
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox processes in Cu-binding proteins: the “in-between” states in intrinsically disordered peptides†
Enrico
Falcone
 ab and
Christelle
Hureau
*c
ab and
Christelle
Hureau
*c
aInstitut de Chimie (UMR 7177), Université de Strasbourg, CNRS, Strasbourg, France
bSchool of Chemistry, University of Nottingham, Nottingham, UK
cLCC-CNRS, Université de Toulouse, CNRS, Toulouse, France. E-mail: christelle.hureau@lcc-toulouse.fr
First published on 13th September 2023
Abstract
We report on a concept that some of us first described a decade ago for pure electron transfer [V. Balland, C. Hureau and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17113]. In the present viewpoint, based on more recent results, we refine and extend this “in-between state” concept to explain the formation of reactive oxygen species by copper ions bound to the amyloid-β (Aβ) peptide involved in Alzheimer's disease. In such intrinsically disordered peptides, the Cu coordination is versatile due to the lack of stable folding and the presence of multiple possible binding anchors. Hence, the Cu(I) and Cu(II) ions do impose their favoured sites, with Cu(I) bound in a linear fashion between two His residues and Cu(II) in a square-based pyramid bound to Asp1 amine and carbonyl groups and two His residues in the equatorial plane. Hence a direct electron transfer is prevented and alternatively an in-between state (IBS) mechanism applies, whose description and analysis with respect to other electron transfer processes is the topic of the present viewpoint.
 Enrico Falcone | Enrico Falcone pursued his undergraduate studies in Chemistry (BSc) and Biomolecular Chemistry (MSc) at the University of Catania (Italy), focusing on copper-amyloids interaction. In 2021, he obtained his PhD at the University of Strasbourg (under the supervision of Prof. Peter Faller), where he developed peptide-based luminescent probes for copper(II) detection. Then, he undertook a post-doc in the same group, studying the reactivity (e.g. ROS production) of copper-based drugs in intracellular-like conditions. He is currently a Post-Doctoral Research Associate at the University of Nottingham (UK), where he works on the development of peptide-based mimics of the copper-dependent enzyme lytic polysaccharide monooxygenase. |
 Christelle Hureau | Christelle Hureau is the head of the “Alzheimer, Amyloids and Bio-Inorganic Chemistry (ALAMBIC, https://hureaulab.wixsite.com/equipeflcc)” group at the Coordination Chemistry Lab in Toulouse. She did her PhD in Orsay (France) on the coordination chemistry of Mn-based structural models of the water-oxidizing centre in the Photosystem II and graduated in 2003. Then she completed three post-docs at the frontier between chemistry, biochemistry and advanced spectroscopy and electrochemistry. She is interested in the impact of metallic ions in Alzheimer's disease (AD) and other amyloid-related diseases, in the design and study of new drug candidates targeting copper ions, and of inorganic compounds able to tune the self-assembly of the Aβ peptide, involved in AD. |
1. Introduction
In most organisms, copper (Cu) represents an essential redox cofactor for electron-transfer proteins (e.g. plastocyanin, azurin) and enzymes, most of which catalyse the activation of dioxygen (e.g. cytochrome C oxidase, superoxide dismutase, monooxygenases).1,2 During the last decades, an increasing interest arose for the interaction of Cu ions with intrinsically disorder peptides/proteins (IDPs, i.e. proteins that are totally or partially devoid of a well-defined three-dimensional structure in their isolated state),3 essentially because both Cu ions and IDPs like amyloid-β (Aβ), α-synuclein (αSyn) and prion protein (PrP) are involved in neurodegenerative diseases such as Alzheimer's (AD), Parkinson's (PD) and prion diseases, respectively.4–7 For instance, in AD, it is well established that Cu is accumulated in the amyloid plaques,5 whose main constituent is Aβ aggregates, i.e. monomeric Aβ that have self-assembled. As Cu-IDP complexes are able to catalyse the production of reactive oxygen species (ROS) in the presence of dioxygen and a reducing agent, it has been reasonably speculated that this reactivity contributes to the oxidative stress observed in neurodegenerative diseases.8–10 More specifically, the redox activity of the Cu-Aβ complexes is responsible for the production of ROS that contribute to the overall oxidative stress observed in AD.8In the last years, we and others have proposed, based on various electrochemical, chemical and computational studies, that ROS formation catalysed by Cu-Aβ proceeds through an intricate and unprecedented mechanism involving a so-called “in-between state” (IBS), whose coordination sphere and geometry is between those of Cu(I) and Cu(II) bound to Aβ.
In this viewpoint, we report on the concept of IBS with respect to other ones applied for Cu sites in redox proteins and bio-inspired models and conclude on the biological relevance of the IBS.
2. Coordination and redox chemistry of Cu: from natural enzymes to synthetic mimics
The main oxidation states of Cu ions in biological systems are Cu(I) and Cu(II). These redox states show very different coordination preferences. First, according to Pearson's hard/soft acid–base theory,11 Cu(I) is a soft acid whilst Cu(II) is an intermediate one. Hence, Cu(I) prefers binding to soft sulphur donors in proteins (cysteine and methionine), whereas Cu(II) to intermediate or hard nitrogen ligands (histidine side chain, amine and amidate). Besides, due to the d10 configuration, Cu(I) has no ligand-field stabilization energy and hence it can acquire quite a different set of coordination geometries, e.g. linear, trigonal, tetrahedral (Td) depending on the coordination number (2 to 4), which is mostly dictated by the valence shell electron pair repulsion theory.12 In contrast, Cu(II) d9 configuration favours four strongly bound equatorial ligands and potentially one or two weaker apical ligands, mainly leading to square planar (SP) and square-based (bi)pyramidal (SBP) coordination geometry in line with the Jahn–Teller distortion.13 Hence, since according to the Marcus theory the rate of electron transfer depends on the reorganization energy associated with the changes in metal–ligand bond length and angles,14 a very sluggish redox cycling would be expected for the Cu(II)/Cu(I) redox couples.Interestingly, Cu-proteins and enzymes, due to their 3D-folding, tackle this issue by binding both redox states in a well-defined high-energy, so-called entatic, site representing a compromise between the two redox sites, both in terms of ligand types and coordination geometry.
In the Cu sites of blue copper proteins (BCPs), such as plastocyanin and azurin, involved in an electron-transfer-only process and where Cu cycles between Cu(I) and Cu(II) via an outer-sphere electron transfer (with no substrate binding), a combination of soft sulphur (Cys, Met) and intermediate nitrogen (His) ligands are found, constrained in a flattened tetrahedral or trigonal (bi)pyramidal (T(B)P) geometries by the protein scaffold (Fig. 1, exemplified with TBP). These geometries represent the best compromise between the Td and SP (or SBP). Thus, BCPs achieve very efficient electron transfer by minimizing the structural reorganization between the two redox states.15
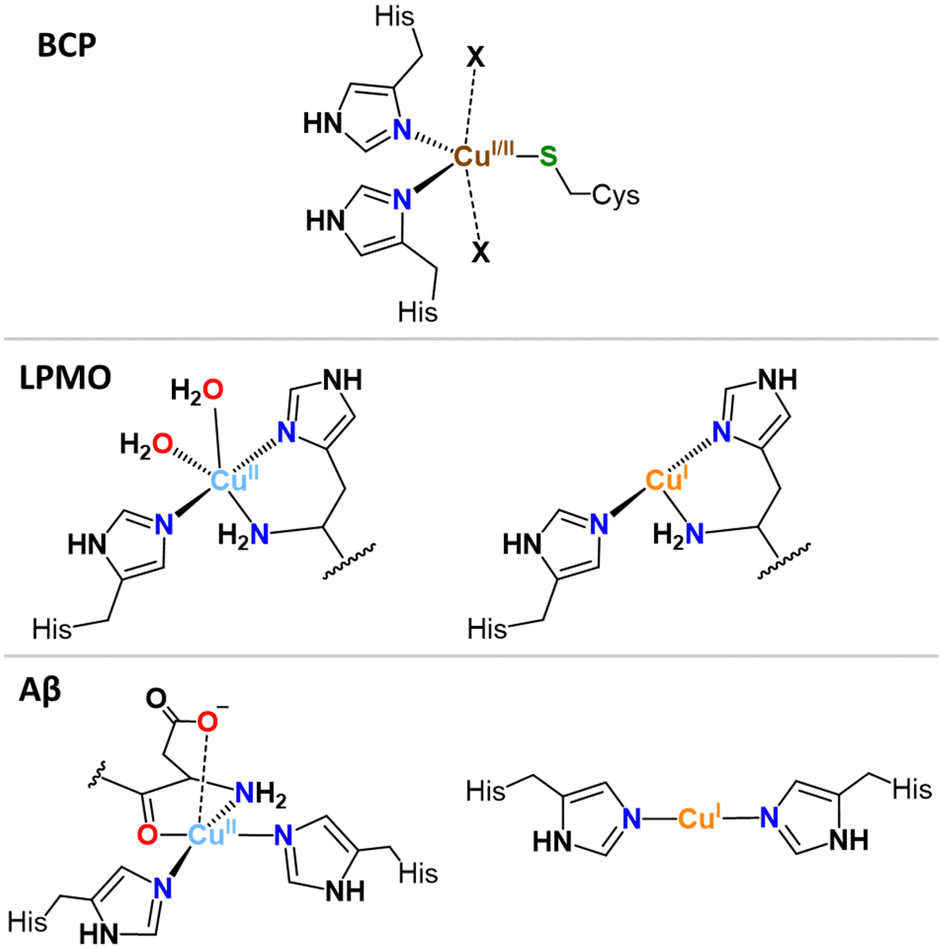 | ||
| Fig. 1 Cu sites in blue copper protein (BCP), lytic polysaccharide monooxygenases (LPMO), and amyloid-β (Aβ). | ||
Likewise, Cu-enzymes, show well-defined Cu sites accommodating both Cu(I) and Cu(II) states with minimal ligand reorganization. For instance, in the histidine brace motif, found in lytic polysaccharide monooxygenases (LPMOs), a couple of His residues bind both Cu(II) and Cu(I) in a T-shaped geometry,16 with one or two additional water molecules binding to Cu(II) state only (Fig. 1), a place that might be occupied by the substrate (O2 or H2O2) under working conditions.17,18
