
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into coverage-affected selective catalytic oxidation of ethylene on Ag(111) from comprehensive microkinetic analyses†
Zhuozheng
Wang
a,
Wenbo
Xie
a,
Yarong
Xu
b,
Menglei
Jia
ac,
Jiayan
Xu
a and
P.
Hu
 *ad
*ad
aSchool of Chemistry and Chemical Engineering, The Queen's University of Belfast, Belfast BT9 5AG, UK. E-mail: p.hu@qub.ac.uk
bResearch Institute of Urumqi Petrochemical Company, Petrochina Company Limited, Urumqi 830019, China
cKey Laboratory for Advanced Materials, Centre for Computational Chemistry and Research Institute of Industrial Catalysis, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
dSchool of Physical Science and Technology, ShanghaiTech University, 393 Middle Huaxia Road, Shanghai 201210, China
First published on 9th May 2023
Abstract
Ethylene epoxidation is one of the most critical industrial reactions. Although there have been many studies on the reaction, the details of the reaction kinetics remain elusive. In this work, an extensive microkinetic modelling based on DFT calculations was conducted to study ethylene epoxidation on Ag(111). Coverage-dependent and coverage-independent microkinetic modellings were performed with rigorous energetics calculated on adsorption and transition states. The detailed comparisons between the two modellings show that the coverage-independent modellings are inadequate even though the coverage is very low at the steady state. It is also found that the activity and selectivity trends from the coverage-dependent modelling are consistent with the experimental values under the experimental conditions. A comprehensive kinetic analysis under industrial conditions was conducted based on the coverage-dependent modelling, which shows that increasing the temperature and pressure can effectively enhance the activity and ethylene epoxide (EO) selectivity.
1. Introduction
Ethylene epoxidation is an essential part of the ethylene industry: ethylene epoxide (EO) is the precursor of many complex chemicals, including plastics, detergents, and bactericides. With the development of global economy, the demand for ethylene epoxide is increasing sharply. Therefore, how to increase the yield of ethylene epoxide has always been an enduring hot topic in the industry. Selective ethylene oxidation based on silver catalysts is the primary approach of industrial production of ethylene epoxide, which has been an important research field experimentally and theoretically.1–7 One of the most concerning issues is improving the activity and selectivity, and some good progress has been made.8–13 For example, some studies have utilized dopants to increase the EO selectivity from 40% to more than 70%.3,14–17 Since the catalytic activity and selectivity are primarily affected by the state of the original crystal facet, it is still highly desirable to study the reaction details on pure metal surfaces as a benchmark.The most stable silver catalyst Ag(111) has been extensively studied theoretically and experimentally.18–24 The mechanism of ethylene epoxidation on the unpromoted Ag(111) surface is relatively straightforward. Firstly, oxygen is adsorbed on the silver surface and dissociated into oxygen atoms. In the past two decades, several research groups have proposed the idea of an initial reaction between ethylene and oxygen atoms to form an oxametallacycle (OME) intermediate.16,22,25–27 Subsequently, two parallel reactions occur from the OME intermediates to generate ethylene epoxide and acetaldehyde (AA), respectively, which are the selectivity-determining steps in the ethylene oxidation reaction.28 Some experiments and theoretical calculations showed that the reaction barriers of EO and AA are almost the same, which indicates that the EO selectivity may be about 50% on the unpromoted Ag(111) surface.18,29,30 Although both parallel reactions are exothermic, it is evident that the EO path is thermally disadvantaged.31–33 Therefore, improving the EO selectivity needs to consider both thermodynamics and kinetics. However, since the coverage effect was not considered and the activation Gibbs free energies of EO and AA under industrial conditions (i.e., high temperature and high pressure) were yet unresolved, the debate on the activity and selectivity still exists in previous theoretical studies.
The coverage effect is one of the critical factors affecting surface reactions. There is often a considerable difference between simulated results that do not consider the coverage effect and experimental values.34–40 Reaction kinetics is an essential part of studying the reaction mechanism in various catalytic processes.41–45 Therefore, incorporating the coverage effects when studying surface reactions is an indispensable step in computational kinetic simulations and implementing the coverage-dependent kinetic model is necessary to obtain accurate kinetic results. Our group has developed more realistic heterogeneous microkinetic simulation models with coverage effects for several systems, such as selective acetylene hydrogenation and nitric oxide oxidation on Pd(111), and with the coverage effect, the simulated activity and selectivity were in good agreement with the experimental values.36–38,46 It is expected that the coverages of Ag catalysts during the ethylene epoxide process are very low due to the fact that the Ag catalysts are generally inert to adsorbates and the temperatures under industrial conditions are also high. Hence, it is not clear whether the coverage effect is still of importance to the accuracy of kinetic simulations.
In previous studies, several simple microkinetic models were discussed for ethylene epoxidation.27,33,47,48 However, there is still a gap when comparing the simulated results to the experimental values, especially lacking simulation data under industrial conditions. Therefore, it is desirable to systematically perform the quantitative microkinetic study of ethylene epoxidation under industrial conditions with the coverage effects. In this work, the Ag(111) surface was utilized to study the kinetic process of ethylene epoxidation with the coverage effect. Quantitative comparisons between coverage-dependent and coverage-independent microkinetic results and experimental values were carried out. Our results of coverage-dependent microkinetic modelling are in good agreement with the experimental values. The effect of adsorbed oxygen on the reaction kinetics under different industrial conditions was systematically studied. There are two reactant feeding approaches (i.e., the oxygen-rich and ethylene-rich) in the EO industrial production, corresponding to the ratios of oxygen partial pressure to ethylene partial pressure (pO2/pEt) being 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (oxygen-rich) and 1:2 (ethylene-rich), respectively.49 Thus, the pO2/pEt range from 3:1 to 1:3 was used in our work, which is sufficient to cover the industrial reaction conditions. Our results provide some significant insights into the industrial EO production process.
:1 (oxygen-rich) and 1:2 (ethylene-rich), respectively.49 Thus, the pO2/pEt range from 3:1 to 1:3 was used in our work, which is sufficient to cover the industrial reaction conditions. Our results provide some significant insights into the industrial EO production process.
2. Computational method
2.1 Energy calculations
All DFT calculations in this work utilize the VASP package50,51 with the generalized gradient approximation (GGA) based on the Perdew–Burke–Ernzerhof (PBE) functional.52 PBE is the most utilized functional for studying catalytic reactions on metal surfaces. Valence electrons were simulated utilizing projector-augmented-wave (PAW) pseudopotentials,51,53 with a cutoff energy of 400 eV. The determination of the transition state utilized the constrained minimization method based on the quasi-Newton algorithm.54–57 The Ag(111) bulk simulation utilized a p(3 × 3) supercell with 2 × 2 × 1 Monkhorst–Pack k-point mesh sampling.58 The vacuum layer was set as 12 Å for all the simulations. The required structural optimization accuracy was reached when the forces on the relaxed atoms were less than 0.05 eV Å−1. The DFT-D3 approximation method was utilized to correct van der Waals (vdW) interactions.59 All the energy utilized were free energies, which were obtained from total energies with thermodynamic corrections. All the gas-phase molecules were optimized in a 10 Å × 10 Å × 10 Å box with the thermodynamic correction calculated by Gaussian 09 (ref. 60) based on the B3LYP functional utilizing ideal gas approximation.61,622.2 Microkinetic models
Ethylene epoxidation on the Ag(111) surface utilizing microkinetic modeling optimized by the coverage effects was systematically studied. Nine elementary steps were utilized to describe the overall process of ethylene epoxidation, as shown in Table 1. In Table 1, ri represents the reaction rate of the i-th step, and ki and k−i represent the forward reaction rate constant and the reverse reaction rate constant. Since the Langmuir–Hinshelwood and Eley–Rideal mechanisms in ethylene epoxidation are still controversial and no unified results have been obtained,63–66 the model in this work includes both mechanisms. The parallel pathways of EO and AA formation (i.e., OME* → EO* and OME* → AA*) are defined as the selective competition steps. The transition state theory was utilized to obtain the reaction rate constant, | (1) |
| Elementary step | Rate equations | |
|---|---|---|
| 1 | O2(g) + 2* ↔ |

|
| 2 | C2H4(g) + * ↔ |
r 2 = k2PC2H4θ* − k−2θC2H4 |
| 3 |
 ↔ 2O* ↔ 2O* |
r 3 = k3θO2 − k−3θO2 |
| 4 |
 + O* ↔ OME* + * + O* ↔ OME* + * |
r 4 = k4θC2H4θO − k−4θOMEθ* |
| 5 | C2H4(g) + O* ↔ OME* | r 5 = k5PC2H4θO − k−5θOME |
| 6 | OME* ↔ EO* | r 6 = k6θOME − k−6θEO |
| 7 | OME* ↔ AA* | r 7 = k7θOME − k−7θAA |
| 8 | EO* ↔ EO(g) + * | r 8 = k8θEO − k−8PEOθ* |
| 9 | AA* ↔ AA(g) + * | r 9 = k9θAA − k−9PAAθ* |
There are nine active sites on the surface of p(3 × 3) Ag(111), which are occupied by adsorbates entirely defined as 100% coverage, that is, one monolayer (1 ML). Except for adsorbed oxygen molecules occupying two active sites, all other surface species occupy only one active site. Therefore, if one adsorbed oxygen atom is present on the surface, the coverage is 0.11 ML, that is, the coverage at which the energetics were obtained in the coverage-independent modelling defined in this work. The adsorption energies under different coverages are
| Gads(θn) = Gn − Gn−1 − Ggas | (2) |
In order to study the relationship between surface structure and adsorption energy under different coverage conditions as well as an in-depth understanding of the ethylene epoxidation reaction mechanism with coverage effects, an optimized two-line model was utilized in this work.37,38,67–69 The two curves describing the interaction between environmental adsorbates and target adsorbates could be written as
 | (3) |
Turnover frequency (TOF) is a commonly used measurement of the catalytic activity and hence can represent the intrinsic activity of the catalyst. A self-consistent iterative method is applied to calculate the TOF values (see Fig. 1) under industrial and experimental conditions. The microkinetic modeling software utilized for data analysis is CATKINAS.70–73 The coverage of species and kinetic data of reactions at the steady state is achieved when the coverage converges. The convergence value X set during the convergence process of the microkinetic modeling is small enough (X <0.001) to make the result reliable.
 | ||
| Fig. 1 Flow chart of the self-consistent process of coverage-dependent microkinetic modeling. | ||
3. Results and discussion
3.1 Traditional ethylene epoxidation mechanism on the Ag(111) surface
On the unpromoted Ag(111) surface, the ethylene epoxidation mechanism consists of nine elementary steps, as shown in Fig. 2(a): firstly, oxygen molecules and ethylene molecules adsorb on the silver surface (reactions (1) and (2), respectively). Then, the adsorbed oxygen molecules dissociate into oxygen atoms through transition state TSOO (reaction (3)). Both the Langmuir–Hinshelwood (reaction (4)) and Eley–Rideal (reaction (5)) pathways to the oxametallacycle (OME) intermediates are considered through transition state 1 (TS1). Subsequently, the OME intermediate decomposes to produce EO through TSEO (reaction (6)) and AA through TSAA (reaction (7)) in parallel, which determines the selectivity of the catalyst. Finally, the adsorbed EO and AA desorb (reactions (8) and (9), respectively) into the gas phase.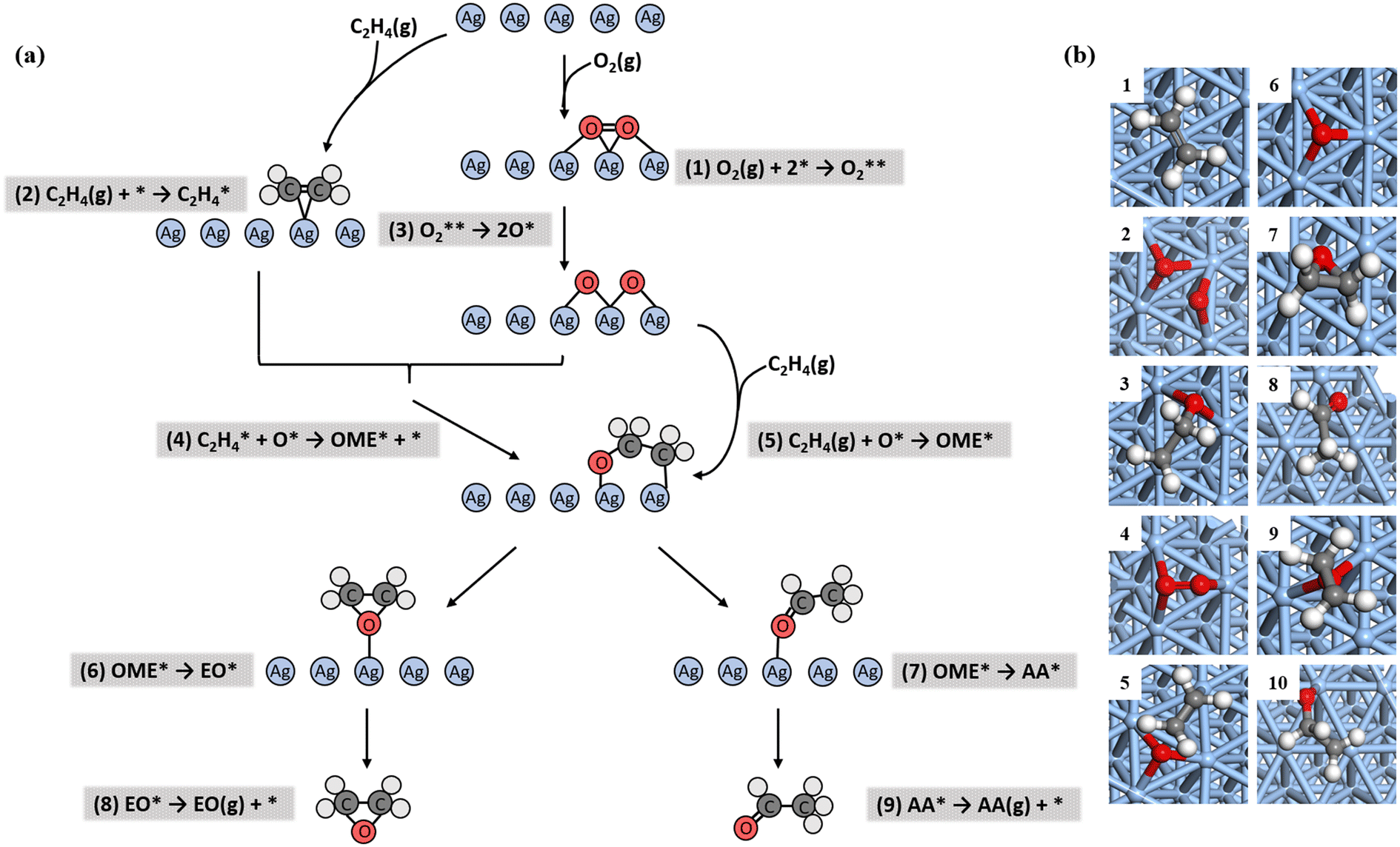 | ||
Fig. 2 (a) Reaction networks of ethylene epoxidation starting from ethylene and oxygen in the gas phase. (b) The top view of surface species during the epoxidation. The first column (from 1 to 5):  , ,  , OME*, TSOO, and TS1. The second column (from 6 to 10): O*, EO*, AA*, TSEO, and TSAA. Color code: red, oxygen; white, hydrogen; dark grey, carbon; blue, silver, which is adopted throughout the paper. , OME*, TSOO, and TS1. The second column (from 6 to 10): O*, EO*, AA*, TSEO, and TSAA. Color code: red, oxygen; white, hydrogen; dark grey, carbon; blue, silver, which is adopted throughout the paper. | ||
The structures of all the species involved in the epoxidation are shown in Fig. 2(b) and S1.† The oxygen atom adsorbs on the fcc site instead of the hcp site because of the stronger adsorption energy on the fcc site, which is consistent with the experimental result.32,74 The adsorption energy of oxygen molecules is very small. However, the activation energy of O2 dissociation to form adsorbed oxygen atoms is distinctly sizeable. This has a considerable impact on the ethylene epoxidation, which will be discussed in detail in the subsequent microkinetic analysis. The ethylene adsorption on the Ag(111) surface is weak and easy to desorb, unlike those on other transition metal surfaces.38,75 The surface intermediate OME is widely present on transition metal catalysts.18,76,77 On the Ag(111) surface, the intermediate is a five-membered ring, as reported by Linic and Barteau.16,27 The –O–C–C– fragments on the surface form a spine-like structure, as shown in Fig. 2(b)-3. The structure of OME is stable because its formation is a significantly exothermic process. The selective competition steps of OME to generate EO and AA have considerable reaction barriers in the reaction network. As a result, the selective competition steps are the crucial steps in determining the reaction selectivity.
3.2 Coverage-dependent microkinetic modelling
 | ||
| Fig. 3 Structures of O*/Oenv at the coverages from 0.11 ML to 1.0 ML. | ||
The structures under different coverages of OME*/Oenv, which reflect the interactions between the environmental species and the adsorbed OME intermediate, are included in Fig. S2.† The curves of adsorption energy changes with the changes of coverages at 490 K are shown in Fig. 4, and detailed diagrams at other temperatures are shown in Fig. S7 and S8.† All the adsorption energies utilized were determined as the formation energies to reduce the simulation errors, the detail of which is illustrated in ESI† S5. Fig. 4(a) and (b) present the self-interactions between the environmental oxygen atoms and the oxygen atom adsorbate and the interactions between the environmental oxygen atoms and the OME intermediate adsorbate, respectively. As the number of environmental species increases, the adsorption of the adsorbate becomes weaker, which can be readily understood, considering the combination of bonding competitions and surface repulsion effects.80–83 With the increase of surface environmental species, the surface becomes more inert and resists new adsorption, leading to a moderate decrease in adsorption strength.
 | ||
| Fig. 4 Variations of adsorption free energies with different coverages at 490 K. (a) O*/Oenv (0.11 ML to 1.0 ML) and (b) OME*/Oenv (0.11 ML to 0.67 ML). The red lines represent the linear curves at low coverages, and the blue lines represent the linear curves at high coverages. | ||
As shown in Fig. 4, the adsorption energy changes smoothly in the low coverage region, beyond which the adsorption energy changes more rapidly. It is apparent from Fig. 4(b) that the differential adsorption energy of OME* is changed by 0.44 eV from 0.11 ML to 0.33 ML, while from 0.44 ML to 0.67 ML, the differential adsorption energy changes significantly by 0.84 eV. The slopes of the regression lines at different temperatures at different coverages are listed in Table 2. It can be clearly seen from the table that all the slopes of high coverages are higher than those of low coverages at all the temperatures, and the trends of adsorption energy changes are similar. The difference in adsorption energy changes in the two coverage regions suggests that the application of the two-line model is necessary and effective.
| O* | Slope (eV ML−1) | OME* | Slope (eV ML−1) | ||
|---|---|---|---|---|---|
| Low coverage | High coverage | Low coverage | High coverage | ||
| 490 K | 2.545 | 2.738 | 490 K | 2.000 | 3.652 |
| 500 K | 2.523 | 2.729 | 500 K | 1.977 | 3.630 |
| 520 K | 2.523 | 2.727 | 520 K | 1.955 | 3.652 |
| 540 K | 2.568 | 2.716 | 540 K | 2.023 | 3.587 |
| 600 K | 2.523 | 2.688 | 600 K | 2.023 | 3.587 |
| Slope (eV ML−1) | ||||
|---|---|---|---|---|
| T | TSOO | TS1 | TSEO | TSAA |
| 490 K | 4.409 | 3.045 | 0.932 | 0.705 |
| 500 K | 4.409 | 3.045 | 0.932 | 0.705 |
| 520 K | 4.432 | 3.023 | 0.955 | 0.682 |
| 540 K | 4.455 | 3.068 | 1.023 | 0.727 |
| 600 K | 4.500 | 3.068 | 1.068 | 0.750 |
The TSOO structures at the coverages of 0.11 ML and 0.89 ML and the TS1 structures at 0.11 ML and 0.67 ML are shown in Fig. 5(a)–(d), respectively. For TS1, the bond distances of O–C1 and Ag–C2 change from 2.049 to 2.103 Å and 2.514 to 2.438 Å, respectively, corresponding to the coverage range of 0.11 ML to 0.67 ML. Meanwhile, the bond distance of O–O gradually increases from 1.880 Å to 1.975 Å with the coverage range of 0.11 ML to 0.89 ML at TSOO. The diagrams show that the distance between O–O becomes more extended, while the distance between oxygen atoms and surface silver atoms becomes shorter at TSOO with the coverage increase. Compared to the TS1 structure at 0.11 ML, the distance between O–C1 is longer, but the distance between Ag–C2 is shorter in the TS1 structure at 0.67 ML, which means that the ethylene molecules are closer to the surface adsorption state at 0.67 ML. In other words, the transition state where the adsorbed ethylene molecule reacts with the oxygen atom is easier to achieve with the coverage increase. Taken together, these results suggest that the transition state structures are closer to the adsorption state structures as the surface coverage increases and the reaction barriers associated with the corresponding transition states decrease as the surface coverage increases along with the structural variations.
 | ||
| Fig. 5 Comparison between (a) the TSOO structure from the coverage-independent model and (b) one from the coverage-dependent model at 0.89 ML. The O–O distance in the oxygen molecule is illustrated (colored light green and deep blue). Comparison between (c) the TS1 structure from the coverage-independent model and (d) one from the coverage-dependent model at 0.67 ML. The two carbon atoms in ethylene are labeled C1 (grey) and C2 (orange). The oxygen atom involved in the reaction is colored bright green. | ||
3.3 Comparison of reaction kinetics between coverage-independent and coverage-dependent models under industrial conditions
In this work, the coverage-independent microkinetic modelling means that the microkinetic simulations were conducted using the energies of all species obtained on Ag(111) at a low coverage (0.11 ML), an approach widely used. All the adsorption and activation energies were obtained from the DFT calculations, and the free energies at different temperatures were acquired by thermodynamic corrections.38,67 It is worth noting that all the molecule adsorption and desorption barriers are very small on the Ag(111) surface. Consequently, the microkinetic model utilized in this work treats all the adsorption and desorption processes as quasi-equilibrium steps. Extensive microkinetic calculations under different temperatures and pressures were systematically carried out, and the conversion rate was set to be 1% to be consistent within the experimental range.30,49 In the coverage-dependent microkinetic modelling, the coverage-dependent adsorption energies and reaction barriers were accurately calculated with the coverage effects and the steady state was achieved self-consistently. All the microkinetic modellings were performed utilizing the microkinetic program CATKINAS developed by our group, mentioned in the Computational method section.Several temperatures (510 K, 540 K, and 580 K) within the experimental temperature range were selected for comparison with the experimental values at pO2 = 20 kPa and pEt = 2.66 kPa, as shown in Fig. 6. The microkinetic modelling results reveal the following kinetic features of ethylene epoxidation. Firstly, the range of log(TOF) values (−1.41 to −2.30) calculated by the coverage-independent modelling is several orders of magnitude smaller than the range of experimental log(TOF) values (2.15–3.17) between 510 K and 580 K, as shown in Fig. 6(a). Instead, the log(TOF) range (2.36–2.67) from the coverage-dependent microkinetic modelling agrees well with the experimental values. Furthermore, the calculated coverage of adsorbed species on the Ag(111) surface is nearly zero from the coverage-independent model, as shown in Fig. 6(b). There is an evident difference between the coverage at the steady state derived from the coverage-independent modelling (near zero) and the coverage of its input (0.11 ML), implying that the neglect of coverage effects leads to significant kinetic distortion. Meanwhile, the coverage-dependent microkinetic modelling gives rise to a 7% coverage of surface oxygen atoms, which is more reasonable for the reaction than the coverage-independent results and is in fact in good agreement with experimental values,30,49 reflecting the accuracy of our microkinetic modelling with the coverage effect.
 | ||
| Fig. 6 (a) TOF values of ethylene epoxidation obtained from experiments, the coverage-dependent modelling, and the coverage-independent modelling at pO2 = 20 kPa, pEt = 2.66 kPa. (b) The calculated surface species distributions from the coverage-dependent modelling and coverage-independent modelling at 540 K, pO2 = 20 kPa, pEt = 2.66 kPa. | ||
Then, rigorous analyses of the microkinetic modelling results under industrial conditions (pO2= pEt = 13.4 kPa, T = 490 K) were performed and the results are shown in Fig. 7(a). The log(TOF) values from the coverage-dependent kinetic modelling fall in the range of 2 to 4, in good agreement with the reported experimental values.30 It is also not surprising that the coverage-independent kinetic modelling results are way off from the experimental values with temperature variations: It decreases more rapidly as the temperature drops. The free energy profiles for both kinetic models are illustrated in Fig. 7(b). It is worth mentioning that the energies in the figure obtained from the coverage-dependent kinetic modelling at the steady state are at 0.051ML. The significant differences between the energy results of the two kinetic modellings are precisely striking. It can be seen from the energy profiles that with the inclusion of the coverage effect, the dissociation energy barrier of adsorbed oxygen molecules drops significantly from 0.41 eV to 0.15 eV. The decline of the reaction barrier allows consequently the epoxidation to be more efficient. In addition, the reaction barriers of the selective competition steps obtained from the coverage-dependent kinetic modelling are higher than those from the coverage-independent kinetic modelling. Compared with the coverage-independent modelling, the selective competition steps affect the reaction rate more strongly than that in the coverage-dependent modelling. With an almost empty surface in the coverage-independent modelling, it is almost impossible for the steps after the O2 dissociation to affect the overall reaction rate. Linic et al. developed a straightforward selectivity indicator expressed as ΔE (TSEO – TSAA), which represents the difference in the reaction barriers of the TSEO and TSAA.84 Herein, the indicator is used to determine the selectivity variations in ethylene epoxidation qualitatively. As can be seen from Fig. 7(c), the reaction barrier of TSAA is gradually higher than that of TSEO as the temperature increases, which illustrates that the dominant reaction pathway shifts from the AA formation step to the EO formation step with the temperature increase.
 | ||
| Fig. 7 Comparisons between the coverage-dependent modelling results and the coverage-independent modelling ones: (a) TOF diagrams for the formation of EO at pO2 = pEt = 13.4 kPa. (b) Free energy profiles of ethylene epoxidation on Ag(111) at 490 K, pO2 = pEt = 13.4 kPa. The black curve represents the energy profile of the coverage-independent model, and the red curve represents the energy profile of the coverage-dependent model at the steady-state. The coverage-dependent data were obtained as follows: for each surface species, including O, OME *, TSOO, TS1, TSEO, and TSAA, under each coverage, all the configurations we could think of were all tried. Then the structure with the lowest energy was selected as the corresponding structure to determine the energy for our kinetic simulations. For clarity, the profiles in (b) contain only the L–H mechanism (step 4) without the E–R mechanism (step 5). Both L–H and E–R mechanisms were considered in the microkinetic modelling, while the profiles of the E–R mechanism pathway are shown in Fig. S13.† (c) The different ΔE (TSEO–TSAA) at temperatures from 490 K to 600 K, pO2 = pEt = 13.4 kPa. | ||
3.4 Comparison between the microkinetic modelling results and experimental data
Although some experimental data have been mentioned in the previous sections, here we specifically discuss the comparison between experimental values, which were obtained from the single-crystal experiment of Campbell and co-workers, and the coverage-dependent microkinetic results as well as the coverage-independent microkinetic ones.30 To determine the effects of different reaction conditions of ethylene epoxidation, Campbell and co-workers carefully compared experimentally the various kinetic properties on Ag(111) and Ag(110), and some valuable experimental data were obtained. As a result, it is feasible to utilize the experimental data of Campbell as the benchmark for our simulations. In addition, Huš and Hellman conducted a series of KMC simulations in which they obtained several sets of kinetic properties of ethylene epoxidation on silver catalysts.33 Therefore, the quantitative microkinetic modelling we developed can be compared to the KMC simulations to evaluate the simulation accuracy.Firstly, the temperature-related TOF results calculated by the coverage-dependent microkinetic modelling are in good agreement with the experimental values, as shown in Fig. 8(a). We built the microkinetic model based on the elementary steps and rate equations in Table 1. The steady state was solved with the reaction energetics and reaction conditions when the surface coverages of intermediates are invariant with time, i.e., dθ/dt = 0. Accordingly, the steady-state reaction rate of each elementary step can be achieved, and here the rate of EO formation is regarded as the TOF and illustrated in Fig. 8(a). These results also corroborate the results of Huš and Hellman, who suggested that the log(TOF) values are located in the range of 0 to 4. In addition to the similarities between the two simulation methods, the TOF values of our coverage-dependent microkinetic modelling are higher than those of KMC values. Interestingly, our coverage-dependent values are closer to the experimental results, whereas the KMC values are consistently lower than the experimental values. In contrast to earlier experimental and theoretical findings, however, the coverage-independent microkinetic modeling results are wildly distorted from the previous results. Taken together, these results suggest that the coverage effect must be taken into account in the microkinetic modelling to obtain accurate kinetic results. Regarding the EO selectivity, the experimentally measured EO selectivity does not change significantly due to temperature and pressure changes but remains around 40%. In general, all the EO selectivity from the coverage-dependent microkinetic models in this work are consistent with the experimental values and trends, as shown in Fig. 8(b)–(d).
 | ||
| Fig. 8 Comparison between the experimental data from Campbell, simulated values obtained from the coverage-dependent/coverage-independent microkinetic modellings, and simulated results from Huš and Hellman.30,33 (a and b) log(TOF) and EO selectivity as a function of temperature at pO2 = 20 kPa and pEt = 2.66 kPa. (c) EO selectivity as a function of oxygen pressure at pEt = 0.55 kPa and T = 490 K. (d) EO selectivity as a function of ethylene pressure at pO2 = 20 kPa and T = 490 K. | ||
Finally, it is worth addressing a general consensus in the field: When a catalyst is inert for adsorption or the reaction temperature is relatively high, the coverage effect could be omitted, and kinetic simulations could be carried out with the energetics calculated from a low coverage. The rationale for this consensus is simple: if the catalyst is relatively inert for adsorption or the reaction is carried out at high temperatures, the coverages of surface species are low under the reaction conditions; then the adsorbate–adsorbate interactions are weak and the energetics from the low coverage would be a good approximation to describe the real reactions. However, our work shows that this is not the case: the coverage-independent microkinetic modelling, in which the energetics were obtained at low coverage (0.11 ML) on the Ag(111) surface, gives rise to inaccurate results even at high temperatures, as discussed above. How can we understand these results? Although the coverages at the steady states from both the coverage-independent microkinetic modelling and the coverage-dependent microkinetic modelling are indeed low, there are two problems in the coverage-independent microkinetic modelling. Firstly, the coverage at the steady state from the coverage-independent microkinetic modelling is 1.0 × 10−4 ML (the output coverage from the kinetic simulation), which is lower than the input coverage (0.11 ML), meaning that the steady state coverage of the coverage-independent microkinetic modelling is inconsistent with the input coverage (0.11 ML). Namely, it is not self-consistent kinetically. Secondly, the energetics of such adsorption energies and reaction barriers were obtained at 0.11 ML, which should be different from those at the steady state, resulting in the errors in the kinetics. Therefore, it is clear that the coverage effect cannot be ignored even under low coverage conditions.
3.5 The impact of industrial conditions on the surface coverage, catalyst activity, and selectivity
In this section, the effects of industrial conditions on ethylene epoxidation will be discussed. The temperatures from 490 K to 600 K were selected in this work, corresponding to the industrial temperature range.30,49 To be consistent with the industrial conditions, the ratios of oxygen and ethylene partial pressures pO2/pEt were chosen from 3:1 to 1:3, corresponding to the experimental pressure range of 13.4 kPa (O2):4.47 kPa (Et) to 13.4 kPa (O2):40.2 kPa (Et). The kinetic simulations of this wide range of industrial conditions should be sufficient enough to provide a comprehensive understanding of the ethylene epoxidation on the Ag(111) surface.
The coverage-dependent microkinetic model was utilized to rigorously calculate the distribution of adsorbed oxygen on the surface at different temperatures and different partial pressures, as shown in Fig. 9(a). It can be seen that as the temperature increases under constant pressure, the coverage of oxygen atoms on the surface maintains almost constant without dramatic changes. For example, the coverage changes by only 0.3% when pO2/pEt = 1:2 and the temperature is increased from 490 K to 600 K. This result may appear to be puzzling but can be understood as follows: as the temperature increases, the rate of O2 dissociation increases, leading to an increase of surface coverage, whereas the rates of the elementary steps after the O2 dissociation also increase (Fig. 9(b)) so that the coverage of adsorbed oxygen atoms reaches almost a dynamic equilibrium. The variation of the coverage with the partial pressure is also illustrated (Fig. 9(a)). Compared with the influence of temperature on the coverage, the partial pressure changes have a more significant influence on the coverage under constant temperature. As the partial pressure of ethylene increases, the coverage of adsorbed oxygen atoms on the surface decreases relatively rapidly, being consistent with experimental results.85 For example, the coverage changes by 1.7% when T = 600 K and the pO2/pEt changes from 3:1 to 1:3. Like the temperature-related effects, more adsorbed oxygen atoms participate in generating EO and AA as the reaction rate (TOF) increases, resulting in a decrease in oxygen coverage. Consequently, the temperature changes have a relatively weak effect, while the variations of partial pressures possess a considerable impact on the oxygen coverage.
 | ||
| Fig. 9 Diagrams of (a) the surface oxygen coverage distributions, (b) the TOF values, and (c) the EO selectivity variations between 490 K and 600 K with pO2/pEt ranging from 3:1 to 1:3. | ||
As can be seen from Fig. 9(b), the TOF value steadily increases with the increase in temperature. Likewise, the effect of oxygen and ethylene partial pressure changes on the ethylene epoxidation activity is distinct: The TOF value improves swiftly when the pO2/pEt changes from 3:1 to 1:3. The TOF change trends with temperature changes are similar to those of pressure changes. The general trend of the ethylene epoxidation activity increases with the increase in temperature and pressure.
As mentioned before, the main by-product of the ethylene epoxidation reaction is acetaldehyde (AA). However, from the perspective of product ratio, ethylene epoxide seems to be the “by-product” since the selectivity of ethylene epoxide on the unpromoted Ag(111) surface is lower than 50%. Consequently, improving the selectivity of ethylene epoxide is a more critical issue in the ethylene epoxidation industry. In this work, the EO selectivity is defined as eqn (4), and the microkinetic modellings were carried out to scrutinize the selectivity.
 | (4) |
Firstly, some significant differences are observed between the selectivities obtained by the coverage-dependent microkinetic modelling and the coverage-independent modelling. For example, when the industrial condition was set to 540 K and pO2:pEt = 13.4 kPa:40.2 kPa, the selectivity obtained by the coverage-dependent microkinetic modelling was 49%, but the coverage-independent result was only 28%. This discrepancy could be attributed to TSEO being more affected by the coverage effect than TSAA, and hence the selectivity calculated by the coverage-independent model was relatively low. The results of the coverage-dependent microkinetic modelling show that the EO selectivity increases from 35% to 54% as the temperature increases from 490 K to 600 K, as illustrated in Fig. 9(c). The temperature induced EO selectivity changes are consistent with the previous discussion in section 3.2. Compared with the effect of temperature changes, the selectivity is not sensitive to pressure changes. Therefore, increasing the reaction temperature is crucial for improving the EO selectivity.
In addition, the degree of rate control (DRC) analysis was also performed to find out which step is the rate-determining step. Through DRC analysis, it is found that oxygen dissociation is the rate-determining step of the reaction. This means that the surface oxygen coverage effect has a crucial impact on the reaction kinetics. Meanwhile, we explain the reason for the EO selectivity change caused by the temperature change from the perspective of kinetics, which is included in the ESI† S11.
4. Conclusions
In this work, the activity and selectivity of ethylene epoxidation on the Ag(111) surface with coverage effects were quantitatively studied. The coverage-dependent microkinetic modelling with consideration of all the major adsorbate–adsorbate interactions including transition states was applied to the reaction kinetic analysis. By the incorporation of the coverage effect, the conflict between the simulation results of the traditional first-principles method and the experimental values was resolved. Our results show that the correct microkinetic modelling could be utilized for quantitative comparison with experimental values. Through the systematic exploration of ethylene epoxidation, some compelling conclusions are obtained as follows:(i) The traditional coverage-independent microkinetic modelling based on DFT calculation was not adequate to describe the activity of ethylene epoxidation. The kinetic data and coverage obtained by the coverage-independent microkinetic modelling illustrated an extinct discrepancy with the experimental results even though the coverages are low on the Ag catalyst.
(ii) The coverage-dependent microkinetic modelling using the energetics from rigorous DFT calculations was established and investigated systematically. The coverage-dependent microkinetic modelling was utilized to compare the experimental data comprehensively. A good agreement between the simulation results from the coverage-dependent microkinetic modelling and the experimental data was achieved.
(iii) The coverage-dependent microkinetic modelling was used to rigorously analyze ethylene epoxidation under industrial conditions. The impacts of industrial temperature range and different partial pressures of oxygen and ethylene on surface coverage, reaction rate, and EO selectivity were systematically examined. The following was found. Firstly, the surface coverage maintains almost a dynamic equilibrium as the temperature increase. Secondly, as the temperature and total pressure increase, the reaction activity moderately increases. Thirdly, the EO selectivity is affected by temperature, but not significantly by pressure.
Although the surface coverage is low under industrial conditions (7%), this does not indicate that the coverage effect is not essential. Remarkable differences were revealed through the quantitative comparison of coverage-dependent modelling and coverage-independent modelling throughout the work. Therefore, including the coverage effect will be pivotal for quantitatively or semi-quantitatively studying ethylene epoxidation activity and EO selectivity.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the NSFC (92045303) and NKRDPC (2021YFA1500700) and the UK Materials and Molecular Modeling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1). We are grateful for computational support from the UK national high-performance computing service ARCHER funded by EPSRC grant ref. EP/P022561/1 and QUB Kelvin HPC service funded by ESPRC (EP/T022175/1).References
- J. W. Medlin and M. A. Barteau, J. Phys. Chem. B, 2001, 105, 10054–10061 CrossRef CAS
.
- J. Greeley and M. Mavrikakis, J. Phys. Chem. C, 2007, 111, 7992–7999 CrossRef CAS
- M. O. Özbek, I. Önal and R. A. vanSanten, ChemCatChem, 2013, 5, 443–451 CrossRef
- T. Pu, H. Tian, M. E. Ford, S. Rangarajan and I. E. Wachs, ACS Catal., 2019, 9, 10727–10750 CrossRef CAS
- W. Diao, C. D. Digiulio, M. T. Schaal, S. Ma and J. R. Monnier, J. Catal., 2015, 322, 14–23 CrossRef CAS
- Y. Xu, J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2005, 127, 12823–12827 CrossRef CAS PubMed
- T. Pu, A. Setiawan, B. Mosevitzky Lis, M. Zhu, M. E. Ford, S. Rangarajan and I. E. Wachs, ACS Catal., 2022, 12, 4375–4381 CrossRef CAS
- D. Torres, F. Illas and R. M. Lambert, J. Catal., 2008, 260, 380–383 CrossRef CAS
- Y. Le, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winansl, W. Elám, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlö'Gl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef PubMed
- D. Ren, H. Xu, J. Li, J. Li and D. Cheng, Mol. Catal., 2017, 441, 92–99 CrossRef CAS
- R. A. Van Santen and H. P. C. E. Kuipers, Adv. Catal., 1987, 35, 265–321 CAS
- P. Christopher and S. Linic, J. Am. Chem. Soc., 2008, 130, 11264–11265 CrossRef CAS PubMed
- T. E. Jones, R. Wyrwich, S. Böcklein, E. A. Carbonio, M. T. Greiner, A. Y. Klyushin, W. Moritz, A. Locatelli, T. O. Menteş, M. A. Niño, A. Knop-Gericke, R. Schlögl, S. Günther, J. Wintterlin and S. Piccinin, ACS Catal., 2018, 8, 3844–3852 CrossRef CAS
- J. T. Jankowiak and M. A. Barteau, J. Catal., 2005, 236, 379–386 CrossRef CAS
- C. T. Campbell and M. T. Paffett, Appl. Surf. Sci., 1984, 19, 28–42 CrossRef CAS
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2002, 124, 310–317 CrossRef CAS PubMed
- T. C. R. Rocha, M. Hävecker, A. Knop-Gericke and R. Schlögl, J. Catal., 2014, 312, 12–16 CrossRef CAS
- D. Chen, P. L. Kang and Z. P. Liu, ACS Catal., 2021, 11, 8317–8326 CrossRef CAS
- A. Michaelides, K. Reuter and M. Scheffler, J. Vac. Sci. Technol., A, 2005, 23, 1487–1497 CrossRef CAS
- M. L. Bocquet, A. Michaelides, D. Loffreda, P. Sautet, A. Alavi and D. A. King, J. Am. Chem. Soc., 2003, 125, 5620–5621 CrossRef CAS PubMed
- A. Michaelides, M. L. Bocquet, P. Sautet, A. Alavi and D. A. King, Chem. Phys. Lett., 2003, 367, 344–350 CrossRef CAS
- S. Linic, H. Piao, K. Adib and M. A. Barteau, Angew. Chem., Int. Ed., 2004, 43, 2918–2921 CrossRef CAS PubMed
- J. Schnadt, A. Michaelides, J. Knudsen, R. T. Vang, K. Reuter, E. Lægsgaard, M. Scheffler and F. Besenbacher, Phys. Rev. Lett., 2006, 96, 4–7 CrossRef PubMed
- L. Zhu, W. Zhang, J. Zhu and D. Cheng, Appl. Catal., A, 2017, 538, 27–36 CrossRef CAS
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2003, 125, 4034–4035 CrossRef CAS PubMed
- G. S. Jones, M. Mavrikakis, M. A. Barteau and J. M. Vohs, J. Am. Chem. Soc., 1998, 120, 3196–3204 CrossRef CAS
- S. Linic and M. A. Barteau, J. Catal., 2003, 214, 200–212 CrossRef CAS
- M. Mavrikakis, D. J. Doren and M. A. Barteau, J. Phys. Chem. B, 1998, 102, 394–399 CrossRef CAS
- H. Li, A. Cao and J. K. Nørskov, ACS Catal., 2021, 11, 12052–12057 CrossRef CAS
- C. T. Campbell, J. Catal., 1985, 94, 436–444 CrossRef CAS
- M. O. Ozbek, I. Onal and R. A. Van Santen, J. Catal., 2011, 284, 230–235 CrossRef CAS
- M. O. Ozbek, I. Onal and R. A. Van Santen, Top. Catal., 2012, 55, 710–717 CrossRef CAS
- M. Huš and A. Hellman, ACS Catal., 2019, 9, 1183–1196 CrossRef
- Z. Yao, C. Guo, Y. Mao and P. Hu, ACS Catal., 2019, 9, 5957–5973 CrossRef CAS
- Z. Yao, J. Zhao, R. J. Bunting, C. Zhao, P. Hu and J. Wang, ACS Catal., 2021, 11, 1202–1221 CrossRef CAS
- C. Guo, Y. Mao, Z. Yao, J. Chen and P. Hu, J. Catal., 2019, 379, 52–59 CrossRef CAS
- Y. Ding, Y. Xu, Y. Song, C. Guo and P. Hu, J. Phys. Chem. C, 2019, 123, 27594–27602 CrossRef CAS
- W. Xie, J. Xu, Y. Ding and P. Hu, ACS Catal., 2021, 11, 4094–4106 CrossRef CAS
- J. Zhao, S. Zha, R. Mu, Z. J. Zhao and J. Gong, J. Phys. Chem. C, 2018, 122, 6005–6013 CrossRef CAS
- K. Frey, D. J. Schmidt, C. Wolverton and W. F. Schneider, Catal. Sci. Technol., 2014, 4, 4356–4365 RSC
- E. Nowicka, T. J. Clarke, M. Sankar, R. L. Jenkins, D. W. Knight, S. Golunski, G. J. Hutchings, D. J. Willock, M. Francisco and S. H. Taylor, Chem. – Eur. J., 2018, 24, 655–662 CrossRef CAS PubMed
- Y. K. Chow, N. F. Dummer, J. H. Carter, R. J. Meyer, R. D. Armstrong, C. Williams, G. Shaw, S. Yacob, M. M. Bhasin, D. J. Willock, S. H. Taylor and G. J. Hutchings, ChemPhysChem, 2018, 19, 402–411 CrossRef CAS PubMed
- J. Cheng and P. Hu, Angew. Chem., Int. Ed., 2011, 50, 7650–7654 CrossRef CAS PubMed
- Y. Giret, P. Guo, L. F. Wang and J. Cheng, J. Chem. Phys., 2020, 152, 1–9 CrossRef PubMed
- P. Chen, Y. Liu, Y. Xu, C. Guo and P. Hu, JACS Au, 2023, 3(1), 165–175 CrossRef CAS PubMed
- W. Xie, J. Xu, J. Chen, H. Wang and P. Hu, Acc. Chem. Res., 2022, 55, 1237–1248 CrossRef CAS PubMed
- M. Huš, M. Grilc, A. Pavlišič, B. Likozar and A. Hellman, Catal. Today, 2019, 338, 128–140 CrossRef
- C. Stegelmann, N. C. Schiødt, C. T. Campbell and P. Stoltze, J. Catal., 2004, 221, 630–649 CrossRef CAS
- J. T. Jankowiak and M. A. Barteau, J. Catal., 2005, 236, 366–378 CrossRef CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- Z. P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS PubMed
- A. Alavi, P. Hu, T. Deutsch, P. L. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650–3653 CrossRef CAS
- A. Michaelides, Z. P. Liu, C. J. Zhang, A. Alavi, D. A. King and P. Hu, J. Am. Chem. Soc., 2003, 125, 3704–3705 CrossRef CAS PubMed
- H. F. Wang and Z. P. Liu, J. Am. Chem. Soc., 2008, 130, 10996–11004 CrossRef CAS PubMed
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 1–19 CrossRef PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- C. Lee, W. Yang and G. R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- L. Petrov, A. Eliyas and D. Shopov, Appl. Catal., 1985, 18, 87–103 CrossRef CAS
- J. R. H. Carucci, V. Halonen, K. Eränen, J. Wärnå, S. Ojala, M. Huuhtanen, R. Keiski and T. Salmi, Ind. Eng. Chem. Res., 2010, 49, 10897–10907 CrossRef CAS
- T. Salmi, J. Hernández Carucci, M. Roche, K. Eränen, J. Wärnå and D. Murzin, Chem. Eng. Sci., 2013, 87, 306–314 CrossRef CAS
- P. C. Borman and K. R. Westerterp, Ind. Eng. Chem. Res., 1995, 34(1), 49–58 CrossRef CAS
- W. Xie and P. Hu, Catal. Sci. Technol., 2021, 11, 5212–5222 RSC
- L. C. Grabow, B. Hvolbæk and J. K. Nørskov, Top. Catal., 2010, 53, 298–310 CrossRef CAS
- N. Yang, A. J. Medford, X. Liu, F. Studt, T. Bligaard, S. F. Bent and J. K. Nørskov, J. Am. Chem. Soc., 2016, 138, 3705–3714 CrossRef CAS PubMed
- J. Chen, M. Jia, P. Hu and H. Wang, J. Comput. Chem., 2021, 42, 379–391 CrossRef CAS PubMed
- J. Chen, M. Jia, Z. Lai, P. Hu and H. Wang, J. Chem. Phys., 2021, 154, 1–8 Search PubMed
- J. F. Chen, Y. Mao, H. F. Wang and P. Hu, ACS Catal., 2016, 6, 7078–7087 CrossRef CAS
- X. Sun, P. Wang, Z. Shao, X. Cao and P. Hu, Sci. China: Chem., 2019, 62, 1686–1697 CrossRef CAS
- W. Li, C. Stampfl and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 1–19 Search PubMed
- P. P. Chen, B. Y. Zhang, X. K. Gu and W. X. Lic, Chin. J. Chem. Phys., 2019, 32, 437–443 CrossRef CAS
- D. Torres, N. Lopez, F. Illas and R. M. Lambert, J. Am. Chem. Soc., 2005, 127, 10774–10775 CrossRef CAS PubMed
- H. Li, A. Cao and J. K. Nørskov, ACS Catal., 2021, 11, 12052–12057 CrossRef CAS
- H. Guo, P. Poths, P. Sautet and A. N. Alexandrova, ACS Catal., 2022, 12, 818–827 CrossRef CAS
- G. Sun and P. Sautet, Acc. Chem. Res., 2021, 54, 3841–3849 CrossRef CAS PubMed
- J. Feibelman, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 12133–12138 CrossRef PubMed
- Z. P. Liu, P. Hu and M. H. Lee, J. Chem. Phys., 2003, 119, 6282–6289 CrossRef CAS
- Z. P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS PubMed
- K. Bleakley and P. Hu, J. Am. Chem. Soc., 1999, 121, 7644–7652 CrossRef CAS
- S. Linic, J. Jankowiak and M. A. Barteau, J. Catal., 2004, 224, 489–493 CrossRef CAS
- R. B. Grant and R. M. Lambert, J. Catal., 1985, 92, 364–375 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01477g |
| This journal is © The Royal Society of Chemistry 2023 |