
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe protometabolic nature of prebiotic chemistry
Noemí
Nogal
 a,
Marcos
Sanz-Sánchez
a,
Sonia
Vela-Gallego
a,
Kepa
Ruiz-Mirazo
bc and
Andrés
de la Escosura
*ad
a,
Marcos
Sanz-Sánchez
a,
Sonia
Vela-Gallego
a,
Kepa
Ruiz-Mirazo
bc and
Andrés
de la Escosura
*ad
aDepartment of Organic Chemistry, Universidad Autónoma de Madrid, Campus Cantoblanco, 28049, Madrid, Spain. E-mail: andres.delaescosura@uam.es
bBiofisika Institute (CSIC, UPV/EHU), University of the Basque Country, Leioa, Spain
cDepartment of Philosophy, University of the Basque Country, Leioa, Spain
dInstitute for Advanced Research in Chemistry (IAdChem), Campus de Cantoblanco, 28049, Madrid, Spain
First published on 19th October 2023
Abstract
The field of prebiotic chemistry has been dedicated over decades to finding abiotic routes towards the molecular components of life. There is nowadays a handful of prebiotically plausible scenarios that enable the laboratory synthesis of most amino acids, fatty acids, simple sugars, nucleotides and core metabolites of extant living organisms. The major bottleneck then seems to be the self-organization of those building blocks into systems that can self-sustain. The purpose of this tutorial review is having a close look, guided by experimental research, into the main synthetic pathways of prebiotic chemistry, suggesting how they could be wired through common intermediates and catalytic cycles, as well as how recursively changing conditions could help them engage in self-organized and dissipative networks/assemblies (i.e., systems that consume chemical or physical energy from their environment to maintain their internal organization in a dynamic steady state out of equilibrium). In the article we also pay attention to the implications of this view for the emergence of homochirality. The revealed connectivity between those prebiotic routes should constitute the basis for a robust research program towards the bottom-up implementation of protometabolic systems, taken as a central part of the origins-of-life problem. In addition, this approach should foster further exploration of control mechanisms to tame the combinatorial explosion that typically occurs in mixtures of various reactive precursors, thus regulating the functional integration of their respective chemistries into self-sustaining protocellular assemblies.
 Noemí Nogal | Noemí Nogal graduated in Chemistry from Universidad Autónoma de Madrid (UAM) in 2017. In 2019, she started her PhD studies under the supervision of Dr Andrés de la Escosura. In 2023, she carried out predoctoral studies as a visiting student under the supervision of Dr Mathieu Surin at Université de Mons, Belgium, working on the preparation and characterization of new supramolecular hybrid systems formed between oligo-DNA templates and phenylazopyrazole molecular photoswitches. Her research interests deal with the prebiotic synthesis of biomolecules that could have given rise to the origin of life in the context of systems chemistry. |
 Marcos Sanz-Sánchez | Marcos Sanz Sánchez is a Graduate Teaching Assistant at the Organic Chemistry Department of Universidad Autónoma de Madrid (UAM). He received his BSc degree in Chemistry from UAM in 2021. Then, he joined the group of Prof. Andrés de la Escosura to carry out his MSc studies in 2022, focusing on the design of chemical networks based on biological building blocks. He is currently working as a PhD student in the same group, with the aim to integrate these systems into replication networks. |
 Sonia Vela-Gallego | Sonia Vela-Gallego graduated in chemistry at Universidad de Castilla-La Mancha, where she worked in the green chemistry group developing the synthesis of new ionic liquids. After this period, she moved to Madrid to carry out predoctoral studies in the group of organic molecular materials at Universidad Complutense de Madrid. During her thesis, she worked under the supervision of Prof. Nazario Martín and Prof. Carmen Atienza, obtaining her PhD degree in 2017. Since 2018, she is a postdoctoral researcher at Universidad Autónoma de Madrid, working in the systems chemistry group led by Prof. Andrés de la Escosura, where she is developing a broad knowledge in thermodynamics and chemical kinetics applied to reaction networks, and their study out of equilibrium. |
 Kepa Ruiz-Mirazo | Kepa Ruiz-Mirazo is a physicist who has devoted most of his research career to study the epistemological foundations of biology, focusing on the problem of origins of life and its non-trivial chemical roots. After completing a PhD in Sciences (Complex Systems) at the UPV/EHU in 2001, he worked as a postdoc at the Centre for Astrobiology (Spain), Institute for Polymers at ETH-Zürich (Switzerland) and D. Biology of the University Roma Tre (Italy). His main scientific contributions have been in the elaboration of protocell models, both in vitro and in silico. Former RyC Fellow, KR-M is permanent research staff of the University of the Basque Country (UPV/EHU) since 2012, PI of the Protocell Biophysics Lab (https://www.biofisika.org/en/research/theoretical-protocell-biophysics-laboratory) at Biofisika Institute (CISC, UPV/EHU), senior member of the IAS-Research Group (https://www.ias-research.net/), and has been recently involved in a European ITN on the origins of metabolism (https://protomet-etn.eu/). |
 Andrés de la Escosura | Andrés de la Escosura (PhD 2005) carried out postdoctoral studies at the Radboud University, in Nijmegen (The Netherlands). He joined Universidad Autónoma de Madrid (UAM) in 2009, first as Assistant Professor and, since February 2012, as a Ramón y Cajal Fellow. Currently, he is Associate Professor at the Organic Chemistry Department of UAM and belongs to the Institute for Advanced Research in Chemistry (IAdChem) at Madrid. He leads the Research Group of Biohybrid Materials and Systems Chemistry (https://syschemdelaescosura.es/). After finding useful properties of protein capsids as nanoreactors and nanocontainers to incorporate different kinds of materials, the group is currently working in areas that include biomedical applications, catalysis, prebiotic systems chemistry and the origins of life. He is currently the coordinator of the European FET-OPEN Network CLASSY, focused on the construction of cell-like molecular assembly lines as game changers in chemical synthesis, and the EIC Pathfinder Network CORENET, dealing with complex reaction networks for different purposes. |
Key learning points(1) The construction of a complete ‘protometabolic’ map will facilitate the task to identify and classify the different control mechanisms that could emerge from subsets of interconnected prebiotic reaction pathways and transformation processes, leading to ‘minimal metabolic systems’.(2) Putting together a protometabolic network from these highly interconnected prebiotic chemistries would require that some reactions are run in both the forward and reverse directions, channelling the exploration of the available chemical space in ways that reinforce complex, non-equilibrium states/mixtures. (3) Non-enzymatic reaction networks could have provided, from a set of central protometabolic cycles, the adequate conditions for the emergence of oligomer/polymer catalysts and replicators. (4) Energetic funnelling, enabled by the coupling of multiple catalytic cycles under dissipative conditions, would represent a transition towards systems and networks with increasing robustness (partly expressed as ‘dynamic kinetic stability’). (5) The establishment of auto- and cross-catalytic loops, together with the self-assembly of non-equilibrium supramolecular structures from some network components, would open evolutionary possibilities towards protocellular assemblies with a higher stability and adaptability. These phenomena could also have striking consequences for the amplification of small enantiomeric excesses of building blocks in the resulting protocells. |
1. Introduction
Unravelling how life could have emerged from a set of interconnected out-of-equilibrium chemistries, on the primitive Earth or somewhere else, will require understanding the constraining mechanisms that enable prebiotic reaction pathways to get organized into self-maintaining systems.1–6 These mechanisms imply different types of self-assembly (e.g., phase separation, spatial heterogeneity, compartmentalization), autocatalysis (template replication, cyclic or network autocatalysis, etc.), the use of chemical fuels (i.e., energy-rich compounds whose exergonic degradation can be coupled to run thermodynamically unfavourable transformations), together with the kinetic control exerted by diverse catalysts.7–9 Normally, such fundamental control functions are executed by complex biomolecules, biopolymers in particular (e.g., proteins and nucleic acids), but it is reasonable to make the hypothesis that precursor species like oligonucleotides and peptides, with the assistance of organic (lipid) interfaces, mineral surfaces and/or cofactors, could have carried them out, as well.10–13 The field of prebiotic chemistry has been dedicated, over decades, to finding synthetic routes to generate those (precursor) biomolecules, including an explanation of their remarkable homochirality. There are nowadays multiple known possibilities to synthesize in the lab most amino acids,14,15 fatty acids,16 simple sugars,17 nucleobases,18,19 nucleosides, nucleotides,20 and core metabolites of extant life,21 under aqueous conditions that are compatible with different early Earth geochemical scenarios.22 Their oligomerization and self-assembly processes have also been extensively studied.23 It could be argued that, although some important synthetic challenges remain (in particular, concerning the efficiency, selectivity and compatibility of the conditions under which the aforementioned processes take place), there is almost certainty about the availability of the main biological molecules at the onset of life, as well as a good number of possible mechanisms by which they could have been enantiomerically enriched.The major bottleneck then seems to be the organization of those building blocks into systems (i.e., non-reducible combinations of molecules and transformation processes) that sustain themselves in non-equilibrium conditions. This requires the establishment of recurrent control and feedback loops (autocatalytic cycles, kinetic and energetic couplings between common intermediates, compartmentalization through membranes or liquid–liquid phase separation to form condensates/aggregates like microdroplets) by which higher-level functional networks and assemblies harness the synthetic pathways that produce their own components.24 As it has been pointed out by Ruiz-Mirazo and co-workers,25 there is in that kind of organization a double-way causal connection, upwards and downwards, between the endogenously created control mechanisms and the pathways to synthesize the system's functional molecules. That basic hierarchical relationship defines the minimal condition for a network of reactions to be considered a metabolism (in its most elementary sense), suggesting that the application of an organizational/metabolic perspective is crucial in the search for prebiotic ‘systems principles’.
Different conceptual frameworks have been proposed to this end,26–31 but often with little support from experiments, remaining at a rather abstract level. Nevertheless, in the last two decades, various experimental lines have been initiated towards the demonstration that central metabolic pathways (e.g., glycolysis, the pentose phosphate cycle, the citric acid cycle, and the glyoxylate scenario) can be run in the absence of enzymes.32 All these recent approaches are setting a solid base for research on the key transition between protometabolic reaction networks and ‘minimal metabolisms’ (relatively more intricate, but still infra-biological self-sustaining systems). Still, the scope of such experimental approaches is limited, to a significant extent, by the complexity of the biochemical pathways they aim to mimic. In addition, this complexity makes the analysis related to the homochirality of the involved molecular species more difficult. Although biological inspiration is obviously necessary to guide research on the origins of life,33,34 extant life features/signatures could also be misleading in certain aspects, as chemical pathways to the first protobiological systems may not need to be identical to those of current biochemistry.
In this regard, it would be useful to build a map, as complete as possible, of the available prebiotic routes towards the main biological monomers, with the aim to identify common intermediates and links of other kinds between them, including the possibility that intermediates of one route act as catalysts or fuels in others, and species from different routes can be grouped together in autocatalytic subsets, to be able to project to subsequent transitions in the process. Finding that type of interconnected feedbacks requires thinking out of the ‘biochemical box’, taking up a bottom-up strategy that relies on the rules of chemistry and the space of molecular structures that they allow to explore, and not only in open aqueous conditions but also in more heterogeneous and dynamic environments – in which the presence of interfaces (soft interfaces, in particular) may enhance and make compatible some of the processes involved.35 In the context of these complex systems, the question about whether homochirality was a requirement for or a result of establishing protometabolic networks becomes really pertinent.36
Theoretical and computational investigations into this problem will be capital, and various cheminformatic tools are starting to be used for the purpose, including simulation platforms that deal with chemistries taking place in diverse reaction domains simultaneously37,38 or the particularly interesting and promising rule-based algorithmic methods.39–41 Yet the purpose of this review is to have a closer look, guided mostly by experimental research, into the earliest stages of the process of origins of biological complexity, stemming from the combination of chemical and physical transformations and constraints. This means, in practice, dealing with compounds of ca. 5–6 carbon atoms maximum, which are at the core of the main prebiotic chemistries (i.e., the formose reaction, HCN oligomerization, formamide chemistry, the cyanosulfidic and the glyoxylate scenarios) that already show multiple possible links and couplings (see Scheme 1). Realizing the high degree of connectivity between these main prebiotic synthetic routes could set the basis for further exploration of control mechanisms that tame the otherwise expected combinatorial explosion of compounds and reactions, helping to regulate their grouping into self-sustaining networks and assemblies.
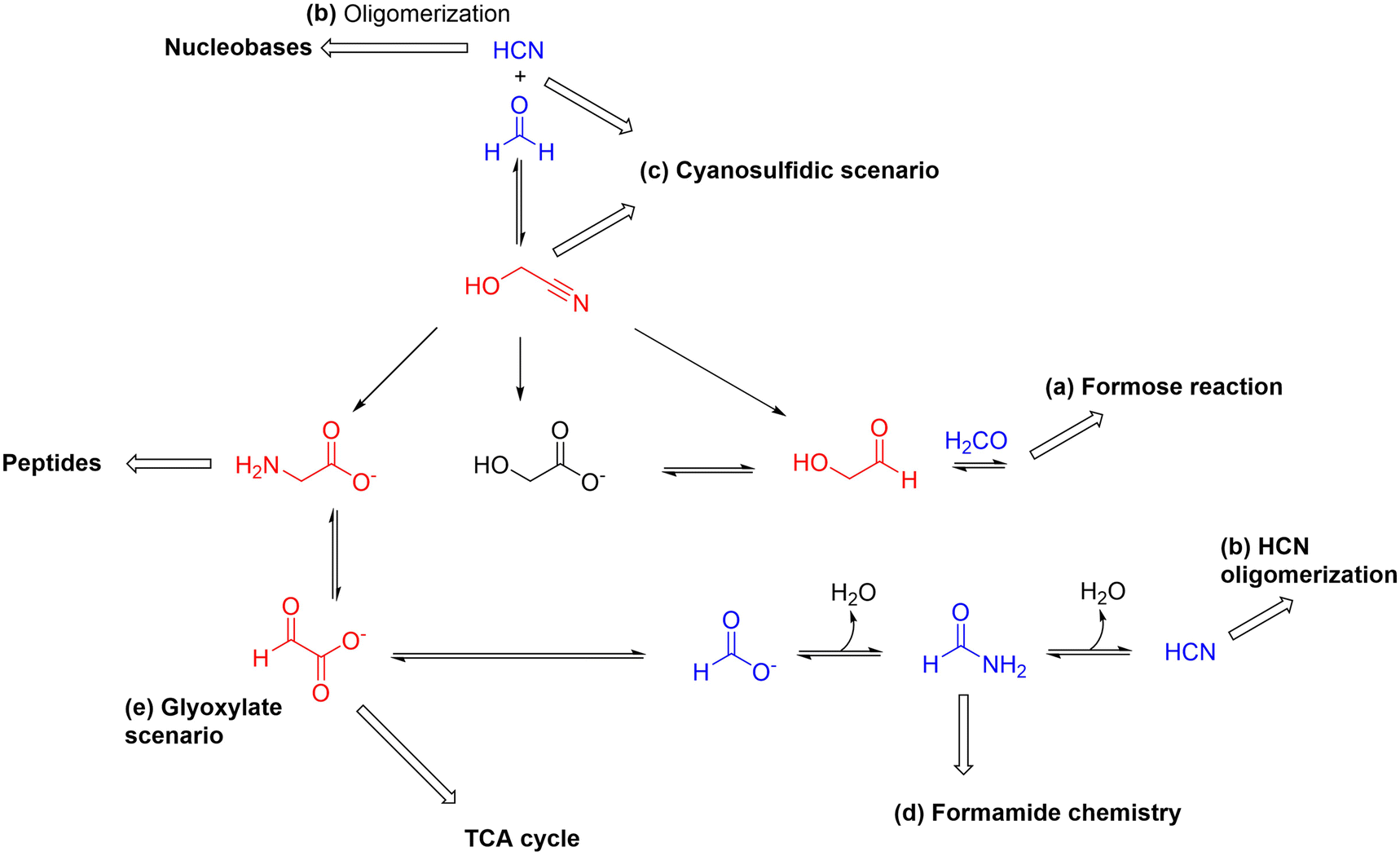 | ||
| Scheme 1 Scheme showing the deep interconnection that could exist between the main prebiotic synthetic routes, most likely through non-enzymatic reactions converting one- and two-carbon atom species with different reactive functional groups into each other. This type of reactions could also be established at higher levels of structural complexity (i.e. with three- to six-carbon atom molecules). Importantly, they may proceed in a reversible manner depending on the conditions and if additional recurrent processes operate in the system (e.g., wetting-drying or freeze–thaw cycles, oscillations in concentration for certain compounds, periodic phase transitions, etc.). Under such circumstances, complex networks could be established where there is a flow of matter towards increasing complexity, provided that there is a supply of energy and substrate materials. In red, we have marked compounds with special relevance as reactive intermediates in possibly ancient protometabolic pathways/cycles, and in blue key one-carbon feedstock molecules of the main prebiotic routes. Note: Some of the processes depicted have been found to occur under prebiotic conditions. Others occur in biology and may have a prebiotic counterpart. In some cases, the selected intermediates could be produced through other pathways. So, this scheme must just be considered as a suggested hypothesis of possible interconnections that require further research in the field. | ||
Thus, the structure of the review is divided in sections that first describe previous work on the main pillars of prebiotic chemistry, and then delineate strategies that combine them into more complex networks and self-sustaining systems. This effort leads to the construction of a ‘prebiotic metabolic map’, which facilitates the task of identifying and classifying the different physicochemical processes and control mechanisms that could emerge from such a set of interlinked reaction pathways. While we proceed, a number of experimental challenges for the field will be highlighted, in particular those related to the emergence of homochirality and non-equilibrium chemical systems in heterogeneous conditions.
