
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogenization of molecular catalysts within porous solids: the case of Ni-catalyzed ethylene oligomerization from zeolites to metal–organic frameworks
Rémy
Rajapaksha
,
Partha
Samanta
 ,
Elsje Alessandra
Quadrelli
and
Jérôme
Canivet
*
,
Elsje Alessandra
Quadrelli
and
Jérôme
Canivet
*
Univ. Lyon, Université Claude Bernard Lyon 1, CNRS, IRCELYON – UMR 5256, 2 Av. Albert Einstein, 69626 Villeurbanne, France. E-mail: jerome.canivet@ircelyon.univ-lyon1.fr
First published on 30th October 2023
Abstract
The last decade has seen a tremendous expansion of the field of heterogenized molecular catalysis, especially with the growing interest in metal–organic frameworks and related porous hybrid solids. With successful achievements in the transfer from molecular homogeneous catalysis to heterogenized processes come the necessary discussions on methodologies used and a critical assessment on the advantages of heterogenizing molecular catalysis. Here we use the example of nickel-catalyzed ethylene oligomerization, a reaction of both fundamental and applied interest, to review heterogenization methodologies of well-defined molecular catalysts within porous solids while addressing the biases in the comparison between original molecular systems and heterogenized counterparts.
 Rémy Rajapaksha | After studying chemistry engineering at CPE Lyon, Rémy Rajapaksha worked during his PhD on two types of porous supports, Metal–Organic Frameworks (MOFs) and Porous Organic Polymers (POPs), with bipyridyl coordination sites, under the direction of Dr Jérôme Canivet and Dr Elsje Alessandra Quadrelli at IRCELYON. His thesis work focused on nickel coordination chemistry at bipyridyl sites within MOF and POP structures in order to obtain isolated active sites. The synthesized catalysts were studied in the ethylene dimerization reaction. After obtaining his PhD from the University of Lyon in 2022, Rémy Rajapaksha joined Elkem Silicones – Lyon, in the Upstream team, as a research engineer. |
 Partha Samanta | Partha Samanta completed his BSc degree in 2011 and MSc degree in 2013 from the University of Calcutta and IIT Kharagpur, respectively. Then, he joined the research group of Dr Sujit K. Ghosh at IISER Pune to pursue his doctoral studies. In 2019, he obtained a PhD degree and then he joined IRCELYON as postdoctoral fellow with Dr Jerome Canivet, where he worked on heterogeneous catalysts for important organic transformations. And currently, he is working with Dr Daniel Maspoch at ICN2 (Barcelona, Spain) as postdoctoral researcher, and his research interest is focused on the Clip-off chemistry of reticular materials. |
 Elsje Alessandra Quadrelli | Elsje Alessandra Quadrelli is director of research in chemistry from the French National Centre for Scientific Research, CNRS, at the IRCELYON laboratory. Her research focuses on organometallic mechanisms for grafting catalytically active transition metals or for growing thin films by atomic layer deposition (ALD) on surfaces (Silica, MOFs and silicon wafer substrates). Her current main target reaction is CO2 reduction. |
 Jérôme Canivet | Jérôme Canivet was appointed CNRS researcher at the IRCELYON in 2010. He works at developing innovative catalytic processes for sustainable fine chemicals and energy. His research topics range from C–C coupling to asymmetry, photocatalysis and green fuels. In 2018, he received the Young Investigator Award from the Catalysis Division of the French Chemical Society for creating trends reducing the gap between homogeneous and heterogeneous catalysis. He further aims at exploiting the confinement of molecular catalytic systems into porous structures for the improvement of their catalytic activity and selectivity, and he is coordinating cooperative projects on this topic. |
1. Introduction
Beyond an historical dichotomy, the frontier is continuously blurring between homogeneous catalysis, where a typically single atom in the same phase as the substrate is responsible of the catalytic transformation, and heterogeneous catalysis, where the cooperation between several surface atoms, in a separate phase than the substrate, is typically at the origin of substrate activation and transformation.Using heterogenized molecular species enables to isolate active sites from each other and, as a consequence, prevents them from aggregating or interacting with one another, which could lead to the catalyst deactivation or undesired reactivity.1–5
Moreover, the site isolation changes the interactions at play during substrate activation on the heterogeneous catalyst, leading to unprecedented reactivities which can be understood in the light of organometallic chemistry mechanisms.6–10
The usefulness of mobilizing organometallic chemistry mechanisms in single-site heterogeneous catalysts is particularly pertinent for catalysts baring organic-based ligand-like moieties in their first coordination sphere since the metal–ligand environment is a key descriptor of the catalyst performance in organometallic chemistry.
The heterogenization of ligand-bearing metal-centered molecular catalysts can be achieved by grafting organometallic precursors on oxide surface. This strategy, also known as Surface Organometallic Chemistry (SOMC, Fig. 1),11–15 gave access to well-defined isolated sites derived from molecular organometallic counterparts. Beyond the simple heterogenization, the SOMC strategy has already proven to lead in some cases to unique reactivity not accessible by any other molecular analogue, including enzymes and organometallics.16
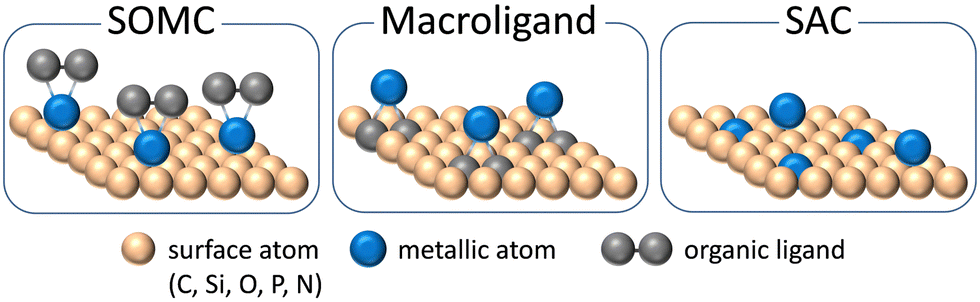 | ||
| Fig. 1 Schematic representation of selected differences in designing heterogeneous single-site catalysts through the concepts of SAC, SOMC and macroligands. | ||
More recently, the concept of porous macroligands proposed a variation of the SOMC approach in the field of single-site catalysts (Fig. 1).17–19 Here a porous organic or hybrid organic–inorganic polymer, made with coordinating monomers like bipyridine derivatives, is defined and used as a solid ligand for the active metal cations. Macroligands include porous solids among periodic mesoporous organosilica (PMO), metal–organic frameworks (MOF) and porous organic polymers (POP).20 The macroligand strategy lies in the derivatization of traditional organic ligand in coordination complexes, like bipyridine or triphenylphosphine, with functional groups such as carboxylic acids or polymerizable vinyl groups for subsequent reticulation in carboxylate-based MOF or in POP, respectively. In macroligands, the molecular catalyst is self-supported in the porous framework, minimizing possible detrimental interactions. Moreover, the nature of porous macroligands allows for an unprecedented site density, in catalytic sites per mass, since no bulk matter remains unused, unlike in traditional grafted siliceous materials.
Porous macroligands like MOF and POP already account for a wide variety of metal–ligand combinations for catalytic applications ranging from fine chemical synthesis to energy.21–27 Such as for homogeneous and heterogeneous catalysts in general, the evolution of the heterogenized molecular catalyst during catalysis has to be scrutinized, especially in the well-documented case of highly reactive palladium complexes prompt to form clusters and particles, as well as prompt to release single-atom from aggregates, and showing high activity even at so-called homeopathic level.28–31 Indeed, the retention of the molecular nature of the active site, i.e. coordination and structure, and the accessibility to substrates molecules when immobilized are key issues to be addressed. If confirmed, this allows for mechanistic studies at the molecular level following both computational and experimental methodologies from molecular science.32–36 Furthermore, the electronic effect of the support on the active site dramatically affects its reactivity.18,19 Therefore, the challenge remains to disentangle the electronic effect resulting from embedding the complex within the macroligand structure from the effect due to the confinement within macroligand's pores.
Since the early 2000s, a novel concept in heterogeneous catalysis appeared with the single atom catalysts (SAC) described as isolated single metal atoms or cations either embedded or supported on solid surfaces (oxide, metal, graphene or polymer, Fig. 1).37–40 At the same time, the SAC concept constitutes a proximal yet distinct class of catalyst with respect to single-site catalysts of interest here if we follow Copéret and Korzyński classification: while single-site catalyst, a concept applicable to homogeneous and heterogeneous catalyses, is centered on the reactivity of well-defined isolated active sites, SAC are mostly defined by textural properties of individual atoms in a surface rather than by the coordination environment or nature of the bonding between the metal and the support (Fig. 1).8
The SAC concept therefore implies no (or weak) influence of other atoms in the first coordination sphere of grafted metal atom beyond the metal to surface bonds (such as M–O or M–C bonds at oxide or carbon surface respectively), while typically at least one coordinated organic molecule, which is absent in SAC systems, directly influences the electronic and/or steric properties of the grafted metal complex in SOMC or macroligand strategies though M–X bonds where X = C, O, N, P, S. Therefore the SAC systems, as well as the similarly ligand free catalysts that can be obtained by the thermolytic molecular precursor method,41,42 are beyond the scope of this review, and will only be sporadically mentioned if they lead to performing materials against whose performance the performance of the SOMC or macroligand-based materials can be compared and contrasted.
In summary, we aim here at focussing on approaches directly influencing the ligand-based coordination environment of heterogenized single-site catalysts. The support described as an extended solid ligand for the active metal would allow direct comparison between heterogenized and homogeneous analogues beyond the practical advantages of using solid catalyst like easy separation from the catalysis medium and recycling. For a clear overview on the scope and limitations of molecular catalyst heterogenization, it is essential to focus on molecular catalytic application that has been extensively studied and documented. Furthermore, this application should be of industrial relevance, with particular attention given to sustainability with the use of earth-abundant active metal catalysts. The overall sustainability of a catalytic process can also be improved by the recycling of the heterogenized catalyst and the easy separation achieved. The absence of active species leaching and the high accessibility of active sites within solid supports can give access to the full productivity of the catalyst without diffusional limitation.43
In this review we aim at highlighting the perspectives offered by heterogenized molecular catalysts compared to homogeneous counterparts using the prism of the nickel-catalyzed ethylene oligomerization as widely studied and industrially relevant reaction.
2. Tailor-made nickel-based molecular catalyst for ethylene oligomerization
Ethylene α-oligomers are typically used in copolymerization with ethylene to modulate the properties of polyethylene. The recent increase of the availability of shale gas makes economically appealing to produce selectively α-oligomers or linear alpha-olefins (LAO) directly from ethylene. Industrial processes of ethylene oligomerization towards LAO rely mainly on titanium-, chromium-, zirconium- and nickel-based catalysis.44,45Two types of mechanisms have been highlighted for ethylene oligomerization:
– a cycle of activation/coordination/insertion/elimination and/or propagation (also called Cossee–Arlman mechanism),46,47
– a cycle of coordination/oxidative coupling/metallacycle intermediate/elimination and/or propagation (also called metallacycle mechanism).48,49
In the case of a Cossee–Arlman mechanism, which has been demonstrated to be the most plausible for the nickel-catalysed reaction,50 either metal halide (like nickel dihalide or titanium chloride) or metal hydride (like nickel hydride) complexes are typically used. Metal hydride complexes readily react with ethylene without prior activation.51 In contrast, metal halide complexes, which are among the easiest to synthesize and the most stable, required an activation step in order to obtain the catalytically active species for ethylene oligomerization. This step typically involves an alkyl-aluminum activator (also called co-catalyst) which subtracts halides and subsequently generates the active metal alkyl species.
The important role of nickel in catalysis and its development as alternative to platinum group metals (PGM) during the last decades was already comprehensively reviewed.52–57
Beyond its acknowledged sustainability due to its large abundance and reduced cost compared to PGM,58 nickel presents advantageous intrinsic electronic properties when dealing with C–C bond formation. Indeed, as compared to palladium and platinum, nickel–carbon bonds show the weakest dissociation energy which makes Ni–C intermediates highly reactive.59,60
The proliferation of catalytic systems based on molecular nickel species benefited to the field of polymer chemistry.61–65 As of interest here, molecular nickel catalysts are well-known to oligomerize ethylene, among other olefins such as propylene and butene.66,67
Linked to its high reactivity, nickel catalysis often proceeds though a wide number of parallel pathways and elementary steps which multiplies the number of active intermediates.68,69
The reactivity of nickel species is highly dependent to their coordination sphere which makes the design of ligand in molecular catalysts crucial to obtained the desired product.70,71 In terms of ligation, the cis-coordination of tetracoordinated Ni(II) sites is shown to favor migratory insertion step.72 Moreover, the hemilability of the ligand coordination, such as with N or P groups, allows for the stabilization of actives species and for the coordination of substrates.
In particular, two types of active nickel species were described though the action of activating agent (cocatalyst) in ethylene oligomerization (Fig. 2):
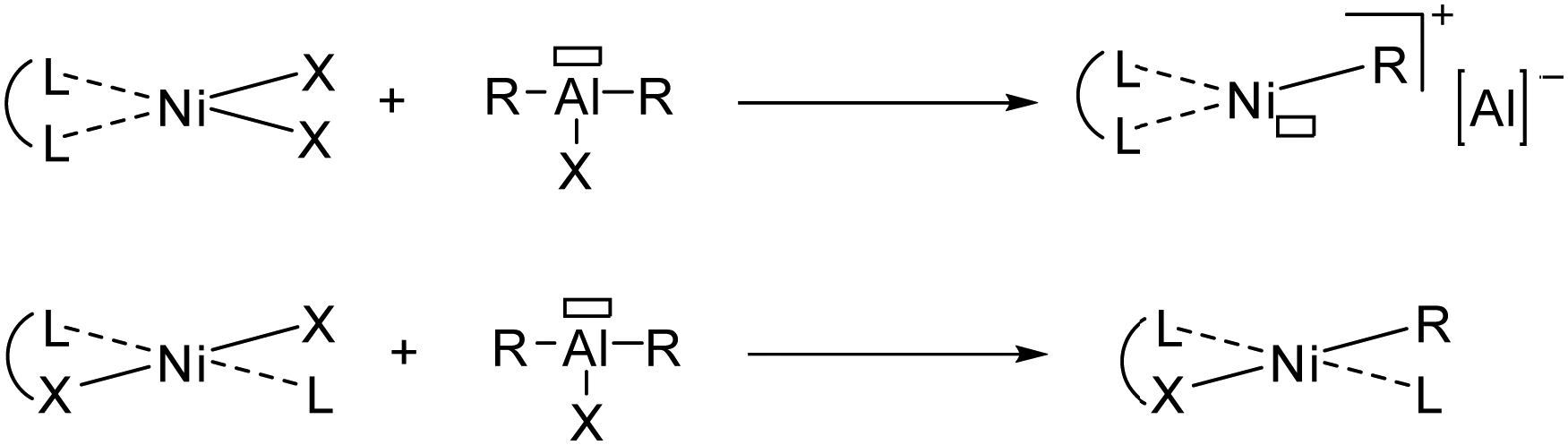 | ||
| Fig. 2 Neutral ligand to cationic catalyst (top) and anionic ligand to neutral catalyst (bottom) upon activation with Al-based cocatalyst. | ||
– cationic species, highly electrophilic, isolated by Wilke et al. in 196673 from neutral ligand;
– neutral species, less electrophilic and isolated by Keim et al. in 197874 from monoanionic ligand.
Among bidentate neutral ligands, the α-diimine ligands developed by Brookhart61 allowed for producing highly electrophilic nickel(II) active species upon activation and favoring olefin insertion through β-H elimination (Fig. 2).75
In contrast, monoanionic ligands give rise to neutral nickel(II) active species (Fig. 2). Highly productive (P,O)-based nickel(II) catalysts pioneered by Keim, known as SHOP catalysts,76 and (N,O)-based phenoxyimine analogues by Grubbs63 were found to produce α-olefins in a broad Schulz–Flory distribution, implying that short oligomers are favored, as well as polyethylene with high molecular weight.
The Shell Higher Olefin Process (SHOP) relies on a nickel-based catalyst with a P–O ligand developed by Keim in 1978 (Fig. 3). This type of catalyst is synthesized with a phosphorus ylide, a [Ni(1,5-cyclooctadiene)2] (Ni(COD)2) complex and a phosphine.77 SHOP-based catalysts with a phosphino-enolate component favors the elimination step rather than the propagation step, leading to LAOs up to twenty carbon atoms. Removing the phosphine part with a scavenger would lead to a polymerization catalyst rather than an oligomerization catalyst.74,78–80 The SHOP process enables to produce a broad range of oligomers but mainly C12 to C20+ olefins and one of the challenge remains to make this type of catalyst more selective towards 1-butene.
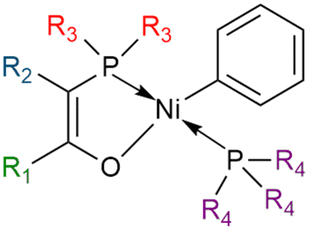 | ||
| Fig. 3 SHOP catalyst with different groups that enable to tune its activity and selectivity (R = alkyl or aryl). | ||
3. Molecular Ni catalysts grafted onto siliceous materials
3.1 Purely inorganic supports
In the field of heterogeneous catalysis, inorganic solids are the most established supports, with siliceous-based materials at the forefront. This section will review the state of the art for ethylene oligomerization by this class of materials.Lallemand et al. tested different types of zeolitic materials (Ni-MCM-36 and Ni-MCM-22) for ethylene oligomerization.81–83 The catalysts are obtained from zeolite structures, by cation exchange. Cations are used during zeolite synthesis in order to ensure charge neutrality within the structure. The pristine materials underwent a treatment with an ammonium nitrate solution in order to replace the cations and then the ammonium ions were replaced by nickel cations. This technique of cation exchange with zeolites (for ethylene oligomerization purposes) enables to obtain an optimal dispersion of the catalytic active sites throughout the support, since they also participate to the structure's charge neutrality and do not require any activation with an aluminum-based cocatalyst. However, the activation of the catalyst requires high temperatures in order to desorb any impurities within the pores and to obtain an optimal porosity for the reaction. The authors postulated that this activation was responsible for the occurrence of acidic sites and the reduction of Ni2+ sites into Ni+ sites. For a reaction time of 30 minutes, under 40 bar of ethylene at 70 °C, the catalysts Ni-MCM-36 and Ni-MCM-22 respectively achieved 1.1 and 10 goligomers gcatalyst−1 h−1 with a selectivity in butene around 81% in both cases. A product distribution close to a Schulz–Flory one is described by the authors and they noticed the higher production of 2-butene, which could be attributed to the acidic sites, decreasing the selectivity in LAOs.
In 2011, with the NiMCM-41 catalyst, the authors observed a Schulz–Flory distribution of the products. This distribution remained steady during seven days at 30 °C and 30 bar of ethylene in continuous flow, with an initial activity of 6.7 goligomers gcatalyst−1 h−1. The production of heavier oligomers, due to acidic sites increasing the dimerization of lighter olefins, can lead to micropores blocking (3.5 nm for NiMCM-41) and the high temperatures (150 °C) can boost coke production. These two phenomena, due to a lack of control on the nickel environment, can lead to the deactivation of the catalyst.
More recently, in 2020, Andrei et al. reported the use of amorphous AlSiO2 and ordered AlSBA-15, mesoporous aluminum silicates.84 The Ni–AlSiO2 and Ni–AlSBA-15 were prepared the same way as described previously, by ammonium nitrate treatment and with nickel nitrate hexahydrate. The catalysts were tested for ethylene oligomerization. Despite initial conversion of 87% and 80%, respectively for Ni–AlSBA-15 and Ni–AlSiO2 with selectivities in butene of 55 wt% and 58 wt%, the authors proved by TGA analysis that heavier products (C8 and higher olefins) were responsible for catalyst deactivation, especially due to acid sites. Ni–AlSiO2 with a wider pore width than Ni–AlSBA-15 underwent a less significant deactivation.
The Ni exchange technique does not allow control over the active site's environment. The exact nature of the active sites is still unknown and the occurring mechanism is not fully understood.50 Acid sites represent also another type of active sites, responsible not only for isomerization reactions but also for dimerization of the products formed in the first place. Combined to the microporosity of some catalysts, the production of heavier products (C8 and more) can lead to pore blocking. Working at higher temperatures also favors catalytic cracking of the products.
Surface organometallic chemistry has been used for the synthesis of two mesoporous silica-grafted catalysts for ethylene oligomerization:
– a tantalum-based catalyst, [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)TaVCl2Me2], by grafting TaMe3Cl2 on SBA-1585
SiO)TaVCl2Me2], by grafting TaMe3Cl2 on SBA-1585
– a niobium-based catalyst, [(SiO)NbMe4], resulting from the grafting and methylation of NbMe2Cl3 on a SiO2 surface dehydroxylated at 700 °C.86
The niobium-based catalyst enabled to achieve 91.9 wt% of butene selectivity with an activity of 1050 molethylene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) consumed molNb−1 h−1 under 50 bars of ethylene at 100 °C during one hour in toluene. In similar conditions (30 minutes of reaction instead of one hour), the tantalum-based catalyst's activity reached 375 molethyleneconsumed molTa−1 h−1 with 82.7 wt% of hexene products. Both catalysts are active without aluminum-based cocatalyst activation. However, they suffered from deactivation by polyethylene production, stemming from the complexes geometry and coordination sphere on the surface.87 Hence, the interactions between the active sites and the free oxide or hydroxide sites might hamper the catalytic activity.6,20,88,89
consumed molNb−1 h−1 under 50 bars of ethylene at 100 °C during one hour in toluene. In similar conditions (30 minutes of reaction instead of one hour), the tantalum-based catalyst's activity reached 375 molethyleneconsumed molTa−1 h−1 with 82.7 wt% of hexene products. Both catalysts are active without aluminum-based cocatalyst activation. However, they suffered from deactivation by polyethylene production, stemming from the complexes geometry and coordination sphere on the surface.87 Hence, the interactions between the active sites and the free oxide or hydroxide sites might hamper the catalytic activity.6,20,88,89
3.2 Hybrid periodic mesoporous silica
Periodic Mesoporous Organosilica (PMOs) is a class of siliceous materials with organized mesoporosity and possibly containing organic moieties.20,90,91 They present high surface areas, high porous volume and structural diversity (MCM-48 with a 3D porous structure or MCM-50 with a 2D structure). Aluminum can be introduced into the structure to tune its acidity.Recently, Shin et al. synthesized a series of eight bpy-SBA-15, with different molar amounts of bipyridyl (bpy) sites metalated with NiCl2·H2O (Fig. 4).92 The design of these catalysts was motivated by the attempt to minimizing polyethylene formation and improving oligomer production, with heterogeneous (bpy)Ni(II)Cl2 catalysts.
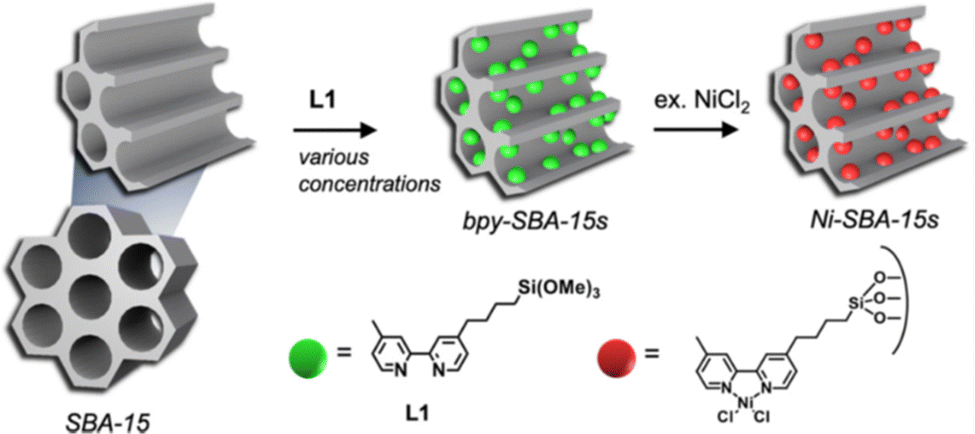 | ||
| Fig. 4 From a synthesized SBA-15 PMO, the design of a series of eight bpy-SBA-15 (bipyridyl sites embedded on the surface and within the walls of the structure) and their metalation with NiCl2·H2O. Reprinted from Appl. Catal., A, 590, Shin et al., A Way to Avoid Polymeric Side Products during the Liquid-Phase Ethylene Oligomerization with SBA-15 Supported (Bpy)Ni(II)Cl2 Heterogeneous Catalyst, p. 117363, Copyright (2020), with permission from Elsevier.92 | ||
Ethylene oligomerization was performed in heptane during 30 minutes with 70 or 700 equivalents of Et2AlCl compared to Ni and under 30 bar of ethylene, at 150 °C. First, the series of catalysts were more active than their homogeneous counterpart Ni(bpy)Cl2 for the given conditions, with 406 mololigomers molNi−1 h−1 and a selectivity in butene of 51%. The authors observed minimal polyethylene formation with the catalyst made with 0.21 wt% of Ni and a 4 nm pore size. It achieved 4422 mololigomers molNi−1 h−1 and a selectivity in butene of 77%, with 700 equivalents of Et2AlCl. The catalyst has been recycled seven times, without nickel leaching, suggesting the successful immobilization of the molecular complex within the large pores of the material. However, catalysts with higher pore sizes (7 and 10 nm) and higher nickel loadings (0.27 wt% and 0.31 wt% of Ni respectively), proved to be less active and less butene-selective in the same conditions. Same conclusions can be drawn for 2.25 wt% and 1.51 wt% nickel loadings.
Different inorganic and non-porous materials have been also synthesized and tested for catalytic purposes, like clays, aluminum sulfates and oxide surfaces, but they will not be developed here since they have not shown particular activity in ethylene oligomerization.81,93–95
4. Molecular Ni catalysts embedded into porous polymeric materials
In contrast to siliceous materials, molecular complexes heterogenized within porous polymers can be embedded within the porous network, with a high site density per volume and per mass of solid. Moreover, the high porosity of the solids allows for high accessibility of the active sites. Porous polymers of hybrid organic–inorganic nature like Metal–Organic Frameworks (MOF),96–104 or purely organic, like Porous Organic Polymers (POP) or Porous Organic Networks (PON) can be made by design using coordinating motif as organic repeating unit.105–113 MOF and POP both allow for a wide versatility in composition and porous network topology by varying organic linkers/monomers (for MOF and POP) and metal nodes (in MOF). The crystalline MOF present well-defined topologies and their extended ordered nature allows for the control over grafted sites environment within the MOF pores and in-depth mechanistic studies using computational chemistry (Fig. 5).114–117 In contrast, POP/PON are amorphous or semi-crystalline and present a more disordered network which however is made by C–C bonds, much more stable than the MOF coordination bonding towards hydrolysis, for example.118 Among porous organic polymers, Porous Aromatic Frameworks (PAF) are rigid frameworks where C–C bonds connect aromatic units,119 Covalent Organic Frameworks (COF) are slightly crystalline layered materials120,121 and covalent triazine frameworks (CTF) are a sub-class of COF having triazine units as node (Fig. 5).122–124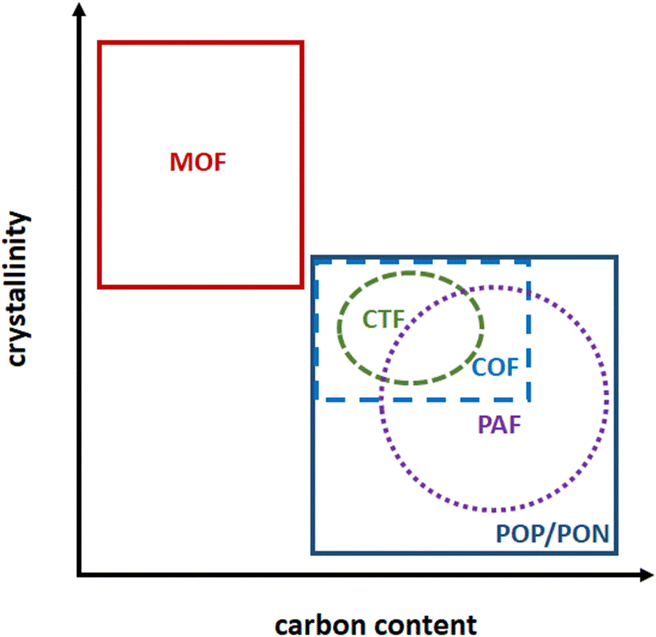 | ||
| Fig. 5 Representation of classification of non-siliceous polymeric porous materials. (MOF: metal–organic frameworks; POP: porous organic polymers; PAF: porous aromatic frameworks; PON: porous organic networks; COF: covalent organic frameworks; CTF: covalent triazine frameworks). | ||
Different methods have been employed to embed isolated active sites for ethylene oligomerization within MOF and POP porous frameworks (Fig. 6):
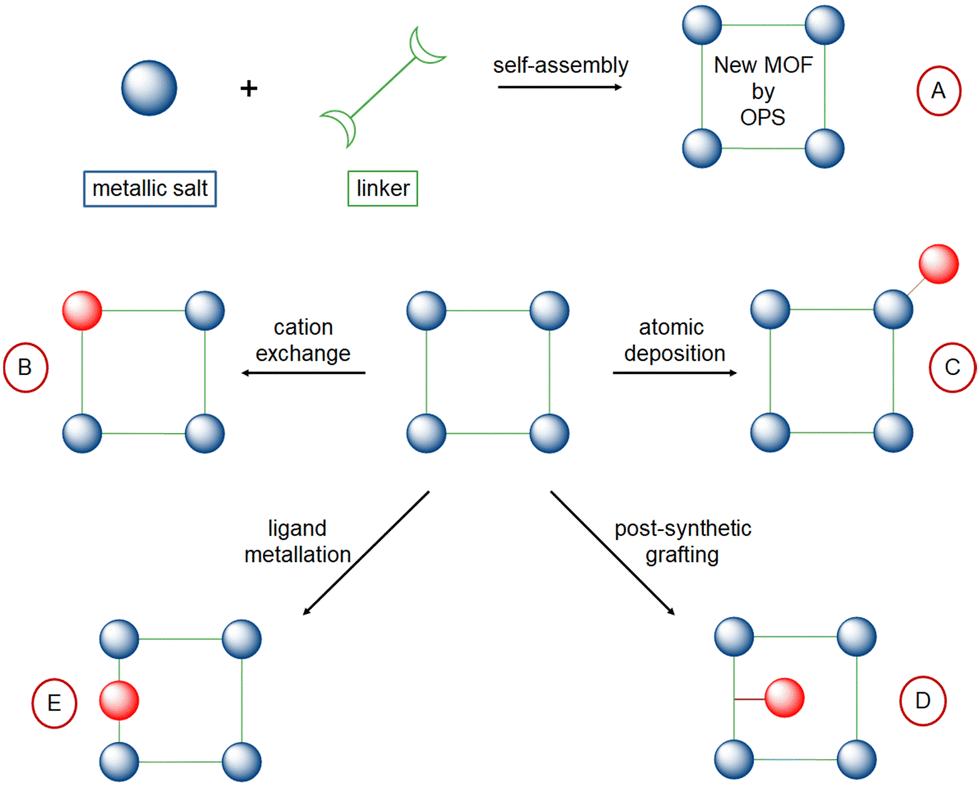 | ||
| Fig. 6 Different synthetic pathways to get isolated active sites within MOF. | ||
– by one-pot synthesis, with Ni active sites already incorporated within the structure of the MOF (route A);125
– by cation exchange in the MOF inorganic node (route B);126,127
– by atomic deposition onto the MOF inorganic node (route C);32,33
– by post-synthetic grafting of pending ligand inside the MOF (route D);128–130
– by coordination of metal cation into porous structures containing vacant coordination sites as building monomers, i.e. bipyridine131–134 or phenoxy-imine (route E).135
4.1 Self-assembled Ni-based MOF (route A)
In the following examples, the MOF are synthesized using nickel salt through one-pot synthesis (OPS). The nickel is thus present either coordinated to oxygens from carboxylates or to nitrogen from imidazolates, either as single atom or as a dimer within a paddle-wheel complex.consumed molNi−1 h−1 with a selectivity in butene of 75.6%. With the same catalyst, a TOF of 3000 molethyleneconsumed molNi−1 h−1 was already achieved after thirty minutes of reaction with a selectivity in butene of 76.6% whereas a lower TOF of 2500 molethyleneconsumed molNi−1 h−1 could be observed after one hour and a half, with an improved selectivity in butene of 90.2%. Atomic Force Microscopy (AFM) analysis enabled the authors to evaluate the thickness of the catalyst nanosheets after four recycling experiments. For one hour of reaction, Ni-UMOFN-190 achieved 4893 molethyleneconsumed molNi−1 h−1 with a 72.1% butene selectivity for a first cycle compared to 3929 molethyleneconsumed molNi−1 h−1 with a 71.4% butene selectivity for a fourth cycle. The stable catalyst activity was attributed to the fact that fresh nickel active sites are continuously exposed to aluminum-based cocatalyst and ethylene.
Later, in 2019, Kaskel and coworkers proposed seven nickel-based MOFs, synthesized by one-pot synthesis for ethylene oligomerization (Fig. 7):137 the CPO-27(Ni) or [Ni2(dhtp)]n from a mixture of a nickel(II) acetate tetrahydrate in 1-butanol with 2,5-dihydroxyterephtalic acid (dhtpH2), the [Ni(bdc)(dabco)]n and [Ni(bdc)(dabco)0.5]n (bdc = 1,4-benzenedicarboxylate and dabco = 1,4-diazabicyclo[2.2.2]octane, and the [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (ndc = 2,6-naphtalenedicarboxylate and DMF = N,N-dimethylformamide). The DUT-8(Ni) or [Ni(ndc)(dabco)0.5]n was obtained, from a mixture of nickel(II) nitrate hexahydrate in DMF with 2,6-naphtalenedicarboxylic acid and dabco, in two forms, [Ni(ndc)(dabco)0.5]n_rigid and [Ni(ndc)(dabco)0.5]n_flexible. The two forms differ by the fact that the flexible DUT-8(Ni) can switch between open and close pore form whereas rigid DUT-8(Ni) remains in the open pore. Each nickel-node represents an isolated active site for ethylene oligomerization. Prior to ethylene oligomerization experiments, the authors evaluated the integrity of the Ni-based MOFs after leaving them to stir overnight with Et2AlCl (Al/Ni = 17) in toluene. Powder X-ray diffraction (PXRD) measurements showed that only CPO-27(Ni), [Ni3(ndc)3(DMF)2((CH3)2NH)2]n, DUT-8(Ni)_rigid and DUT-8(Ni)_flexible remained structurally unchanged, compared to the other MOFs.
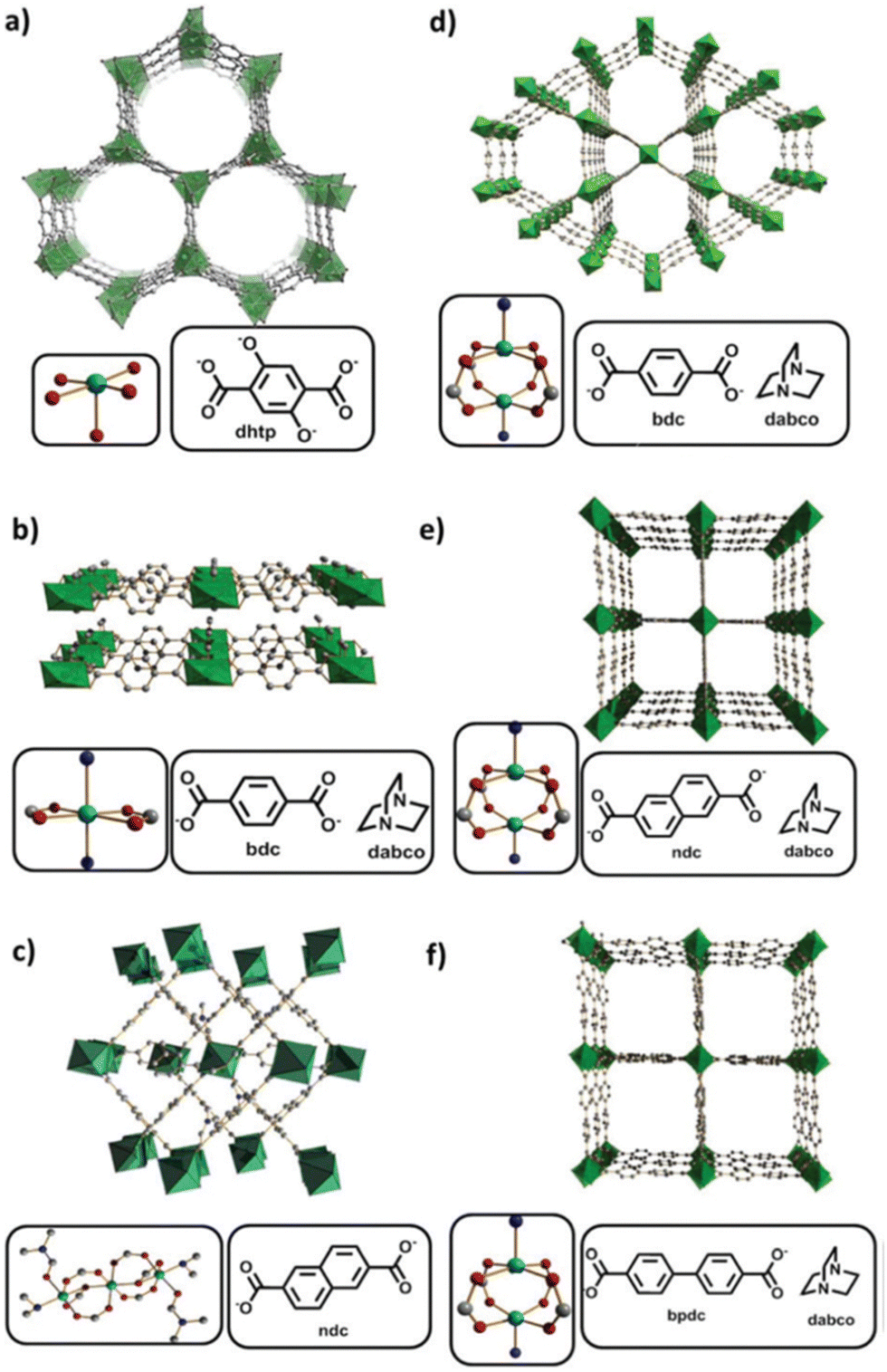 | ||
| Fig. 7 (a) CPO-27(Ni); (b) [Ni(bdc)(dabco)]n; (c) [Ni3(ndc)3(DMF)2((CH3)2NH)2]n; (d) [Ni(bdc)(dabco)0.5]n; (e) DUT-8(Ni); (f) DUT-128. Reproduced from ref. 138 with permission from the Royal Society of Chemistry. | ||
The authors tested these seven catalysts for ethylene oligomerization in toluene, activated with 17 equivalents of Et2AlCl at 21 °C and 10 bars of continuous ethylene feed. The catalytic activities ranged from 1 mololigomers molNi−1 h−1 (100% C4 selectivity) with the CPO-27(Ni) to 49 mololigomers molNi−1 h−1 with [Ni3(ndc)3(DMF)2((CH3)2NH)2]n (50% selectivity in C4) and DUT-128 (47% selectivity in C4). It is noteworthy that with these nickel-based MOFs no polymeric product could be observed.
More recently, another nickel carboxylate MOF, Ni-MIL-77,139 has been synthesized by Wang and coworkers, using glutaric acid as linker. This MOF can present distinct 1D or 3D topology as confirmed by PXRD, however without a clear evidence of porosity.140 Under 10–20 bars pressure range of ethylene and using between 80 to 210 equivalent of Al-based cocatalyst, the 1D Ni-MIL-77 was found to perform the best with a TOF of 5544 h−1 and a selectivity of 98% for butenes. Unfortunately, the solid catalysts showed a drastic loss of crystallinity after reaction and could not be efficiently reused. Indeed, the authors found that hydrolysis products from the Al-based cocatalyst cover the MOF's surface leading to catalyst deactivation.
900 equivalents), under 30 bars of ethylene and at 35 °C.
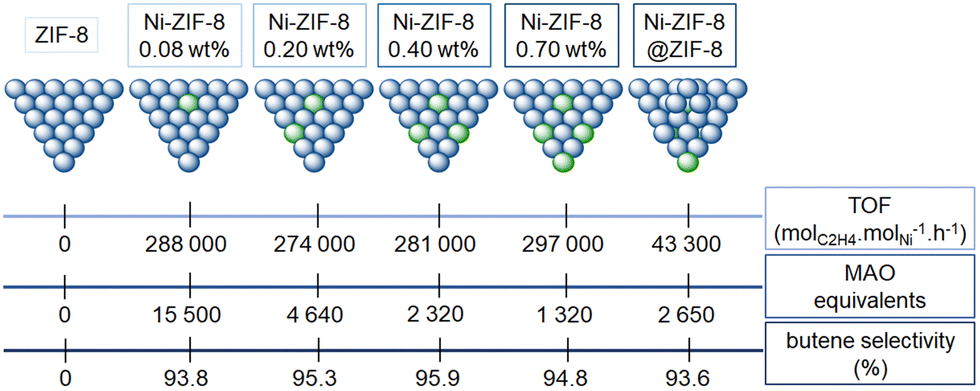 | ||
| Fig. 8 Representation of the ethylene oligomerization TOF (top), of the MAO equivalents used (in toluene, at 35 °C and 30 bar of ethylene; middle) and of the butene selectivity (bottom) with ZIF-8, four different catalysts obtained from ZIF-8 and exhibiting different nickel loadings (0.08, 0.20, 0.40 and 0.70 wt%) and Ni-ZIF-8@ZIF-8.125 | ||
First, the ZIF-8 itself (with only Zn sites) was not active whereas all the other Ni-ZIF-8 catalysts generate oligomers (whatever the loading or the catalytic conditions), indicating the necessity of nickel sites in order to observe a catalytic activity. Besides, for given conditions, the Ni-ZIF-8 was seven times more active than the Ni-ZIF-8@ZIF-8, indicating that only some of the nickel active sites on the surface have been deactivated by the ZIF-8 growth. No polyethylene was reported by any Ni-ZIF-8 catalyst, with butene selectivities above 90%. Among nickel-based catalyst discussed so far, Ni-ZIF-8 catalyst seems to be more active and selective towards butene than other self-assembled nickel-based MOF catalysts, for the given conditions in each work. The catalytic activity enhancement of the Ni-ZIF-8 catalyst could stem from the fact that the macroligand is made of imine-like moieties. The effects of the thousand-fold excess of alkyl aluminum seems poorly understood (from 1320 to 15500 equivalents to Ni, see Fig. 8).
As an alternative to ZIF-8, Dong and coworkers recently reported Ni-ZIF-L as highly active ethylene dimerization catalyst.141 ZIF-L present similar composition and chemical environment than ZIF-8 but with different topology, with quite lower surface area of ca. 160 m2 g−1, which allows the insertion of nickel atoms only at the surface of the MOF's particles. According to the authors, the 2D leaf-like shape of ZIF-L would allow for higher external accessible surface compared to ZIF-8. Using MAO as cocatalyst under 30 bars of ethylene and 30 °C in toluene, the NI-ZIF-L allowed accessing ethylene dimerization with a TOF of 330230 h−1 with a selectivity in 1-butene higher than 90%. Unfortunately, after one hour of reaction, the measured selectivity in 1-butene slightly dropped from 95% to 77% (overall C4 selectivity constant from 97% to 94% with same TOF), likely due to butene isomerization by Lewis acid sites either from the MOF or the cocatalyst, and the possible recycling of the catalyst was not demonstrated.
4.2 Single sites by Ni cation exchange in MOF (route B)
Following the above-described examples of Ni-based MOF, nickel sites can be introduced post-synthetically by replacing metal cations natively present at the MOF node, thus generating isolated active sites for ethylene oligomerization.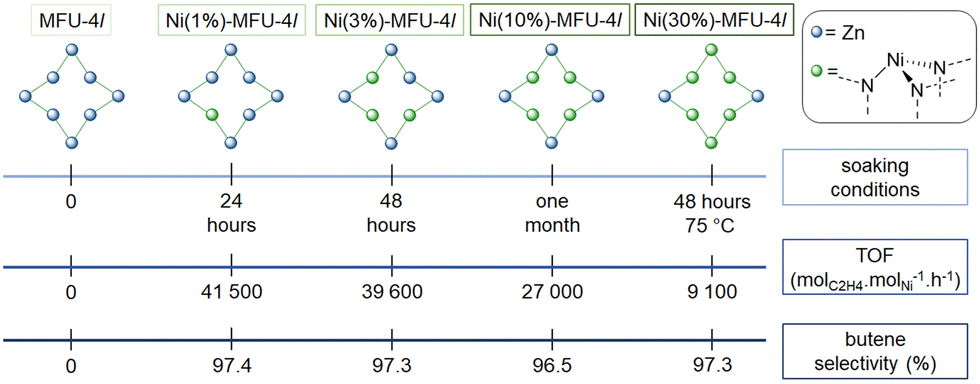 | ||
| Fig. 9 Representation of the effect of soaking conditions for MFU-4l in a Ni(NO3)2·6H2O DMF solution on the number of Zn2+ replaced by Ni2+ (top), of the nickel loading on the ethylene oligomerization TOF (led in toluene, at 25 °C and 50 bars of ethylene and activated with 500 equivalents of MAO) (middle) and on the butene selectivity (bottom). | ||
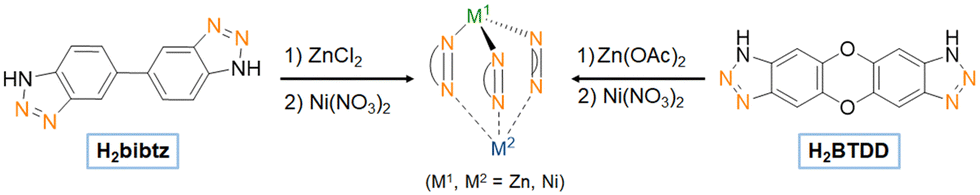 | ||
| Fig. 10 Representation of how the H2bibtz (1H,1′H-5,5′-bibenzo[d][1,2,3]triazole)) linker and the H2BTDD linker, used respectively to synthesize CFA-1 and MFU-4l enable to obtain the same scorpionate-like configuration with three coordinating nitrogen atoms embedded within the MOF structure. | ||
Ethylene oligomerization was performed in toluene with 50 to 2000 equivalents of MMAO-12, under 50 b of ethylene at 22 °C. Under similar conditions, Ni(1%)-CFA-1 achieved 1800 molethyleneconverted molNi−1 h−1 with a 88.6% butene selectivity whereas Ni(1%)-MFU-4l enabled to achieve 41500 molethyleneconverted molNi−1 h−1, with a 97.4% butene selectivity when activated with 500 equivalents of MAO.
4.3 Single sites by atomic Ni deposition on MOF (route C)
So far, this methodology relies on the use of zirconium oxo clusters of Zr-based MOF as a model inorganic surface to graft nickel precursors. | ||
| Fig. 11 Ethylene oligomerization catalytic activity obtained with Ni-AIM-NU-1000, Ni-Facac-AIM-NU-1000 and Ni-Acac-AIM-NU-1000 in a packed reactor under continuous flow of 100% ethylene gas feed at 2 bar and 45 °C, activated with Et2AlCl. | ||
4.4 Post-synthetic grafting of molecular Ni complexes within MOFs (route D)
In this section, defined nickel molecular complexes are introduced stepwise within the MOF cavity by immobilizing vacant coordinating ligands which are then metallated with a nickel salt.formed molNi−1 h−1, with a selectivity in 1-butene around 94% in both cases), proving that the catalytic sites are accessible (Fig. 12). It can also be assumed that, up to a certain nickel loading, the reaction is not limited by diffusion. It is noteworthy that no catalytic activity could be observed with only (Fe)-MIL-101-NH2 or with (Fe)-MIL-101-NH2 put into solution with either NiCl2 or PyCHO and with the filtrated catalytic solution (leaching experiment) re-activated with Et2AlCl. Catalytic activities of 10Ni@(Fe)-MIL-101 and 30Ni@(Fe)-MIL-101 originate from the nickel-isolated active sites within the framework. In this study, the most competitive result was obtained under mild conditions (25 °C and 30 bar of ethylene, with 70 equivalents of Et2AlCl) to reach 10455 mololigomers molNi−1 h−1 with a 1-butene selectivity above 90%.
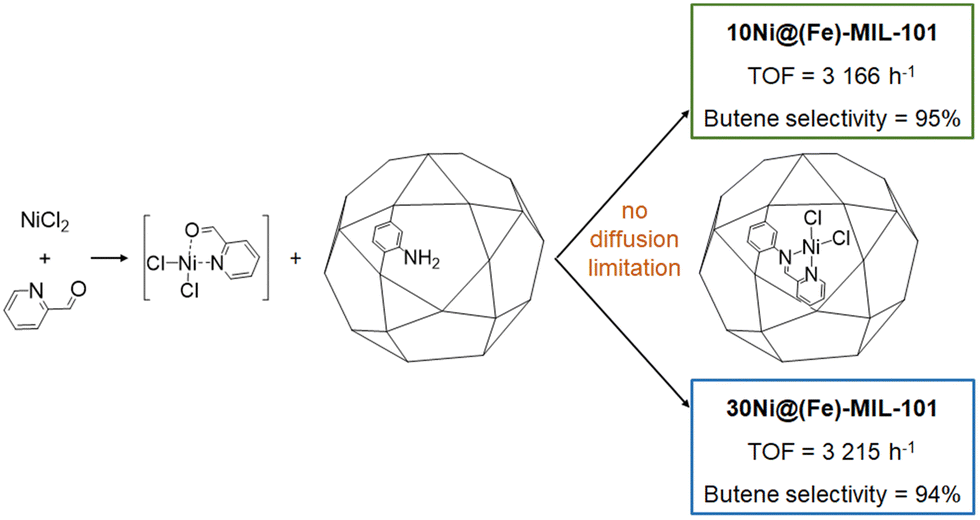 | ||
| Fig. 12 10Ni@(Fe)-MIL-101 and 30Ni@(Fe)-MIL-101 catalytic activity under 15 bars of ethylene at 10 °C with 70 equivalents of Et2AlCl per nickel. | ||
| MOF catalyst | NH2 contenta (10−4mol g−1) | surface area before PSMb (m2 g−1) | surface area after PSMb (m2 g−1) | Ni contentc (10−3mol g−1) | catalytic activity (104 goligomers molNi−1 h−1) | Butene selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a As determined by elemental analysis. b As determined from nitrogen physisorption isotherm using BET method. c As determined by ICP analysis. | |||||||
| MixMOFs-Ni-a | 5.95 | 1080 | 262 | 0.65 | 5.8 | 71.3 | 129 |
| MixMOFs-Ni-b | 8.12 | 1213 | 176 | 1.07 | 6.9 | 79.5 | 129 |
| MixMOFs-Ni-c | 10.95 | 1671 | 125 | 1.22 | 5.6 | 79.5 | 129 |
| IRMOF-3-Ni-a | 36.81 | 1086 | 8 | 0.17 | 6.3 | 35 | 129 |
| 30Ni@(Fe)MIL-101 | 42.86 | 1884 | 155 | 1.12 | 58.5 | 94 | 128 |
| Ni@(Ti)MIL-125-NH2 | 36.29 | 1164 | 1083 | 0.23 | 18.0 | 27 | 130 |
4.5 Nickelation of MOFs and POPs macroligands (route E)
In contrast to the previously described methods, here the MOFs and POPs are not used as support to graft molecular complex at the pore surface but are envisioned as porous macroligands, i.e. solids acting as the organic ligand in the molecular complex. The MOFs and POPs described below are made using coordinating linkers/monomers which are derivatized from known efficient organic ligands in molecular complexes.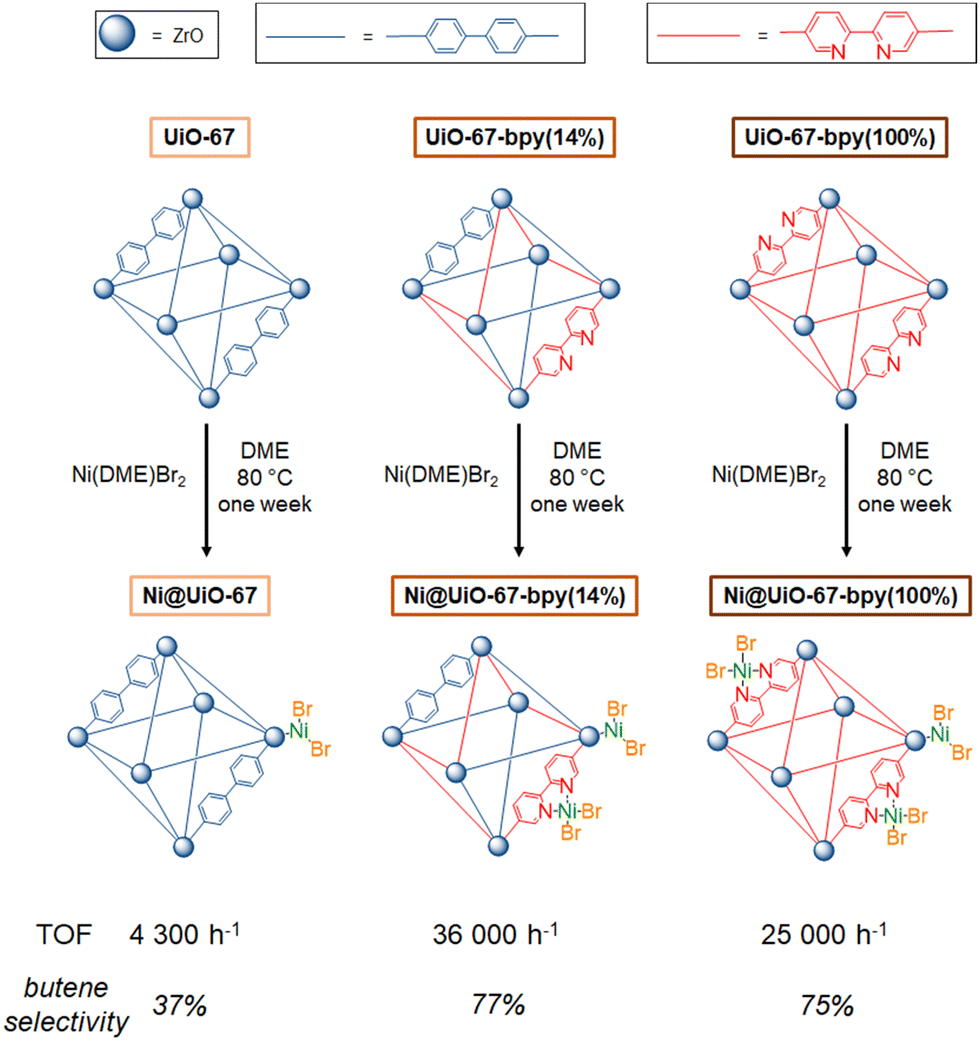 | ||
| Fig. 13 Metalation procedure of UiO-67, UiO-67(bpy14%) and UiO-67(bpy100%) with Ni(DME)Br2 and TOFs and selectivity under the following conditions: 59 bars of ethylene at 55 °C during one hour and with 100 equivalents of Et2AlCl per nickel. | ||
These results showed that, in this case, higher metal loadings might lead to detrimental steric hindrance around the active sites consequently leading to polyethylene production. Furthermore, the authors cannot rule out a catalytic contribution from wild nickel sites at the MOF node with nevertheless a negligible impact on the overall productivity and selectivity.
In 2020, Kaskel and coworkers synthesized two imine-based MOFs, PCN-161 and PCN-164, and one phenoxy-imine novel MOF, DUT133.135 For PCN-161 and PCN-164, the imine condensation between 4-formylbenzoic acid and 4-aminobenzoic acid and p-phenylenediamine, respectively, in presence of ZrCl4, enables to achieve imine-based MOFs. The authors also proposed the DUT-133 framework, isoreticular to PCN-161, where the imine condensation of 4-formyl-3-hydroxybenzoic acid with 4-aminobenzoic acid, in presence of ZrCl4, enables to achieve a phenoxy-imine based MOF. The nitrogen physisorption measurements highlighted the microporosity of the material, making it suitable for the immobilization of a molecular complex within its structure. After metallation with NiCl2, ethylene oligomerization has been carried out in an autoclave with 1,2-dichlorobenzene and 51 equivalents of Et2AlCl at 21 °C and under 15 bars of ethylene. The authors also synthesized a NiCl2@UiO-67(bpydc) catalyst, similar to the one developed by Long and coworkers.131 The authors compared the 63%NiCl@DUT-133 (63% Ni loading verified by ICP-OES) to NiCl2@UiO-67(bpydc) (with a 5.3% Ni loading verified by ICP-OES) under the same catalytic conditions described before. NiCl2@UiO-67(bpydc) proved to be two times more active and less butene-selective (25 mololigomers molNi−1 h−1 with 28% of butene) than 63%NiCl@DUT-133 (12 mololigomers molNi−1 h−1 with 100% butene). It could be concluded that, within Zr-MOF, a Ni active site coordinated to a phenoxy-imine would be less active and more selective towards butenes than a Ni complex coordinated to a bipyridyl site. The phenoxy-imine seems to favor β-H elimination over propagation step whereas the bipyridyl one favors propagation step, for the given catalytic conditions.
converted molNi−1 h−1, with respective selectivities in butene of 58%, 59% and 54%. Lower TOFs and selectivities are obtained with Ni-based COFs, compared to bpy-based MOFs and POPs.
More recently, Li et al. reported two Schiff-base CTF made by condensation of melamine with either 2,3-butanedione, MABD-COF, or p-phthalaldehyde, MAPA-COF.147 The subsequent metalation with nickel dichloride allowed obtaining two catalysts Ni@MAPA-COF and Ni@MABD-COF with Ni loading of 4.53 wt% and 7.58 wt%, respectively. The coordination of nickel to N atoms from imine-CTF was assessed by FT-IR analysis. The Ni-loaded CTF were evaluated for ethylene oligomerization using MAO as cocatalyst (Al/Ni = 500 to 700) and the best activity were found in cyclohexane as solvent under at 25 °C with a productivity of 7.62 × 104 g molNi−1 h−1 and selectivity of 76% for mainly 1-butene using Ni@MABD-COF and a productivity of 15.68 × 104 g molNi−1 h−1 and a selectivity of 57% mainly 1-butene using Ni@MAPA-COF. The higher activity of Ni@MAPA-COF was attributed to its larger pore size and accessible surface area compared to Ni@MABD-COF, 192.6 and 6.8 m2 g−1 respectively, as determined by nitrogen physisorption. The two catalysts were reused three times with a decrease in both the Ni content inside the CTF, indicative for a leaching of active species in the liquid phase, and in the activity, the selectivity remaining the same. In addition, the crystallinity of both catalysts was also altered during the reaction, with a drastic loss of crystallinity for Ni@MABD-COF.
In summary, the literature shows several attempts to obtain very high ethylene oligomerization activity and selectivity towards specific oligomers, by using different methods to obtain isolated active sites. Isolating active sites with materials already containing coordination sites, like bipyridine, present high activity and selectivity towards butenes. However, the side-production of polyethylene remains an issue yet unsolved.
5. Assessing the stability of heterogenized molecular Ni catalysts for reuse
Aiming at high production rate and increased sustainability, the catalyst stability, including both solid integrity and metal leaching, and its subsequent recyclability remains a key issue which can be systematically addressed by assessing the textural, spectroscopic and elemental analyses of the used solid.128 However, beyond the catalyst integrity, the active site can itself evolve during catalytic runs, leading to a change in reactivity.In 2015, Farha and coworkers observed that the reuse of NU-1000-bpy-NiCl2 catalyst for a second cycle led to the enhancement of the intrinsic activity for butenes from 1950 to 6040 molbutenes molNi−1 h−1 with a constant selectivity in butene around 93%.146 Powder X-Ray diffraction together with the analysis of products and microscopy allowed to evidence that a polymer layer was created around the MOF crystals during the first cycle. The authors assumed that this polymer layer was increasing the local solubility of ethylene during the second cycle at the origin of the enhancement in intrinsic activity for butenes production. When the catalyst was washed carefully with anhydrous ethanol before using it for two more cycles, the observed activity remained the same from 3166 to 2972 mololigomers molNi−1 h−1.
In 2017, with the POP-based catalyst Ni(II)-POP-1, Kim et al. also confirmed that the “white insoluble polymer around the POP particles” was polyethylene.132 Authors also recovered the catalyst for two more cycles and, each time, washed it with dichloromethane before activating it under super critical CO2. Despite the fact that the catalyst remained active for ethylene dimerization, polyethylene continued to cover Ni(II)-POP-1 particles during each cycle, hampering the accessibility of the active sites and hence, decreasing the activity.
Finally, in 2019, Long and coworkers observed that the ethylene consumption of the molecular catalyst [NiCl2(bpy)] (bpy = 2,2′-bipyridine) and of the MOF [Ni3(ndc)3(DMF)2((CH3)2NH)]n (ndc = 2,6-naphtalenedicarboxylate; DMF = N,N-dimethylformamide), under 10 bars of ethylene at 21 °C, in toluene, with 17 eq. of Et2AlCl, during one hour, gradually increased in the first minute of each cycle of a five-cycle experiment.138 This phenomenon was partly attributed to the increasing solubility of ethylene during the recycling experiments due to the presence of oligomers from the previous cycle. In addition, in 2019, Hu et al. recycled the Ni-UMOFN catalyst for four consecutive runs, using 10 bars of ethylene at 25 °C, in toluene, with 500 eq. of Et2AlCl, during one hour, without any significant loss of activity and butene selectivity, by washing the recovered catalyst with dry ethanol before each new cycle.136
Thus, in most of the cases reported above, the solid catalyst encountered textural modifications. Most of these modifications occurred during catalysis with polymer formation leading to pore blocking. This detrimental polymerization might be avoided learning lessons from molecular catalysis with better suited combination of active metal and co-catalyst (i.e. aluminium salt).67,148–150 Also large porous system should allow fast diffusion of both C2 reactant and C4 products to limit further reaction towards higher oligomers and polymers. Furthermore, the high confinement of Ni sites in small pores, with strong steric constraints, would reduce the probability for β-H elimination and favour further olefin insertion and, consequently, detrimental pore blocking.67,76 In addition, layered solids like COF seem less suitable due to their 3D structure collapse through exfoliation under catalytic conditions, concomitant with active site leaching in solution.151 Indeed, the spacing between sheets within the COF structure might be affected by the growth of interlayered oligomers at the COF-supported nickel sites, leading to a loss of the COF structure.
Finally, at the molecular level, the coordination of nickel single-sites to the porous macroligand might be further tuned. The most reported here are Ni–N species due to the wide availability of N-functionalized building blocks for macroligands, like imidazoles or bipyridines.17 However, new macroligands based on phosphine and N-heterocyclic carbenes (NHC) should be investigated considering that, in Ni–P, phosphine coordination strength and basicity greatly impacts the selectivity by favouring β-H termination for short oligomers77 and, in Ni–NHC, stabilization provided by the carbene ligand prevents the catalyst decomposition.148
6. Comparing heterogenized molecular catalysts with homogeneous analogues
The embedding of nickel sites into MOFs and POPs macroligands described in the Fig. 6 proved to be an efficient strategy to design heterogeneous catalysts for ethylene oligomerization inspired by molecular counterparts.However, in self-assembled and cation exchanged Ni-MOF catalysts (routes A and B in the Fig. 6), the Ni–O2 (from carboxylate) or Ni-N2 (from imidazolate) species are constitutive of the MOF nodes and relevant comparisons can hardly be made with mono- or dinuclear molecular analogues (Fig. 14, blue). Indeed, a single molecular nickel bis-imidazolate complex, envisioned as analogue for Ni-ZIF MOF, can be hardly compared to an extended MOF structure because the electronic effect of both (i) the two other imidazolates on the nickel and (ii) the second nickel atom coordinated to the imidazolate is impossible to mimic using organic/organometallic functionalization. Considering the atomic deposition (route C in the Fig. 6) using the MOF node as model inorganic surface, the electronic effect of the oxide node on the complex and the effect of the MOF pore microenvironment can also be hardly distinguished. In contrast, supported molecular nickel complexes grafted onto or embedded within the porous network (routes D and E in the Fig. 6) might be more easily compared to homogenous molecular analogues. However the reported heterogenized molecular catalysts often lack comparisons with homogeneous counterparts under the same reaction conditions.152,153
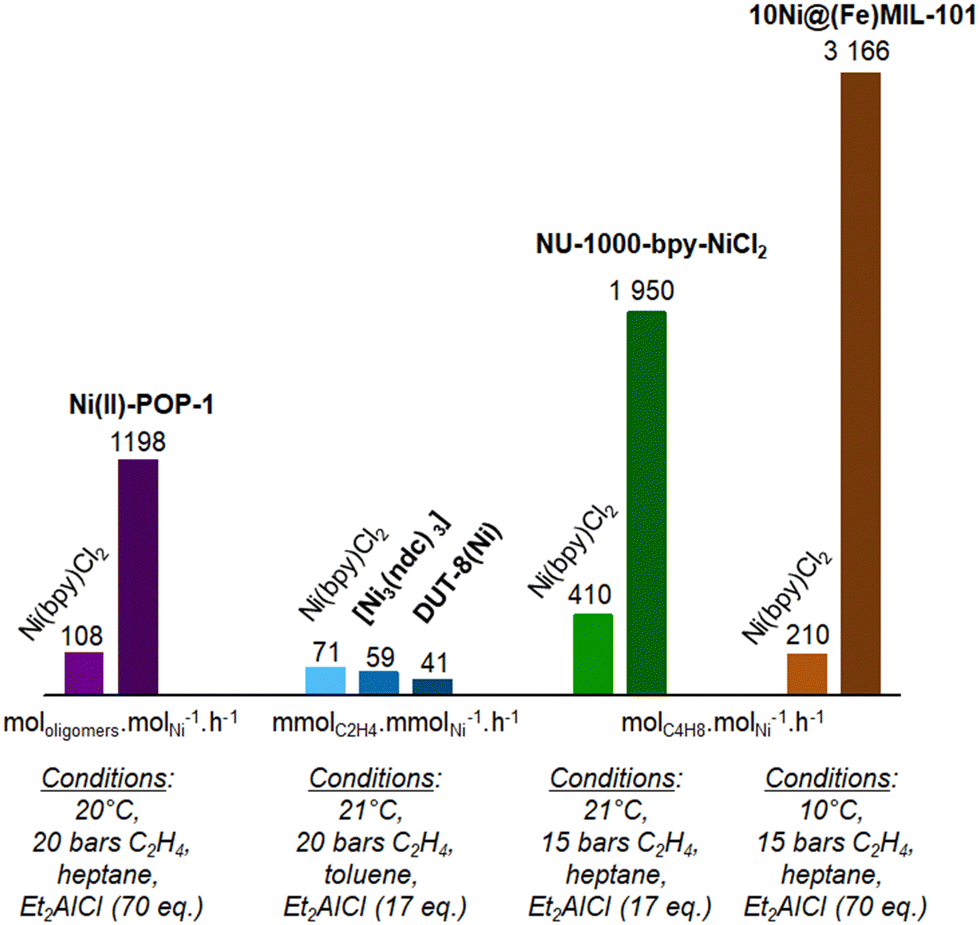 | ||
| Fig. 14 Comparison of the activity reported for MOF and POP heterogeneous catalysts with homogeneous counterpart for liquid phase ethylene oligomerization under similar conditions: metallatated POP-1 (violet),132 self-assembled Ni-MOF (blue), and post-synthetically grafted NU-1000 (green)146 and MIL-101 (brown).128 Data are given for similar amount of Ni used under mentioned conditions. | ||
In the case of ethylene oligomerization, some of the heterogeneous catalysts developed referred to homogeneous catalysis data previously reported, but often under different conditions.125,127,131,154 Some other studies compared the catalytic activity of the newly designed heterogeneous catalyst with homogeneous counterparts and under the same conditions (Fig. 14).128,132,138,146
The Ni@(Fe)-MIL-101128 and the Ni(II)-POP-1,132 respectively a MOF and a POP, proved to be ten times more active than the Ni(bpy)Cl2 molecular complex, under the given conditions.
In the case of Ni@(Fe)-MIL-101, a kinetic model showed that the reaction mechanism of the complex confined within the MOF was similar to that of the molecular complex and included the same elementary steps.128 According to this kinetic model, there was a small contribution of the solubility of ethylene to the variation of activity as a function of pressure and temperature. The model was based on a reaction mechanism involving the chemisorption of two ethylene molecules on an active nickel site followed by an oligomerization step to 1-butene.
If the choice of organic homogeneous analogue to the MOF and POP solid macroligand is driven by its synthetic or commercial availability, like for bipyridine, a further parameter to be considered is the electronic effect of the organic ligand on the nickel site within the porous solid. Wisser et al. demonstrated that the Hammett parameter is an adequate descriptor of electronic effects on the heterogenized molecular active site in the case of rhodium catalysts for reduction reactions in both homogeneous and heterogenized systems.19,155 The authors showed a linear correlation between the intrinsic catalytic activity and the Hammett parameter calculated for molecular ligands as well as for MOF- and POP-based macroligands, highlighting that the molecular nature of the complexes remained after heterogenization and also the absence of diffusion limitations in the selected solid platforms. For example, organic ester does not account for the same σ value of the Hammett parameter than a zirconium or aluminum carboxylate usually found in bpy-based MOFs. Thus, beyond textural effects due to the confinement into a porous system, the electronic effect of reticulation nodes within porous supports, i.e. inorganic units in MOFs or organic bonding in POPs, should not be neglected when comparing MOF and POP catalysts with homogeneous analogues. Such comparison has not been considered in the reported examples.
Diffusion limitations into porous system remain the main aspect addressed – if discussed at all – while comparing homogenous and heterogenized catalysts. In reality, liquid phase/triphasic catalytic systems, such as the case of ethylene oligomerization, implies technical biases that are often not considered such as the local concentration of substrate molecules, especially in the case of different gases solubility between the porous system and the bulk liquid and which will have huge impact on reaction kinetics.
Finally, the most important methodological bias remains the heterogenization of low performing homogeneous catalysts, which is indeed the case of Ni(bpy) species for the ethylene oligomerization, widely studied and discussed here. If a homogeneous catalyst gives outstanding productivity and selectivity, the gain reached by its heterogenization within somehow expensive porous solid becomes much less relevant, even considering its potential recyclability. The immobilization of performing catalysts within a solid porous system thus seems only appealing and valuable if it allows accessing a unique reactivity, impossible to reach in homogeneous phase.
7. Conclusion and perspectives
With the explosion of new families of hybrid porous materials embedding organic ligands for molecular complexes, the heterogenization of molecular catalysts attracts a continuously growing attention. In principle, heterogenized molecular catalyst should benefit for both molecular-level control and advantageous solid-substrate interface interactions. Among catalytic applications, the ethylene oligomerization is a widely and already comprehensively studied reaction in both homogeneous and heterogeneous catalysis and this makes this reaction an example of choice to critically discuss the interest in molecular catalyst heterogenization, as a part of the ever-growing single-site catalyst family.In the cases summarized here for nickel complexes heterogenization, metal–organic frameworks and porous organic polymers appear to be highly appealing supports because of their high porosity and site density, their infinite variety of design and possible post-synthetic modifications.
For ethylene oligomerization, many MOFs have already been developed, most of them employing Ni as catalytic active site for its high activity and selectivity towards 1-butene. So far, only one bipyridyl-based POP, metallated with a Ni molecular complex, has been developed and tested for ethylene oligomerization. However, a recurrent pitfall is the uncontrolled production of polyethylene, leading to catalyst deactivation.
Beyond obvious technical advantages lying on the easy catalyst separation from the product and its subsequent recycling, the advantages of developing such heterogenized molecular catalysts using sophisticated and costly supports has to be demonstrated. Notwithstanding the wide variety of catalytic conditions reported, this comparison can hardly be done since the organic ligand in the homogeneous complex seldom reproduces the exact same electronic environment as the one in the solid-embedded analogue. Furthermore, the lack of evidence demonstrating the true heterogeneity of the catalysts, particularly from hot filtration and recycling tests, impedes a comprehensive understanding of their nature.
Single-site catalysts, which bridge the gap between homogeneous and heterogeneous catalysis, offer an ideal platform to elucidate the structure–activity relationship using molecular chemistry mechanisms and through the integration of computational chemistry, transient spectroscopy, and advanced diffraction techniques. To facilitate accurate comparison and full comprehension, the geometry of the active site must be unravelled by employing a combination of extensive analytical techniques such as in situ X-ray absorption spectroscopy techniques supported by computational chemistry.156–159 Meanwhile, computational chemistry was demonstrated to be mature enough to give access to molecular mechanisms and even predict reactivity for homogeneous catalysis but is still at its infancy for heterogeneous counterpart.160 The use of single-site catalyst strategy would allow benefiting from knowledge from homogeneous catalysis while considering the solid support as traditional ligand. However computational methodologies have to be carefully designed on purpose considering the nature of the support, including rigid organic–inorganic hybrids or highly flexible organic networks.
Finally, an appealing objective of the transfer from homogeneous to heterogeneous phase catalysis is the implementation of heterogenized catalysts in fixed-bed reactor, for either liquid- or gas-phase reactions. However, this often requires prior shaping of the solid catalyst with non-trivial material- and application-dependent strategies in order to avoid critical pressure drop,161–164 the gas phase testing in fixed-bed reactor being consequently scarcely reported for MOF and POP-based catalysts.165–167 Even with adequate shaping methodologies linked to gas-phases applications, the fast discovery of high-performing MOF and POP-based catalysts by high-throughput testing remains hindered for sophisticated hybrid solids. One main obstacle in this context is their lab-scale synthesis which offers limited options for upscaling considering their synthetic conditions and associated costs which include raw materials, monomers and metal-based active sites, as well as solvents, often toxic, waste produced and number of synthetic steps.
Thus, the knowledge required to understand and design new generations of heterogenized catalysts could come from the study at the molecular level of the catalyst configuration and interactions at the solid's interface. Moreover, macroscopic behavior plays a crucial role in heterogenous catalysis, making essential to study mechanisms of adsorption, diffusion and kinetics using operando techniques.
This is only by combining the efforts from molecular chemistry, materials science and heterogeneous catalysis that heterogenized molecular catalysis will find its place in today's catalytic processes in order to discover new synthetic pathways while increasing their sustainability.
List of abbreviations
| Acac | Acetylacetonate |
| AFM | Atomic force microscopy |
| AIM | Atomic layer deposition in MOFs |
| ALD | Atomic layer deposition |
| Bdc | 1,4-Benzenedicarboxylate |
| Bipy | Bipyridyl |
| Bpdc | 4,4′-Biphenyldicarboxylate |
| Bpydc | 2,2′-Bipyridine-5,5′-dicarboxylate |
| COF | Covalent–organic framework |
| CPO | Coordination polymer of Oslo |
| CTF | Covalent triazine framework |
| CVD | Chemical vapor deposition |
| Dabco | 1,4-Diazabicyclo[2.2.2]octane |
| DFT | Density functional theory |
| DUT | Dresden University of Technology |
| Dme | Dimethoxyethane |
| DMF | N,N-Dimethylformamide |
| Facac− | Hexafluoroacetylacetonate |
| H2bibtz | 1H,1′H-5,5′-Bibenzo[d][1,2,3]triazole |
| H2BTDD | Bis(1H-1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin |
| ICP-OES | Inductively Coupled Plasma-Optical Emission Spectrometry |
| IL | Ionic liquids |
| IL-PON | Imine-linked-porous organic network |
| IRMOF | IsoReticular MOF |
| LAO | Linear alpha olefins |
| MAO | Methylaluminoxanes |
| MCM | Mobil composition material |
| MFU | Metal–Organic Framework Ulm-University |
| MIL | Matériaux of Institut Lavoisier |
| MMAO | Modified methylaluminoxane |
| MOF | Metal–organic framework |
| Ndc | 2,6-Naphtalenedicarboxylate |
| NHC | N-heterocyclic carbene |
| NMR | Nuclear magnetic resonance |
| NU | Northwestern University |
| OPS | One-pot synthesis |
| PAF | Porous aromatic framework |
| PCN | Periodic coordination network |
| PMO | Periodic mesoporous organosilica |
| POP | Porous organic polymer |
| PXRD | Powder X-ray diffraction |
| SAC | Single-atom catalyst |
| SIM | Solvothermal deposition in MOFs |
| SOMC | Surface organometallic chemistry |
| TGA | Thermogravimetric analysis |
| THF | Tetrahydrofuran |
| TOF | Turnover frequency |
| UiO | University of Oslo |
| UMOFN | Ultrathin metal–organic framework nanosheets |
| Wt% | Weight percent |
| ZIF | Zeolite imidazole frameworks |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Agence Nationale de la Recherche, through the grants no. ANR-18-CE07-0025 (Pomac project) and no. ANR-21-CE07-0028 (Flips project), and from the European Union's Horizon 2020 research and innovation program under grant agreement no. 814557 (C123 project). R. R., E. A. Q. and J. C. are thankful to Université Claude Bernard Lyon 1 for R. R. doctoral fellowship.References
- B. Pugin, J. Mol. Catal. A: Chem., 1996, 107, 273–279 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- J. A. Moulijn, A. E. van Diepen and F. Kapteijn, Appl. Catal., A, 2001, 212, 3–16 CrossRef CAS.
- A. Z. Fadhel, P. Pollet, C. L. Liotta and C. A. Eckert, Molecules, 2010, 15, 8400–8424 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2015, 115, 127–150 CrossRef CAS PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef PubMed.
- M. D. Korzyński and C. Copéret, Trends Chem., 2021, 3, 850–862 CrossRef.
- S. R. Docherty, L. Rochlitz, P.-A. Payard and C. Copéret, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC.
- C. Copéret, Z. J. Berkson, K. W. Chan, J. de, J. Silva, C. P. Gordon, M. Pucino and P. A. Zhizhko, Chem. Sci., 2021, 12, 3092–3115 RSC.
- M. K. Samantaray, E. Pump, A. Bendjeriou-Sedjerari, V. D’Elia, J. D. A. Pelletier, M. Guidotti, R. Psaro and J.-M. Basset, Chem. Soc. Rev., 2018, 47, 8403–8437 RSC.
- J.-M. Basset, F. Lefebvre and C. Santini, Coord. Chem. Rev., 1998, 178–180, 1703–1723 CrossRef CAS.
- C. Copéret and J.-M. Basset, Adv. Synth. Catal., 2007, 349, 78–92 CrossRef.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- J. M. Basset and A. Choplin, J. Mol. Catal., 1983, 21, 95–108 CrossRef CAS.
- P. Avenier, M. Taoufik, A. Lesage, X. Solans-Monfort, A. Baudouin, A. de Mallmann, L. Veyre, J.-M. Basset, O. Eisenstein, L. Emsley and E. A. Quadrelli, Science, 2007, 317, 1056–1060 CrossRef CAS PubMed.
- F. M. Wisser, Y. Mohr, E. A. Quadrelli and J. Canivet, ChemCatChem, 2020, 12, 1270–1275 CrossRef CAS.
- F. M. Wisser, P. Berruyer, L. Cardenas, Y. Mohr, E. A. Quadrelli, A. Lesage, D. Farrusseng and J. Canivet, ACS Catal., 2018, 8, 1653–1661 CrossRef CAS.
- F. M. Wisser, Y. Mohr, E. A. Quadrelli, D. Farrusseng and J. Canivet, ChemCatChem, 2018, 10, 1778–1782 CrossRef CAS.
- F. M. Wisser, Y. Mohr, E. A. Quadrelli and J. Canivet, ChemCatChem, 2020, 12, 1270–1275 CrossRef CAS.
- S. Daliran, A. R. Oveisi, Y. Peng, A. López-Magano, M. Khajeh, R. Mas-Ballesté, J. Alemán, R. Luque and H. Garcia, Chem. Soc. Rev., 2022, 51, 7810–7882 RSC.
- T. Drake, P. Ji and W. Lin, Acc. Chem. Res., 2018, 51, 2129–2138 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, ChemCatChem, 2020, 12, 4732–4753 CrossRef CAS.
- S. Abednatanzi, M. Najafi, P. Gohari Derakhshandeh and P. Van Der Voort, Coord. Chem. Rev., 2022, 451, 214259 CrossRef CAS.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. L. i Xamena, V. V. Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- A. Alimardanov, L. Schmieder-van de Vondervoort, A. H. M. de Vries and J. G. de Vries, Adv. Synth. Catal., 2004, 346, 1812–1817 CrossRef CAS.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- L. Kuai, Z. Chen, S. Liu, E. Kan, N. Yu, Y. Ren, C. Fang, X. Li, Y. Li and B. Geng, Nat. Commun., 2020, 11, 48 CrossRef PubMed.
- A. S. Galushko, D. A. Boiko, E. O. Pentsak, D. B. Eremin and V. P. Ananikov, J. Am. Chem. Soc., 2023, 145, 9092–9103 CrossRef CAS PubMed.
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. “Bean” Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS PubMed.
- V. Bernales, A. B. League, Z. Li, N. M. Schweitzer, A. W. Peters, R. K. Carlson, J. T. Hupp, C. J. Cramer, O. K. Farha and L. Gagliardi, J. Phys. Chem. C, 2016, 120, 23576–23583 CrossRef CAS.
- J. Canivet, E. Bernoud, J. Bonnefoy, A. Legrand, T. K. Todorova, E. A. Quadrelli and C. Mellot-Draznieks, Chem. Sci., 2020, 11, 8800–8808 RSC.
- H.-Y. Zhuo, X. Zhang, J.-X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- A. Nandy, H. Adamji, D. W. Kastner, V. Vennelakanti, A. Nazemi, M. Liu and H. J. Kulik, ACS Catal., 2022, 12, 9281–9306 CrossRef CAS.
- M. K. Samantaray, V. D’Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS PubMed.
- A. K. Datye and H. Guo, Nat. Commun., 2021, 12, 895 CrossRef CAS PubMed.
- S. Mitchell and J. Pérez-Ramírez, Nat. Commun., 2020, 11, 4302 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- I. B. Moroz, A. Lund, M. Kaushik, L. Severy, D. Gajan, A. Fedorov, A. Lesage and C. Copéret, ACS Catal., 2019, 9, 7476–7485 CrossRef CAS.
- Z. Chen, S. R. Docherty, P. Florian, A. Kierzkowska, I. B. Moroz, P. M. Abdala, C. Copéret, C. R. Müller and A. Fedorov, Catal. Sci. Technol., 2022, 12, 5861–5868 RSC.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- G. P. Belov and P. E. Matkovsky, Pet. Chem., 2010, 50, 283–289 CrossRef.
- O. L. Sydora, Organometallics, 2019, 38, 997–1010 CrossRef CAS.
- P. Cossee, Tetrahedron Lett., 1960, 1, 12–16 CrossRef.
- E. J. Arlman, J. Catal., 1964, 3, 89–98 CrossRef CAS.
- J. Petit, L. Magna and N. Mézailles, Coord. Chem. Rev., 2022, 450, 214227 CrossRef CAS.
- R. Robinson, D. S. McGuinness and B. F. Yates, ACS Catal., 2013, 3, 3006–3015 CrossRef CAS.
- R. Y. Brogaard and U. Olsbye, ACS Catal., 2016, 6, 1205–1214 CrossRef CAS.
- U. Müller, W. Keim, C. Krüger and P. Betz, Angew. Chem., Int. Ed. Engl., 1989, 28, 1011–1013 CrossRef.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19–30 CrossRef CAS.
- E. Richmond and J. Moran, Synthesis, 2018, 499–513 CrossRef CAS.
- S. Bhakta and T. Ghosh, Adv. Synth. Catal., 2020, 362, 5257–5274 CrossRef CAS.
- N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed.
- V. P. Ananikov, D. G. Musaev and K. Morokuma, Organometallics, 2005, 24, 715–723 CrossRef CAS.
- S. A. Macgregor, G. W. Neave and C. Smith, Faraday Discuss., 2003, 124, 111–127 RSC.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664–11665 CrossRef CAS.
- C. Wang, S. Friedrich, T. R. Younkin, R. T. Li, R. H. Grubbs, D. A. Bansleben and M. W. Day, Organometallics, 1998, 17, 3149–3151 CrossRef CAS.
- T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef CAS PubMed.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS.
- P.-A. R. Breuil, L. Magna and H. Olivier-Bourbigou, Catal. Lett., 2015, 145, 173–192 CrossRef CAS.
- H. Olivier-Bourbigou, P. A. R. Breuil, L. Magna, T. Michel, M. F. Espada Pastor and D. Delcroix, Chem. Rev., 2020, 120, 7919–7983 CrossRef CAS PubMed.
- J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- J.-B. Liu, X. Wang, A. M. Messinis, X.-J. Liu, R. Kuniyil, D.-Z. Chen and L. Ackermann, Chem. Sci., 2021, 12, 718–729 RSC.
- R. Lhermet, E. Moser, E. Jeanneau, H. Olivier-Bourbigou and P.-A. R. Breuil, Chem. – Eur. J., 2017, 23, 7433–7437 CrossRef CAS PubMed.
- F. Speiser, P. Braunstein and L. Saussine, Inorg. Chem., 2004, 43, 4234–4240 CrossRef CAS PubMed.
- G. Wilke, B. Bogdanović, P. Hardt, P. Heimbach, W. Keim, M. Kröner, W. Oberkirch, K. Tanaka, E. Steinrücke, D. Walter and H. Zimmermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 151–164 CrossRef CAS.
- W. Keim, F. H. Kowaldt, R. Goddard and C. Krüger, Angew. Chem., Int. Ed. Engl., 1978, 17, 466–467 CrossRef.
- D. H. Camacho and Z. Guan, Chem. Commun., 2010, 46, 7879–7893 RSC.
- M. Peuckert and W. Keim, Organometallics, 1983, 2, 594–597 CrossRef CAS.
- P. Kuhn, D. Sémeril, D. Matt, M. J. Chetcuti and P. Lutz, Dalton Trans., 2007, 515–528 RSC.
- U. Klabunde, R. Mulhaupt, T. Herskovitz, A. H. Janowicz, J. Calabrese and S. D. Ittel, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1989–2003 CrossRef CAS.
- G. Wilke, Angew. Chem., Int. Ed. Engl., 1988, 27, 185–206 CrossRef.
- W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235–244 CrossRef.
- A. Finiels, F. Fajula and V. Hulea, Catal. Sci. Technol., 2014, 4, 2412–2426 RSC.
- M. Lallemand, O. A. Rusu, E. Dumitriu, A. Finiels, F. Fajula and V. Hulea, Appl. Catal., A, 2008, 338, 37–43 CrossRef CAS.
- M. Lallemand, O. A. Rusu, E. Dumitriu, A. Finiels, F. Fajula and V. Hulea, Studies in Surface Science and Catalysis, Elsevier, 2008, vol. 174, pp. 1139–1142 Search PubMed.
- R. D. Andrei, E. Borodina, D. Minoux, N. Nesterenko, J.-P. Dath, C. Cammarano and V. Hulea, Ind. Eng. Chem. Res., 2020, 59, 1746–1752 CrossRef CAS.
- Y. Chen, E. Callens, E. Abou-Hamad, N. Merle, A. J. P. White, M. Taoufik, C. Copéret, E. Le Roux and J.-M. Basset, Angew. Chem., Int. Ed., 2012, 51, 11886–11889 CrossRef CAS PubMed.
- A. Hamieh, R. Dey, B. Nekoueishahraki, M. K. Samantaray, Y. Chen, E. Abou-Hamad and J.-M. Basset, Chem. Commun., 2017, 53, 7068–7071 RSC.
- Y. Chen, R. Credendino, E. Callens, M. Atiqullah, M. A. Al-Harthi, L. Cavallo and J.-M. Basset, ACS Catal., 2013, 3, 1360–1364 CrossRef CAS.
- J. M. Basset and A. Choplin, J. Mol. Catal., 1983, 21, 95–108 CrossRef CAS.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- D. Y. Shin, J. H. Yoon, H. Baik and S. J. Lee, Appl. Catal., A, 2020, 590, 117363 CrossRef CAS.
- A. Aid, R. D. Andrei, S. Amokrane, C. Cammarano, D. Nibou and V. Hulea, Appl. Clay Sci., 2017, 146, 432–438 CrossRef CAS.
- M. Fallahi, E. Ahmadi and Z. Mohamadnia, Appl. Organomet. Chem., 2019, 33, e4975 CrossRef.
- A. Al Khudhair, K. Bouchmella, R. D. Andrei, A. Mehdi, P. H. Mutin and V. Hulea, Microporous Mesoporous Mater., 2021, 322, 111165 CrossRef CAS.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- T. Zhang and W. Lin, in Metal-Organic Frameworks for Photonics Applications, ed. B. Chen and G. Qian, Springer, Berlin, Heidelberg, 2014, pp. 89–104 Search PubMed.
- Y. Wen, J. Zhang, Q. Xu, X.-T. Wu and Q.-L. Zhu, Coord. Chem. Rev., 2018, 376, 248–276 CrossRef CAS.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- C. Xu, R. Fang, R. Luque, L. Chen and Y. Li, Coord. Chem. Rev., 2019, 388, 268–292 CrossRef CAS.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819–7832 RSC.
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- Q. Sun, Z. Dai, X. Meng, L. Wang and F.-S. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS.
- C. Mellot-Draznieks and A. K. Cheetham, Nat. Chem., 2017, 9, 6–8 CrossRef CAS PubMed.
- P. Mialane, C. Mellot-Draznieks, P. Gairola, M. Duguet, Y. Benseghir, O. Oms and A. Dolbecq, Chem. Soc. Rev., 2021, 50, 6152–6220 RSC.
- J. Sauer, Acc. Chem. Res., 2019, 52, 3502–3510 CrossRef CAS PubMed.
- Y. J. Colón and R. Q. Snurr, Chem. Soc. Rev., 2014, 43, 5735–5749 RSC.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1–19 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- P. Kuhn, A. Forget, J. Hartmann, A. Thomas and M. Antonietti, Adv. Mater., 2009, 21, 897–901 CrossRef CAS.
- C. Chen, M. R. Alalouni, X. Dong, Z. Cao, Q. Cheng, L. Zheng, L. Meng, C. Guan, L. Liu, E. Abou-Hamad, J. Wang, Z. Shi, K.-W. Huang, L. Cavallo and Y. Han, J. Am. Chem. Soc., 2021, 143, 7144–7153 CrossRef CAS PubMed.
- E. D. Metzger, R. J. Comito, Z. Wu, G. Zhang, R. C. Dubey, W. Xu, J. T. Miller and M. Dincă, ACS Sustainable Chem. Eng., 2019, 7, 6654–6661 CrossRef CAS.
- E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dincă, ACS Cent. Sci., 2016, 2, 148–153 CrossRef CAS PubMed.
- J. Canivet, S. Aguado, Y. Schuurman and D. Farrusseng, J. Am. Chem. Soc., 2013, 135, 4195–4198 CrossRef CAS PubMed.
- B. Liu, S. Jie, Z. Bu and B.-G. Li, RSC Adv., 2014, 4, 62343–62346 RSC.
- L. Chen, Y. Jiang, H. Huo, J. Liu, Y. Li, C. Li, N. Zhang and J. Wang, Appl. Catal., A, 2020, 594, 117457 CrossRef CAS.
- M. I. Gonzalez, J. Oktawiec and J. R. Long, Faraday Discuss., 2017, 201, 351–367 RSC.
- M. J. Kim, S. Ahn, J. Yi, J. T. Hupp, J. M. Notestein, O. K. Farha and S. J. Lee, Catal. Sci. Technol., 2017, 7, 4351–4354 RSC.
- K. Kyogoku, C. Yamada, Y. Suzuki, S. Nishiyama, K. Fukumoto, H. Yamamoto, S. Indo, M. Sano and T. Miyake, J. Jpn. Pet. Inst., 2010, 53, 308–312 CrossRef CAS.
- E. Rozhko, A. Bavykina, D. Osadchii, M. Makkee and J. Gascon, J. Catal., 2017, 345, 270–280 CrossRef CAS.
- U. S. F. Arrozi, V. Bon, S. Krause, T. Lübken, M. S. Weiss, I. Senkovska and S. Kaskel, Inorg. Chem., 2020, 59, 350–359 CrossRef CAS PubMed.
- Y. Hu, Y. Zhang, Y. Han, D. Sheng, D. Shan, X. Liu and A. Cheng, ACS Appl. Nano Mater., 2019, 2, 136–142 CrossRef CAS.
- U. S. F. Arrozi, V. Bon, C. Kutzscher, I. Senkovska and S. Kaskel, Dalton Trans., 2019, 48, 3415–3421 RSC.
- U. S. F. Arrozi, V. Bon, C. Kutzscher, I. Senkovska and S. Kaskel, Dalton Trans., 2019, 48, 3415–3421 RSC.
- N. Guillou, C. Livage, M. Drillon and G. Férey, Angew. Chem., Int. Ed., 2003, 42, 5314–5317 CrossRef CAS PubMed.
- C. Wang, G. Li and H. Guo, Mol. Catal., 2022, 524, 112340 CrossRef CAS.
- C. Chen, M. R. Alalouni, P. Xiao, G. Li, T. Pan, J. Shen, Q. Cheng and X. Dong, Ind. Eng. Chem. Res., 2022, 61, 14374–14381 CrossRef CAS.
- E. D. Metzger, R. J. Comito, Z. Wu, G. Zhang, R. J.-C. Dubey, W. Xu, J. T. Miller and M. Dinca, ACS Sustainable Chem. Eng., 2019, 7, 6654–6661 CrossRef CAS.
- J. Liu, J. Ye, Z. Li, K. Otake, Y. Liao, A. W. Peters, H. Noh, D. G. Truhlar, L. Gagliardi, C. J. Cramer, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2018, 140, 11174–11178 CrossRef CAS PubMed.
- B. Yeh, S. P. Vicchio, S. Chheda, J. Zheng, J. Schmid, L. Löbbert, R. Bermejo-Deval, O. Y. Gutiérrez, J. A. Lercher, C. C. Lu, M. Neurock, R. B. Getman, L. Gagliardi and A. Bhan, J. Am. Chem. Soc., 2021, 143, 20274–20280 CrossRef CAS PubMed.
- S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568–7576 CrossRef CAS PubMed.
- S. T. Madrahimov, J. R. Gallagher, G. Zhang, Z. Meinhart, S. J. Garibay, M. Delferro, J. T. Miller, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 6713–6718 CrossRef CAS.
- D. Li, L. Guo, F. Li, J. Huang, J. Li, M. Li and C. Li, Microporous Mesoporous Mater., 2022, 338, 111979 CrossRef CAS.
- D. S. McGuinness, W. Mueller, P. Wasserscheid, K. J. Cavell, B. W. Skelton, A. H. White and U. Englert, Organometallics, 2002, 21, 175–181 CrossRef CAS.
- D. E. Ortega, D. Cortés-Arriagada, O. S. Trofymchuk, D. Yepes, S. Gutiérrez-Oliva, R. S. Rojas and A. Toro-Labbé, Chem. – Eur. J., 2017, 23, 10167–10176 CrossRef CAS PubMed.
- K. N. Tayade, M. V. Mane, S. Sen, C. N. Murthy, G. L. Tembe, S. M. Pillai, K. Vanka and S. Mukherjee, J. Mol. Catal. A: Chem., 2013, 366, 238–246 CrossRef CAS.
- Y. Tao, W. Ji, X. Ding and B.-H. Han, J. Mater. Chem. A, 2021, 9, 7336–7365 RSC.
- Q. Sun, Z. Dai, X. Meng, L. Wang and F.-S. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS.
- U. I. Kramm, R. Marschall and M. Rose, ChemCatChem, 2019, 11, 2563–2574 CrossRef CAS.
- E. D. Metzger, R. J. Comito, Z. Wu, G. Zhang, R. C. Dubey, W. Xu, J. T. Miller and M. Dincă, ACS Sustainable Chem. Eng., 2019, 7, 6654–6661 CrossRef CAS.
- F. M. Wisser, P. Berruyer, L. Cardenas, Y. Mohr, E. A. Quadrelli, A. Lesage, D. Farrusseng and J. Canivet, ACS Catal., 2018, 8, 1653–1661 CrossRef CAS.
- N. M. Sackers, A. Iemhoff, P. Sautet and R. Palkovits, Catal. Sci. Technol., 2023, 13, 2652–2655 RSC.
- N. V. Velthoven, S. Waitschat, S. M. Chavan, P. Liu, S. Smolders, J. Vercammen, B. Bueken, S. Bals, K. P. Lillerud, N. Stock and D. E. D. Vos, Chem. Sci., 2019, 10, 3616–3622 RSC.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 CrossRef PubMed.
- J. P. Reid and M. S. Sigman, Nat. Rev. Chem., 2018, 2, 290–305 CrossRef CAS.
- S. Das, T. Ben and S. Qiu, Polymer, 2020, 207, 122928 CrossRef CAS.
- A. Abramova, N. Couzon, M. Leloire, P. Nerisson, L. Cantrel, S. Royer, T. Loiseau, C. Volkringer and J. Dhainaut, ACS Appl. Mater. Interfaces, 2022, 14, 10669–10680 CrossRef CAS PubMed.
- Y. Khabzina, J. Dhainaut, M. Ahlhelm, H.-J. Richter, H. Reinsch, N. Stock and D. Farrusseng, Ind. Eng. Chem. Res., 2018, 57, 8200–8208 CrossRef CAS.
- B. Yeskendir, J.-P. Dacquin, Y. Lorgouilloux, C. Courtois, S. Royer and J. Dhainaut, Mater. Adv., 2021, 2, 7139–7186 RSC.
- C. Wang, B. An and W. Lin, ACS Catal., 2019, 9, 130–146 CrossRef CAS.
- R. A. Peralta, M. T. Huxley, J. D. Evans, T. Fallon, H. Cao, M. He, X. S. Zhao, S. Agnoli, C. J. Sumby and C. J. Doonan, J. Am. Chem. Soc., 2020, 142, 13533–13543 CrossRef CAS PubMed.
- D. P. Zhuchkov, M. V. Nenasheva, M. V. Terenina, Yu. S. Kardasheva, D. N. Gorbunov and E. A. Karakhanov, Pet. Chem., 2021, 61, 1–14 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |