
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective formation of boron-stereogenic organoboron derivatives
Amel
Abdou-Mohamed
,
Clara
Aupic
,
Corentin
Fournet
,
Jean-Luc
Parrain
,
Gaëlle
Chouraqui
 * and
Olivier
Chuzel
*
* and
Olivier
Chuzel
*
Aix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: gaelle.chouraqui@univ-amu.fr; olivier.chuzel@univ-amu.fr
First published on 16th June 2023
Abstract
Four-coordinate organoboron derivatives present interesting chemical, physical, biological, electronical, and optical properties. Given the increasing demand for the synthesis of smart functional materials based on chiral organoboron compounds, the exploration of stereoselective synthesis of boron-stereogenic organo-derivatives is highly desirable. However, the stereoselective construction of organoboron compounds stereogenic at boron has been far less studied than other elements of the main group due to configurational stability concerns. Nowadays, these species are no longer elusive and configurationally stable compounds have been highlighted. The idea is to show the potential of the stereoselective building of the four-coordinate boron centre and encourage future endeavors and developments in the field.
 Amel Abdou-Mohamed | Amel Abdou Mohamed studied chemistry at Aix Marseille University where she obtained her MSc degree in 2017. She obtained her PhD degree in 2020 at Aix-Marseille University under the supervision of Dr J.-L. Parrain and O. Chuzel working on the synthesis of new tools for organic and organometallic catalysis. |
 Clara Aupic | Clara Aupic began her education as an organic chemist in 2010 at the Université Joseph Fournier (Grenoble, France). She obtained her MSc degree in 2015 from Université Claude Bernard, Lyon and then moved to the South of France. She received her PhD degree in organic chemistry in 2019 from Aix-Marseille University under the guidance of Dr J.-L. Parrain and O. Chuzel, working on the design, synthesis, and reactivity of novel chiral boron scaffolds. She is now a Senior Scientist in a contract research organization in Groningen. |
 Corentin Fournet | Corentin Fournet studied chemistry at University of Rennes 1 where he obtained his bachelor's degree in 2019, followed by a Master diploma in organic synthesis in the same university. After his graduation, he moved to Aix-Marseille University where he is currently working towards his PhD under the supervision of Dr Gaëlle Chouraqui, and Dr Olivier Chuzel. His research interests focus on the development of new original FLP-based catalysts designed for hydrogenation reactions. |
 Jean-Luc Parrain | Jean-Luc Parrain obtained his PhD in Chemistry at the University of Nantes (France) under the supervision of Professor Jean-Paul Quintard. After post-doctoral studies in the laboratory of Professor Steve Davies at the University of Oxford (UK), he was appointed a CNRS tenured researcher at the University of Nantes. In 1995, he moved to the University of Marseille and was appointed CNRS “directeur de recherches” in 2001. He was the director of the “Fédération des Sciences Chimiques de Marseille” from 2008 to 2017. He is currently regional delegate for research and innovation for the Provence Alpes Côte d’Azur region. He was awarded, the Clavel Lespiau Distinction (2019) from the French Academy of Sciences. His research interests include new catalytic reactions toward new synthetic methods, development of new organotin, silicon, boron reagents, total synthesis of natural compounds and more recently surface functionalization at nanometric scale. |
 Gaëlle Chouraqui | Gaëlle Chouraqui obtained her PhD in 2003, from Université Pierre et Marie Curie (Paris – France), where she worked under the guidance of Prof. Max Malacria and Dr Corinne Aubert. After a two year-stay as a Postdoctoral Research Fellow with Prof. James H. Rigby at Wayne State University, she returned to Europe and completed a further year as a Postdoctoral Research Fellow in Prof. Ian Paterson's laboratory at the University of Cambridge. She was appointed CNRS Tenured-Researcher in 2007 at Aix-Marseille University (France) and her research interests are centered on: the development of novel methodologies to reach molecular complexity, the reactivity of donor–acceptor cyclopropanes, and main group catalysis. |
 Olivier Chuzel | Olivier Chuzel obtained his PhD degree in 2006 from Université Catholique de Louvain (UCL, Belgium) with Professors O. Riant and J.-P. Soumillion, based mainly on photochemistry and metal catalysis. Its PhD was completed by two post-doctoral trainings in E. Schulz's group (ICCMO, Paris-Saclay University) and in V. Vidal's group (Chimie ParisTech – PSL) where he was involved in new methodologies in organometallic enantioselective catalysis. He was then hired as an Assistant Professor in 2008 at Aix Marseille University. His research interests deal with the development of new catalytic and enantioselective processes, with a deep interest in boron chemistry, and growing applications in physical organic chemistry and at the interface activities with physics. |
1. Introduction
Boron-containing molecules are important from an academic and industrial point of view with widespread applications in organic synthesis, biologically active agents, and functional molecules.1 Historically, organoboron compounds proved to be strong and reliable key synthons within a diverse range of organic and organometallic transformations. A vacant p-orbital on the group 13 element provides the organoboron derivatives with Lewis acidic properties.2 In the presence of a Lewis base or a nucleophile, the substrates thus readily interconvert from trivalent to tetravalent and this ability has been used for decades now for the synthesis of optically active molecules; to name only a few examples, asymmetric hydroboration,3 asymmetric reduction, asymmetric aldol reaction,4 and asymmetric allyl-and crotylboration.5 Boron chemistry at that time provided a new perspective for asymmetric synthesis and they all employed chiral organoboron reagents.As just stated, boron can also form tetravalent compounds.6 When a Lewis base or a nucleophile coordinates to the trivalent main group element, the resultant tetracoordinate boron adopts a tetrahedral geometry.7 When the four substituents attached to the boron are different, the boron center becomes stereogenic. However, due to their labile nature and consequently their lower configurational stability, the stereoselective construction of organoboron compounds stereogenic at boron has been far less studied than other elements of the main group, like S,8 Si,9 P10 or N11 for instance.
Thanks to the recently received boost, these species are no longer elusive and stable compounds have been highlighted. The enantiomerization barrier of the stereogenic center can be correlated to the Lewis acidity of the boron atom. The higher the barrier, the higher the acidity of the boron atom. However, it should be noted that the configurational stability does not only depend on the bond strength of the ligands to the boron atom, but also on different factors, such as the electronegativity and steric effect of the substituents.12 These parameters directly influence the Lewis acid character of the boron atom, and also the geometry around the boron atom. The latter has been linked to a useful parameter: the tetrahedral character (THC), which is calculated from bond angles at a boron atom. There is a correlation between the Lewis base–Boron bond length and the THC value. The stronger the donor–boron interaction is, the more tetrahedral the boron surrounding is (Scheme 1).
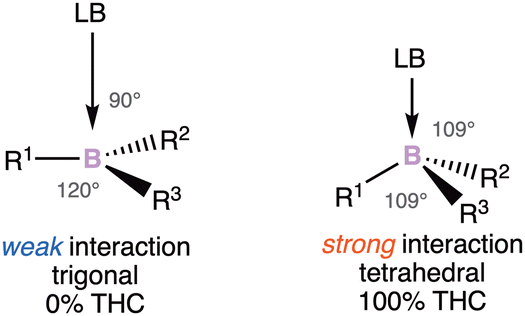 | ||
| Scheme 1 The tetrahedral character (THC) of the boron atom. | ||
Four-coordinate organoboron derivatives present interesting chemical, physical, biological, and stereoelectronic properties. For instance, from an enantioselective catalysis point of view, the development of alternative to rare, toxic, and costly transition metal is required. In this respect, boron which allows the activation of various chemical bonds offers great potential. Main-group element organo-derivatives have been employed as a useful tool for activation of small molecules in place of transition-metal complexes.13 In a recent review, Taniguchi stated that “the substituent effects of tetracoordinate boryl groups are unique.7 They are strong electron-rich groups that greatly influence reactive intermediates such as cations, radicals and π-conjugation systems.” On the other hand, given the increasing demand for the synthesis of smart functional-materials based on chiral organoboron compounds,14 the exploration of stereoselective synthesis of boron-stereogenic organo-derivatives is highly desirable.
Historically, the synthesis of boron-stereogenic organo-compounds relied on the optical and kinetic resolution with chiral auxiliaries.15 Despite the success of such approaches, they are limited in substrate scope along with low efficiency and poor atom-economy. On the other hand, Vedejs16 took advantage of the configurational lability of the boron atom and performed Crystallization-Induced Asymmetric Transformation (CIAT).17 The in situ epimerization of the boron centre and diastereoselective crystallization allowed the formation of a single diastereomer in high yield. However, this process requires crystalline products and epimerizable undesired stereoisomers at temperatures below the product melting point. Thus, the development of synthetic methods toward boron-stereogenic derivatives based on asymmetric synthesis has attracted more attention but has proved to be very challenging. In this perspective, we will take a glimpse at the recent progress in the stereoselective construction of boron-stereogenic centers.
Three recently published reviews reported the influence of tetracoordinate boryl groups in organic synthesis.6,7,18 However, it is worth underlining that only the most recent one18 mentions, in a non-comprehensive way, the stereoselective formation of boron-stereogenic organoboron derivatives. The intent of this review is to give complementary information on the matter. Namely, we wish to provide insights into the configurational stability of the boron element and to highlight the methods at disposal for its stereogenic construction. This review presents articles collected up until April 2023 and we wish to critically discuss the strategies, scope, applications, and the potential or future development of the stereoselective construction of the tetravalent boron center.
The preparation of original scaffolds in a stereoselective manner offers new perspectives not only in the field of materials science but also in medicinal chemistry. Due to its unique shape and charge distribution, exploration of the chemical space around the stable stereogenic tetravalent boron element could open new avenues in early phase drug discovery19 and in the development of functional materials. To date and as far as our knowledge goes, only three recent examples reported the enantioselective preparation of such compounds.20 Otherwise, the common approaches rely on a racemic diastereoselective synthesis or a diastereoselective synthesis starting from an enantiopure compound.
2. Racemic diastereoselective synthesis of boron-stereogenic compounds
2.1. Thermal- or photo-promoted skeletal rearrangements
The development of chiral N,C-chelate organoborate derivatives embedded in rigid π-conjugated structures is of interest for the development of smart materials.21 The stability of the corresponding stereogenic boron atom offers perspectives in the field of optical materials that exhibit circular dichroism (CD) or circular polarized luminescence (CPL). Those promising properties triggered several groups worldwide and they all showed that irradiation of chelate boron compounds stabilized by a Lewis base (e.g. NHC,22 Py,23 Pyrazole,24 phenylazolyl,25 carbonyl,…26), leads to impressive rearrangements of the initial skeleton of the molecule.27 For instance, Wang has developed a methodology allowing the chemoselective and diastereospecific intramolecular insertion of the boron atom into the π-bond of an aromatic ring. The two-stage photoisomerization and the subsequent reversible photo-rearrangement of the four-coordinate boron derivative 1 deliver a substituted borirane 2 stereogenic at boron (Scheme 2).28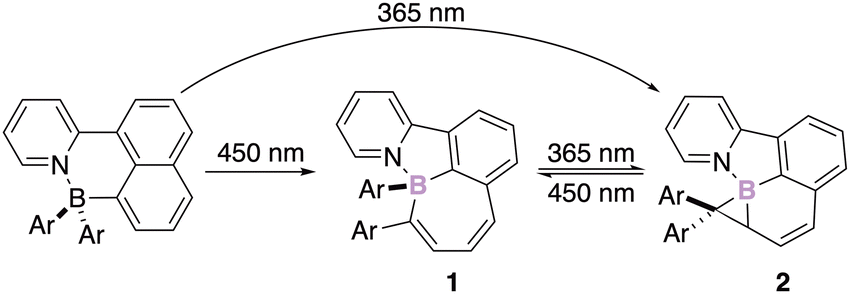 | ||
| Scheme 2 Photoisomerization from naphthalenylpyridine to borirane via borepin. | ||
Interestingly, the regioselectivity of the transformation was demonstrated thanks to the photoisomerization/C–C bond formation of a chiral organoboron derivative substituted by two different aryl groups.29 The discrimination between bulky and nonbulky aryl substituents seemed to indicate that steric factors govern the reaction pathway. However, when aryls of similar sizes were employed, Wang clearly demonstrated that the isomerization occurred on the more delocalized substituent therefore involving the electronic nature of the substituents.30 Mechanistic evidence supports an excited-state reactivity of N,C-chelates organoborates,31 and provides rationalization for the observed regioselectivity.32
In 2020, Curran, Liu and Taniguchi reported the thermal rearrangement of an unprecedented diastereomerically pure NHC–ligated borepin 3 into the corresponding NHC–boranorcaradiene 4 at 150 °C for 12 hours, in 67% yield and 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 diasteromeric ratio.33 The mechanism and the retention of the diastereoselectivity of this rearrangement has been investigated by means of DFT and supported a 6π-electrocyclic reaction followed by a 1,5-boron shift. Interestingly, the stereoselective construction of the starting material was achieved thanks to a double radical trans-hydroboration of cyclic diyne 5 with NHC–borane 6. The expected adduct was isolated as a single diastereomer in 31% yield together with the product of mono hydroboration 7 in 28% yield (Scheme 3). There are only few examples of isolated boranorcaradiene compared to the more stable borepin. Deepening the knowledge on the synthesis of the boron analogs of norcardiene should give insights into the chemistry of this unusual scaffold.
:3 diasteromeric ratio.33 The mechanism and the retention of the diastereoselectivity of this rearrangement has been investigated by means of DFT and supported a 6π-electrocyclic reaction followed by a 1,5-boron shift. Interestingly, the stereoselective construction of the starting material was achieved thanks to a double radical trans-hydroboration of cyclic diyne 5 with NHC–borane 6. The expected adduct was isolated as a single diastereomer in 31% yield together with the product of mono hydroboration 7 in 28% yield (Scheme 3). There are only few examples of isolated boranorcaradiene compared to the more stable borepin. Deepening the knowledge on the synthesis of the boron analogs of norcardiene should give insights into the chemistry of this unusual scaffold.
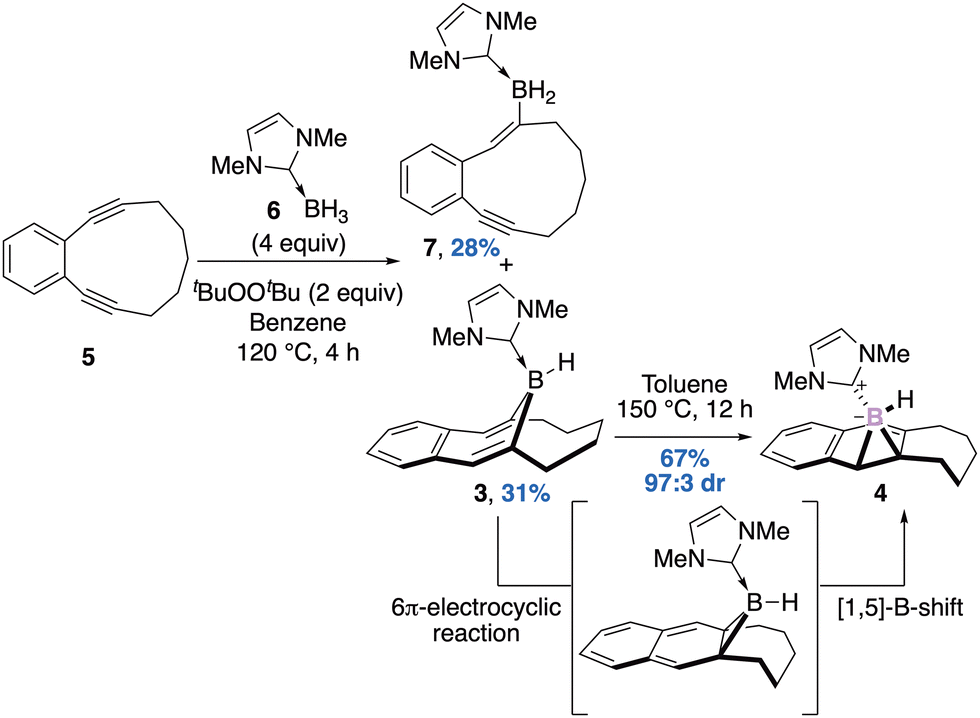 | ||
| Scheme 3 Stereoselective construction of borepin 3 and its thermal rearrangement. | ||
Another photo-promoted skeletal rearrangement of a phosphine–borane Frustrated Lewis Pair (FLP) 8 this time, occurred via a boranorcaradiene 9 followed by migration of the methyl group at the ring junction and provided a stereogenic boron derivative 10.34 The authors demonstrated that irradiation for an extended period resulted in an additional Me-migration from boron to phosphorus and provided a single diastereomer of 11 in 28% yield (Scheme 4). From the prospect of developments that are likely to emerge from FLP chemistry in the future, the photoreactivity reported here leading to the diastereoselective formation of the borate species, should open new avenues in synthetic organic chemistry.
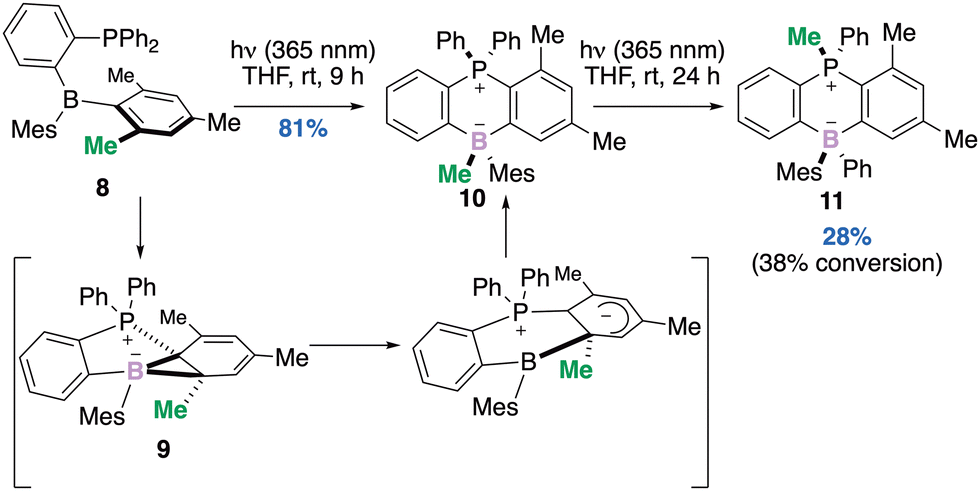 | ||
| Scheme 4 Conversion of a Me-substituent on an arene to an anionic Me-borate to a cationic Me-phosphonium. | ||
It is worth adding that the four-coordinate boron molecules with N,C- or C,C-chelate are often photoresponsive systems.22 The stereocontrol of the stereogenic boron atom and therefore the construction of 3D defined geometries of this kind of structure is an appealing topic and could find application in memory molecular devices and switches.
2.2. Reduction of double bonds (B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) B or BC)
B or BC)
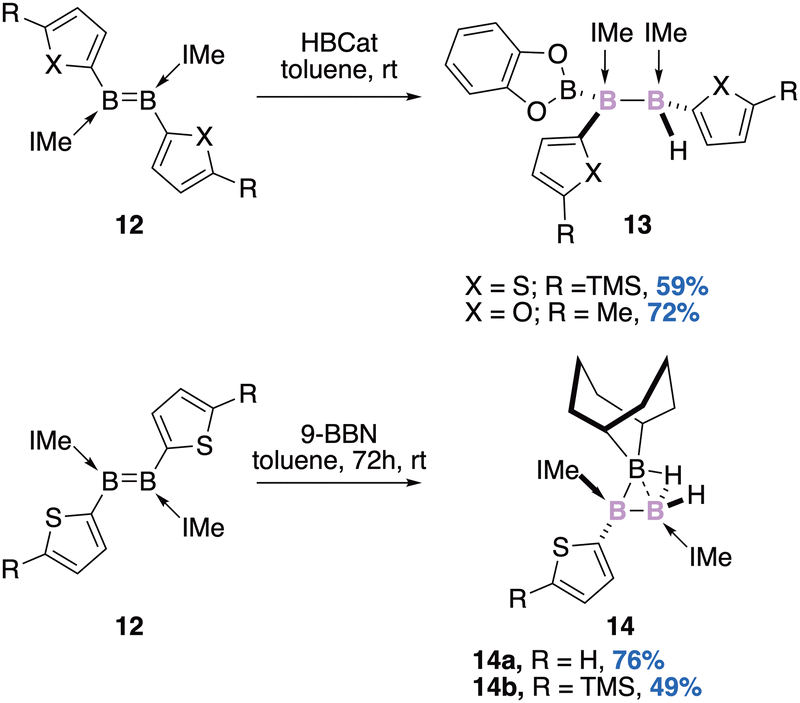
Since the first report of a Lewis base stabilized diborene by Robinson,35 the B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B double bond has attracted considerable attention.36 Diborenes are both isolobal and isoelectronic with olefins and due to their nucleophilic properties, their role as the ligand of transition metal complexes has been considered. Braunschweig used this nucleophilic feature to perform a hydroboration reaction of diborene 12 with catecholborane (Scheme 5, top).37
B double bond has attracted considerable attention.36 Diborenes are both isolobal and isoelectronic with olefins and due to their nucleophilic properties, their role as the ligand of transition metal complexes has been considered. Braunschweig used this nucleophilic feature to perform a hydroboration reaction of diborene 12 with catecholborane (Scheme 5, top).37
 | ||
| Scheme 5 Hydroboration of a base-stabilized diborene (IMe: 1,3-dimethylimidazole-2-ylidene, HBCat: catecholborane). | ||
The reaction is diastereospecific (syn-addition) and delivers a triborane derivative 13 with two stereogenic boron atoms as a single diastereomer.
In an attempt to perform hydroboration of the same base-stabilized diborene 12 in the presence of the sterically demanding 9-borabicyclo[3.3.1]nonane (9-BBN), Braunschweig also showed that a non-classical B3 core 14 could instead be diastereoselectively isolated in decent to good yields (Scheme 5, bottom).38
The hydrofunctionalization of BC bonds is another strategy to perform the diastereoselective construction of the stereogenic boron atom via a syn-addition. Martin and coworkers reported the hydroalkylation, hydroarylation, hydroalkynylation, hydroamination, hydroalkoxyalkylation, and hydration of borataalkene 15 and therefore a versatile approach to tetracoordinate stereogenic boron centre 16 (Scheme 6).39
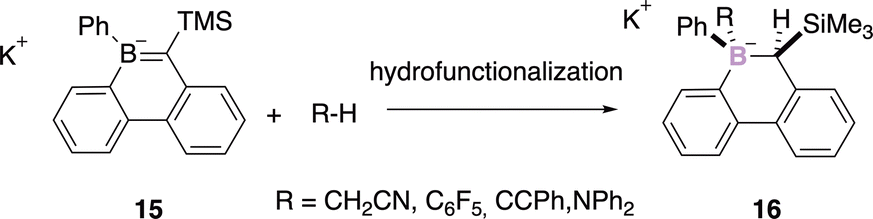 | ||
| Scheme 6 Hydrofunctionalization of borataalkene 16. | ||
The generation of a boron stereogenic centre via the addition on BX systems is quite rare mostly due to the lack of methods to make the aforementioned species.40,41 It is expected that the growing interest for diborene derivatives would result in an increased number of methods at disposal.
2.3. Miscellaneous
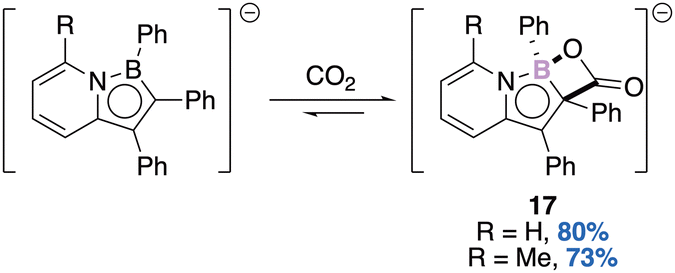 | ||
| Scheme 7 CO2 activation by a boraazaindenide anion. | ||
:1 diasteromeric ratio (Scheme 8).43 Three stereogenic centres are generated, the two carbon ones result from the syn-addition of borylene onto the aromatic substituent of the starting borane (diastereospecific reaction). With an excess of KC8 (0.6 M in toluene), each diastereomer leads diastereospecifically to the corresponding diborene compounds E and Z (3:1 dr).
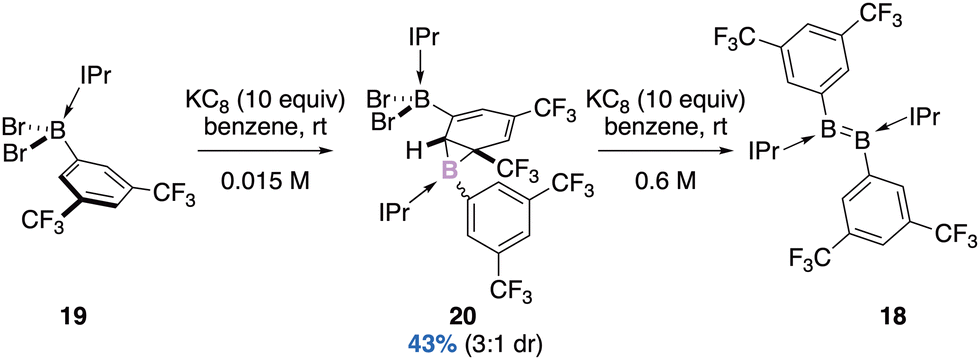 | ||
| Scheme 8 A diastereoselective reduction to borirane (IPr: 1,3-diisopropylimidazole-2-ylidene). | ||
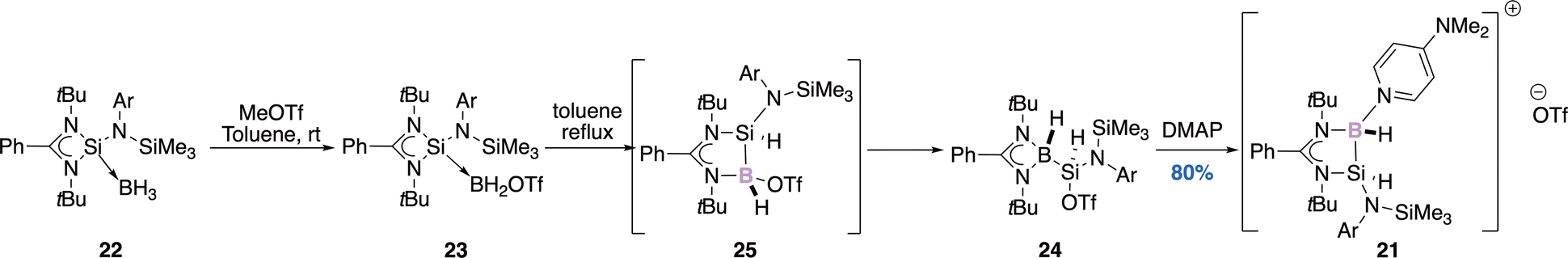 | ||
| Scheme 9 B–H bond activation. | ||
:13 dr (trans:cis). The 1,3-nucleophilic addition of HCl across the B–C bond leads to a ring-opened enolate intermediate. After tautomerization of the transient species, the diastereoselective ring closure accompanied with chloride displacement gives the expected boralactones.
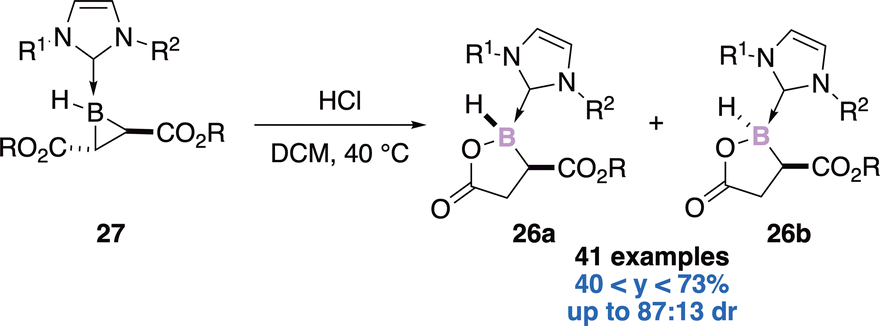 | ||
| Scheme 10 Borirane ring-opening. | ||
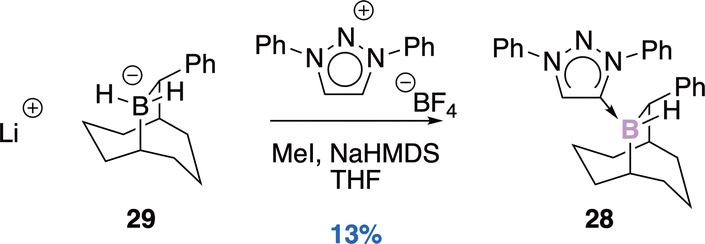 | ||
| Scheme 11 Carbene addition. | ||
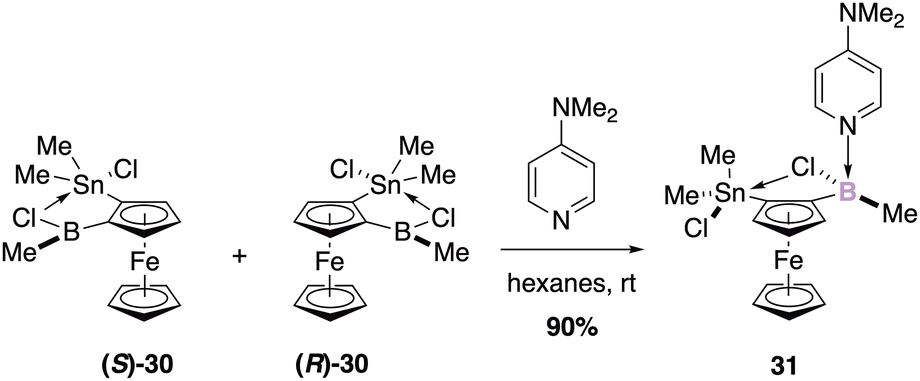 | ||
| Scheme 12 Chemo- and diastereoselective binding of 4-DMAP to borane. | ||
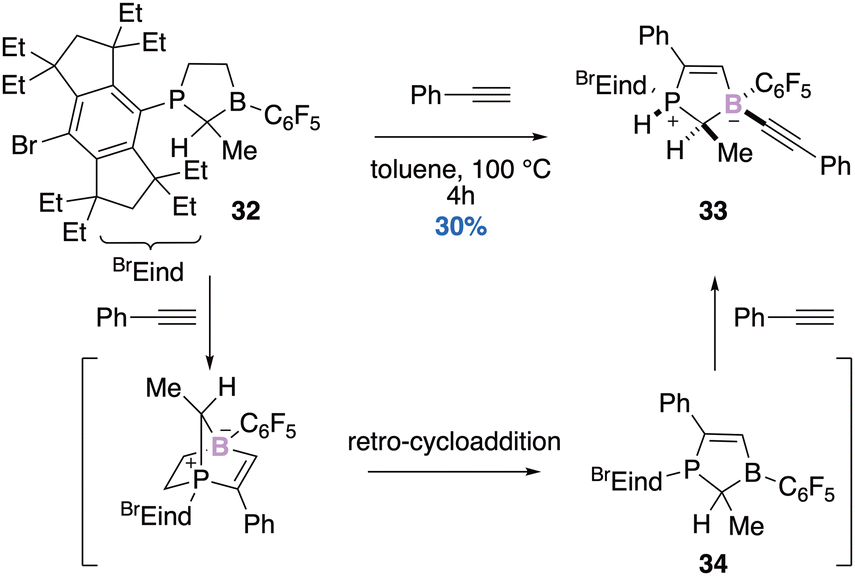 | ||
| Scheme 13 Formal Alder–Rickert cycloaddition reaction. | ||
The approach disclosed by Erker and coworkers could be exploited for the design of novel FLPs since this formal Alder–Rickert cycloaddition leads to an intermediate FLP 34. The surrounding geometry after the split of small molecules like dihydrogen for instance, could play an important role in stereoselective reduction.
It can be concluded that, for the moment, intricate and/or strained structures seems to be required for the diastereoselective control of the stereogenic boron. It is to be expected that the increasing interest for the development of stable tetracoordinate organoboron derivatives and approaches towards elusive species like borylenes or boralkenes would allow considerable progress in this field.
3. Diastereoselective synthesis of boron-stereogenic compounds, starting from an enantiopure substrate
As already mentioned, methods for the enantioselective construction of the four-coordinated boron atom are scarce.21 Racemic resolution or preferred recrystallization methods of a diastereomer is costly from a time and economic perspective. Asymmetric induction, which refers to the control of stereoselectivity exerted by an existing enantiopure starting material, aims at controlling the diastereoselectivity. Accordingly, if the reaction of an enantiopure substrate onto a boron derivative is highly diastereoselective, the absolute configuration of the boron atom will then be perfectly controlled.To this end, several authors utilized the chiral pool like L-proline, or prolinol,50 aminoacids,51 ephedrine,52 in the presence of boronic acid, to access chiral boron chelates. The tridentate imino-ligand on the boron atom strategy has found several applications.
For instance, Braun53 was able to synthesize a family of boronates 35a–c in a diastereoselective manner (Scheme 14). Starting from an enantiopure imine 36, the corresponding boron complex is formed exclusively as a single diastereomer of absolute configuration (RC,RB)-35 (determined by DRX). No epimerization product is observed, indicating high thermodynamic stability of these stereoisomers. Next the authors synthesized racemic boron complexes with the boron atom as the only stereogenic center, to answer the question of kinetic stability. The tetrahedral character (THC) of the boron atom calculated is about 69%. The enantiomeric pairs were separated by chiral HPLC and their Circular Dichroism (CD) spectra were obtained.
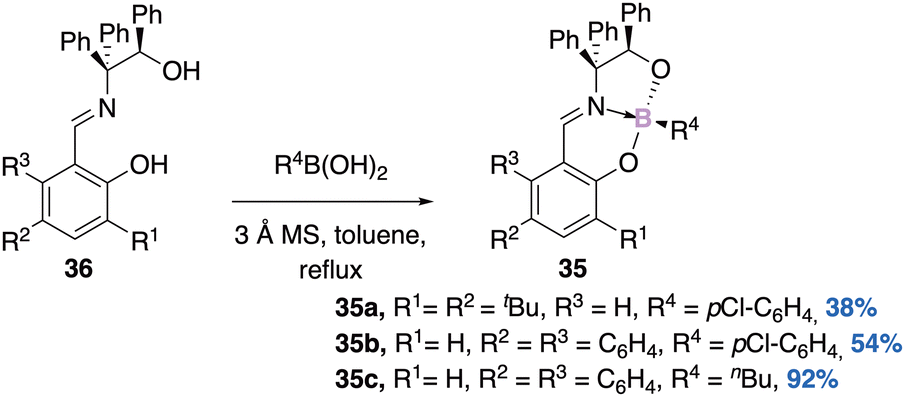 | ||
| Scheme 14 A stable stereogenic center in imine complexes of aryl- & alkyl-boronates. | ||
Finally, the configurational stability of the boron atom was evaluated. The racemization barrier increases slightly with the electron-withdrawing character of the substituents, as the latter increases the B–N bond strength and shortens it. The same authors used the boronate–imine and -amine complexes as chiral dopants for nematic liquid crystals.54
In parallel, Hutton's group reported a similar approach for the diastereoselective preparation of a boronate complex 37.55 The one-pot, three-component reaction between the chiral base 38 and glyoxylic acid could provide the transient imine which could thereafter react with the phenylboronic acid, thus leading to the boronate complex 39. A further tautomerization could provide the boron–imine complex 37, solely stereogenic at boron, in 78% yield with >99% enantiomeric excess (Scheme 15). Three main factors could explain the observed configurational stability of the boron atom (rate of racemization <0.5% in boiling toluene for 24 h): (i) the tridentate nature of the ligands on the boron atom, (ii) the coordinating acyl group, and (iii) the conformational rigidity of the naphthylphenylimine substituent. Interestingly, benzylation of compound 37 leads to the introduction of an extra stereogenic centre 40 in 60% yield (based on the recovered starting material) with a 10.5:1 diastereomeric ratio.
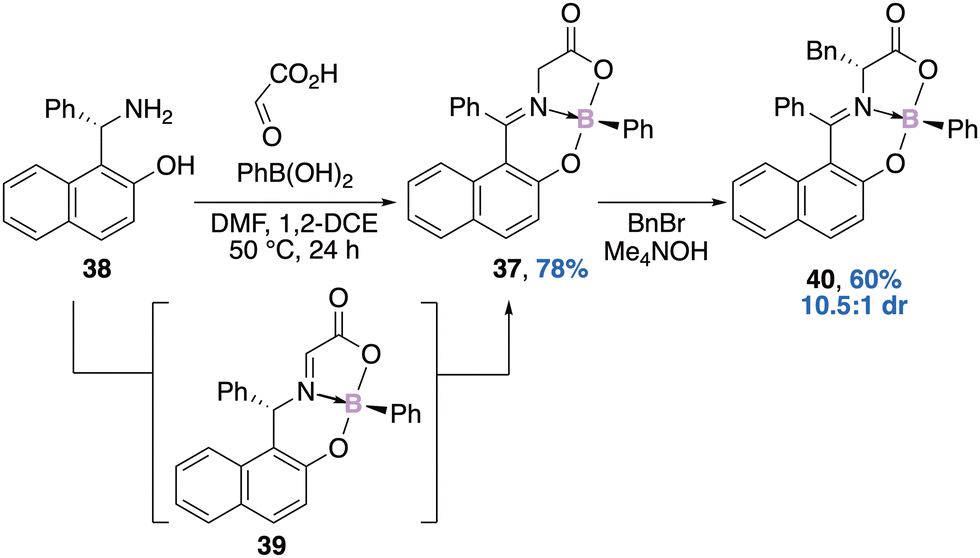 | ||
| Scheme 15 A chirality transfer process for the preparation of an enantiopure boron–imine complex 37 stereogenic at boron only. | ||
Knight, Herrebout and Harriman recently reported the synthesis of configurationally stable chiral tert-leucine iminoborate derivatives obtained thanks to a three-component reaction as single diastereomers (Scheme 16).56 The enantiopure analogous N,O,O- and N,N,O-boron chelates present dissimilar photophysical properties.
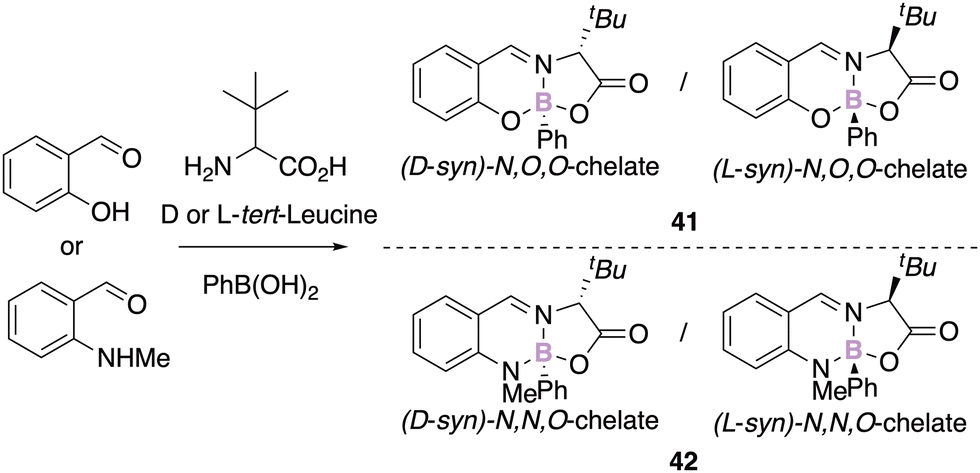 | ||
| Scheme 16 tert-Leucine chiral iminoborates. | ||
While the N,O,O salicylaldehyde derivative 41 is mainly deactivated by an intramolecular charge transfer process, the corresponding N,N,O-aminobenzaldehyde compound 42 fluoresces in the visible region. Interestingly, the latter is more fluorescent in the solid state, and represents another example of aggregation-induced emission phenomenon.
The multicomponent strategy was also applied to the synthesis of boron analogs of 5-oxofuro[2,3-b]furan motifs and led to the assembly of N–B heterocyclic compounds with natural product-like scaffolds, thus expanding the chemical space.
Gois’ approach relied on a one-pot four-components reaction to reach the natural product like-bicyclic system 43.57 The authors reported 25 examples in yields up to 95% and up to >97% diastereomeric excess (Scheme 17). They further used this strategy to build libraries of potentially bioactive molecules and discovered new HNE inhibitors with IC50 up to 1.1 μM58 or novel modulators of human phenylalanine hydroxylase.59 Finally, the rapid, reversible, and selective N-terminal cysteine functionalization, under mild aqueous conditions, thanks to this approach was described, therefore highlighting the potential to build well-defined bioconjugates.60
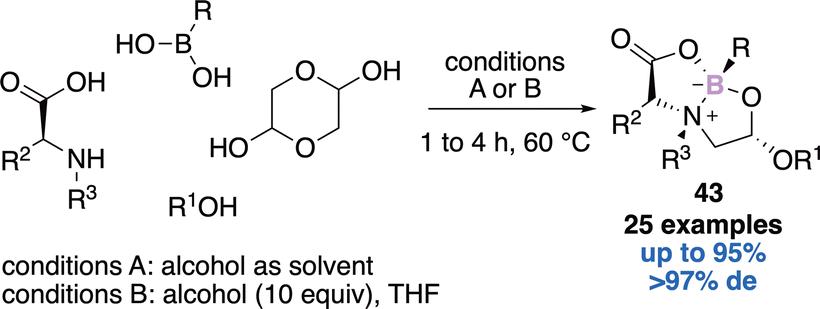 | ||
| Scheme 17 A one-pot four-component reaction. | ||
Another one-pot four-components reaction reported by Nielsen allowed the construction of dioxadiazaborocines 44 (Scheme 18).61 First a Petasis three-component reaction occurs in the presence of a readily available hydrazide, an α-hydroxy aldehyde and a first boronic acid. Then the condensation of a second orthogonally reactive boronic acid delivers the expected bicyclic architecture 44 in a diastereo and enantiomerically pure fashion.
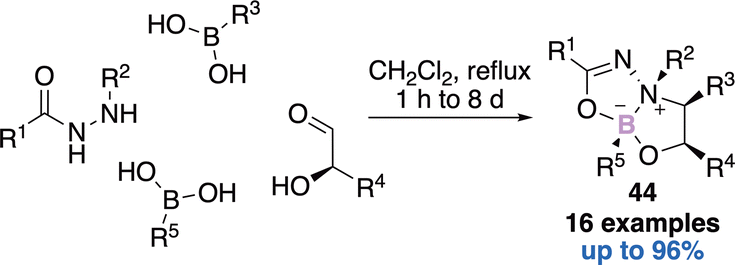 | ||
| Scheme 18 Construction of dioxadiazaborocines. | ||
The configurational stability of boron(III) chelate was also utilized by Iwasawa and his team as a circular dichroism probe to determine the absolute configuration of chiral alcohols. The addition of a chiral racemic alcohol to a 1,2-dihydro-1-hydroxy-2,3,1-benzo-diazaborine 45 generates a chiral borate 46 containing a stereogenic boron atom (Scheme 19).62
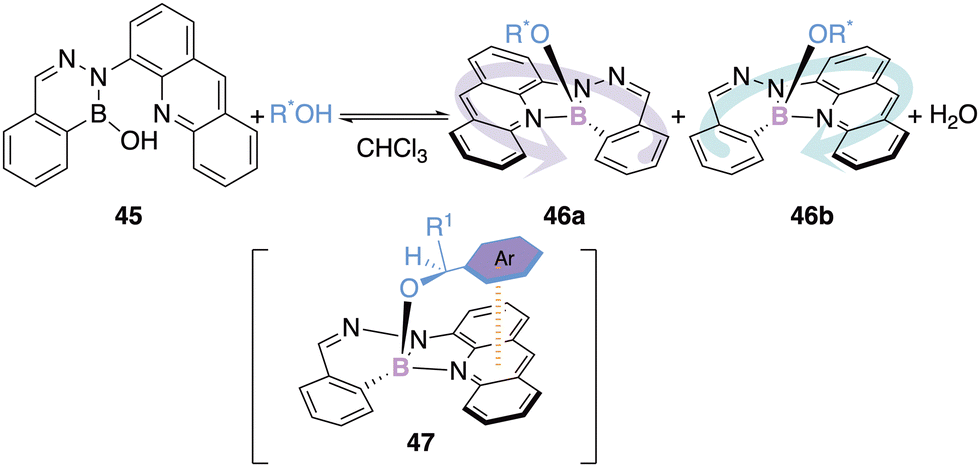 | ||
| Scheme 19 Circular dichroism probe for the determination of absolute configuration of mono-alcohols. | ||
The addition of a chiral alcohol containing a π-system allows a secondary π–π interaction due to the presence of the aza-anthracenyl group of the borate (see 47). Twenty-five examples could be synthesized with diastereomeric excess ranging from 1.8 to 19%.
In 2019, our team reported the diastereoselective preparation of chiral NHC–boranes 48 stereogenic at the boron atom thanks to a chlorination/arylation sequence.63 The chlorinated NHC borane 49 (1:1 dr), with a fixed stereochemistry at the α-carbon center and obtained quantitatively from the enantioenriched NHC–BH250 undergoes a diastereoselective functionalization at boron, in the presence of a simple Grignard reagent (Scheme 20). The diastereoselectivity has been rationalized through a plausible SRN1 mechanism and was supported by EPR observations and DFT calculations. Another interesting finding is the excellent diastereoselective regeneration of the boron stereogenic centre, from a borenium intermediate with a hydride. This represents a promising strategy for metal-free enantioselective reduction catalysis, particularly in FLP-type reduction reactions.
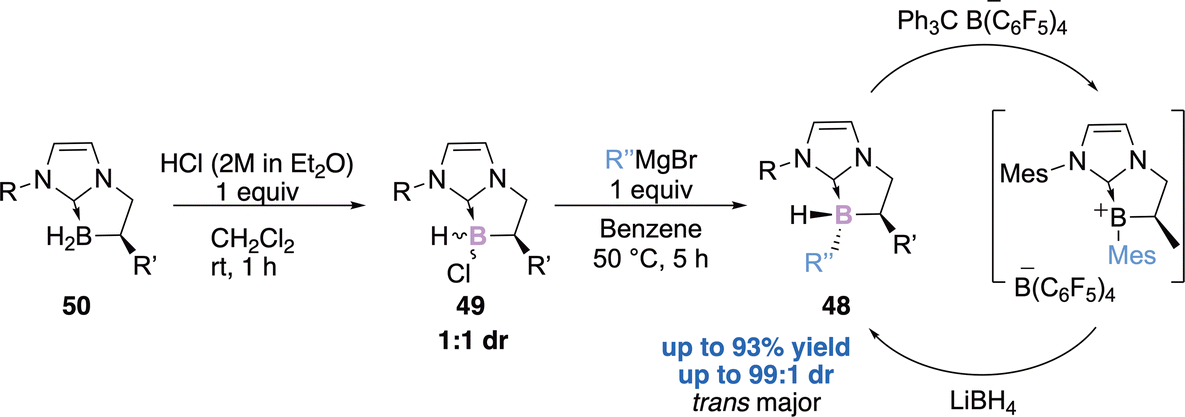 | ||
| Scheme 20 A two-step sequence for the diastereoselective preparation of NHC–boranes stereogenic at the boron atom. | ||
Asymmetric induction for the construction of the additional stereogenic boron is effective. The type of scaffolds is heavily dominated by iminoboronates. Interestingly, heterocyclic natural product-like derivatives are likely to provide access to new bioactive compounds, probes for chemical biology or catalysts for asymmetric synthesis. On the other hand, the diastereoselective construction of NHC–borane compounds offer new perspectives, and applications in the field of main-group elements catalysis. Finally, the photophysical properties imparted by these novel chiral objects could find applications in functional, smart, optical materials.
Going beyond would consist of the construction of tetracoordinate boron derivatives in an enantioselective fashion.
However, given the labile nature of ligands attached to the boron atom, this approach is a challenge to synthetic organic chemists. As the following section will show, this is a challenge recently and successfully addressed by two different groups.21
4. Enantioselective synthesis of boron-stereogenic compounds
In 2021, He and coworkers elegantly tackled the challenge of creating a tetrahedral boron-stereogenic center in an enantioselective manner,21a thanks to a copper-catalyzed azide–alkyne cycloaddition (CuAAC) desymmetrization reaction (Scheme 21). In the presence of two equivalents of azide, two equivalents of DABCO, 10 mol% of copper catalyst [Cu(CH3CN)4PF6] and 12 mol% of the indane-fused BOX chiral ligand L1, the authors were able to obtain the desired N,N-π conjugated scaffolds 51 in yields ranging from 57 to 91% and up to 99% enantiomeric excess (ee). The scope of this desymmetrization is large (41 examples) and the novel chiral building blocks exhibit bright fluorescence and CPL (Circularly Polarized Luminescence) signals. These chiral compounds have therefore potential applications as chiral fluorescent probes.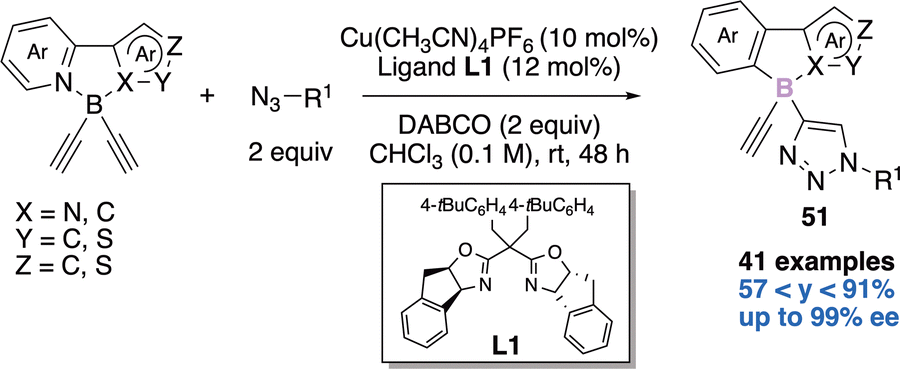 | ||
| Scheme 21 First enantioselective construction of a boron-stereogenic center. | ||
The second example reports an enantioselective copper-catalyzed desymmetric B–H bond insertion reaction of 2-arylpyridine-boranes 52 with diazo derivatives 53a–b to access boron-stereogenic centres 54a–b (Scheme 22).21b Computational investigations were reported and explain the excellent enantioselectivity and diastereoselectivity observed.
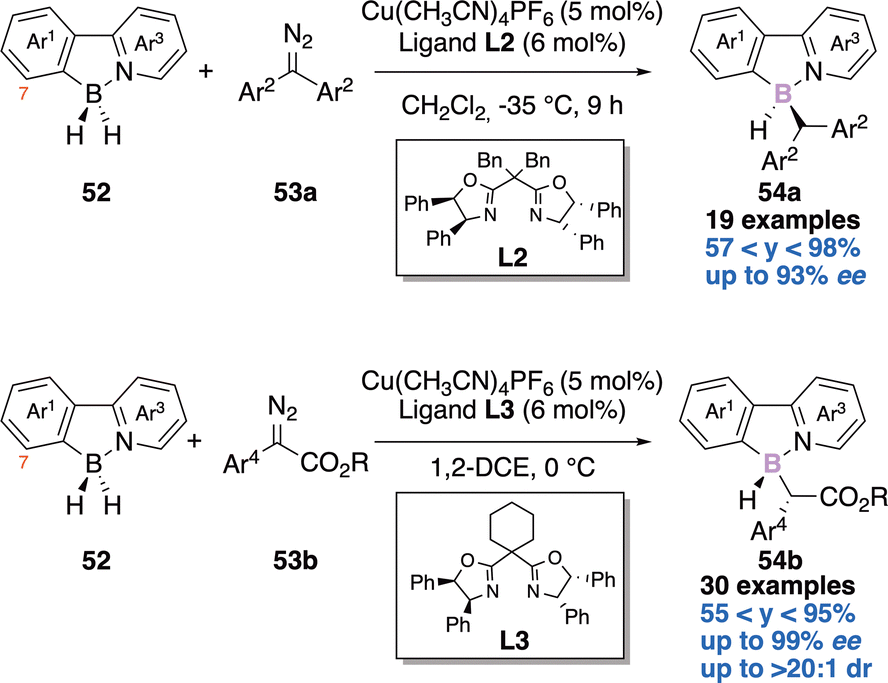 | ||
| Scheme 22 Second enantioselective construction of a boron-stereogenic centre. | ||
Mechanistically, the borane could transfer its hydride to the carbene centre leading to a transient borenium. A subsequent SE2 process could deliver the observed product 54a–b. Notably, introduction of a substituent at the C7-position of the 2-arylpyridyl improves the stereocontrol (Scheme 22).
The third and last example disclosed the desymmetrisation of the equivalent hydrogens Ha and Hb on the pyrrole units of the BODIPY core 55.21c The Pd-mediated desymmetric intramolecular C–H arylation reaction using a TADDOL-based phosphonite ligand L4 delivered a large scope of 6- to 9-membered ring BODIPY scaffolds 56 and the enantiomeric excesses reported are good to excellent (Scheme 23). The authors next studied the photophysical properties of their molecules and found out that they display yellow to bright fluorescence and the quantum yields range from 0.7 to 48.4%. Very interestingly they further functionalized these valuable molecules and demonstrated their use as chiral fluorescent probes.
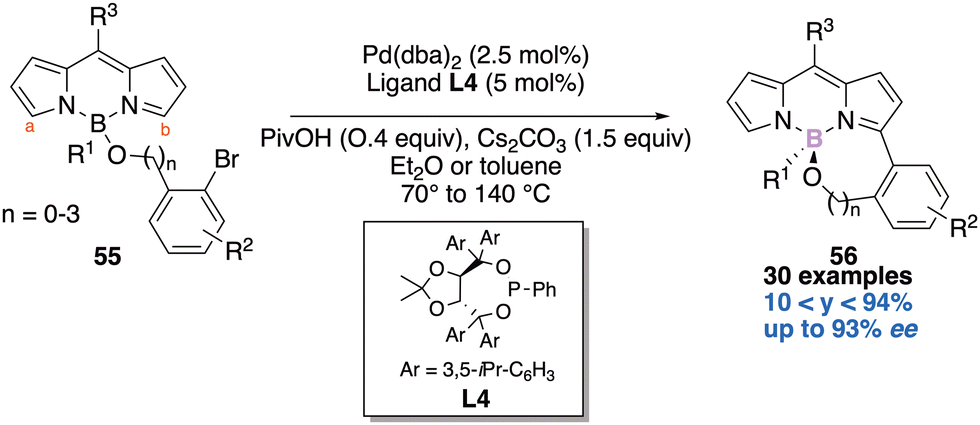 | ||
| Scheme 23 Third enantioselective construction of a boron-stereogenic centre. | ||
Due to configurational stability concerns, examples reporting the enantioselective construction of the tetravalent stereogenic boron element are scarce and limited. Accordingly, the approaches (i) rely on a desymmetrization strategy and (ii) utilize similar π-conjugated starting scaffolds. These three examples are clearly important breakthroughs in this field and elegantly expand the diversity of chiroptical objects at disposal. A step forward would consist in enlarging the scope of methods at disposal to access a broader range of different chiral building blocks.
5. Conclusions
Four-coordinate organoboron derivatives cover a broad spectrum of scientific applications and thanks to the development of more stable species, significant progress has been made in recent years for the stereoselective construction of the boron atom. This review intended to give an overview of the state of the art. Obviously, there is a lack of a general method for the enantioselective approach, and we believe that the full potential of the stereoselective building of the four-coordinate boron center is yet to come. To conclude, this highlight clearly illustrates that tetracoordinate organoboron species stereogenic at boron has become a mainstay and is very likely to play a greater role in the future.Author contributions
Writing – original draft: AAM, GC and OC. Writing – review & editing: AA, CA, CF, JLP, GC and OC. Funding acquisition: JLP, GC and OC. The final manuscript has been read and approved by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Agence National de la Recherche (ANR-16-CE07-0002), Novachim, Aix Marseille Université, the CNRS and the “Provence-Alpes-Côte d’Azur” Council for financial support.Notes and references
- (a) H. A. Ali, V. M. Dembitsky and M. Srebnik, Contemporary Aspects of Boron Chemistry and Biological Applications (Studies in Organic Chemistry 22), Elsevier, Amsterdam, 2005 Search PubMed; (b) D. G. Hall, Boronic Acids: Preparation and Applications, in Organic Synthesis and Medicine, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd, Completely Revised edn, 2011 Search PubMed; (c) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31 CrossRef CAS; (d) B. Su and R. Kinjo, Synthesis, 2017, 2985 CAS; (e) G. F. S. Fernandes, W. A. Denny and J. L. Dos Santos, Eur. J. Med. Chem., 2019, 179, 791 CrossRef CAS PubMed; (f) Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat’ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800 CrossRef CAS PubMed; (g) L. Kong and C. Cui, Synlett, 2021, 1316 CrossRef CAS.
- (a) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75 CrossRef CAS; (b) J. R. Lawson and R. L. Melen, Recent developments and applications of acidic boron reagents, Organometallic Chemistry, The Royal Society of Chemistry, 2017, vol. 41, pp. 1–27 Search PubMed.
- See for instance: (a) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695 CrossRef CAS; (b) S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877 RSC.
- See for instance: (a) C. J. Cowden and I. Paterson, Org. React., 1997, 51, 1–200 CAS; (b) A. Abiko, Acc. Chem. Res., 2004, 37, 387 CrossRef CAS PubMed.
- P. Kaur, G. L. Khatik and S. K. Nayak, Curr. Org. Synth., 2017, 14, 665 CAS.
- K. Yang and Q. Song, Acc. Chem. Res., 2021, 54, 2298 CrossRef CAS PubMed.
- T. Taniguchi, Chem. – Eur. J., 2022, 28, e202104333 CAS.
- S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147 CrossRef CAS PubMed.
- M. Oestreich, Synlett, 2007, 1629 CrossRef CAS.
- (a) M. J. Johansson and N. C. Kann, Mini-Rev. Org. Chem., 2004, 1, 233 CrossRef CAS; (b) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS.
- (a) D. Qian and J. Sun, Chem. – Eur. J., 2019, 25, 3740 CrossRef CAS PubMed; (b) F. A. Davis and R. H. Jenkins Jr., Synthesis and utilization of compounds with chiral nitrogen centers, in Asymmetric synthesis, ed. J. D. Morrison and J. W. Scott, Academic Press, San Diego, 1984, vol. 4 Search PubMed.
- S. Toyota, F. Ito, N. Nitta and T. Hakamata, Bull. Chem. Soc. Jpn., 2004, 77, 2081 CrossRef CAS.
- (a) M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231 CrossRef PubMed; (b) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS PubMed.
- (a) J. Q. Shi and F. W. Peng, Dyes Pigm., 2022, 204, 110283 Search PubMed; (b) S. Shah, P. Marandi and P. P. Neelakandan, Front. Chem., 2021, 9, 708854 CrossRef CAS PubMed.
- (a) G. E. Ryschkewitsch and J. M. Garrett, J. Am. Chem. Soc., 1968, 90, 7234 CrossRef CAS; (b) T. Imamoto and H. Morishita, J. Am. Chem. Soc., 2000, 122, 6329 CrossRef CAS.
- E. Vedejs, S. C. Fields, S. Lin and M. R. Schrimpf, J. Org. Chem., 1995, 60, 3028 CrossRef CAS.
- N. G. Anderson, Org. Process Res. Dev., 2005, 9, 800 CrossRef CAS.
- X. Li, G. Zhang and Q. Song, Chem. Commun., 2023, 59, 3812 RSC.
- See for instance: C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC.
- (a) B. Zu, Y. Guo and C. He, J. Am. Chem. Soc., 2021, 143, 16302 CrossRef CAS PubMed; (b) G. Zhang, Z. Zhang, M. Hou, X. Cai, K. Yang, P. Yu and Q. Song, Nat. Commun., 2022, 13, 2624 CrossRef CAS PubMed; (c) B. Zu, Y. Guo, L.-Q. Ren, Y. Li and C. He, Nath. Synth., 2023, 2, 564 CrossRef.
- (a) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416 RSC; (b) D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290 CrossRef CAS PubMed; (c) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537 RSC; (d) X. Chen, D. Tan and D.-T. Yang, J. Mater. Chem. C, 2022, 10, 13499 RSC.
- Y.-L. Rao, L. D. Chen, N. J. Mosey and S. Wang, J. Am. Chem. Soc., 2012, 134, 11026 CrossRef CAS PubMed.
- (a) D.-T. Yang, S. K. Mellerup, J.-B. Peng, X. Wang, Q.-S. Li and S. Wang, J. Am. Chem. Soc., 2016, 138, 11513 CrossRef CAS PubMed; (b) Y.-L. Rao, H. Amarne, S.-B. Zhao, T. M. McCormick, S. Martic, Y. Sun, R.-Y. Wang and S. Wang, J. Am. Chem. Soc., 2008, 130, 12898 CrossRef CAS PubMed.
- Y.-L. Rao, C. Hörl, H. Braunschweig and S. Wang, Angew. Chem., Int. Ed., 2014, 53, 9086 CrossRef CAS PubMed.
- Y.-L. Rao, H. Amarne, L. D. Chen, M. L. Brown, N. J. Mosey and S. Wang, J. Am. Chem. Soc., 2013, 135(9), 3407 CrossRef CAS PubMed.
- G.-F. Hu, H.-J. Li, C. Zeng, X. Wang, N. Wang, T. Peng and S. Wang, Org. Lett., 2019, 21, 5285 CrossRef CAS PubMed.
- (a) Photoresponsive Organoboron Systems, S. K. Mellerup and S. Wang, in Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, 2017, ch. 3 Search PubMed; (b) S. K. Mellerup and S. Wang, Sci. China Mater., 2018, 61, 1249 CrossRef CAS.
- Z.-C. He, S. K. Mellerup, L. Liu, X. Wang, C. Dao and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 6683 CrossRef CAS PubMed.
- (a) S. K. Mellerup, C. Li, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 6093 CrossRef CAS PubMed; (b) C. Li, S. K. Mellerup, X. Wang and S. Wang, Organometallics, 2018, 37, 3360 CrossRef CAS; (c) S. K. Mellerup, L. Häfele and A. Lorbach, Org. Lett., 2017, 19, 3851 CrossRef CAS PubMed; (d) S. K. Mellerup, C. Li, J. Radtke, X. Wang, Q.-S. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 9634 CrossRef CAS PubMed.
- S. K. Mellerup, C. Li, X. Wang and S. Wang, J. Org. Chem., 2018, 83, 11970 CrossRef CAS PubMed.
- (a) S. K. Mellerup, G. Yousefalizadeh, S. Wang and K. G. Stamplecoskie, J. Phys. Chem. A, 2018, 122, 9267 CrossRef CAS PubMed; (b) Q.-S. Li and X.-Y. Du, Chem. – Eur. J., 2020, 26, 12891 CrossRef PubMed.
- F.-P. Li, H.-Y. Zhu, Q.-S. Li and Z.-S. Li, Phys. Chem. Chem. Phys., 2019, 21, 8376 RSC.
- M. Shimoi, I. Kevlishvili, T. Watanabe, K. Maeda, S. J. Geib, D. P. Curran, P. Liu and T. Taniguchi, Angew. Chem., Int. Ed., 2020, 59, 903 CrossRef CAS PubMed.
- T. Ito, N. Iwasawa and J. Takaya, Angew. Chem., Int. Ed., 2020, 59, 11913 CrossRef CAS PubMed.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, III, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412 CrossRef CAS PubMed.
- R. Borthakur, K. Saha, S. Kar and S. Ghosh, Coord. Chem. Rev., 2019, 399, 213021 CrossRef CAS.
- H. Braunschweig, R. D. Dewhurst, C. Hörl, A. K. Phukan, F. Pinzner and S. Ullrich, Angew. Chem., Int. Ed., 2014, 53, 3241 CrossRef CAS PubMed.
- H. Braunschweig and C. Hörl, Chem. Commun., 2014, 50, 10983 RSC.
- T. A. Bartholome, J. J. Martinez, A. Kaur, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Organometallics, 2021, 40, 1966 CrossRef CAS.
- For boraalkenes examples, see: (a) H. Klusik and A. Berndt, Angew. Chem., Int. Ed. Engl., 1983, 22, 805 CrossRef; (b) X. Jie, C. Chen, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2023, 62 DOI:10.1002/anie.202214700; (c) S. Helm and H. Nöth, Angew. Chem., Int. Ed. Engl., 1988, 27, 1331 CrossRef; (d) J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer and J. Mies, Angew. Chem., Int. Ed., 2015, 54, 13801 CrossRef PubMed.
- For borataalkenes example, see: (a) M. W. Rathke and R. Kow, J. Am. Chem. Soc., 1972, 94, 6854 CrossRef CAS; (b) C.-W. Chiu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2007, 46, 6878 CrossRef CAS PubMed; (c) K. Watanabe, A. Ueno, X. Tao, K. Škoch, X. Jie, S. Vagin, B. Rieger, C. G. Daniliuc, M. Letzel, G. Kehr and G. Erker, Chem. Sci., 2023, 11, 7349 RSC.
- M. M. Morgan, J. M. Rautiainen, W. E. Piers, H. M. Tuononen and C. Gendy, Dalton Trans., 2018, 47, 734 RSC.
- A. Hermann, M. Arrowsmith, D. E. Trujillo-Gonzalez, J. O. C. Jiménez-Halla, A. Vargas and H. Braunschweig, J. Am. Chem. Soc., 2020, 142, 5562 CrossRef CAS PubMed.
- S. Khoo, Y.-L. Shan, M.-C. Yang, Y. Li, M.-D. Su and C.-W. So, Inorg. Chem., 2018, 57, 5879 CrossRef CAS PubMed.
- W. Dai, S. J. Geib and D. P. Curran, J. Am. Chem. Soc., 2019, 141, 3623 CrossRef CAS PubMed.
- J. Lam, B. Guenther, J. Farrell, P. Eisenberger, B. Bestvater, P. D. Newman, R. Melen, C. Crudden and D. W. Stephan, Dalton Trans., 2016, 45, 15303 RSC.
- A. Z. González, J. G. Román, E. Alicea, E. Canales and J. A. Soderquist, J. Am. Chem. Soc., 2009, 1269 CrossRef PubMed.
- R. Boshra, A. Sundararaman, L. N. Zakharov, C. D. Incarvito, A. L. Rheingold and F. Jäkle, Chem. – Eur. J., 2005, 11, 2810 CrossRef CAS PubMed.
- S. Dong, C. G. Daniliuc, G. Kehr and G. Erker, Chem. – Eur. J., 2020, 745 CrossRef CAS PubMed.
- B. Gawdzik, A. Wzorek, J. Mattay and W. Iwanek, Org. Prep. Proced. Int., 2009, 41, 217 CrossRef CAS.
- (a) H. I. Beltrán, S. J. Alas, R. Santillan and N. Farfán, Can. J. Chem., 2002, 80, 801 CrossRef; (b) H. I. Beltrán, L. S. Zamudio-Rivera, T. Mancilla, R. Santillan and N. Farfán, J. Organomet. Chem., 2002, 657, 194 CrossRef.
- A. Rosendo Rico, M. Tlahuextl, A. Flores-Parra and R. Contreras, J. Organomet. Chem., 1999, 581, 122 CrossRef CAS.
- M. Braun, S. Schlecht, M. Engelmann, W. Frank and S. Grimme, Eur. J. Org. Chem., 2008, 5221 CrossRef CAS.
- S. Schlecht, W. Frank and M. Braun, Eur. J. Org. Chem., 2010, 3721 CrossRef CAS.
- P. F. Kaiser, J. M. White and C. A. Hutton, J. Am. Chem. Soc., 2008, 130, 16450 CrossRef CAS PubMed.
- A. Harriman, J. G. Knight, P. G. Waddell, N. Algoazy, J. K. G. Karlsson, H. H. T. Al-Sharif, W. Herrebout and R. Aerts, Chem. – Eur. J., 2020, 5246 Search PubMed.
- F. Montalbano, N. R. Candeias, L. F. Veiros, V. André, M. T. Duarte, M. R. Bronze, R. Moreira and P. M. P. Gois, Org. Lett., 2012, 14, 988 CrossRef CAS PubMed.
- F. Montalbano, P. M. S. D. Cal, M. A. B. R. Carvalho, L. M. Gonçalves, S. D. Lucas, R. C. Guedes, L. F. Veiros, R. Moreira and P. M. P. Gois, Org. Biomol. Chem., 2013, 11, 4465 RSC.
- F. Montalbano, J. Leandro, G. D. V. F. Farias, P. R. Lino, R. C. Guedes, J. B. Vicente, P. Leandro and P. M. P. Gois, RSC Adv., 2014, 4, 61022 RSC.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052 RSC.
- T. Flagstad, M. T. Petersen and T. E. Nielsen, Angew. Chem., Int. Ed., 2015, 54, 8395 CrossRef CAS PubMed.
- S. Shimo, K. Takahashi and N. Iwasawa, Chem. – Eur. J., 2019, 25, 3790 CrossRef CAS PubMed.
- C. Aupic, A. Abdou Mohamed, C. Figliola, P. Nava, B. Tuccio, G. Chouraqui, J.-L. Parrain and O. Chuzel, Chem. Sci., 2019, 10, 6524 RSC.
| This journal is © The Royal Society of Chemistry 2023 |