2. Main prebiotic synthetic pillars
Most prebiotic chemistries originate from simple one-carbon atom feedstock precursors, including formaldehyde, hydrogen cyanide (HCN) and formamide.22 Apart from these highly reactive carbon sources, ammonia and hydrogen sulfide could be considered as the ultimate sources of nitrogen and sulfur, respectively, while water mediates in most prebiotic reactions. With these six reactive species in hand, and some inorganic phosphate source,42,43 it should be possible to build most of the structural complexity comprising the main biological molecular components. In particular, the formose reaction (a in Scheme 1),17 which uses formaldehyde as starting material, allows generating dozens of sugars, in what has been called an ‘oxygenous’ chemistry. The ‘nitrogenous’ oligomerization of HCN, in turn, can lead to purine and pyrimidine nucleobases (b in Scheme 1).44 Both reactions present, however, similar problems: the formation of intractable mixtures, yielding an extremely complex mixture of tetroses, pentoses and hexoses, with little abundance of the most interesting ones (e.g., ribose), in the former, or large amounts of an insoluble polymer and little of the nucleobases, in the latter.If both types of chemistries are combined, for example through the Strecker reaction of aldehydes (the simplest one being formaldehyde) with cyanide (the conjugate base of HCN) in the presence of ammonia, aminonitrile precursors of amino acids are obtained,14 in a process that is likely behind the high abundance of these protein constituents in Miller-type experiments (based on the application of high-energy spark discharges to gas atmospheres simulating that of the prebiotic Earth)15 and carbonaceous meteorites (i.e., chondritic meteorites that have shown to contain a significant proportion of organic compounds).45 From the simple idea of merging both oxygen- and nitrogen-based highly reactive substrates, in combination with the reducing power of hydrogen sulfide and the assistance of phosphate buffering in some key steps, the group of Sutherland,20,46 and later that of Powner,47 have built a complex set of synthetic routes towards both purine and pyrimidine activated nucleotides, plus a number of amino acids, sugars and lipid precursors (c in Scheme 1). A similar scenario emerges from the chemistry of formamide, which is indeed related with HCN through a dehydration process, and can be a source of carbon monoxide and ammonia, and of formaldehyde, through different catalytic degradation processes.48 This explains why the chemistry of formamide (d in Scheme 1), in the presence of mineral surfaces, has led to amino acids, acyclonucleosides, sugars and biologically relevant carboxylic acid derivatives, as it will be shortly revised below. In addition to the above approaches, there is an important body of work that proposes synthetic routes towards di- and tricarboxylic acids, which constitute core metabolites of the tricarboxylic acids (TCA/rTCA) cycle, also called the (reductive) Krebs cycle (e in Scheme 1).49,50 These pathways seem to start from glyoxylate and pyruvate, which in turn can be transformed through reductive amination or transamination reactions into the amino acids glycine and alanine, respectively.51
Besides the obvious common products that are shared by the above synthetic routes, one could hypothetically expand the available prebiotic chemical space by deconstructing them into their basic constituent transformations, and letting each transformation operate over any product that is a compatible substrate for it in such space. On this premise, in this review we have classified the ‘toolbox’ of possible prebiotic reactions into two sets. The first one accounts for so called ‘constructive reactions’, that is, transformations that contribute to increase the structural complexity of molecules through elongation of their skeleton (Scheme 2). The set of ‘interconversion reactions’, on the other hand, contribute to connect the constructive synthetic pathways through functional group transformations (Scheme 3). Both groups of reactions may be seen as a more detailed expansion of the 6 main types of reactivities that were considered by Vijayasarathy and Morowitz to explain the origins of metabolism.52 Establishing a proto- or minimal metabolic network out of these highly interconnected chemistries would require, however, that they can in principle be run in both the forward and reverse directions, so that exploration of the available chemical space leads to amplification of the most favourable ones. Of course, the conditions are what determines in which direction (forwards or backwards) each reaction should progress at a particular moment, and not all them could occur in a certain environment. In this context, please note that there are plenty of possible prebiotic scenarios involving conditions that range from very mild (e.g., a warm little pond, as claimed by Darwin) to quite harsh (e.g., hydrothermal vents), and different physical processes and interactions between those environments that could help cycling them. Here, favourable then does not necessarily imply higher thermodynamic stability of the products. Instead, it could refer to some sort of ‘dynamic kinetic stability’53 or, more precisely, the emergence of certain functional behaviours, provided that some cyclic organization is established within the mixture (by means of autocatalytic subsets, oscillatory networks, etc.).12
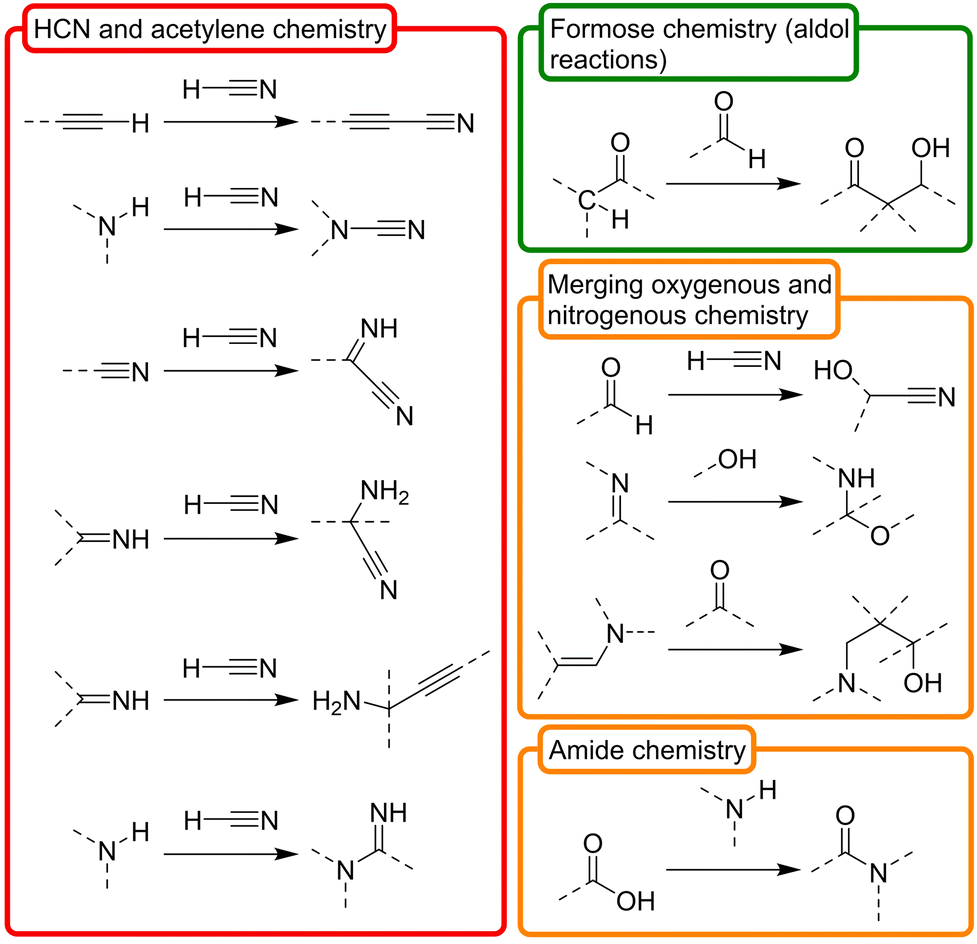 | ||
| Scheme 2 Set of ‘constructive reactions’ that contribute to increase the structural complexity of the skeleton of molecules, inferred from the main prebiotic synthetic routes pointed out in Scheme 1. To favour complexity growth, they should be favourable in the forward direction, yet the backwards reaction is also possible under suitable conditions. In a prebiotic context, the ultimate starting materials for these reactions are HCN, formaldehyde, ammonia and acetylene. In the chemical structures, dashed lines represent (single or double) bonds with H or C (or N or O if they are on C atoms). The three dimensional view of the structures is not represented. The involved chemistries have been classified, in turn, in three main subgroups, depending on whether they are based on nitrogenous (red), oxygenous (green) or mixed compounds (orange). Of course, these are just selected examples, and there are other possible transformations with prebiotic relevance. | ||
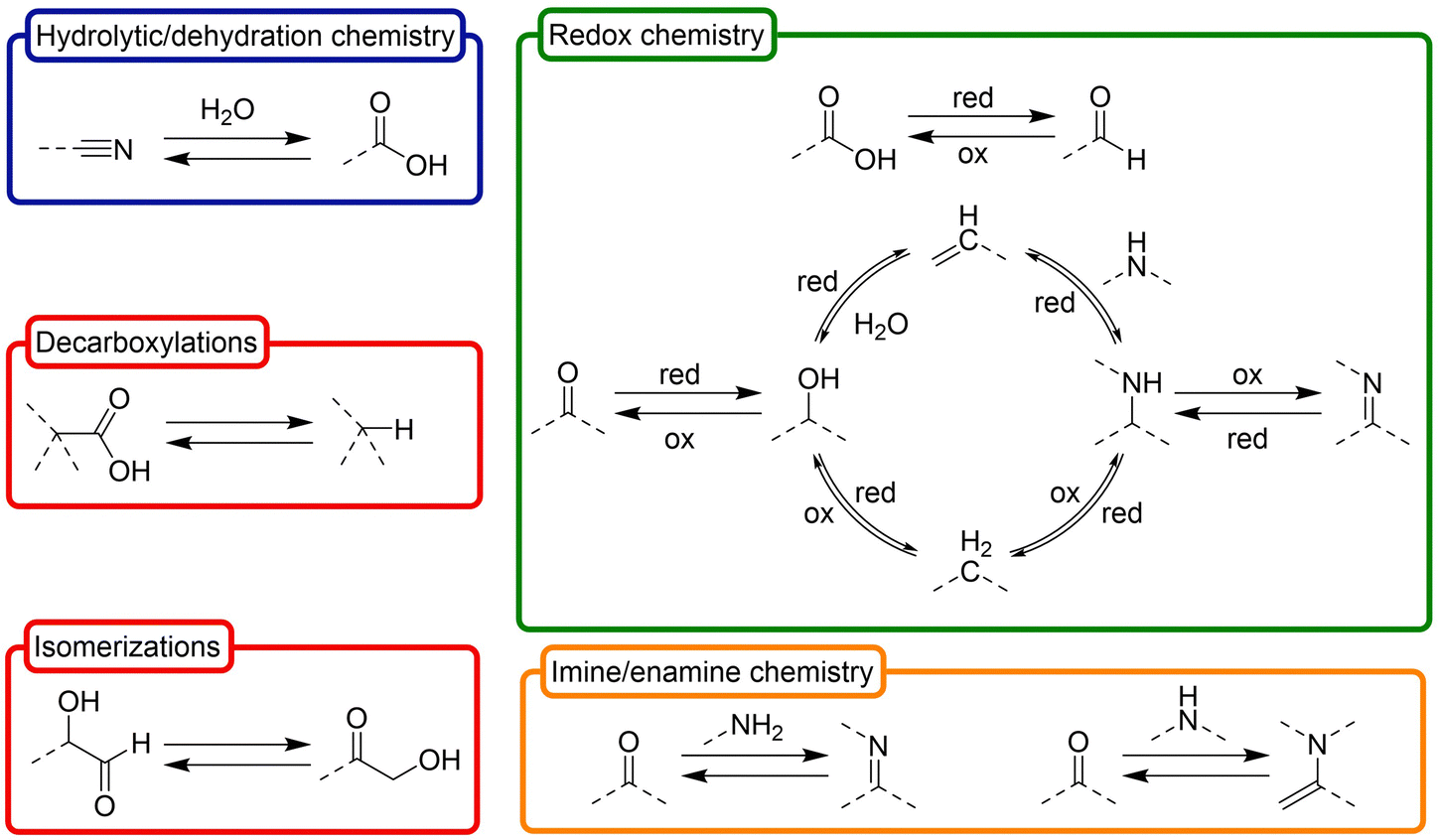 | ||
| Scheme 3 Set of ‘interconversion reactions’ that contribute to connect different constructive synthetic pathways through interchange of functional groups, inferred from the main prebiotic synthetic routes indicated in Scheme 1. These reactions are preferably considered in both directions, as they are normally reversible (depending on the conditions). In the chemical structures, dashed lines represent (single or double) bonds with H or C (or N or O if they are on C atoms). The involved chemistries have been classified, in turn, in four main subgroups: hydrolytic/dehydration processes (blue), oxidation/reduction transformations (green), imine/enamine chemistry, and other important reactions (red). Of course, these are just selected examples, and there are other possible transformations with prebiotic relevance. | ||
Indeed, equilibrium disruption by cyclic (recursive) processes is the most promising way to assemble chemical systems in the out-of-equilibrium dynamic state that characterizes biological phenomena.54,55 This includes chemical replication but also other physical–chemical processes, such as compartmentalization, phase separation, wetting-drying or freezing-thawing cycles. Some of the reactions needed for this purpose imply, for example, interconversion between functional groups with different oxidation states (see Scheme 3), for which a crucial issue would be to recycle in the reaction medium the redox agents that allow those transformations. Different species, both organic and inorganic, could play this role, including iron sulfide minerals, hydrogen peroxide as an oxidant, hydrogen sulfide as a reducing agent, organic cofactors, or just the network molecular components, which have different redox levels and will present oscillating concentrations due to the recursivity of some physical processes. Other reactions that could transform core metabolites of Scheme 1 are hydrolytic or dehydration reactions. Their occurrence, and the concomitant alternation between different states of those equilibria, would be highly favoured in the presence of alternating wetting-drying events, which could act as a recurrent cycling force driving the network out of equilibrium. Similar processes could enable carboxylations (e.g, with formate or formaldehyde in oxidative conditions) and decarboxylations, or aminations (with ammonia) and deaminations, coupled in many cases to redox events (e.g., oxidative decarboxylations, reductive aminations, etc.). In addition, a more heterogeneous reaction medium (highly probable in natural prebiotic conditions) including soft, organic interfaces, liquid–liquid phase separation processes or the presence of lipid domains (like micelles or vesicles) could also contribute to enhance chemical reactions that would be highly improbable in open aqueous solution, at the same time as it provides a natural scaffolding for the spatial organization of the system.
In sum, the hypothesis underlying the present perspective review is that all those chemistries and physicochemical constraints are interconnected, and that such interconnection is key to promote the establishment of protometabolic relationships leading to both (i) the production of functional biomolecules and (ii) their collective organization into non-reducible complex systems that distinguish themselves from the environment and, at the same time, couple with it to self-maintain in non-equilibrium conditions. On these lines, having introduced the general context of prebiotic systems chemistry, in the next subsections we will briefly revise its five main pillars, illustrating with specific, carefully chosen examples some of the processes sketched in the above paragraphs and their potential for developing functional constraints (control mechanisms) in emergent protometabolic systems.
As a matter of fact, some of the physicochemical processes mentioned above (e.g., the establishment of some form of autocatalysis together with crossed inhibition events or non-equilibrium self-assembly processes) could have assisted in the amplification of either deterministic or stochastic imbalances generated by certain chiral forces or chemical polarizations. The topic of absolute asymmetric synthesis has been extensively revised in the past,56–60 and it obviously connects to the contents of the present tutorial review. A number of individual reactions have been investigated under this perspective (see Section 4.3), yet with rather limited prebiotic relevance. In the context of chemical networks, their analyses are usually carried out with chromatographic techniques that do not allow looking at enantiomeric excesses of the molecules obtained. This is the reason why, in spite of its importance, most of the studies revised in Sections 2 and 3 do not pay much attention to the problem of homochirality. In the schemes within these two sections, we have thus opted for representing most structures without drawing their precise stereochemistry (with the exception of those in which it was specified in the original works).
2.1. The formose reaction
Sugars are an essential part of extant life, as we know it on Earth. Formaldehyde is their most plausible prebiotic precursor,61 due to its capacity to yield a variety of carbohydrates in a one-pot process known as the formose reaction, discovered by Butlerow in 1861.62 This simple precursor can be synthesized by electric discharges or photoreduction of CO2.63 The formose reaction then takes place under alkaline conditions, in presence of catalysts that accelerate the polymerization of formaldehyde at moderate temperature. Hydroxides of divalent metal ions (e.g., Ca2+) are the typical catalysts for this transformation, and result in a combinatorial explosion where dozens of linear and branched aldoses and ketoses are generated.64 Hexoses are normally the major products, although different minerals and clays can be used to drive the formose network towards amplification of key pentoses, ribose among them.65–67Concerning kinetics, the reaction is rather complex, and possess an autocatalytic nature.68,69 The first step, i.e., the dimerization of formaldehyde into glycolaldehyde (compound 1 in Scheme 4A), implies an umpolung of the former, which makes it very slow. When glycolaldehyde is condensed, however, a cascade of other transformations is initiated, where homologation occurs mostly through aldol additions and aldose–ketose isomerizations, accelerated by both the alkaline medium that assists in the enolate generation, through proton abstraction, and coordination of the intermediate enolates by the divalent metal ions. Importantly, the alkaline medium also favours retroaldol reactions (like that from compounds 5/6 in Scheme 4A), feeding back the network with the lowest molecular weight sugars (mostly glycolaldehyde and its enolate), in autocatalytic cycles like the one depicted at the centre of Scheme 4A (the Breslow's cycle). Apart from these general considerations, different mechanistic insights into the reaction have been elucidated recently.17,70
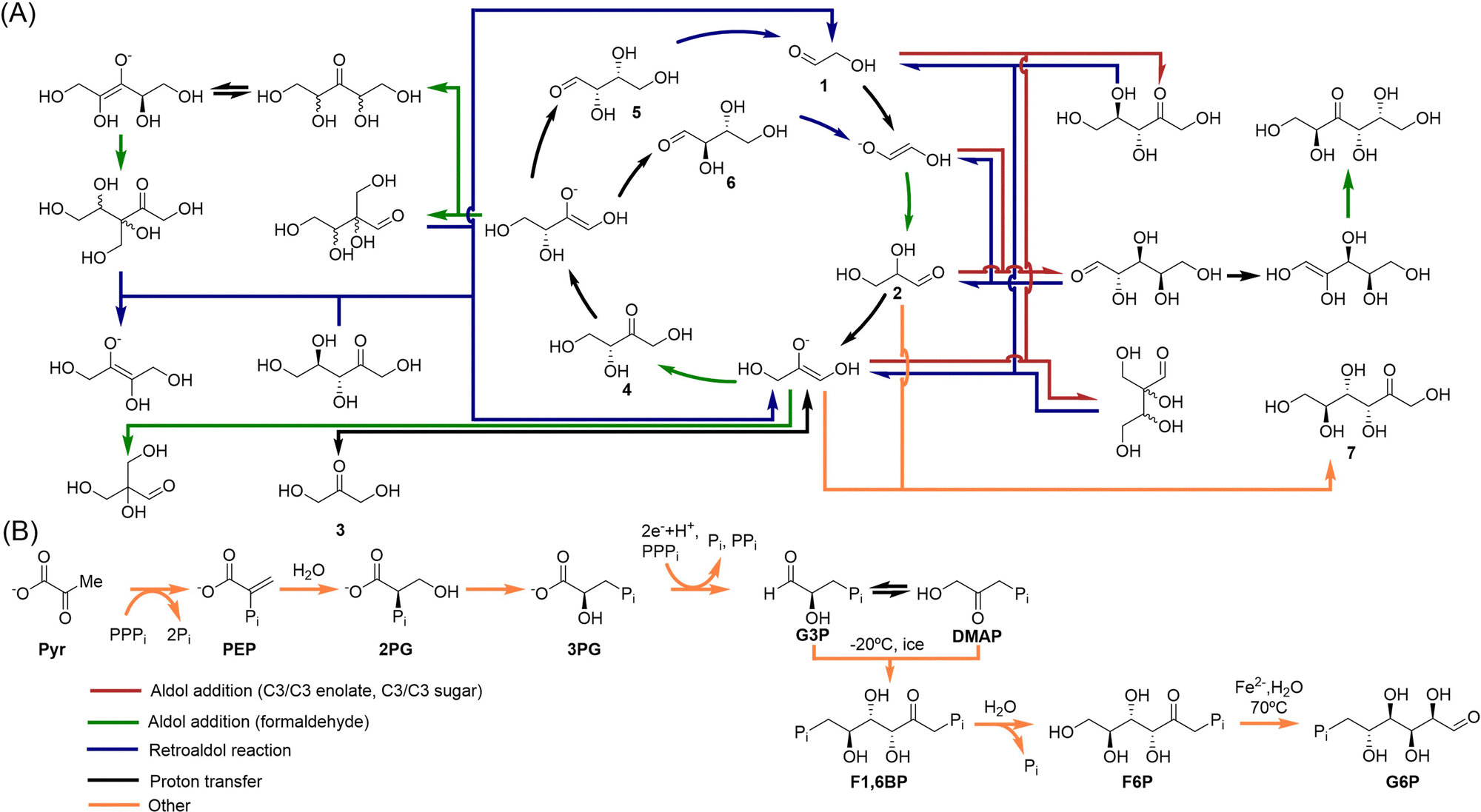 | ||
| Scheme 4 (A) A subset of reaction pathways that account for the majority of the formose observed behaviour. The Breslow's autocatalytic cycle is at the centre, and serves as a continuous source of small sugars (mostly two- and three-carbon ones), while various dominant formaldehyde chain-growth reaction pathways arise from the cycle components. Only key compounds, such as those constituting the Breslow cycle, dihydroxyacetone (3) and fructose (7), have been numbered. Adapted with permission from ref. 72. Copyright 2022 Nature Publishing Group. (B) Alternative route to biological sugars, analogous to that of gluconeogenesis, that could be subject to occur through non-enzymatic reactions. From glucose-6-phosphate, it would be possible to generated ribose-5-phosphate through an analogous version of the pentose phosphate pathway (not shown). Compounds G3P, DMAP, F1,6BP and F6P in panel B are phosphorylated versions of compounds 2, 3 and 7 in panel A. Adapted with permission from ref. 32. Copyright 2020 American Chemical Society. | ||
An autocatalytic cycle is a set of reactions and intermediates that form a cycle, in such a way that when the cycle operates over the substrates at the required stoichiometric ratios the amount of at least one of the intermediates increases over time. The presence of autocatalytic cycles has important implications with regards to the potential for self-organization of this sugar chemistry. Indeed, it was recently shown that its inherent high complexity could be tamed via recursion with mineral environments.71 In a later paper, the group of Huck demonstrated that the formose process actually leads to a network-like organization that allows the control of the generated chemical complexity in response to changes in environmental conditions such as feedstock molecules (in this case, formaldehyde, glycoaldehyde and dihydroxyacetone) and catalysts availability.72 In principle, the recursive iteration of the limited number of reactions involved in the formose chemistry can lead to a wide set of synthetic pathways and product compositions. However, by applying graph-based clustering methods for pathway analysis, Huck and coworkers found that, depending on the environmental conditions, the process is directed to only a subset of all possible pathways. This ‘selection’ of reaction pathways results in a restriction of the typical combinatorial explosion that otherwise leads to tar mixtures. On the contrary, well-defined compositions of a limited number of products can be obtained under certain conditions. Scheme 4A, shows the dominant pathways, as revealed by time-resolved analysis of the propagation of periodically changing environmental inputs, pointing out that the Breslow's cycle is crucial among them, as it restores the C2 building blocks while off-cycle formaldehyde chain-growth reaction pathways start from its other constituents.
The above results demonstrate that the formose network can display a collective response to environmental conditions. Of course, it is not known for sure whether this chemistry could be connected with the current metabolic route for sugars synthesis (i.e., gluconeogenesis).73 Gluconeogenesis starts from pyruvate (Pyr in Scheme 4B), and it is therefore connected to the acetyl-CoA pathway (the most ancient CO2 fixation route, which produces pyruvate) and the rTCA cycle (where pyruvate is one of the starting products).32 Then, it progresses through two phosphorylated glycerate intermediates to give glyceraldehyde-3-phosphate (G3P) and its isomer dihydroxyacetone phosphate (DMAP), whose non-phosphorylated versions are present in the formose reaction (compounds 2 and 3 in panel A). Some authors sustain that non-enzymatic primordial versions of these transformations could be possible (see Scheme 4B).74 The Ralser's group has for example found a primitive version of the aldol reaction between G3P and DMAP (both unstable) that proceeds at freezing conditions (−20 °C), yielding fructose-1,6-bisphosphate (F1,6BP) and from there fructose-6-phosphate (F6P), both core metabolites of that anabolic pathway.75 There are indeed catalytic versions of this reaction with glycine and lysine as catalysts. In any case, no matter which sugar intermediates were involved in the synthesis of pentoses and hexoses at the prebiotic era, it seems clear that their chemistry should have been subject to the establishment of self-organized autocatalytic networks like those described for the formose process, for them to become sustainable under constantly changing environmental conditions.
2.2. The HCN oligomerization
HCN is ubiquitous in the universe, as it has been detected in different kinds of extraterrestial environments (interstellar clouds, interplanetary dust, meteorites, comets, the atmosphere of some solar system satellites, etc.).76 On the Earth, it is abundant in volcanic eruptions and hydrothermal vents, so it is assumed to have been also present in the prebiotic times. Interestingly, there is an extensive literature about its oligo- and polymerization in aqueous alkaline solutions,18,77,78 preferably between pH 8 and 10, meaning that in most cases the monomer is cyanide, rather than its conjugate acid.79 In any case, such reactions can occur under a variety of conditions, the main requirement being that the monomer concentration is above 0.1 M, otherwise hydrolytic processes compete with oligomerization (between 0.01 and 0.1 M) or are the dominant pathways (below 0.01 M).19As a result of that, HCN oligomers and polymers constitute a variety of highly complex, pretty much insoluble organic substances, with colours ranging from yellow to black depending on the degree of polymerization and cross-linking, which upon hydrolysis can yield different groups of biomolecules.80 Key intermediates in this process are the HCN trimer aminomalononitrile, the open-chain tetramer diaminomaleonitrile (DAMN) (and its geometrical stereoisomer diaminofumaronitrile), and the two possible cyclic tetramer derivatives aminoimidazole carbonitrile (AICN) and aminoimidazole carboxamide (AICA).81–83 From them, it is possible to obtain nucleobases, both purines and pyrimidines, certain amino acids (e.g., glycine, from the HCN trimer, through hydrolysis and decarboxylation), precursors of cofactors such as pteridines, long chain carboxylic acids and other carboxylic derivatives that are members of the rTCA cycle (see Scheme 5). All this chemistry has been extensively explored and is well-reviewed in ref. 19 and 78–80.
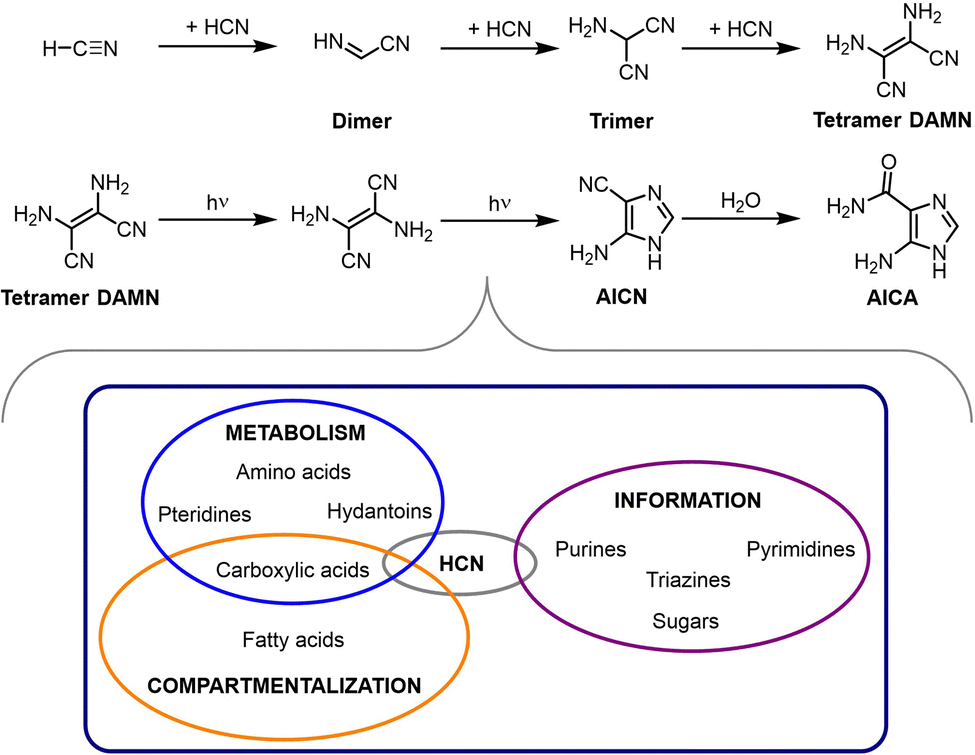 | ||
| Scheme 5 Main intermediates in the route for HCN oligomerization, which after hydrolytic processes lead to a variety of molecular components of the three main subsystems (for compartmentalization, information storage/processing and metabolism) of living entities.80 | ||
The main problem with this chemistry, however, is the enormous complexity and insoluble character of the polymers that can be generated, which has not been fully characterized yet. This is in part due to the absence of oxygen-containing functional groups in their structures. The most obvious way to incorporate oxygen into this chemistry is through hydrolysis of the obtained polymeric materials. The simplest possible hydrolytic product from the HCN chemistry is formamide, which indeed also leads to a very rich prebiotic chemistry,84 worthy of analysis before inspection of more complex scenarios (see next section). Interestingly, a computational exploration of the chemical space of HCN oligomerizations, carried out by Andersen et al. with a model based on graph grammar rules, allowed to identify various subnetworks that are autocatalytic precisely in formamide.85 The existence of autocatalytic sets when this chemistry involves oxygenous compounds would certainly help alleviating the combinatorial explosion that is inherent to the chemistry of this prebiotic monomer.
2.3. The formamide chemistry
As prebiotic building blocks, formamide and HCN are intimately related, both being one-carbon precursors that differ by the presence of an oxygen atom and two additional hydrogens in the former. Their methods of synthesis, abundance in the universe and reactivities are actually similar, mostly because they are interconnected through a hydrolytic/dehydration process (see Scheme 6),86 which could have allowed alternating between both through some kind of cycling physical process (e.g., wetting-drying cycles as a consequences of geological events). Formamide presents, however, specific physical properties and stability features that makes it especially suitable as a universal prebiotic starting material.84,87 First, unlike HCN, it already contains in its structure the four universal atomic elements that constitute living organisms: H, C, N and O. Second, it allows different mechanisms for concentration in aqueous solution, which is crucial to favour condensation/oligomerization reactions rather than hydrolysis.88 For example, the high boiling point of formamide (>200 °C), with no formation of an azeotrope in combination with water, enables its concentration through evaporation. Further concentration can be favoured by different mineral surfaces, e.g., through adsorption in the interstitial layers of montmorillonite and other clays. Indeed, the chemistry of formamide at high temperatures and in the presence of mineral surfaces has led to the production of biological building blocks ranging from amino acids, sugars, acyclonucleosides, condensing agents, long chain carboxylic acids and core metabolic intermediates of the glyoxylate and TCA cycles (Scheme 6, compounds outside the dashed circle).89,90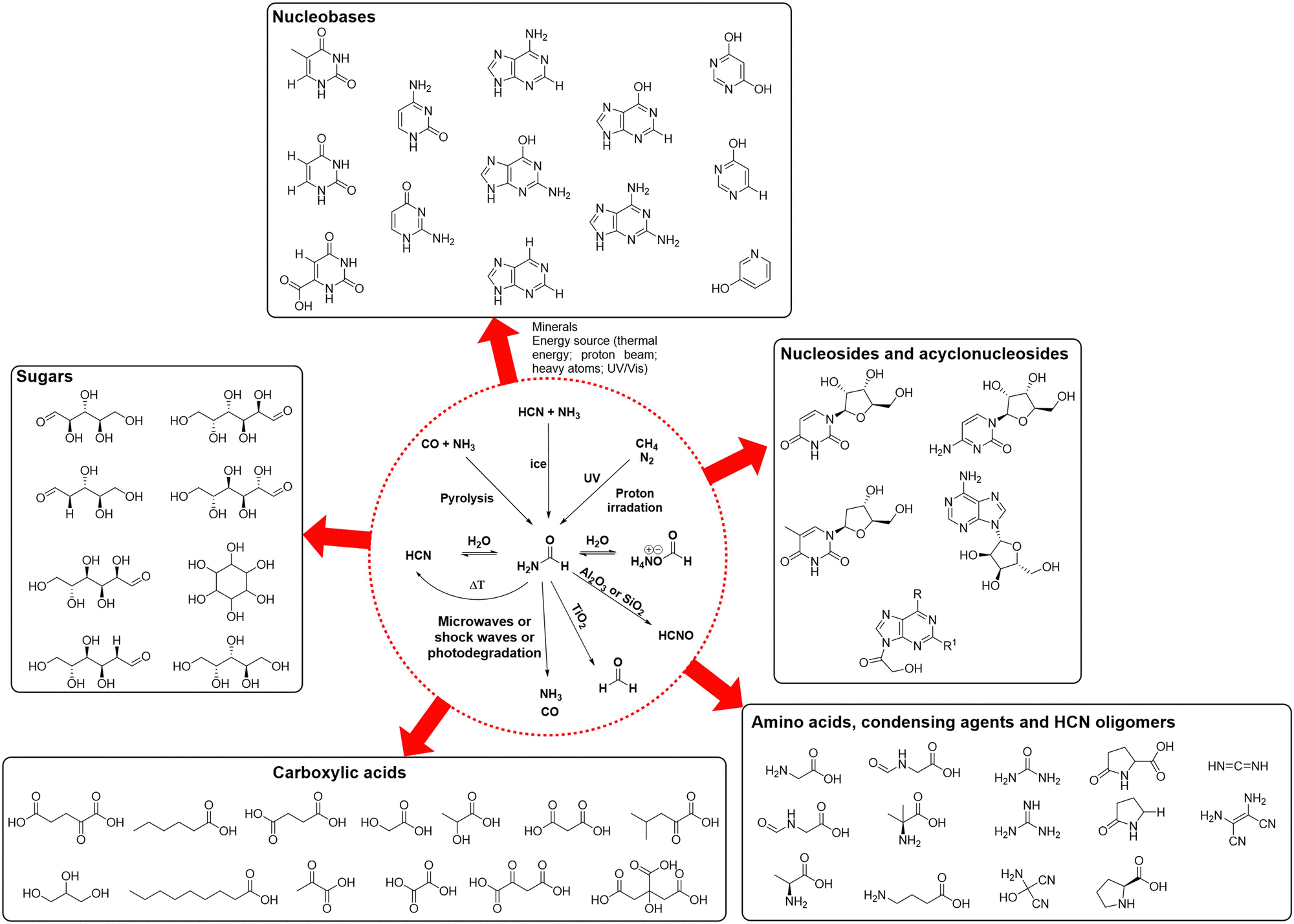 | ||
| Scheme 6 Main prebiotic syntheses of formamide (top reactions within the dashed red circle), its reversible interconnections with HCN and formate salts (middle line within the circle), and its main degradation pathways towards prebiotic compounds that can then lead (bottom reactions within the dashed red circle), under hydrothermal conditions and different mineral catalysts, to a wide variety of biomolecules (outside the circle).87,89 | ||
In comparison to the chemistry of HCN, the reasons why formamide yields a wider variety of prebiotic biomolecules are diverse.84–89 The presence of oxygen in its structure allows an interplay of oxygenous and nitrogenous reactive species that is not possible in the HCN oligomerization alone. The hydrolysis of formamide leads for instance to formic acid and its ammonium salt,91 which could engage in other synthetic pathways to increase structural complexity. With the assistance of metal oxides at high temperature, formamide can also dehydrate into HCN, closing the potentially reversible linear pathway of hydrolysis/dehydration events that interconnect the species at the middle row of Scheme 6 (inside the dashed circle).86 Other mineral-assisted degradative transformations of formamide can generate carbon monoxide and ammonia, formaldehyde, and highly reactive species such as HCNO (Scheme 6, bottom reactions inside the circle).84 The formamide-minerals scenario can thus afford, through decomposition, the necessary substrates that are required to establish classical prebiotic transformations such as the formose reaction towards sugars, the Strecker synthesis of amino acids, the HCN oligomerization or the Fischer–Tropsch process to give biogenic fatty acids, explaining why formamide lies at a central position (yet with certain debate) in the efforts to find routes towards the main biological components under the same compatible conditions.
The formamide framework lacks, however, a proper mechanistic explanation of how all those pathways are interconnected, probably because its reactions normally take place at high temperatures (most commonly, around 160 °C), which makes difficult to detect and identify low molecular weight intermediates. Overall, the chemistry of formamide supports the occurrence of various interconnected pathways leading to a pool of stable, biologically relevant structures. These processes can be fine-tuned with different mineral catalysts but, on top of that, purine is always obtained, no matter which conditions are employed. This fact suggests that its formation may involve some kind of autocatalytic loop in formamide,92 of a similar type as that of Breslow's cycle in the formose reaction, which has actually been predicted through computational analysis of the chemical space of HCN–polymer hydrolysis (see previous section).85 A proper understanding of this phenomenon is however still missing, likely due to the harsh conditions employed in this chemistry. In the early 2000s, an alternative scenario started to arise, that involves reactions at much milder conditions, and which therefore enables to study the whole complexity of intermediate steps leading to the main biomonomers from HCN and formaldehyde.
2.4. The cyanosulfidic scenario
It has been discussed in the literature why, in spite of the fact that Miller-type experiments produce, together with multiple amino acids, feedstock molecules that could lead to the assembly of ribonucleotides, the latter were never found in those experiments.14 The reasons for having separated chemistries leading to ribose and nucleobases are broad, but the major issue is probably the implausible formation of the glycosidic bond that allows connecting them.93 The severe problems that characterize the formose reaction and the HCN oligomerization have also been discussed abundantly, and makes many researchers question their prebiotic relevance.94 The merging of their feedstock substrates, formaldehyde and HCN, into a productive chemistry that leads to both types of biological components under the same conditions is not obvious, mostly because they react readily to yield glyconitrile (8), in a process that is fast and thermodynamically favoured (Scheme 7, red transformations).95 Glyconitrile then polymerizes but not into the products arising from the separate formose and HCN chemistries.96 Other difficulty that arise in this context is the possible intramolecular imidolactone formation (10) by larger cyanohydrins (from parent triose, tetrose and pentose sugars; e.g., 9), which conduces to their consumption and hydrolysis into the homologous carboxylic acids (12) (Scheme 7, inbox).97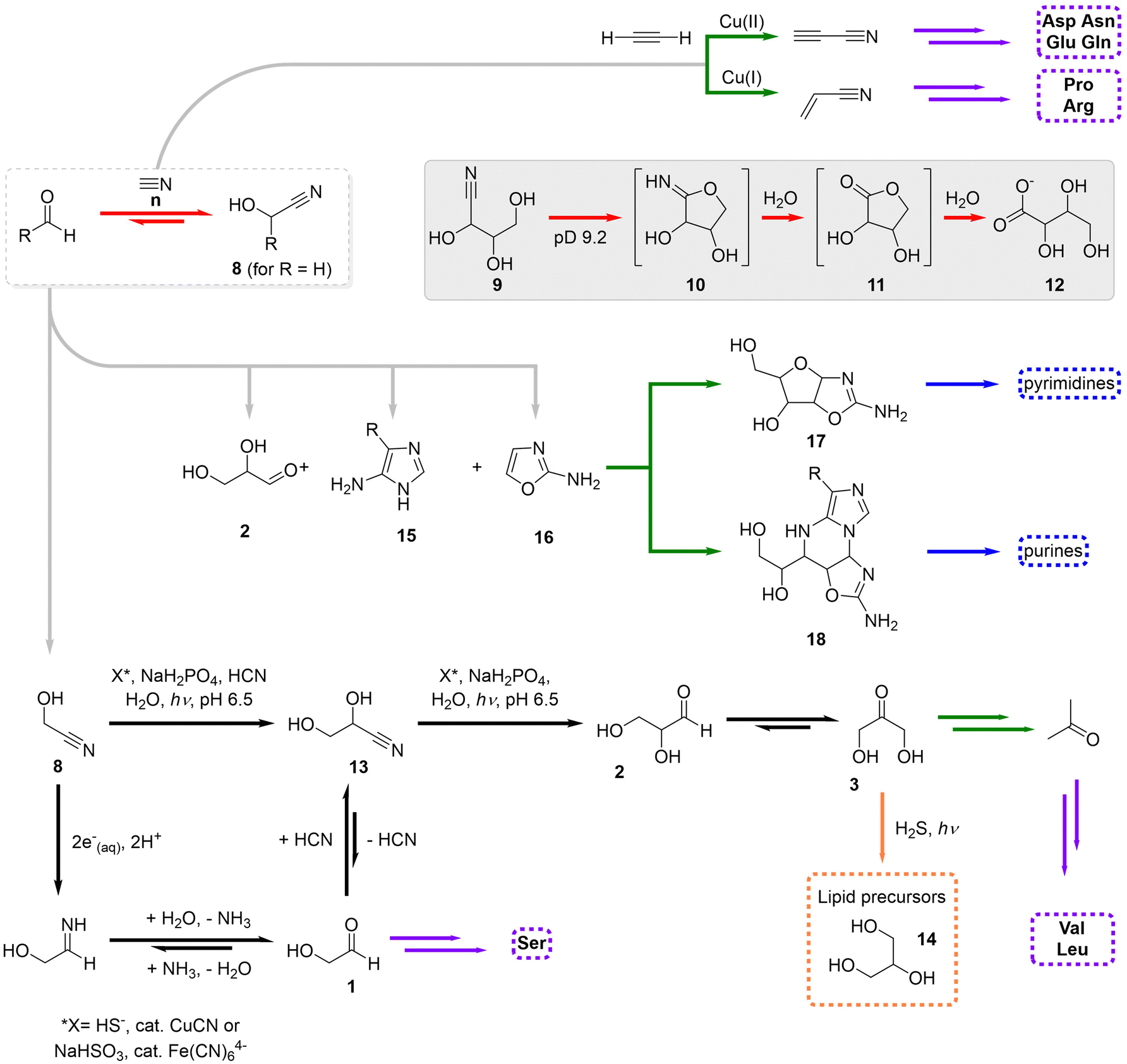 | ||
| Scheme 7 Schematic overview of the cyanosulfidic scenario, highlighting its initiation through glyconitrile formation and associated reactions (red transformations), the core homologation steps of this scenario (black), and the most important routes towards purine and pyrimidine ribonucleotides (blue), lipid precursors (orange), amino acids (purple) and other key intermediates (green).14,20,99 Only the key intermediate compounds have been numbered. | ||
In order to overcome these limitations, Powner, Sutherland and coworkers have developed during the last 15 years an alternative chemistry that supports the geochemical scenario so called cyanosulfidic.20,46,97,98 This scenario relies on two main monomers, formaldehyde and HCN, to produce glyconitrile, plus the action of schreibersite ((Fe, Ni) 3P), hydrosulfide (HS−) as reducing agent, copper and ultraviolet light, to promote the homologation to higher cyanohydrins (e.g., 13 in Scheme 7, black transformations).99 These successive homologation steps occur by photochemical reduction of the nitrile group into the corresponding aldehyde, making use of hydrated electrons (e−(aq)) and/or H˙, followed by addition of another HCN moiety.100 The reduction reaction is equally efficient when taking place with bisulfite as reducing agent and ferrocyanide mineral as catalyst.101 In any case, the consequence is that glycoaldehyde and glyceraldehyde can be produced through an umpoled equivalent of the otherwise very unfavourable formaldehyde dimerization.
One of these aldehydes, glyceraldehyde, is an important precursor not only in the formose reaction but also in the synthesis of glycerine (14), a lipid precursor,102 for instance through an isomerization step followed by reduction with SH2 (Scheme 7, orange transformation). Glyceraldehyde is also the starting product in a sequence of reactions that produce through intermediates 15–18 both purine and pyrimidine (deoxy)ribonucleosides (Scheme 7, green transformations).103 Importantly, phosphate buffering is crucial to tame the formation of intractable mixtures of cytidine-like products in this scenario, and to allow phosphorylation of the nucleoside precursors,104 while UV light irradiation is necessary in the last step to photodegrade wrong stereoisomers.105 Finally, this chemistry is compatible with the production of a wide variety of amino acids (purple in Scheme 7), as the cyanohydrins produced from all possible α-hydroxyaldehydes are, if ammonia is present in the medium, in equilibrium with their aminonitriles, and the irreversible hydrolysis of the latter can shift the process towards the monomers of current peptides and proteins.106
3. Protometabolic scenarios
The cyanosulfidic scenario represents an impressive synthesis of all previous prebiotic pathways towards different groups of biomolecules, i.e., sugars, nucleobases, amino acids and carboxylic acid derivatives. In a way, it shows the realm of what is possible to make with an organic chemistry-based approach, yet it is still strongly based on the typical workflow in most synthetic labs: a succession of reaction and isolation/purification steps. How all this intricate network of reactions would operate under the same environment is for the moment unknown, and so this chemistry cannot be really invoked as protometabolic. It is indeed possible that various feedback and inhibition events could happen when running the whole set of reactions that constitute the cyanosulfidic chemistry simultaneously in ‘one pot’, which could actually funnel the mixture towards formation of only certain subgroups of molecules, based on either their relative stabilities or more complex organizational principles (autocatalytic loops, amplification by templated processes, etc.).The first one to point in this direction was Eschenmoser,107 suggesting the likely existence of cooperative interactions when combining HCN and aldehydes as starting prebiotic products. For example, the production of the HCN-tetramer DAMN could be catalysed by aldehydes,108 defining a catalytic cycle that could in principle be iterated, enabling multiple current metabolites and biological building blocks to arise from it through functional group transformations (mostly hydrolytic and redox processes). It was proposed by Eschenmoser that the main connection between that type of cycles and the set of biomolecules arising from this chemistry would be α-ketoacids, the simplest one being glyoxylate.109 This prebiotic scheme was thus coined the ‘glyoxylate scenario’, and has been extensively researched with experiments by the group of Krishnamurthy at Scripps.110 In the next paragraphs, we then try to schematize the glyoxylate scenario in a different way, showing in a single network multiple reactions that can interlink, at least formally, the members of one of those catalytic cycles (purple in Scheme 8) to different biomolecular transformations. Even if some of these processes are not possible under the same conditions, the resulting analysis evidences the high interconnection between the pillars of prebiotic chemistry that have been analysed in the previous section, and helps understanding the efforts carried out later on by the groups of Krishnamurthy and Moran to experimentally reconstruct protometabolic cycles (see next section).
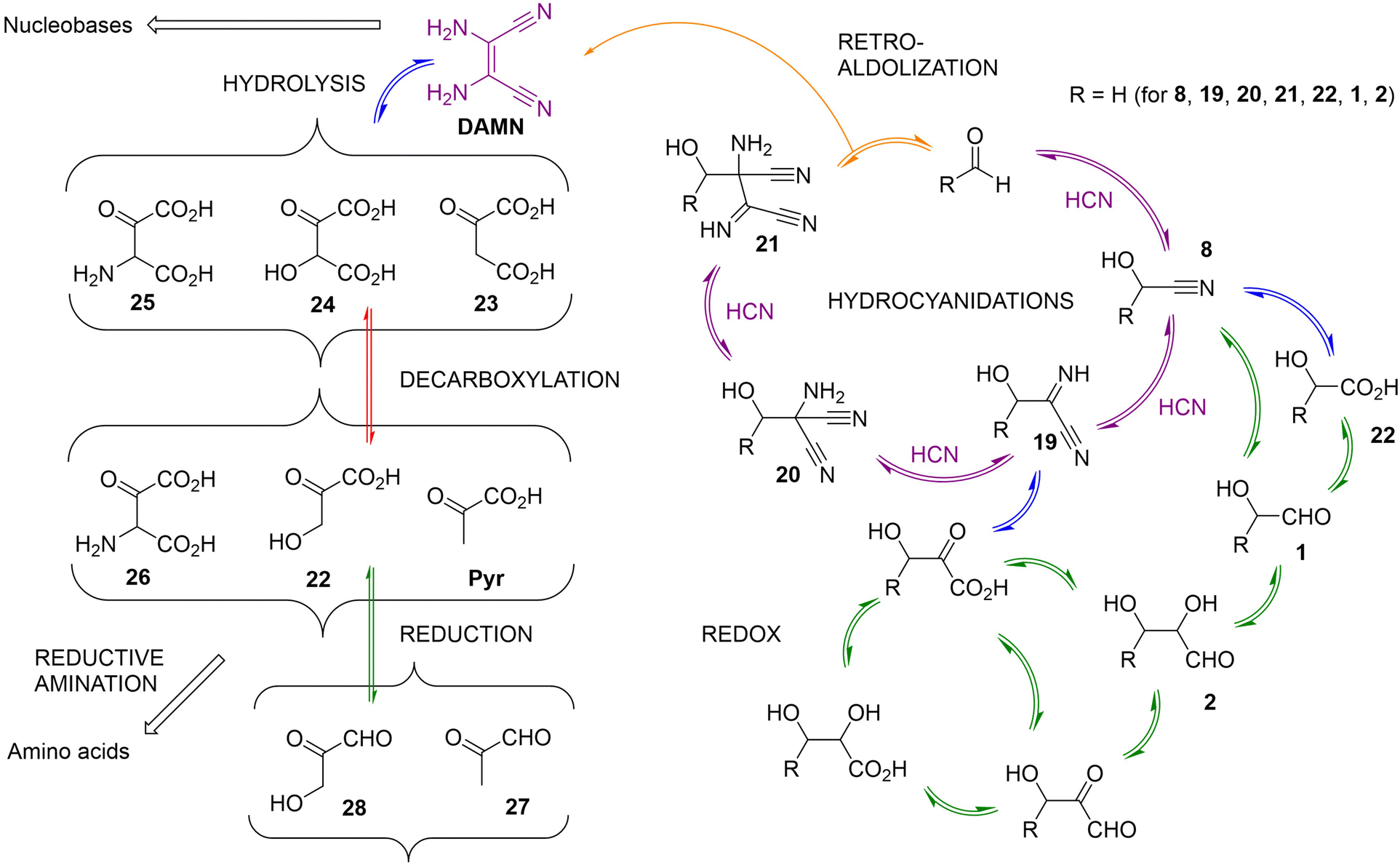 | ||
| Scheme 8 Schematic representation of the catalytic cycle (depicted with purple arrows) by which aldehydes (herein, formaldehyde) catalyse the formation of a HCN tetramer (DAMN). According to Koch et al., larger aldehyde derivatives are also catalysts of this process.108 Hence, if some of those aldehydes can be produced through hydrolytic (marked with blue arrows)/redox (marked with green arrows) reactions from DAMN or some of the cycle intermediates (e.g., 19, 20 or 21), as proposed by Eschenmoser and shown in the side pathways arising from the catalytic cycle, different autocatalytic loops could be established.107,109 Some of the potential aldehyde catalysts for this chemistry could be generated by the network itself (acetaldehyde, glycoaldehyde, glyceraldehyde), and analogues with 4 and 5 carbon atoms would also be possible to obtain from intermediates 20 and 21. Other reactions are also included in the scheme, such as decarboxylations or retro-aldolizations, which could be responsible for reducing the number of carbon atoms of some network members. | ||
For the case of the simplest possible aldehyde substrate (formaldehyde) as an example, the first (heterodimer) intermediate would be glyconitrile (8), the thermodynamically favoured product of the reaction between HCN and formaldehyde. A subsequent hydrocyanidation would lead to the α-iminonitrile 19. In both intermediates, one can formulate successive hydrolysis steps of the nitrile group, to render the corresponding amide (not shown) and carboxylic acid derivatives. In the case of 8, glycolic acid (22) would be the product, which could be converted into glycoaldehyde (1) through reduction of the carboxylic group, or by direct photochemical reduction from glyconitrile under conditions characteristic of the cyanosulfidic scenario (see above). Hydrolysis could also transform the imine group of 19 into its ketone, and the nitrile group into the carboxylic acid. Further reduction of the ketone into an alcohol and/or the carboxylic/nitrile group into an aldehyde would also be possible under certain conditions, defining additional cycles. This chemistry should in principle be considered as reversible, provided that adequate oxidative/reducing power is present in the medium. One could even expect cyclic chemical or physical processes by which hydrolysis/dehydration and oxidation/reduction events could be alternated.
