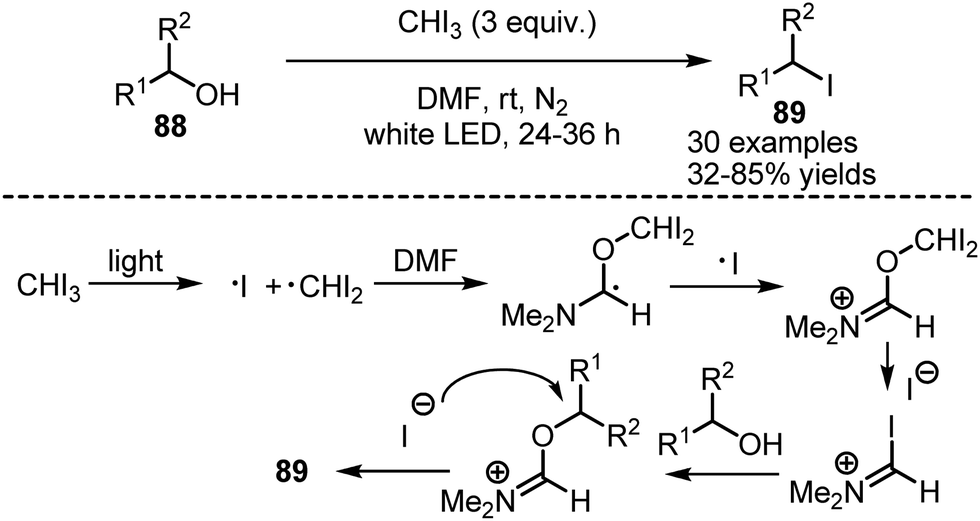
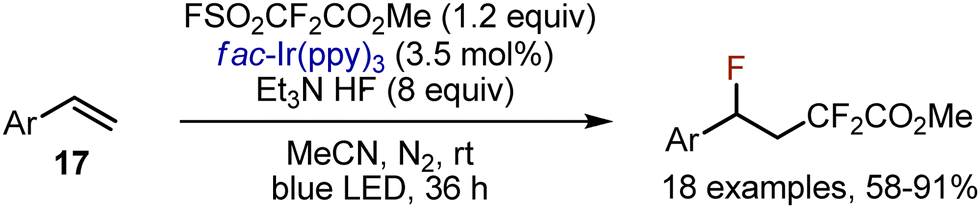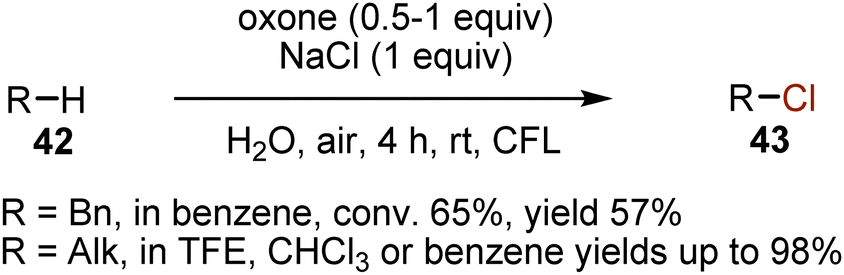DOI:
10.1039/D3CS00366C
(Review Article)
Chem. Soc. Rev., 2023,
52, 8678-8698
Visible light-mediated halogenation of organic compounds
Received
31st August 2023
First published on 17th November 2023
Abstract
The use of visible light and photoredox catalysis emerged as a powerful and sustainable tool for organic synthesis, showing high value for distinctly different ways of bond creation. Halogenated compounds are the cornerstone of contemporary organic synthesis: it is almost impossible to develop a route towards a pharmaceutical reagent, agrochemical, natural product, etc. without the involvement of halogen-containing intermediates. Moreover, the halogenated derivatives as final products became indispensable for drug discovery and materials science. The idea of this review is to understand and summarise the impact of visible light-promoted chemistry on halogenation and halofunctionalisation reactions.

Alexey A. Festa
| Alexey A. Festa was born in Moscow, Russia in 1988. After graduating from the Chemistry Department of the Lomonosov Moscow State University in 2010, he defended his PhD thesis under the guidance of Professor L. G. Voskressensky at RUDN University in 2013. He has worked as Assistant at the Organic Chemistry Department, RUDN University since 2013 and as Assistant Professor – since 2015. His area of scientific interests includes the chemistry of heterocyclic compounds, domino reactions in organic synthesis, photochemistry. |

Olga A. Storozhenko
| Olga A. Storozhenko is an Assistant Professor at the Organic Chemistry Department, RUDN University. She received a master's degree from the Peoples’ Friendship University of Russia in 2015. She defended her PhD thesis in group of Professor L. G. Voskressensky in 2019. She worked as an Assistant at the Organic Chemistry Department, RUDN University from 2019 to 2021. Her area of research includes heterocyclic chemistry, radical transformations of allenes and related compounds. |

Leonid G. Voskressensky
| Leonid G. Voskressensky received his PhD-degree in Organic Chemistry from the Peoples’ Friendship University of Russia in 1999. In 2001 he joined the Prof. Cosimo Altomare (Universita Degli Studi di Bari, Italy) group as a Post-Doc Fellow (Medicinal Chemistry). In 2013 he became Full Professor of the Organic Chemistry department of the RUDN. Since 2013, he has been the Dean of the Science Faculty of the RUDN University. His scientific interests mainly focus on heterocyclic chemistry, domino reaction methodology, new MCR reactions as well as medicinal chemistry. Prof. Voskressensky has a vast experience in the field of heterocyclic chemistry. |

Erik V. Van der Eycken
| Erik V. Van der Eycken is Full Professor Organic Chemistry and head of the Division Molecular Design & Synthesis (>100 people), as well as head of the Laboratory for Organic & Microwave-Assisted Chemistry at the University of Leuven (KU Leuven), Belgium. He is also Visiting Professor at Peoples’ Friendship University of Russia, Moscow (RUDN University). His research focuses on the application of microwave irradiation, transition metal catalysis, C–H activation, multicomponent reactions (MCRs), post-MCR modifications, and solid phase organic synthesis. Also Flow Chemistry and Photoredox Chemistry have been recently addressed. Until now 32 PhD-students performed their research under his guidance. |
1. Introduction
The introduction of a halogen atom into an organic molecule – a hydrocarbon, an aromatic compound, or a heterocycle – is a frequently employed process.1,2 The halogens are valuable as leaving group in i.a. substitution reactions and cross-couplings, and as property-defining groups in bioactive molecules or various materials.3–6 Halogenation is often the starting point in the synthesis of natural products, pharmaceuticals or agrochemicals. Nowadays, it is hard to find a process not involving halogen-containing compounds. UV-light was historically employed for free radical halogenation, and the deep mechanistic understanding of those processes helps the contemporary research. Still, these methods lack selectivity and are usually unsuitable for late-stage functionalization.
Visible light photocatalysis has shown an immense growth since the success of pioneering works in 2008.7,8 This field emerged as a novel paradigm for bond creation in organic synthesis.9–12 Numerous reactions, novel in their concept and formidable in their applicability, have been developed since then. This review aims at assessing the impact of photoredox catalysis on halogenation and halofunctionalisation reactions, and it compares new opportunities with conventional approaches.
2. Fluorination
2.1. Fluorination of Csp3–H bonds
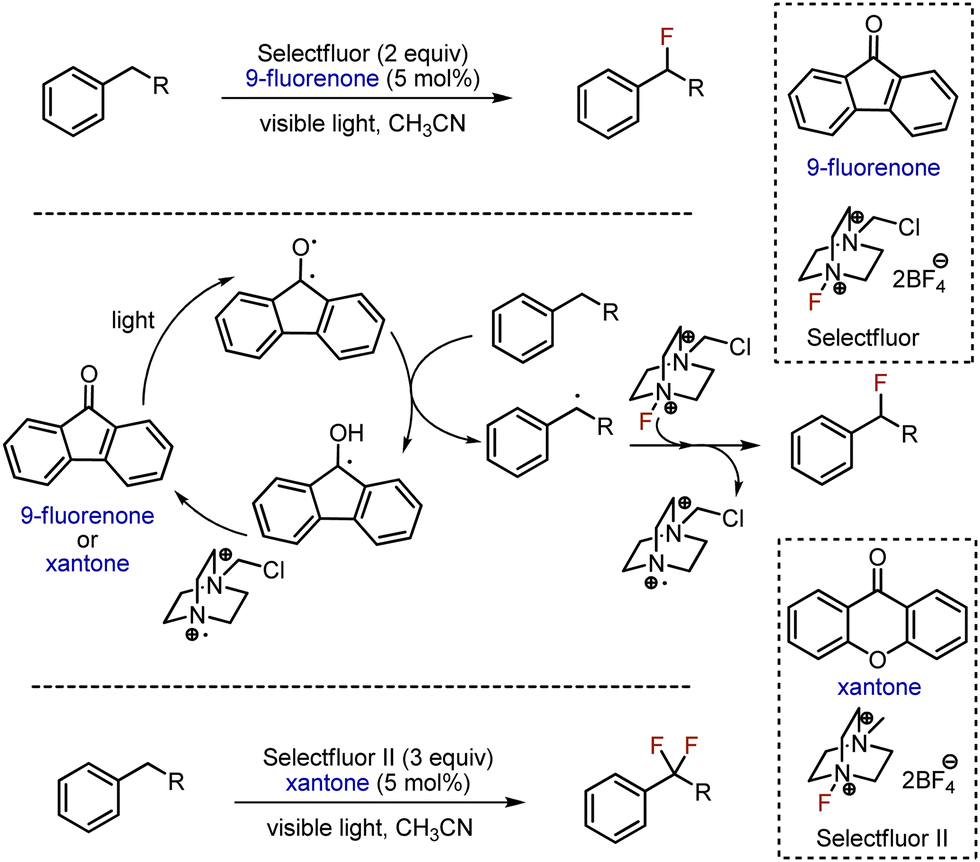
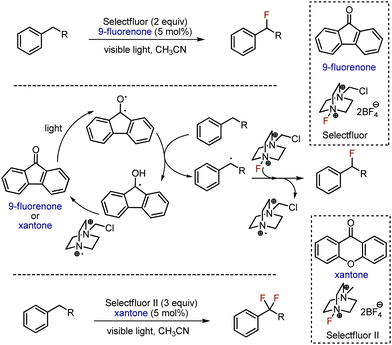
Direct fluorination of hydrocarbons using gaseous fluorine is unsafe and poses significant challenges, prompting only a limited number of laboratories around the world to be authorized to work with this reagent. Typically, safer alternatives such as Et3N-nHF or Selectfluor, are used instead. Combined with the mild and sustainable energy source as visible light, these reagents were successfully employed for alkane fluorination. The first fluorination of an aliphatic C–H bond under visible light irradiation was achieved in 2013 (Scheme 1).13 The benzylic position was fluorinated with Selectfluor as fluorine source and fluorenone-9 as photocatalyst, under the irradiation with visible light. Interestingly, the reaction of ethylbenzene was faster than of toluene (primary carbon) or cumene (tertiary carbon). Using Selectluor II and xantone, two benzylic protons could be replaced by fluorines. Photoexcited aromatic ketone abstracted hydrogen atom from benzylic position, and the generated radical reacted with Selectfluor, giving fluorinated product. The photocatalyst was recycled by cation-radical from Selectfluor. Alternatively, benzylic fluorination was achieved in 2017 employing acridinium salt as photocatalyst.14
 |
| | Scheme 1 Fluorination of benzylic C–H bonds. | |
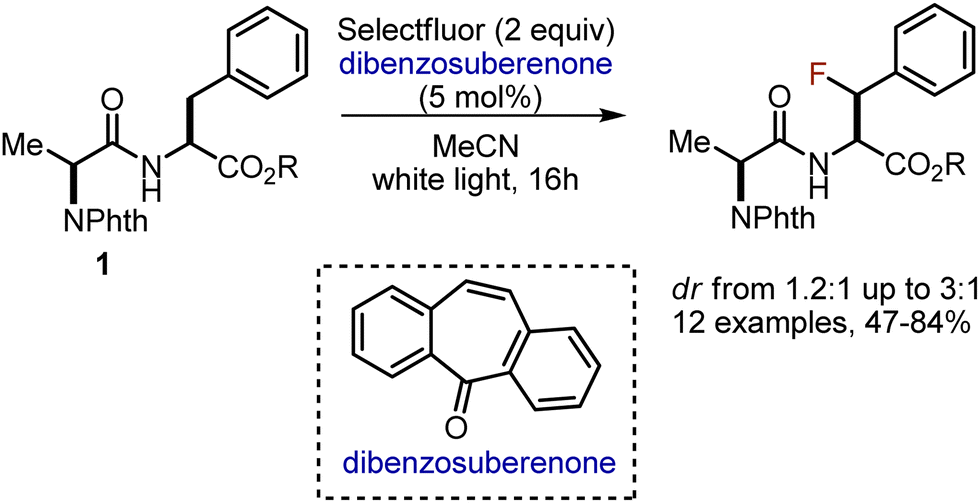
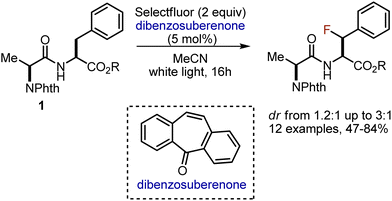
Later, the approach was adopted to functionalise benzylic positions of Phe- and Tyr-containing dipeptides 1, employing dibenzosuberenone as photosensitiser (Scheme 2).15
 |
| | Scheme 2 Fluorination of dipeptides. | |
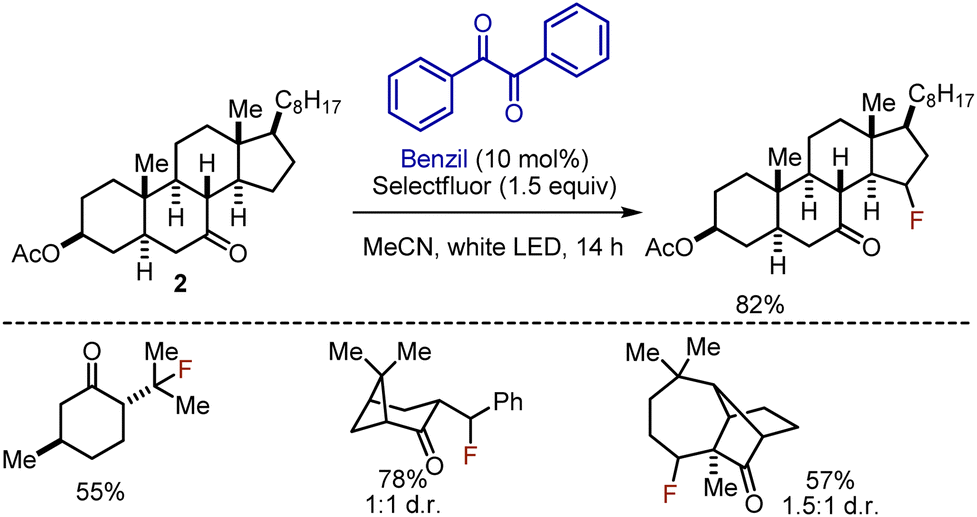
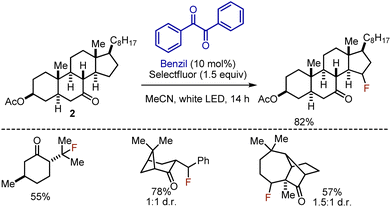
The polar effect of the carbonyl group could be utilized to perform selective photoinduced β- or γ-fluorinations on rigid cyclic ketones.16 Ketone substrates 2 were irradiated with white LED in the presence of Selectfluor as fluorine source and benzil as photosensitizer in acetonitrile for 14 h (Scheme 3). The reaction has been prevalently employed for steroidal ketones, but other mono-, di-, and tricyclic ketones could be fluorinated as well. Continuous flow approach was later developed.17
 |
| | Scheme 3 Organophotoredox fluorination of ketones. | |
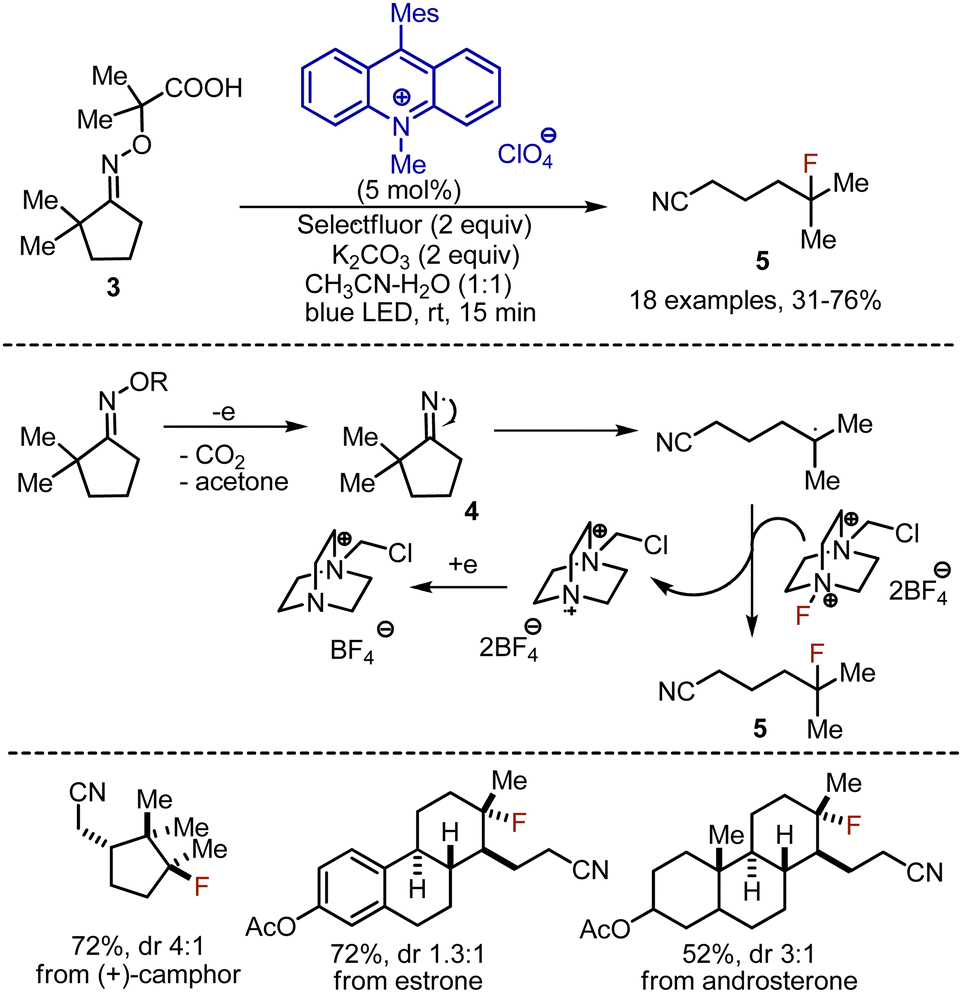
Distal fluorination via 1,5-hydrogen atom transfer (HAT) approaches has been developed for various compound classes.18 In 2018 Leonori et al. reported a cascade of oxidation-ring opening-fluorination reactions of oximes 3 under visible light-mediated conditions (Scheme 4).19 Acridinium salt was used as photocatalyst, which oxidised the carboxylic group into an oxime, leading to the release of CO2 and acetone. The thus formed iminyl radical 4 underwent ring opening reaction to give a nitrile moiety and a C-centered radical. The latter abstracted fluorine from Selectfluor, forming the product 5. The catalytic cycle was closed by single electron transfer (SET) reduction of a cation radical from Selectfluor with the reduced form of the acridinium catalyst. Short-lived radical chain propagation processes were found to be acting along with the photoredox pathway according to the quantum yield of the reaction measurements (Φ = 2.8). Oximes derived from cyclobutanones reacted smoothly, whereas oximes derived from cyclopentanones, cyclohexanones, and cycloheptanones required an aromatic substituent in the α-position to stabilize the resulting radical. Interestingly, various natural products, like (+)-camphor, estrone, androsterone, etc. could be used.
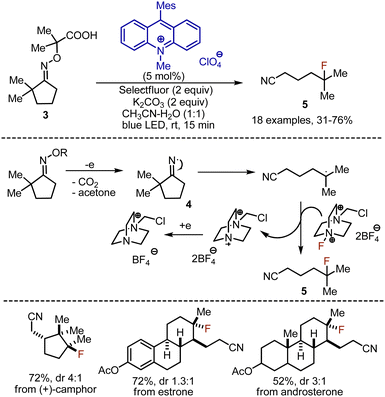 |
| | Scheme 4 Fluorination through iminyl radical formation. | |
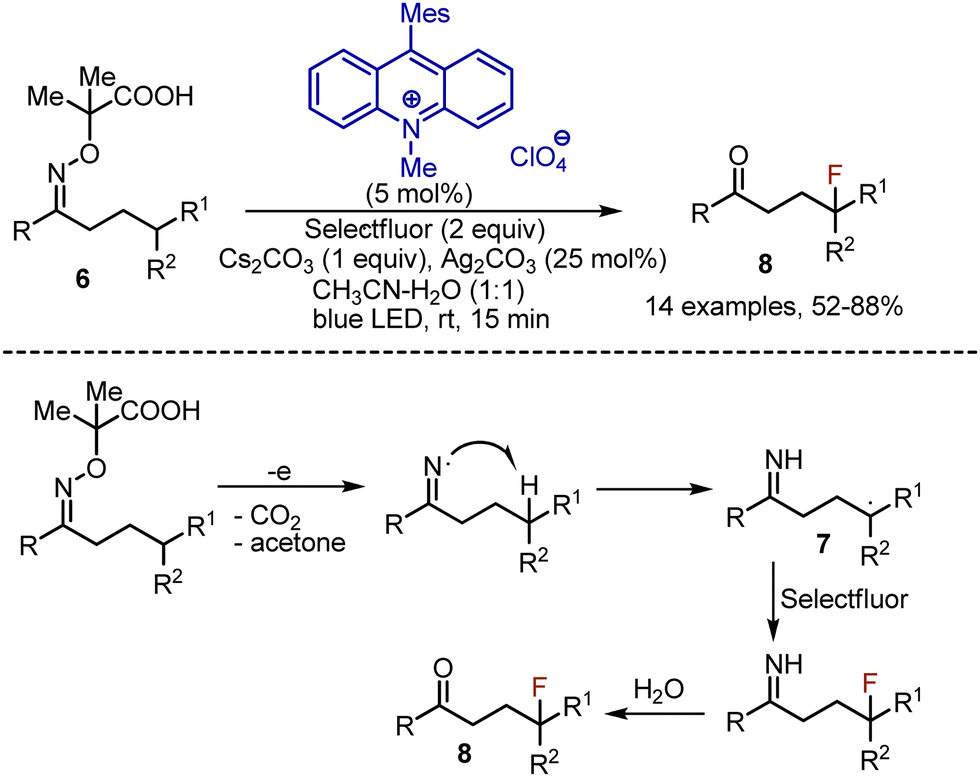
Linear oxime derivatives 6 could be used as well, which were transformed into imines 7 instead of nitriles, and eventually hydrolyzed into ketones 8 (Scheme 5). This process was compatible with the substrates, delivering tertiary carbon-radicals only due to the small difference in bond dissociation energies (BDE) of iminyl N–H and C–H bonds. If Selectfluor was replaced by NCS, chlorination took place, but hydrolysis of the imine did not occur.
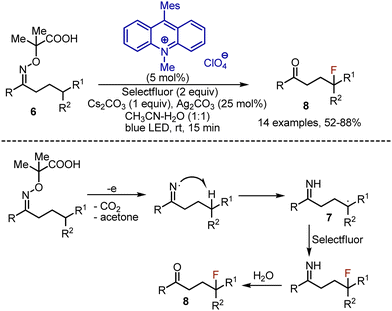 |
| | Scheme 5 Fluorination-imine hydrolysis through iminyl radical formation. | |
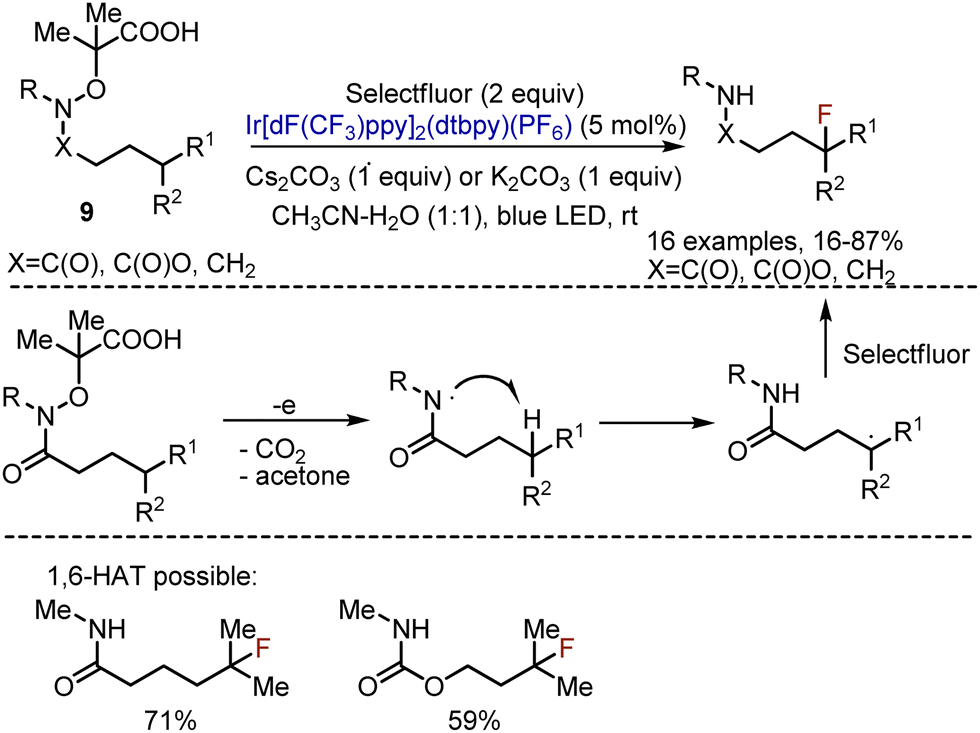
To overcome the energetical limitation of iminyl radicals, the analogous fluorination of secondary carbon centers was performed with the help of amidyl radicals, formed from amides, carbamates or amines 9 (Scheme 6).20 The process was operating through 1,5-HAT from C–H to the amidyl radical. The latter was formed analogously by the oxidation of the carboxylic group in the corresponding precursor via a photoredox cycle. In a standard procedure the substrate, Selectfluor, Ir-catalyst, and a base were irradiated with blue LED at rt in an CH3CN–water mixture. As far as amidyl radicals were involved, secondary substrates were reactive (R1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H). Protected amines participated in the reaction as well (9, XCH2, R = Boc or Ts). Despite 1,5-HAT was usually favoured, the 1,6-transposition could also be achieved for the substrates, bearing substituents at the corresponding positions (Scheme 6). Later, N-sulfonyl protected amines were fluorinated under the action of hypervalent iodine(III) reagent21 or with the use of a cheaper Ru(bpy)3Cl2 catalyst.22
H). Protected amines participated in the reaction as well (9, XCH2, R = Boc or Ts). Despite 1,5-HAT was usually favoured, the 1,6-transposition could also be achieved for the substrates, bearing substituents at the corresponding positions (Scheme 6). Later, N-sulfonyl protected amines were fluorinated under the action of hypervalent iodine(III) reagent21 or with the use of a cheaper Ru(bpy)3Cl2 catalyst.22
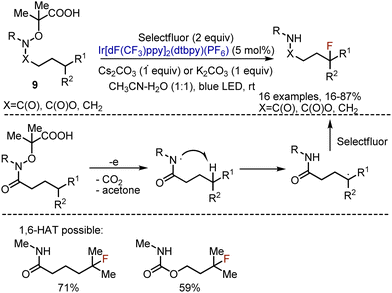 |
| | Scheme 6 Fluorination through amidyl radical formation. | |
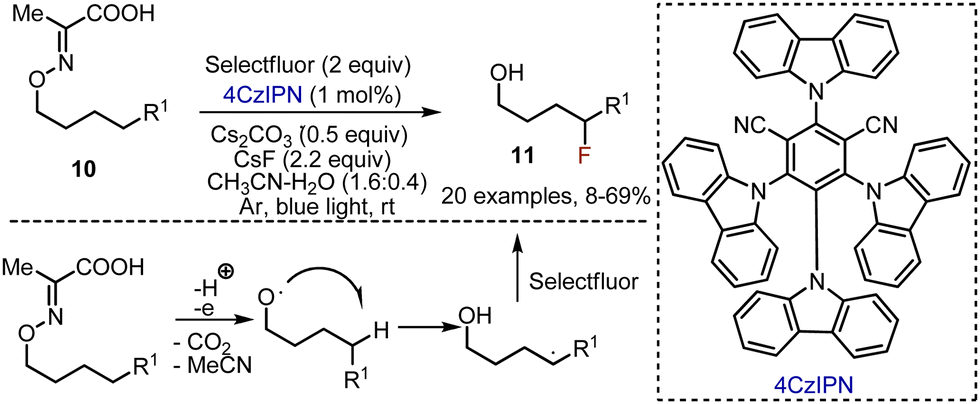
The approach with 1,5-radical translocation was interestingly used for the synthesis of δ-fluorinated alcohols by Yu et al.23 Various oxime derivatives of pyruvic acid 10 were irradiated in the presence of Selectfluor and (4s,6s)-2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) photocatalyst in an acetonitrile–water mixture at rt under argon atmosphere (Scheme 7). Caesium fluoride and carbonate were found to be a crucial mixture of bases to maintain the acceptable pH of the reaction mixture. To ease the isolation of the alcohols 11, many of the products were in situ converted into the corresponding carbamates by the addition of 4-nitrophenyl isocyanate to the reaction mixture. The reaction was wide in scope, and worked smoothly to deliver fluorinated alcohols with aromatic, heterocyclic, cyclic, azide moieties, etc.
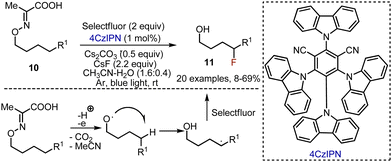 |
| | Scheme 7 Visible light-mediated synthesis of δ-fluorinated alcohols. | |
To involve non-activated alkanes in fluorination, shorter wavelength sources (375–400 nm) were used together with acetophenone,24 tetra-n-butylammonium decatungstate (TBADT),25 1,2,4,5-tetracyanobenzene,26 anthraquinone,27 or uranyl compounds28–30 as photosensitisers.
2.2. Fluorination of unsaturated compounds
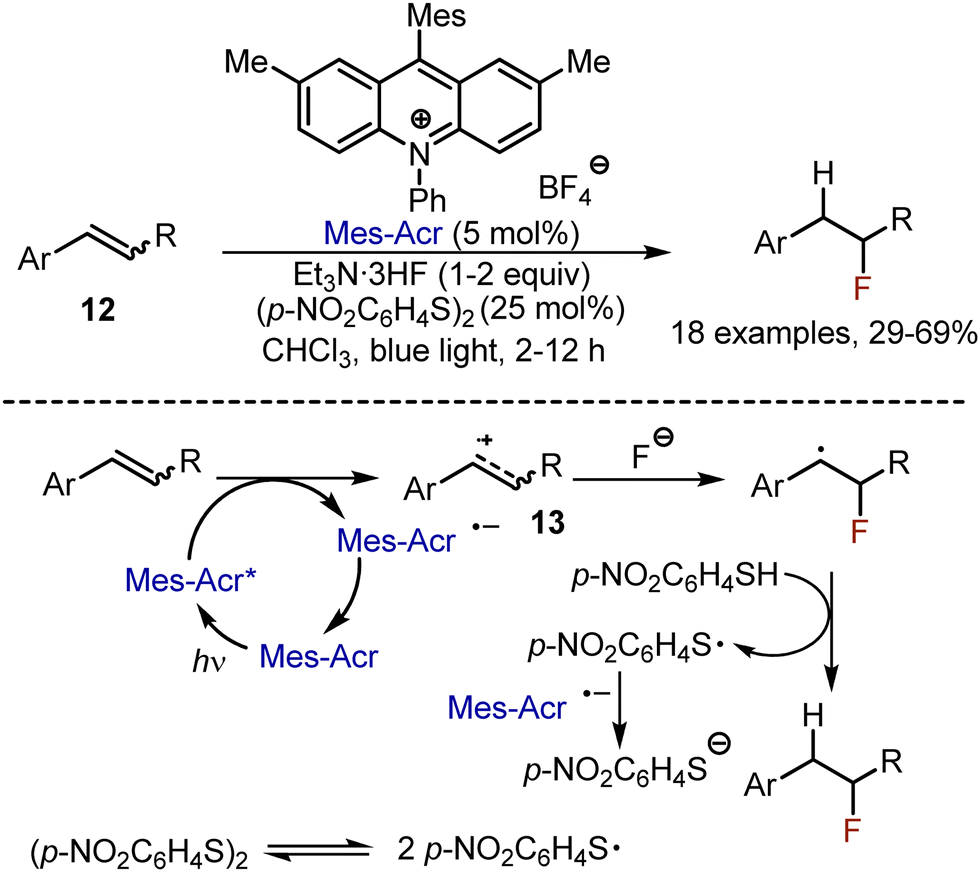
Photoredox-catalysed anti-Markovnikov hydrofluorination of alkenes 12 has been developed by Nicewicz in 2014.31 Styrenes were irradiated by blue light in the presence of Et3N·3HF (1–2 equiv.), 4-nitrophenyl disulfide (25 mol%) in chloroform, employing 9-mesityl-2,7-dimethyl-10-phenylacridinium salt (Mes-Acr) as photocatalyst (Scheme 8). The reaction started with the oxidation of the alkene by the photo-excited catalyst, delivering a cation radical 13, which could react with a fluoride anion. The recovery of the catalyst was achieved by reducing the phenylsulfide radical to its anion. The latter could get protonated and participate in a HAT process, quenching the alkyl radical. The reaction was limited to aryl and thienyl conjugated alkenes.
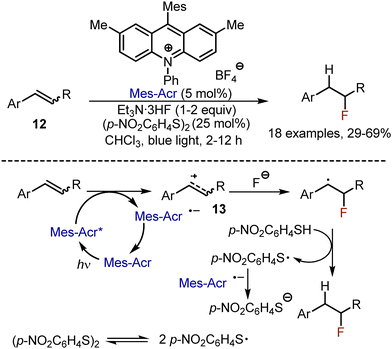 |
| | Scheme 8 Visible light-mediated hydrofluorination of alkenes. | |
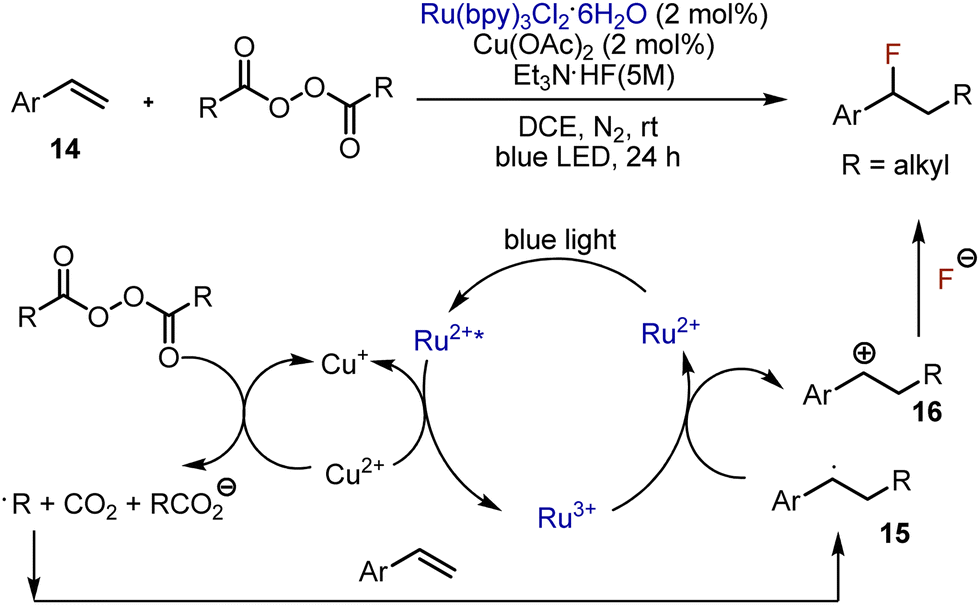
Various visible light-mediated fluorofunctionalizations are available for aryl alkenes. Alkylfluorination of styrenes was developed in 2018 by Li, Bao et al.32 The combination of photocatalyst Ru(bpy)3Cl2 and copper acetate was used to generate the alkyl radical from an acyl peroxide (Scheme 9). The alkyl radical reacted with styrene 14, forming a benzyl radical 15 which was subsequently oxidized by Ru3+ to produce the corresponding carbocation 16. The latter reacted with fluoride anion, delivering the final product. The proposed mechanism was supported by radical trapping and radical clock experiments. Moreover, when the reaction was performed in the presence of methanol, the respective ether was formed as a by-product, confirming the involvement of carbocationic species. The reaction worked smoothly with various styrenes bearing electron-donating or -withdrawing substituents. Disubstituted alkenes worked as well. Interestingly, thiazolyl-, and alkynyl-conjugated alkenes worked too. The scope of acyl peroxides was limited to aliphatic derivatives.
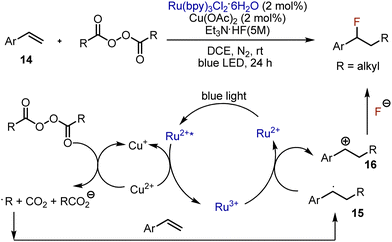 |
| | Scheme 9 Alkylfluorination of styrenes. | |
Fluorodifluoroacetylation of styrenes 17 was achieved by the action of Chen's reagent FSO2CF2CO2Me in the presence of an Ir-complex (Scheme 10).33
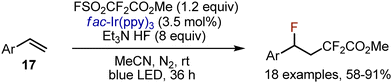 |
| | Scheme 10 Visible light-mediated fluorodifluoroacetylation of styrenes. | |
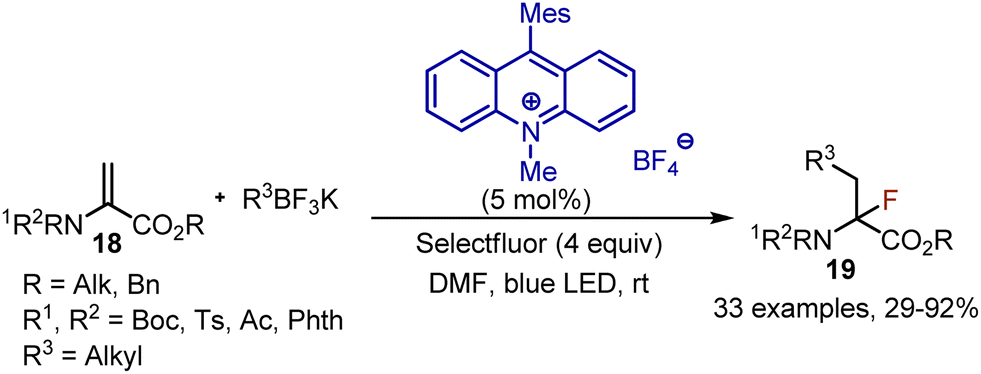
Synthesis of fluorinated α-amino acid derivatives was developed through a photoredox-catalysed alkylfluorination of dehydroaminoacids 18.34 When dehydroalanine and potassium alkyltrifluoroborate were subjected to irradiation with blue LED in the presence of acridinium salt as a photocatalyst, the desired α-fluoro-α-amino acid derivatives 19 were isolated with moderate to excellent yields (Scheme 11). Initially, the photoexcited acridinium salt oxidized trifluoroborate to generate an alkyl radical. The alkyl radical is added to the double bond of dehydroalanine, and the resulting radical can then abstract fluorine from Selectfluor to complete the transformation. The recyclization of the photocatalyst was achieved by the reaction of Acr-Mes with Selectfluor cation radical.
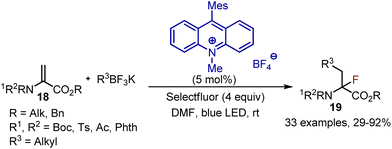 |
| | Scheme 11 Fluoroalkylation with alkyltrifluoroborates. | |
Visible light-mediated fluorination of non-activated alkenes has not yet been developed.
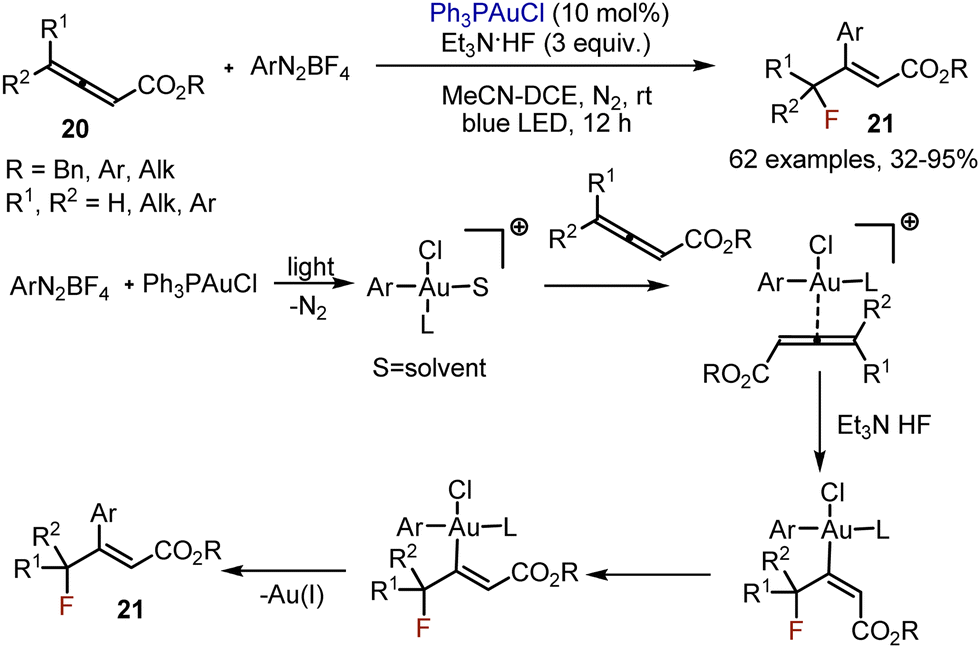
Fluorofunctionalisation of allenic esters 20 was achieved with the help of gold-complexes.35 Aryldiazonium salts were used as aryl source, while Et3N·3HF was employed as fluorine source (Scheme 12). The following mechanism was proposed. The reaction started with the photomediated oxidative addition between the gold-complex and the aryldiazonium salt. The resultant gold-species coordinated with an allenic ester. This was followed by hydrogen bonding assisted nucleophilic addition of a fluoride anion. Isomerization of the double bond was followed by reductive elimination, giving the desired product 21, and regenerating the Au(I)-catalyst.
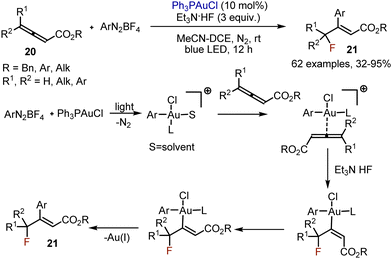 |
| | Scheme 12 Visible light-mediated fluoroarylation of allenes. | |
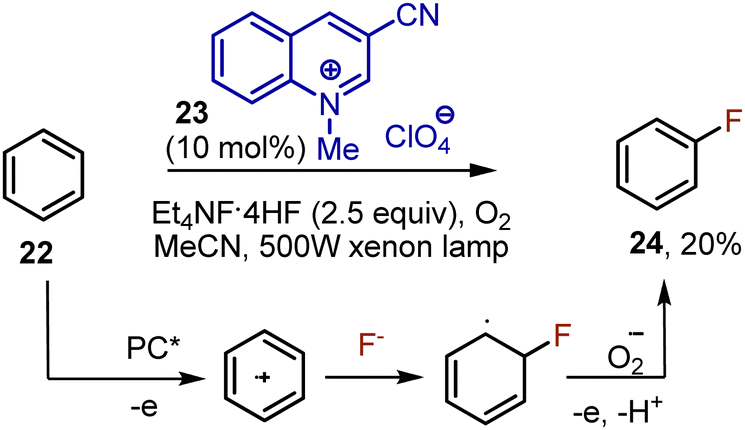
The direct visible light-mediated fluorination of aromatic Csp2–H bonds is still an underdeveloped area. In 2013, Fukuzumi reported the fluorination of benzene 22 catalyzed by quinolinium 23.36 Tetraethylammonium hydrofluoride was used as the source of fluoride, resulting in the formation of fluorobenzene 24 with a 20% yield. (Scheme 13). The irradiation with a powerful xenon lamp was needed. The photoexcited catalyst oxidized benzene, giving a cation radical, which reacted with a fluoride anion. Oxygen recyclized the photocatalyst being transformed into an oxygen anion radical, which was further aromatizing the intermediate. The fluorination of halogenated benzenes was less effective, bromo- and chlorobenzene were para-fluorinated with only 6% and 7% yields, respectively.
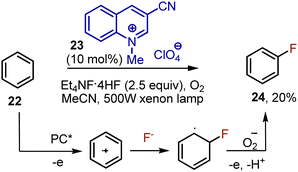 |
| | Scheme 13 Quinolinium-catalysed fluorination of benzene. | |
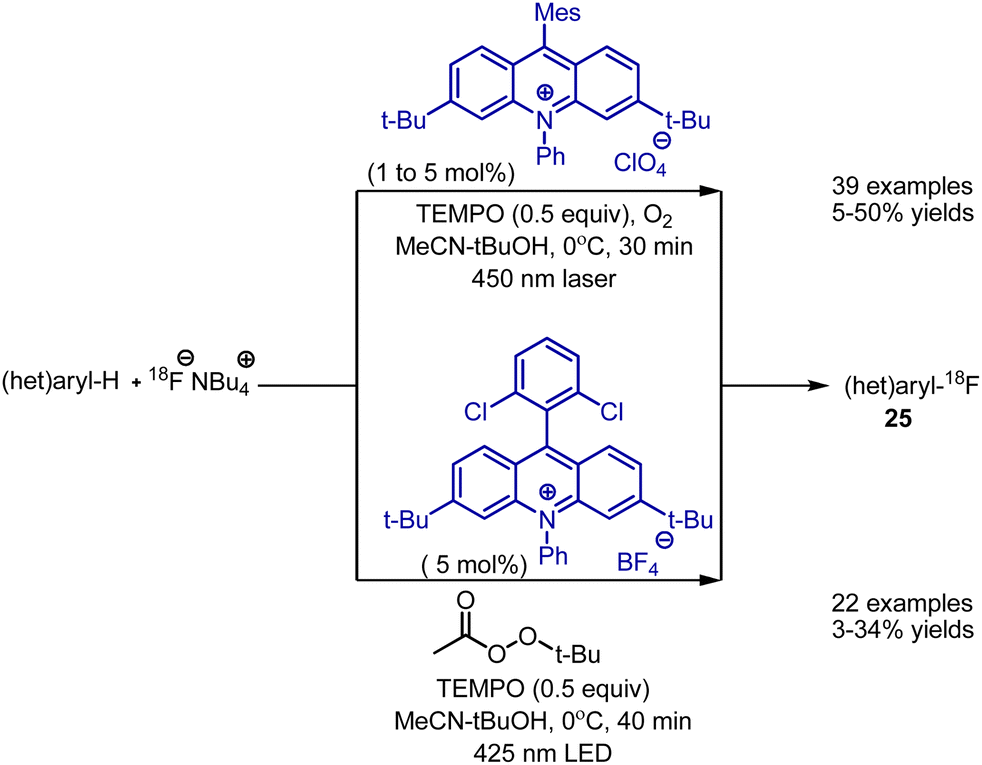
Direct radiofluorination with nucleophilic fluoride was developed by Nicewicz et al. In the original report, 18FNBu4, acridinium catalyst, TEMPO and laser irradiation was employed to generate radio-labelled fluorobenzenes 25 (Scheme 14).37 The approach was also based on SET oxidation of an aromatic compound to the corresponding radical cation, further reacting with a fluoride anion. The process worked with electron-rich and electron-poor aromatic systems, heterocycles, and bioactive molecules. Moreover, the reaction was employed on positron emission tomography (PET) tracers. Later, the procedure was optimized to the use of simple 425 nm LED irradiation, as well as tert-butyl peroxyacetate as an oxidant instead of oxygen.38 Importantly, this approach was also realized in continuous flow.
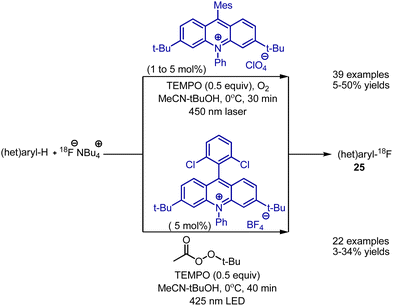 |
| | Scheme 14 Visible light-mediated radiofluorination of aromatic compounds. | |
2.3. Decarboxylative fluorination
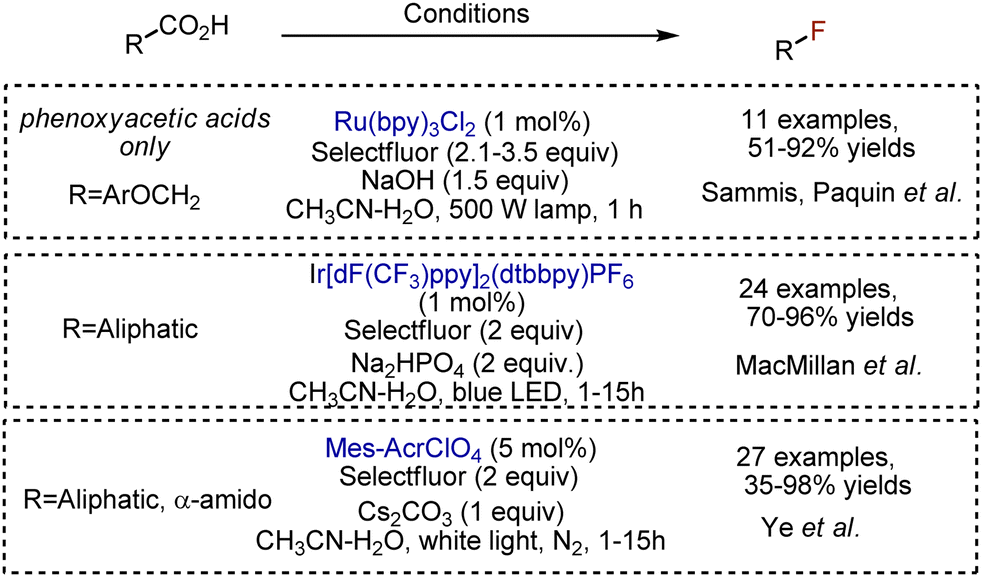
A carboxylic acid is an abundant group, and many decarboxylative functionalisations are described.39,40 Fluorination is not an exception and could be achieved under visible light. Single-electron oxidation of a carboxylate anion to a carboxylic radical is followed by extrusion of CO2 and the formation of an alkyl radical, capable to abstract fluorine from Selectfluor. Sammis, Paquin et al. reported the modification of arylacetic acids, catalyzed by Ru(bpy)3Cl2 photocatalyst (Scheme 15).41 The presence of an oxygen atom could facilitate the initial oxidation and starts the process. Aliphatic acids were fluorodecarboxylated with broad scope by MacMillan et al., employing an Ir-catalyst.42 In this transformation, the photoexcited Ir-complex was quenched by Selectfluor, delivering strongly oxidizing Ir[dF(CF3)ppy]2(dtbbpy)2+, capable to oxidize the carboxylate. The reaction worked with various acids, and primary, secondary, and tertiary fluorides were synthesized with 70–96% yields. Analogous fluorination worked with acridinium salts.43 The addition of bases is needed to form the carboxylate anion, which is easier oxidized. Alternatively, phthalimide esters of carboxylic acids could be used for visible light-mediated Ir-catalysed fluorinations.44
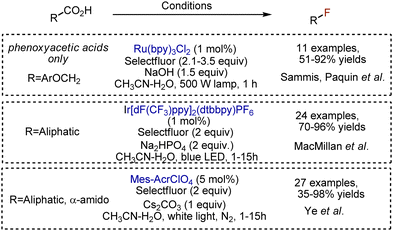 |
| | Scheme 15 Decarboxylative fluorinations. | |
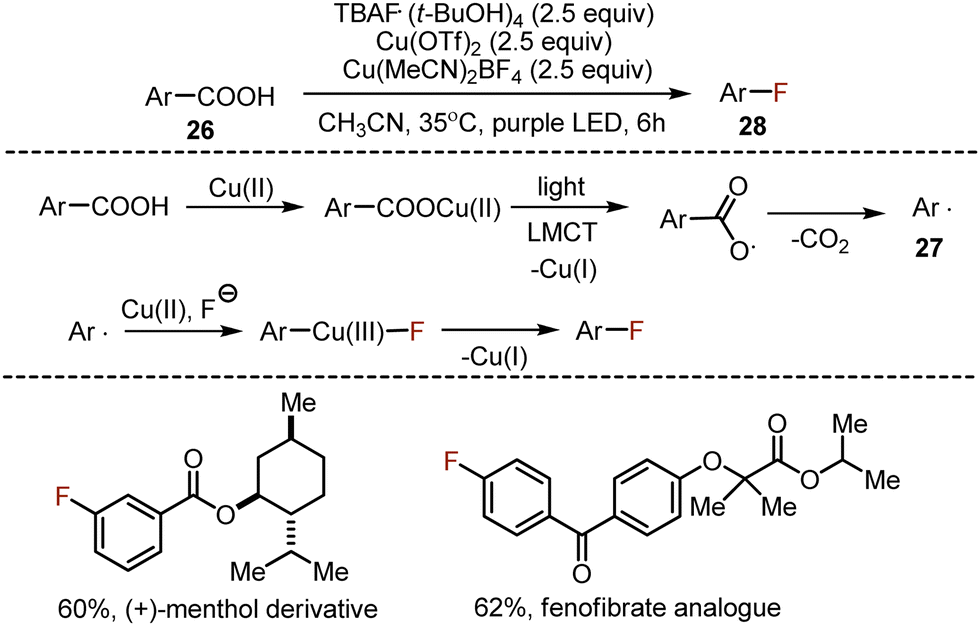
The decarboxylative fluorination of benzoic acids is deteriorated by a low nucleophilicity of the aryl radical, and the consequent reluctance of the latter to react with electrophilic fluoride sources. An interesting and conceptually novel approach was suggested for the decarboxylative halogenation of aromatic carboxylic acids 26, based on a photoinduced ligand-to-metal charge transfer mechanism (LMCT) (Scheme 16).45 Presumably, copper(II) formed a salt with the acid, which underwent photoexcitation, leading to a single electron transfer from the carboxylic anion to copper(II), giving the carboxylate radical and Cu(I). The carboxylate radical was capable of decarboxylation to form an aryl radical 27. The latter could be trapped by Cu(II), and in the presence of fluoride anions aryl copper(III) fluoride species were formed. The reductive elimination from Cu(III) coordination compound delivered the desired aryl fluoride 28. This transformation was wide in scope, and the opportunities for late stage functionalization were successfully demonstrated by the syntheses of drug-like molecules.
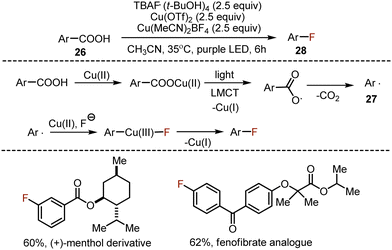 |
| | Scheme 16 Decarboxylative fluorination via a ligand-to-metal charge transfer mechanism. | |
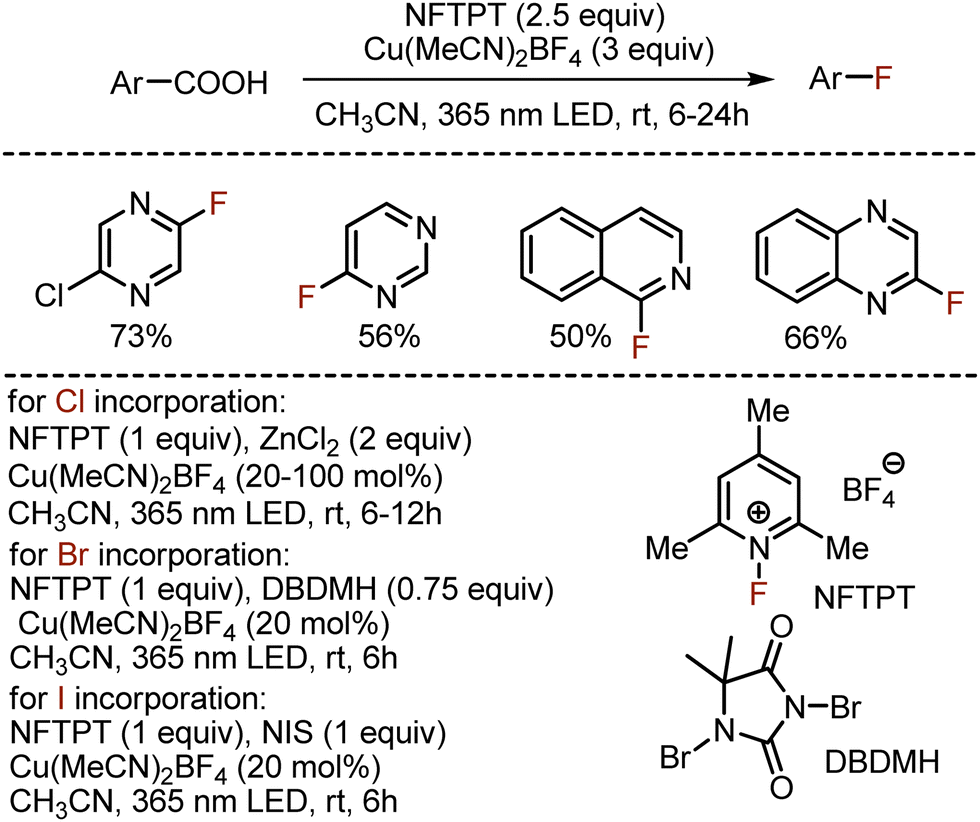
Later, MacMillan could extend the approach to all other halogenations (Scheme 17).46 Instead of a nucleophilic fluorine source, the electrophilic 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT) was used, also playing the role of oxidant. The method worked smoothly not only for aromatic carboxylic acids, but various heterocyclic derivatives could be readily modified. Chlorination smoothly occurred with zinc chloride as chlorine source, while use of 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) led to bromination products. The iodination was achieved with N-iodosuccinimide (NIS).
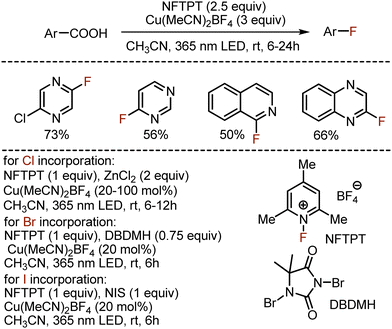 |
| | Scheme 17 General decarboxylative halogenation approach. | |
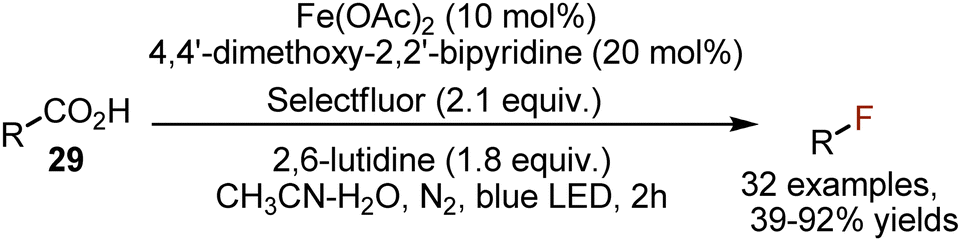
A cheaper non-noble iron catalyst was recently used for decarboxylative fluorination, based on an LMCT mechanism (Scheme 18).47 This transformation was applicable for aliphatic acids 29 only.
 |
| | Scheme 18 Iron-catalysed decarboxylative fluorination. | |
2.4. Deoxygenative fluorination
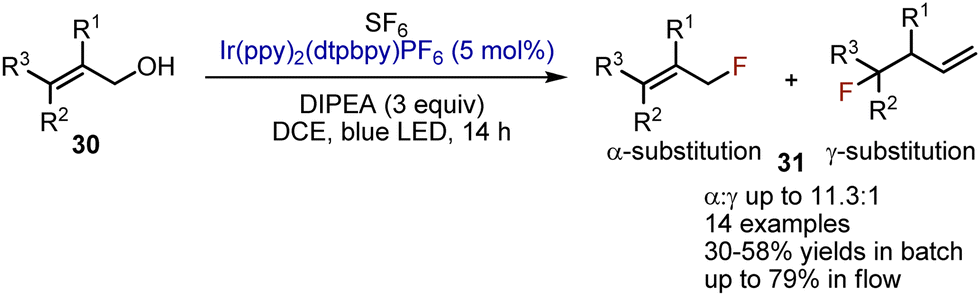
The use of safe and inexpensive, but inert SF6 for deoxygenative fluorination has been reported by Jamison in 2016.48 Allylic alcohols 30 could be transformed into allyl fluorides 31 with moderate yields when irradiated with blue LED-light in DCE in the presence of SF6, Ir(ppy)2(dtbbpy)PF6 (5 mol%), and DIPEA (3 equiv.) (Scheme 19). Interestingly, when performed under continuous flow conditions, the yields were improved.
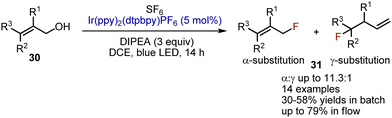 |
| | Scheme 19 Deoxygenative fluorination of allylic alcohols. | |
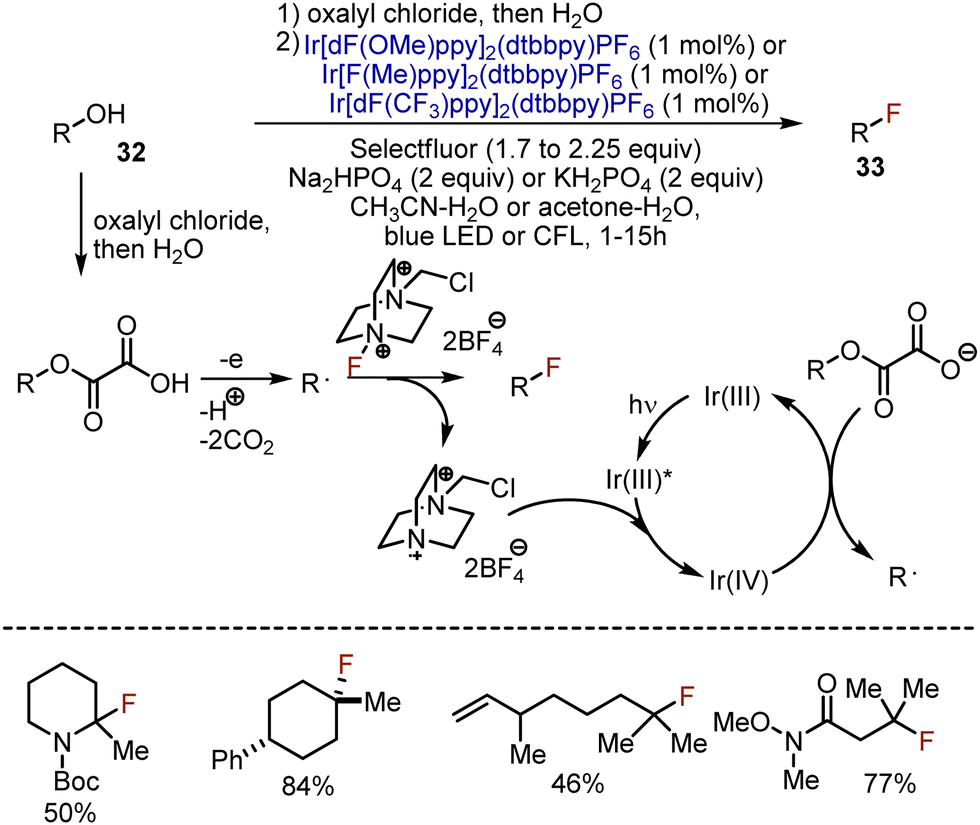
Replacement of the hydroxyl group in primary and secondary alcohols was established through SN2-type reactions, while tertiary alcohols were still challenging substrates. On the other hand, photoredox conditions employ free radical reactivity compatible with tertiary substrates. MacMillan et al. took advantage of this chemistry and developed a visible light-mediated deoxygenative fluorination for a broad scope of alcohols 32 (Scheme 20).49 Initially, oxalic esters of alcohols were used in the reactions with an electrophilic fluorine source – Selectfluor – and various Ir photocatalysts. The addition of an inorganic base was crucial for the reaction to generate salts from oxalate derivatives. Importantly, the prefunctionalisation of an alcohol with oxalyl chloride could be performed in a one-pot fashion, and the isolation of the ester was not needed. The reaction started with the acylation of the alcohol with oxalyl chloride. The photoexcited Ir(III)* reduced the Selectfluor cation radical, delivering Ir(IV) which was capable of oxidizing the carboxylate anion. The single electron oxidation of the carboxylate led to the extrusion of CO2 (2 equiv.) and the formation of an alkyl radical, which abstracted the fluorine atom from Selectfluor to generate the final product 33. The most valuable aspect of the transformation was the possibility to smoothly involve tertiary alcohols to give fluorinated derivatives with good to excellent yields.
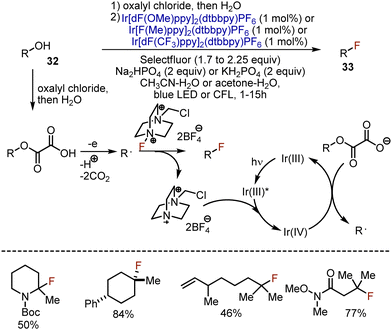 |
| | Scheme 20 Deoxygenative fluorination through initial oxalyl chloride acylation. | |
3. Chlorination
3.1. Chlorination of Csp3–H bonds
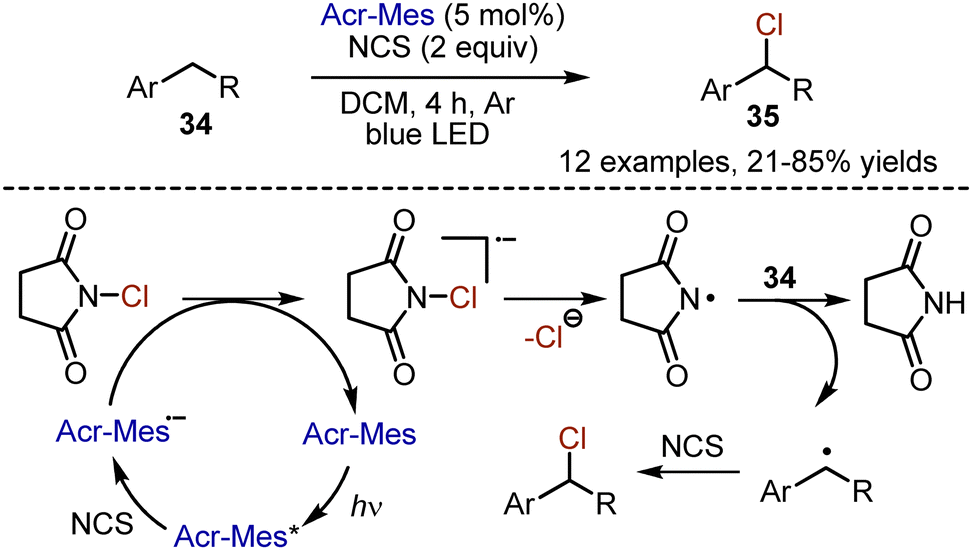
Selective chlorination with molecular chlorine is a challenging task due to the high reactivity of this halogen. Moreover, its toxicity and corrosive character complicate the transformations. A safer alternative for chlorination employs oxidation of chlorides, but the use of stoichiometric oxidants is needed. Visible light-mediated approaches suggest a more sustainable way for radical generation. In 2020, Wu et al. reported a visible light-mediated procedure for the chlorination of the benzylic position with NCS (Scheme 21).50 Alkylbenzene 34 was irradiated with blue LED-light for 4 h in DCM solution, in the presence of acridinium salt as photocatalyst. The benzyl chlorides 35 were generated with 21–85% yields. It is proposed that the photoexcited acridinium salt oxidizes NCS and is transformed into a strongly reducing species, which gives an electron to NCS to generate the NCS anion radical. Subsequently this succinyl radical, abstracts a hydrogen atom from the alkyl benzene. This benzyl radical abstracts chlorine from NCS to give the final product.
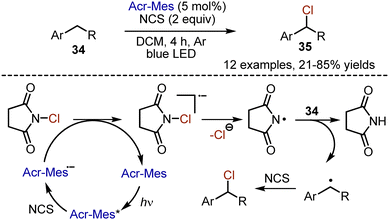 |
| | Scheme 21 Chlorination of alkylbenzenes with NCS. | |
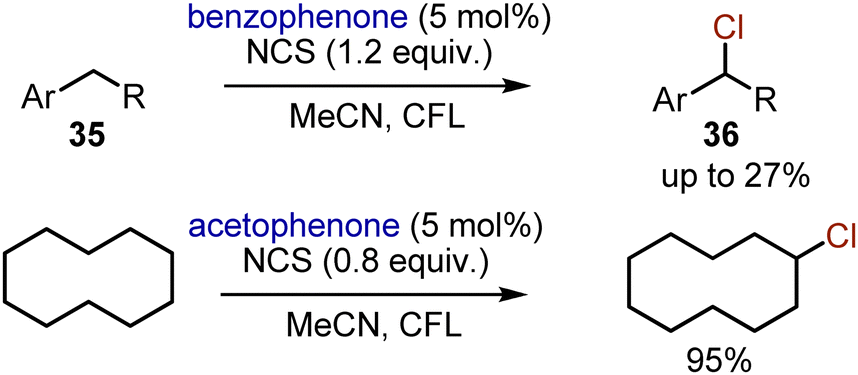
When alkylbenzenes 35 were subjected to chlorination with NCS employing aromatic ketones as photocatalysts, the yields of the benzyl chlorides 36 were not exceeding 27%.51 Moreover, double chlorination could occur. Still, this approach could be very useful for visible light-mediated chlorination of non-activated alkanes. For instance, cyclodecane could be mono-chlorinated in MeCN with 95% yield with NCS (0.8 equiv.) and acetophenone as a photocatalyst under irradiation with white CFL light (Scheme 22).
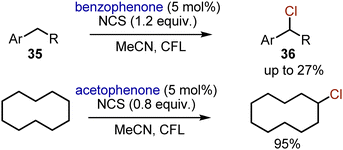 |
| | Scheme 22 Aromatic ketones sensitized chlorination of alkanes. | |
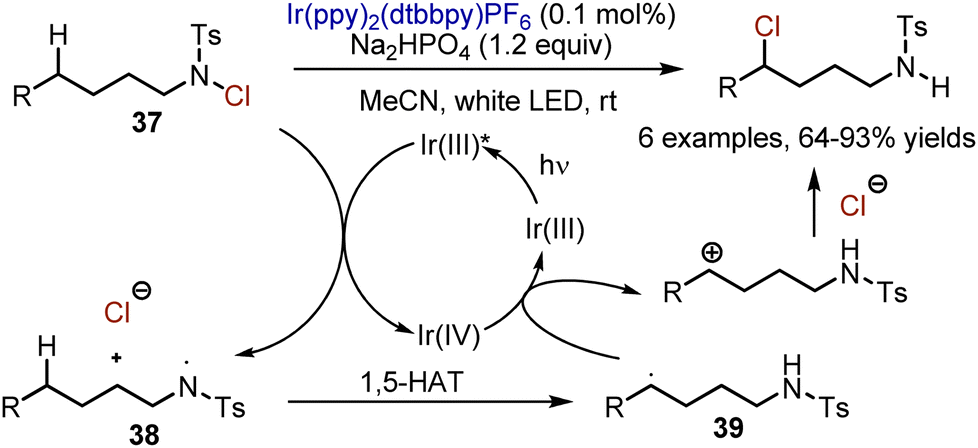
UV-light-mediated chlorination, followed by intramolecular chloride substitution with nitrogen is known as a Hofmann–Löffler–Freytag (HLF) reaction, operating through 1,5-hydrogen atom transfer from Csp3–H to an amidyl radical. In 2015, Yu and Qin developed an interrupted HLF reaction under visible light irradiation in the presence of Ir(ppy)2(dtbbpy)PF6 catalyst (Scheme 23).52 Photoexcited Ir(III) reduced N-chlorotosylamide 37 so it was transformed into an amidyl radical 38 and a chloride anion. The amidyl radical 38 could undergo intramolecular 1,5-HAT to deliver a carbon radical 39. This radical could be oxidized by Ir(IV) into a carbocation, which was quenched by a chloride.
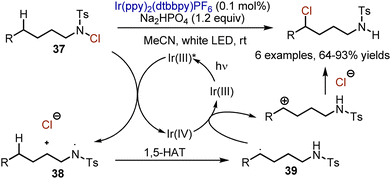 |
| | Scheme 23 Intramolecular 1,5-chlorination in N-chlorosulfonamides. | |
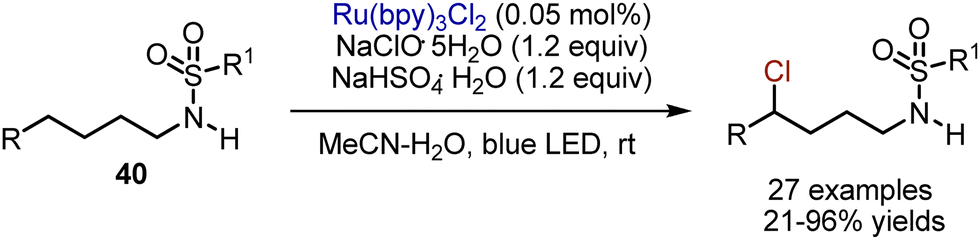
Later, preliminary non-chlorinated sulfonamides 40 could be involved in an analogous transformation (Scheme 24).53 The initial N-chlorination took place in situ in the presence of NaClO·5H2O, while further amidyl radical generation-1,5-HAT and carbocation formation were promoted by Ru(bpy)3Cl2 photocatalyst under blue LED irradiation. Analogous transformation could also be performed with t-BuOCl as chlorine source, and Mn2(CO)10 as a photocatalyst.54
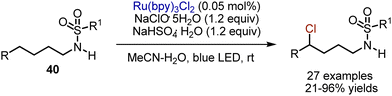 |
| | Scheme 24 Visible light-mediated chlorination of sulfonamides. | |
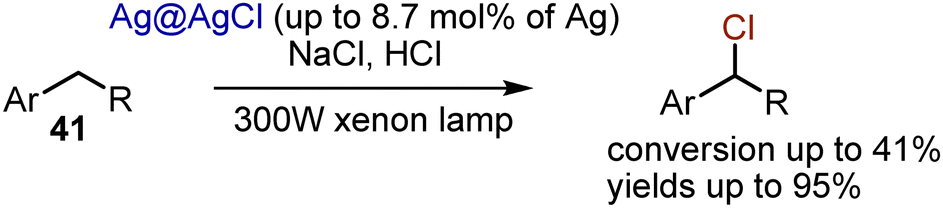
N-Chlorosuccinimide and analogous reagents are abundant chlorine sources. Still, the use of various chlorides, like NaCl, could be considered beneficial due non-toxic and low-cost qualities. Few methods for visible light-mediated chlorination with NaCl are known. For instance, alkylbenzenes 41 could be chlorinated by NaCl in the presence of a nano composite Ag@AgCl catalyst and irradiation with a xenon lamp, equipped with a UV cut-off filter (Scheme 25).55 Although good selectivity with excellent chemical yields were achieved, the conversions were not high (up to 41%). Later, it was shown that the addition of a phase transfer catalyst like NBu4Cl improved the performance of the nano composite catalyst.56
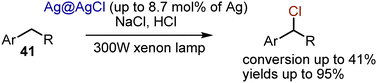 |
| | Scheme 25 Nano composite Ag catalyst for visible light-induced alkane chlorination. | |
Higher conversions were demonstrated for oxone-mediated chlorinations with NaCl (Scheme 26).57 However, as electrophilic aromatic substitution could take place under these conditions, the solvent choice was crucial for the selectivity. This approach was successful for the chlorination of non-activated alkanes 42 as well. The role of oxone (2KHSO5·KHSO4·K2SO4) was to oxidize chloride to generate chlorine Cl2. Without light, the yields of the products 43 were not higher than 4%, showing the need for the irradiation.
 |
| | Scheme 26 NaCl-oxone mediated chlorination of alkanes. | |
Ferrous chloride was also used as a source of chlorine in visible light-mediated chlorination of cyclohexane.58
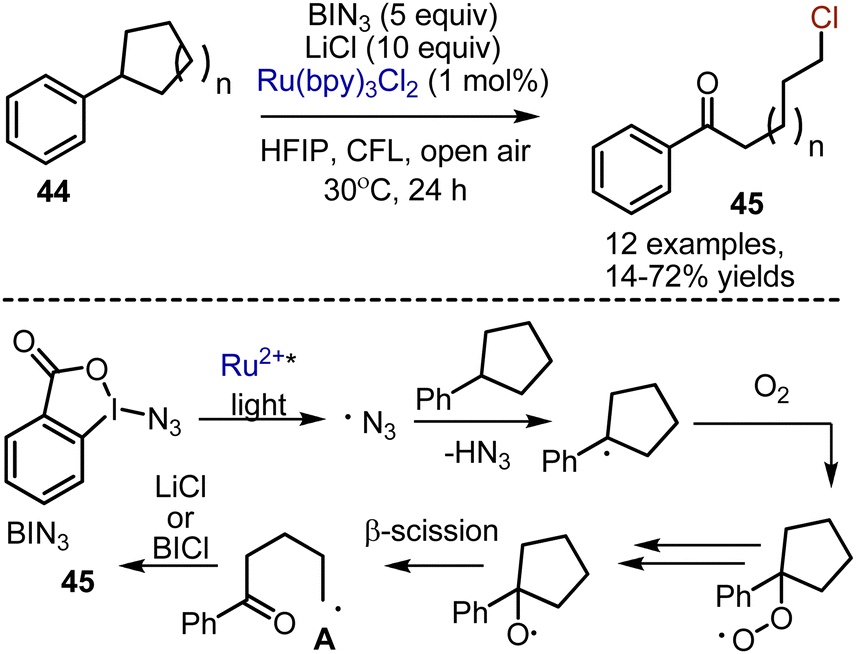
Visible light-mediated deconstructive chlorination of aryl cycloalkanes 44 was developed recently, leading to the formation of chlorinated arylketones 45 (Scheme 27).59 The reactions of aryl cycloalkane were performed in the presence of Zhdankin reagent BIN3 – hyper valent benziodoxolone, – Ru(bpy)3Cl2 as a photocatalyst and LiCl as a chlorine source. Along with the desired chlorinated products, azidation usually took place. The following pathway for the transformation was suggested. Benzyl radical was generated by azido radical, which was produced from BIN3 by SET reduction with photoexcited Ru2+*. Subsequent interaction with oxygen was followed by HAT and hydroperoxide homolysis to give an alkoxy radical. The latter underwent β-scission to deliver intermediate A, and the chlorination took place. It was suggested that the chlorine atom could have been finally abstracted from in situ formed BICl species. The formation of BICl was confirmed by mixing BIN3 with LiCl. Alternatively, the oxidation of A by Ru3+ into a carbocation, followed by ionic reaction with LiCl could be proposed.
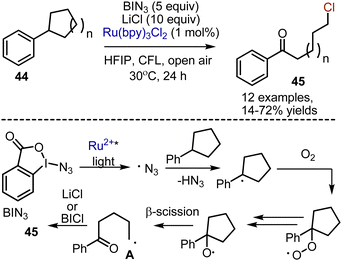 |
| | Scheme 27 Visible light-mediated deconstructive chlorination of aryl cycloalkanes. | |
3.2. Chlorination of unsaturated compounds
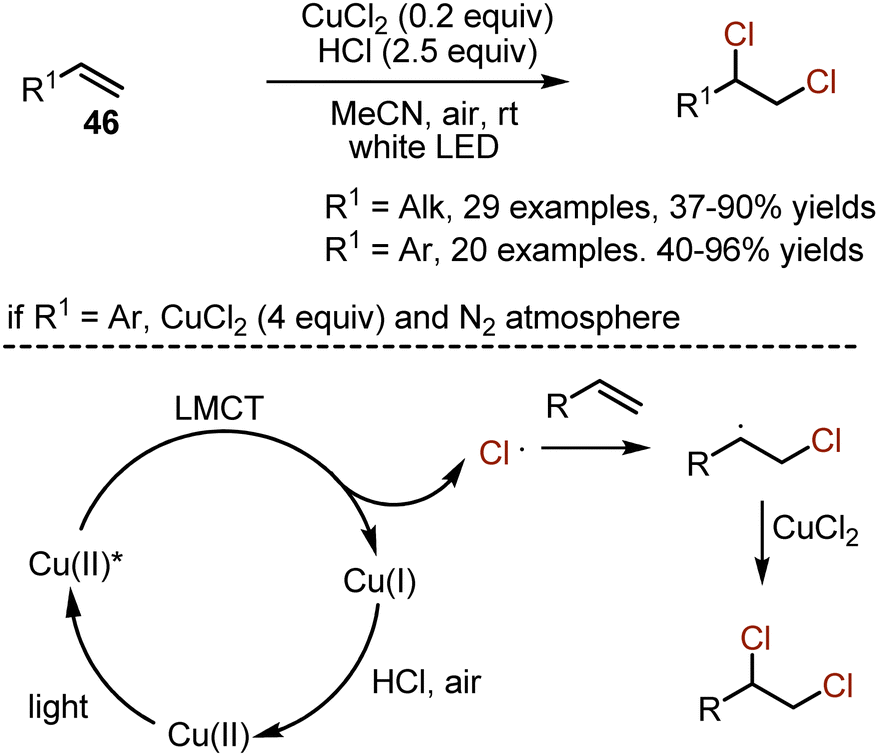
Alkenes are reactive organic species, and the chlorination with molecular chlorine occurs without any additional conditions. However, toxicity, corrosiveness, and lack of selectivity of molecular chlorine are the challenges to overcome. Various methods have been developed, including the use of benign chloride salts with expensive oxidants, used in stoichiometric amounts. The recent employment of a visible light-induced ligand-to-metal charge transfer approach allowed to conquer the listed drawbacks (Scheme 28).60 Aliphatic alkenes 46 could be dichlorinated by the action of a cheap CuCl2 salt in the presence of HCl. The reaction was performed under irradiation with white LED and on air. The method was adapted for dichlorination of styrenes as well. Due to numerous side reactions, the transformations of styrenes were performed under N2 atmosphere and with excess amounts of CuCl2. The following mechanism was proposed. As a result of irradiation with light, copper(II) chloride underwent ligand-to-metal charge transfer, leading to the formation of CuCl and a chloride radical. The latter added to a double bond to form a carbon centered radical, which abstracted chlorine from CuCl2. The HCl and air recycled Cu(I) to Cu(II) chloride.
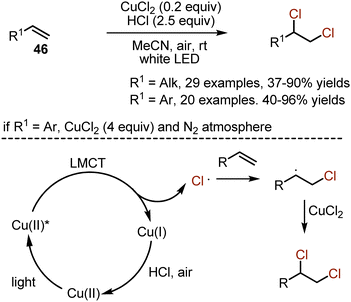 |
| | Scheme 28 Visible light-mediated copper-catalysed chlorination of alkenes. | |
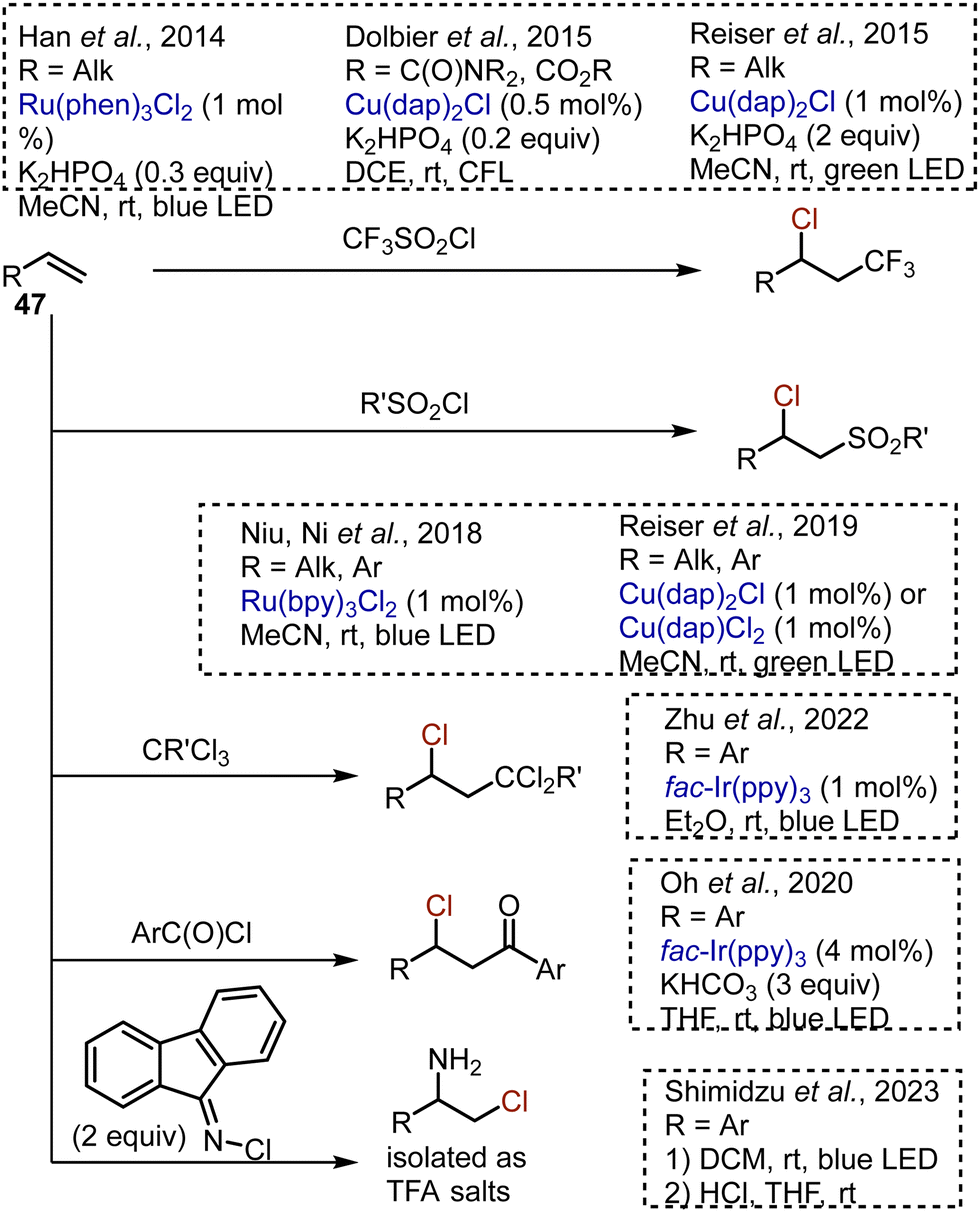
Many visible light-mediated reactions of alkene 47 difunctionalisation were developed (Scheme 29). The chlorotrifluoromethylation of unactivated alkenes took place under the action of CF3SO2Cl in the presence of Ru(phen)3Cl261 or Cu(dap)2Cl62 photocatalysts. Unsaturated amides and esters, as well as other electron deficient alkenes, could be chlorotrifluoromethylated as well.63 Recently, the eosin Y-catalysed chlorotrifluoromethylation of terminal alkenes was reported.64 The use of other sulfonyl chlorides allowed the chlorosulfonylation processes without SO2 extrusion.65,66 These reactions were compatible with both aryl and alkyl alkenes. Styrenes could be chloroacylated by benzoyl chlorides67 or chlorodichloromethylated by chloroform,68 using an Ir-catalyst. Chloroacylation of alkylidenecyclopropanes was reported as well.69 Aminochlorination was achieved by irradiating alkenes with fluorenone-derived N-chloroimine, followed by a one-pot imine hydrolysis.70
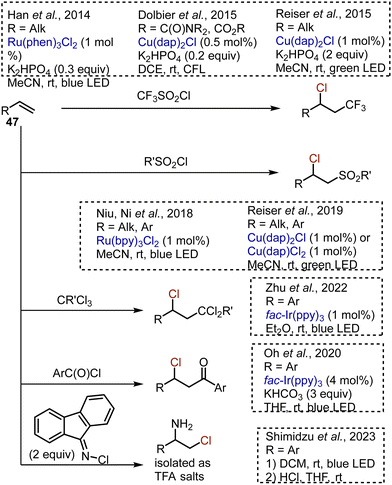 |
| | Scheme 29 Visible light-mediated procedures for alkene difunctionalisation. | |
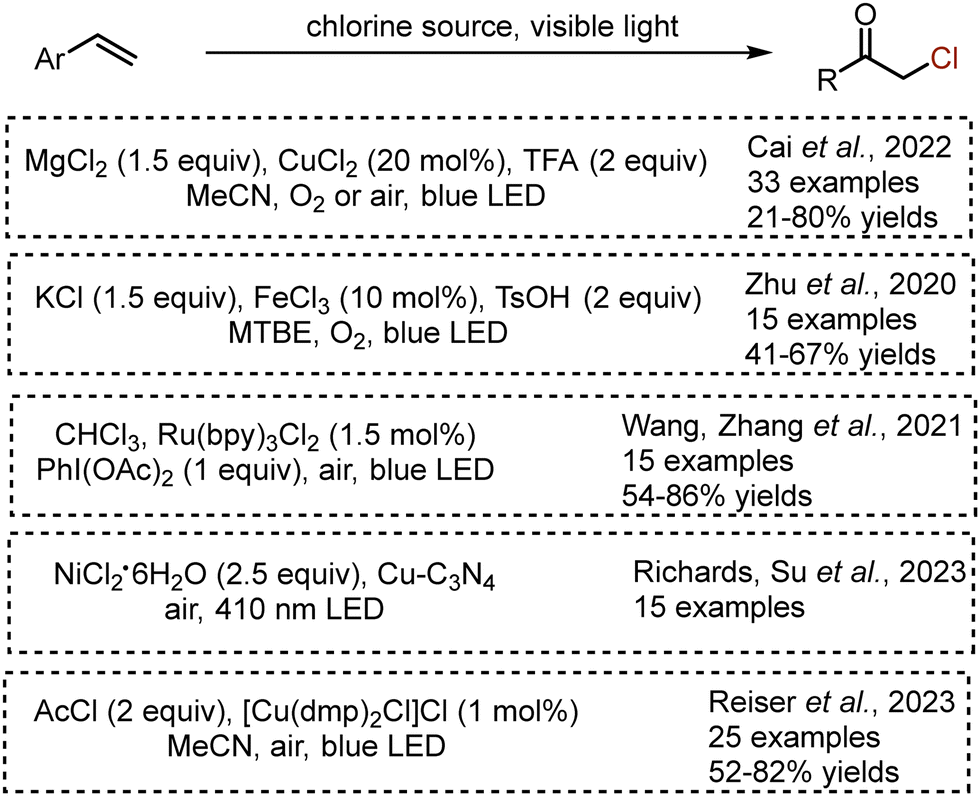
Moreover, several methods for alkene oxochlorination with various chlorine sources have been reported (Scheme 30). Oxochlorination of aryl alkenes was developed based on an LMCT approach – the combination of MgCl2, CuCl2, TFA and oxygen delivered α-chloroarylethanones with high yields through the formation of hydroperoxyl and chlorine atom radicals.71 The use of ferrous(III) chloride and KCl under blue LED irradiation delivered desired oxochlorination products with moderate yields.72 Another interesting source of chlorine – chloroform – was elegantly used in the combination with Ru(bpy)3Cl2 photocatalyst and stoichiometric amounts of PhI(OAc)2.73 Oxochlorination could be also achieved under heterogeneous photocatalysis by copper-modified graphitic carbon nitride (Cu–C3N4).74 Recently, Reiser et al. succeeded in the use of acetyl chloride and Cu-complex for alkene oxochlorination.75 Importantly, the latter method has been successfully employed for late stage functionalisation of bioactive molecules – estrone, fenofibrate, (−)-menthol, theophylline.
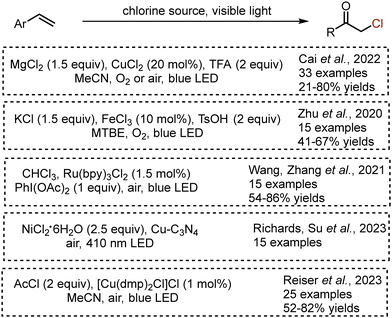 |
| | Scheme 30 Visible light-mediated procedures for alkene oxochlorination. | |
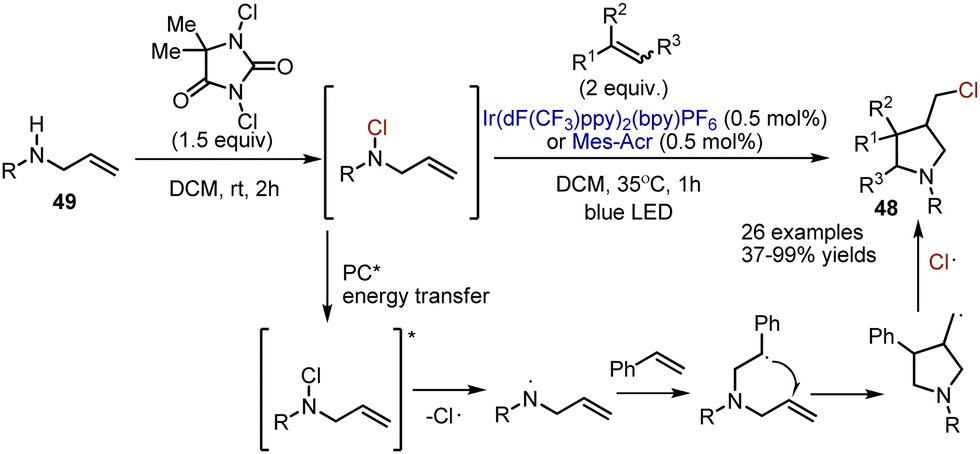
Free radical annulation of chlorinated pyrrolidine 48 was achieved by Crespin et al.76 Initially, alkenyl-containing N-sulfonylamide 49 was transformed into an N–Cl derivative by the action of 1,3-dichloro-5,5-dimethylhydantoin in DCM at rt (Scheme 31). This derivative was used without purification, and the addition of alkene and photocatalyst led to a cascade cyclization. The authors suggest a pathway involving energy transfer from the photoexcited catalyst to N–Cl sulfonamide. The latter underwent homolytic cleavage into an amidyl radical and chlorine atom radical. Further, the amidyl radical added to the alkene double bond to produce a C-centered radical, which underwent an intramolecular cyclization. The reaction sequence was finalized by recombination with a chlorine radical.
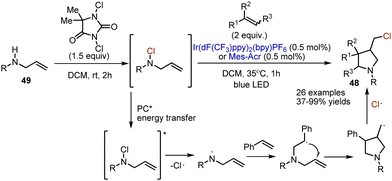 |
| | Scheme 31 Visible light-mediated synthesis of chlorinated pyrrolidines. | |
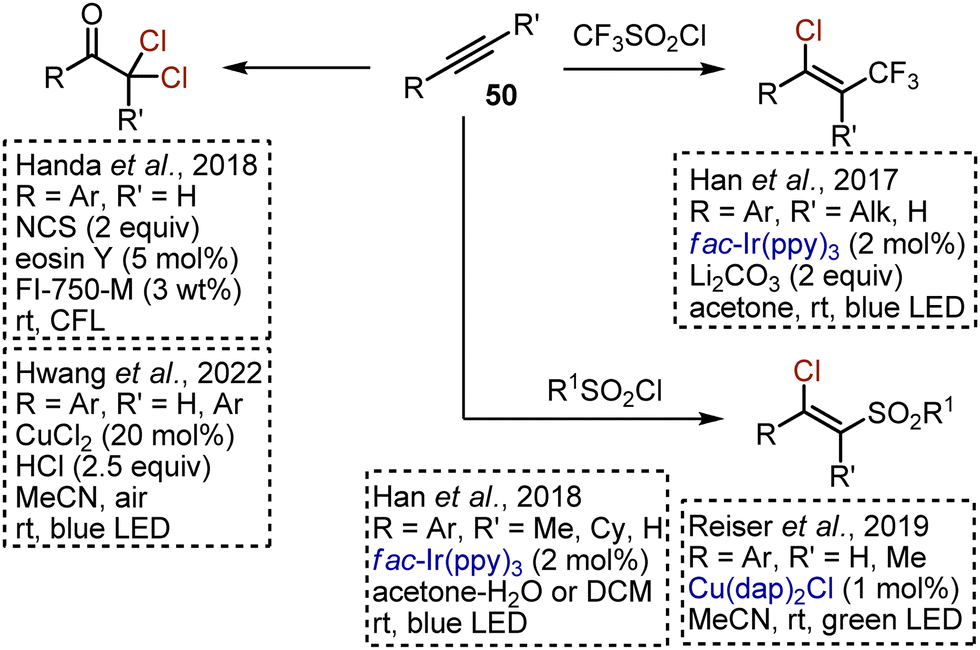
Alkynes 50 present an appealing substrate class for the synthesis of various chlorinated derivatives through free radical addition reactions under visible light irradiation (Scheme 32). Chlorotrifluoromethylation of aryl alkynes was reported by Han in 2017.77 Trifluoromethylsulfonyl chloride was used as a source of CF3 radicals and chlorine, while an Ir-catalyst was needed to perform the reaction. Other sulfonyl chlorides were found to undergo chlorosulfonylation reactions. Extrusion of SO2 did not take place. These sulfonylative transformations were realized under Ir-catalysis,78 and under Cu-catalysis.66 Eosin Y-catalysed oxydichlorination of aryl alkynes with NCS was developed in 2018.79 The reaction was facilitated by the use of proline-based surfactant FI-750-M. Later, an analogous transformation was developed under LMCT-conditions, using cheap CuCl2 and HCl as chlorine sources.80
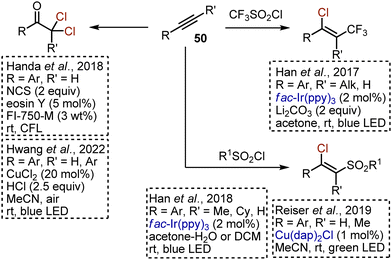 |
| | Scheme 32 Visible light-mediated chlorofunctionalisation of alkynes. | |
Chlorotrifluoromethylative cyclization of enynes 51 was a useful approach towards chlorinated pyrrolidines 52 (Scheme 33).81 Trifluoromethylsulfonyl chloride was used as a well-established source of trifluoromethyl radicals. It underwent a single electron reduction by a photoexcited acridinium salt, followed by the decomposition into CF3 radical, SO2, and chloride anion. It was proposed that addition of ˙CF3 to the double bond resulted in the formation of a C-centered radical, that readily cyclized onto the triple bond. The formed vinyl radical abstracted chlorine atom from CF3SO2Cl, propagating the chain process.
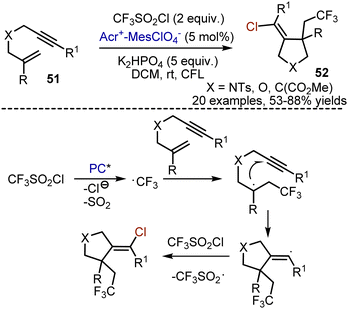 |
| | Scheme 33 Acridinium dye-catalysed cyclative chlorotrifluoromethylation. | |
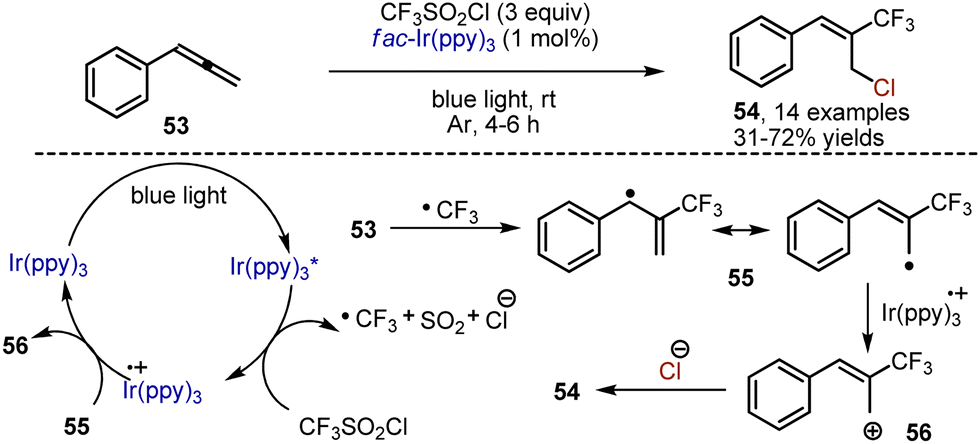
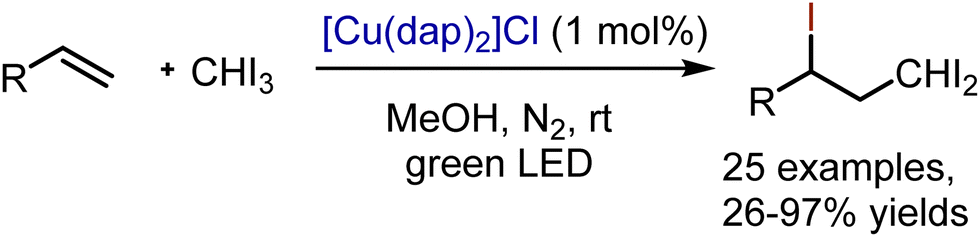
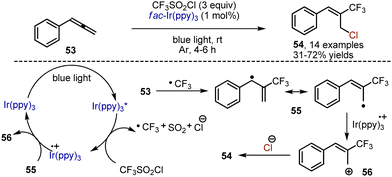
Visible light mediated transformations of allenes are yet underdeveloped. Chlorotrifluoromethylation of arylallenes 53 has been recently reported by Festa, Voskressensky et al.82 Valuable building blocks – trifluoromethylated allyl chlorides 54 – have been synthesized by treatment of arylallenes with trifluoromethylsulfonyl chloride, which is an inexpensive source of a trifluoromethyl radical. The reaction has been performed in the presence of Ir(ppy)3 catalyst in acetone under argon atmosphere (Scheme 34). The following mechanism is proposed. Initially, the photoexcited Ir-catalyst reduces CF3SO2Cl to give an anion radical, decomposing into Cl−, SO2 and a CF3 radical. The addition of the latter to an allene system delivers an allyl-type radical 55, capable of undergoing oxidation to cation 56 with the Ir4+-species. The interaction of the chloride anion with B furnishes the resulting trifluoromethylated allyl chloride. Importantly, these allyl chlorides were successfully employed for the preparation of a trifluoromethylated analog of the drug flunarizine.
 |
| | Scheme 34 Visible light-mediated chlorotrifluoromethylation of arylallenes. | |
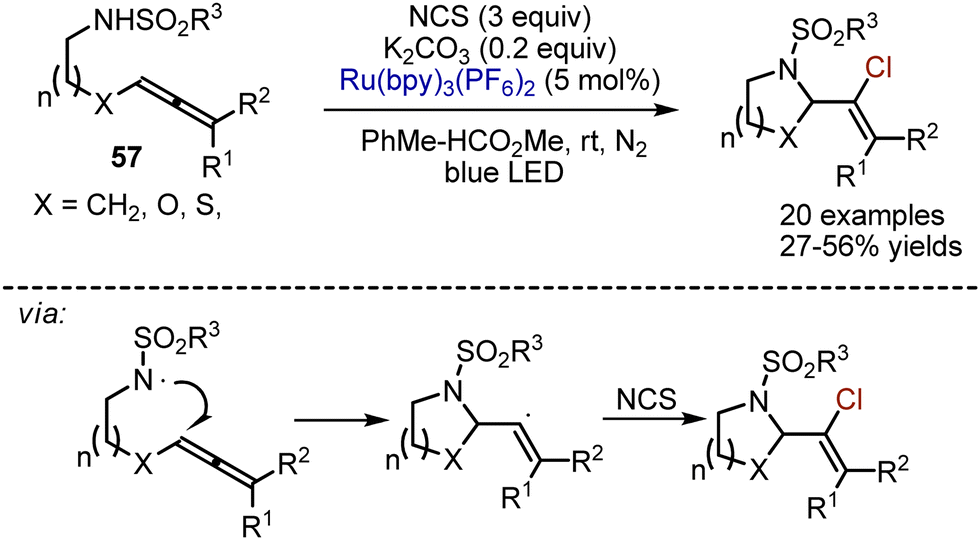
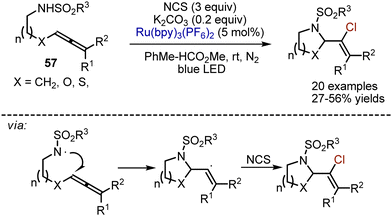
Another visible-light induced method for allene transformation relies on an intramolecular cyclative aminochlorination.83 Allenes 57, containing a sulfonamide moiety, could participate in a Ru(bpy)3(PF6)2-catalysed reaction with NCS in a toluene-methylformate mixture under nitrogen atmosphere (Scheme 35). Potassium carbonate was used in catalytic amounts.
 |
| | Scheme 35 Photoredox-catalysed intramolecular aminochlorination of allenes. | |
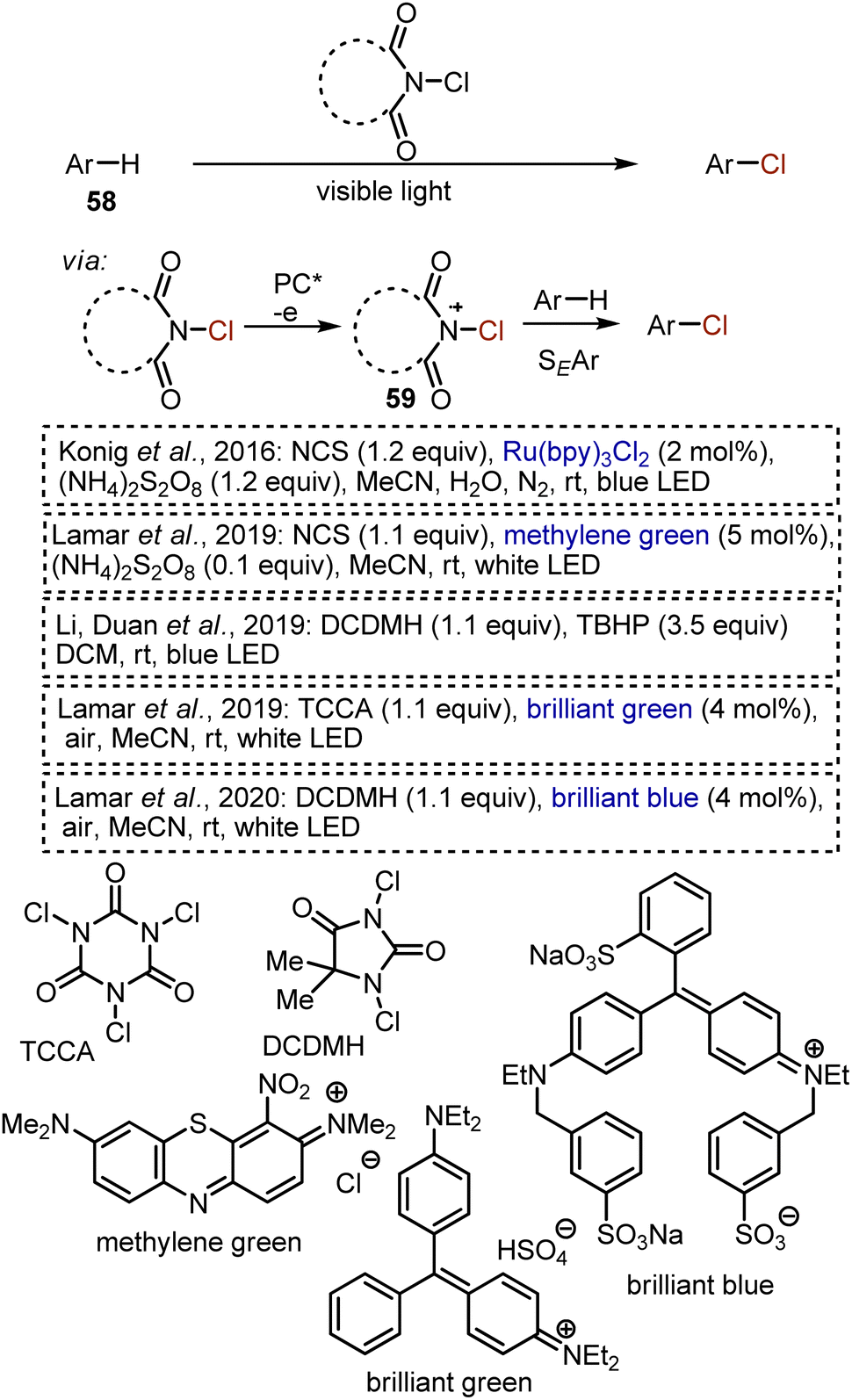
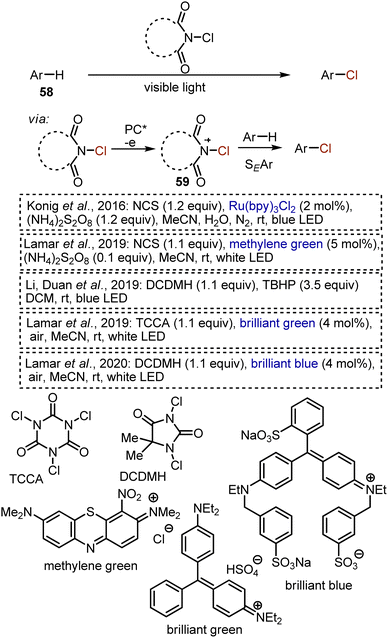
Some methods for chlorination of aromatic substrates 58 were developed under visible light irradiation (Scheme 36). Firstly, a series of works reported on visible light-mediated processes, employing NCS or analogous reagents as chlorine sources. It is believed that the role of a photocatalyst in these transformations is usually to generate an active NCS cation radical 59, which is more readily participating in aromatic electrophilic substitutions.84–88 The chlorination with NCS without light irradiation is a well-established process under the action of protic acids, Lewis acids, or transition metals. Visible-light mediated reactions benefit from the use of benign catalysts (like organic dyes) and mild conditions i.e. neutral medium and ambient temperature. Electron-rich and electron-neutral aromatic compounds, as well as π-excessive heterocycles are competent substrates.
 |
| | Scheme 36 Visible light-mediated chlorination of aromatic compounds. | |
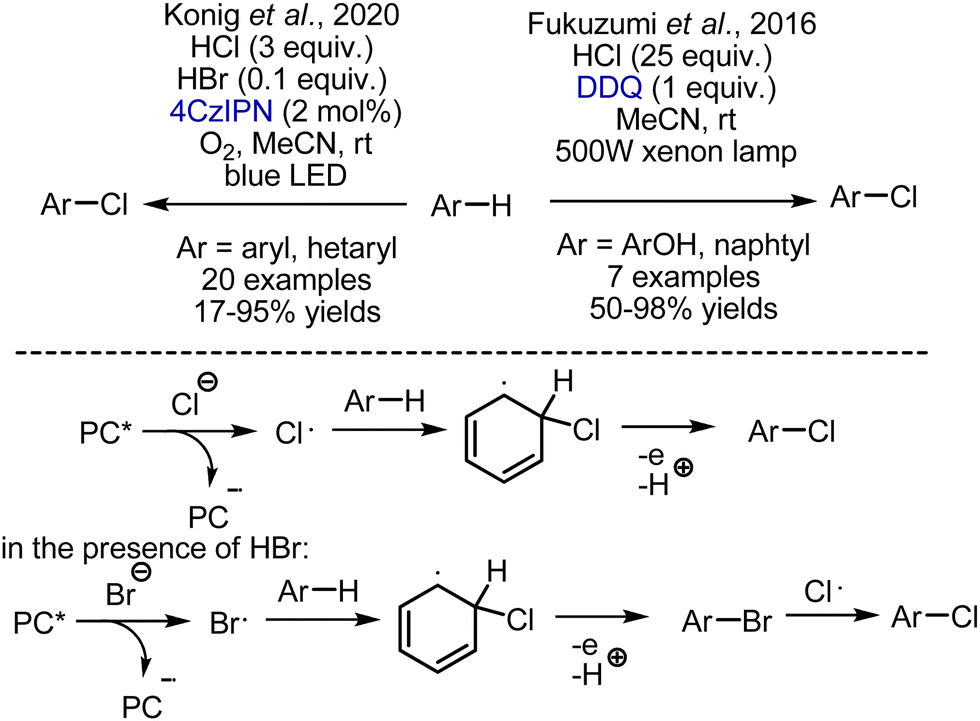
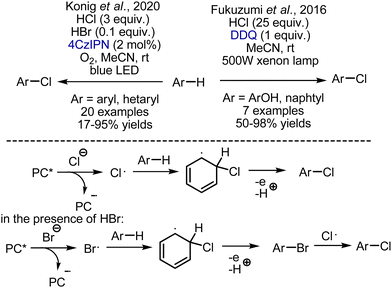
The approaches that employ HCl as a source of chlorine are more intriguing. (Scheme 37). Fukuzumi et al. developed chlorination of phenols by a DDQ-HCl system under visible light irradiation.89 Photoexcited DDQ was capable of chloride oxidation to generate a chlorine atom radical, which reacted with phenol. The reaction worked smoothly with phenols, while naphthalene was the only electron-neutral substrate. Another reaction, employing HCl as a chlorine source was developed by König et al.90 The generation of chlorine atom radicals was performed by oxidation with photoexcited 4CzIPN organic photocatalyst, while oxygen was regenerating the catalyst. It was shown that bromination with HBr was occurring faster, but bromo-derivatives were undergoing bromine substitution by chlorine.
 |
| | Scheme 37 Photochemical chlorination of aromatic compounds with HCl as chlorine source. | |
3.3. Decarboxylative chlorination
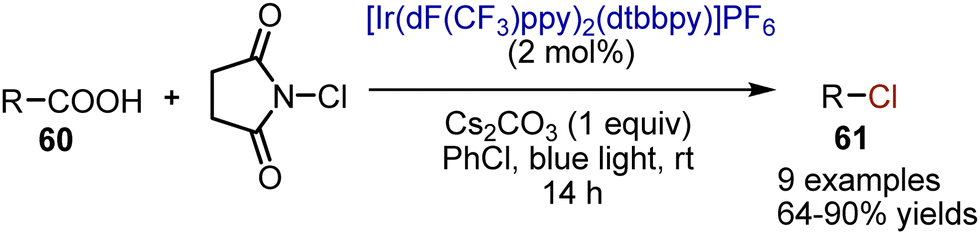
Aliphatic carboxylic acids 60 were smoothly halogenated with loss of the carboxylic group.91 The reaction was performed by irradiating an acid and N-chlorosuccinimide (NCS) in chlorobenzene in the presence of Cs2CO3 with blue light, employing [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as photocatalyst (Scheme 38). Primary, secondary, and tertiary acids were reacting smoothly, delivering the desired products 61 with 64–90% yields. The protocol's utility has been effectively demonstrated through the synthesis of costly 1-bromoheptadecane using affordable stearic acid found in nature. The reaction is believed to initiate with the oxidation of cesium carboxylate by the photoexcited Ir-complex, resulting in the generation of an alkyl radical that seizes a chlorine atom from NCS. The catalytic cycle concludes by the reduction of the succinimidyl radical by Ir2+, thereby closing the cycle. Besides chlorination, the developed protocol also allows a decarboxylative bromination (diethyl bromomalonate instead of NCS), as well as iodination (NIS instead of NCS).
 |
| | Scheme 38 Decarboxylative chlorination. | |
4. Bromination
4.1. Bromination of Csp3–H bonds
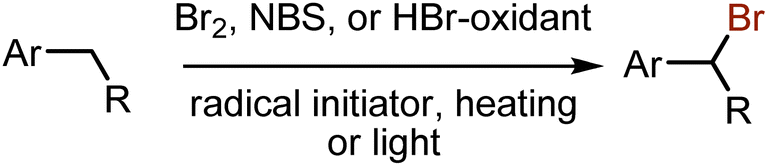
Benzylic bromination with bromine is a well-established method (Scheme 39). Heating or light has been used for a long time to initiate the bromine homolytic cleavage into bromine radicals.92–94 Being a toxic and corrosive reagent, molecular bromine is often replaced by NBS. Radical initiators,95 heating, or visible light96 are needed for NBS-mediated benzylic brominations. More recently brominations could be performed in water,97 perfluoroalkane,98 CH3CN,99 or without solvent.100 More benign systems like HBr-oxidant or MBr-oxidant101–104 were also used with visible light irradiation. A photoredox process for benzylic bromination, employing Ir-complex as a photocatalyst and CBr4 as a bromine source, has been developed as well.105 Interestingly, oxybromination could be achieved for ethylarenes by subjecting them to aerobic visible light-mediated reactions with HBr in H2O–EtOAc.106,107
 |
| | Scheme 39 Benzylic bromination. | |
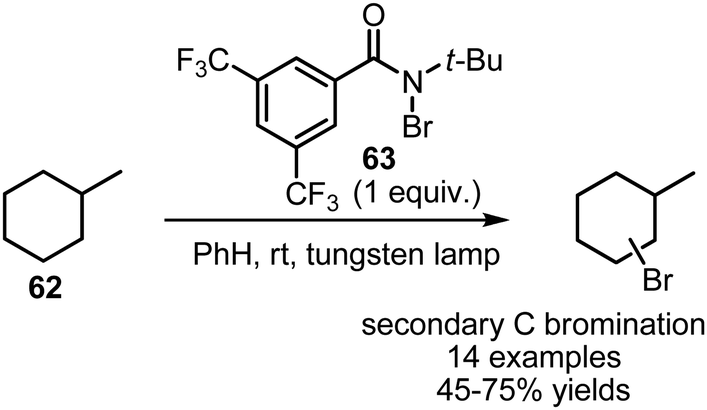
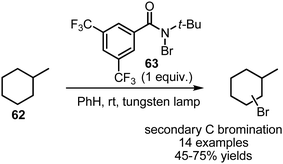
Several interesting processes for visible-light mediated bromination of non-activated alkanes 62 have been developed. Alexanian et al. have shown that bulky bromoamide 63 can present an advanced selectivity in visible light-mediated bromination of alkanes, favoring secondary C–H bonds over tertiary, which is very uncommon for free radical reactions (Scheme 40).108
 |
| | Scheme 40 Visible light-mediated bromination of secondary carbons. | |
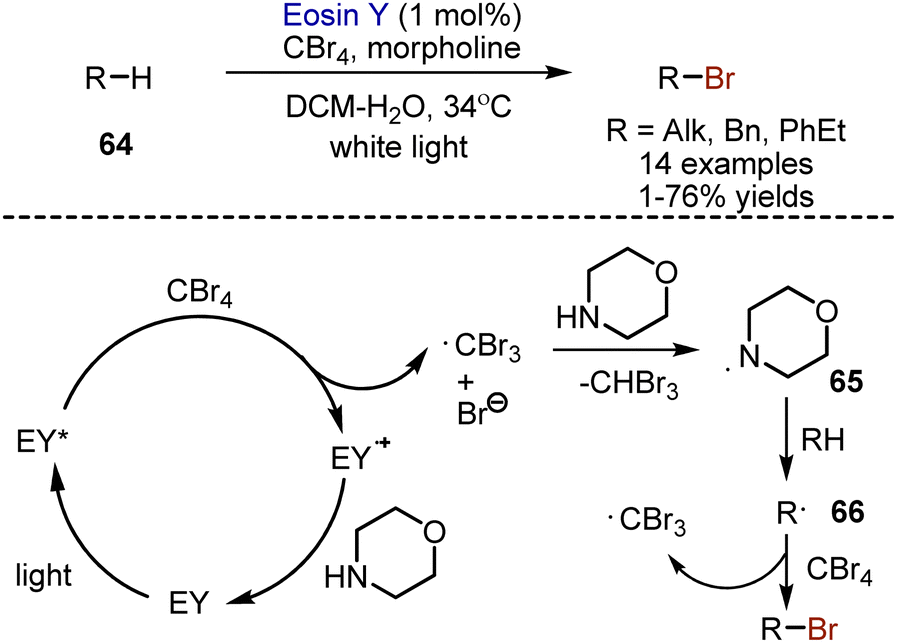
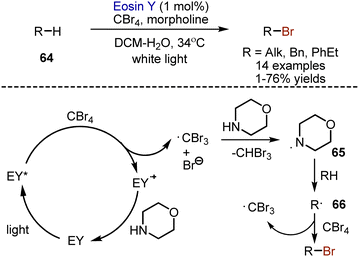
Non-activated alkanes, as well as alkylarenes 64 could be brominated with CBr4 in the presence of eosin Y photocatalyst and morpholine (Scheme 41).109 It was suggested that photoexcited eosin Y reduces CBr4 to give an anion radical, which decomposes into a bromide anion and ˙CBr3. Hydrogen atom transfer from morpholine to the tribromomethyl radical occurred, generating aminyl radical 65, which could, in it's turn, abstract hydrogen from alkane. The resulting carbon-centered radical 66 reacted with CBr4 to produce the desired compound. The photocatalytic cycle could be closed by oxidation of morpholine. The process allowed bromination with moderate yields, but good selectivities.
 |
| | Scheme 41 Eosin Y-catalysed bromination with carbon tetrabromide. | |
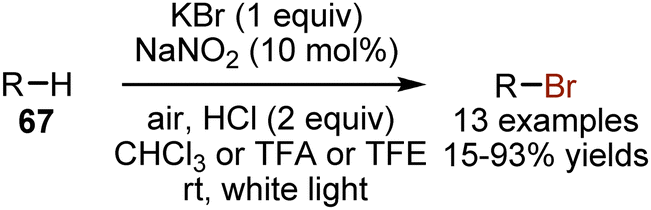
Visible light has been found to enhance the bromination of non-activated alkanes 67 with potassium bromide (Scheme 42).110 A catalytic amount of NaNO2 oxidizes the bromide anion into Br2, which undergoes a free-radical reaction under white light irradiation. The reoxidation of NO to NO2 occurs on ambient air. The visible light mediated bromination of alkanes has also been achieved with CBr4 as a bromine source.111
 |
| | Scheme 42 Visible light-induced bromination with KBr. | |
4.2. Bromination of unsaturated compounds
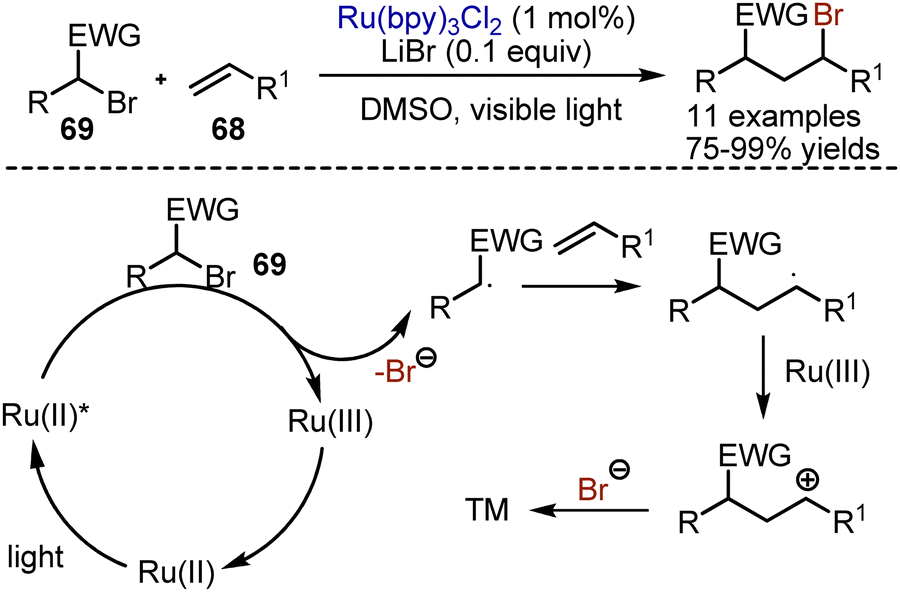
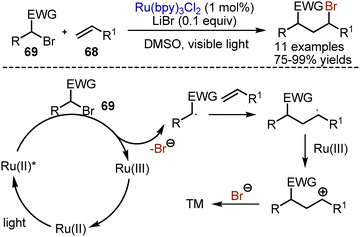
Bromination of alkenes or alkynes with molecular bromine does not need visible light to proceed. On the contrary, atom transfer radical addition reactions of alkyl bromides are going smoothly under photoredox-catalytic conditions. In 2012, Stephenson reported a general haloalkylation of alkenes 68, employing Ru(bpy)3Cl2 as photocatalyst in DMSO with the addition of a catalytic amount of LiBr (Scheme 43).112 The mechanism implied initial reduction of alkyl bromide 69 with the photoexcited catalyst, which led to the decomposition of the resultant anion radical into the bromide anion and a carbon-centred radical. The addition of the alkyl radical to a double bond formed another carbon–centred radical. The oxidation of the latter by Ru(III) closed the catalytic cycle and formed a cation, quenched by the bromide anion. This approach worked with various alkyl bromides, containing electron-withdrawing groups, as well as with polyhalomethanes.
 |
| | Scheme 43 Bromoalkylation of alkenes. | |
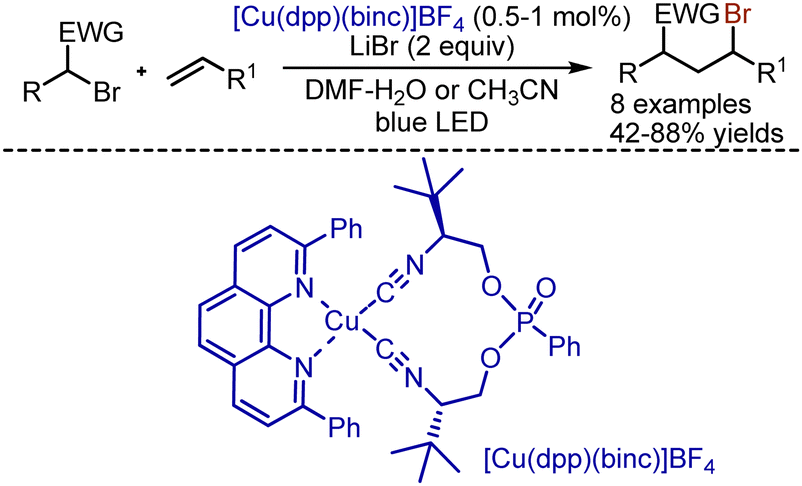
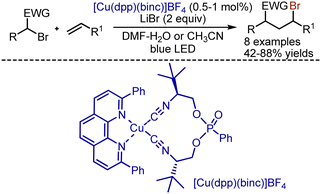
Analogous bromoalkylation was realized with the help of Cu-complexes by Reiser group (Scheme 44).113 It was found that various activated alkyl bromides reacted smoothly with alkenes under irradiation in the presence of [Cu(dpp)(binc)]BF4 (dpp = 2,9-diphenyl-1,10-phenanthroline; binc = bis(2-isocyanophenyl) phenylphosphonate). Except for bromomalonates, benzyl bromides with nitro groups could be employed, but larger amounts of catalyst were needed, as well as excess amounts of alkenes.
 |
| | Scheme 44 Bromoalkylation of alkenes with Cu-complex. | |
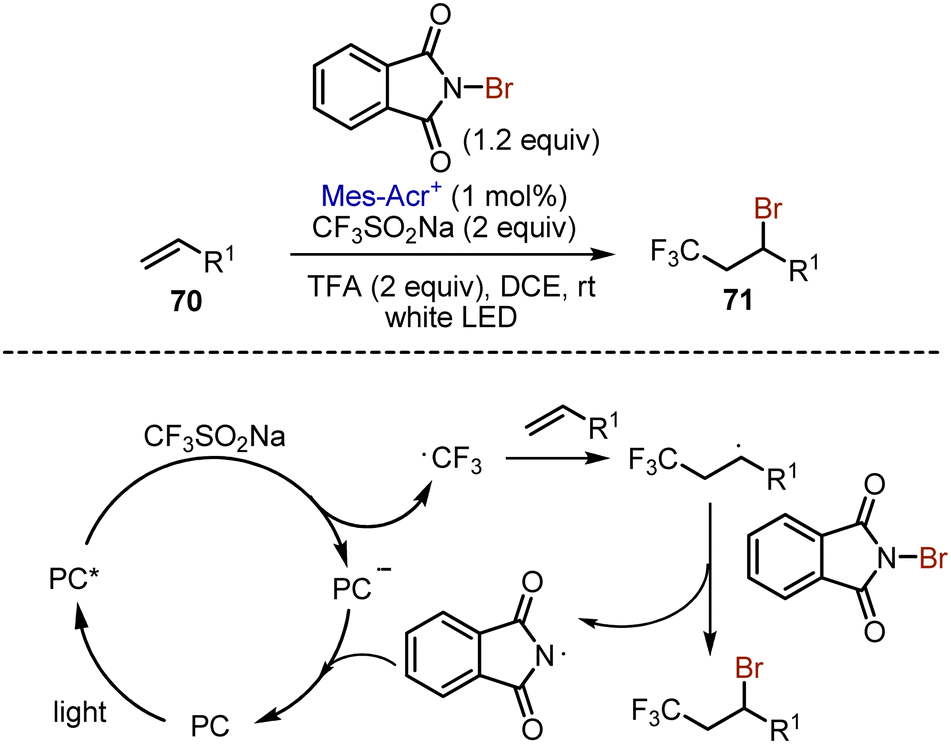
Later, organophotoredox approach was developed for alkene 70 bromotrifluoromethylation (Scheme 45).114 The photoexcited acridinium salt oxidized Langlois’ reagent to generate a trifluoromethyl radical, which added to the alkene double bond. The resulting radical abstracted a bromine atom from N-bromophthalimide, giving the target molecule 71.
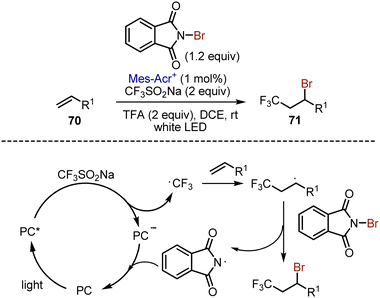 |
| | Scheme 45 Photoredox-catalysed bromotrifluoromethylation of alkenes. | |
Copper-catalysed bromotribromomethylation with CBr4 could be also realized (Scheme 46).115
 |
| | Scheme 46 Photoredox-catalysed bromotrifluoromethylation of alkenes. | |
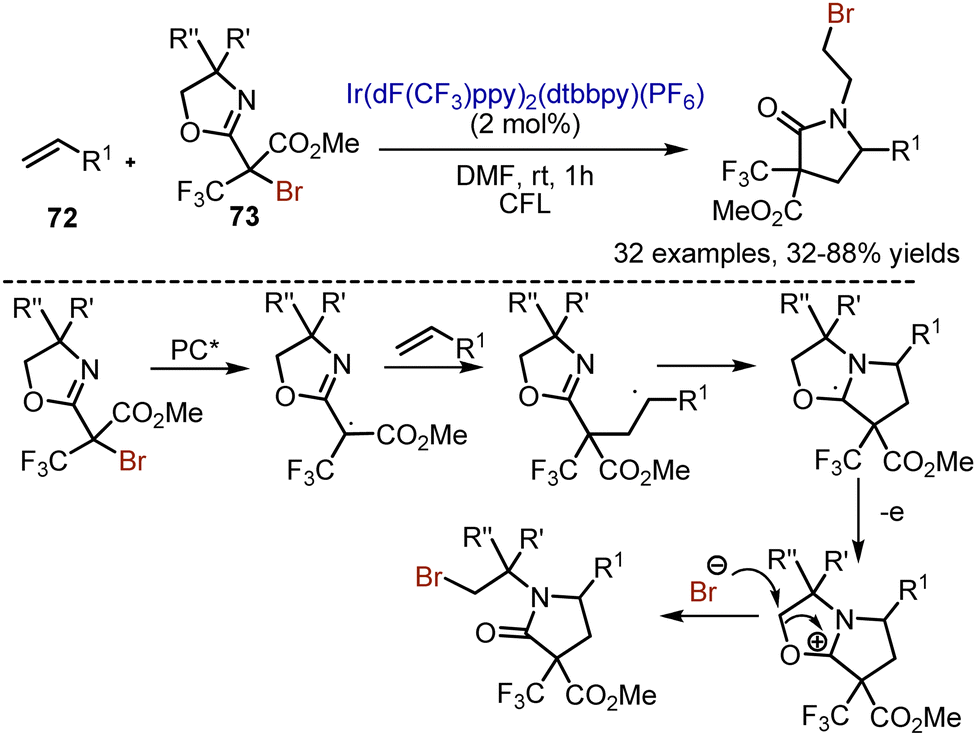
When alkenes 72 were reacted with oxazolidine-containing alkyl bromides 73 under visible light irradiation, an interesting cleavage of the oxazolidine ring took place (Scheme 47).116 The reaction was performed with the Ir(dF(CF3)ppy)2(dtbbpy)(PF6) photocatalyst in DMF. The initial steps were analogous to described above methods: SET-reduction of alkyl bromide, formation of alkyl radical, and addition to the double bond. The carbon-centered radical interacted with nitrogen of oxazolidine, and subsequent oxidation led to cation formation. The attack of the bromide anion cleaved the positively charged ring and finalized the sequence. Various aryl and alkyl alkenes, as well as enol ethers reacted smoothly under the reaction conditions.
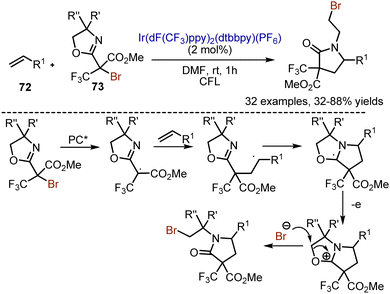 |
| | Scheme 47 Visible light-mediated synthesis of pyrrolidones through oxazolidine ring cleavage. | |
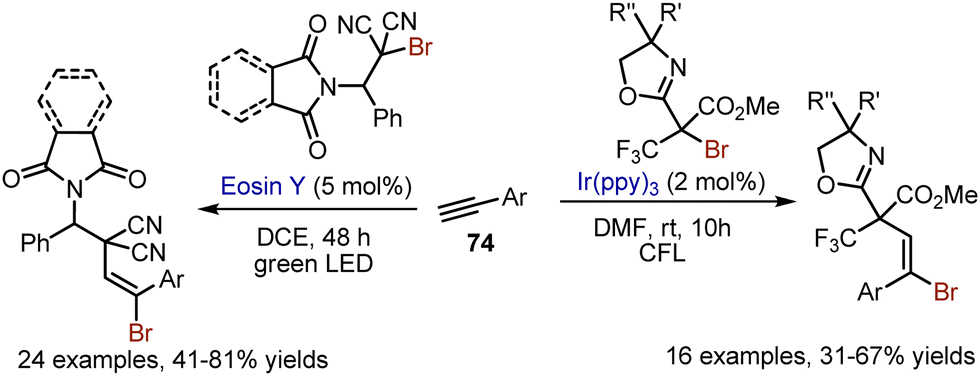
This approach was amenable for alkyne 74 bromoalkylation too, but the oxazolidine ring was preserved (Scheme 48). Another bromoalkylation of alkynes was developed under eosin Y catalysis.117
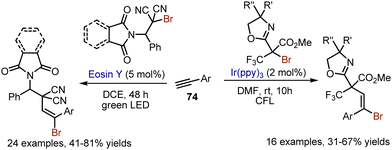 |
| | Scheme 48 Bromoalkylation of alkynes. | |
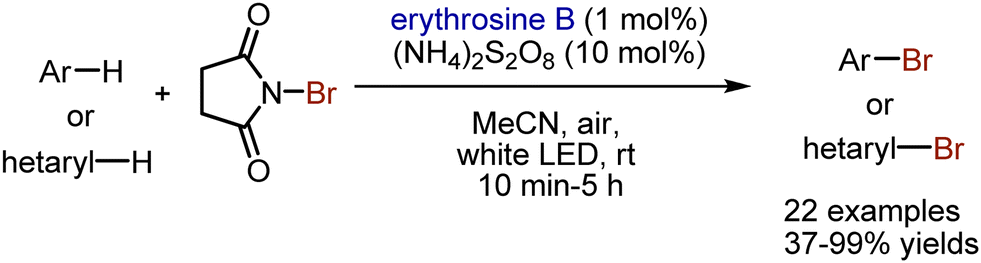
Bromination of aromatic compounds under visible light-mediated conditions has been achieved by Lamar et al.118 Xanthene dye erythrosine B has been used as photocatalyst in the bromination of arenes with NBS (Scheme 49). Although ammonium peroxodisulfate was not found essential according to some control experiments, its’ presence facilitated the process. The conditions were compatible with bromination of heteroarenes. In the suggested mechanism, the activation of NBS was achieved via SET oxidation by a photoexcited dye.
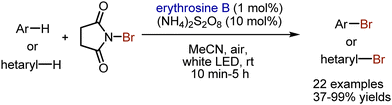 |
| | Scheme 49 Bromination of aromatic compounds. | |
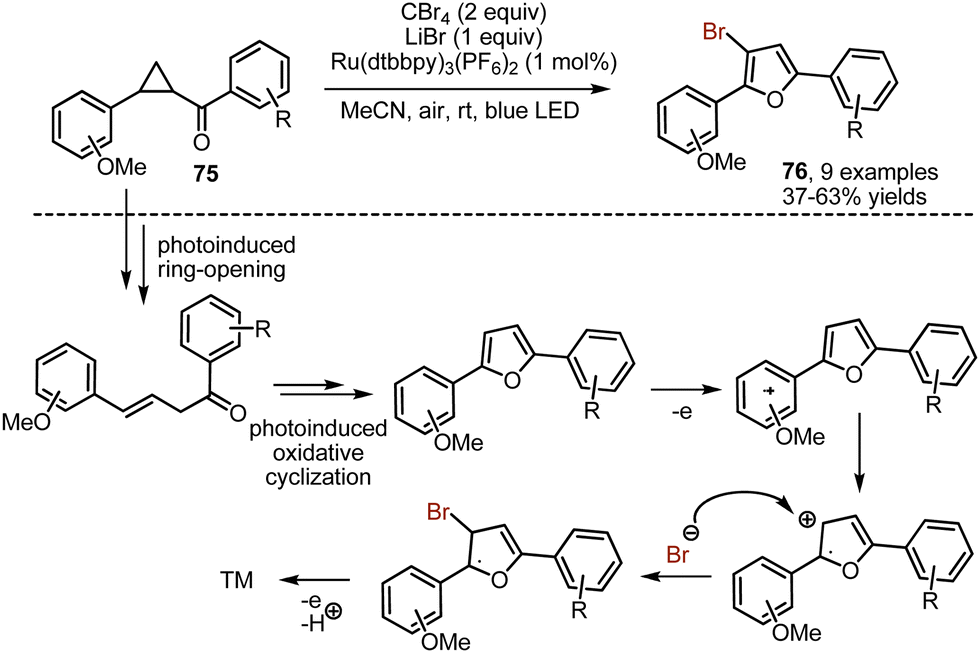
While investigating oxidative transformations of donor–acceptor cyclopropanes 75 into furans under visible light irradiation, Xia and-workers discovered that taking CBr4 in an excess amounts led to the formation of brominated furans 76 (Scheme 50).119 The reaction was regioselectively producing 3-bromofurans. Carbon tetrabromide could be also exploited for the bromination of 8-aminoquinolines.120
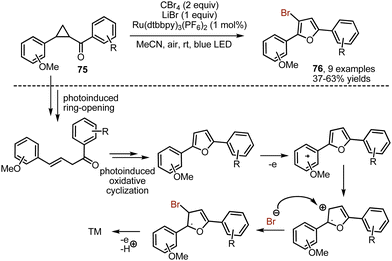 |
| | Scheme 50 Visible light-mediated synthesis of 3-bromofurans from cyclopropanes. | |
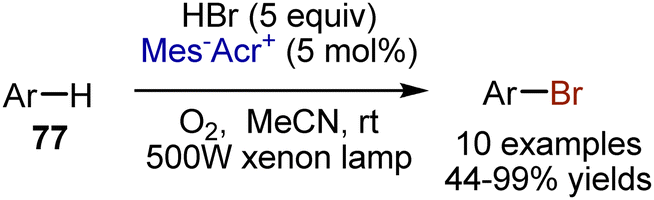
The more sustainable bromine source HBr, is usually employed for bromination under oxidative conditions, which can be performed with a photoredox catalyst. Fukuzumi reported an acridinium dye-catalysed bromination of electron-rich arenes 77 employing HBr and oxygen-saturated MeCN (Scheme 51).121 According to the mechanistic studies, the photoexcited acridinium salt oxidized the aromatic compound into a cation radical, which reacted with the bromide anion. Oxygen was recycling the photocatalyst. Later, a microporous organic polymer-catalysed bromination was reported. The polymer was based on 4,7-diphenylbenzo[c][1,2,5]thiadiazole photoactive units bridged with formaldehyde dimethyl acetal. An important advantage of the polymer catalyst is its high reusability potential.
 |
| | Scheme 51 Acridinium dye-catalysed bromination of electron-rich arenes. | |
4.3. Deoxygenative bromination
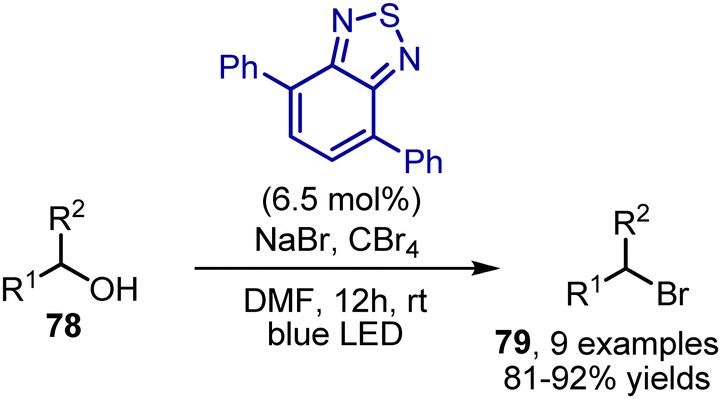
Aliphatic alcohols 78 could be converted into bromides 79 when subjected to tetrabromomethane and NaBr in DMF under blue light irradiation in the presence of a benzothiadiazole catalyst (Scheme 52).122
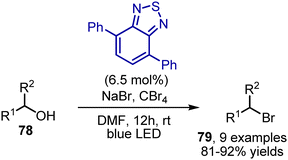 |
| | Scheme 52 Deoxygenative bromination. | |
5. Iodination
5.1. Iodination of unsaturated compounds
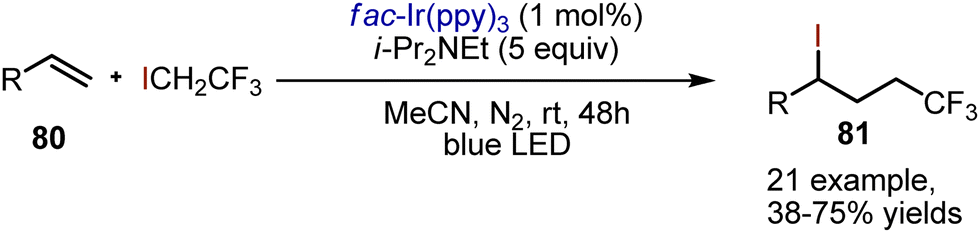
Atom transfer radical addition reactions to alkenes are applicable for iodofunctionalisations, as described earlier in other sections. Guo et al. have reported a photoredox-catalysed iodotrifluoroethylation (Scheme 53).123 Various terminal non-conjugated alkenes 80 have been evaluated, giving addition products 81 with moderate yields. Diisopropylethylamine has been used as a reductive quencher of photoexcited Ir(ppy)3 to generate strongly reducing Ir(II). The latter interacted with 1,1,1-trifluoro-2-iodoethane, reducing it to an anion radical, which subsequently decomposed into an alkyl radical and iodide. Addition of the trifluoroethyl radical to an alkene was giving another carbon radical. The final iodination presumably took place by abstracting iodine from 1,1,1-trifluoro-2-iodoethane.
 |
| | Scheme 53 Photoredox-catalysed alkene iodotrifluoroethylation. | |
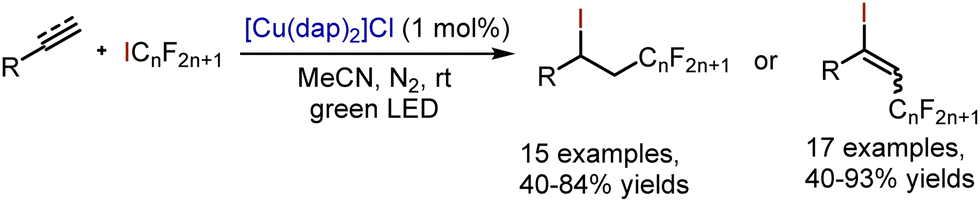
Iodoperfluoroalkylation of styrenes and aryl acetylenes has been accomplished through copper-catalysed ATRA reactions by Reiser et al. (Scheme 54).124
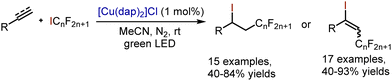 |
| | Scheme 54 Copper-catalysed alkene iodoperfluoroalkylation. | |
Analogously, addition of iodoform could occur under copper catalysis (Scheme 55).125
 |
| | Scheme 55 Copper-catalysed alkene iodoperfluoroalkylation. | |
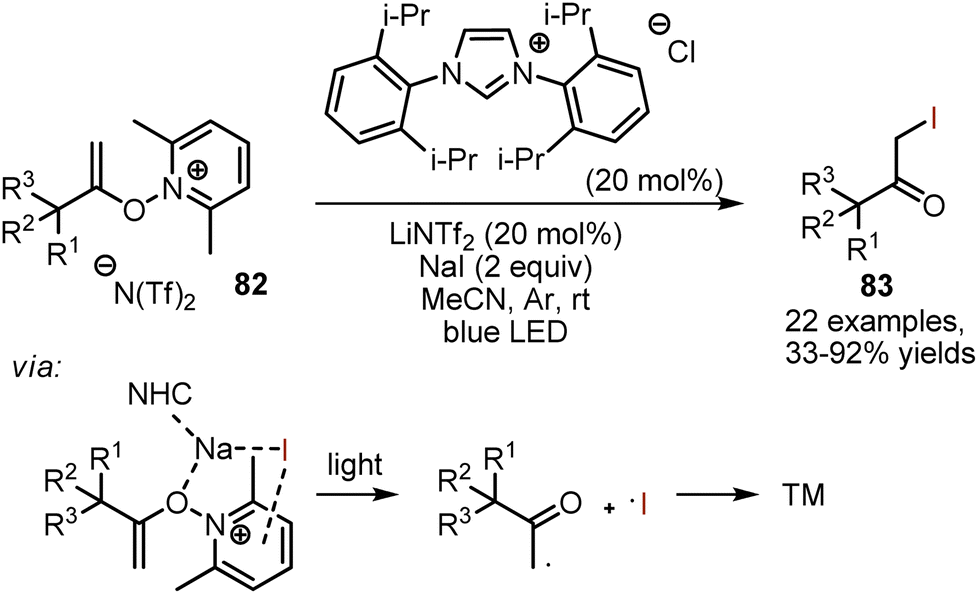
Visible light-mediated iodination of N-alkenoxypyridinium salts 82 was giving α-iodo ketones 83 under N-heterocyclic carbene catalysis (Scheme 56).126 According to the mechanistic studies, the pyridinium salt was forming a photoactive electron donor–acceptor (EDA) complex with sodium iodide. Excitation with light led to the formation of an alkyl radical, an iodine radical and 2,6-lutidine. It was suggested that the role of NHC was to facilitate the EDA-complex formation through electrostatic interaction with NaI. Base was needed to generate NHC from the imidazolium salt.
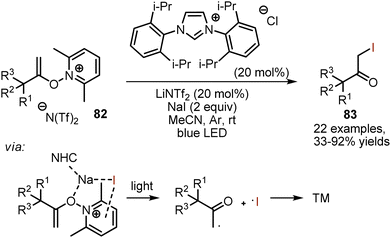 |
| | Scheme 56 Visible light-mediated NHC-catalysed iodination of N-alkenoxypyridinium salts. | |
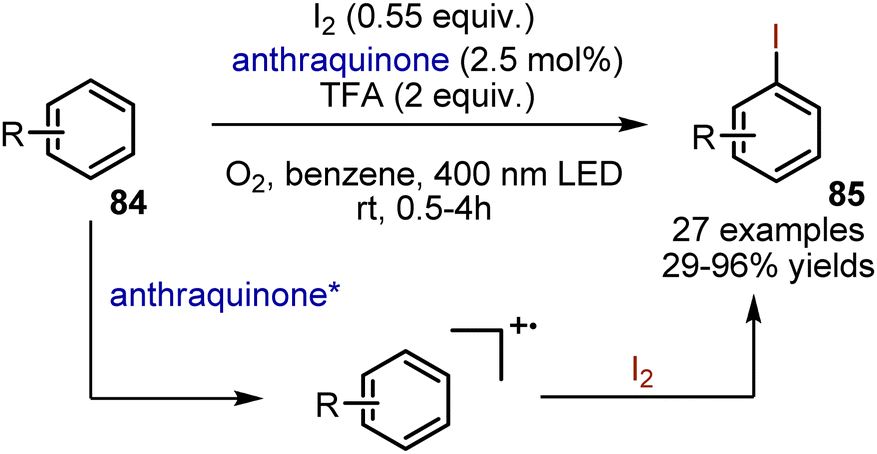
Iodination of aromatic compounds typically involves the use of terminal oxidants. Classical methods utilize a combination of I2 with HNO3 or KMnO4. König et al. have described a visible light-mediated iodination method under mild conditions, utilizing oxygen as the terminal oxidant. (Scheme 57).127 Electron-rich aromatic compounds 84 were irradiated in benzene under oxygen atmosphere in the presence of iodine, TFA and anthraquinone. The desired compounds 85 were obtained with 29–96% yields. Anisole was iodinated with high para-selectivity. No iodination of the benzylic position of alkylbenzenes was observed. Mechanistic studies suggested that the interaction of iodine with an arene radical cation takes place.
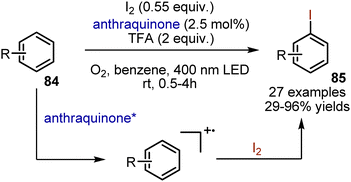 |
| | Scheme 57 Visible light-mediated iodination of aromatic compounds. | |
5.2. Decarboxylative iodination
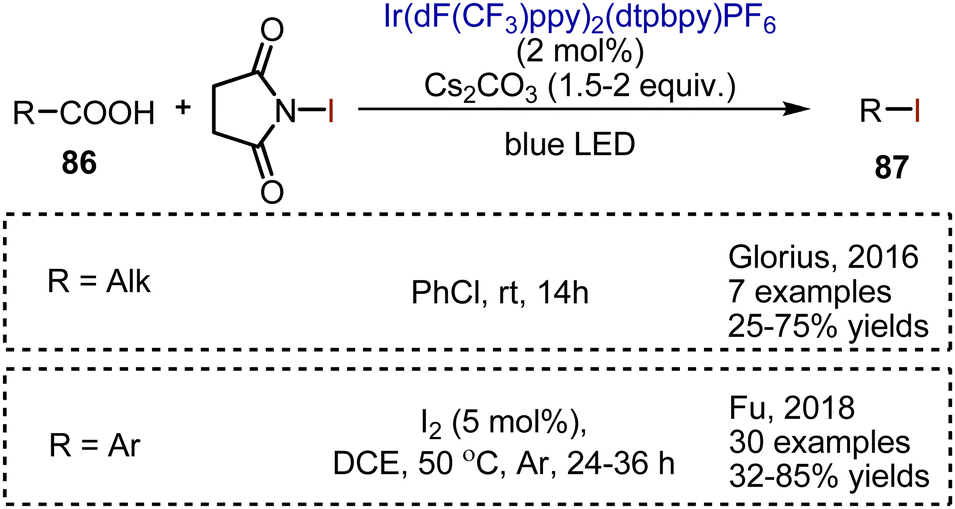
Carboxylic acids 86 could be decarboxylatively iodinated employing an Ir-complex-catalyzed visible light-mediated reaction (Scheme 58). NIS has been used as iodine source, while cesium carbonate is needed to generate the cesium salt of the carboxylic acid. For the iodination of aliphatic carboxylic acids, the reaction was performed at rt in PhCl.128 Aromatic acids are iodinatively decarboxylated on heating in DCE in the presence of a catalytic amount of molecular iodine.129 The optimized conditions allowed the syntheses of alkyl and aryl iodides 87 with a broad substrate scope and moderate to very good yields.
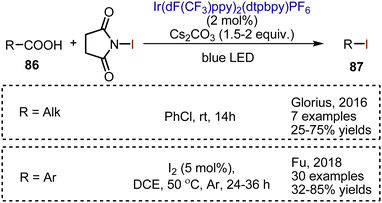 |
| | Scheme 58 Decarboxylative iodination with NIS. | |
5.3. Deoxygenative iodination
A photocatalyst-free protocol for the deoxygenative iodination of alcohols 88 has been developed by Zhao and Antonietti.130 A solution of the alcohol and iodoform in anhydrous DMF was irradiated with white LED-light under nitrogen atmosphere, to deliver the corresponding iodides 89 with good to excellent yields (Scheme 59). It has been found that iodoform is capable of homolytic cleavage under visible light irradiation. The formed diiodomethyl radical interacted with DMF, forming a Vilsmeier type reagent, that iodinates the alcohol. The reaction worked smoothly with primary and secondary alcohols.
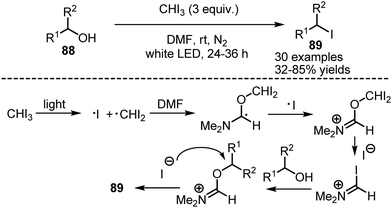 |
| | Scheme 59 Deoxygenative iodination with iodoform. | |
6. Conclusion
Visible light has become a useful tool for organic chemists. Numerous reactions for halogenation under visible light irradiation were recently developed. These reactions are usually performed under milder conditions, allowing to avoid high temperatures and non-selective UV-light. Benign halogenation reagents could be used with visible light, and some classical reactions were enhanced under these conditions. Major breakthroughs were developed in the field of difunctionalisation reactions, where other functionalities along with halogen were introduced under excellent control and with remarkable selectivity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The paper has been supported by the RUDN University Strategic Academic Leadership Program.
Notes and references
- D. Benedetto Tiz, L. Bagnoli, O. Rosati, F. Marini, L. Sancineto and C. Santi, New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use, Molecules, 2022, 27, 1643, DOI:10.3390/molecules27051643.
- R. S. Malykhin and A. Yu Sukhorukov, Nucleophilic Halogenation of Heterocyclic N-Oxides: Recent Progress and a Practical Guide, Adv. Synth. Catal., 2021, 363, 3170–3188, DOI:10.1002/adsc.202100284.
- M. Z. Hernandes, S. M. Cavalcanti, D. R. Moreira, W. F. de Azevedo Junior and A. C. Leite, Halogen atoms in the modern medicinal chemistry: hints for the drug design, Curr. Drug Targets, 2010, 11, 303–314, DOI:10.2174/138945010790711996.
- P. Jeschke, The unique role of halogen substituents in the design of modern agrochemicals, Pest Manage. Sci., 2010, 66, 10–27, DOI:10.1002/ps.1829.
- G. Berger, P. Frangville and F. Meyer, Halogen bonding for molecular recognition: new developments in materials and biological sciences, Chem. Commun., 2020, 56, 4970–4981, 10.1039/D0CC00841A.
- M. L. Tang and Z. Bao, Halogenated Materials as Organic Semiconductors, Chem. Mater., 2011, 23, 446–455, DOI:10.1021/cm102182x.
- D. A. Nicewicz and D. W. C. Macmillan, Merging Photoredox Catalysis with Asymmetric Alkylation of Aldehydes, Science, 2008, 322, 77–80, DOI:10.1126/science.1161976.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions, J. Am. Chem. Soc., 2008, 130, 12886–12887, DOI:10.1021/ja805387f.
- N. Holmberg-Douglas and D. A. Nicewicz, Photoredox-Catalyzed C–H Functionalization Reactions, Chem. Rev., 2022, 122, 1925–2016, DOI:10.1021/acs.chemrev.1c00311.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363, DOI:10.1021/cr300503r.
- J. Xuan and W.-J. Xiao, Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2012, 51, 6828–6838, DOI:10.1002/anie.201200223.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Visible light photoredox-controlled reactions of N-radicals and radical ions, Chem. Soc. Rev., 2016, 45, 2044–2056, 10.1039/C5CS00655D.
- J. Xia, C. Zhu and C. Chen, Visible Light-Promoted Metal-Free C–H Activation: Diarylketone-Catalyzed Selective Benzylic Mono- and Diffluorination, J. Am. Chem. Soc., 2013, 135, 17494–17500, DOI:10.1021/ja410815u.
- M. Xiang, Z. K. Xin, B. Chen, C. H. Tung and L. Z. Wu, Exploring the Reducing Ability of Organic Dye (Acr + -Mes) for Fluorination and Oxidation of Benzylic C(Sp3)–H Bonds under Visible Light Irradiation, Org. Lett., 2017, 19(11), 3009–3012, DOI:10.1021/acs.orglett.7b01270.
- D. D. Bume, C. R. Pitts, R. T. Jokhai and T. Lectka, Direct, Visible Light-Sensitized Benzylic CeHFl Uorination of Peptides Using Dibenzosuberenone: Selectivity for Phenylalanine-like Residues, Tetrahedron, 2016, 72(40), 6031–6036, DOI:10.1016/j.tet.2016.08.018.
- E. Article, D. D. Bume, C. R. Pitts, F. Ghorbani, S. A. Harry, J. N. Capilato, M. A. Siegler and T. Lectka, Chem. Sci., 2017, 6918–6923, 10.1039/c7sc02703f.
- D. D. Bume, S. A. Harry, C. R. Pitts and T. Lectka, Sensitized Aliphatic Fluorination Directed by Terpenoidal Enones: A “ Visible Light” Approach, J. Org. Chem., 2018, 83, 1565–1575, DOI:10.1021/acs.joc.7b02807.
- E. Nobile, T. Castanheiro and T. Besset, Radical-Promoted Distal C−H Functionalization of C(Sp3) Centers with Fluorinated Moieties, Angew. Chem., Int. Ed., 2021, 60(22), 12170–12191, DOI:10.1002/anie.202009995.
- E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Photoinduced Remote Functionalisations by Iminyl Radical Promoted C–C and C–H Bond Cleavage Cascades, Angew. Chem., Int. Ed., 2018, 57(3), 744–748, DOI:10.1002/anie.201710790.
- S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Photoinduced Remote Functionalization of Amides and Amines Using Electrophilic Nitrogen Radicals, Angew. Chem., Int. Ed., 2018, 57(39), 12945–12949, DOI:10.1002/anie.201807941.
- D. Bafaluy, Z. Georgieva and K. Muñiz, Iodine Catalysis for C(Sp3)–H Fluorination with a Nucleophilic Fluorine Source, Angew. Chem., Int. Ed., 2020, 59(34), 14241–14245, DOI:10.1002/anie.202004902.
- Z. Deng, Z. Zhao, G. He and G. Chen, Photoredox-Mediated Mono- and Di Fl Uorination of Remote Unactivated Methylene C(Sp3)–H Bonds of N-Alkyl Sulfonamides, Org. Lett., 2021, 23, 3631–3635, DOI:10.1021/acs.orglett.1c01020.
- A. N. Herron, D. Liu, G. Xia and J. Q. Yu, C–H Mono- A Nd Dihalogenation of Alcohols, J. Am. Chem. Soc., 2020, 142(6), 2766–2770, DOI:10.1021/jacs.9b13171.
- J. Xia, C. Zhu and C. Chen, Visible Light-Promoted Metal-Free Sp3–C–H Fluorination, Chem. Commun., 2014, 50, 11701–11704, 10.1039/c4cc05650g.
- S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, A Convenient Photocatalytic Fluorination of Unactivated C–H Bonds, Angew. Chem., Int. Ed., 2014, 53, 4690–4693, DOI:10.1002/anie.201400420.
- S. Bloom, J. L. Knippel and T. Lectka, A photocatalyzed aliphatic fluorination, Chem. Sci., 2014, 5, 1175–1178, 10.1039/c3sc53261e.
- C. W. Kee, K. F. Chin, M. W. Wong and C. Tan, Selective Fluorination of Alkyl C–H Bonds via Photocatalysis, Chem. Commun., 2014, 50, 8211–8214, 10.1039/c4cc01848f.
- J. G. West, T. A. Bedell and E. J. Sorensen, The Uranyl Cation as a Visible-Light Photocatalyst for C(sp3)−H Fluorination, Angew. Chem., Int. Ed., 2016, 55, 8923–8927, DOI:10.1002/anie.201603149.
- L. Wu, X. Cao, X. Chen, W. Fang and M. Dolg, Visible-Light Photocatalysis of C(Sp3)–H Fluorination by the Uranyl Ion: Mechanistic Insights, Angew. Chem., Int. Ed., 2018, 57, 11812–11816, DOI:10.1002/anie.201806554.
- X. Zhang, P. Li, M. Krzyaniak, J. Knapp, M. R. Wasielewski and O. K. Farha, Stabilization of Photocatalytically Active Uranyl Species in a Uranyl − Organic Framework for Heterogeneous Alkane Fluorination Driven by Visible Light, Inorg. Chem., 2020, 59, 16795–16798, DOI:10.1021/acs.inorgchem.0c00850.
- D. J. Wilger, J. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Acids to Styrenes, Nat. Chem., 2014, 6, 720–726, DOI:10.1038/nchem.2000.
- W. Deng, W. Feng, Y. Li and H. Bao, Merging Visible-Light Photocatalysis and Transition-Metal Catalysis in Three-Component Alkyl-Fluorination of Olefins with a Fluoride Ion, Org. Lett., 2018, 20, 4245–4249, DOI:10.1021/acs.orglett.8b01658.
- S. Zou, X. Luo, C. Chen and C. Xi, Photoredox-Catalyzed Fluorodifluoroacetylation of Alkenes with FSO2CF2CO2Me and Et3N·3HF, Org. Biomol. Chem., 2022, 20, 3726–3730, 10.1039/D2OB00488G.
- J. Sim, M. W. Campbell and G. A. Molander, Synthesis of α-Fluoro-α-Amino Acid Derivatives via Photoredox-Catalyzed Carbofluorination, ACS Catal., 2019, 9, 1558–1563, DOI:10.1021/acscatal.8b04284.
- H. J. Tang, X. Zhang, Y. F. Zhang and C. Feng, Visible-Light-Assisted Gold-Catalyzed Fluoroarylation of Allenoates, Angew. Chem., Int. Ed., 2020, 59(13), 5242–5247, DOI:10.1002/anie.201916471.
- K. Ohkubo, K. Mizushima and S. Fukuzumi, Oxygenation and Chlorination of Aromatic Hydrocarbons with Hydrochloric Acid Photosensitized by 9-Mesityl-10-Methylacridinium under Visible Light Irradiation, Res. Chem. Intermed., 2013, 39(1), 205–220, DOI:10.1007/s11164-012-0643-5.
- W. Chen, Z. Huang, N. E. S. Tay, B. Giglio, M. Wang, H. Wang, Z. Wu, D. A. Nicewicz and Z. Li, Direct Arene C–H Fluorination with 18F−via Organic Photoredox Catalysis, Science, 2019, 364(6446), 1170–1174, DOI:10.1126/science.aav7019.
- L. Wang, A. R. White, W. Chen, Z. Wu, D. A. Nicewicz and Z. Li, Direct Radio Fluorination of Arene C–H Bonds via Photoredox Catalysis Using a Peroxide as the Terminal Oxidant, Org. Lett., 2020, 22, 7971–7975, DOI:10.1021/acs.orglett.0c02815.
- X.-Q. Hu, Z.-K. Liu, Y.-X. Hou and Y. Gao, Single Electron Activation of Aryl Carboxylic Acids, iScience, 2020, 23, 101266, DOI:10.1016/j.isci.2020.101266.
- Z. Zeng, A. Feceu, N. Sivendran and L. J. Gooßen, Decarboxylation-Initiated Intermolecular Carbon-Heteroatom Bond Formation, Adv. Synth. Catal., 2021, 363, 2678–2722, DOI:10.1002/adsc.202100211.
- M. Rueda-Becerril, O. Mahe, M. Drouin, M. B. Majewski, J. G. West, M. O. Wolf, G. M. Sammis and J.-F. Paquin, Direct C–F Bond Formation Using Photoredox Catalysis, J. Am. Chem. Soc., 2014, 136, 2637–2641, DOI:10.1021/ja412083f.
- S. Ventre, F. R. Petronijevic and D. W. C. Macmillan, Decarboxylative Fluorination of Aliphatic Carboxylic Acids via Photoredox Catalysis, J. Am. Chem. Soc., 2015, 137, 5654–5657, DOI:10.1021/jacs.5b02244.
- X. Wu, C. Meng, X. Yuan, X. Jia, X. Qian and J. Ye, Transition-metal-free visible-light photoredox catalysis at room-temperature for decarboxylative fluorination of aliphatic carboxylic acids by organic dyes, Chem. Commun., 2015, 51, 11864–11867, 10.1039/c5cc04527d.
- E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, Nucleophilic (Radio)Fluorination of Redox-Active Esters via Radical-Polar Crossover Enabled by Photoredox Catalysis, J. Am. Chem. Soc., 2020, 142(20), 9493–9500, DOI:10.1021/jacs.0c03125.
- P. Xu, P. López-Rojas and T. Ritter, Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination, J. Am. Chem. Soc., 2021, 143(14), 5349–5354, DOI:10.1021/jacs.1c02490.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A. M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. Macmillan, A Unified Approach to Decarboxylative Halogenation of (Hetero)Aryl Carboxylic Acids, J. Am. Chem. Soc., 2022, 144, 8296–8305, DOI:10.1021/jacs.2c02392.
- Y. Zhang, J. Qian, M. Wang, Y. Huang and P. Hu, Visible-Light-Induced Decarboxylative Fluorination of Aliphatic Carboxylic Acids Catalyzed by Iron, Org. Lett., 2022, 24, 5972–5976, DOI:10.1021/acs.orglett.2c02242.
- T. A. Mcteague and T. F. Jamison, Photoredox Activation of SF6 for Fluorination, Angew. Chem., Int. Ed., 2016, 55, 15072–15075, DOI:10.1002/anie.201608792.
- J. Mir, J. L. Jeffrey and D. W. C. Macmillan, Photoredox-Catalyzed Deoxyfluorination of Activated Alcohols with Selectfluor®, Tetrahedron, 2023, 141, 133494, DOI:10.1016/j.tet.2023.133494.
- M. Xiang, C. Zhou, X. Yang, B. Chen, C. Tung and L. Wu, Visible Light-Catalyzed Benzylic C–H Bond Chlorination by a Combination of Organic Dye (Acr + -Mes) and N-Chlorosuccinimide, J. Org. Chem., 2020, 85, 9080–9087, DOI:10.1021/acs.joc.0c01000.
- L. Han, J. B. Xia, L. You and C. Chen, Ketone-Catalyzed Photochemical C(Sp3)–H Chlorination, Tetrahedron, 2017, 73(26), 3696–3701, DOI:10.1016/j.tet.2017.05.008.
- Q. Qin and S. Yu, Visible-Light-Promoted Remote C(Sp3)–H Amidation and Chlorination, Org. Lett., 2015, 17(8), 1894–1897, DOI:10.1021/acs.orglett.5b00582.
- Y. Zhu, J. J. Wang, D. Wu and W. Yu, Visible-Light-Driven Remote C−H Chlorination of Aliphatic Sulfonamides with Sodium Hypochlorite, Asian J. Org. Chem., 2020, 9(10), 1650–1654, DOI:10.1002/ajoc.202000354.
- R. Z. Liu, J. Li, J. Sun, X. G. Liu, S. Qu, P. Li and B. Zhang, Generation and Reactivity of Amidyl Radicals: Manganese-Mediated Atom-Transfer Reaction, Angew. Chem., Int. Ed., 2020, 59(11), 4428–4433, DOI:10.1002/anie.201913042.
- S. Liu, Q. Zhang, H. Li, Y. Yang, X. Tian and A. Whiting, A Visible-Light-Induced α-H Chlorination of Alkylarenes with Inorganic Chloride under NanoAg@AgCl, Chem. – Eur. J., 2015, 21(27), 9671–9675, DOI:10.1002/chem.201501439.
- S. Liu, Q. Zhang, X. Tian, S. Fan, J. Huang and A. Whiting, Highly Selective Halogenation of Unactivated C(Sp3)–H with NaX under Co-Catalysis of Visible Light and Ag@AgX, Green Chem., 2018, 20(20), 4729–4737, 10.1039/c8gc02628a.
- M. Zhao and W. Lu, Visible Light-Induced Oxidative Chlorination of Alkyl Sp3 C–H Bonds with NaCl/Oxone at Room Temperature, Org. Lett., 2017, 19(17), 4560–4563, DOI:10.1021/acs.orglett.7b02153.
- W. Wu, Z. Fu, X. Wen, Y. Wang, S. Zou, Y. Meng, Y. Liu, S. R. Kirk and D. Yin, Light-Triggered Oxy-Chlorination of Cyclohexane by Metal Chlorides, Appl. Catal., A, 2014, 469, 483–489, DOI:10.1016/j.apcata.2013.08.045.
- Y. Wang, N. Wang, J. Zhao, M. Sun, H. You, F. Fang and Z. Q. Liu, Visible-Light-Promoted Site-Specific and Diverse Functionalization of a c(Sp3)–c(Sp3) Bond Adjacent to an Arene, ACS Catal., 2020, 10(12), 6603–6612, DOI:10.1021/acscatal.0c01495.
- P. Lian, W. Long, J. Li, Y. Zheng and X. Wan, Visible-Light-Induced Vicinal Dichlorination of Alkenes through LMCT Excitation of CuCl2, Angew. Chem., Int. Ed., 2020, 59(52), 23603–23608, DOI:10.1002/anie.202010801.
- S. H. Oh, Y. R. Malpani, N. Ha, Y. S. Jung and S. B. Han, Vicinal Difunctionalization of Alkenes: Chlorotrifluoromethylation with CF3SO2Cl by Photoredox Catalysis, Org. Lett., 2014, 16(5), 1310–1313, DOI:10.1021/ol403716t.
- D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Trifluoromethylchlorosulfonylation of Alkenes: Evidence for an Inner-Sphere Mechanism by a Copper Phenanthroline Photoredox Catalyst, Angew. Chem., Int. Ed., 2015, 54(24), 6999–7002, DOI:10.1002/anie.201501880.
- X. Tang and W. R. Dolbier, Efficient Cu-Catalyzed Atom Transfer Radical Addition Reactions of Fluoroalkylsulfonyl Chlorides with Electron-Deficient Alkenes Induced by Visible Light, Angew. Chem., Int. Ed., 2015, 54, 4246–4249, DOI:10.1002/anie.201412199.
- V. R. Rao, K. T. Patil, D. Kumar, S. Sebastian, M. K. Gupta and D.-S. Shin, Facile metal-free visible-light-mediated chlorotrifluoromethylation of terminal alkenes, Monatsh. Chem., 2022, 153, 495–500, DOI:10.1007/s00706-022-02930-y-2005.
- T. F. Niu, D. Lin, L. S. Xue, D. Y. Jiang and B. Q. Ni, Visible-Light-Induced Chemoselective Synthesis of α-Chloro and Vinyl Sulfones by Sulfonylation of Alkenes, Synlett, 2018,(3), 364–368, DOI:10.1055/s-0036-1590925.
- A. Hossain, S. Engl, E. Lutsker and O. Reiser, Visible-Light-Mediated Regioselective Chlorosulfonylation of Alkenes and Alkynes: Introducing the Cu(II) Complex [Cu(Dap)Cl2] to Photochemical ATRA Reactions, ACS Catal., 2019, 9(2), 1103–1109, DOI:10.1021/acscatal.8b04188.
- D. V. Patil, H. Y. Kim and K. Oh, Visible Light-Promoted Friedel–Crafts-Type Chloroacylation of Alkenes to β-Chloroketones, Org. Lett., 2020, 22(8), 3018–3022, DOI:10.1021/acs.orglett.0c00788.
- C. Wu, X. Hui, D. Zhang, M. Zhang, Y. Zhu and S. Wang, Visible Light-Mediated Polychlorination of Alkenes: Via the Dichloromethyl Radical Generated by Chloroform and Chlorides, Green Chem., 2022, 24(3), 1103–1108, 10.1039/d1gc04109f.
- C. Ding, P.-F. Huang, B. Xiong, K.-W. Tang and Y. Liu, Visible-Light-Induced Difunctionalization of the C–C Bond of Alkylidenecyclopropanes with Acyl Chlorides, Catalysts, 2023, 13(6), 919, DOI:10.3390/catal13060919.
- E. Mejri, K. Higashida, Y. Kondo, A. Nawachi, H. Morimoto, T. Ohshima, M. Sawamura and Y. Shimizu, Visible-Light-Induced Aminochlorination of Alkenes, Org. Lett., 2023, 25(24), 4581–4585, DOI:10.1021/acs.orglett.3c01645.
- X. X. He, H. H. Chang, Y. X. Zhao, X. J. Li, S. A. Liu, Z. L. Zang, C. H. Zhou and G. X. Cai, CuCl2-Catalyzed α-Chloroketonation of Aromatic Alkenes via Visible-Light-Induced LMCT, Chem. – Asian J., 2023, 18(1), 1–5, DOI:10.1002/asia.202200954.
- Z. Luo, Y. Meng, X. Gong, J. Wu, Y. Zhang, L.-W. Ye and C. Zhu, Facile Synthesis of α-Haloketones by Aerobic Oxidation of Olefins Using KX as Nonhazardous Halogen Source, Chin. J. Chem., 2020, 38, 173–177, DOI:10.1002/cjoc.201900372.
- Z. Wang, L. Wang, Z. Wang, P. Li and Y. Zhang, A practical synthesis of α-bromo/iodo/chloroketones from olefins under visible-light irradiation conditions, Chin. Chem. Lett., 2021, 32, 429–432, DOI:10.1016/j.cclet.2020.02.022.
- F. Han, D. Zhang, S. Salli, J. Ye, Y. Li, F. Rosei, X.-D. Wen, H. Niemantsverdriet, E. Richards and R. Su, Copper Cocatalyst Modulated Radical Generation for Selective Heterogeneous Photosynthesis of α-Haloketones, ACS Catal., 2023, 13, 248–255, DOI:10.1021/acscatal.2c05189.
- T. Mandal, N. Katta, H. Paps and O. Reiser, Merging Cu(I) and Cu(II) Photocatalysis: Development of a Versatile Oxohalogenation Protocol for the Sequential Cu(II)/Cu(I)-Catalyzed Oxoallylation of Vinylarenes, ACS Org. Inorg. Au, 2023, 3, 171–176, DOI:10.1021/acsorginorgau.3c00011.
- L. N. S. Crespin, A. Greb, D. C. Blakemore and S. V. Ley, Visible-Light-Mediated Annulation of Electron-Rich Alkenes and Nitrogen-Centered Radicals from N-Sulfonylallylamines: Construction of Chloromethylated Pyrrolidine Derivatives, J. Org. Chem., 2017, 82(24), 13093–13108, DOI:10.1021/acs.joc.7b02146.
- H. S. Han, Y. J. Lee, Y. S. Jung and S. B. Han, Stereoselective Photoredox-Catalyzed Chlorotrifluoromethylation of Alkynes: Synthesis of Tetrasubstituted Alkenes, Org. Lett., 2017, 19(8), 1962–1965, DOI:10.1021/acs.orglett.7b00470.
- P. Chakrasali, K. Kim, Y. S. Jung, H. Kim and S. B. Han, Visible-Light-Mediated Photoredox-Catalyzed Regio- and Stereoselective Chlorosulfonylation of Alkynes, Org. Lett., 2018, 20(23), 7509–7513, DOI:10.1021/acs.orglett.8b03273.
- L. Finck, J. Brals, B. Pavuluri, F. Gallou and S. Handa, Micelle-Enabled Photoassisted Selective Oxyhalogenation of Alkynes in Water under Mild Conditions, J. Org. Chem., 2018, 83(14), 7366–7372, DOI:10.1021/acs.joc.7b03143.
- V. Pramod Charpe, M. Gupta and K. Chu Hwang, Visible-Light-Induced Oxidative α-Keto-Dichlorination of Arylalkynes by CuCl2 at Room Temperature, ChemSusChem, 2022, 15(17), e202200957, DOI:10.1002/cssc.202200957.
- H. Hou, D. Tang, H. Li, Y. Xu, C. Yan, Y. Shi, X. Chen and S. Zhu, Visible-Light-Driven Chlorotrifluoromethylative and Chlorotrichloromethylative Cyclizations of Enynes, J. Org. Chem., 2019, 84(11), 7509–7517, DOI:10.1021/acs.joc.9b00842.
- O. A. Storozhenko, A. A. Festa, V. A. Zolotareva, V. B. Rybakov, A. V. Varlamov and L. G. Voskressensky, Photoredox-Catalyzed Chlorotrifluoromethylation of Arylallenes: Synthesis of a Trifluoromethyl Building Block, Org. Lett., 2023, 25, 438–442, DOI:10.1021/acs.orglett.2c04214.
- E. Azzi, G. Ghigo, L. Sarasino, S. Parisotto, R. Moro, P. Renzi and A. Deagostino, Photoinduced Chloroamination Cyclization Cascade with N-Chlorosuccinimide: From N-(Allenyl)Sulfonylamides to 2-(1-Chlorovinyl)Pyrrolidines, J. Org. Chem., 2023, 88(10), 6420–6433, DOI:10.1021/acs.joc.2c01963.
- T. Hering and B. König, Photocatalytic Activation of N-Chloro Compounds for the Chlorination of Arenes, Tetrahedron, 2016, 72(48), 7821–7825, DOI:10.1016/j.tet.2016.06.028.
- Q. Shu, Y. Li, T. Liu, S. Zhang, L. Jiang, K. Jin, R. Zhang and C. Duan, Visible Light Induced Regioselective C5 Halogenation of 8-Aminoquinolines with 1,3-Dihalo-5,5-Dimethylhydantoin in Continuous Flow, Tetrahedron, 2019, 75(26), 3636–3642, DOI:10.1016/j.tet.2019.05.037.
- D. A. Rogers, J. M. Gallegos, M. D. Hopkins, A. A. Lignieres, A. K. Pitzel and A. A. Lamar, Visible-Light Photocatalytic Activation of N-Chlorosuccinimide by Organic Dyes for the Chlorination of Arenes and Heteroarenes, Tetrahedron, 2019, 75(36), 130498, DOI:10.1016/j.tet.2019.130498.
- D. A. Rogers, A. T. Bensalah, A. T. Espinosa, J. L. Hoerr, F. H. Refai, A. K. Pitzel, J. J. Alvarado and A. A. Lamar, Amplification of Trichloroisocyanuric Acid (TCCA) Reactivity for Chlorination of Arenes and Heteroarenes via Catalytic Organic Dye Activation, Org. Lett., 2019, 21(11), 4229–4233, DOI:10.1021/acs.orglett.9b01414.
- D. A. Rogers, M. D. Hopkins, N. Rajagopal, D. Varshney, H. A. Howard, G. Leblanc and A. A. Lamar, U.S. Food and Drug Administration-Certified Food Dyes as Organocatalysts in the Visible Light-Promoted Chlorination of Aromatics and Heteroaromatics, ACS Omega, 2020, 5(13), 7693–7704, DOI:10.1021/acsomega.0c00631.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, Aromatic Monochlorination Photosensitized by DDQ with Hydrogen Chloride under Visible-Light Irradiation, Chem. – Asian J., 2016, 11(7), 996–999, DOI:10.1002/asia.201600083.
- S. J. S. Düsel and B. König, Oxidative Photochlorination of Electron-Rich Arenes via in Situ Bromination, Eur. J. Org. Chem., 2020,(10), 1491–1495, DOI:10.1002/ejoc.201900411.
- L. Candish, E. A. Standley, A. Gómez-suμrez, S. Mukherjee and F. Glorius, Catalytic Access to Alkyl Bromides, Chlorides and Iodides via Visible Light-Promoted Decarboxylative Halogenation, Chem. – Eur. J., 2016, 22, 9971–9974, DOI:10.1002/chem.201602251.
- G. H. Coleman and G. E. Honeywell, P-NITROBENZYL BROMIDE, Org. Synth., 1936, 16, 54, DOI:10.15227/orgsyn.016.0054.
- R. L. Shriner and F. J. Wolf, Phthalaldehydic Acid, Org. Synth., 1943, 23, 74, DOI:10.15227/orgsyn.023.0074.
- E. F. M. Stephenson, O-Xylylene dibromide, Org. Synth., 1954, 34, 100, DOI:10.15227/orgsyn.034.0100.
- A. Kalir, O-Nitrobenzaldehyde, Org. Synth., 1966, 46, 81, DOI:10.15227/orgsyn.046.0081.
- I. A. Koten and R. J. Sauer, 3-Bromophthalide, Org. Synth., 1962, 42, 26, DOI:10.15227/orgsyn.042.0026.
- A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Visible Light Induced “on Water” Benzylic Bromination with N-Bromosuccinimide, Tetrahedron Lett., 2006, 47(7), 1097–1099, DOI:10.1016/j.tetlet.2005.12.040.
- A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Association of Fluorous “Phase-Vanishing” Method with Visible-Light Activation in Benzylic Bromination by Bromine, Eur. J. Org. Chem., 2006,(2), 483–488, DOI:10.1002/ejoc.200500355.
- S. Mart, C. F. Marcos and A. G. Neo, A Safe and Green Benzylic Radical Bromination Experiment, J. Chem. Educ., 2020, 97, 582–585, DOI:10.1021/acs.jchemed.9b00020.
- M. Jereb, M. Zupan and S. Stavber, Visible-Light-Promoted Wohl–Ziegler Functionalization of Organic Molecules with N-Bromosuccinimide under Solvent-Free Reaction Conditions, Helv. Chim. Acta, 2009, 92(3), 555–566, DOI:10.1002/hlca.200800308.
- R. Mestres and J. Palenzuela, High Atomic Yield Bromine-Less Benzylic Bromination, Green Chem., 2002, 4(4), 314–316, 10.1039/b203055a.
- W. B. Yu, D. P. Yu, M. M. Zheng, S. T. Shan, Y. J. Li and J. R. Gao, Catalyst and Solvent-Free Bromination of Toluene Derivatives by HBr–H2O2 with Visible-Light Photocatalysis Using a Continuous-Flow Micro Reactor, J. Chem. Res., 2012, 36(5), 258–260, DOI:10.3184/174751912X13333614619977.
- A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Environmentally Benign Electrophilic and Radical Bromination “on Water”: H2O2–HBr System versus N-Bromosuccinimide, Tetrahedron, 2009, 65(22), 4429–4439, DOI:10.1016/j.tet.2009.03.034.
- M. Zhao, M. Li and W. Lu, Visible-Light-Driven Oxidative Mono- and Dibromination of Benzylic Sp3 C–H Bonds with Potassium Bromide/Oxone at Room Temperature, Synth., 2018, 50(24), 4933–4939, DOI:10.1055/s-0037-1610651.
- T. Hou, P. Lu and P. Li, Visible-Light-Mediated Benzylic Sp3 C–H Bond Functionalization to C–Br or C–N Bond, Tetrahedron Lett., 2016, 57(21), 2273–2276, DOI:10.1016/j.tetlet.2016.04.036.
- N. Tada, K. Ban, S. I. Hirashima, T. Miura and A. Itoh, Direct Synthesis of α-Bromoketones from Alkylarenes by Aerobic Visible Light Photooxidation, Org. Biomol. Chem., 2010, 8(20), 4701–4704, 10.1039/c0ob00101e.
- N. Tada, K. Ban, T. Ishigami, T. Nobuta, T. Miura and A. Itoh, Tandem Oxidation/Bromination of Ethyl Aromatics to α,α-Dibromoacetophenones with Molecular Oxygen under Visible Light Irradiation, Tetrahedron Lett., 2011, 52(29), 3821–3824, DOI:10.1016/j.tetlet.2011.05.077.
- V. A. Schmidt, R. K. Quinn, A. T. Brusoe and E. J. Alexanian, Site-Selective Aliphatic C–H Bromination Using N-Bromoamides and Visible Light, J. Am. Chem. Soc., 2014, 136(41), 14389–14392, DOI:10.1021/ja508469u.
- C. W. Kee, K. M. Chan, M. W. Wong and C.-H. Tan, Selective Bromination of sp3 C–H Bonds by Organophotoredox Catalysis, Asian J. Org. Chem., 2014, 3, 536–544, DOI:10.1002/ajoc.201300169.
- M. Zhao and W. Lu, Catalytic Bromination of Alkyl Sp3C–H Bonds with KBr/Air under Visible Light, Org. Lett., 2018, 20, 5264–5267, DOI:10.1021/acs.orglett.8b02208.
- Y. Nishina, B. Ohtani and K. Kikushima, Bromination of Hydrocarbons with CBr4, Initiated by Light-Emitting Diode Irradiation, Beilstein J. Org. Chem., 2013, 9, 1663–1667, DOI:10.3762/bjoc.9.190.
- C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts, J. Am. Chem. Soc., 2012, 134(21), 8875–8884, DOI:10.1021/ja300798k.
- M. Knorn, T. Rawner, R. Czerwieniec and O. Reiser, [Copper(phenanthroline)(bisisonitrile)] + -Complexes for the Visible-Light-Mediated Atom Transfer Radical Addition and Allylation Reactions, ACS Catal., 2015, 5, 5186–5193, DOI:10.1021/acscatal.5b01071.
- J. Fang, Z. Wang, S. Wu, W. Shen and G. Ao, Photoredox-Catalysed Chloro-, Bromo- and Trifluoromethylthio-Trifluoromethylation of Unactivated Alkenes with Sodium Triflinate, Chem. Commun., 2017, 7638–7641, 10.1039/c7cc01903c.
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, [Cu(dap)2Cl] As an Efficient Visible-Light-Driven Photoredox Catalyst in Carbon–Carbon Bond-Forming Reactions, Chem. – Eur. J., 2012, 18, 7336–7340, DOI:10.1002/chem.201200967.
- F. Huan, Q. Y. Chen and Y. Guo, Visible Light-Induced Photoredox Construction of Trifluoromethylated Quaternary Carbon Centers from Trifluoromethylated Tertiary Bromides, J. Org. Chem., 2016, 81(16), 7051–7063, DOI:10.1021/acs.joc.6b00930.
- K. Wang, L. G. Meng and L. Wang, Visible-Light-Initiated Na2-Eosin y Catalyzed Highly Regio- and Stereoselective Difunctionalization of Alkynes with Alkyl Bromides, J. Org. Chem., 2016, 81(16), 7080–7087, DOI:10.1021/acs.joc.6b00973.
- D. A. Rogers, R. G. Brown, Z. C. Brandeburg, E. Y. Ko, M. D. Hopkins, G. Leblanc and A. A. Lamar, Organic Dye-Catalyzed, Visible-Light Photoredox Bromination of Arenes and Heteroarenes Using N-Bromosuccinimide, ACS Omega, 2018, 3(10), 12868–12877, DOI:10.1021/acsomega.8b02320.
- L. Feng, H. Yan, C. Yang, D. Chen and W. Xia, Visible-Light Induced Direct Synthesis of Polysubstituted Furans from Cyclopropyl Ketones, J. Org. Chem., 2016, 81(16), 7008–7022, DOI:10.1021/acs.joc.6b00436.
- B. Ma, F. Lu, H. Yang, X. Gu, Z. Li, R. Li and H. Pei, Visible Light Mediated External Oxidant Free Selective C5 Bromination of 8-Aminoquinoline Amides under Ambient Conditions, Asian J. Org. Chem., 2019, 8, 1136–1140, DOI:10.1002/ajoc.201900293.
- K. Ohkubo, K. Mizushima, R. Iwataa and S. Fukuzumi, Selective Photocatalytic Aerobic Bromination with Hydrogen Bromide via an Electron-Transfer State of 9-Mesityl-10-Methylacridinium Ion, Chem. Sci., 2011, 2(4), 715–722, 10.1039/c0sc00535e.
- R. Li, D. W. Gehrig, C. Ramanan, P. W. M. Blom, F. F. Kohl, M. Wagner, K. Landfester and K. A. I. Zhang, Visible Light-Mediated Conversion of Alcohols to Bromides by a Benzothiadiazole-Containing Organic Photocatalyst, Adv. Synth. Catal., 2019, 361, 3852–3859, DOI:10.1002/adsc.201900416.
- M. Huang, L. Li, Z. G. Zhao, Q. Y. Chen and Y. Guo, Visible-Light-Induced Photocatalysis of 1,1,1-Trifluoro-2-Iodoethane with Alkylalkenes and Silyl Enol Ethers, Synth., 2015, 47(24), 3891–3900, DOI:10.1055/s-0035-1560260.
- T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, The Different Faces of Photoredox Catalysts: Visible-Light-Mediated Atom Transfer Radical Addition (ATRA) Reactions of Perfluoroalkyl Iodides with Styrenes and Phenylacetylenes, ACS Catal., 2018, 8, 3950–3956, DOI:10.1021/acscatal.8b00847.
- S. Engl and O. Reiser, Copper Makes the Difference: Visible Light-Mediated Atom Transfer Radical Addition Reactions of Iodoform with Olefins, ACS Catal., 2020, 10, 9899–9906, DOI:10.1021/acscatal.0c02984.
- H. Sheng, Q. Liu, X. D. Su, Y. Lu, Z. X. Wang and X. Y. Chen, Visible-Light-Triggered Iodinations Facilitated by Weak Electrostatic Interaction of N-Heterocyclic Carbenes, Org. Lett., 2020, 22(18), 7187–7192, DOI:10.1021/acs.orglett.0c02523.
- R. Narobe, S. J. S. Düsel, J. Iskra and B. König, Photocatalytic Oxidative Iodination of Electron-Rich Arenes, Adv. Synth. Catal., 2019, 361, 3998–4004, DOI:10.1002/adsc.201900298.
- L. Candish, E. A. Standley, A. Gómez-Suárez, S. Mukherjee and F. Glorius, Catalytic Access to Alkyl Bromides, Chlorides and Iodides via Visible Light-Promoted Decarboxylative Halogenation, Chem. – Eur. J., 2016, 22(29), 9971–9974, DOI:10.1002/chem.201602251.
- M. Jiang and M. Jiang, Visible-Light-Induced Decarboxylative Iodination of Aromatic Carboxylic Acids, Synlett, 2018, 1572–1577, DOI:10.1055/s-0037-1610188.
- Y. Zhao and M. Antonietti, Visible-Light-Driven Conversion of Alcohols into Iodide Derivatives with Iodoform, ChemPhotoChem, 2018, 2, 720–724, DOI:10.1002/cptc.201800084.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Olga A.
Storozhenko
a,
Leonid G.
Voskressensky
a,
Olga A.
Storozhenko
a,
Leonid G.
Voskressensky