In bio-inspired enzyme mimics, the constraints imposed by the 3D-folding of the proteins are missing, and copper redox processes are slow and associated reactivity is low. To overcome this issue, several strategies have been reported. A selection of key examples is given here while the interested reader can refer to ref. 19 for an extensive review. A straight example of an “entatic-like” state is obtained with Cu complexes in which two ligands impose a Cu(I)-like Td geometry due to steric hindrance of the ligand preventing the formation of the square-based Cu(II)-like geometries. This includes phenanthroline- and quinoline-based ligands (Fig. S1 and S2, ESI†).20 Another example is the bispidine ligand family, which has been developed to impose a constrained, ligand-controlled coordination site for metal ions, including Cu(I) that lies in a Cu(II)-like coordination (Fig. S3, ESI†). Supramolecular control of the Cu coordination site was also proposed. The first example is the use of calixarene ligands.21 Depending on their exact chemical nature, they can favour a classical or an entatic-like mechanism. In the former case, the Cu(II) is first reduced, followed by a structural change to a Td geometry. In the latter one, the structural change to a Td geometry is observed first, promoted by the insertion of a nitrile-based solvent in the calixarene cavity and is followed by the Cu(II) reduction (Fig. S4, ESI†).21 A second example has been described with a ligand and its glyco-derivative, where the added sugar moiety induces long-range strains on the Cu centre thus increasing its TBP character (versus SP) (Fig. S5, ESI†).22 The last illustration is given by ligands built on de novo designed 3-helix bundles peptide/protein that can create a site closer to those reported in enzymes (i.e. with amino acids) and in which pre-folding versus pre-organization concepts were illustrated.23 In the former case, a tertiary structure, where the metal-chelating side chains are present, is formed but the metal ion guides the ligand coordination, while in the latter the ligands are well prepositioned to directly host the metal ion.
3. Coordination and redox chemistry of Cu in IDPs: the IBS concept
In contrast to folded proteins, IDPs are very flexible and hence can easily adapt to the requirements of the redox-state-dependent Cu coordination. In addition, more binding anchors than necessary are present thus affording a great diversity in the nature of the binding sites. Hence, unlike enzymes, they bind Cu(II) and Cu(I) in sites with different geometry and partially distinct ligands. As the archetypical example, Aβ binds Cu(I) via two His in a digonal geometry, whereas Cu(II) is bound in a distorted SBP geometry, (Fig. 1). In particular, the main (i.e. with the lowest energy and hence most populated) “resting state” (RS) of Cu(II)-Aβ at pH 7.4 is made of the N-terminal amine nitrogen and the carbonyl oxygen from Asp1, the imidazole rings of His6, and of either His13 or His14 with about the same occurrence.5,24 A water molecule or Asp1 side-chain can bind apically. Other sites can exist, for instance with the His13 and His14, but not His6, bound. The exchange between the possible sites involves the transient formation of higher energy coordination species where for instance the three His can be simultaneously bound to the Cu(II) in addition to the N and/or O from Asp1. For Cu(I)-Aβ, the main RS are composed of two among the three possible His residues in a digonal fashion, with the most populated His couple being His6–His13 (Fig. 1).5,25,26 These RS states are in fast exchange and a state where the three His are involved could be formed transiently as well. Therefore, not only the geometry but also the ligands are different between Cu(II) and Cu(I) resting states, suggesting that a large reorganization would be required to switch between those RS.As discussed above, in order to have an entatic state very prone to fast redox reactions, a protein scaffold is needed to impose the same strained coordination of Cu in the two redox states. As this is not possible for IDPs, the coordination sites of the Cu(I) and Cu(II) in IDPs are driven by the oxidation state of the metal centre and are thus very different. Nevertheless, some of the Cu-IDP complexes were revealed to be quite competent to do redox reactions. To explain such an unexpected trend, the in-between state(s) (IBS) concept was introduced. Such IBS have geometries in-between those of Cu(I) and Cu(II) and are accessible due to the flexible nature of IDPs and the fast dynamics of exchange between several energetically equivalent structures. The importance of this flexibility in redox chemistry was initially realized a decade ago, when Cu(I)- and Cu(II)-Aβ1–16 were characterized at room temperature by NMR, X-ray absorption and electrochemistry.25 A subsequent detailed electrochemical study on Cu-Aβ1-16, proposed the electrochemical IBS (eIBS, also called pre-organized state in this seminal report), whose population was estimated to be about 0.1%, and which can undergo electron transfer very rapidly (estimated redox potential of 0.3 V vs. NHE).27 The concept of the (e)IBS is schematized in Fig. 2, together with two extreme cases encountered in Cu(I)/Cu(II) redox processes. The case shown on the left corresponds to Cu(I) and Cu(II) in their respective preferred geometry (for instance, A = Td and B = SBP). Hence there is no overlap between the two environments of Cu(I) and Cu(II) complexes and to get the Cu(II)A reduced into Cu(I)B an EC (Electrochemical–Chemical) mechanism is required, in which a Cu(I)A is first formed followed by rearrangement to Cu(I)B. The transition state corresponds to Cu(I)A. Vice-versa, to get the Cu(I)B oxidized into Cu(II)A, a EC mechanism is required where the Cu(II)B is first formed followed by rearrangement to Cu(II)A. On the right, the entatic case is shown, where due to external constrains the coordination sphere of both Cu(I) and Cu(II) are very close; hence there is an important overlap in the conformation diagram. The imposed constraints induce an energization of the RS of Cu(II) and Cu(I) and no chemical reaction is associated with the redox process.19,28,29 In the middle, is the IBS case: the RS are in fast equilibrium with the IBS in which the redox processes occur. This corresponds to the overlap of minor states (IBS) of higher energy co-present with the main states (RS) of lower energy. The redox process thus occurs according to a CEC (chemical–electrochemical–chemical) mechanism. Beyond the entatic state, which is now textbook, the ecstatic state's principle was recently introduced. It can be seen as the formation of a distribution of environments around the Cu(I) and Cu(II) ions (without coordination bonds being broken), large enough to have some overlay between the two redox states that allow electron transfer to proceed (Fig. S6, ESI†).30 In the ecstatic case, there is a unique set of binding atoms, whose position (i.e. distance, angles) with respect to the Cu centre can be significantly distributed. In contrast, in the IBS case, several discrete coordination sets of similar energy are formed through equilibria with the various RS in which coordination bonds break and reform.
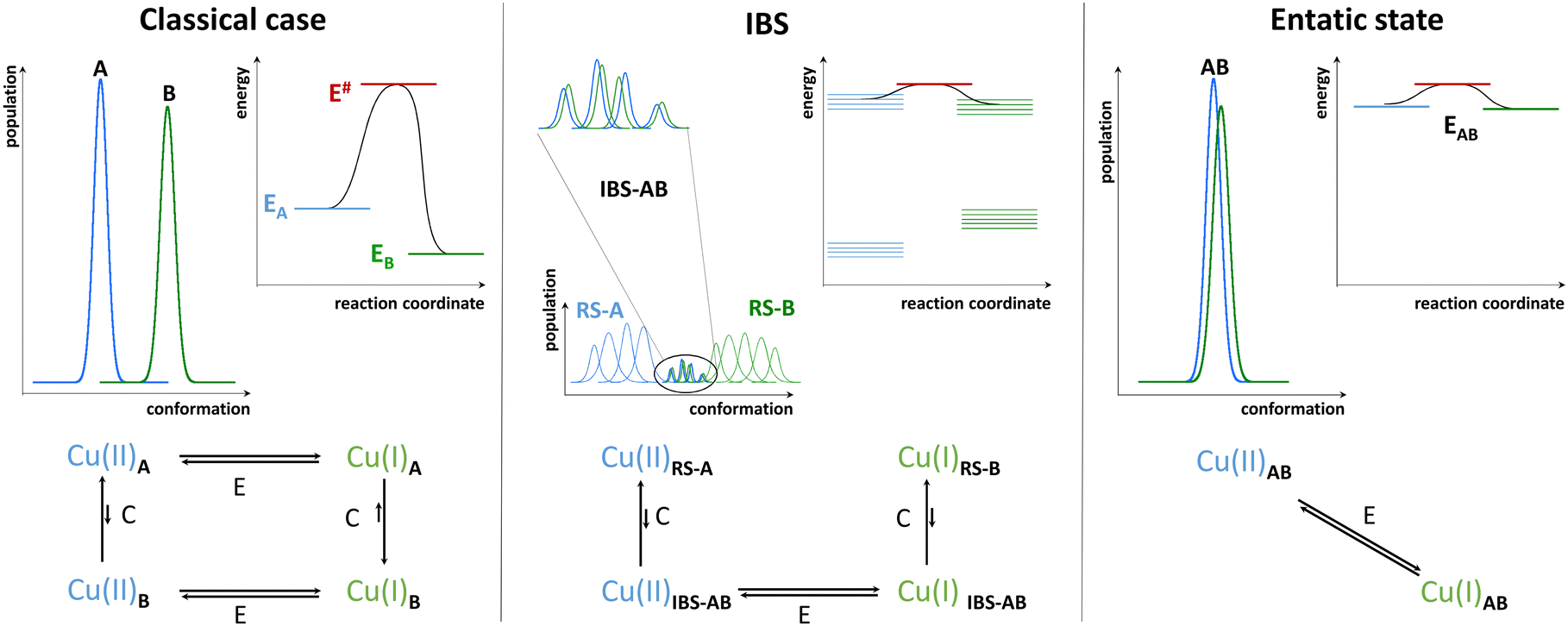 | ||
| Fig. 2 Different cases of Cu(I)/Cu(II) redox process in copper complexes. Three data are given: the population as a function of the conformation, the energy as a function of a reaction coordinate and the chemical–electrochemical scheme describing the whole reaction and the corresponding energy diagram. Subscripts A and B correspond to the preferred environment for Cu(I) and Cu(II), respectively. AB to a geometry intermediate between A and B, RS to the resting state, IBS to the in-between state, and # to the transition state, E to electrochemical reaction and C to chemical reaction. | ||
Besides these studies on an outer-sphere electrochemical mechanism, the redox reactivity of Cu-Aβ was then investigated with chemical substrates, mainly ascorbate as reductant and O2 or H2O2 as oxidants, leading to the formation of O2˙− and H2O2, or HO˙, respectively (Fig. 3). The nature of the IBS site was identified based on the assumption that only the IBS state(s) are able to catalyze efficiently the production of ROS and that the very reactive HO˙ attacks the nearest ligands of the IBS site. Indeed, the oxidative damage by HO˙ was not in line with the resting state of Cu(I), the species that forms HO˙, as Asp1 was also affected.31,32 This clearly indicated that it is not the resting state of Cu(I) in Aβ to be accountable for the reaction with H2O2 (reaction iv, Fig. 3). Further investigations were done with a multitude of modified Aβ peptides. The rates of ascorbate oxidation were measured with the rationale that by mutating a potential ligand, if (i) the rate becomes slower, the mutated ligand is involved in the IBS, (ii) the rate becomes faster, the mutated ligand stabilizes a non-IBS state, and (iii) no effect is observed, the mutated residue is not relevant for the IBS. This led to the proposition of a so-called catalytic IBS (cIBS), to distinguish it from the eIBS, implicating three peptide ligands: N-terminus and carboxylate oxygen from Asp1 side chain and one of the His (Fig. 3).33,34 Interestingly, some of us recently showed that the X-ray absorption spectroscopy spectra of a species generated by X-ray induced photoreduction of Cu(II)-Aβ followed by partial thermal relaxation were well reproduced using such a model of the IBS.35 These data represent the first spectroscopic characterization of an IBS.
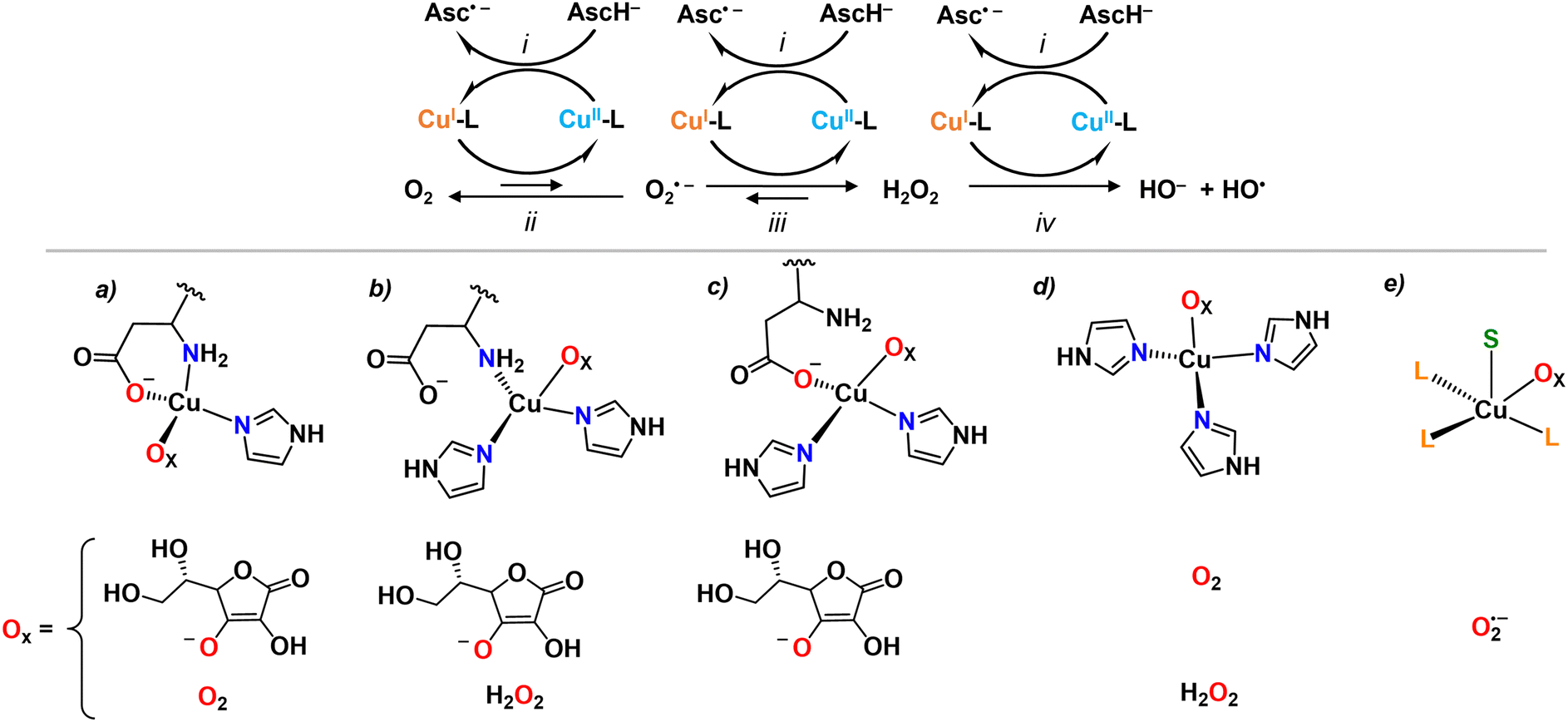 | ||
| Fig. 3 Schematic mechanism of ROS production by Cu-Aβ in the presence of ascorbate (AscH−) as a reducing agent (top) and proposed structures of cIBS (bottom). The Ox atom can belong to AscH− ((a)–(c) for reaction i), O2 ((a) and (d) for reaction ii), O2˙− ((e), for reaction iii), or H2O2 ((b) and (d) for reaction iv); in (e), S represents the substrate (e.g. ascorbate). | ||
Overall, most experimental studies focused on the reaction of Cu-Aβ with ascorbate and O2. It is likely that only the IBS of the rate-determining step is accessible by measuring the rate of ascorbate oxidation and thus it is still not yet clear from experimental work if the same cIBSs is at play for O2˙− and H2O2 reduction as well as ascorbate oxidation, and whether the ascorbate and/or O2 are bound to the Cu in the cIBS. In other words, we do not know yet whether the chemical mechanism follows an inner- or outer-sphere electron transfer or, otherwise stated, whether the eIBS is the same as the cIBS. In this regard, theoretical studies addressed the nature of the cIBS for reaction with ascorbate (i), O2 (ii), O2˙− (iii) and H2O2 (iv).36–39 For all reactions an inner-sphere mechanism was proposed, i.e. binding of ascorbate, O2, O2˙− or H2O2 to the Cu ion, and several suited IBS resulted to be possible (Fig. 3). Interestingly, for reactions i and ii, the propositions included the experimentally suggested IBS (Fig. 3(a)). However, different IBS (Fig. 3(b) and (d)) were suggested for reaction iv as the side-chain carboxyl group from Asp1 implies a higher energy barrier towards H2O2 reduction.37 A penta-coordinated cIBS, where Cu is bound to three peptide ligands and simultaneously to O2˙− and AscH (S in Fig. 3e), has been recently suggested for reaction iii.39 There is currently no experimental evidence about when the substrates bind. It is possible that either the substrate binds to a pre-formed IBS, or that the substrate binds in another state and then the IBS is reached with the bound substrate, following an induced-fit mechanism.
4. Conclusions and perspectives
To conclude, we gather some considerations about the exact definition of the IBS with respect to other concepts. Our best (and current) view of the IBS is one, or more likely several, high-energy, low-populated, states where both the Cu(I) and Cu(II) can highly efficiently make redox transfer due to a very low energy gap between the two redox states (as in an entatic state). Such IBS states can be reached due to the flexibility of the peptide and fast ligand exchange around the Cu centre. However, in contrast to the entatic state, there are no constraints applied by the biomolecular scaffold towards a unique optimized environment. Instead, there are equilibria between several environments, including those of the resting states, and among the various possible environments some are fully appropriate for electron transfer. Hence, it is highly possible that a set of IBSs exists for the same reaction, and each IBS of the set could be differently populated and yield different reaction rates. In addition, the set of IBS for one reaction could be different from the set for another one (Fig. 2).40 Although Cu(I) and Cu(II) have drastically different coordination spheres, the ROS production is thus made possible due to the high versatility of peptides as ligands (flexibility and multiple binding anchors and sites) and make Cu-IDP complexes a unique redox system throughout the chemical and biological spaces. Beyond the case of Aβ discussed here, other IDPs may share this ability, such as truncated Aβ peptides also involved in AD,41 the α-Syn involved in Parkinson's and the PrP involved in Prion-related diseases, despite thorough studies have not been reported yet for these IDPs. This may also be true for non-pathogenic IDPs such as the salivary histatins and the N-terminal region of the human Cu transporter Ctr1. These sequences possess two distinct Cu(I) and Cu(II) sites: the Cu(I) is bound to a bis-His motif (and potential additional ligands) while the Cu(II) is bound by an ATCUN (Amino-Terminal Cu and Ni binding) motif (Fig. S7, ESI†).42,43 Nevertheless, the formation of the rigid ATCUN site is anticipated to make the access to the IBS more difficult than with Aβ when starting from the Cu(II) species, as reported for truncated Aβ peptides with analoguous Cu(I) and Cu(II) sites.41 With respect to AD, two main consequences of the IBS mechanism can be foreseen. Firstly, the constraints imposed in the Aβ aggregates can be responsible for the lower ROS production ability of Cu bound to Aβ aggregates versus Cu bound to monomer or small oligomers of Aβ44 by preventing the formation of the IBS states, although the Cu binding domain remains flexible in most models of Aβ fibrils. Secondly, the IBS mechanism points to species other than the RS as therapeutic targets. Further approaches may thus consider the possibility of precluding the formation of the redox active IBS and/or promoting the formation of redox-inert (and non-toxic by other ways) aggregates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. H. and E. F. warmly thank Prof. P. Faller (University of Strasbourg) for his input into the seminal version of the review and for inspiring discussions. C. H. thanks Drs C. Esmieu and L. Berthonnaud for fruitful discussions and experiments on that topic (to be further published). E. F. thanks Dr Luisa Ciano (University of Nottingham) for helpful discussions. C. H. acknowledges ERC StG 638712 ‘aLzINK’ for financial support. E. F. acknowledges financial support from the French National Research Agency (ANR) through the ANR-17-EURE-0016 and CHAPCOP-ANR-19-CE44-0018 programs.References
- R. A. Festa and D. J. Thiele, Curr. Biol., 2011, 21, R877 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659 CrossRef CAS PubMed.
- R. van der Lee, M. Buljan, B. Lang, R. J. Weatheritt, G. W. Daughdrill, A. K. Dunker, M. Fuxreiter, J. Gough, J. Gsponer, D. T. Jones, P. M. Kim, R. W. Kriwacki, C. J. Oldfield, R. V. Pappu, P. Tompa, V. N. Uversky, P. E. Wright and M. M. Babu, Chem. Rev., 2014, 114, 6589 CrossRef CAS PubMed.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, Annu. Rev. Biophys., 2008, 37, 215 CrossRef CAS PubMed.
- E. Atrian-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 375, 38 CrossRef PubMed.
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014, 47, 2252 CrossRef CAS PubMed.
- N. D'Ambrosi and L. Rossi, Neurochem. Int., 2015, 90, 36 CrossRef PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450 CrossRef CAS PubMed.
- C. Wang, L. Liu, L. Zhang, Y. Peng and F. Zhou, Biochemistry, 2010, 49, 8134 CrossRef CAS PubMed.
- L. Liu, D. Jiang, A. McDonald, Y. Hao, G. L. Millhauser and F. Zhou, J. Am. Chem. Soc., 2011, 133, 12229 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- R. J. Gillespie and R. S. Nyholm, Q. Rev., Chem. Soc., 1957, 11, 339 RSC.
- H. A. Jahn, E. Teller and F. G. Donnan, Proc. R. Soc. London, Ser. A, 1937, 161, 220 CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966 CrossRef CAS.
- H. B. Gray, B. G. Malmström and R. J. P. Williams, JBIC, J. Biol. Inorg. Chem., 2000, 5, 551 CrossRef CAS PubMed.
- G. R. Hemsworth, E. J. Taylor, R. Q. Kim, R. C. Gregory, S. J. Lewis, J. P. Turkenburg, A. Parkin, G. J. Davies and P. H. Walton, J. Am. Chem. Soc., 2013, 135, 6069 CrossRef CAS PubMed.
- K. K. Meier, S. M. Jones, T. Kaper, H. Hansson, M. J. Koetsier, S. Karkehabadi, E. I. Solomon, M. Sandgren and B. Kelemen, Chem. Rev., 2018, 118, 2593 CrossRef CAS PubMed.
- A. Paradisi, M. J. Steward, P. Lindley, G. J. Davies and P. H. Walton, Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, p. 500 Search PubMed.
- J. Stanek, A. Hoffmann and S. Herres-Pawlis, Coord. Chem. Rev., 2018, 365, 103 CrossRef CAS.
- J. Heck, F. Metz, S. Buchenau, M. Teubner, B. Grimm-Lebsanft, T. P. Spaniol, A. Hoffmann, M. A. Rübhausen and S. Herres-Pawlis, Chem. Sci., 2022, 13, 8274 RSC.
- N. Le Poul, Y. Le Mest, I. Jabin and O. Reinaud, Acc. Chem. Res., 2015, 48, 2097 CrossRef CAS PubMed.
- L. Garcia, F. Cisnetti, N. Gillet, R. Guillot, M. Aumont-Nicaise, J.-P. Piquemal, M. Desmadril, F. Lambert and C. Policar, J. Am. Chem. Soc., 2015, 137, 1141 CrossRef CAS PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495 CrossRef CAS PubMed.
- C. Hureau and P. Dorlet, Coord. Chem. Rev., 2012, 256, 2175 CrossRef CAS.
- C. Hureau, V. Balland, Y. Coppel, P. L. Solari, E. Fonda and P. Faller, J. Biol. Inorg. Chem., 2009, 995 CrossRef CAS PubMed.
- G. De Gregorio, F. Biasotto, A. Hecel, M. Luczkowski, H. Kozlowski and D. Valensin, J. Inorg. Biochem., 2019, 195, 31 CrossRef CAS PubMed.
- V. Balland, C. Hureau and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17113 CrossRef CAS PubMed.
- P. Comba, Coord. Chem. Rev., 2000, 200–202, 217 CrossRef CAS.
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651 CrossRef CAS PubMed.
- W. R. Hagen, Metallomics, 2019, 11, 1768 CrossRef CAS PubMed.
- L.-E. Cassagnes, V. Hervé, F. Nepveu, C. Hureau, P. Faller and F. Collin, Angew. Chem., Int. Ed., 2013, 52, 11110 CrossRef CAS PubMed.
- C. Cheignon, C. Hureau and F. Collin, Inorg. Chim. Acta, 2018, 472, 111 CrossRef CAS.
- C. Cheignon, M. Jones, E. Atrian-Blasco, I. Kieffer, P. Faller, F. Collin and C. Hureau, Chem. Sci., 2017, 8, 5107 RSC.
- E. Atrian-Blasco, M. Del Barrio, P. Faller and C. Hureau, Anal. Chem., 2018, 90, 5909 CrossRef CAS PubMed.
- E. Falcone, G. Nobili, M. Okafor, O. Proux, G. Rossi, S. Morante, P. Faller and F. Stellato, Angew. Chem., Int. Ed., 2023, e202217791 CAS.
- A. Mirats, J. Alí-Torres, L. Rodríguez-Santiago, M. Sodupe and G. La Penna, Phys. Chem. Chem. Phys., 2015, 17, 27270 RSC.
- T. Prosdocimi, L. De Gioia, G. Zampella and L. Bertini, JBIC, J. Biol. Inorg. Chem., 2016, 21, 197 CrossRef CAS PubMed.
- F. Arrigoni, T. Prosdocimi, L. Mollica, L. De Gioia, G. Zampella and L. Bertini, Metallomics, 2018, 10, 1618 CrossRef CAS PubMed.
- F. Arrigoni, C. Di Carlo, A. Rovetta, L. De Gioia, G. Zampella and L. Bertini, Eur. J. Inorg. Chem., 2022, e202200245 CrossRef CAS.
- C. Bacchella, S. Dell’Acqua, S. Nicolis, E. Monzani and L. Casella, Inorg. Chem., 2021, 60, 606 CrossRef CAS PubMed.
- C. Esmieu, G. Ferrand, V. Borghesani and C. Hureau, Chem. – Eur. J., 2021, 27, 1777 CrossRef CAS PubMed.
- M. J. Pushie, K. Shaw, K. J. Franz, J. Shearer and K. L. Haas, Inorg. Chem., 2015, 54, 8544 CrossRef CAS PubMed.
- S. E. Conklin, E. C. Bridgman, Q. Su, P. Riggs-Gelasco, K. L. Haas and K. J. Franz, Biochemistry, 2017, 56, 4244 CrossRef CAS PubMed.
- J. T. Pedersen, S. W. Chen, C. B. Borg, S. Ness, J. M. Bahl, N. H. Heegaard, C. M. Dobson, L. Hemmingsen, N. Cremades and K. Teilum, J. Am. Chem. Soc., 2016, 138, 3966 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cs00443k |
| This journal is © The Royal Society of Chemistry 2023 |