From compound 19, it is possible to assume two further HCN additions that yield 20 and 21 in consecutive steps. The catalytic cycle could then be closed by a retroaldolization from 21, which allows recovering the aldehyde catalyst while producing the HCN tetramer DAMN. Interestingly, DAMN is a precursor of both purine and pyrimidine nucleobases, as shown in Scheme 5. Moreover, DAMN and the cycle intermediates 20 and 21 are susceptible to the same type of redox and hydrolysis chemistries as shown in the right part of Scheme 8, opening the way to subsequent layers of molecular complexity with 3–5 carbon-atom derivatives. In this scenario, going upwards through the layers implies hydrocyanidation reactions, while going down occurs through retroaldolizations. Interestingly, in the new layers of larger structures, upon hydrolysis of the nitrile groups, there are compounds that present at least two carboxylic acid functionalities, potentially linking this chemistry to the constituents of the TCA cycle. Examples of some possible α-ketoacid products that would be formed in this way (from the hydrolysis of DAMN) are shown on the left side of Scheme 8, as well as the subsequent decarboxylation, reduction and reductive amination processes that could take place from them, leading to important 2- and 3-carbon-atoms metabolites of extant living organisms (acetate, glyoxylate, glycoaldehyde, pyruvate, glyceraldehyde). Reductive reactions could also lead to different aldehydes with a number of carbons between 2 and 5 (e.g., compounds 1, 2, 27 and 28). Since these aldehydes are also catalysts of the central purple cycle depicted in Scheme 8, their formation from the cycle intermediates, or from DAMN, may lead to autocatalytic loops.
All the above suggests that the merging of HCN chemistry with that of aldehydes (the presence of which is ensured by the copper-catalyzed photochemical reductions of nitriles, which was later demonstrated by Sutherland)20,100 could have facilitated the establishment of autocatalytic cycles, allowing to navigate a wide chemical space comprised by the most preeminent 2–5 carbon-atoms prebiotic compounds. However, this view has not yet been sufficiently embraced by the most important experimental research lines that are being conducted to elucidate the prebiotic origins of current central metabolic pathways, as we are summarizing them in the next section.
3.1. Experimental reconstruction of protometabolic cycles
Modern approaches postulate that non-enzymatic reaction networks could have provided, from this type of central protometabolic cycles, an environment from which polymer replicators could then emerge.32,111 One of the main lines of research to understand the metabolic origins of life focuses on the chemistry of the citric acid cycle (TCA), also known as the Krebs cycle.49 In this regard, Moran and coworkers have reported that pyruvate and glyoxylate react in warm iron-rich water to produce most of the intermediates and biological pathways of the TCA cycle, in addition to some amino acids.21 The reactions were carried out at 70 °C under an inert atmosphere to simulate a mild hydrothermal environment. This context has a prebiotic geochemical relevance, as it is well known that ferrous iron (Fe2+) was abundant in the Archaean crust and waters.112 Samples generated under such conditions were analysed by GC-MS and NMR, revealing a significant production of acetate, pyruvate, malate, fumarate, succinate, α-ketoglutarate, isocitrate and aconitate (Scheme 9). In the presence of hydroxylamine, as a prebiotically plausible nitrogen source, and metallic iron (Fe0) under the same reaction conditions, four amino acids were also detected: glycine, alanine, aspartate and glutamate. These amino acids could be generated through transamination51 or reductive amination113 from the corresponding α-ketoacids. The presence of aspartate and malonate also points to the formation of oxaloacetate in the mixtures, maybe transiently, as it was not detected by GC-MS/NMR. Indeed, the same group has shown that hydrogen drives part of the Krebs cycle, from oxaloacetate to succinate, assisted by rhodium or meteorite catalysis.114 Overall, this work has led to the formation of 9 out of 11 TCA intermediates and 5 universal metabolic precursors. Such molecular complexity has emerged from a limited set of reaction types: aldol/retro-aldol, hydrations/dehydrations, oxidative decarboxylations and reductions/oxidations.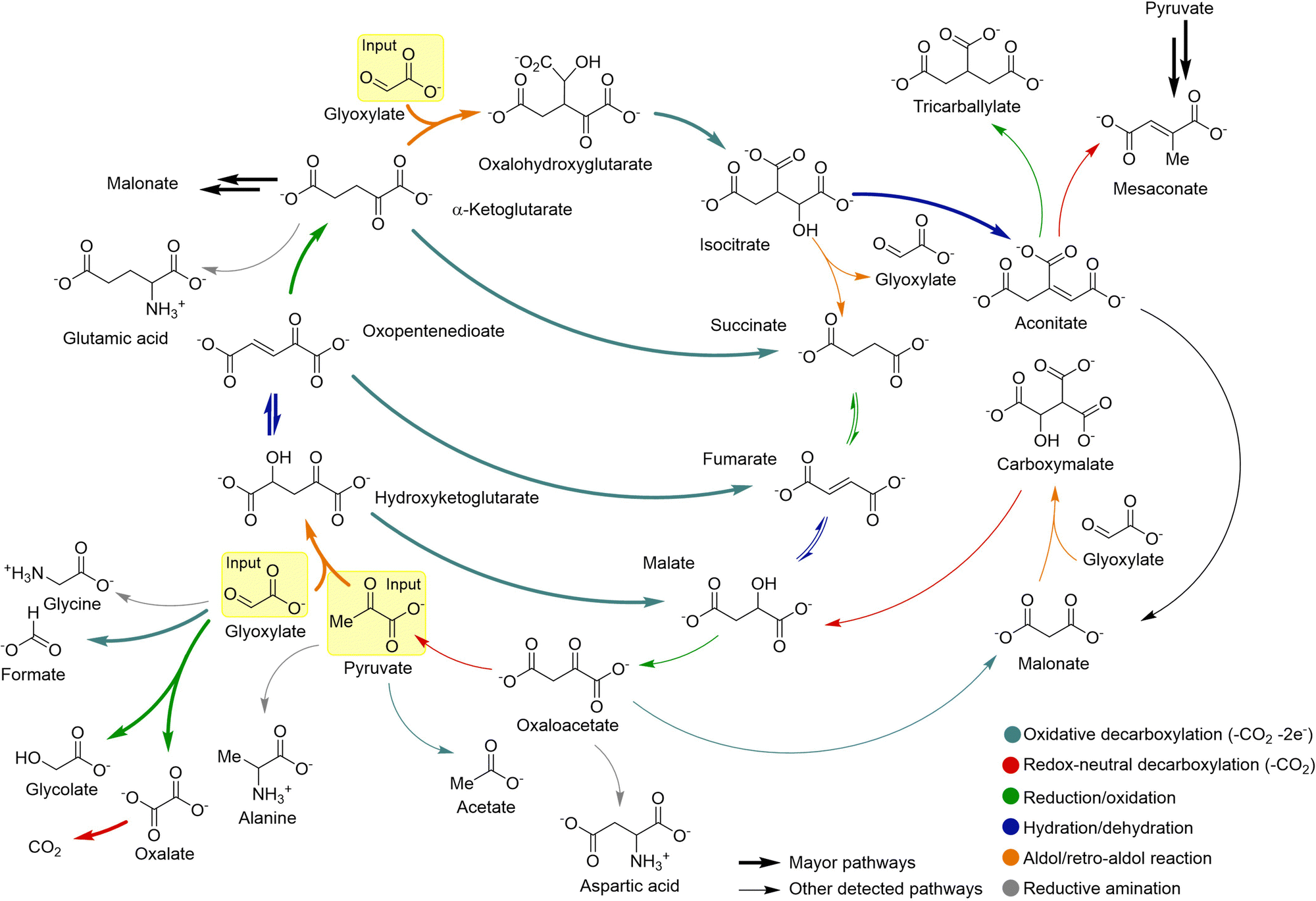 | ||
| Scheme 9 Fe2+-promoted synthesis and breakdown of 9 out of 11 members of the TCA cycle, and of the 5 universal metabolic precursors, as reported by Moran's group. Adapted with permission from ref. 21. Copyright 2019 Nature Publishing Group. | ||
The group of Krishnamurthy has reported, in turn, on two similar abiotic cycles, the HKG and malonate cycles, which are connected through a common intermediate, oxaloacetate, and together constitute the glyoxylate scenario.50 Importantly, both cycles share some common di- and tricarboxylic acid derivatives with the TCA cycle,110,115 as well as the occurrence of similar reactions, including the decarboxylation of β-ketoacids and oxidative decarboxylations of α-ketoacids, all under plausible prebiotic conditions (Scheme 10). Both abiotic cycles use glyoxylate as carbon source, and hydrogen peroxide to favour oxidative steps, while they operate in a pH range between 7.0 to 8.5 at a temperature ≤50 °C. This chemistry alternates aldol-type additions of key metabolites (pyruvate, oxaloacetate, malonate) to glyoxylate with the decarboxylation of oxalomalate and oxidative decarboxylations from HKG, malate, oxaloacetate and 3-carboxymalate.116,117 Additionally, in presence of ammonia the malonate cycle leads, through the carbinolamine of glyoxylate, to the formation of aspartate and glutamate, in a reductive amination process from the corresponding α-ketoacids.118
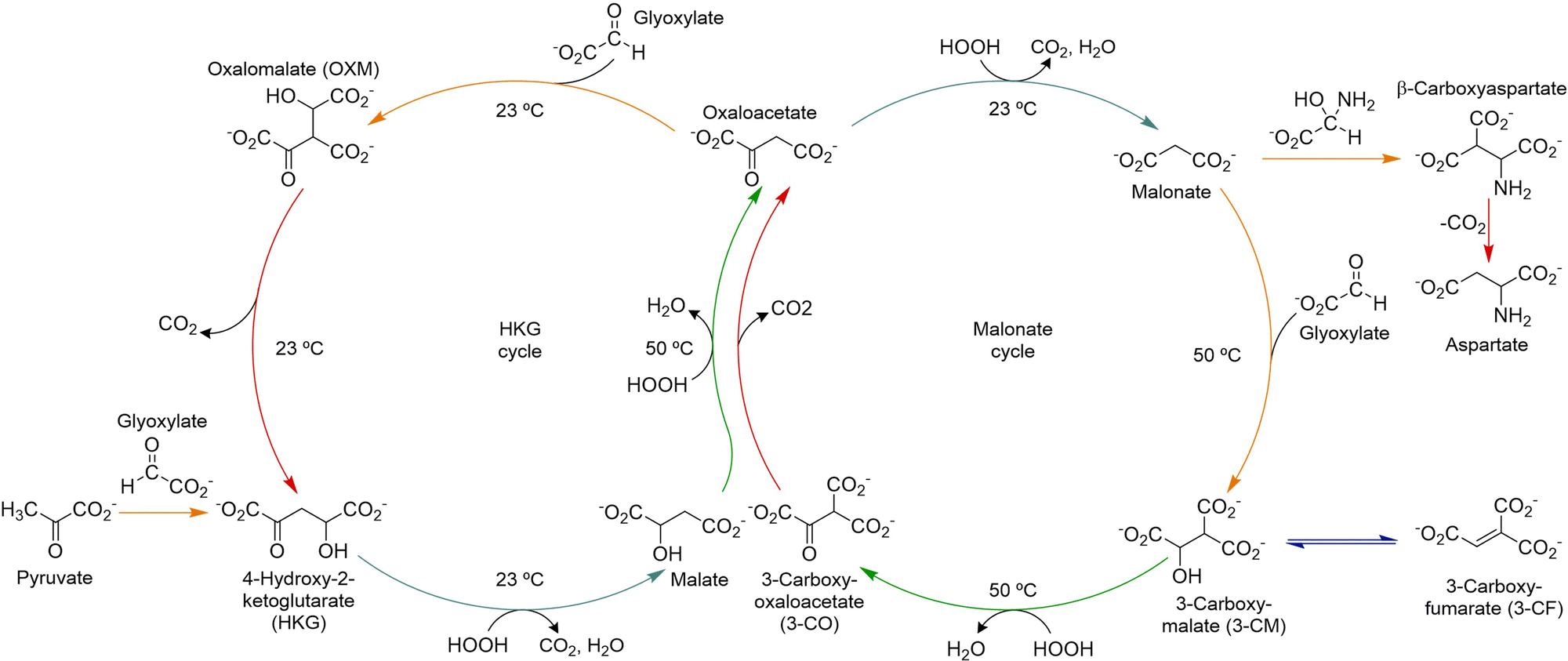 | ||
| Scheme 10 The HKG and malonate abiotic cycles proposed by Krishnamurthy's group. Adapted with permission from ref. 50. Copyright 2018 Nature Publishing Group. | ||
What the above two approaches have in common is that they find plausible routes to synthesize, under mild conditions, most of the molecular constituents of existing metabolic cycles, or analogues, which have been proposed that could be ancient based on phylogenetic analyses and other top-down strategies. Similar non-enzymatic versions of the glycolysis and pentose phosphate pathways have been investigated by Ralser's75,119,120 and Powner's121,122 groups, yet in this case they would not lead to a protometabolic cycle but to linear anabolic routes. All these efforts point to the likelihood that, if the molecular species of those ancient cycles were readily available on the prebiotic Earth, the establishment of protometabolic networks should have been feasible and may have played a key role in supporting other chemistries – like the emergence of peptide and oligonucleotide replicators, or of new catalysts to couple and make more efficient the required reactions (see below, Section 4). There are, however, a series of thermodynamic and kinetic bottlenecks that still need to be overcome if this proto-metabolic chemistry is to be turned into a feasible (minimal) metabolism, and these cornerstones are revised in the following section.
3.2. Robustness in transition: combining kinetics and thermodynamics
Although the above experimental lines to synthesize the constituents of potential protometabolic cycles have brought prebiotic chemistry relatively close to some of the main extant metabolic pathways, being able to form the cycle intermediates is not the same as making the cycle operate in the right sense, either with an anabolic or a catabolic purpose (i.e, to build up further molecular complexity or to liberate high-energy molecules, which could, in turn, act as chemical fuels to drive endergonic processes). An additional requirement in this regard would be that the protometabolic cycle becomes a self-sustaining system. This means that, with the help of fuelling molecules to promote the endothermic steps, together with catalysts to facilitate the slowly running ones, the kinetics and thermodynamics of the different reactions constituting the cycle should be coupled, in such a way that allows adapting the matter and energy balance of the system to the state of its close environment.12,24 In this direction, a revolution is taking place in the capacity of chemists to manage out-of-equilibrium self-assembled systems and networks,123–127 which makes this challenge potentially solvable in the future. A pertinent question, however, is whether the complexity of the TCA or the glyoxylate cycles would enable achieving an efficient dynamic coupling of their constituent transformations, in order to be run out of equilibrium. In the paragraphs below, we use the scheme applied in ref. 126 to the study of out-of-equilibrium chemical systems for the analysis of how metabolic cycles could be similarly fuelled.According to Prins and coworkers,126 the key for being able to populate a high-energy non-equilibrium state (step a in Fig. 1, left top cycle) from a lower one is to use a chemical fuel (F) that templates the reaction of the initially stable substrate A (in a low-energy state) into its activated form B* (step b). This activated species can undergo a transformation process (schematized in the figure as step c) that could not happen through pathway a, yielding C*. The asterisk (*) in this scheme means that the compound is in an energy-rich state and therefore remains reactive. From C*, final detachment of the fuelling fragment, through a process that converts it into a waste (W), renders the initially target lower-entropy structure (D, step d). These consecutive steps constitute a thermodynamic cycle that, when run in the right sense (anticlockwise), is able to dissipate energy (see the right diagram in Fig. 1), using it to maintain the thermodynamically unfavoured product D. Such control in the cycle directionality can be enabled through establishment of a kinetic asymmetry in certain steps. The occurrence of low energy barriers for the template coupling in step b and the waste liberation in step d, and high energy barriers for the reverse processes, would make those steps kinetically favoured in the forward direction and the cycle operative in an anticlockwise sense. Importantly, the occurrence of reactions between B* and the waste, or D and the fuel, would make the cycle run in the opposite direction, and so they need to be assumed kinetically hindered (light grey arrows in the figure).
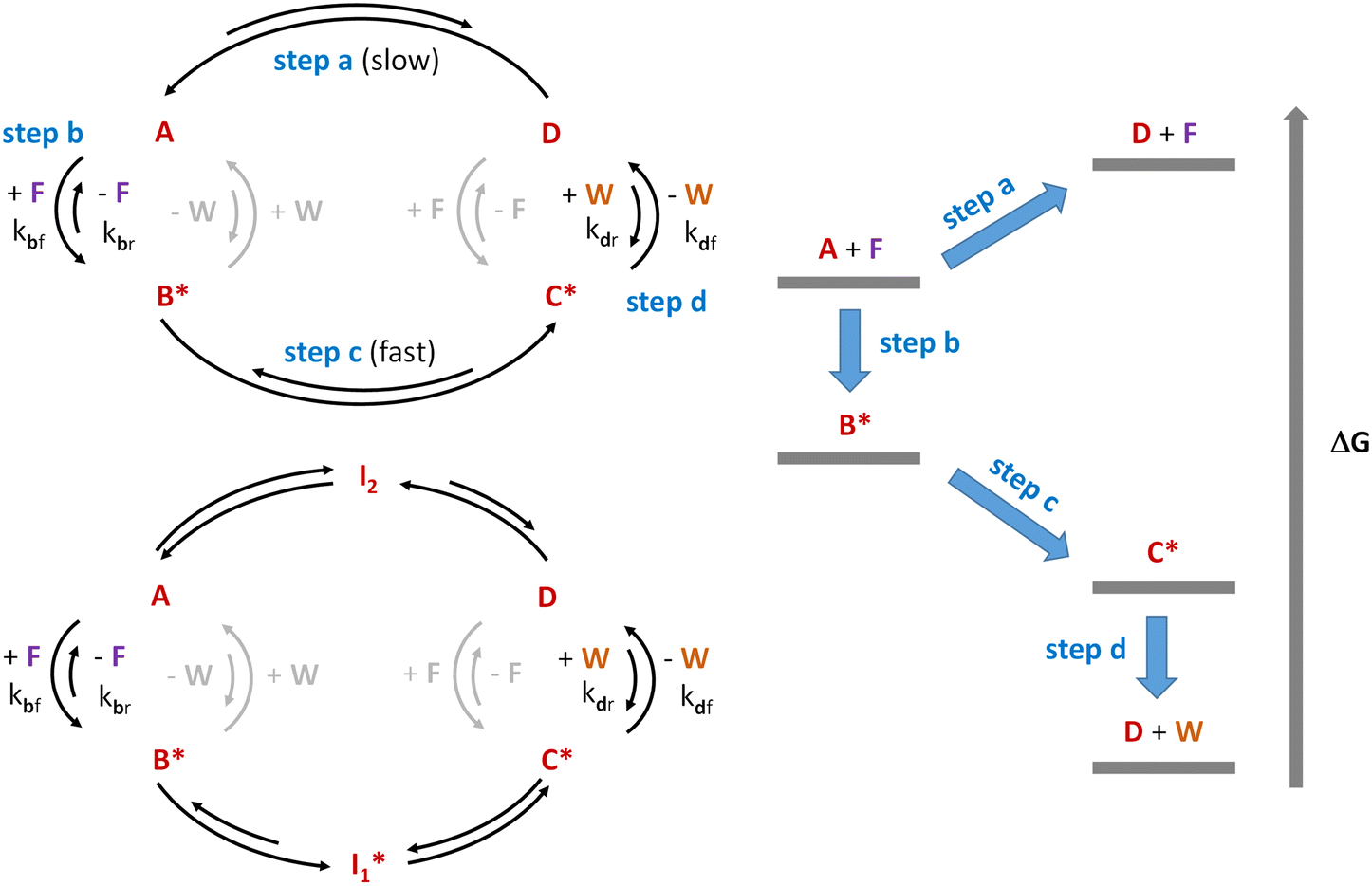 | ||
Fig. 1 Dissipative cycle of reactions where the thermodynamically unfavourable transformation of substrate A into product D occurs through a kinetically asymmetric cycle, sustained through the consumption of a chemical fuel (F) with the concomitant production of waste (W). B* and C* are reactive species produced by activation of A with the fuel. The left top panel shows the smallest possible cycle of this kind, as inspired by Prins and coworkers.126 The left bottom cycle shows how that type of cycle could be expanded by incorporation of intermediates 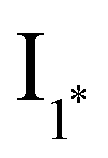 and I2, formed from B* and D through rapid equilibria. Finally, the right panel presents the different energetic levels involved in the top left cycle. and I2, formed from B* and D through rapid equilibria. Finally, the right panel presents the different energetic levels involved in the top left cycle. | ||
This type of kinetically controlled energy-dissipating cycles (i.e., cyclic chemical processes that consume energy to be driven in the thermodynamically unfavourable direction) have been proposed for the oligomerization of amino acids and nucleotides,128 the self-assembly of dynamic micellar/vesicular compartments129 or DNA nanostructures,130 and the establishment of autocatalytic networks.131 In general, the experimental approaches towards this end have involved molecules with a sufficiently complex structure, so that their spontaneous prebiotic emergence cannot be taken for granted. On a theoretical basis, the same scheme (thermodynamic cycles running out of equilibrium through dissipation of a chemical fuel) could be applied to protometabolic cycles, precisely to trigger their unfolding into more complex systems/organizations. From this perspective, the TCA, HKG or malonate cycles that are currently being investigated in relation to present day central metabolic pathways appear to be too large and complex and, thus, the rather fine-tuned kinetic asymmetry that would be needed to run them under dissipative conditions in heterogeneous prebiotic mixtures (i.e., starting from scratch) does not look too tenable as a work-hypothesis. The alternative scenario we put forward here favours simpler (proto-metabolic) reaction cycles that get soon 'functionalized' (in a naturalized sense: i.e., they autonomously develop functional parts), bringing about systems in which chemical transformations and control mechanisms of diverse types (kinetic, spatial, energetic) are coupled, in line with some of our previous hypotheses.12
The underlying assumption is that the TCA cycle, or related ones, were probably selected for the robustness of the chemical pathways leading to their intermediates, perhaps based on their relative stabilities compared to other derivatives, but in order to be viable from a kinetic point of view, they should have been initially supported by a network of interconnected smaller dissipative cycles. For example, from the cycle depicted in the left top panel of Fig. 1, a reasonable hypothesis is that it could be enlarged by integration of fast and reversible intermediate steps between b and d (Fig. 1, left top cycle), after formation of either the activated species B* (leading to intermediate I1*) or of the product D (leading to intermediate I2). Various of those steps could indeed be added, provided that they are all faster equilibrium processes than the forward reactions in b and d, leading to energy-dissipating cycles of gradually increasing sizes.
Being able to establish multiple interconnected (ideally catalytic) cycles, under dissipative conditions like those described above, would provide a means for the potential transition towards networks with increasing dynamic kinetic stability (DKS).53 This, combined with the heterogeneity of the reaction medium, self-assembly and phase separation processes, together with compartmentalization events, would enrich the space of possibilities for the emergence of the first self-sustaining autonomous systems (in a biological/metabolic relevant sense – see again ref. 12 and 24). In the context of this tutorial review, where various small cycles with prebiotic relevance have been discussed (see Schemes 4, 8 and 10), the networks resulting from their integration could be seen as a funnel that drives energy from abundant, constantly produced and highly reactive one-carbon precursors (formaldehyde, HCN, formamide) into self-sustaining protometabolic cycles of gradually increasing complexities (Fig. 2). This scenario also implies that the most robust cycles, considering both the synthetic yields of their member species and their turnover kinetics, would finally be favoured. This could be the case for the ancient and biologically widespread TCA cycle.
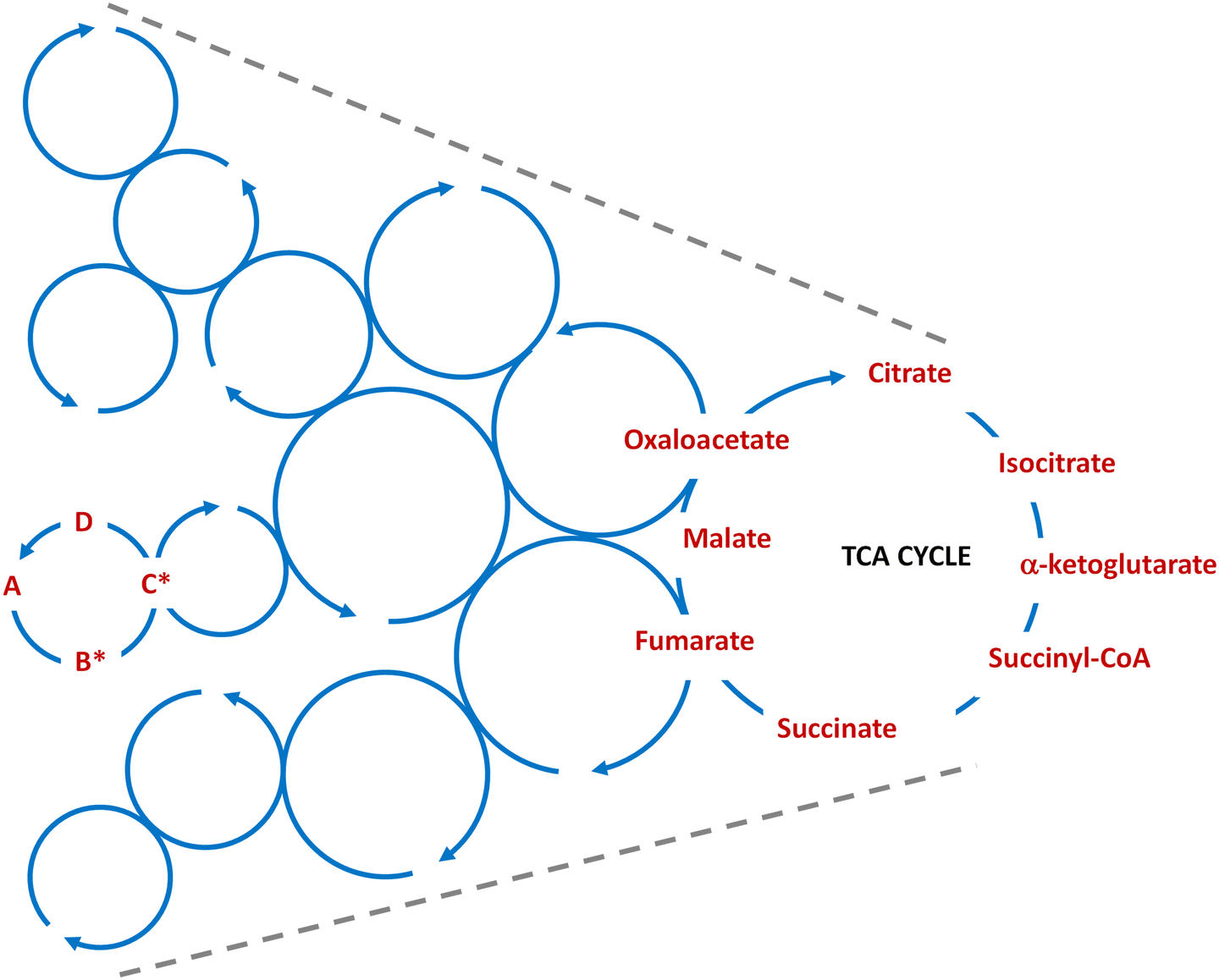 | ||
| Fig. 2 Schematic cartoon of how the coupling of multiple small dissipative cycles, like the ones depicted in Fig. 1, would allow funnelling energy for the sustainment of larger cycles with increasing complexity, leading to the construction of protometabolic networks where the cycles that are more robust, with regards to both the prebiotic synthesis of their members and from the kinetic perspective, would finally be favoured (for instance, the TCA cycle). | ||
To make the scenario depicted in Fig. 2 a possible one, a cornerstone would be to ensure that enough energy-rich carbon-based substrate materials are provided to the smaller cycles, to thermodynamically drive the complexity enhancement associated with the population of increasingly larger cycles. This requires some kind of carbon fixation mechanism, as it occurs in modern archaeal and bacterial cells with the acetyl-CoA pathway.132 The first step in this pathway is the endergonic reduction of CO2, to yield successively formate, acetate and pyruvate. Some researchers are working on an abiotic analogue and possibly evolutionary predecessor of the acetyl-CoA pathway, utilizing for that purpose iron-based mineral solids as catalysts under alkaline hydrothermal-vent conditions.133–136 Submarine hydrothermal vents are geochemical reactive habitats that hold complex microbial communities.137 It has been proposed that a crucial feature of such rich life-sustaining environments is their capacity to generate electrochemical gradients across different iron mineral structures.138 Serpentinization is one of those ancient geochemical processes in which water reacts with iron minerals and produces hydrogen. Various research groups have studied how hydrogen generation in those conditions can be coupled with CO2 fixation, utilizing minerals such as greigite (Fe3S4), magnetite (Fe3S4),135 awaruite (Ni3Fe),136 iron oxyhydroxyde (FeO(OH))134 or Fe(Ni)S precipitates,133 in the latter case aided by a pH gradient. These processes lead to different organics (from one- to three-carbon-atoms) that could feed the cycles schematized in Fig. 2. Besides, other researchers are working on possible links between acetyl-CoA pathway intermediates and the prebiotic synthesis of ATP, through iron-catalysed phosphorylation of AMP with acetyl phosphate,139 and in the generation of pH-gradients in synthetic protocells.140 All these approaches evidence the importance of looking into the energetics sustaining a potential primordial protometabolism, yet much research is still required to completely clarify how they could be coupled with the reaction network underlying it. In the next section we explore some of the aspects involved to this end.
4. Towards ‘minimal metabolisms’
The possible emergence of auto- and cross-catalytic feedback loops (i.e., systems in which a product catalyses its own formation or two products mutually catalyse each other's synthesis, respectively) in the – relatively simple but increasingly complex – reaction networks resulting from the chemistries reviewed in previous sections, combined with the self-assembly of non-equilibrium supramolecular structures formed from some of the network components, is bound to reinforce their trend towards functionally integrated, self-sustaining systems. One likely consequence of the establishment of those types of cycles and self-assembly events is the amplification of small chiral imbalances, mostly through Frank-type mechanisms.141 Yet, for these complexification steps to be accomplished, a number of different processes and molecular components should co-develop within the same prebiotic setting (thus, be amenable to ‘one-pot chemistry’ approaches). More specifically, the assistance of short oligomers (peptides and/or oligonucleotides) with enhanced catalytic properties should facilitate a more efficient control of the kinetic asymmetry that cycles – like the ones shown in Fig. 1 – may run into when they operate out-of-equilibrium, opening the route to larger ensembles with higher robustness (see Fig. 2). The possibility that these oligomers became enantiomerically enriched at this stage would have certainly improved efficiency in their synthesis (e.g., avoiding cross-inhibition during the oligomerization process) and functionality. In addition, it is quite reasonable to consider that the self-assembly of supramolecular structures and phase separation processes would lead to a diversity of soft interfaces and reaction domains in which water can be partially excluded, providing micro-environments (compartmentalized spaces) where those asymmetries can be further extended, and where spatial control mechanisms may start coordinating the diverse transformations in progress.One of our main hypotheses of work is precisely that diverse synergetic effects should appear in that kind of situation. In particular, the synthesis and amplification of oligomeric catalysts, if it is to have any biological relevance, cannot take place without a network of interconnected and dissipative catalytic cycles, including the self-assembly processes supporting them, which would also favour the reinforcement of homochirality. Similarly, quasi-equilibrium supramolecular structures cannot transform into growing and potentially reproducing systems unless they are coupled with chemical reaction networks, which take those structures further away from equilibrium. Therefore, although in this section of the review our attention will be focused on previous approaches to the prebiotic synthesis of peptides and oligonucleotides, on their potential replication dynamics, and on diverse compartmentalization and global-system reproduction processes, the final ‘take-home message’ will be that the most promising way of dealing with such ‘complexification chemistries’ should not be to try them in parallel but in various combinations. In other words, we reinforce here a view of ‘minimal metabolism’ that goes beyond that of a network of reactions that ‘mysteriously‘ acquires the capacity to self-sustain (typically considering the network in homogeneous solution conditions), conceiving it, rather, as a set of reactions intimately linked to the phenomena and physicochemical aspects (physical heterogeneity, self-assembly, compartmentalization, energy gathering) that actually assist in its configuration and sustenance on the way to cellular organization. In the sub-sections below we revise some of those aspects that can help bringing a protometabolic reaction network into a minimal metabolism.
4.1. A systems perspective on the prebiotic synthesis of peptides and oligonucleotides
Peptide bond formation is unfavorable in aqueous solution under reasonable conditions, for which the polymerization of amino acids in water only yields ridiculously small amounts of short peptides (di- and tripeptides) and dimeric cyclic anhydrides (diketopiperazines).22 A similar argument can be used for the oligomerization of ribonucleotides to give phosphodiester bonds and the concomitant nucleic acids. Often, a ‘catalytic‘ effect of mineral surfaces is invoked to overcome this conundrum, but this is misleading: the problem is not only kinetic but also thermodynamic.111,142,143 In this respect, mineral surfaces have been shown to promote the formation of both peptides and oligonucleotides, thanks to the adsorption free energy that overcomes the energetic cost required to link their monomers in the presence of water.144,145 Montmorillonite, in particular, and other clays such as layered double hydroxydes, comprise the most interesting minerals with regards to prebiotic chemistry, because they have shown to promote different processes, including the ligation of activated nucleotides/amino acids, and of their dimers/trimers, as well as the self-assembly of vesicles as protocell models.146 Key in these processes is that the clay can concentrate, align and act as adsorption template of the monomers (Fig. 3A),147 subsequently promoting bond formation if sufficient energy is provided to the system.148 In combination with mineral surfaces, the use of energetically activated forms of the monomers (e.g., amino acid N-carboxyanhydrides –NCAs– or aminoacyl intermediates for the synthesis of peptides, and 5′-phosphorimidazolide, phosphoramidate ester or 2′–3′ cyclic nucleoside monophosphate derivatives for the synthesis of RNAs) is common.149–151 Prebiotically plausible coupling agents such as cyanimide, carbonyl sulfide, 2-aminoimidazole or pyrophosphate are typical energy-rich species that could react with amino acids and nucleosides, making them more reactive towards the polymerization reaction.152,153 Quite remarkably, using these approaches, either with mineral surface catalysts and/or activating agents to promote condensation reactions, some researchers have also reported the stereoselectivities obtained in the synthesis of oligopeptides154,155 and oligonucleotides.156–158 An interesting alternative for chirogenesis in the oligomerization of peptides was that proposed by Lahav and occurring on supramolecular β-sheet templates.159,160 Overall, all these approaches have been extensively reviewed in the literature and, thus, the present review will not go through them in detail. On the contrary, the aim here is to focus on recent work that helps to start looking at the problem from a ‘systems-metabolic’ perspective.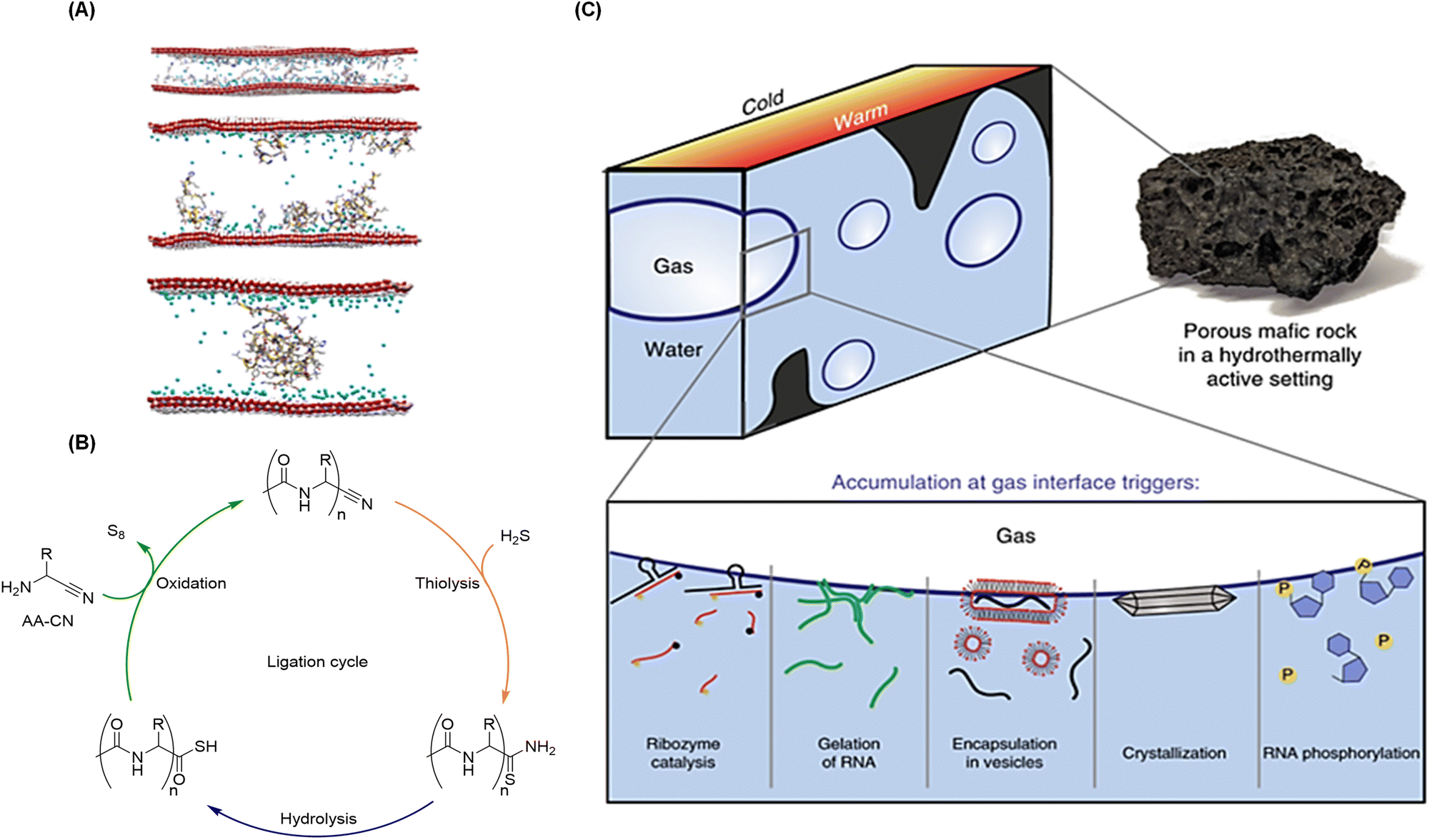 | ||
| Fig. 3 Examples of physical and chemical methods to produce biopolymers under plausible prebiotic conditions, and to impart recursivity in their production. (A) Models of how amino acids (top), short (middle) and long (bottom) peptides adsorb on double-layered hydroxide clays, which promotes their oligomerization and elongation. Reproduced with permission from ref. 147. Copyright 2017 Nature Publishing Group. (B) Iterative aminonitrile ligation cycle to give N-acetyl peptide nitriles. Reproduced with permission from ref. 105. Copyright 2019 Nature Publishing Group. (C) Accumulation of molecules through continuous capillary flow at the gas–water interface of microbubbles, generated in heated mafic rocks within wet environments, enables different chemical processes related to the origins of life and its first informational polymers. Reproduced with permission from ref. 163. Copyright 2019 Nature Publishing Group. | ||
A crucial aspect to be considered is how the resulting peptides and oligonucleotides could get assembled into chemical systems that self-maintain in the non-equilibrium dynamic state that characterizes biology. We assume that mechanisms for equilibrium disruption through recurrent/periodic environmental processes should be quite prominent in this context. We mean physical–chemical processes such as wetting-drying cycles55 or freezing-thawing cycles,161 periodic changes in pH, or osmolarity, oscillatory reaction networks, phase separation, etc. One interesting example is the iterative ligation cycle developed by Powner and coworkers, which in the same process allows a chemoselective activation of chain-growing α-aminonitriles into α-aminothioacids (through thiolysis and hydrolysis) followed by subsequent coupling with another α-aminonitrile monomer (Fig. 3B).106 In a later development, this reaction was adapted to the case of cysteine, whose aminothiol is in principle not compatible with nitriles, leading to a process where different cysteine derivatives are at the same time the substrate and the catalyst for peptide ligation.162 Another way to reach non-equilibrium conditions is through thermal gradients in mineral rock environments, which can produce heated gas bubbles in rock pores or dew on surfaces.163–165 This type of physical phenomena has shown to enrich certain prebiotic processes by subjecting all kinds of building blocks (RNA precursors, lipids, ribozymes and oligonucleotides) to wetting-drying cycles. Among the viable consequences of this recursivity are the occurrence of RNA phosphorylation, the increase of ribozymes activity, and the formation of supramolecular assemblies such as hydrogels, crystals, coacervates and vesicles that can undergo subsequent fission (Fig. 3C).163 The group of Braun has also suggested that dew on rock surfaces could drive the first stages of Darwinian evolution, promoting the replication of long DNA and RNA strands over short, faster replicating ones.165
Concerning all these recent approaches with a ‘systems’ motivation, it is important to point out that, whatever the mechanism, chemical recursion has the potential to impose a selection pressure on the network of reactions involved, as well as on the consequently changing boundary conditions, imparting a historical character to the successive generations of cyclic reactions: in other words, the result of one cycle will always be influenced by the previous ones.55 In absence of such ‘programmed chemical historicity’, a combinatorial explosion would occur, given that each species in the network may react with the others in more than one possible way. The establishment of feedback loops, most likely through auto- and cross-catalysis, has actually been demonstrated with relatively simple sets of peptides and nucleic acids. Since these would be crucial to induce functionality and evolutionary capabilities in the transition from chemical protometabolic networks towards biological minimal metabolisms, we now continue with a closer look into experimental works that have reported replication capacities of those types of molecules, before addressing a possible origin for homochirality and, finally, the question of compartmentalization and reproduction of more complex chemical organizations (protocells).
4.2. Peptide and oligonucleotide replication networks
Replication networks, as they have been described in the literature,166–171 may be interpreted as reminiscent of a protometabolism, in the sense that they allow selecting for certain features and could in principle become self-sustaining if the necessary substrates, chemical energy and compartmentalization are provided. However, experiments up to now were based on the external supply of ingredients to sustain the network, given that current replication systems lack the kinetic, energetic and spatial control functions to actually provide them endogenously. When designing replication systems, the use of peptides or oligonucleotides is widely spread because of their inherent biological (and pre-biological) interest, which includes their potential to implement a variety of additional roles (not just replication, per se), which could help precisely to transition in the direction of developing further capacities within the network.172 In this sense, peptide-based systems23,169,170 show potential for a high diversity of functions and accessible structures (e.g., catalytic activity and the capacity for specific molecular recognition), great kinetic stability due to amide bonds, and were prebiotically available from amino acids. Oligonucleotides,166 in turn, have a simplified DNA/RNA-like structure that allows them to behave as templates, precursor carriers of information (through base pairing) and, although their prebiotic plausibility is not so widely recognized, important advances were made in that sense in the recent past (see, for instance, Sections 2.4 and 4.1). These characteristics have led both types of molecules to be central components of protobiological replication networks. Some researchers are also working with hybrid systems that possess components from both kinds of biopolymers.168,171 Different network topologies can be achieved by combining these biomolecular components into synthetic replicators, as it is illustrated in Fig. 4. Importantly, in all these examples (and others revised below), homochiral systems were employed, clearly because that maximizes their functional role (e.g., the potential for well-defined self-assembly and/or catalysis). Of course, that cannot be given for granted at this stage of chemical evolution. Whether homochirality emerged prior to the establishment of self-replication capacities with prebiotic oligomers or not (i.e., whether it is actually a consequence of such evolutionary implementation) remains a critical open question, which will be addressed in the next section, once the different mechanisms that exist to establish replication networks are considered. | ||
| Fig. 4 Different network topologies that have been achieved with nucleic acid analogues or peptide-based synthetic replicators. (A). Schematic representation of an assembly-driven self-replication of peptide-based disulfide macrocycles, and their performance in a replication-destruction regime. Adapted with permission from ref. 177. Copyright 2021 Wiley-VCH. (B) Coupled redox-replication cycles that afford bistability within a non-enzymatic network of thiodepsipeptide replicators. Adapted with permission from ref. 170. Copyright 2019 Nature Publishing Group. (C) Topology and operative catalytic cycles that enable selection of the fittest in a replication network of nucleopeptide structures.168 (D) Network topology that would allow collective adaptability in a set of cystine-based minimal nucleobase sequences. Adapted with permission from ref. 171. Copyright 2022 Royal Society of Chemistry. | ||
An interesting function to implement in replicating systems would be the ability to catalyze multiple processes at the same time. Otto's group has developed, for example, synthetic peptide-based building blocks that form hexameric macrocycles upon oxidation.173 These macrocycles are capable of catalyzing their own formation through self-assembly, and in parallel two other chemical transformations that are different from the replication reaction, that is, the system shows catalytic promiscuity. Apart from this replicator, other less complex architectures based on the same type of building blocks arise in the system, resulting in a competing population of replicators. Nevertheless, if the system is fed with a chemical fuel, only the most complex structure (i.e., the hexamers) prevails, even in the case when its formation process is slower (Fig. 4A).170 Another feature that can be added to these networks is a cofactor that accelerates the rate limiting process of replicator formation (the oxidation of monomers), for instance through the action of light.174 This use of an external energy source to synthesize the precursors of the self-replicating species from certain substrate molecules could be seen as a form of protometabolism, although it still lacks a way to store that energy and to use it in the promotion of an endergonic reaction that is useful for the system.
