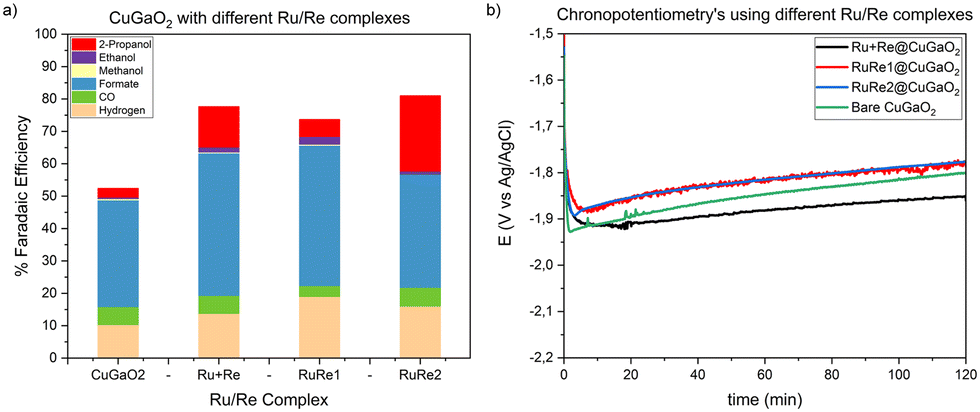 | ||
| Fig. 14 a) Faradaic efficiencies of gas and liquid products produced in 120 min chronopotentiometry tests at −1.5 mA cm−2, under continuous simulated sunlight irradiation in a 0.3 M BMI·TfO CO2 saturated acetonitrile solution using different Ru and Re complexes. b) 120 min chronopotentiometry recorded curves applying −1.5 mA cm−2 under the same conditions as those in a) using different Ru and Re complexes. | ||
Moreover, the hybrid materials synthesized in this work not only enhanced the faradaic efficiency of the process but also showed higher stability than the bare CuGaO2. Finally, the stability of the hybrid materials was corroborated by analyzing the electrodes after the CP experiments by FESEM-EDX. A similar morphology of the hybrid materials was observed after the CP experiments, indicating a positive effect of the immobilization of the molecular complexes (see the ESI†). Additionally, the semi-quantitative results obtained by EDX (Table 2) suggested the similar loading of the Ru and Re species before and after the CP experiments, thus confirming the stability of these systems under the reported reaction conditions.
| Electrode | Ratio of Cu:Ru |
Ratio of Cu:Re |
Ratio of Ru:Re |
|---|---|---|---|
| Ru + Re@CuGaO 2 | 39:1/40:1 |
19:1/22:1 |
1:2/1:1.8 |
| RuRe1@CuGaO 2 | 1.6:1/2.3:1 |
1.5:1/2.5:1 |
1:1.1/1:1.1 |
| RuRe2@CuGaO 2 | 26:1/33:1 |
23:1/43:1 |
1:1.1/1:1.3 |
Experimental section
Synthesis and functionalization of CuGaO2
The synthesis of the CuGaO2 metal oxide was carried out using a solid state reaction procedure adapted from the literature.14 A mixture of Cu2O (1.76 mmol, 251.9 mg, Sigma-Aldrich, 99.99% purity) and Ga2O3 (1.76 mmol, 330.0 mg, Sigma-Aldrich, 99.99% purity) powders was heated up from room temperature to 1100 °C at 5 °C min−1 under a N2 flow. Then the mixture was maintained for 15 h at 1100 °C and finally cooled down to room temperature under a N2 flow.The synthesized CuGaO2 was then functionalized with vinyltriethoxysilane (VTES) by adding 0.1 g of CuGaO2 to a 20 mL solution of isopropanol containing 10 mM vinyltriethoxysilane. The dispersion was stirred overnight at room temperature, then filtered, and finally washed with isopropanol.
Synthesis of molecular Ru and Re complexes
Four new complexes with 2,2′-bipyridine containing vinyl groups in the para-position (bpy-V) were synthesized in this work: (i) Re CO2 reduction co-catalyst (ReCAT); (ii) Ru visible light absorber (RuVLA); (iii) supramolecular Ru–Re complex (RuRe1) and (iv) the binuclear Ru–Re (RuRe2) complex. The synthesis of the ReCAT complex was adapted from a reported procedure26 but using the bpy-V ligand. The synthesis of the RuVLA, RuRe1 and RuRe2 materials was instead performed according to another procedure.18,27 The detailed synthetic procedures for these complexes are described in the ESI.†RuRe@CuGaO2 photocathode preparation
The electrodes were manufactured by airbrushing a catalytic ink onto a porous carbon support (Toray carbon paper 060, from FuelCellStore).28 The catalytic ink was constituted by various components: (i) the CuGaO2-VTES catalyst in powder form; (ii) NAFION® (dispersion, 5 wt% in water and 1-propanol, from Sigma Aldrich) as a binder for the particles and ionomer, and (iii) ethanol (from Sigma Aldrich), as a carrier for the ink deposition, since the ink must be fluid to be uniformly spread on the area of interest. Each CuGaO2-VTES electrode was prepared with a geometric area of 1 cm2 and a catalyst loading of 1 mg cm−2. The deposition process was performed by placing carbon paper on a heating plate at 50 °C to ensure complete solvent evaporation. A pressure of 1.5 bar for the carrier gas (nitrogen) in the airbrush inlet was selected to have a continuous ink flow, avoiding undesired liquid drops. All the electrodes were then kept on the heating plate for 15 min before their use.Next, the prepared electrodes containing CuGaO2-VTES were subjected to electropolymerization of the corresponding molecular complexes.15 The electropolymerization of the complexes was performed employing a three electrode set-up with CuGaO2-VTES, a platinum wire and Ag/AgNO3 as the working, counter, and reference electrodes, respectively. The electrodes were submerged in an argon degassed acetonitrile solution containing the corresponding complex at a 0.5 mM concentration and tetraethylammonium hexafluorophosphate (Et4N·PF6, 0.1 M) as an electrolyte. Cyclic voltammetry measurements were performed from 0 to −1.9 V vs. Ag/AgNO3 and the applied voltage was scanned multiple times with a scan rate of 100 mV s−1. After 60 scans, the electrode was removed and washed with acetonitrile. Three different photoelectrodes were prepared by immobilizing a series of homogeneous complexes onto CuGaO2-VTES: (i) Ru visible light absorber complex and Re CO2 reduction catalyst complex co-immobilized (Ru + Re@CuGaO2), (ii) supramolecular Ru–Re complex (RuRe1@CuGaO2) and (iii) binuclear Ru–Re complex (RuRe2@CuGaO2).
Characterization
The physicochemical properties of the synthesized CuGaO2 catalysts and of the electrodes prepared by electropolymerization were analyzed by X-ray diffraction (XRD, Panalytical X'Pert PRO diffractometer) and with Field Emission Scanning Electron Microscopy (FESEM) with Energy Dispersive X-ray Spectroscopy (ZEISS Auriga, equipped with an OXFORD X-MAX EDS detector). The electrodes were analyzed before and after the CO2RR tests to confirm the presence of Ru and Re and to determine the morphology and the approximate content of the complexes. UV-vis reflectance spectra (F(R) = (1 − R)2/2R: Kubelka–Munk function) of the prepared electrodes were collected with a spectrophotometer (Varian Cary 5000 spectrophotometer).The molecular complexes were characterized by 1H and 13C NMR (Varian Mercury VX 400), HRMS (Agilent Time-of-Flight 6210 using ESI-TOF), FT-IR (Bruker Vertex-70 instrument with attenuated total reflectance) and UV-vis (UV-1800 Shimadzu apparatus).
Photoelectrochemical measurements
The photoelectrochemical (PEC) measurements were performed in an H-type cell (made of quartz) with a three-electrode configuration. In a typical test, the anodic chamber was equipped with 28 mL of a 0.1 M KOH aqueous solution and a Pt mesh as the counter electrode. The cathodic chamber was instead equipped with 50 mL of 0.3 M BMIM·TfO acetonitrile solution (except when another electrolyte is specified), Ag/AgCl (KCl sat.) as the reference electrode and a CuGaO2-based electrode as the working electrode. The cathodic and anodic chambers were separated by a Fumasep® FBM bipolar exchange membrane (from FUMATECH BWT GmbH). Prior to each measurement, the electrolyte solution was purged for 20 min with a nitrogen (N2) flow rate of 20 mL min−1 to remove the oxygen present in the solution. Afterwards, the solution was saturated for 20 min under a CO2 flow rate of 20 mL min−1. During the experiments, a constant CO2 flow rate of 20 mL min−1 was maintained.The PEC measurements were carried out with a Voltalab potentiostat (from Radiometer Analytical SAS). Linear sweep voltammetry (LSV) and cyclic voltammetry curves were recorded in the dark and under continuous or chopped simulated solar light illumination conditions, in the range between −0.20 and −2.2 V vs. Ag/AgCl (KCl sat.) (with a sweep rate of 20 mV s−1). Chronoamperometry (CA) measurements were performed at −2.0 V vs. Ag/AgCl (KCl sat.) over 1 min darkness and 1 min simulated irradiation intervals. Chronopotentiometry (CP) measurements were performed both in the dark and under continuous simulated solar light irradiation conditions by using a Newport 450 W Xe lamp equipped with an AM 1.5 G filter. The H-type cell was illuminated from the cathodic side maintaining the intensity of the light at 1000 W m−2 by adjusting the distance between the light source and the PEC cell.
The gas products (H2 and CO) were determined continuously during all the experiments using a micro gas-chromatograph (Varian 490-GC from Agilent) directly connected to the cell. Samples of the liquid products were analyzed at the beginning and at the end of each experiment, by means of high-performance liquid chromatography (Shimadzu HPLC) and a gas chromatograph (Perkin Elmer GC with a mass spectrometer) with a headspace for the quantification of formate and alcohols, respectively.
Conclusions
A series of four molecular complexes was synthesized and characterized. Three different hybrid materials were prepared with the synthesized molecular complexes and their photoelectrocatalytic CO2 reduction performance was tested in the BMIM·TfO ionic liquid. An electropolymerization process was used for the first time for the immobilization of supramolecular Ru–Re complexes on VTES-modified CuGaO2. A comparison of the co-immobilization procedure of the separate RuVLA and ReCAT complexes with the supramolecular RuRe1 and the binuclear RuRe2 complexes demonstrated an enhanced photovoltage generation and electron transfer when the supramolecular and binuclear complexes were used. The potential of the approach of anchoring molecular complexes onto the CuGaO2 photo-electrocatalyst to boost both its carrier transport and catalytic activity with respect to the bare semiconductor material was shown. Furthermore, various electrolytes were evaluated and the use of ionic liquids was demonstrated to enhance both the CO2 reduction selectivity (by suppressing the H2 evolution reaction) and the stability of the photo-electrocatalytic performance. The combination of the use of the IL and the hybrid materials at the photocatalyst surface increased the semiconductor stability towards its restructuration, which resulted in a 30% increase of the total faradaic efficiency vs. the bare CuGaO2, and in an enhanced formic acid production (from 30% to 45%) as well as C2+ alcohol formation. Further investigations will be devoted to investigating the long-term stability and to elucidating the role of the supramolecular and binuclear complexes, and of the IL, in co-catalyzing multi-electron CO2 reduction reaction pathways under photo-electrochemical conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the European Union's Horizon 2020 Research and Innovation Programme under grant agreement no. 862192 (SUNCOCHEM project), the Ministerio de Economia y Competividad (PID2019-104427RB- I00) and Eurecat's “Vicente López” PhD grant program for funding.Notes and references
- A. Rafiee, K. Rajab Khalilpour, D. Milani and M. Panahi, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- S. Lian, C. Song, Q. Liu, E. Duan, H. Ren and Y. Kitamura, J. Environ. Sci., 2021, 99, 281–295 CrossRef CAS PubMed.
- D. R. Macfarlane, M. Forsyth, P. C. Howlett, J. M. Pringle, J. Sun, G. Annat, W. Neil and E. I. Izgorodina, Acc. Chem. Res., 2007, 40, 1165–1173 CrossRef CAS PubMed.
- J. Feng, S. Zeng, J. Feng, H. Dong and X. Zhang, Chin. J. Chem., 2018, 36, 961–970 CrossRef CAS.
- J. Medina-Ramos, J. L. Dimeglio and J. Rosenthal, J. Am. Chem. Soc., 2014, 136, 8361–8367 CrossRef CAS PubMed.
- J. Medina-Ramos, R. C. Pupillo, T. P. Keane, J. L. Dimeglio and J. Rosenthal, J. Am. Chem. Soc., 2015, 137, 5021–5027 CrossRef CAS PubMed.
- Z. Zhang, M. Chi, G. M. Veith, P. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Zhu, ACS Catal., 2016, 6, 6255–6264 CrossRef CAS.
- S. N. V. K. Aki, B. R. Mellein, E. M. Saurer and J. F. Brennecke, J. Phys. Chem. B, 2004, 108, 20355–20365 CrossRef CAS.
- M. G. Freire, C. M. S. S. Neves, I. M. Marrucho and A. P. Coutinho, J. Phys. Chem. A, 2010, 114, 3744–3749 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- T. T. Li, B. Shan and T. J. Meyer, ACS Energy Lett., 2019, 4, 629–636 CrossRef CAS.
- R. Kamata, H. Kumagai, Y. Yamazaki, M. Higashi, R. Abe and O. Ishitani, J. Mater. Chem. A, 2021, 9, 1517–1529 RSC.
- M. Lee, D. Kim, Y. T. Yoon and Y. Il Kim, Bull. Korean Chem. Soc., 2014, 35, 3261 CrossRef CAS.
- R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS PubMed.
- J. W. Lekse, M. K. Underwood, J. P. Lewis and C. Matranga, J. Phys. Chem. C, 2012, 116, 1865–1872 CrossRef CAS.
- C. Y. Toe, Z. Zheng, H. Wu, J. Scott, R. Amal and Y. H. Ng, Angew. Chem., Int. Ed., 2018, 57, 13613–13617 CrossRef CAS PubMed.
- J. Choi, T. M. Benedetti, R. Jalili, A. Walker, G. G. Wallace and D. L. Officer, Chem. – Eur. J., 2016, 22, 14158–14161 CrossRef CAS PubMed.
- A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Chem. Commun., 2018, 54, 11630–11633 RSC.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- H. Guzm, F. Salomone, S. Bensaid, M. Castellino, N. Russo and S. Hern, ACS Appl. Mater. Interfaces, 2022, 14, 517–530 CrossRef PubMed.
- D. Yang, Q. Zhu and B. Han, Innovation, 2020, 1, 100016 Search PubMed.
- M. Braumüller, M. Schulz, M. Staniszewska, D. Sorsche, M. Wunderlin, J. Popp, J. Guthmuller, B. Dietzek and S. Rau, Dalton Trans., 2016, 45, 9216–9228 RSC.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- H. Guzmán, F. Zammillo, D. Roldán, C. Galletti, N. Russo and S. Hernández, Catalysts, 2021, 11, 482 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01523d |
| This journal is © The Royal Society of Chemistry 2023 |