Another functional aspect that has been accomplished in synthetic replication networks is bistability, which provides cells in nature with a mechanism for long-term memory storage, namely the ability to integrate a transient molecular stimulus into a sustained molecular response.175 In a bistable dynamic regime, a change in internal variables, organization parameters or external conditions can lead a system to switch, abruptly, between two (stable) states. This typically provides cells with a mechanism for memory storage, namely the ability to integrate a transient molecular stimulus into a sustained molecular response. In practice, the Ashkenasy lab designed a dynamic reaction network that, depending on initial thiodepsipeptide concentrations, leads to one of two distinct steady-state concentration distributions.176 By applying physical (heat) or chemical stimuli, the system can be switched from one state to the other. Nevertheless, a continuously fueled reducing environment is crucial for preventing the bistable system from falling into less defined states (Fig. 4B).177 Interestingly, coupling this type of dynamic bistable network to the formation and self-assembly of gold nanoparticles has enabled to mimic an additional network-building biological function, i.e., signaling.178
With regards to DNA/RNA replication, function is inherent to nucleic acid structures but, so far, two issues have limited the non-enzymatic replication of these molecules. First, the template inhibition problem, related to the impossibility of binding to existing product strands of new substrate molecules, due to the high duplex stability, which blocks the template for further replication cycles. Second, the higher survival of the shortest sequences in mixtures of competing replicators because of their faster kinetics, which causes a loss of information. On these matters, progress has been made in the recognition of potential prebiotic scenarios where both replication and prevalence of longer oligonucleotides could have taken place.179 Hud's group, for example, proposed that thermal heating cycles combined with a viscous environment could have provided a mechanism for denaturing the formed nucleic acid duplex into its two strands.180 Later on, they showed that concentrated solutions of urea and acetamide in water (both plausibly prebiotic) may have assisted in this task.181 In addition, Braun's group put forward submerged rocks with open pores as ancient sorting machines capable of excluding short oligonucleotides from a constant size-mixture flow, setting up the system for replication of longer strands.182 This machinery would have worked with the aid of a heat flux across it, which could let the molecules be swept along by the flow inside the pore (via thermophoresis), causing the largest ones to accumulate in it while the smallest ones leave. In this way, a common setting on the early Earth like an open pore could provide an interesting non-equilibrium habitat for the recursive feeding, replication and positive length selection of genetic polymers.
Concerning nucleic acid replication, we would like to finish clarifying that longer sequences of nucleic acid analogues with ribozyme/polymerase activity have also been developed for exploring the chemistry of genetic information storage and propagation,183,184 yet they are not revised here, as their length or backbone structure usually get out of what could be considered prebiotically plausible, and so that type of approaches would fit better as possible routes of early biological evolution.
A critical issue when designing replication networks is whether some of the components can be selected based on the environmental non-equilibrium conditions surrounding the system, and how such conditions affect the network composition. This issue somehow provides a way to assess the protometabolic character of a given network, as it is known that there are always ecological and evolutionary consequences of environmental changes on metabolisms.185 Interestingly, the Ashkenasy and de la Escosura groups reported on a network of nucleopeptide systems where nucleobase interactions between complementary sequences facilitate control on different auto- and cross-catalytic pathways (Fig. 4C).168 When the system is set out of equilibrium in a CSTR flow reactor, the network is biased, through functional synergy between the nucleobase sequences and the peptide fragment inducing self-assembly, pushing the systems towards the most prominent replicator. Semenov's group, in turn, has described a network based on dimeric thioesters of tripeptides displaying non-selective autocatalysis, which can be driven by simple mechanisms such as the formation of strongly nucleophilic species (e.g., thiolate) or catalytically active ligands.167 Braun's group has also studied mixtures composed of short oligonucleotides that form longer ones by random templated ligation, leading to the simultaneous elongation and sequence selection of oligomers.186 The product strands showed highly structured sequence motifs, which inhibited self-folding and built templated reaction networks. By reduction of the sequence space, the kinetics of duplex formation increased and led to a faster replication through the ligation process.
These findings imply that the elementary binding properties of nucleotides can lead to an early selection of sequences even before the onset of Darwinian evolution. An important question relates, indeed, to whether this type of collective network behaviors could appear with simpler sequence replicators. In this respect, the group of de la Escosura has shown that collective adaptability would be possible in a replication network of synthetic minimal nucleobase sequences – yet not based on canonical nucleic acid structures (Fig. 4D).171
4.3. A plausible protometabolic origin of homochirality
During this tutorial review, the topic of how molecules constituting protometabolic/replication networks could have been enantiomerically enriched has been mentioned, but left quite aside. This is not due to the lack of importance of this problem, but to the lack of a solid body of evidences and knowledge to explain this phenomenon in a prebiotic context. In this section, we try to summarize some of the approaches that have been examined around the question of the emergence of homochirality. Our revision of such topic does not intend to be comprehensive but focussed on examples and models that present special relevance in the context of protometabolic networks. For an in-depth insight, there is in the literature a number of reviews that cover the substantial amount of work that has dealt with processes and mechanisms accounting for the accumulation and amplification of small initial chiral imbalances into homochiral systems.56–60,159,187,188The role of chiral forces and chemical polarizations to induce spontaneous mirror symmetry breaking (SMSB),59 generating small enantiomeric excesses from racemic mixtures, and the subsequent chiral amplification of those excesses, is well known from a theoretical perspective.37,60,189 The classical and most compelling (Frank-type) models to explain the asymmetric amplification of an initial imbalance, either stochastic or caused by deterministic chiral forces, normally involve autocatalytic reaction systems.190Fig. 5A shows for instance the system originally proposed by Frank,191 which involves the autocatalyzed synthesis of a chiral compound, in its two enantiomeric forms, from an achiral one, together with an irreversible reaction that destroys such chiral product and an implicit flow of matter required to maintain the symmetry breaking. Frank's model constitutes a minimalistic system from which other related models were later derived, in order to account for chiral amplification in different experimental systems.190–196 In the original model the involved reactions were considered irreversible, but they can also be described as reversible, which allows the system to evolve back to the equilibrium symmetric state when the flow of matter is disrupted.195 For a review on this topic and for deeper mathematical insights about it, see ref. 190 and 197, respectively. In any case, there is scarce evidence about the abiotic scenarios that could facilitate this type of processes, the stage of chemical evolution at which symmetry breaking took place, and its actual relevance for the emergence of life.198
 | ||
| Fig. 5 (A) Schematic representation of the processes implicit in Frank's original model:191 in which the autocatalytic formation of a chiral product (from an achiral substrate) leads, in an open stationary system, to the amplification of one of the enantiomers. (B) Scheme showing the asymmetric amplification via autocatalytic self-replication and ‘mutual antagonism’ that occurs in Frank-type models. (C) Scheme of an example of the Soai autocatalytic reaction.201 (D) Coupled catalytic cycles proposed by Blackmond, in which amines and aldehydes can act as both substrates and catalysts, possibly allowing for the establishment of autocatalytic replication and subsequent amplification of enantiomeric excesses. Adapted with permission from ref. 60. Copyright 2020 American Chemical Society. | ||
Two requirements are crucial for a chemical process to lead to homochirality according to Frank-type models: (i) that each enantiomer is able to catalyse its own production, and (ii) that they also inhibit the other enantiomer formation (what Frank called ‘mutual antagonism’ and can occur through different types of reactions/interactions), enabling a non-linear growth during the autocatalytic reaction they emerge from (Fig. 5A).141 Another necessary feature is that these non-linear growth dynamics must occur in non-equilibrium steady states. It is the continuous flow of energy that maintains the system out of equilibrium what actually drives the ongoing flow of reactions, allowing several possible steady states. When the matter flow becomes important enough (i.e., above a critical value), and thanks to the autocatalytic and mutual destruction reactions, the racemic state can be destabilized. The system then spontaneously evolves into one of the two possible non-racemic states through a process like the scheme depicted in Fig. 5B. Such state remains stable as long as the non-equilibrium regime persists, and can counteract different destructive phenomena, thus resisting against the ‘calamity of racemization’.190 Interestingly, we have seen that autocatalysis out-of-equilibrium, in its different forms (autocatalytic reactions, cyclic or network autocatalysis, and template replication),199 is a fundamental feature for chemical evolution to proceed towards protometabolic systems with the capacity to self-sustain and replicate themselves. Consequently, herein we assume the hypothesis, suggested by Ribó, Hochberg et al.,200 that the emergence of homochirality would have run in parallel to the establishment of networks and assemblies with those catalytic, self-sustaining and replication features.
The problem is that, leaving aside crystallization and supramolecular polymerization processes, the number of autocatalytic covalent reactions that behave according to Frank-type models is very limited. The asymmetric autocatalytic dialkylzinc alkylation of certain aromatic aldehydes, known as the Soai reaction (Fig. 5C), is actually the only autocatalytic transformation that fulfils the above criteria, and it has no prebiotic relevance.201,202 Strong efforts have been made to find examples that are meaningful in a prebiotic context, mostly among reactions that present a sigmoidal kinetic behaviour,203 albeit with no real success. The Breslow's cycle, for instance, produces glycoaldehyde autocatalytically in the formose reaction, but this substrate is achiral and so it cannot lead to chiral amplification (see Scheme 4A),68 while only modest enantioselectivities in glyceraldehyde are obtained when the formose reaction is run in presence of chiral amino acids as asymmetric catalysts.58,204,205 The synthesis of amino acids by hydration of aminonitriles reported by Commeyras and coworkers, on the other hand, is not truly autocatalytic but a product-autoinductive process, since the catalyst is a carbonyl compound (acetaldehyde or acetone) formed by degradation of the starting aminonitrile.206 Thus, given that the catalyst total concentration is limited by the amount of starting material in the reaction, it cannot increase exponentially with the product – so the asymptotic emergence of homochirality predicted by Frank-type models is not feasible.
In order to overcome this elusive search for prebiotic reactions that present autocatalysis/replication with chiral amplification, as suggested by Blackmond,60 the key may lie in the establishment of self-organized connected catalytic cycles,207i.e., in the type of protometabolic networks reviewed above. Indeed, there are various experimental findings of aldehydes catalyzing reactions from substrates that involve amines (e.g., the hydrolysis of HCN oligomers and aminonitriles in Strecker-type syntheses)107 or amines (mostly from aminoacids) catalyzing reactions with aldehydes as substrates (e.g., proline-catalyzed Mannich reactions),208 in which significant enantiomeric excesses are obtained (e.g., when asymmetric amino acids catalyse the formose reaction).13,22,23 All this indicates that amines and aldehydes can act as both substrates and catalysts, and so they could be coupled in potentially autocatalytic cycles and networks through the formation of hemiaminal intermediates (Fig. 5D).60 Introducing asymmetry into these systems may allow Frank-type behaviours for enantiomeric enrichment throughout subsequent turnover of the cycles.
The previous scenario may have provided building blocks with moderate but sufficient ee (enantiomeric excess) values for the onset of informational polymers capable to establish template replication mechanisms, somehow initiating a rudimentary version of Darwinian evolution.36 Ribó et al., however, have raised objections to the assumption that the assembly of informational polymer replicators would have required solving the problem of homochirality at a previous stage, arguing that such a singular event scenario lacks the mutualistic and competitive character of evolution, even at its chemical stage.200 How to prevent for example the problem of racemization, necessarily significant when long geological timescales are considered? In contrast to that, they propose a gradual model for chiral enrichment in life-like systems, which runs parallel to chemical evolution, from the simplest molecular building blocks of life to the complexity of protocellular systems (Fig. 6). According to such a scheme, SMSB would have occurred at the stage of formation of condensation polymers, for which they could facilitate an ever more efficient chiral amplification mechanism at different evolutionary phases. For instance, Viedma deracemization209 should have been possible during the initial random polymerization events.210 The onset of template replication could have enhanced the amplification of chirality through the establishment of mutualistic networks (see previous section).211 Finally, fully homochiral systems would have been the result of an engagement of replicators in a sort of instructed metabolism,212 thanks to the emergence of new catalytic functions enabling different channeling pathways.
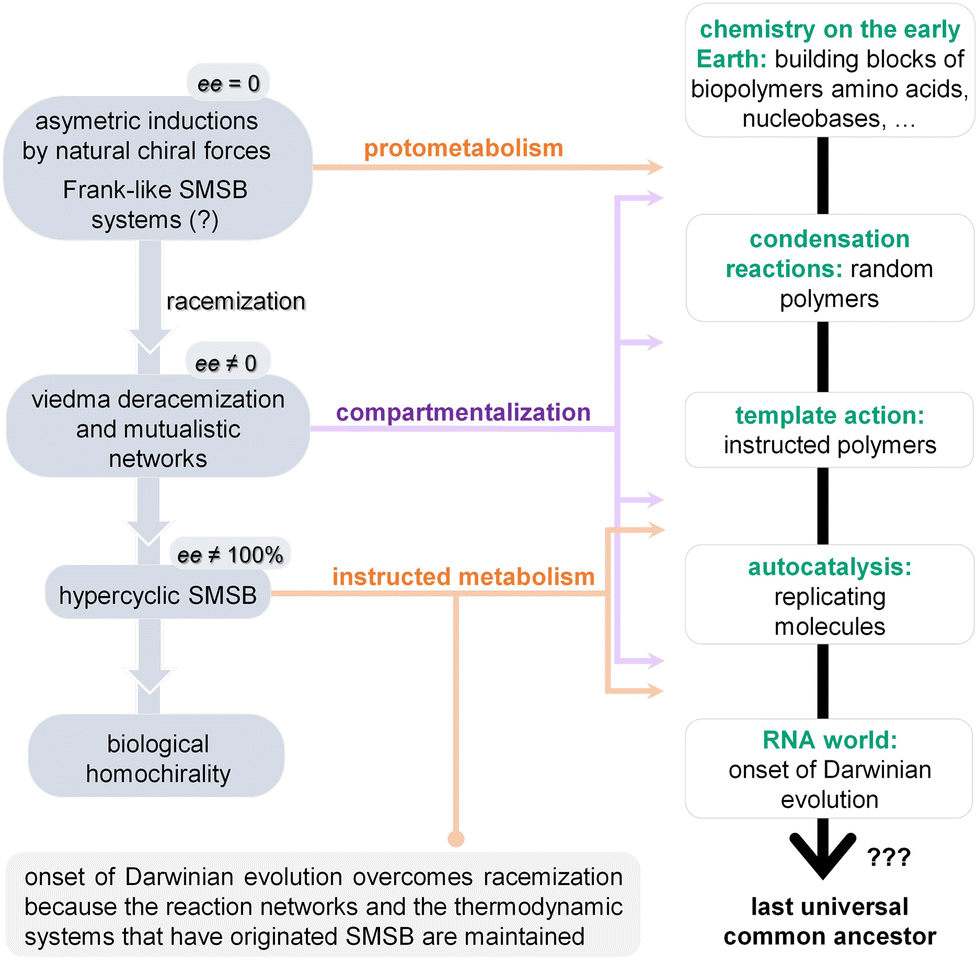 | ||
| Fig. 6 Scheme representing the parallel coevolution of protobiological systems from their simplest molecular components and the chirality problem, with a progressive increase in the enantiomeric excess of molecules within those systems, as it can be amplified through various mechanisms at the different stages of chemical evolution. Adapted with permission from ref. 200. Copyright 2017 Royal Society of Chemistry. | ||
Within this scenario, Frank-like mechanisms would have operated at early-stages of chemical evolution, amplifying small ee values from asymmetric inductions over simple reactions213e.g., when monomers get integrated into functional polymers. This view is clearly a ‘systems’ one, in the sense that it considers the problem of the emergence of homochirality intimately linked to the mechanisms by which the first minimal metabolisms could get assembled from self-sustaining networks of simple replicators and protocellular structures. Considering the importance of compartmentalization for the functioning of protometabolic networks,214–219 in general, and for chiral enrichment itself,214 next we summarize how all these aspects could be integrated into protocellular assemblies.
4.4. Compartmentalized chemistries: coupling reaction networks with protocell compartments
Although it is in principle an open research question whether ‘minimal metabolisms’ could be implemented as reaction networks in open and homogeneous (well-mixed) aqueous solution, compartmentalization events, which could be triggered through direct phase separation (e.g., coacervation) or supramolecular self-assembly of amphiphilic molecules (e.g., vesicle or micelle formation) seem quite important, since they involve spatial constraints and mechanisms that potentially control the diffusion/transport of molecules, in and out of the system, leading to individual protocellular organizations (for a definition of protocell, see the glossary at the end of the review).217 All known metabolisms, in fact, take place under cytoplasmic conditions, which are very complex environments, spatially speaking: apart from condensates and lipid membranes, ‘molecular crowding’ effects have been demonstrated as very relevant for many aspects of biochemistry and cell physiology.218,219 Furthermore, the energetic viability of any extant metabolism depends both on soluble ‘molecular currencies’ (like ATP or GTP) and ‘chemiosmotic’ ones (electrochemical gradients of H+ or Na+), so the vectorial character of metabolic processes220,221 could be a critical aspect to consider also in their first emergence.222,223The scenario that we are putting forward in this review as the most tenable prebiotic setting provides ample room for the presence of soft interfaces and heterogeneity in the reaction medium, with changes in the temperature, ionic strength, pH, etc., as natural driving forces already operating during the first steps of the origins-of-life process, so this is perfectly coherent with an early development of protocellular systems. One should be aware that this added initial complexity can nevertheless be a source of difficulties, not only for the researcher (the analysis of samples in messy or colloidal conditions is typically harder) but also for the actual chemistry to be explored. Many chemical reactions that proceed without any problem in water solution tend to be perturbed – or even totally inhibited – if the aqueous medium loses homogeneity.35 For instance, oscillatory behavior is marginal in real metabolisms (except for rare cases, like glycolytic oscillations) probably due to the heterogeneity of the cytoplasmic environment.224 Furthermore, when some chemistry is encapsulated by lipid membranes (e.g., within vesicles), the access of fresh reactants into the internal micro-environment becomes an issue, so diverse transformation processes could be suffocated simply by scarcity of nutrients – unless the bilayer is permeable enough.225 However, at the same time, chemistries that look dull or would hardly run in homogeneous water solution, may become much richer and dynamic in a heterogenous context, especially if they couple with self-assembly processes, or if they are part of a volume-changing compartment.226 The key will probably be, as usual, finding a suitable combination of reactive components and boundary conditions that opens an unexpected window of possibilities, in terms of complex dynamic behavior. If the phenomenon that emerges from there contains aspects of biological significance, the search should continue on similar lines.
A number of authors within the so-called ‘protocell camp’ have tried to overcome the nuances of heterogeneous media and pushed in this direction in the last decades. The motivation behind has been to deal with the problem of individuation and encapsulation at a stage when it is hard, but still tractable.227,228 In this context, the advent of ‘liposome technologies’, after the pioneer work of Bangham and colleagues,229 was instrumental to move beyond the first approaches to prebiotic compartments, based on Oparin's ‘coacervate’ hypothesis,230 and to start implementing more elaborate ‘vesicle models’ in which the properties of simplified bio-membranes (e.g., fatty acid bilayers) and their potential prebiotic role could be more directly addressed.231 Nevertheless, recent discoveries highlighting the importance of liquid–liquid phase separation and biomolecular condensates for various cell functions, have revived the coacervate idea, as perhaps a simpler (two-phase) compartmentalized system of relevance for the origins of life.232 Most of the recent literature on these different experimental protocell models (vesicle and coacervate systems, including tentative ‘hybrids’233) has been extensively reviewed in specialized articles.22,234,235 A large part of that work, though, has focused on demonstrating that relatively complex biomolecules (polymers, typically) and biochemical processes of different kinds can operate in vitro, under those simplified compartmentalized conditions.
The lines of protocell research that best combine with a metabolic/organizational way of approaching the initial stages of the process of origins of life are those that concentrate on how relatively simple chemistries could couple with compartment self-assembly processes. Supramolecular structures in biological systems are much more dynamic than the quasi-equilibrium aggregates that they constitute under standard laboratory conditions (where they tend to lack, precisely, the coupling with an underlying chemistry that would take them away from equilibrium, inducing a much more dynamic behavior – like growth and potential reproduction). Yet, the investigations started by Luisi's group at the end of last century236,237 established a tradition in the field that tackles this issue upfront (a tradition that was later followed – in part, at least – by Szostak and coworkers238 and, more recently, by groups like Devaraj's239 or Fletcher's240,241). In particular, Fletcher's lab has recently described a replication cycle that works autonomously and leads to an oscillatory behaviour, playing precisely with the following four ingredients: (i) an autocatalytic reaction that leads to a synthetic lipid amphiphile (29) from the disulfide exchange between a reactive hydrophilic disulfide (30) and a lipophilic thiol (31); (ii) the self-assembly of this amphiphile into transient micelles that catalyze the amphiphile synthesis; (iii) the micelles decay due to degradation of the amphiphile, and (iv) the regeneration of disulfide 30 with an oxidant chemical fuel (H2O2) (Fig. 7).242 Interestingly, the reaction occurs in two phases, with rapid stirring, which is crucial to adjust the kinetics of both the autocatalytic micelles formation and their destruction. The latter process is actually slowed down due to phase separation, as the lipidic thiol 31 that depletes disulfide 29 constitutes the organic phase and is not easily mixed with the micelles and compounds that are dispersed in the aqueous medium. Therefore, this work represents an excellent example of how investigations from the bottom-up can lead to processes of complexification, illustrating one of our main themes in this review. The implementation of various couplings within the same experimental setting leads to a chemical system in which a number of key biologically-relevant features are successfully combined: replication (variability control), adjustment by stirring and self-assembly/phase separation (spatial control) of the reaction rates of the different steps in the replication cycle (kinetic control), and the fueling with hydrogen peroxide to regenerate the reactive disulfide substrate (energetic control), all contributing to the maintenance of the system out of equilibrium.
 | ||
| Fig. 7 Schematic representation of the oscillatory self-replication cycle in which an amphiphilic disulfide replicator (29) is built from the lipophilic thiol 31 and the hydrophilic disulfide 30. The amphiphile forms micelles the uptake thiol and accelerate their own formation, leading to a replication process. The rate of decay of the micelles by reaction with the lipophilic thiol (in its thiolate form) is in principle avoided through phase separation, but it can be adjusted by rapid stirring. The initial hydrophilic disulfide is then regenerated from 32 using hydrogen peroxide as fuel, which allows maintaining the system out of equilibrium. Adapted with permission from ref. 242. Copyright 2022 Nature Publishing Group. | ||
Let us finish this section (and, somehow, prepare the terrain for the conclusions) by noting that the advantage of coupling reaction networks and compartments in synergetic ways (like it is achieved in the works above) is not simply the triggering of growth dynamics and reproduction events that involve the whole system, but the actual constitution of a ‘self’: i.e., an ‘individual’ which is different from its environment and can build its own components, including the components of its boundary. Indeed, this transition from ‘self-assembly’ (of some molecular building blocks into a supramolecular structure) to ‘self-production’ (of a system that performs the synthesis of its own components, establishes a clear ‘in/out’ asymmetry and has potential for growth and reproduction as such a system) is an excellent opportunity to consider whether the ‘network complexity’ that we usually associate with the idea of metabolism (when we have in mind extant living organisms) could be significantly reduced at prebiotic stages, by focusing on much simpler reaction networks, but provided that the latter are closely linked to the constitution and dynamic properties of the system's boundary. Anyway, the complete implementation of ‘proto-organisms’ (understood as ‘basic autonomous systems’),12,24 may still require additional functionalities (in particular, endogenously synthesized energy control mechanisms – like the ones being explored by Mansy's group243 – more tightly integrated with the spatial and kinetic ones). Therefore, it calls for further research, ideally making use of prebiotically plausible components.
5. Summary and conclusions
In this tutorial review, we have carried out an in-depth analysis of the main pillars of prebiotic chemistry, demonstrating the multiple interconnections that exist among them and the great possibilities that these provide to establish a solid and highly promising research program towards the bottom-up reconstruction of metabolic systems, taken as a central part of the origins-of-life problem. The theoretical framework we suggest for that challenging task distinguishes between ‘proto-metabolisms’ (initial sets of coupled physical–chemical processes and non-equilibrium boundary conditions leading to biologically relevant synthetic pathways) and ‘minimal metabolisms’ (more complicated chemical systems that synthesize their own functional components and put together a spatial, temporal and energetically viable organization). This key conceptual distinction is aimed to guide the strongly-needed empirical exploration of control mechanisms that, being an endogenous product of the reaction system, should tame the combinatorial explosion that would otherwise happen in such a complex, compositionally diverse and heterogeneous context. Those control mechanisms would also allow the functional integration of the involved molecular ingredients and transformation processes, which should bring forward a collection of ‘self-sustaining’ (and potentially ‘self-reproducing’) systems, and with them the amplification of small chiral imbalances into homochiral systems.Towards that end, the construction of a complete protometabolic map of prebiotic reactions will facilitate the task of identifying and classifying the different feedback and control mechanisms that could come up from subsets of interconnected reaction pathways. Establishing a protometabolic network out of these highly engaged chemistries would require, however, that some key reactions could be run in both the forward and reverse directions, so that the exploration of the available chemical space leads to amplification of the most favorable pathways. In this context, energetic funneling, enabled by the establishment of interlinked catalytic cycles under dissipative conditions, must have brought forward a transition to systems and networks with increasing dynamic kinetic stability. It is likely that this type of non-enzymatic reaction networks provided, from a set of central protometabolic cycles, an environment from which polymer replicators and catalysts could thrive. Homochirality stands out as another probable consequence of the emergence of this type of polymeric systems. Finally, auto- and cross-catalytic loops in those networks, together with the self-assembly of non-equilibrium supramolecular structures from some of the network components, would open evolutionary possibilities towards protocellular assemblies with increasingly higher robustness.
The difficulties of this research program are of course many, not just finding the right chemical precursors and the conditions under which those phenomena may emerge, but also developing the adequate analytical tools to perform a precise and scientifically sound characterization of the systems implemented and their complex dynamics. In this regard, the design of experimental setups in which non-equilibrium chemistries can be thoroughly explored, both in aqueous and more heterogeneous (soft, interface-rich) reaction environments, including time-resolved analyses of specific features like the chirality of the species involved or their catalytic effects, looks rather critical. In addition, theoretical modelling and computer simulations should complement empirical research (each giving continuous and mutual input/feedback to the other – as we commented in the introduction), but the territory to investigate is so foreign and arduous that experiments should lead the way (among other things, because we lack knowledge about potential ‘first principles’ that could be operating in non-equilibrium and non-homogenous conditions).
Finally, it is very important to highlight, before concluding, that we were concentrating here only on the early stages of the problem. In other words, the origins-of-life question would not be solved even if we managed to implement in the lab genuinely autonomous protocell populations (i.e., in the terminology we used above, families of ‘minimal metabolisms’). The road from that stage to the first living organisms (i.e., cells with a ‘genetically-instructed’ metabolism) could be long and winding, with very narrow bottlenecks that only natural selection and evolutionary dynamics taking place at much larger timescales would allow to overcome. We are quite convinced that the minimal ‘complexity threshold’ for life involves systems roughly similar to LUCA (or to the top-down constructs that some research groups have been able to obtain in the lab),244 which would thus require the bottom-up development of a genetic code and all the translation apparatus (a huge problem that we did not cover, at all, in this contribution). Nevertheless, our basic work assumption on these lines is that genetic mechanisms would lack any reason for existence without a metabolic context that provided not only the material and energy resources for their highly demanding synthesis and maintenance, but also the framework within which molecular sequences may have some relevant biological meaning. Therefore, in line with some hypotheses already put forward in the literature,245,246 we consider that the transition towards cells based on DNA and proteins, linked through a translation code, was relatively late in the context of abiogenesis and should be conceived as a ‘co-evolution’ process between the synthetic pathways of minimal metabolisms and the establishment of reliable hereditary mechanisms. In any case, we believe that the research approach to the problem proposed herein, which is significantly different from previously considered ones, will shed light on novel links between top-down and bottom-up strategies that study the interfaces between chemistry and biology, and will definitely span various subareas including not only origins-of-life research but also systems chemistry and synthetic biology.
Glossary
Dissipative networks/assemblies – Systems that consume energy from their environment to maintain their internal organization in a dynamic steady state out of equilibrium.Control – (Including feedback mechanisms but also other dynamic/structural constraints) – that part of the molecular/material composition or internal organization of a chemical network that brings forward/maintains a system property or steady state associated with it, despite eventual perturbations.
Energy-dissipating cycles – Cyclic chemical processes that consume energy to be driven in the thermodynamically unfavourable direction.
Chemical fuel – Energy-rich compound whose exergonic degradation can be coupled to run thermodynamically unfavourable transformations and self-assembly processes.
Autocatalytic cycles – Set of reactions and intermediates that form a cycle, in such a way that when the cycle operates over the substrates at the required stoichiometric ratios the amount of at least one of the intermediates increases over time.
Protocell – A self-assembled compartment (typically a supramolecular structure, like a lipid vesicle) linked to chemical processes taking place around or within it, aimed at explaining how more complex biological cells or alternative forms of cellular organization may come about.
Protometabolism – A set of reaction and transformation processes that need to couple in order to maintain themselves in non-equilibrium conditions.
Minimal metabolism – A set of physical–chemical transformation processes that synthesize diverse material constraints (molecular mechanisms) that control their behaviour and ensure their dynamic robustness and adaptability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Spanish Agencia Estatal de Investigación (PID2020-119306GB-100 (AdlE); PID2019-104576GB-100 (KRM)) and the European Union (CLASSY project, Horizon 2020 research and innovation programme, Grant Agreement No. 862081 (AdlE); CORENET project, supported by the Swiss State Secretariat for Education, Research and Innovation (SERI), Grant Agreement No. 101046294 (AdlE); and ProtoMet project, Grant Agreement No. 813873 (KRM). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Innovation Council and SME’s Executive Agency (EISMEA). Neither the European Union nor the granting authority can be held responsible for them. KRM also acknowledges support from the Basque Government (IT1668-22) and the John Templeton Foundation (grant 62220).References
- F. J. Dyson, Origins of Life, Cambridge University Press, Cambridge, 1985 Search PubMed.
- C. De Duve, Blueprint for a Cell: The Nature and Origin of Life, Neil Patterson Publishers, Burlington, North Carolina, 1991 Search PubMed.
- S. Kauffman, Investigations, Oxford University Press, Oxford, 2000 Search PubMed.
- F. M. Harold, The Way of the Cell: Molecules, Organisms and the Order of Life, Oxford University Press, Oxford, 2001 Search PubMed.
- P. L. Luisi, The Emergence of Life, Cambridge University Press, Cambridge, 2006 Search PubMed.
- L. Cronin and S. I. Walker, Science, 2016, 352, 1174–1175 CrossRef CAS PubMed.
- R. Krishnamurthy and N. V. Hud, Chem. Rev., 2020, 120, 4613–4615 CrossRef CAS PubMed.
- S. Otto, Acc. Chem. Res., 2021, 55, 145–155 CrossRef PubMed.
- D. Deamer, Microbiol. Mol. Biol. Rev., 1997, 61, 239–261 CAS.
- J. W. Szostak, Philos. Trans. R. Soc., B, 2011, 366, 2894–2901 CrossRef CAS PubMed.
- J. W. Szostak, Angew. Chem., Int. Ed., 2017, 56, 11037–11043 CrossRef CAS PubMed.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Open Biol., 2017, 7, 170050 CrossRef PubMed.
- D. Kroiss, G. Ashkenasy, A. B. Braunschweig, T. Tuttle and R. V. Ulijn, Chem, 2019, 5, 1917–1920 CAS.
- S. Islam and M. W. Powner, Chem, 2017, 2, 470–501 CAS.
- S. L. Miller, Science, 1953, 117, 528–529 CrossRef CAS PubMed.
- T. M. McCollom, G. Ritter and B. R. T. Simoneit, Origins Life Evol. Biospheres, 1999, 29, 153–166 CrossRef CAS PubMed.
- S. A. Benner, H. J. Kim and M. A. Carrigan, Acc. Chem. Res., 2012, 45, 2025–2034 CrossRef CAS PubMed.
- J. Oro and A. P. Kimball, Arch. Biochem. Biophys., 1962, 96, 293–313 CrossRef CAS PubMed.
- R. A. Sanchez, J. P. Ferbis and L. E. Orgel, J. Mol. Biol., 1967, 30, 223–253 CAS.
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma and J. Moran, Nature, 2019, 569, 104–107 CrossRef CAS PubMed.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 CrossRef CAS PubMed.
- Y. Bai, A. Chotera, O. Taran, C. Liang, G. Ashkenasy and D. G. Lynn, Chem. Soc. Rev., 2018, 47, 5444–5456 RSC.
- A. de la Escosura, Life, 2019, 9, 66 CrossRef CAS PubMed.
- N. Lauber, C. Flamm and K. Ruiz-Mirazo, BioEssays, 2021, 43, e2100103 CrossRef PubMed.
- T. Ganti, BioSystems, 1975, 7, 15–21 CrossRef CAS PubMed.
- H. Maturana and F. J. Varela, Autopoiesis and Cognition: The Realization of the Living, Reidel Publishing Company, Dordrecht, 1980 Search PubMed.
- S. A. Kauffman, J. Theor. Biol., 1986, 119, 1–24 CrossRef CAS PubMed.
- R. Rosen, Life Itself: A Comprehensive Inquiry into the Nature, Origin and Fabrication of Life, Columbia University Press, New York, 1991 Search PubMed.
- W. Fontana and L. W. Buss, Bull. Math. Biol., 1994, 56, 1–64 Search PubMed.
- H. J. Morowitz, J. D. Kostelnik, J. Yang and G. D. Cody, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7704–7708 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma and J. Moran, Chem. Rev., 2020, 120, 7708–7744 CrossRef CAS PubMed.
- J. Pereto, Chem. Soc. Rev., 2012, 41, 5394–5403 RSC.
- S. A. Harrison and N. Lane, Nat. Commun., 2018, 9, 5176 CrossRef CAS PubMed.
- S. Serrano-Luginbühl, K. Ruiz-Mirazo, R. Ostaszewski, F. Gallou and P. Walde, Nat. Rev. Chem., 2018, 2, 306–327 CrossRef.
- M. Wu, S. I. Walker and P. G. Higgs, Astrobiology, 2012, 12, 818–829 CrossRef CAS PubMed.
- P. V. Coveney, J. B. Swadling, J. A. D. Wattis and H. C. Greenwell, Chem. Soc. Rev., 2012, 41, 5430–5446 RSC.
- F. Mavelli and K. Ruiz-Mirazo, Phys. Biol., 2010, 7, 036002 CrossRef PubMed.
- J. L. Andersen, C. Flamm, D. Merkle and P. F. A. Stadler, A Software Package for Chemically Inspired Graph Transformation, Springer International Publishing, Austria, 2016 Search PubMed.
- A. Wolos, R. Roszak, A. Zadlo-Dobrowolska, W. Beker, B. Mikulak-Klucznik, G. Spolnik, M. Dygas, S. Szymkuc and B. Grzybowski, Science, 2020, 369, eaaw1955 CrossRef CAS PubMed.
- S. M. Marshall, C. Mathis, E. Carrick, G. Keenan, G. J. T. Cooper, H. Graham, M. Craven, P. S. Gromski, D. G. Moore, S. I. Walker and L. Cronin, Nat. Commun., 2021, 12, 3033 CrossRef CAS PubMed.
- A. W. Schwartz, Philos. Trans. R. Soc., B, 2006, 361, 1743 CrossRef CAS PubMed.
- J. D. Toner and D. C. Catling, Proc. Natl. Acad. Sci. U. S. A., 2020, 17, 883–888 CrossRef PubMed.
- M. Ruiz-Bermejo, J. L. de la Fuente, C. Rogero, C. Menor-Salvan, S. Osuna-Esteban and J. A. Martin-Gago, Chem. Biodiversity, 2012, 9, 25–40 CrossRef CAS PubMed.
- S. Pizzarello, Acc. Chem. Res., 2006, 39, 231–237 CrossRef CAS PubMed.
- M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed.
- S. Islam, D. K. Bucar and M. W. Powner, Nat. Chem., 2017, 9, 584–589 CrossRef CAS.
- R. Saladino, E. Di Mauro and J. M. Garcia-Ruiz, Chem. – Eur. J., 2019, 25, 3181–3189 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma, E. Chevallot-Beroux, L. Lethuillier-Karl, G. Li and J. Moran, Nat. Ecol. Evol., 2017, 1, 1716–1721 CrossRef PubMed.
- G. Springsteen, J. R. Yerabolu, J. Nelson, C. J. Rhea and R. Krishnamurthy, Nat. Commun., 2018, 9, 91 CrossRef PubMed.
- R. J. Mayer, H. Kaur, S. A. Rauscher and J. Moran, J. Am. Chem. Soc., 2021, 143, 19099–19111 CrossRef CAS PubMed.
- V. Vijayasarathy and H. J. Morowitz, Biol. Bull., 2009, 217, 232 Search PubMed.
- R. Pascal, A. Pross and J. D. Sutherland, Open Biol., 2013, 3, 130156 CrossRef PubMed.
- H. W. H. van Roekel, B. J. H. M. Rosier, L. H. H. Meijer, P. A. J. Hilbers, A. J. Markvoort, W. T. S. Huck and T. F. A. de Greef, Chem. Soc. Rev., 2015, 44, 7465–7483 RSC.
- D. Doran, Y. M. Abul-Haija and L. Cronin, Angew. Chem., Int. Ed., 2020, 58, 11253–11256 CrossRef PubMed.
- M. Avalos, R. Babiano, P. Cintas, J. L. Jiménez, J. C. Palacios and L. D. Barron, Chem. Rev., 1998, 98, 2391–2404 CrossRef CAS PubMed.
- B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef PubMed.
- J. E. Hein and D. G. Blackmond, Acc. Chem. Res., 2012, 45, 2045–2054 CrossRef CAS PubMed.
- T. Buhse, J. M. Cruz, M. E. Noble-Terán, D. Hochberg, J. M. Ribo, J. Crusats and J. C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed.
- D. G. Blackmond, Chem. Rev., 2020, 120, 4831–4847 CrossRef CAS PubMed.
- I. Cleaves and H. James, Precambrian Res., 2008, 164, 111–118 CrossRef.
- A. Butlerow and J. Liebigs, Ann. Chem., 1861, 120, 295–298 CrossRef.
- G. Schlesinger and S. L. Miller, J. Mol. Evol., 1983, 19, 383–390 CrossRef CAS PubMed.
- D. Delidovich, A. N. Simonov, O. P. Taran and V. N. Parmon, ChemSusChem, 2014, 7, 1833–1846 CrossRef PubMed.
- H. J. Kim and S. A. Benner, Science, 2010, 329, 902 CrossRef CAS PubMed.
- H. J. Kim, A. Ricardo, H. I. Illankoong, M. J. Kim, M. A. Carrigan, F. Frye and S. A. Benner, J. Am. Chem. Soc., 2011, 133, 9457–9468 CrossRef CAS PubMed.
- M. Haas, S. Lamour, S. B. Christ and O. Trapp, Chem. Commun., 2020, 3, 140 CrossRef CAS PubMed.
- R. Breslow, Tetrahedron Lett., 1959, 1, 22–26 CrossRef.
- A. N. Simonov, O. P. Pestunova, L. G. Matvienko and V. N. Parmon, Kinet. Catal., 2007, 48, 261–270 Search PubMed.
- C. Appayee and R. Breslow, J. Am. Chem. Soc., 2014, 136, 3720–3723 CrossRef CAS PubMed.
- S. Colon-Santos, G. J. T. Cooper and L. Cronin, ChemSystChem, 2019, 1, e1900014 CrossRef CAS.
- W. E. Robinson, E. Daines, P. van Duppen, T. de Jong and W. T. S. Huck, Nat. Chem., 2022, 14, 623–631 CrossRef CAS PubMed.
- A. Berg, D. Kockelkorn, W. H. Ramos-Vera, R. F. Say, J. Zarzycki, M. Hugler, B. E. Alber and G. Fuchs, Nat. Rev. Microbiol., 2010, 8, 447–460 CrossRef PubMed.
- R. F. Say and G. Fuchs, Nature, 2010, 464, 1077–1081 CrossRef CAS PubMed.
- C. B. Messner, P. C. Driscoll, G. Piedrafita, M. F. L. de Volder and M. Ralser, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7403–7407 CrossRef CAS PubMed.
- H. Cleaves, Evol.: Educ. Outreach, 2012, 5, 342–360 Search PubMed.
- J. Oro and A. P. Kimball, Arch. Biochem. Biophys., 1962, 96, 293–313 CrossRef CAS PubMed.
- M. Yadav, R. Kumar and R. Krishnamurthy, Chem. Rev., 2020, 120, 4766–4805 CrossRef CAS PubMed.
- A. Fernandez, M. Ruiz-Bermejo and J. L. de la Fuente, Phys. Chem. Chem. Phys., 2018, 20, 17353–17366 RSC.
- M. Ruiz-Bermejo, J. L. de la Fuente, C. Perez-Fernandez and E. Mateo-Marti, Processes, 2021, 9, 597 CrossRef CAS.
- S. Hudson, J. F. Eberle, R. H. Vachhani, L. C. Rogers, J. H. Wade, R. Krishnamurthy and G. A. Springsteen, Angew. Chem., Int. Ed., 2012, 51, 5134–5137 CrossRef PubMed.
- E. Boulanger, A. Anoop, D. Nachtigallova, W. Thiel and M. Barbatti, Photochemical Steps in the Prebiotic Synthesis of Purine Precursors from HCN, Angew. Chem., Int. Ed., 2013, 52, 8000–8003 CrossRef CAS PubMed.
- C. Grundke, C. Kong, C. J. Kampf, F. Gupton, D. T. McQuade and T. Opatz, J. Org. Chem., 2021, 86, 10320–10329 CrossRef CAS PubMed.
- R. Saladino, G. Botta, S. Pino, G. Costanzo and E. Di Mauro, Chem. Soc. Rev., 2012, 41, 5526–5565 RSC.
- J. L. Andersen, T. Andersen, C. Flamm, M. M. Hanczyc, D. Merkle and P. F. Stadler, Entropy, 2013, 15, 4066–4083 CrossRef CAS.
- F. Pietrucci and A. M. Saitta, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15030–15035 CrossRef CAS PubMed.
- R. Saladino, C. Crestini, S. Pino, G. Costanzo and E. Di Mauro, Phys. Life Rev., 2012, 9, 84–104 CrossRef PubMed.
- D. Niether, D. Afanasenkau, J. K. G. Dhont and S. Wiegand, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4272–4277 CrossRef CAS PubMed.
- R. Saladino, E. Di Mauro and J. M. Garcia-Ruiz, Chem. – Eur. J., 2019, 25, 3181–3189 CrossRef CAS PubMed.
- B. M. Bizzarri, R. Saladino, I. Delfino, J. M. Garcia-Ruiz and E. Di Mauro, Int. J. Mol. Sci., 2021, 22, 917 CrossRef PubMed.
- B. Wang and Z. Cao, J. Phys. Chem. A, 2010, 114, 12918–12927 CrossRef CAS PubMed.
- V. Enchev and S. Slavova, Phys. Chem. Chem. Phys., 2021, 23, 19043–19053 RSC.
- L. E. Orgel, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 99–123 CrossRef CAS PubMed.
- A. Eschenmoser and E. Loewenthal, Chem. Soc. Rev., 1992, 21, 1–16 RSC.
- G. Schlesinger and S. L. Miller, J. Am. Chem. Soc., 1973, 95, 3729–3735 CrossRef CAS.
- T. Arrhenius, G. Arrhenius and W. Paplawsky, Origins Life Evol. Biospheres, 1994, 24, 1–17 CrossRef CAS PubMed.
- S. Islam, D. K. Bucar and M. W. Powner, Nat. Chem., 2017, 9, 584–589 CrossRef CAS.
- D. D. Sasselov, J. P. Grotzinger and J. D. Sutherland, Sci. Adv., 2020, 6, eaax3419 CrossRef CAS PubMed.
- D. J. Ritson, C. Battilocchio, S. V. Ly and J. D. Sutherland, Nat. Commun., 2018, 9, 1821 CrossRef PubMed.
- D. J. Ritson and J. D. Sutherland, Angew. Chem., Int. Ed., 2013, 52, 5845–5847 CrossRef CAS PubMed.
- J. Xu, D. J. Ritson, S. Ranjan, Z. R. Todd, D. D. Sasselov and J. D. Sutherland, Chem. Commun., 2018, 54, 5566–5569 RSC.
- C. Bonfio, C. Caumes, C. D. Duffy, B. H. Patel, C. Percivalle, M. Tsanakopoulou and J. D. Sutherland, J. Am. Chem. Soc., 2019, 141, 3934–3939 CrossRef CAS PubMed.
- J. Xu, V. Chmela, N. J. Green, D. A. Russel, M. J. Janicki, R. W. Gora, R. Szabla, A. D. Bond and J. D. Sutherland, Nature, 2020, 582, 60–66 CrossRef CAS PubMed.
- J. Xu, N. J. Green, C. Gibard, R. Krishnamurthy and J. D. Sutherland, Nat. Chem., 2019, 11, 457–462 CrossRef CAS PubMed.
- J. Xu, N. J. Green, D. A. Russel, Z. W. Liu and J. D. Sutherland, J. Am. Chem. Soc., 2021, 143, 14482–14486 CrossRef CAS PubMed.
- P. Canavelli, S. Islam and M. W. Powner, Nature, 2019, 571, 546–549 CrossRef CAS PubMed.
- A. Eschemoser, Chem. Biodiversity, 2007, 4, 554–573 CrossRef PubMed.
- K. Koch, W. B. Schweizer and A. Eschenmoser, Chem. Biodiversity, 2007, 4, 541–553 CrossRef CAS PubMed.
- A. Eschenmoser, Tetrahedron, 2007, 63, 12821–13040 CrossRef CAS.
- R. T. Stubbs, M. Yadav, R. Krishnamurthy and G. Springsteen, Nat. Chem., 2020, 12, 1016–1022 CrossRef CAS PubMed.
- G. Danger, R. Plasson and R. Pascal, Chem. Soc. Rev., 2012, 41, 5416–5429 RSC.
- E. Camprubi, S. F. Jordan, R. Vasiliadov and N. Lane, IUBMB Life, 2017, 69, 373–381 CrossRef CAS PubMed.
- R. J. Mayer and J. Moran, Angew. Chem., Int. Ed., 2022, 61, e202212237 CrossRef CAS PubMed.
- S. A. Rauscher and J. Moran, Angew. Chem., Int. Ed., 2022, 61, e202212932 CrossRef CAS PubMed.
- S. Pulletikurti, M. Yadav, G. Springsteen and R. Krishnamurthy, Nat. Chem., 2022, 14, 1142–1150 CrossRef CAS PubMed.
- V. N. Sagi, V. Punna, F. Hu, G. Meher and R. Krishnamurthy, J. Am. Chem. Soc., 2012, 134, 3577–3589 CrossRef CAS PubMed.
- C. Butch, E. D. Cope, P. Pollet, L. Gelbaum, R. Krishnamurthy and C. L. Liotta, J. Am. Chem. Soc., 2013, 135, 13440–13445 CrossRef CAS PubMed.
- M. Yadav, S. Pulletikurty, J. R. Yerabolu and R. Krishnamurthy, Nat. Chem., 2022, 14, 170–178 CrossRef CAS PubMed.
- M. Ralser, Biochem. J., 2017, 475, 2577–2592 CrossRef PubMed.
- A. Keller, A. Zylstra, C. Castro, A. V. Turchyn, J. L. Griffin and M. Ralser, Sci. Adv., 2016, 2, e1501235 CrossRef PubMed.
- A. J. Coggins and M. W. Powner, Nat. Chem., 2017, 9, 310–317 CrossRef CAS PubMed.
- A. F. Magalhaes and M. W. Powner, Chem. Commun., 2022, 58, 13519–13522 RSC.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- A. Walther, Adv. Mater., 2020, 32, 1905111 CrossRef CAS PubMed.
- B. Riess, R. K. Grotsch and J. Boekhoven, Chem, 2020, 6, 552–578 CAS.
- K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- A. Chatterjee, A. Reja, S. Pal and D. Das, Chem. Soc. Rev., 2022, 51, 3047–3070 RSC.
- A. Mariani and J. D. Sutherland, Angew. Chem., Int. Ed., 2017, 56, 6563–6566 CrossRef CAS PubMed.
- C. Wanzke, A. Jussupow, F. Kohler, H. Dietz, V. R. I. Kaila and J. Boekhoven, ChemSystemsChem, 2020, 2, e1900044 CrossRef CAS.
- E. Del Grosso, E. Franco, L. J. Prins and F. Ricci, Nat. Chem., 2022, 14, 600–613 CrossRef CAS PubMed.
- B. J. Cafferty, A. S. Y. Wong, S. N. Semenov, L. Belding, S. Gmur, W. T. S. Huck and G. M. Whitesides, J. Am. Chem. Soc., 2019, 141, 8289–8295 CrossRef CAS PubMed.
- M. Preiner and W. F. Martin, Nature, 2021, 592, 688–689 CrossRef CAS.
- R. Hudson, R. de Graaf, M. Strandoo-Rodin, A. Ohno, N. Lane, S. E. McGlynn, Y. M. A. Yamada, R. Nakamura, L. M. Barge, D. Braun and V. Sojo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22873–22879 CrossRef CAS PubMed.
- L. M. Barge, E. Flores, M. M. Baum, D. G. VanderVelde and M. J. Russell, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4828–4833 CrossRef CAS PubMed.
- M. Preiner, K. Igarashi, K. B. Muchowska, M. Yu, S. J. Varma, K. Kleinermanns, M. K. Nobu, Y. Kamagata, H. Tuysuz, J. Moran and W. F. Martin, Nat. Ecol. Evol., 2020, 4, 534–542 CrossRef PubMed.
- T. Beyazay, K. S. Belthle, C. Fares, M. Preiner, J. Moran, W. F. Martin and H. Tuysuz, Nat. Commun., 2023, 14, 570 CrossRef CAS PubMed.
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Rev. Microbiol., 2008, 6, 805–814 CAS.
- L. M. Barge, Y. Abedian, M. J. Russell, I. J. Doloboff, J. H. E. Cartwright, R. D. Kidd and I. Kanik, Angew. Chem., Int. Ed., 2015, 54, 8184–8187 CrossRef CAS PubMed.
- S. Pinna, C. Kunz, A. Halpern, S. A. Harrison, S. F. Jordan, J. Ward, F. Werner and N. Lane, PLoS Biol., 2022, 20, e3001437 CrossRef CAS PubMed.
- E. Altamura, F. Milano, R. R. Tangorra, M. Trotta, O. H. Omar, P. Stano and F. Mavelli, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3837–3842 CrossRef CAS PubMed.
- J. Crusats, D. Hochberg, A. Moyano and J. M. Ribó, ChemPhysChem, 2009, 10, 2123–2131 CrossRef CAS PubMed.
- B. J. Cafferty and N. V. Hud, Curr. Opin. Chem. Biol., 2014, 22, 146–157 CrossRef CAS PubMed.
- S. C. Kim, D. K. O’Flaherty, C. Giurgiu, L. J. Zhou and J. W. Szostak, J. Am. Chem. Soc., 2021, 143, 3267–3279 CrossRef CAS PubMed.
- J. F. Lambert, Origins Life Evol. Biospheres, 2008, 38, 211–242 CrossRef CAS PubMed.
- J. P. Ferris, Mol. Geomicrobiol., 2005, 59, 187–210 CAS.
- M. M. Hanczyc, S. M. Fujikawa and J. W. Szostak, Science, 2003, 302, 618–622 CrossRef CAS PubMed.
- V. Erastova, M. T. Degiacomi, D. G. Fraser and H. C. Greenwell, Nat. Commun., 2017, 8, 2033 CrossRef PubMed.
- P. C. Joshi, M. F. Aldersley, J. W. Delano and J. P. Ferris, J. Am. Chem. Soc., 2009, 131, 13369–13374 CrossRef CAS PubMed.
- D. Jovanovic, P. Tremmel, P. S. Pallan, M. Egli and C. Richert, Angew. Chem., Int. Ed., 2020, 59, 20154–20160 CrossRef CAS PubMed.
- S. J. Zhang, D. Duzdevich and J. W. Szostak, J. Am. Chem. Soc., 2020, 142, 14810–14813 CrossRef CAS PubMed.
- S. J. Roberts, Z. Liu and J. D. Sutherland, J. Am. Chem. Soc., 2022, 144, 4254–4259 CrossRef CAS PubMed.
- A. C. Fahrenbach, C. Giurgiu, C. P. Tam, L. Li, Y. Hongo and J. W. Szostak, J. Am. Chem. Soc., 2017, 139, 8780–8783 CrossRef CAS PubMed.
- P. Tremmel, H. Griesser, U. E. Steiner and C. Richert, Angew. Chem., Int. Ed., 2019, 58, 13087–13092 CrossRef CAS PubMed.
- M. Gleiser and S. M. Walker, Origins Life Evol. Biospheres, 2009, 39, 479–493 CrossRef CAS PubMed.
- G. Danger, R. Plasson and R. Pascal, Astrobiology, 2010, 10, 651–662 CrossRef CAS PubMed.
- P. C. Joshi, M. F. Aldersley and J. P. Ferris, Origins Life Evol. Biospheres, 2011, 41, 213–236 CrossRef CAS PubMed.
- J. E. Sponer, A. Mladek and J. Sponer, Phys. Chem. Chem. Phys., 2013, 15, 6235–6242 RSC.
- O. Doppleb, J. Bremer, M. Bechthold, C. Sanchez-Rico, D. Gohringer, H. Griesser and C. Richert, Chem. – Eur. J., 2021, 27, 13544–13551 CrossRef CAS PubMed.
- I. Weissbuch, R. A. Illos, G. Bolbach and M. Lahav, Acc. Chem. Res., 2009, 42, 1128–1140 CrossRef CAS PubMed.
- N. Wagner, B. Rubinov and G. Ashkenasy, ChemPhysChem, 2011, 12, 2771–2780 CrossRef CAS PubMed.
- S. J. Zhang, D. Duzdevich, D. Ding and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2116429119 CrossRef CAS PubMed.
- C. S. Foden, S. Islam, C. Fernandez-Garcia, L. Maugeri, T. D. Sheppard and M. W. Powner, Science, 2020, 370, 865–869 CrossRef CAS PubMed.
- M. Morasch, J. Liu, C. F. Dirscherl, A. Ianeselli, A. Kuhnlein, K. L. V. P. Schwintek, S. Islam, M. K. Corpinot, B. Scheu, D. B. Dingwell, P. Schwille, H. Mutschler, M. W. Powner, C. B. Mast and D. Braun, Nat. Chem., 2019, 11, 779–788 CrossRef CAS PubMed.
- A. Ianaselli, D. Tetiker, J. Stein, A. Kuhnlein, C. B. Mast, D. Braun and T. Y. D. Tang, Nat. Chem., 2022, 14, 32–39 CrossRef PubMed.
- A. Ianeselli, M. Atienza, P. W. Kudella, U. Gerland, C. B. Mast and D. Braun, Nat. Phys., 2022, 18, 579–585 Search PubMed.
- S. Arsene, S. Ameta, N. Lehman, A. D. Griffiths and P. Nghe, Nucleic Acids Res., 2018, 46, 9660–9666 CrossRef CAS PubMed.
- X. Miao, A. Paikar, B. Lerner, Y. Diskin-Posner, G. Shmul and S. N. Semenov, Angew. Chem., Int. Ed., 2021, 60, 20366–20375 CrossRef CAS PubMed.
- A. K. Bandela, N. Wagner, H. Sadihov, S. Morales-Reina, A. Chotera-Ouda, K. Basu, R. Cohen-Luria, A. de la Escosura and G. Ashkenasy, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2015285118 CrossRef CAS PubMed.
- B. Liu, J. Wu, M. Geerts, O. Markovitch, C. G. Pappas, K. Liu and S. Otto, Angew. Chem., Int. Ed., 2022, 61, e202117605 CrossRef CAS PubMed.
- S. Yang, G. Schaeffer, E. Mattia, O. Markovitch, K. Liu, A. S. Hussain, J. Ottele, A. Sood and S. Otto, Angew. Chem., Int. Ed., 2021, 60, 11344–11349 CrossRef CAS PubMed.
- S. Vela-Gallego, Z. Pardo-Botero, C. Moya and A. de la Escosura, Chem. Sci., 2022, 13, 10715–10724 RSC.
- P. Adamski, M. Eleveld, A. Sood, A. Kun, A. Szilagyi, T. Czaran, E. Szathmary and S. Otto, Nat. Rev. Chem., 2020, 4, 386–403 CrossRef PubMed.
- J. Ottele, A. S. Hussain, C. Mayer and S. Otto, Nat. Catal., 2020, 3, 547–553 CrossRef CAS.
- G. Monreal-Santiago, K. Liu, W. R. Browne and S. Otto, Nat. Chem., 2020, 12, 603–607 CrossRef CAS PubMed.
- J. E. Ferrell, Curr. Opin. Cell Biol., 2002, 14, 140–148 CrossRef CAS PubMed.
- R. Mukherjee, R. Cohen-Luria, N. Wagner and G. Ashkenasy, Angew. Chem., Int. Ed., 2015, 54, 12452–12456 CrossRef CAS PubMed.
- I. Maity, N. Wagner, R. Mukherjee, D. Dev, E. Peacock-Lopez, R. Cohen-Luria and G. Ashkenasy, Nat. Commun., 2019, 10, 4636 CrossRef PubMed.
- I. Maity, D. Dev, K. Basu, N. Wagner and G. Ashkenasy, Angew. Chem., Int. Ed., 2021, 60, 4512–4517 CrossRef CAS PubMed.
- L. Zhou, D. K. O’Flaherty and J. W. Szostak, Angew. Chem., Int. Ed., 2020, 59, 15682–15687 CrossRef CAS PubMed.
- C. He, I. Gallego, B. Laughlin, M. A. Grover and N. V. Hud, Nat. Chem., 2017, 9, 318–324 CrossRef CAS PubMed.
- A. Lozoya-Colinas, B. E. Clifton, M. A. Grover and N. V. Hud, ChemBioChem, 2022, 23, e202100495 CrossRef CAS PubMed.
- M. Kreysing, L. Keil, S. Lanzmich and D. Braun, Nat. Chem., 2015, 7, 203–208 CrossRef CAS PubMed.
- J. T. Sczepanski and G. F. Joyce, Nature, 2014, 515, 440–442 CrossRef CAS PubMed.
- G. Houlihan, S. Arangundy-Franklin and P. Holliger, Acc. Chem. Res., 2017, 50, 1079–1087 CrossRef CAS PubMed.
- T. Norin and N. B. Metcalfe, Philos. Trans. R. Soc., B, 2019, 374, 20180180 CrossRef PubMed.
- P. W. Kudella, A. V. Tkachenko, A. Salditt, S. Maslov and D. Braun, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018830118 CrossRef CAS PubMed.
- L. C. Sögütoglu, R. R. E. Steendam, H. Meekes, E. Vlieg and G. P. J. T. Rutjes, Chem. Soc. Rev., 2015, 44, 6723–6732 RSC.
- K. Soai, T. Kawasaki and A. Matsumoto, Acc. Chem. Res., 2014, 47, 3643–3654 CrossRef CAS PubMed.
- R. Plasson and H. Bersini, J. Phys. Chem. B, 2009, 113, 3477–3490 CrossRef CAS PubMed.
- R. Plasson, D. K. Kondepudi, H. Bersini, A. Commeyras and K. Asakura, Chirality, 2007, 19, 589–600 CrossRef CAS PubMed.
- F. C. Frank, Biochim. Biophys. Acta, 1953, 11, 459–463 CrossRef CAS PubMed.
- M. Calvin, Chemical evolution, Oxford University Press, Oxford, 1969 Search PubMed.
- P. Decker, J. Mol. Evol., 1973, 2, 137–143 CrossRef CAS PubMed.
- D. K. Kondepudi and G. W. Nelson, Phys. Rev. Lett., 1983, 50, 1023–1026 CrossRef CAS.
- D. K. Kondepudi and K. Asakura, Acc. Chem. Res., 2001, 34, 946–954 CrossRef CAS PubMed.
- K. Iwamoto, Phys. Chem. Chem. Phys., 2003, 5, 3616–3621 RSC.
- M. Stich, J. M. Ribó, D. G. Blackmond and D. Hochberg, J. Chem. Phys., 2016, 145, 074111 CrossRef PubMed.
- Q. Sallembien, L. Bouteiller, J. Crassous and M. Raynal, Chem. Soc. Rev., 2022, 51, 3436–3476 RSC.
- A. J. Bissette and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 12800–12826 CrossRef CAS PubMed.
- J. M. Ribo, D. Hochberg, J. Crusats, Z. El-Hachemi and A. Moyano, J. R. Soc., Interface, 2017, 14, 20170699 CrossRef PubMed.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS.
- T. Kawasaki, Y. Matsumura, T. Tsutsumi, K. Suzuki, M. Ito and K. Soai, Science, 2009, 324, 492–495 CrossRef CAS PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2009, 48, 386–390 CrossRef CAS PubMed.
- D. G. Blackmond, A. Moran, M. Hughes and A. Armstrong, J. Am. Chem. Soc., 2010, 132, 7598–7599 CrossRef CAS PubMed.
- J. E. Hein and D. G. Blackmond, Acc. Chem. Res., 2012, 45, 2045–2054 CrossRef CAS PubMed.
- R. Pascal, J. Taillades and M. Commeyras, Tetrahedron, 1978, 34, 2275–2281 CrossRef CAS.
- M. Mauksch and S. B. Tsogoeva, ChemPhysChem, 2008, 9, 2359–2371 CrossRef CAS PubMed.
- J. W. Yang, C. Chandler, M. Stadler, D. Kampen and B. List, Nature, 2008, 452, 453–455 CrossRef CAS PubMed.
- C. Viedman, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed.
- C. Blanco, M. Stich and D. Hochberg, J. Phys. Chem. B, 2017, 121, 942–955 CrossRef CAS PubMed.
- J. M. Ribó, J. Crusats, Z. El-Hachemi, A. Moyano and D. Hochberg, Chem. Sci., 2017, 8, 763–769 RSC.
- P. Schuster, Chemistry on the early Earth, 2011, https://www.tbi.univie.ac.at/~pks/Presentation/heidelberg-11.pdf Search PubMed.
- N. A. Hawbaker and D. G. Blackmond, Nat. Chem., 2019, 11, 957–962 CrossRef CAS PubMed.
- J. Hu, W. G. Cochrane, A. X. Jones, D. G. Blackmond and B. M. Paegel, Nat. Chem., 2021, 13, 786–791 CrossRef CAS PubMed.
- T. Hitz and P. L. Luisi, Biopolymers, 2000, 55, 381–390 CrossRef CAS PubMed.
- T. Hitz, M. Blocher, P. Walde and P. L. Luisi, Macromolecules, 2001, 34, 2443–2449 CrossRef CAS.
- K. Ruiz-Mirazo, Protocell, Encyclopedia of Astrobiology, Springer, Germany, 2011 Search PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- H.-X. Zhou, G. Rivas and A. P. Minton, Annu. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS PubMed.
- P. Mitchell, Nature, 1961, 191, 144–148 CrossRef CAS PubMed.
- V. P. Skulachev, Eur. J. Biochem., 1992, 208, 203–209 CrossRef CAS PubMed.
- F. M. Harold, Microbiol. Mol. Biol. Rev., 2005, 69, 544–564 CrossRef CAS PubMed.
- J. J. Spitzer and B. Poolman, Microbiol. Mol. Biol. Rev., 2009, 73, 371–388 CrossRef CAS PubMed.
- N. Lauber, O. Tichacek, K. Narayanankutty, D. De Martino and K. Ruiz-Mirazo, Collective catalysis under spatial constraints: phase separation and size-scaling effects on mass action kinetics, Phys. Rev. E, in press.
- G. Piedrafita, K. Ruiz-Mirazo, P. A. Monnard, A. Cornish-Bowden and F. Montero, PLoS One, 2012, 7, e39480 CrossRef CAS PubMed.
- B. Shirt-Ediss, R. V. Sole and K. Ruiz-Mirazo, Life, 2015, 5, 181–211 CrossRef PubMed.
- J. W. Szostak, J. Syst. Chem., 2012, 3, 2 CrossRef CAS.
- One should realize that, in case proto-metabolic reaction networks developed by themselves in open solution, becoming faster and more efficient (e.g., thanks to the emergence of prebiotic catalysts that could act as the first ‘kinetic controllers’ of such networks), the problem of compartmentalization would become much more acute – simply due to the fact that the rate at which nutrients should access the internal volume of the compartment ought to be much higher.
- A. D. Bangham, M. W. Hill and N. G. A. Miller, Preparation and use of Liposomes as Models of Biological membranes, Springer, Boston MA, Boston, 1974 Search PubMed.
- A. I. Oparin, The Origin of Life, MacMillan, New York, 1938 Search PubMed.
- M. Hanczyc, The Early History of Protocells: the search for the recipe of life, MIT Press, Cambridge MA, Cambridge, 2008, p. 3 Search PubMed.
- C. Donau, F. Späth, M. Sosson, B. A. K. Kriebisch, F. Schnitter, M. Tena-Solsona, H.-S. Kang, E. Salibi, M. Sattler, H. Mutschler and J. Boekhoven, Nat. Commun., 2020, 11, 5167 CrossRef CAS PubMed.
- T.-Y. D. Tang, C. R. C. Hak, A. J. Thompson, M. K. Kuimova, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2014, 6, 527–533 CrossRef PubMed.
- I. A. Chen and P. Walde, Cold Spring Harbor Perspect. Biol., 2010, 2, a002170 CrossRef PubMed.
- I. Gözen, E. S. Köksal, I. Poldsalu, L. Xue, K. Spustova, E. Pedrueza-Villalmanzo, R. Ryskulov, F. Meng and A. Jesorka, Small, 2022, 18, 2106624 CrossRef PubMed.
- P. A. Bachmann, P. L. Luisi and J. Lang, Nature, 1992, 357, 57–59 CrossRef CAS.
- P. Walde, R. Wick, M. Fresta, A. Mangone and P. L. Luisi, J. Am. Chem. Soc., 1994, 116, 11649–11654 CrossRef CAS.
- K. Adamala and J. W. Szostak, Nat. Chem., 2013, 5, 495–501 CrossRef CAS PubMed.
- M. D. Hardy, J. Yang, J. Selimkhanov, C. M. Cole, L. S. Tsimring and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8187–8192 CrossRef CAS PubMed.
- A. H. J. Engwerda, J. Southworth, M. A. Lebedeva, R. J. H. Scanes, P. Kukura and S. P. Fletcher, Angew. Chem., Int. Ed., 2020, 59, 20361–20366 CrossRef CAS PubMed.
- E. A. J. Post and S. P. Fletcher, Chem. Sci., 2020, 11, 9434–9442 RSC.
- M. G. Howlett, A. H. J. Engwerda, R. J. H. Scanes and S. P. Fletcher, Nat. Chem., 2022, 14, 805–810 CrossRef CAS PubMed.
- C. Bonfio, E. Godino, M. Corsini, F. Fabrizi de Biani, G. Guella and S. S. Mansy, Nat. Catal., 2018, 1, 616–623 CrossRef CAS.
- C. A. Hutchison, R. Y. Chuang, V. N. Noskov, N. Assad-Garcia, T. J. Deerinck, M. H. Ellisman, J. Gill, K. Kannan, B. J. Karas, L. Ma, J. F. Pelletier, Z. Q. Qi, R. A. Richter, E. A. Strychalski, L. Sun, Y. Suzuki, B. Tsvetanova, K. S. Wise, H. O. Smith, J. I. Glass, C. Merryman, D. G. Gibson and J. C. Venter, Science, 2016, 351, aad6253 CrossRef PubMed.
- T.-F. Wong, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1909–1912 CrossRef PubMed.
- M. Di Giulio, J. Theor. Biol., 1998, 191, 191–196 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |