
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycomimetics for the inhibition and modulation of lectins
Steffen
Leusmann
 abc,
Petra
Ménová
d,
Elena
Shanin
ef,
Alexander
Titz
*abc and
Christoph
Rademacher
*ef
abc,
Petra
Ménová
d,
Elena
Shanin
ef,
Alexander
Titz
*abc and
Christoph
Rademacher
*ef
aChemical Biology of Carbohydrates (CBCH), Helmholtz-Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research, 66123 Saarbrücken, Germany. E-mail: alexander.titz@helmholtz-hzi.de
bDepartment of Chemistry, Saarland University, 66123 Saarbrücken, Germany
cDeutsches Zentrum für Infektionsforschung (DZIF), Standort Hannover-Braunschweig, Germany
dUniversity of Chemistry and Technology, Prague, Technická 5, 16628 Prague 6, Czech Republic
eDepartment of Pharmaceutical Sciences, University of Vienna, Josef-Holaubek-Platz 2, 1090 Vienna, Austria. E-mail: christoph.rademacher@univie.ac.at
fDepartment of Microbiology, Immunobiology and Genetics, Max F. Perutz Laboratories, University of Vienna, Biocenter 5, 1030 Vienna, Austria
First published on 26th May 2023
Abstract
Carbohydrates are essential mediators of many processes in health and disease. They regulate self-/non-self- discrimination, are key elements of cellular communication, cancer, infection and inflammation, and determine protein folding, function and life-times. Moreover, they are integral to the cellular envelope for microorganisms and participate in biofilm formation. These diverse functions of carbohydrates are mediated by carbohydrate-binding proteins, lectins, and the more the knowledge about the biology of these proteins is advancing, the more interfering with carbohydrate recognition becomes a viable option for the development of novel therapeutics. In this respect, small molecules mimicking this recognition process become more and more available either as tools for fostering our basic understanding of glycobiology or as therapeutics. In this review, we outline the general design principles of glycomimetic inhibitors (Section 2). This section is then followed by highlighting three approaches to interfere with lectin function, i.e. with carbohydrate-derived glycomimetics (Section 3.1), novel glycomimetic scaffolds (Section 3.2) and allosteric modulators (Section 3.3). We summarize recent advances in design and application of glycomimetics for various classes of lectins of mammalian, viral and bacterial origin. Besides highlighting design principles in general, we showcase defined cases in which glycomimetics have been advanced to clinical trials or marketed. Additionally, emerging applications of glycomimetics for targeted protein degradation and targeted delivery purposes are reviewed in Section 4.
 Steffen Leusmann | Steffen Leusmann studied pharmacy at Saarland University and obtained the licensure to practise pharmacy in 2021. After receiving an MSc in pharmacy from Saarland University in 2022, he started his doctoral studies in the group of Alexander Titz at the Helmholtz Institute for Pharmaceutical Research Saarland. His work focusses on the design of non-carbohydrate glycomimetics of calcium-dependent lectins. |
 Petra Ménová | Petra Ménová received her BSc in drug synthesis and production and MSc in organic chemistry from the University of Chemistry and Technology, Prague. She obtained her PhD (2014) in bioorganic chemistry of nucleic acids jointly at UCT Prague and Czech Academy of Sciences. In 2015 she joined the Peter Seeberger group at the Max Planck Institute of Colloids and Interfaces as Marie Skłodowska-Curie postdoctoral fellow. There, she focussed on the synthesis of semi-synthetic carbohydrate vaccines. She established her own research group at UCT Prague in 2017. Her main interests are medicinal chemistry, carbohydrate chemistry and small-molecule lectin ligands. She was awarded the Alfred Bader Prize for Bioorganic Chemistry in 2020. |
 Elena Shanin | Elena Shanin studied medicine at the Saratov State Medical University (Russia, 2007–2009) and received her BSc (2015) and MSc (2017) in biochemistry from the Free University of Berlin (Germany). She carried out her doctoral research with Christoph Rademacher (2018–2022) at the Max Planck Institute of Colloids and Interfaces in the Department of Biomolecular Systems led by Peter H. Seeberger. The focus of her research was the design of non-carbohydrate glycomimetic inhibitors of bacterial lectins and the application of glycomimetics to modulate the immune cell responses. |
 Alexander Titz | Alexander Titz studied chemistry in Darmstadt, Bordeaux, and wrote his diploma thesis at Novartis, Basel. Following his doctorate in 2008 from the University of Basel, he was a postdoc at ETH Zürich. After research as a fellow of the Zukunftskolleg, University of Konstanz, he became head of the group ‘Chemical Biology of Carbohydrates’ at the Helmholtz Institute for Pharmaceutical Research Saarland and part of the German Centre for Infection Research (DZIF) in 2013. Since 2020, he has also held the Professorship for Organic and Pharmaceutical Chemistry at Saarland University. He received the Klaus-Grohe Prize for medicinal chemistry, an ERC Starting Grant, the Innovation Award in Medicinal/Pharmaceutical Chemistry, as well as the EFMC Prize for a Young Medicinal Chemist. Research interests comprise the development of glycomimetics as antiinfectives. |
 Christoph Rademacher | Christoph Rademacher received his BSc, MSc and doctoral degrees in Molecular Life Sciences from the University of Lübeck, Germany. In 2009–2011 he conducted his postdoctoral training at The Scripps Research Institute in La Jolla, USA and in 2011 became group leader at the Max Planck Institute of Colloids and Interfaces, Potsdam, Germany. There he was awarded Emmy-Noether Research Group Leader and received an ERC Starting Grant. Since 2020 he has been full professor at the University of Vienna and the Max F. Perutz Laboratories and in 2021 became Novartis NIBR Global Scholarship Fellow. His research interests include the development and application of glycomimetics for cell specific targeting. He is co-founder of Cutanos GmbH. |
1. Introduction
Carbohydrates are a class of natural products with wide-ranging functions in nature. As a central energy source they empower life and serve as an integral constituent of bacterial, fungal & plant cell walls and of exoskeletons in insects and crustaceans. Additionally, recognition of carbohydrates plays an important role in a diverse set of intra- and intercellular processes in health and disease.1The main characteristic of carbohydrates, especially oligosaccharides, is their three-dimensional complexity. Monomers vary in ring size and stereochemistry and the synthesis of oligosaccharides leads to linear or branched products as the glycosidic linkage can occur at one or multiple hydroxy groups of a monomer. The coding spatial information of carbohydrates is further complicated by two possible isomers at the glycosidic linkage (anomers) and possible additional modifications such as sulfation, methylation or acetylation. Cumulatively, a staggering structural diversity can be achieved despite only a limited number of monomers being employed.2
All living cells are decorated with a matrix of glycoproteins and glycolipids collectively referred to as the ‘glycocalyx‘, that differs between tissue and cell types in multicellular organisms.3,4 Glycoproteins and glycolipids are conjugation products of carbohydrates and proteins or lipids, which can be further divided based on their linkage. N- and O-glycoproteins are most common, in which the carbohydrate is linked to the asparagine side chain and hydroxy groups of serine/threonine, respectively. On the other hand, phosphate-linked glycans or C-mannosides belong to the rare glycosylation types.5,6 As a further form of glycosylation glycosylphosphatidylinositol (GPI) anchors are used for extracellular presentation of proteins.7,8 A GPI anchor consists of an inositol phospholipid linked to a glucosamine, followed by a trisaccharide and an ethanolamine phosphate, to which the C-terminus of the protein is bound via an amide bond. After transport, the modified protein is presented extracellularly, being anchored to the cell via the phospholipid of the GPI. Apart from the role of carbohydrates as recognition motif, glycosylation of macromolecules is involved in protein folding, stability and activity regulation.9,10
As glycosylation, and therefore composition of the glycocalyx, is a complex, non-templated process sensitive to changes in the metabolic and biosynthetic microenvironment of cells, diseases are frequently associated with an altered glycocalyx.11 While the resulting aberration may aggravate or contribute to the progression of the disease, it also presents an opportunity for diagnosis and therapy at the same time. A prominent example is cancer, in which abnormal expression of glycans, especially O-glycans, serves as prognostic marker and plays a role in tumour growth and metastasis.12–14
The recognition of glycans by carbohydrate-binding proteins, the so-called lectins, is a key mechanism in biology. Due to the abundance of lectins, different classification systems based on ligand specificity, protein structure and localisation have been established.15 In mammals, inter alia calcium-dependent C-type lectins (CTLs), calcium-independent I-type lectins, named based on their homology to the immunoglobulin superfamily, and galectins represent lectin families with significant importance in the context of disease and therapy.
A vital intercellular system that strongly relies on lectin-mediated processes is the immune system. Of significant importance in the innate immune response and early stages of an adaptive immune response is the CTL DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin, CD209).16 DC-SIGN is involved in antigen uptake and presentation in dendritic cells (DCs), as well as regulation of toll-like receptors in a subpopulation of macrophages.17 Another calcium-dependent lectin with similar function of pathogen recognition and antigen presentation is langerin, found in a subset of epidermal DCs in skin Langerhans cells.18 Once an immune response has been triggered, another class of CTLs, the selectins (CD62-E, CD62-L, CD62-P) assist migration of immune cells to the site of infection19 and they are also crucial for inflammatory processes and cancer metastasis.20 Furthermore, the CTLs langerin and the asialoglycoprotein receptor (ASGPR), an uptake receptor for glycoproteins expressed on hepatocytes, have attracted growing interest as targets for drug delivery in recent years.21
Of outstanding interest among the I-type lectins is the family of Siglecs (sialic acid-binding immunoglobulin-type lectins), which mediate regulation of the immune response. Consequently, Siglec-1 (CD169), Siglec-2 (CD22), Siglec-3 (CD33), Siglec-4 (myelin associated glycoprotein/MAG), Siglec-8, and Siglec-15 have been in the focus of glycomimetic drug development.
The family of the galectins differs from the previously mentioned membrane-bound lectins with respect to localization and function. Besides membrane-bound galectins, there are also soluble galectins found in the nucleus, cytosol or extracellular space. Diverse functions such as regulation of the immune system, pre-mRNA splicing, cell signalling, apoptosis, cell adhesion, wound healing as well as cancer progression and metastasis have been reported for members of the galectin family.22 Clinically relevant targets include Gal-1, -3, -8, and -9.
Equally important, the recognition of glycans is also exploited by pathogens for infection. For example, viruses such as HIV,23 hepatitis C,24 SARS-CoV-225 or Ebola26 possess heavily glycosylated capsids and rely on recognition by host lectins, e.g. DC-SIGN, to facilitate host cell entry. The clinical relevance of lectins as drug targets is, however, not limited to mammalian lectins. Bacteria and viruses frequently employ own lectins for adhesion to host cells, determining their host cell tropism.27 Binding to host cell glycans and subsequent cell entry allows the pathogens to evade the immune system while providing the machinery or a nutrient rich environment for replication. Additionally, bacterial lectins are often involved in biofilm formation, a resistance mechanism shielding bacteria against the immune response and antibiotics.28 Consequently, these lectins significantly contribute to the virulence of pathogens and their inhibition with drugs is a promising approach to anti-infective drug research.29 The progress of drug development against the bacterial lectins FimH and FmlH of pathogenic Escherichia coli, LecA and LecB from Pseudomonas aeruginosa, BambL, BC2L-A and BC2L-C of Burkholderia species, as well as recent advances for inhibitors of Influenza A and C hemagglutinins are covered in this review.
Additionally, many bacterial and plant toxins (including ricin, cholera toxin, Shiga toxin as well as tetanus and botulinum neurotoxins) rely on lectin subunits to enter cells, where they exert their detrimental effects.30–32 Therefore, some of these lectin-containing toxins have also served as drug targets, e.g. Shiga toxin.33–36
Targeting lectins is therefore a highly promising, yet underexplored strategy to develop new therapies against a wide array of pathophysiological conditions ranging from autoimmune diseases and cancer to infections and neutralization of toxins. Additional therapeutic value can be gained by exploiting lectins for targeted delivery of imaging agents, drugs or vaccines.
While there is an undoubted importance of carbohydrate–lectin interactions in disease, only a limited number of drugs targeting these processes have been approved so far. One reason for this are the challenges during drug design posed by lectins themselves and their native ligands (Fig. 1). In general, lectins show a low affinity for carbohydrates in a micromolar to millimolar range.37–39 A major setback to high-affinity binding are the often shallow and solvent-exposed carbohydrate-binding sites (CBSs), as well as the hydrophilic natural ligands requiring costly desolvation upon binding. Cabani et al. estimated that desolvation of a single ligand hydroxy group requires 26 kJ mol−1, although carbohydrates may profit from a reduced desolvation penalty of vicinal hydroxy groups due to a shared hydrogen bond network (17 kJ mol−1).40 This cost of free energy is only partially compensated for by the formation of a single new hydrogen bond, which yields approximately 18 kJ mol−1 of free Gibbs energy.41,42 Consequently, each hydroxy group of carbohydrates must engage in more than one H bond with the protein to contribute positively to binding. Considering the high directionality of hydroxy groups and their interactions, as well as the close proximity of several OH groups in carbohydrates, these requirements for high-affinity binding are rarely met. However, the defined steric requirements for favourable interactions of the numerous OH groups also grant selectivity.
 | ||
| Fig. 1 Interactions between lectins and carbohydrates represented by binding of a GalNAc derivative to ASGPR (PDB code 6YAU). Binding of carbohydrates is governed by H bonds (black), calcium ion complexation (yellow) and CH–π-interactions (red arrows). | ||
An additional feature of CBSs is an increased prevalence of aromatic amino acids, especially a 9-fold increased presence of tryptophan.43 Furthermore, it was shown that more electropositive C–H bonds of pyranosides engage more frequently in CH–π interactions. Because the electronic properties of specific CH bonds depend on the general stereochemistry of the monosaccharide, CH–π interactions may also contribute to selectivity of lectins. Nevertheless, the desolvation penalty associated with the high polarity of carbohydrate ligands offsets the gain of binding strength by hydrophobic interactions. High-affinity binding of carbohydrates is further hampered by an entropic penalty originating from a reduction of conformational freedom upon binding. Overall, lectins are generally considered challenging targets with a low druggability index.44
A further problem of targeting carbohydrate-binding proteins is selectivity. As many different lectins recognize the same minimal binding motif, e.g. mannose,44 a poorly designed carbohydrate-based drug may bind not only to the intended target, but also to off-targets. To prevent adverse drug reactions from the off-target effect, generation of specific lectin–ligand interactions is key and special attention should be paid to target selectivity during drug development.
The targeting of lectins is not only demanding because of the low affinity interactions of lectins and native ligands, but also due to the binding kinetics and global pharmacokinetic properties of native ligands. In general, carbohydrate–lectin interactions suffer from slow association (kon) and fast dissociation (koff) kinetics. While optimisation of kon is necessary to improve binding affinity, optimisation of koff should not be ignored. High off rates equal to reduced residence times at the target, contributing to limitation of the effective drug duration and may result in adverse drug effects due to lower selectivity.45 A 2018 study by Fernández-Montalván showed a significant discrepancy in the residence time of drugs under development and FDA-approved drugs with longer residence times for approved drugs, highlighting the importance of koff for drug development.46 Additionally, the unfavourable pharmacokinetic properties of native carbohydrates are a major obstacle for carbohydrate-based drugs.44 In accordance with Lipinski's ‘rule of 5′,47 they often lack oral bioavailability by passive diffusion across membranes due to their polarity and active uptake in the small intestine is not guaranteed. Once systemic availability is achieved via parenteral routes of administration, degradation by glycosidases or fast renal elimination can severely limit the circulation half-life of native carbohydrates.44
In Nature, multivalency is frequently used to overcome the challenges of low affinity.48–50 Spatial clustering of receptors or expression of oligomeric lectins allows the simultaneous binding of multivalent ligands. In particular, the apparent binding affinity to each single binding site is increased via statistical rebinding or chelate effects.51 A striking example for the benefit of multivalency is represented by the human lectin DC-SIGN. While mannose as one of its monomeric ligands binds monomeric DC-SIGN in the millimolar range (KD = 3.5 mM),52 multivalent binding of the viral glycoprotein gp120 of HIV to DC-SIGN-expressing cells occurs with nanomolar affinity.53 Binding of oligo- and polysaccharides may also profit from interactions with sites adjacent to the carbohydrate-recognition domain (CRD), leading to improved affinity and specificity, as demonstrated by a 130-fold affinity increase of the oligosaccharide Man9GlcNAc2 for DC-SIGN compared to monomeric mannose.54
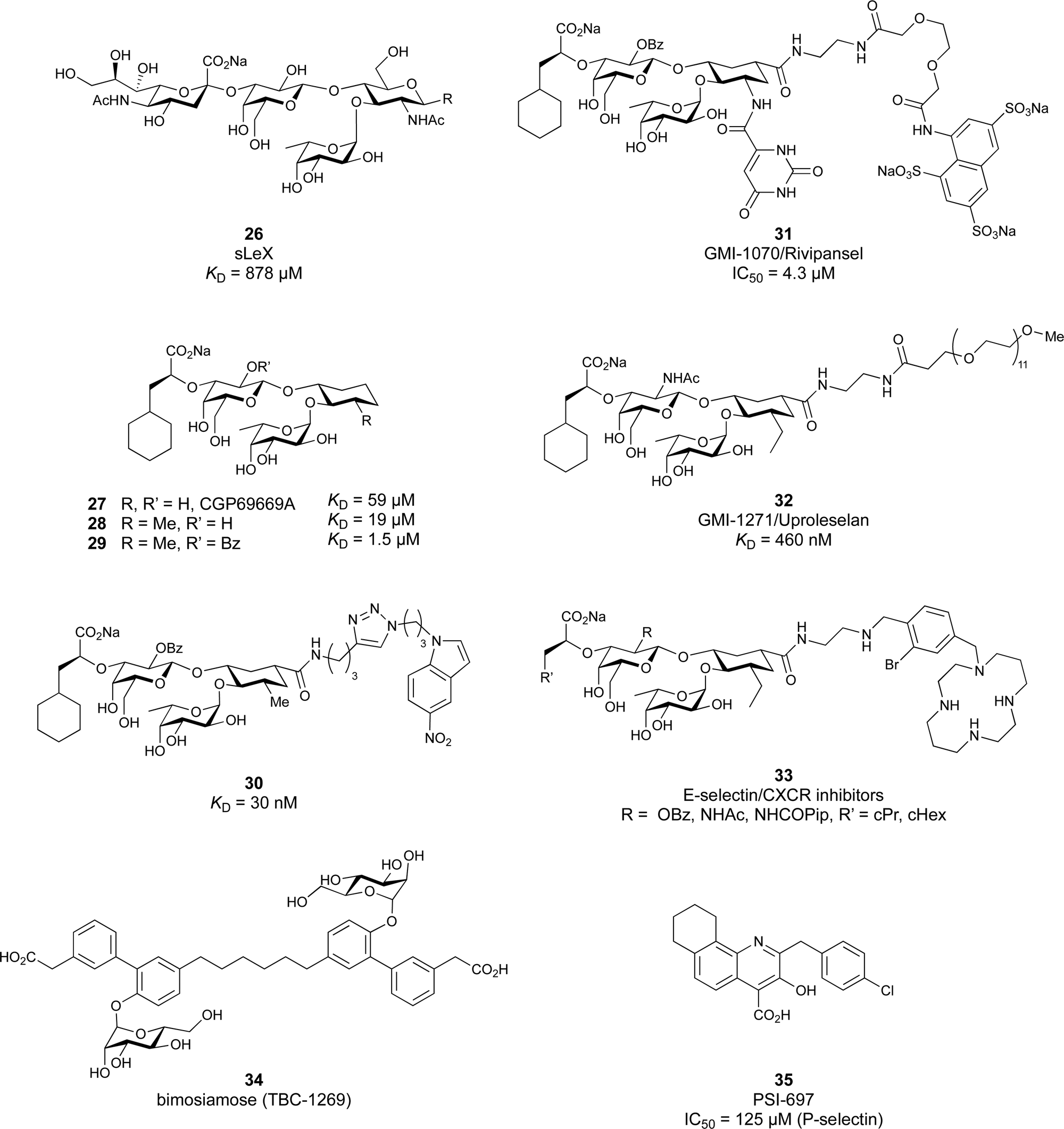
Despite the mentioned drawbacks of native carbohydrates as pharmaceutical agents, a growing number of carbohydrate-based drugs is receiving approval.55Fig. 2 shows selected examples of carbohydrate-based drugs in clinical use. On the one hand, carbohydrates are common constituents of natural products. In these, carbohydrates may be encountered as modification of a large aglycon, e.g. in cardiac glycosides such as digoxin (1),56 or represent the major component, for example in aminoglycoside antibiotics, such as tobramycin (2).57 On the other hand, modification of native carbohydrates during drug design allows the development of novel carbohydrate-based drugs. The resulting compounds, referred to as ‘glycomimetics’, generally show improved drug-like properties such as affinity, selectivity or bioavailability. A class of glycomimetics with great importance are nucleoside/nucleotide analogues used in treatment of cancer and viral infections (gemcitabine (3)58 and remdesivir (4)59 as examples). The diagnostic potential of glycomimetics is exemplified by [18F]fluorodeoxyglucose (5) frequently used as probe in positron emission tomography (PET).60 In some glycomimetics, systemic distribution is not required, such as in acarbose61 (6) and zanamivir62 (7) due to a desired local effect. In others, a sufficient therapeutic effect is achieved after parenteral administration due to improved metabolic stability, for which the heparin glycomimetic fondaparinux (8) serves as example.63 However, even oral bioavailability can be achieved for glycomimetics, as proven by oseltamivir (9),64 topiramate (10),65 miglitol (11),66 and the class of gliflozine antidiabetic drugs (e.g., dapagliflozin (12)).67 Although not lectin-binders, these compounds seeing widespread use in therapy nicely illustrate that carbohydrates are suitable starting points for drug development. In this review, we focus on the progress of the development of glycomimetics targeting lectins in the last 20 years.
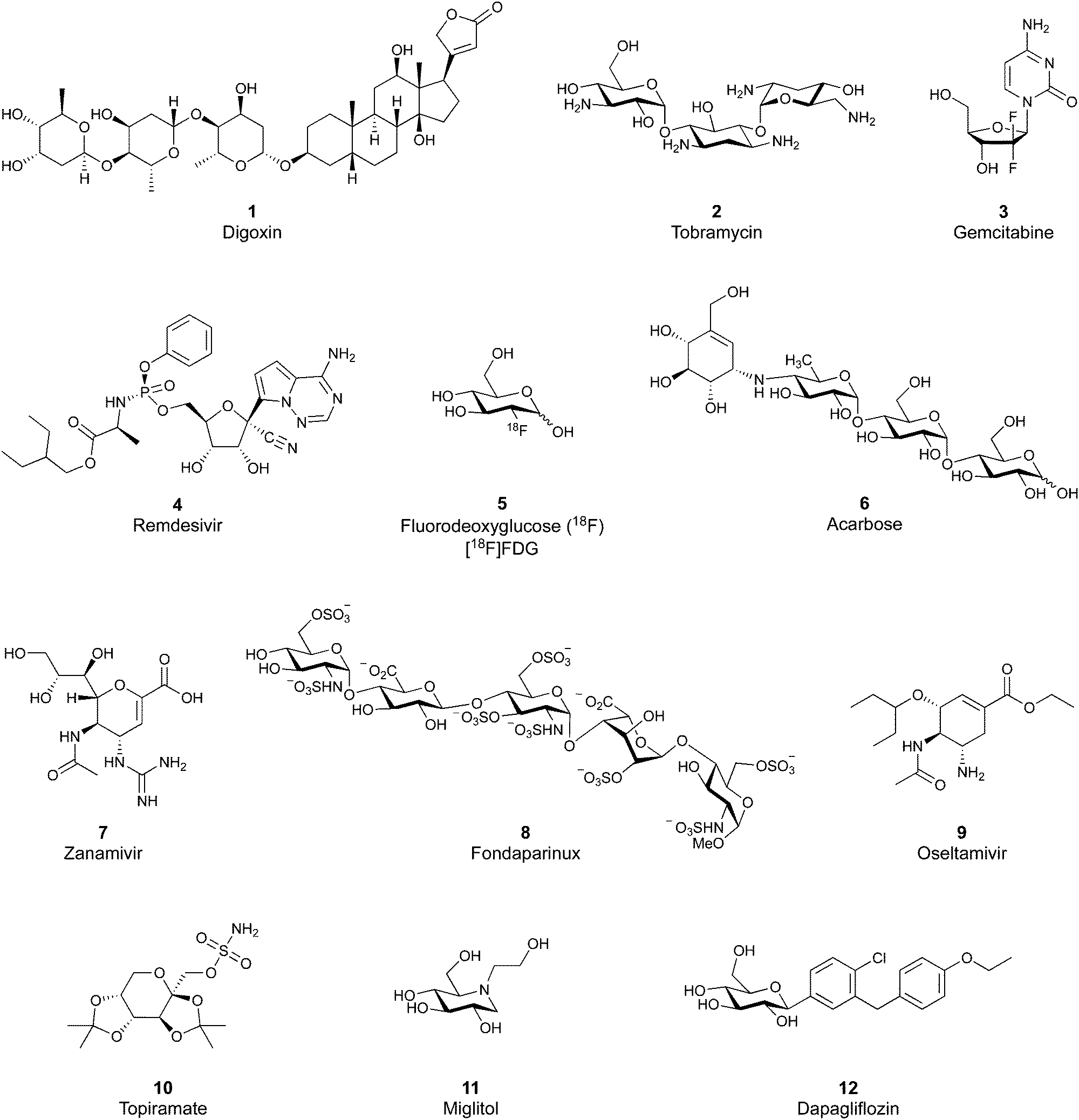 | ||
| Fig. 2 Examples of carbohydrate-based drugs in clinical use. These compounds do not interact with lectins as their primary mode of action. | ||
2. Design principles for glycomimetics
Glycomimetics are structural and functional mimics of carbohydrates that can replace native carbohydrates in their interactions with target proteins. They are designed to show enhanced chemical and enzymatic stability, improved drug-like properties (bioavailability) and the same or possibly better affinity and selectivity for the target. The most efficient lectin antagonists reported to date typically contain a natural carbohydrate or carbohydrate-like scaffold, which serves as an anchor to direct the ligand to the lectin CRD. One common modification of the carbohydrate scaffold is deoxygenation, in which the oxygen atom or hydroxy groups not essential for binding are removed or replaced by a different atom or group. These modifications often lead to changes in polarity, stability, conformation, ring flexibility and hydrogen bond patterns. Importantly, deoxygenation leads to a reduction of polar surface area, which may in turn enhance binding affinity by generation of new hydrophobic interactions with the protein and reduction of the enthalpic cost of ligand desolvation. The carbohydrate or carbohydrate-like scaffold is very often further decorated with other non-carbohydrate moieties that contribute to additional interactions with the target, thus enhancing the ligand's binding affinity and specificity for the target lectin. Furthermore, these additional scaffolds also decrease ligand polarity, and thus improve its drug-like properties. Another concept frequently used in the design of glycomimetics is conformational preorganization of a molecule, which has been shown to significantly reduce entropic penalty associated with ligand binding.68,69In this section, we will classify glycomimetics according to their structural features and briefly mention their general characteristics. Some approaches to the design and synthesis of glycomimetics have been reviewed recently by Bernardi,70,71 Janetka,72 Hevey,73,74 Vidal,75 and others. While these reviews focus on selected lectin targets only, in the following sections, we aim at providing a comprehensive overview of the design and application of glycomimetics for most known clinically relevant lectin targets.
2.1 Modification of the O-glycosidic linkage
Since native O-glycosides are prone to chemical and enzymatic hydrolysis in vivo, a commonly used strategy to improve pharmacokinetic properties such as bioavailability and serum half-life is replacement of oxygen by an atom, which would form a more stable linkage, such as nitrogen, carbon, sulfur or selenium (Fig. 3A). Acylated N-glycosides are extensively used in the glycosylation of peptides, both natural and synthetic.76–78 In contrast, N-glycosidic bonds as an N,O-aminal between two carbohydrate units are relatively labile and therefore only scarcely described.79 While C-glycosides80 and C-acylglycosides (reviewed in ref. 70) are hydrolytically stable glycoside mimics, the introduction of the carbon atom leads to a loss of the exo-anomeric effect and can induce undesirable conformational changes. Thioglycosides serve as more stable analogues of O-glycosides owing to the fact that sulfur is less basic than oxygen and thus, an S-glycosidic bond is more resistant towards hydrolysis. Apart from showing various biological activities,81–83 selenoglycosides have been used as tools for the crystallographic investigation of carbohydrate–protein interactions.84–88 In addition, both seleno- and thioglycosides are used as glycosyl donors in the synthesis of oligosaccharides.89–92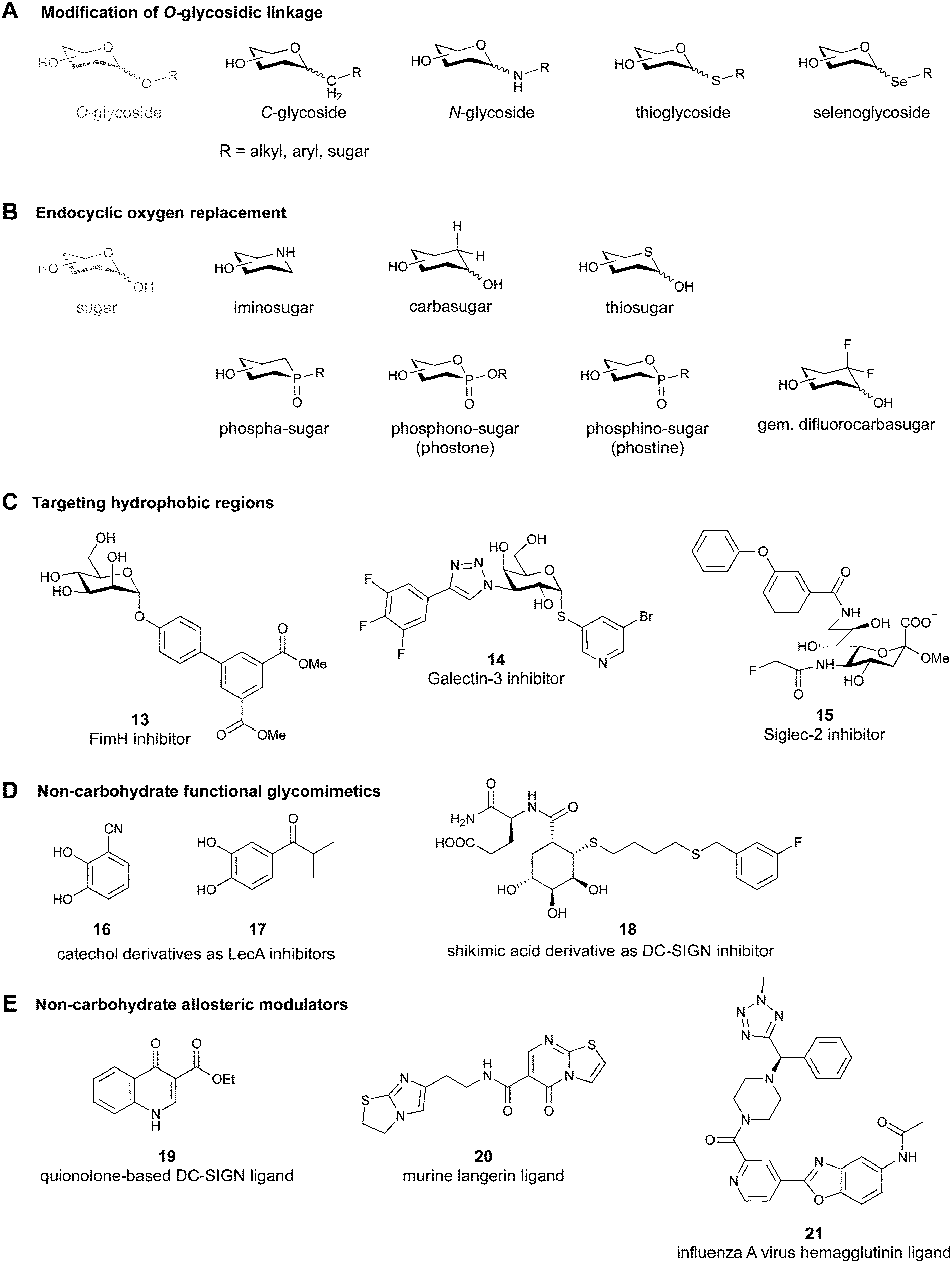 | ||
| Fig. 3 Common modifications of the native carbohydrate structure (A and B) and examples of lectin-binding glycomimetics following the different design strategies (C–E) discussed in Section 2. | ||
2.2 Endocyclic oxygen replacement
Replacement of the endocyclic oxygen atom with nitrogen, carbon, sulfur, and phosphorus leads to imino-, carba-, thio-, and phosphorous-based glycomimetics, respectively (Fig. 3B).Iminosugars are the largest group of monosaccharide mimics reported so far,93,94 they occur in nature and can be found in different plants and microorganisms.95,96 Iminosugars can be divided into two groups: monocyclic (pyrrolidines, piperidines, azepanes) and bicyclic (pyrrolizidines, indolizidines, nortropanes). At physiological pH, the endocyclic nitrogen atom is positively charged; iminosugars can hence mimic the charged oxocarbenium transition state of processing enzymes and have found clinical use as glycosylhydrolase and -transferase inhibitors.97
Carbasugars (cyclitols) lack the typical anomeric reactivity, which leads to their increased metabolic stability towards glycosidases and glycosyltransferases.98,99 Moreover, replacement of the endocyclic oxygen with carbon prevents the anomeric effect and changes the hydrogen-bond pattern, flexibility and conformation of the ring.100 Thiosugars101,102 are more hydrophobic in nature than their oxo-counterparts and can sometimes show enhanced affinity through hydrophobic interactions with the protein.103 Phosphorus-based glycomimetics contain a phosphorus atom in place of the anomeric carbon or in place of the endocyclic oxygen.104,105 Three main classes of such compounds can be distinguished: phospha-, phosphono- (phostones), and phosphino-sugars (phostines, 1,2-oxaphosphinanes). The phosphinolactone group (O–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in phostones and phostines serves as a bioisostere of hemiacetals (O–C–OH) with phosphorus replacing the anomeric carbon. Fluorinated glycomimetics are also attracting ever increasing attention. Recently, it has been shown that replacing the endocyclic oxygen with a CF2 group can imitate the anomeric effect.106,107
O) in phostones and phostines serves as a bioisostere of hemiacetals (O–C–OH) with phosphorus replacing the anomeric carbon. Fluorinated glycomimetics are also attracting ever increasing attention. Recently, it has been shown that replacing the endocyclic oxygen with a CF2 group can imitate the anomeric effect.106,107
2.3 Replacement of OH functional groups
Deoxygenation, i.e. OH to H transformation, leads to a reduction of polar surface area and thus, favours desolvation and may establish new hydrophobic contacts with the protein. Additionally, the removal of an electron-withdrawing group increases electron density of the scaffold and may make other OH groups more nucleophilic, and even strengthen interactions such as metal coordination or hydrogen bonding.108The OH group can be replaced by its bioisosteres, such as F, OCH3, SH, SeH, and NH2. The high electronegativity of fluorine results in a high polarization of the C–F bond. The presence of a fluorine atom can increase lipophilicity,109 decrease pKa values of neighbouring OH groups, and modulate the hydrogen-bond donor/acceptor properties.70 Additionally, fluorine atoms as electronegative substituents destabilize the oxocarbenium transition state, which is present in enzymatic glycosidic bond hydrolysis.74,110 Etherification of an OH group is widely used to assess the binding requirements for ligand interactions and can sometimes even be a requirement for recognition by the lectin.111 Since sulfur and selenium are larger and more polarizable than oxygen, their introduction leads to enhanced lipophilicity, weaker H-bond donor properties and better π-interactions. However, thiol or selenol replacement is rare due to their challenging synthesis and redox instability. Finally, the amino group becomes positively charged at physiological pH, and therefore poorly mimics the neutral OH group.
2.4 Addition of lipophilic fragments
When designing glycomimetics, targeting peripheral regions of the binding site with lipophilic fragments has been often successfully exploited to generate new hydrophobic interactions with the protein surface. Representative examples are FimH antagonists targeting a tyrosine gate (e.g.13),112–114 Siglec-2 inhibitors binding a hydrophobic area through modification at C-9 (e.g.15),115 and Gal-3 inhibitors (e.g.14) interacting with arginine residues in the binding site (Fig. 3C)116,117 and will be discussed in detail below.To this end, biphenyl moieties have become a popular scaffold in many glycomimetics following the anticipation that the biphenyl motif is a replacement for a disaccharide. However, the biphenyl substituent is frequently deeply buried in a hydrophobic cleft (e.g. Siglec-1, -2, DC-SIGN and FimH), being engaged in π-stacking and hydrophobic interactions that increase binding affinity. Nevertheless, in the case of FimH the biphenyl residue indeed occupies a binding site in the tyrosine gate that is normally occupied by mannose residues, although in a slightly different conformation.112,114 The biphenyl residue has also found application in FmlH glycomimetics, where it extends interactions within the binding pocket, thereby increasing potency.118,119 Furthermore, biphenyls have been introduced into MAG antagonists as scaffolds to position two Neu5Ac residues in their bioactive conformation.120
2.5 Other approaches
Non-carbohydrate functional glycomimetics are an interesting alternative to traditional glycomimetics. These are compounds that do not contain a carbohydrate scaffold, but still bind to the carbohydrate binding site and functionally mimic carbohydrates. Representative examples include catechols (e.g.16, 17) and hydroxamic acids as LecA inhibitors,121,122 and shikimic acid derivatives as DC-SIGN inhibitors (e.g.18, Fig. 3D).123Another strategy to circumvent the drawbacks of carbohydrate-based drugs is targeting allosteric sites with higher druggability using a better suited chemical scaffold. Upon binding, these allosteric ligands modify the CBS, thereby affecting carbohydrate recognition. Recent experimental data suggest existence of such druggable secondary sites, especially in mammalian C-type lectins and galectins.124–128 Examples of allosteric inhibitors include 4-quinolones129 as DC-SIGN inhibitors (e.g.19), thiazolopyrimidines (e.g.20)126 as murine langerin inhibitors and pyridinylbenzoxazols (e.g.21) as stabilizers of the prefusion state of hemagglutinin from Influenza A130 (Fig. 3E).
A further strategy frequently used, especially in targeting of bacterial and viral lectins, is multivalency, although mostly terminal native mono- or oligosaccharides are employed. Multivalent glycosides (e.g., glycodendrimers, glycoclusters, glycopolymers, glyconanoparticles) contain multiple sugar moieties and are designed to bind either one lectin at multiple binding sites or several lectins of the same type, promoting receptor clustering or aggregation. While these compounds usually show high affinity,51,131,132 they can also suffer from pharmacokinetic drawbacks due to their large size, high polarity, lack of oral bioavailability, and high likelihood of off-target effects and eliciting an unwanted immune response.133 Consequently, in this review we focus mainly on monovalent glycomimetics and only highlight those multivalent ligands that are of interest for our subjective focus. For details on multivalency, the reader is further referred to other reviews in this issue and elsewhere.51,131,132
3. Inhibitors of lectin function
Lectins are attractive targets for chemical biology and medicinal chemistry and consequently a large number of approaches for the inhibition and modulation of various animal, bacterial and viral lectins have been pursued during the last 40 years. The most common approach is the development of glycomimetics based on the native carbohydrate ligand of the targeted lectin, and the more recent advances of the last two decades will be reviewed in Section 3.1. However, recent research has also led to the discovery of novel glycomimetic scaffolds devoid of a carbohydrate motif as direct binders of the carbohydrate recognition site and these are described in Section 3.2. Finally, allosteric modulators have now also been reported for lectins and their development is summarized in Section 3.3.The various lectins of interest from animal, bacterial and viral sources will be introduced in the following sections when they are first described. Not surprisingly, a number of lectins are addressed by multiple approaches. For example, carbohydrate-derived glycomimetics and novel glycomimetic scaffolds have been developed for bacterial lectins, and C-type lectin receptors proved suitable for allosteric modulators in addition to carbohydrate-derived glycomimetics.
3.1 Progress towards carbohydrate-derived glycomimetic drugs over the last 20 years
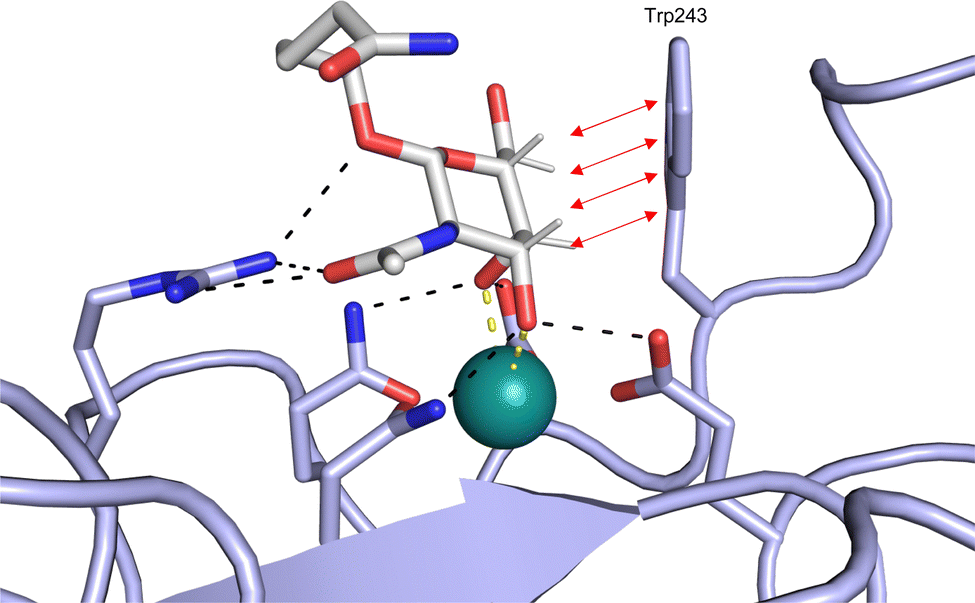
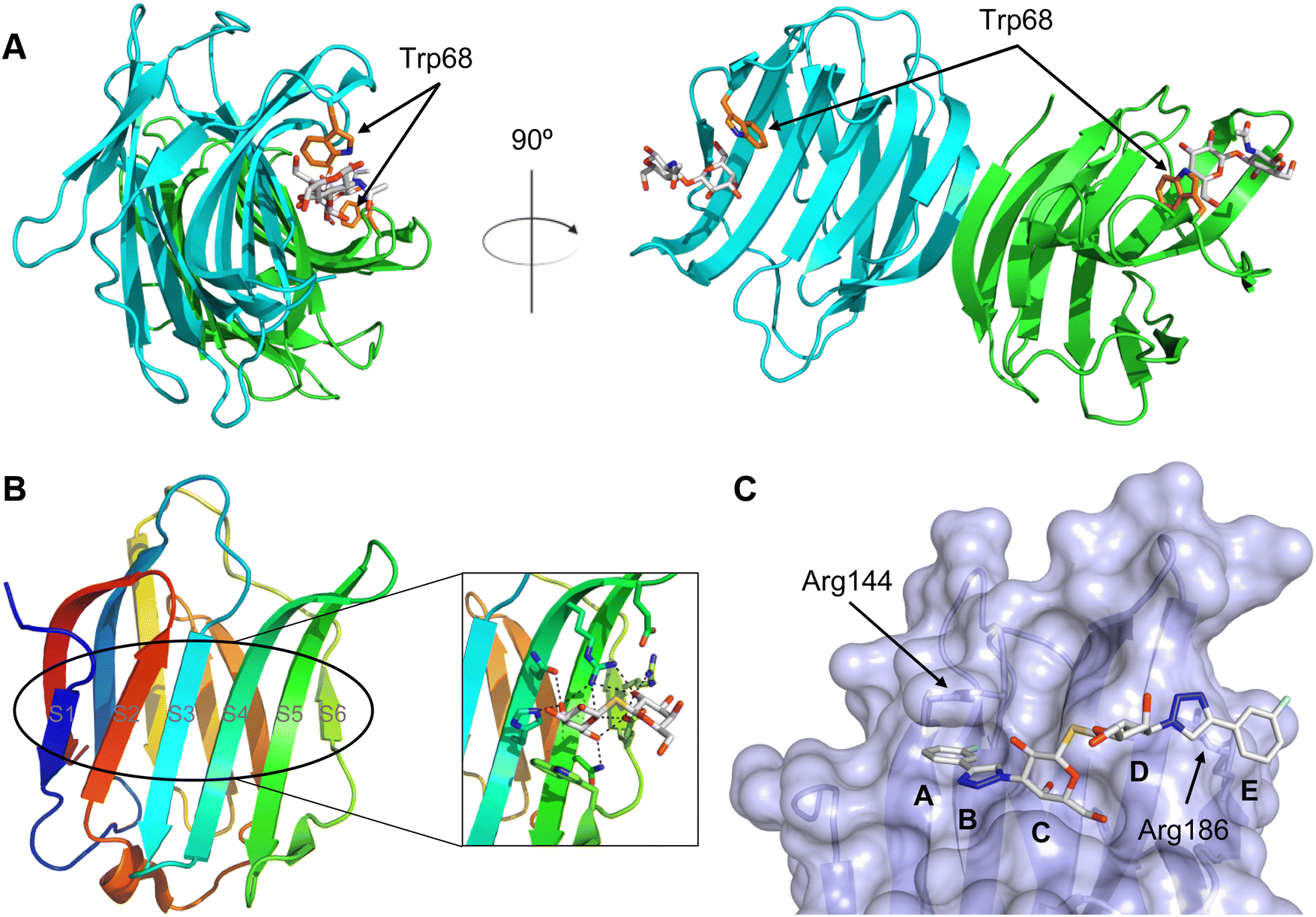
All CTLs share the C-type lectin domain (CTLD),140–143 a looped structure with N- and C-terminal antiparallel β-sheets connected by two flanking α-helices (Fig. 4). The hydrophobic centre of this domain is built by a three-stranded antiparallel β-sheet, stabilized by at least two pairs of highly conserved disulfide bridges. The core harbours a remarkably high number of tryptophans, one of them being part of the conserved ‘WIGL’ motif,140,141 providing stability to the fold.142 The majority of CTLs bind carbohydrates by coordinating their hydroxy groups via a name-giving central Ca2+ ion. The ‘WND’ motif, located in the β-sheet, couples the canonical Ca2+ cage to a hydrophobic core of the CTLD.142 Besides this central Ca2+ cage, the CTLD can host up to three additional Ca2+ ions. Overall, all four Ca2+ sites are located in the upper loop and are referred to as Ca2+ sites 1 to 4, with Ca2+-2 site being located in the carbohydrate site. Taken together, these stabilizing elements generate a high hydrophobicity of the core, while the disulfide bridges and the Ca2+ cages provide remarkable stability to the CTLD. This leads to a high tolerance for a sequence variation, a necessity for immune cell receptors coevolving with pathogens.144
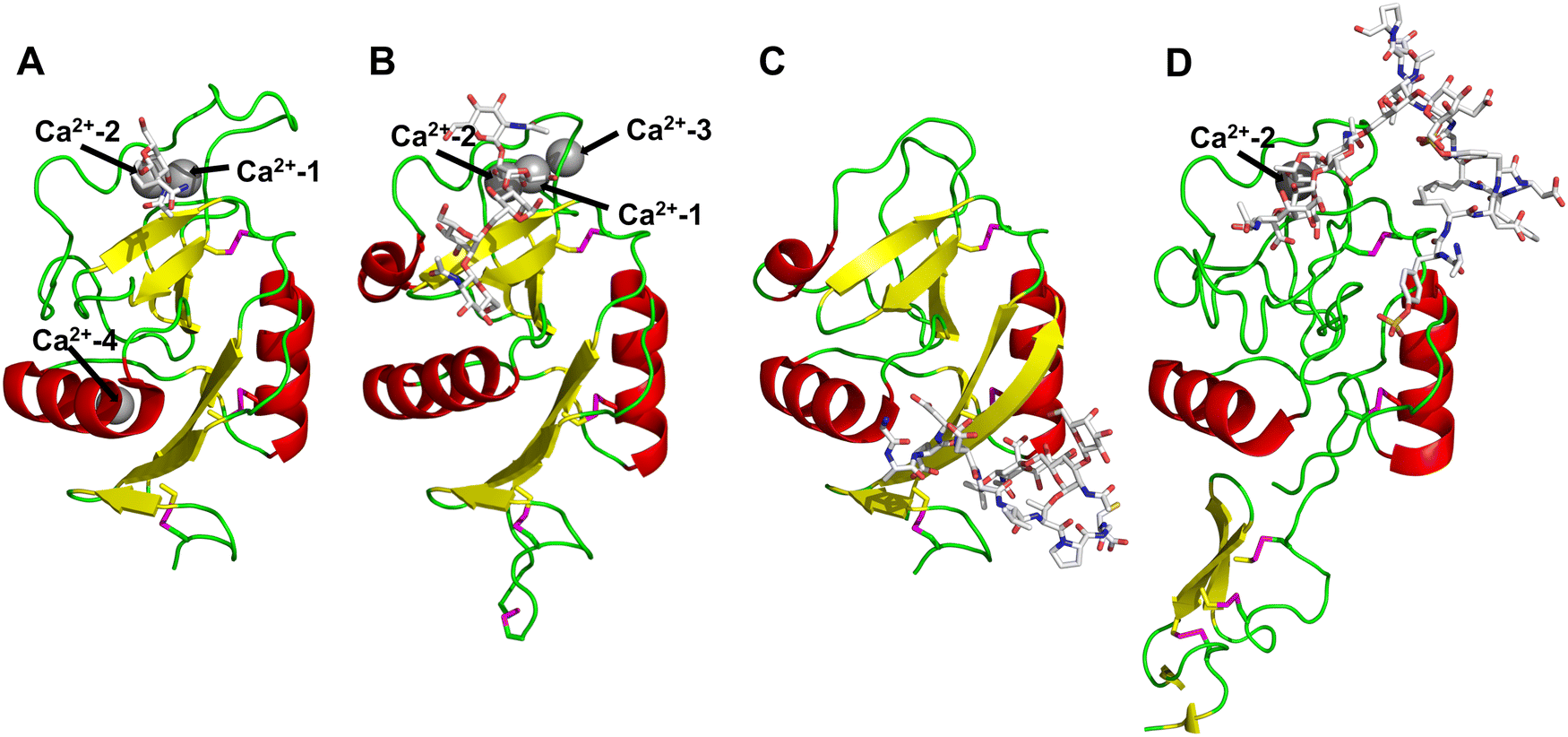 | ||
| Fig. 4 C-type lectin domain fold. Calcium ions are shown as grey spheres and named according to Zelensky and Gready.142 (A) ASGPR in complex with GalNAc derivative 22 (PDB code 6YAU, see Fig. 43) showing only the resolved part of the trivalent ligand. (B) A complex of DC-SIGN/GlcNAc2Man3 highlights the extended secondary site for larger oligosaccharides. Additional contacts beyond the Ca2+ coordination play an important role for oligosaccharide specificity and affinity (PDB code 1K9I). (C) CLEC-2 in complex with a sialylated glycopeptide revealing a non-canonical carbohydrate recognition site (PDB code 3WSR). (D) Complex of PSGL-1 peptide and P-selectin showing the extended binding site (PDB code 1G1S). Disulfide bridges are highlighted in magenta. | ||
CTLs recognize their carbohydrate ligands by coordinating two vicinal hydroxy groups of a central monosaccharide motif (Fig. 5). The canonical Ca2+-2 site is either embedded in an EPN or QPD amino acid sequence motif, dictating mannose/fucose/glucose or galactose specificity, respectively.134,145 This motif is located in the evolutionary and structurally variable long loop. The two hydroxy groups of the carbohydrate ligand can be coordinated by the Ca2+ in two orientations of the core scaffold being 180° flipped. However, the monosaccharide recognition alone provides neither sufficient affinity nor specificity in a biological context. Therefore, oligosaccharide specificity arises from secondary sites close to the canonical carbohydrate binding site. These sites allow for the recognition of extended oligosaccharide structures,146 a principle found in many lectins that originate from convergent evolution.147 For efficient design of glycomimetics, this important feature was used during the design of selectin antagonists. This circumvented the low carbohydrate affinities coming from high desolvation costs of the hydrophilic recognition site, and entropic costs originating from loop rigidification upon binding.148,149
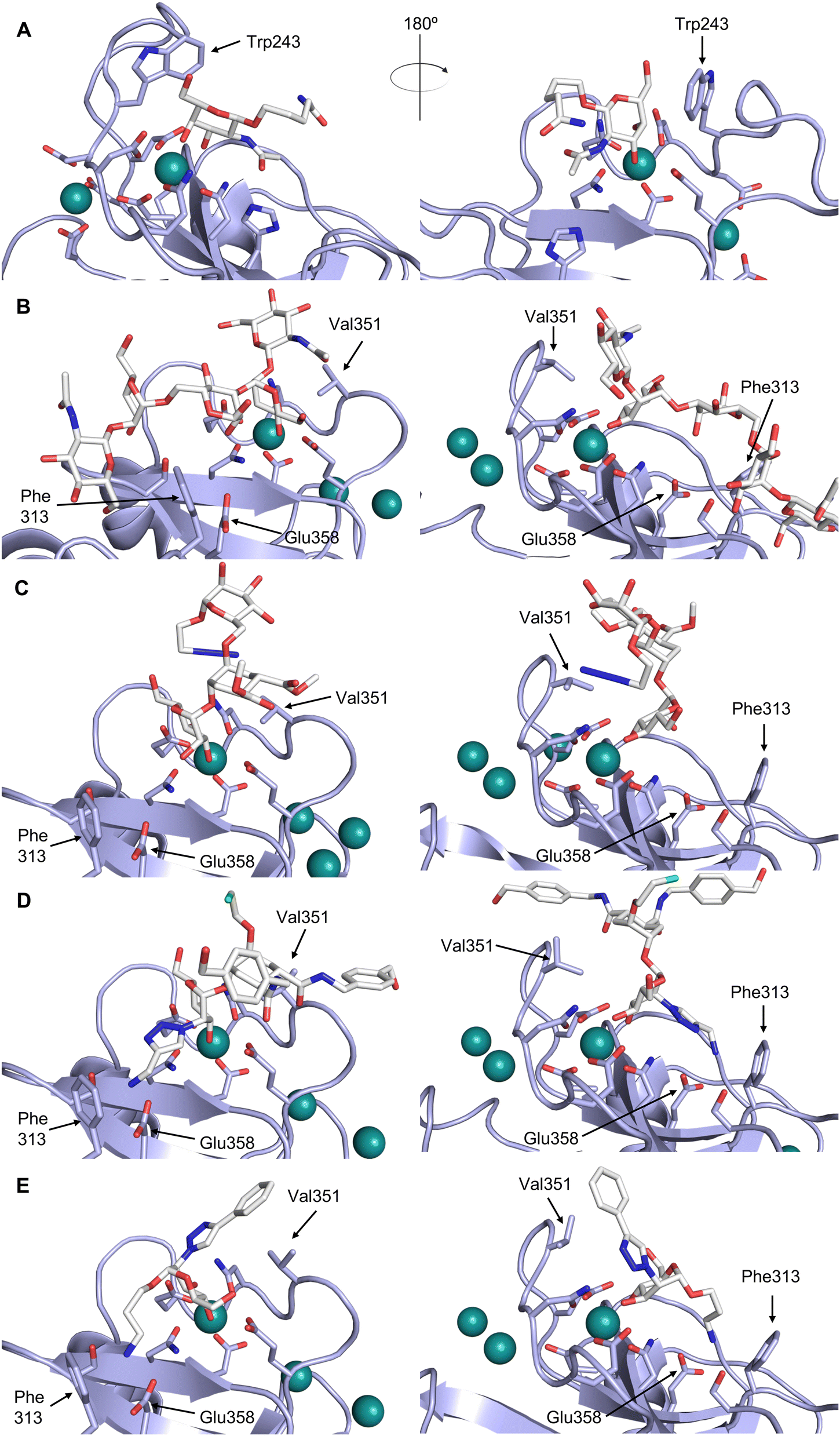 | ||
| Fig. 5 C-type lectin canonical carbohydrate binding site. (A) ASGPR in complex with GalNAc derivative 22 emphasizes the affinity gain by CH–π interaction with Trp243 (PDB code 6YAU). (B–E) DC-SIGN in complex with (B) GlcNAc2Man3 (PDB code 1K9I), (C) pseudo-trisaccharide 23 (PDB code 2XR6), (D) pseudo disaccharide 24 (PDB code 6GHV), and (E) glycomimetic 25 (PDB code 7NL7). As a common design principle, a significant gain of affinity and specificity is achieved by interactions with Phe313 and Val351 in close proximity to the carbohydrate recognition site. | ||
For endocytic CTLs, the Ca2+ has not only a stabilizing function for the CTLD, but also provides means for ligand release in the endosome since it is also the structural element generating pH sensitivity for release of the cargo.136,150,151 At low pH and active Ca2+ export of the endosomal environment, some CTLs may recycle back to the plasma membrane with a remarkably high speed and efficiency.152,153 This is an important feature of this receptor class making them attractive for targeted delivery approaches using glycomimetics (see Section 4).
To the best of our knowledge, there are only a few exceptions of CTL binding carbohydrates in a non-canonical site, in the absence of Ca2+ coordination: dectin-1 and CLEC-2.154,155 Some CTLs provide secondary carbohydrate sites in addition to their canonical primary site. Examples are SIGN-R1 carrying a binding site for repetitive microbial polysaccharides and dextran sulfate on the opposite side of the CRD156 and langerin recognizing larger glycosaminoglycans in a remote site.157,158 With respect to the development of glycomimetics, these secondary sites might play an important role as they provide opportunity to target sites not amenable to traditional medicinal chemistry.44,159–162
Further mammalian lectin targets related to health and disease include the proinflammatory CLEC9A receptor group163 as well as DEC205 (CD205), a receptor responsible for self-antigen uptake, especially in the context of cell apoptosis.164
3.1.1.1 Selectins. The protein family of CTLs has been focus of very active drug research for many years, spearheaded by early attempts to target the subfamily of the selectins, which has resulted in glycomimetics advancing into Phase III clinical trials.
Selectins are a subfamily of CTLs consisting of P-, E- and L-selectin. They are homing receptors involved in cell adhesion and leukocyte trafficking and are conserved between species. They have first been identified on epithelia, platelets and leukocytes, and thus, named E-, P-, and L-selectin or CD62E, -P, and -L. These proteins are essential for the inflammatory response and mediate the recruitment of leukocytes and lymphocytes into inflamed tissue. Thus, the selectins have been identified as potential drug targets in all diseases with excessive inflammatory response.44 One indication where glycomimetic selectin antagonists are under development is sickle cell disease, with anti-selectin antibodies already in clinical use.165 Furthermore, the selectins also mediate cancer metastasis opening new anti-cancer treatment options.166
These type I transmembrane proteins carry a CRD directly connected to an epidermal growth factor (EGF)-like domain. The minimal binding motif of E-, P- and L-selectin is the tetrasaccharide sialyl LewisX (sLeX, 26, Fig. 6) binding to the canonical carbohydrate binding site, while a secondary site in P-selectin binds sulfated tyrosines generating specificity and affinity for PSGL1 (Fig. 4-D), its natural glycoprotein ligand.
 | ||
| Fig. 6 Sialyl LewisX (26) and its glycomimetic analogues. While major modifications or replacement of the sialic acid and GlcNAc motifs of sLeX are viable strategies in the design of selectin antagonists only minor modifications to the fucose and galactose are tolerated. Indicated KDs are for the binding to E-selectin if not stated otherwise. | ||
This is supported by a crystal structure of E-selectin with sialyl LewisX which originated from soaking experiments (Fig. 7A).167 Moreover, a PSGL-1 glycopeptide fragment bound to P-selectin induced an extended conformation suggesting a conformational coupling of the secondary and the primary sites (see Section 3.3.3).167 However, a co-crystal structure of a four-domain fragment comprising the CRD, the EGF and two additional short consensus repeat (SCR) domains in presence of sialyl LewisX shows the extended conformation with conformational stretching of E-selectin (Fig. 7B). This extended conformation is essential for the catch-bond underlying the molecular recognition responsible for leukocyte recruitment.149 Strikingly, in the new ligated structure the long loop changes its conformation, in particular Gln85 is relocated by about 10 Å. Consequently, the low-affinity structure of E-selectin, present in solution, recognizes the carbohydrate.149,167 This recognition allows the transition to a high affinity, extended state, already in the absence of flow conditions, suggesting a two-state model.149
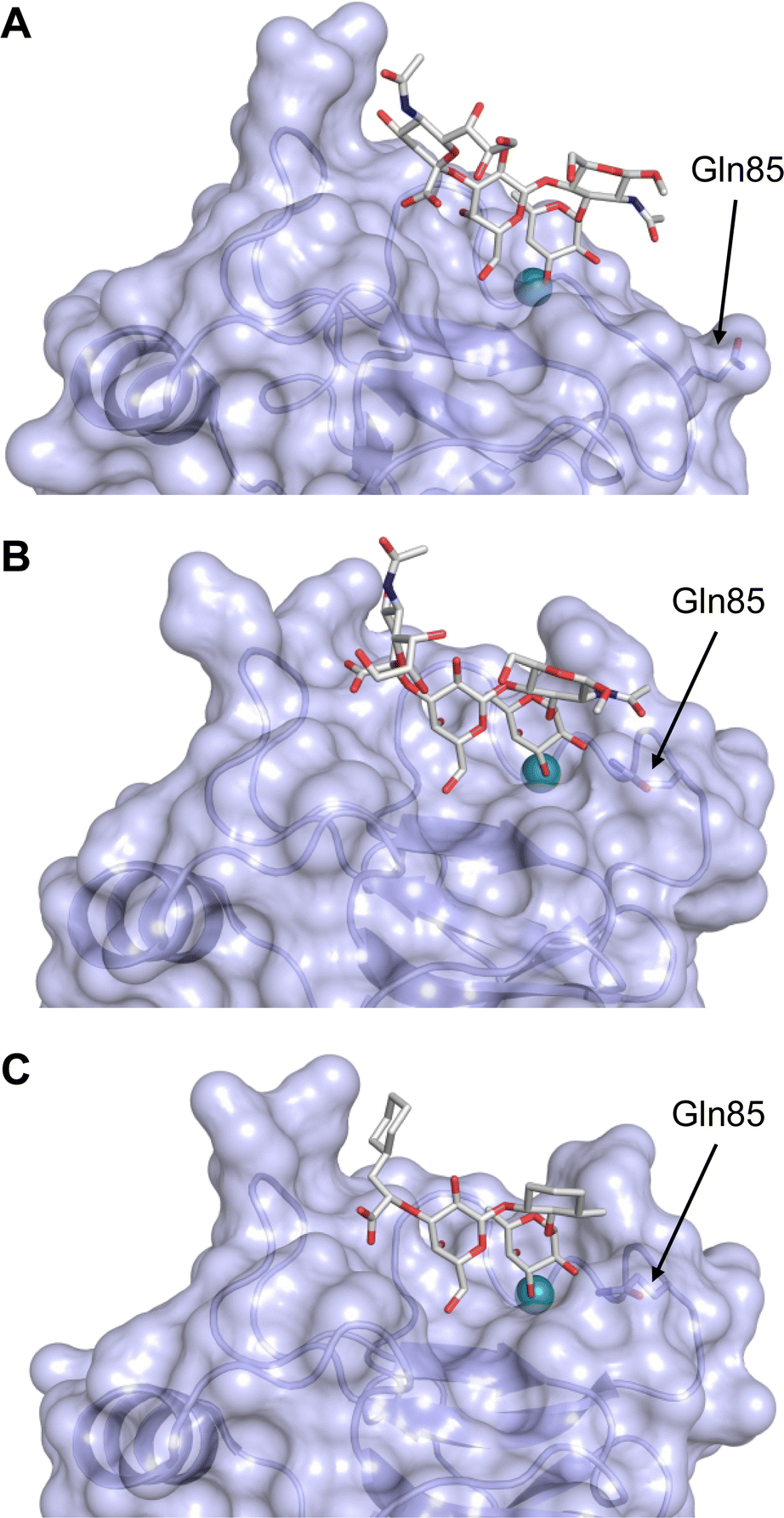 | ||
| Fig. 7 E-selectin in complex with sLeX (A and B) and glycomimetic 28 (C). The structures of the tetrasaccharide sLeX soaked into crystals (A, PDB code 1G1T) or cocrystallised with E-selectin (B, PDB code 4CSY) reveal drastic conformational differences of protein and ligand highlighted by relocation of Gln85 shown as stick. Glycomimetic 28 cocrystallised with E-selectin (C, PDB code 4C16). | ||
Because all three proteins share sLeX as minimal binding epitope, this tetrasaccharide motif has served as a lead for the development of new glycomimetics as pan-selectin antagonists (Fig. 6).44 sLeX binds to the selectins’ rather flat carbohydrate binding site through its sialic acid, galactose and fucose residues.149,167 Ernst and co-workers reported that the binding of sLeX to E-selectin is fully entropy-driven and provided the concerted displacement of several protein bound water molecules by the ligand's hydroxy groups as a rationale.168
Development of selectin-inhibiting glycomimetics started in the mid-1990s and has flourished owing to the fact that numerous major pharma companies were active in the field, e.g. Hoechst/Sanofi, Ciba/Novartis and Wyeth.169,170 The essential pharmacophores of sLeX were identified and it became evident that its fucose and galactose residues as well as the carboxylic acid group of sialic acid are essential for selectin binding, while the rest of the sialic acid and the GlcNAc could be replaced.44 The bioactive conformation of this molecule has been determined by NMR spectroscopy171 and it was reasoned that inhibitors must have pre-organized pharmacophores in order to be effectively bound by the selectins under the prevailing flow in blood vessels.68
In the early lead molecule CGP69669A (27), (S)-cyclohexyllactic acid was chosen as a sialic acid substitute and the GlcNAc residue was replaced with cyclohexanediol.68 This molecule was over 10-fold more active than sLeX and showed efficacy on disrupting leukocyte rolling in vivo.172 The introduction of additional substituents on the cyclohexanediol adjacent to the fucose moiety further improved activity, e.g. in compound 28, which was assigned to an increased steric compression of the molecule leading to lower entropy costs upon binding.69,173 The crystal structure of 28 in complex with E-selectin has recently been solved and demonstrates the interactions of the cyclohexyllactic acid with the protein (Fig. 7C). By keeping the 2-hydroxy group of the galactose residue benzoylated, affinity was further increased 10-fold, resulting in the single digit micromolar selectin antagonist 29. Finally, a carboxylate substituent was introduced at the cyclohexanediol,173 which allowed the addition of second site binders resulting in the 30 nM E-selectin inhibitor 30.159 This development finally led to the synthesis of GMI-1070 (31, Rivipansel), which possesses an orotamide substituent adjacent to the fucose and a distant naphthalene trisulfonate.174 The latter pharmacophore is necessary to target the additional anion recognition site in P-selectin (Fig. 4D), which efficiently binds to its native ligand PSGL-1 through the sLeX carbohydrate epitope and an additional sulfate present in PSGL-1. GMI-1070 was moved to clinical development for the treatment of vaso-occlusive crises in sickle cell anaemia, but failed at the latest stage, in Phase III clinical trials for lack of efficacy to meet its primary endpoint,175 although beneficial tendencies were observed according to Magnani.176
Recently, Ernst and co-workers addressed the pharmacokinetic properties of 27 and 28 in a prodrug approach aiming at oral bioavailability of the active substance.177 To this end, several linear, branched and cyclic alkyl esters have been synthesized and evaluated. This modification indeed led to an increase in permeability through artificial membranes and across Caco-2 cells in vitro, but oral bioavailability could still not be achieved in mice. On the down side, the authors also noted that the promising cyclohexylmethyl ester of 28 was rather quickly metabolized by liver microsomes and the resulting hydroxylated derivative was a poor substrate for the esterase cleavage. The latter is however essential to liberate the active selectin inhibitor 28.
The company Glycomimetics Inc. has two further sialyl LewisX mimetics in development with distinct traits for specific indications. The first compound GMI-1687 (structure undisclosed) is a 2.4 nM E-selectin binding compound with s.c. bioavailability and efficacy in an animal model of thrombosis at 40 μg kg−1 administered twice daily.178 This trait improves its route of administration from the clinics to also outside the hospital and should help patients at an earlier stage of sickle cell disease. The compound will enter first in human trials in 2022.179
The second drug candidate is Uproleselan (32, GMI-1271), a selective E-selectin antagonist (IC50 = 2.4 μM) devoid of binding to P-selectin up to 10 mM and 5 mM binding to L-selectin. This compound is designed to address E-selectin function in cancer.180,181 The structure is also derived from CGP69669A with the 2-O-benzoylated galactose residue now replaced by a GalNAc and the cyclohexanediol residue further conformationally stabilized by an equatorial ethyl group; furthermore, an 11 mer-PEG chain is attached to the carboxylic acid to improve serum half-life and reduce plasma protein binding.181 Presumably, the lack of P-selectin binding results from loss of the naphthalenetrisulfonate residue that is present in the pan-selectin inhibitor Rivipansel (GMI-1070). Uproleselan was active in a mouse model of acute myeloid leukaemia (AML).182 GMI-1271 is currently in Phase III clinical trials for the treatment of AML (NCT05054543) and Phase I/II trials for COVID-19 pneumonia (NCT05057221).
As an advancement in the field, bifunctional inhibitors of E-selectin and the chemokine receptor CXCR4 have been developed to synergistically counteract osteosarcoma and a patent reports the conjugation of a CGP69669A derivative with cyclam,183 a tetraaza-crown ether that binds to carboxylates within CXCR4.184 GMI-1359 belongs to this class of bifunctional compounds (exact structure undisclosed, general structure 33) and has proven effective in animal models of bone metastasis, pancreatic cancer and AML when co-administered with chemotherapy.185–187 GMI-1359 has been studied in a Phase I clinical trial (NCT04197999) for the treatment of breast cancer and results remain to be disclosed.
Another glycomimetic molecule, bimosiamose (34, TBC-1269) originating from Texas Biotech Corporation, was under development as pan-selectin antagonist. This molecule is a symmetric dimer of a sLeX mimetic where the sialic acid is replaced by an acetic acid linked to a biphenyl, the latter serving as a spacer to a mannose residue that mimics the fucose in sLeX.188 Interestingly, this sLeX mimic showed no effect on leukocyte rolling in vivo suggesting a distinct mechanism of action.189 TBC-1269 was tested under the lead of Revotar in three Phase II clinical trials focusing on human allergen challenge model of asthma,190 reducing inflammation in COPD,191 and on ozone-induced airway inflammation.192 However, bimosiamose's current development status is unknown and Revotar was terminated in 2017.
Furthermore, the naphthalene carboxylate PSI-697 (35) originated from a screening campaign followed by optimisation and has been developed as a P-selectin antagonist.193 This molecule showed inhibition of P-selectin binding to immobilized PSGL-1 by SPR in vitro (IC50 = 125 μM) and inhibited leukocyte rolling in murine blood vessels. Interestingly, the mode of action of PSI-697 with P-selectin is unknown. It remains to be elucidated if this compound that completely lacks any carbohydrate character is a direct competitor to sLeX, acts on the secondary site bound by the sulfated tyrosine, acts as an allosteric inhibitor, or has another mode of action. Further, PSI-697 showed some efficacy in mouse models of atherogenesis and vascular injury193 as well as in thrombosis.194 The molecule was then moved to Phase I clinical trials in inflammatory diseases, and administered per orally in scleritis (NCT00367692) in 2006 and, furthermore, in a 2008 study by Pfizer for platelet aggregation with monocytes in smokers (NCT03860506). In the latter study, the results revealed the inactivity of PSI-697 on the inhibition of platelet aggregation whereas a P-selectin antibody was effective.195 These data urge for the structural determination of the mode of action of PSI-697 with P-selectin and, furthermore, demonstrate the challenges associated with the development of glycomimetics.
3.1.1.2 DC-SIGN. One of the most studied members of the CTL family is DC-SIGN.196,197 This homotetrameric receptor has a broad expression profile and can be found on monocytes, dermal DCs, macrophages and platelets amongst others.198,199 The latter finding renders the in vivo application of high affinity, multivalent DC-SIGN ligands likely risky for the induction of thrombosis. Since its discovery as an uptake receptor for HIV allowing the hijacking of the mucosal DCs for dissemination of the virus and subsequently transinfecting CD4+ T cells, DC-SIGN became a prime target for the development of antagonists.44,200,201 However, a clear experiment for target validation in vivo is missing, because of the absence of a suitable animal model.138 Many pathogens have been found to be recognized by DC-SIGN such as Mycobacterium tuberculosis, Leishmania, Ebola virus, and Candida albicans.26,202–204 Recently, DC-SIGN has also been identified as a co-receptor for SARS-CoV-2, together with a number of other CTLs L-SIGN, LSECtin, ASGPR1, and CLEC10A, recognizing the spike protein.25,205 Lectin engagement with the virus induces proinflammatory responses and this early event during infection is correlated with COVID-19 severity. These findings have again spurred interest limiting the lectin-induced hyperactivation by the application of lectin inhibitors as a potential target for COVID-19 therapy.
Two routes have been followed to generate glycomimetic ligands for DC-SIGN. For targeting the primary carbohydrate binding site, carbohydrate-based ligands were derived starting from a monosaccharide or disaccharide unit. Alternatively, there are several reports on non-carbohydrate ligands, likely targeting secondary sites that may even allosterically modulate the primary carbohydrate site (see Section 3.3).13,24–26
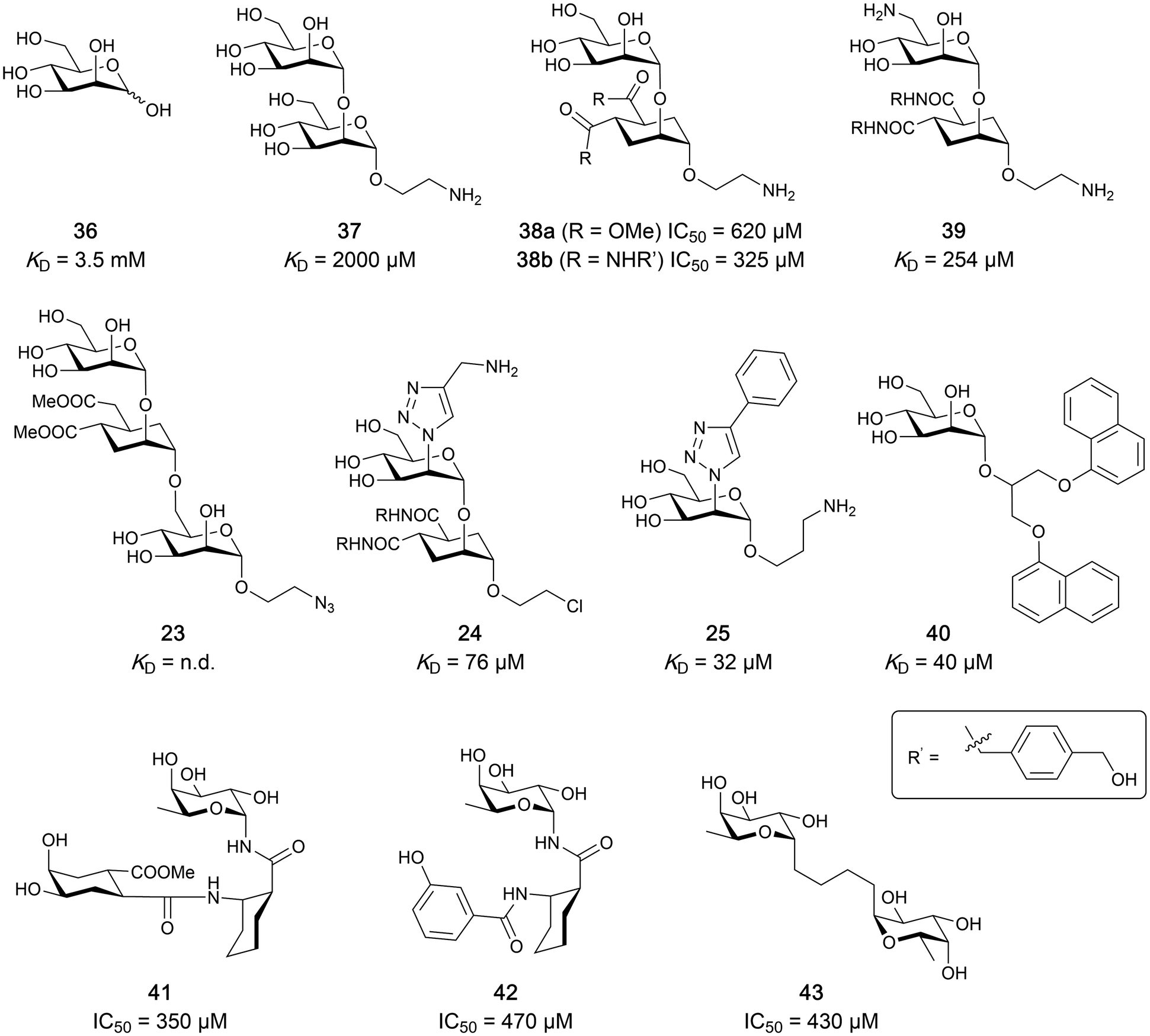
Carbohydrate-derived glycomimetic inhibitors for DC-SIGN have been developed showing affinities in the lower two-digit micromolar range.206–213 The development highlights the challenges and opportunities during the process of designing inhibitors for mammalian lectins due to their shallow and hydrophilic binding sites. Moreover, starting from a promiscuous starting point such as a monosaccharide, keeping potential off-targets in mind is a prime concern. Mannose (36) has low affinity as monosaccharide (KD = 3.5 mM) and fucose is only slightly better.197,214–216
One of the most advanced routes for the development of a carbohydrate-based glycomimetic inhibitor is based on the ethanolamine modified Manα(1,2)Man disaccharide 37, recently nicely summarized elsewhere.138,139 Briefly, disaccharide 37 does not provide the basis for specific recognition by DC-SIGN over other CTLs such as langerin, dectin-2, mannose receptor, and mannose binding lectin. Consequently, additional modifications were introduced to increase specificity (Fig. 5 and 8). The substitution of the reducing end mannose substructure with a cyclohexane in 38a led to improvement of specificity and at the same time increased affinity with positioning the aliphatic ring onto Val351 (Fig. 5C and 8).208,210,217 Next, rational design led to the expansion of the cyclohexane by additional substituents leading in 38b to a two-fold affinity increase, but strikingly to even higher selectivity over langerin, one of the potential off-target receptors (Fig. 8).211,212,218 With additional substituents at the non-reducing end mannose, an additional affinity and selectivity gain was achieved (39, 24).210Fig. 5D shows 24 in complex with DC-SIGN. This was a critical step for biophysical and biochemical data analysis, since additional substitutions at the mannose core have previously been reported to enhance protein aggregation in activity assays and consequently mislead the mode of action analysis of such inhibitors.213 This was accounted for using appropriate controls such as analytical ultracentrifugation to carefully monitor protein aggregation.210
 | ||
| Fig. 8 Carbohydrate-based glycomimetic DC-SIGN inhibitors. The ligands are derivatives of mannose (25, 40), mannobiose (24, 37–39) and mannotriose (23) and fucose (41–43). | ||
In a similar design approach, Ernst and co-workers explored the periphery of mannose with a focused library, also making use of the hydrophobic interactions with Val351, a charge-assisted hydrogen bond to Glu358 and cation–π contact with Phe313, as proposed earlier (Fig. 5E and 8).210,219 Taken together, this led to 25 with an affinity of 32 μM for DC-SIGN and full selectivity over langerin.219 Earlier studies tried to make use of the hydrophobic environment of Phe313 starting from the anomeric position of mannose as a scaffold209 and achieved similar affinities (40, KD = 40 μM). However, clear evidence for the orientation of 40 in the binding site is still missing, in particular taking into account aggregation effects213 and secondary sites.160,162 Along the same lines, in an elegant approach using phage display, primary site binding mannose was used to anchor a secondary site-binding peptide yielding high specificity and a 15-fold increase in affinity over the initial monosaccharide.220 Yet, despite the progress made in the development of carbohydrate-based DC-SIGN inhibitors, only multivalent display of the presented compounds demonstrated inhibitory constants sufficient for the successful application as anti-infectives in preclinical models, such as inhibition of trans-infection of ACE2+ cells by SARS-CoV-2 spike protein-expression vesicular stomatitis virus or SARS-CoV-2 isolates.219,221
It is interesting to note that albeit fucose has a higher affinity for DC-SIGN compared to mannose,216,222,223 it has less often been explored for carbohydrate-based glycomimetic design. Chemical tractability is likely one of the main reasons for this. Building on the LewisX trisaccharide several fucose-based glycomimetics such as 41 and later 42 were developed.216,222 Capitalizing on statistical rebinding, a divalent structure 43 was evaluated as potent DC-SIGN inhibitor among a C-glycoside series of mannose and fucose derivates.223
To this date, a total of 16 mammalian galectins have been identified.227 They are defined by a conserved CRD and a common structural fold.226 Galectins are categorized into three groups according to the organization of their CRD: prototype galectins (Gal-1, -2, -5, -7, -10, -11, -13, -14, -15, -16) contain one CRD and form homodimers in solution via noncovalent interaction through their CRDs; tandem-repeat galectins (Gal-4, -6, -8, -9, -12) comprise two distinct CRDs in their N- and C-termini that are tethered by a linker of variable length (5–50 amino acids), and the chimera galectin Gal-3 has a CRD at the C-terminus and a short non-lectin peptide motif at the N-terminus, rich in proline, glycine, and tyrosine residues through which it can form oligomers.
Galectins play various roles in diverse biological processes, such as cell–cell adhesion,228 cell signalling,229 regulation of apoptosis230,231 or cellular activation and mitosis.75,232 In addition, some galectins are involved in pathological processes, such as inflammation,233–235 tumour progression,236–239 and cancer cell migration.240,241 The ability of galectins to modulate different events in tumorigenesis and metastasis makes them attractive targets for cancer therapy.242 Two Gal-3 inhibitors are currently in clinical trials: Olitigaltin (55) is undergoing a Phase IIb trial for idiopathic pulmonary fibrosis and GB1211 (14) Phase I/IIa trials for non-alcoholic steatohepatitis (NASH). In addition, Gal-1 inhibitor OTX008 (62) completed a Phase I trial as inhibitor of angiogenesis.
A number of crystal structures of various galectins in complexes with natural glycan ligands are known, e.g. complexes of lactose with Gal-1 (PDB code 3M2M), Gal-2 (PDB code 5DG2), Gal-3 (PDB code 3ZSJ), Gal-4N (PDB code 5DUV), Gal-4C (PDB code 4YM3), Gal-8N (PDB code 2YXS), Gal-9N (PDB code 3LSE) and many others. In these structures, the galectin CRD is arranged in a slightly bent β-sandwich. Its concave side comprises six- and the convex side five-stranded antiparallel β-sheets and the carbohydrate is bound to the concave side.243 In most dimeric proteins, such as Gal-1 and -2, the subunits are related by a two-fold rotational axis perpendicular to the plane of the β-sheets. The glycan binding sites are located at opposite ends of the dimer (Fig. 9A).
 | ||
| Fig. 9 Galectin-1 and -3. (A) Gal-1 in complex with LacNAc (PDB code 4XBL).244 The essential tryptophan of the carbohydrate binding site is shown as stick. (B) Gal-3 CRD and in complex with a thiodigalactoside (PDB code 4JC1) (see lit.245). (C) Complex of Gal-3 with TD139 (55, PDB code 5H9P). While the carbohydrate core of TD139 interacts with the subsite C and D as seen for native sugar ligands, the hydrophobic substituents interact favourably with neighbouring subsites, increasing affinity and specificity. | ||
The galectin CRDs can be formally divided into five subsites A–E (Fig. 9C).243 Subsite C is the galactose binding site and the neighbouring subsite D accommodates a further carbohydrate moiety, for example N-acetylglucosamine (GlcNAc) in the case of LacNAc. The binding of a galactose residue in site C is the most conserved feature of galectin binding, whereby six out of eight amino acids interact with the Gal residue via hydrogen bonds. In addition, van der Waals interactions through ring stacking between Gal and a highly conserved Trp residue are key interactions in galectin–ligand recognition and are characteristic for the galectin family.
Because the CRD of mammalian galectins is highly conserved, with only minor variations between members of the family, designing selective glycomimetics is particularly challenging. A common practice is to introduce new moieties into a carbohydrate scaffold to extend the interactions to other sites (B or E), thereby increasing affinity and selectivity (Fig. 9C).246 The interaction of galectins with β-galactosides involves a CH–π stacking of the galactose α-face with a conserved Trp moiety and hydrogen-bonding interactions through its 4 and 6 OH groups. Therefore, positions 1, 2 and 3 are available for further modification.
Considering the numerous functions of galectins, it comes as no surprise that these proteins have become prominent targets in drug discovery. Among the 16 known galectins, Gal-1 and Gal-3 have been studied most.247–249 In recent years, Gal-9 and Gal-8 have also gained attention, since inhibitors of these galectins have the potential for the treatment of cancer and fibrosis.
3.1.2.1 Galectin-3. In the galectin family, the pro-inflammatory Gal-3 is unique in its structure as the only chimeric galectin in humans, which is typically present as a monomer in solution, but assembles into a pentamer in the presence of multivalent ligands, in a process mediated through the N-terminal domain.250 At low concentrations, Gal-3 is present as monomer and inhibits adhesion, but at high concentrations it forms large complexes that promote adhesion. This makes Gal-3 an attractive therapeutic target. In particular, there is a tight correlation between Gal-3 expression levels and various types of fibrosis. Gal-3 is a key mediator of transforming growth factor β (TGF-β), a central mediator of fibrogenesis, through which Gal-3 plays an important role in the fibrosis in numerous organs, such as liver,251 kidney,252 and lungs.253,254 Gal-3 is thus involved in non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), and cirrhosis. Moreover, Gal-3 is overexpressed in many tumours and supports tumour proliferation and metastasis. Since many cancers use Gal-3 to avoid immune recognition, the inhibition of Gal-3 is considered supportive in restoring the immune system's ability to fight cancer.226
With respect to pharmacological intervention, Gal-3 inhibitors can be divided into three categories: peptide-derived inhibitors, carbohydrate-derived multivalent inhibitors and carbohydrate-derived monovalent inhibitors. The first two categories are beyond the scope of this review and have been reviewed elsewhere.247 Here, we only mention representative compounds from both categories and concentrate on the third category, monovalent glycomimetic antagonists.
Peptides and proteins. G3-C12 is a Gal-3 binding peptide with an outstanding affinity (KD = 88 nM).255 Its i.v. administration significantly reduced metastatic cell deposition in several mice models.256 It is also being studied as targeting molecule to deliver peptide–polymer–drug conjugates to cancer cells.257–259
Polysaccharide-based multivalent inhibitors. Some of the earliest Gal-3 inhibitors are based on the structure of pectin, a complex plant polysaccharide rich in anhydrogalacturonic acid, galactose, and arabinose that binds Gal-3 in a multivalent manner.260
GCS-100 (developed by La Jolla) is a modified citrus pectin derivative that was evaluated in several clinical trials.261,262 In 2009, a Phase II safety study in patients suffering from relapsed chronic lymphocytic leukaemia was conducted (NCT00514696).263 A Phase IIb study (NCT02312050) conducted in 2014 in patients with chronic kidney disease caused by diabetes showed significant improvement of the kidney function.264 Although GCS-100 performed well in the studies, its development was discontinued, the main reason being its chemical composition complexity and unknown mechanism of action.
Belapectin (44, GR-MD-02, developed by Galectin Therapeutics) is a 50 kDa galactoarabino-rhamnogalacturonan polysaccharide265,266 and a good inhibitor of Gal-3 and Gal-1 (KD = 2.8 and 8.0 μM, respectively).245 It is currently in clinical trials for the treatment of liver fibrosis and resulting portal hypertension; Phase II was successfully completed in 2017 (NCT02462967)267 and Phase IIb/III is ongoing (NCT04365868). A Phase IIa study using belapectin for the treatment of psoriasis was also conducted (NCT02407041). In parallel, belapectin is currently undergoing two clinical trials for a combination drug with immune checkpoints inhibitors for the treatment of metastatic melanoma: the anti-PD-1 mAb pembrolizumab (Phase Ib study, NCT02575404)268 and the anti-CTLA-4 mAb ipilimumab (Phase I, NCT02117362, was completed in 2018).
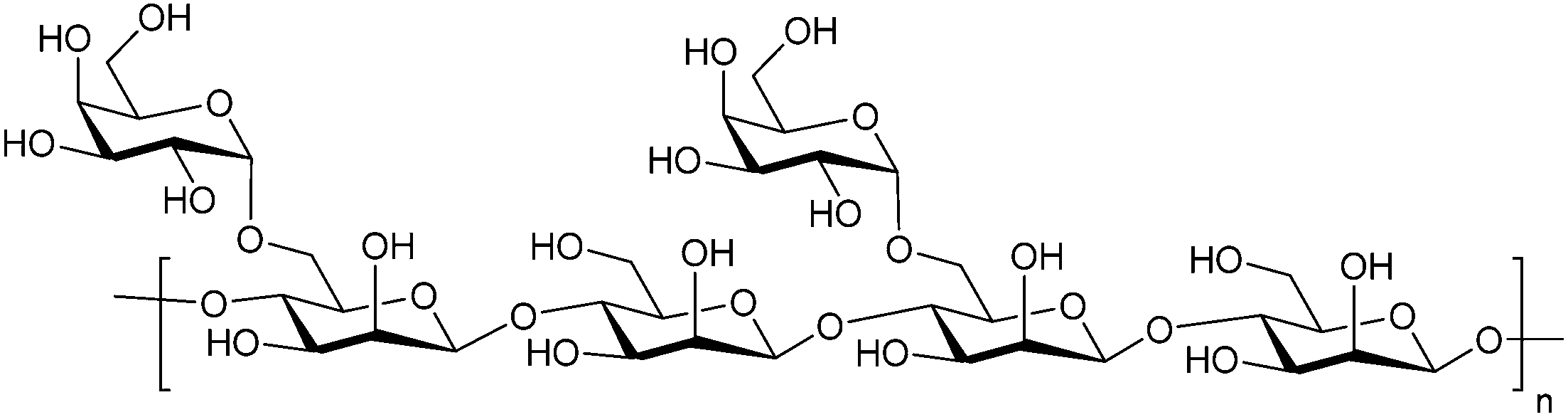
Besides pectin derivatives, β-D-(1,4)-galactomannans have also been studied for galectin inhibition. Davanat (45, GM-CT-01, invented by Pro-Pharmaceuticals, developed by Galectin Therapeutics)269 is a natural galactomannan with an average molecular weight of up to 60 kDa in which the polymannoside backbone is branched with galactose residues. It was developed for co-administration with 5-fluorouracil (5FU) (NCT00110721) and later on with 5FU, leucovorin (folinic acid) and bevacizumab as third- or fourth-line therapy for metastatic colorectal cancer (NCT00388700). Its development was discontinued in 2011 due to financial constraints and a decline in the use of 5FU.
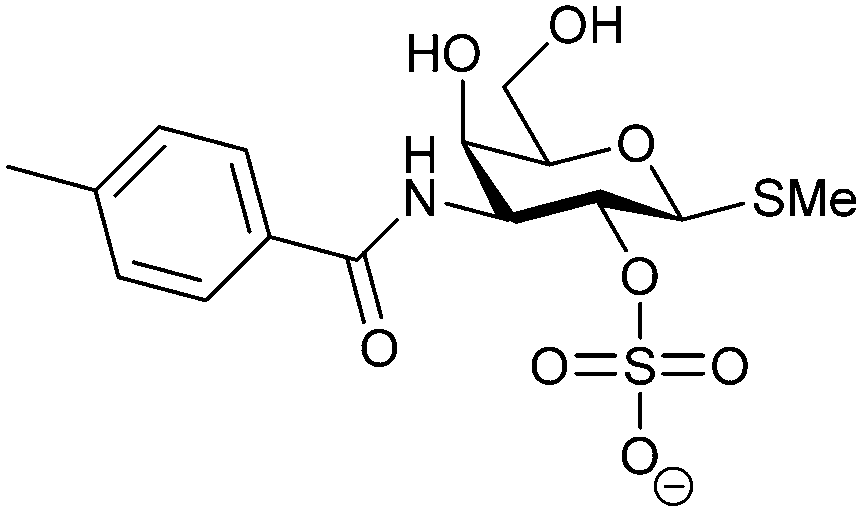
Carbohydrate-derived monovalent inhibitors. In the initial development of carbohydrate-based monovalent Gal-3 inhibitors, chemical modifications were introduced into the structure of natural galectin ligands, such as the disaccharides lactose (Lac) and N-acetyllactosamine (LacNAc). The Nilsson group first introduced LacNAc derivatives bearing aryl carboxamides at C3 of galactose. The best compound from the series, amide 46, had an IC50 of 4.4 μM, rendering this compound 50 times more potent than LacNAc.270 According to the crystal structure of an inhibitor–Gal-3 complex, the increase in binding affinity resulted from interactions between the aromatic benzamido moiety and the Arg144 side chain in the binding site.271 This interaction is entropically favourable by the displacement of a water molecule. Based on this finding, a second generation of LacNAc derivatives carrying aromatic amides at galactose-C3 was synthesized. The most potent compound 47, bearing 3-carboxy-2-naphthamide, showed a KD of 320 nM.
Another strategy developed by the Nilsson group was to modify galactose by the introduction of a benzamido group to position 3 and an anionic substituent to position 2 (H-phosphonate, benzyl phosphate, sulfate), both substituents acting as tweezers for Arg144. These compounds displayed moderate affinities for Gal-3, with sulfate being superior to the other O2 substituents (compound 48, KD = 87 μM).272 In a follow-up study, the same compound was studied for affinity for other galectin receptors (KD = 370 μM for Gal-1, 1000 μM for Gal-7, 900 μM for Gal-8N, and 370 μM for Gal-9N, the N-terminal domain of Gal-9).273 Although this strategy proved successful for target binding, it should be noted that anionic compounds typically do not possess optimal pharmacological properties and the design of neutral prodrugs might be necessary.
Introducing a hydrophobic aromatic moiety to position 2 of LacNAc (e.g. compound 49)274,275 or Lac (e.g. compound 50)276 also led to compounds with low micromolar affinity for Gal-3. Molecular modelling showed beneficial interactions between the aromatic ester/amide moiety and arginine guanidinium groups present in both Gal-1 and Gal-3. The positively charged guanidinium and its poor solvation make it an ideal interaction partner for aromatic systems. Such arene–guanidinium interactions have been intensely studied and it was shown that they can be as strong as cation–anion interactions.277 Interestingly, the modification of galactose C3 with further bulky aromatic groups led to improved selectivity for Gal-3 over Gal-1 (compound 51).275
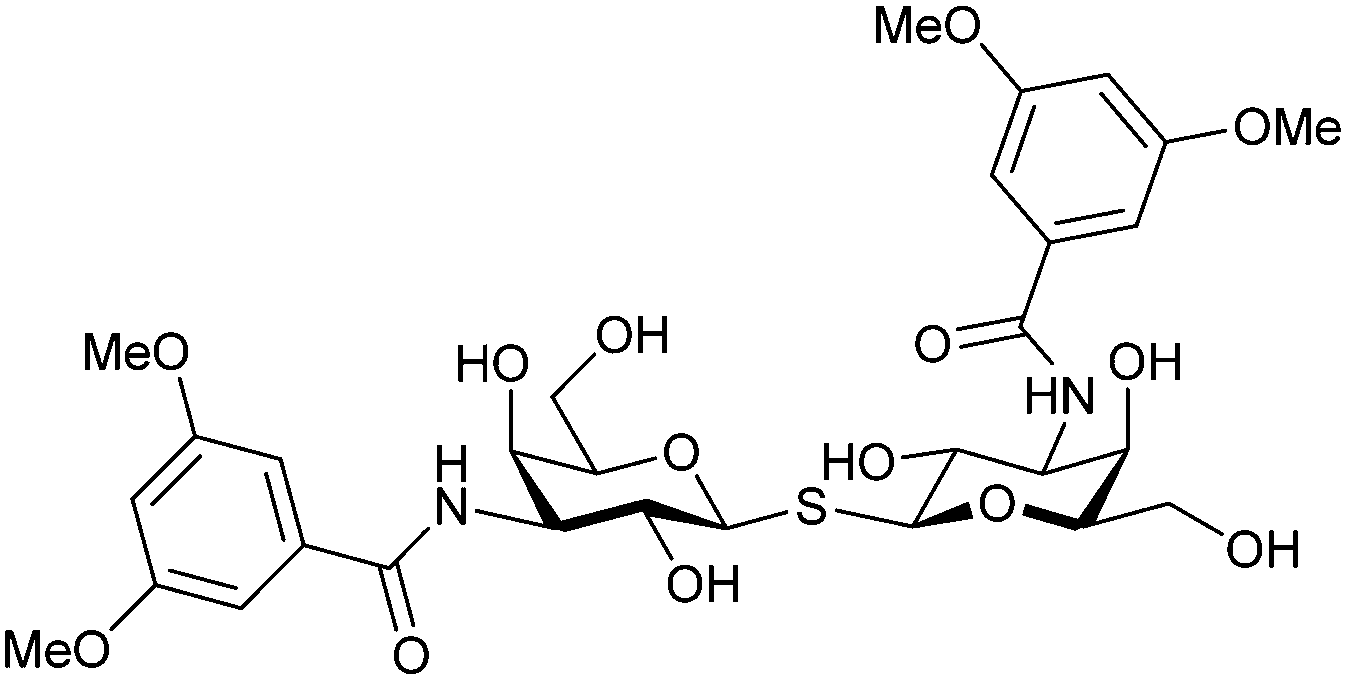
In 2005, the Nilsson group introduced a non-natural, hydrolytically stable thiodigalactoside (TDG) scaffold and its derivatives as some of the most prominent small-molecule inhibitors of Gal-3 (compound 52).278 The compounds are C2-symmetric and possess aromatic amides at C3 of galactose. One of these amides interacts with Arg144 and the other with Arg186, thereby further increasing the affinity for Gal-3 through double arginine–arene interactions (Fig. 9 and 10). Thiodigalactosides bind to subsites C and D, thus mimicking the interaction with lactosides while being hydrolytically more stable. Compound 53 was studied also with other members of the galectin family: while it showed a remarkable affinity for Gal-3 (KD = 46 nM) and medium affinities for Gal-1, Gal-7 and Gal-9N (KD = 4.7, 17, 0.9 μM, respectively), its affinity for Gal-8N was very weak (KD > 1000 μM). This can be explained by the fact that Gal-7 and Gal-9N both contain arginine residues in both relevant positions, Gal-1 contains only one arginine corresponding to Arg186 and Gal-8N only the arginine corresponding to Arg144, while an arginine corresponding to Arg186 is missing.117
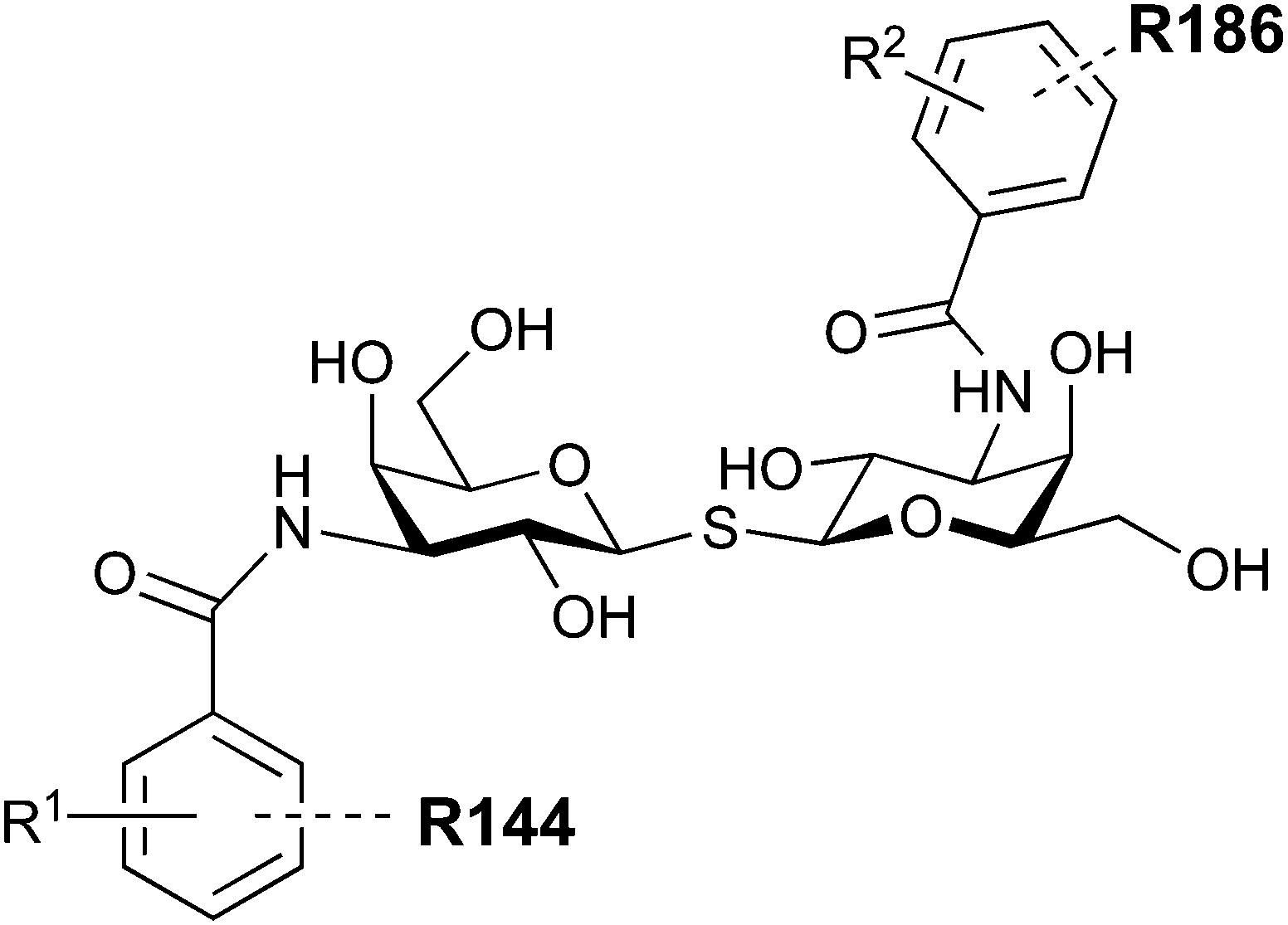 | ||
| Fig. 10 Cation–π-interaction of 3,3′-diamido-thiodigalactoside derivatives with arginine side chains of Gal-3. See also Fig. 9C where the positions of the two arginines, Arg144 and Arg186 are highlighted. | ||
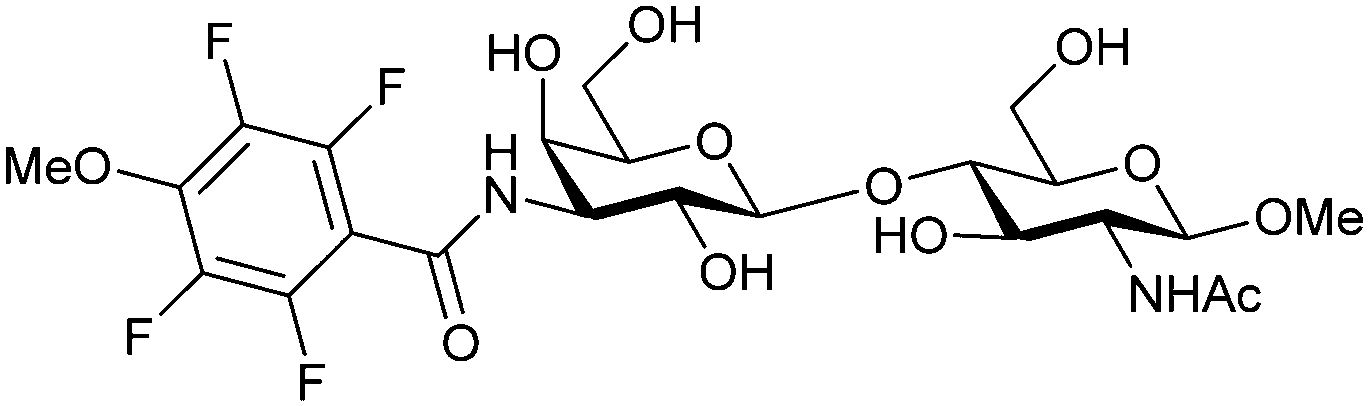
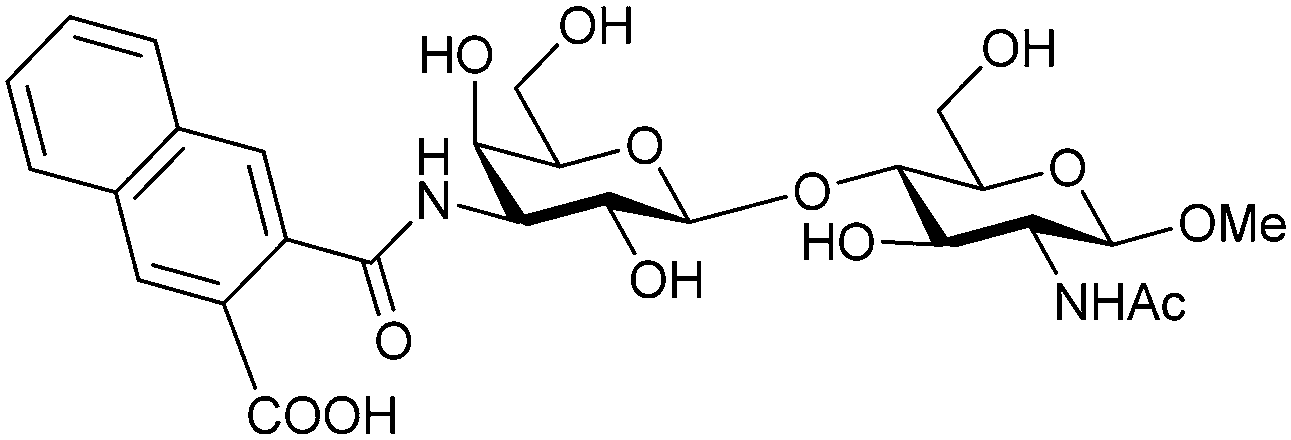
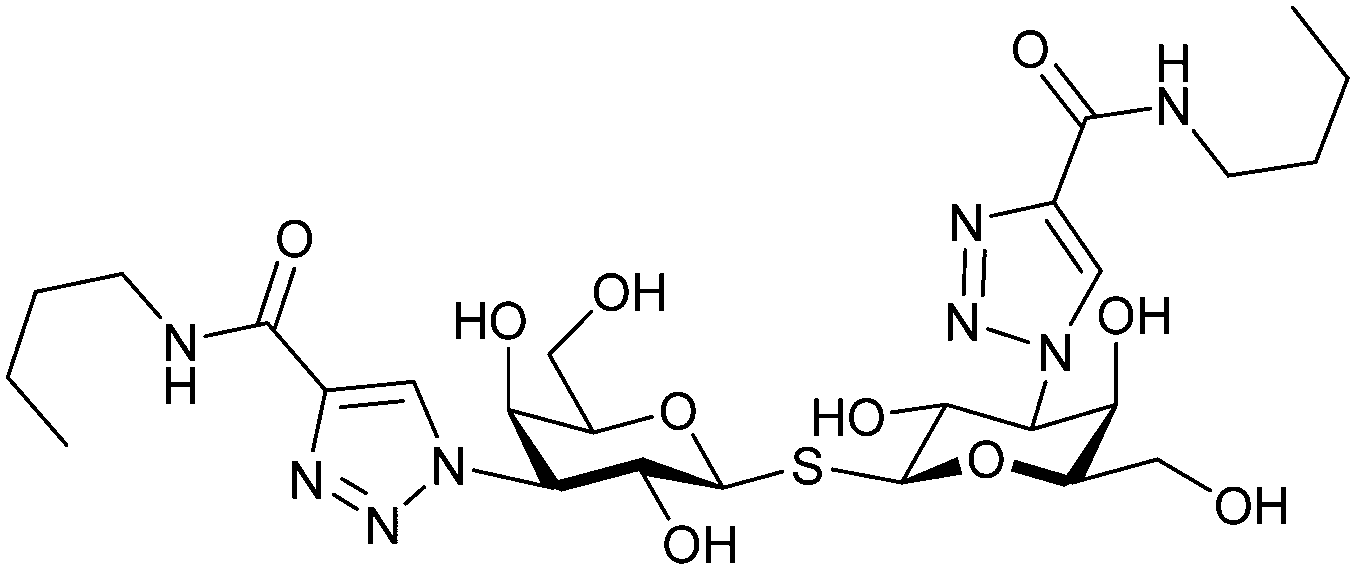
Next, substituents on C3 were introduced through azide–alkyne cycloaddition forming a C3-triazole with affinities comparable to aromatic C3 amides, yet easier to synthesize (e.g. compound 54).279 The compounds also showed high selectivity over other studied galectins (Gal-7, 8N, 9N). Substitution of 4-alkylcarbamoyl groups on the triazol with 4-aryl further improved affinity for Gal-3 (and Gal-1) and selectivity over other galectins (Gal-2, 4N, 4C, 7, 8N, 8C, 9N, 9C). The most potent compound from the series, compound 55, bears a 3-fluorophenyl moiety. X-Ray structural analysis of a complex of compound 55 with Gal-3, as well as a study on Gal-3 mutant, revealed that both the aryltriazolyl moieties and fluoro substituents are involved in key interactions responsible for extraordinary affinity for Gal-1 and -3. Compound 55280 (known initially under the code TD139 and later renamed GB0139 and Olitigaltin) has been studied as a potential drug candidate against idiopathic pulmonary fibrosis (IPF), an irreversible and ultimately fatal lung disease characterized by progressive decline in lung function. The compound is now under development by Galecto Biotech (currently called Galecto Inc., after the merge of Galecto Biotech and PharmAkea). Phase I/IIa clinical studies, administering TD139 as dry powder via inhalers, were conducted in 2014 and 2015 and showed that TD139 is safe and well tolerated. Furthermore, it effectively engages with Gal-3 in the alveolar space and leads to improvement in several markers of inflammation.281,282 A Phase IIb study with GB0139 in individuals with IPF started in February 2019 (GALACTIC-1: NCT03832946). It is also being further evaluated in a Phase IIa trial as an experimental medicine against COVID-19 (NCT04473053).283
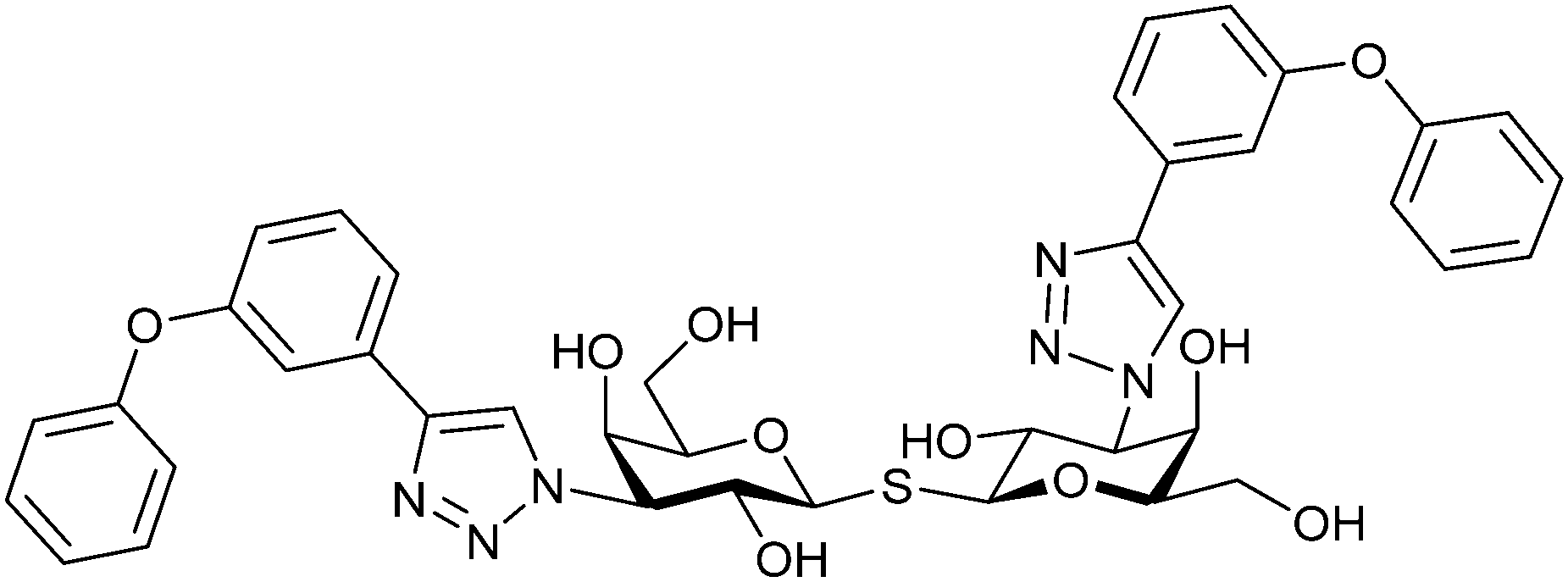
One flaw of GB0139 is that it is not specific for Gal-3 and binds to Gal-1 with equal potency. Interestingly, the introduction of 4-phenoxyphenyl as a triazol substituent provided compound 56 with 230-fold higher affinity for Gal-3 over Gal-1, though the affinity for Gal-3 was 25 times lower than that of compound 55.284
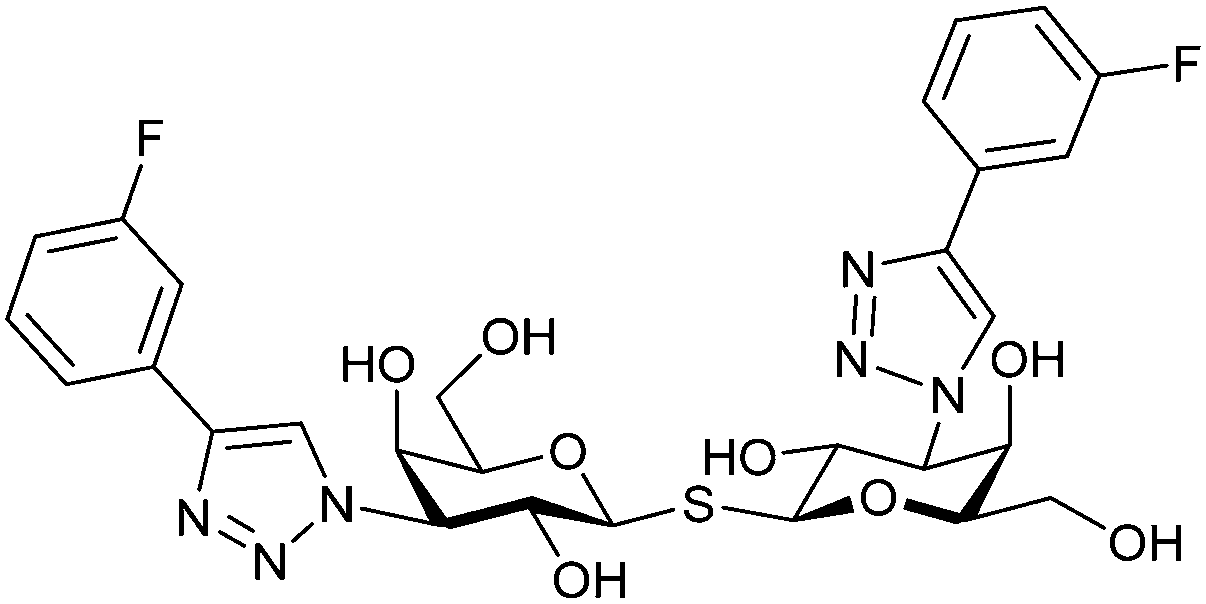
Since its discovery, the TDG scaffold has been further decorated with a number of substituents to increase affinity and selectivity for Gal-3 or other members of the galectin family. The modifications studied and their effects on affinity and selectivity have recently been reviewed elsewhere.245,285
A different high-affinity scaffold for Gal-3 antagonists is based on α-D-galactopyranoside as a monosaccharide in compound 14.286 Its high affinity and selectivity for Gal-3 stems from the utilization of several types of non-covalent interactions: fluorine–amide, phenyl–arginine, sulfur–π, and halogen bonds, spread across subsites B, C and D (Fig. 9 and 11). Compound 14 (known as GB1211, introduced by Galecto Biotech)287 is a substituted pyridinyl α-D-thiogalactopyranoside which demonstrated an anti-cancer effect and antifibrotic activity in multiple preclinical models.288,289 GB1211 is now tested as an orally available drug candidate for non-alcoholic steatohepatitis (NASH), characterized by inflammation of the liver with concurrent fat accumulation, leading to liver cirrhosis. In a Phase I trial, GB1211 was well-tolerated and showed dose-dependent pharmacokinetics (NCT03809052). A Phase I/IIa clinical trial (GULLIVER-2: NCT05009680) was initiated in September 2021, with expected end in April 2023. In 2021, Galecto Biotech announced entering an agreement with Roche for a Phase IIa trial of GB1211 in combination with a PD-1/-L1 checkpoint inhibitor atezolizumab for the treatment of cancer (Tecentriq, start in 2022, GALLANT-1: NCT05240131).290
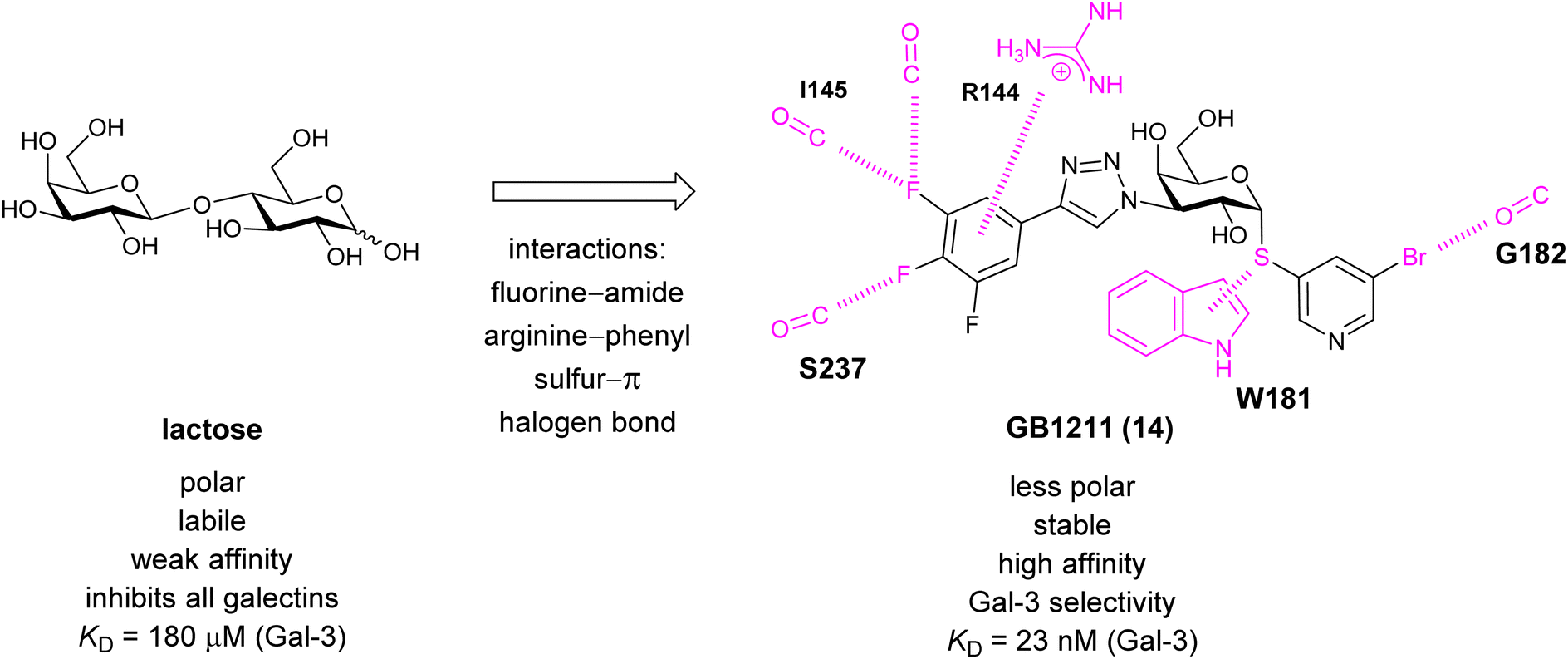 | ||
| Fig. 11 Comparison of lactose and GB1211 (14) and depiction of the Gal-3–GB1211 interactions contributing to its high affinity. | ||
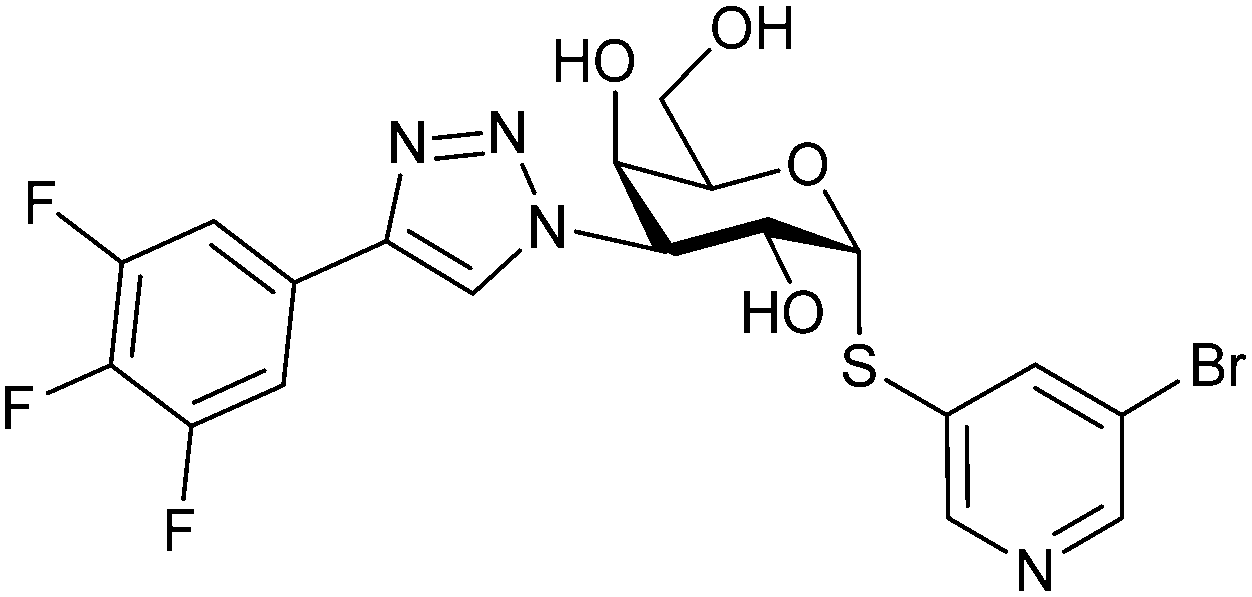
Recently, Bristol Myers Squibb reported novel triazolyl-substituted monosaccharide derivatives, represented by 57, that were identified through molecular modelling and optimized by an in-depth SAR study.291 The binding mode for these Gal-3 ligands was suggested by molecular modelling and experimentally confirmed in an X-ray co-crystal structure with human Gal-3 (PDB code 7XFA). The compounds bind to both mouse and human Gal-3 and show high selectivity for Gal-3 over Gal-1 and Gal-9. The affinity of 57 is comparable to that of 55 and the compound also has high oral bioavailability. For an overview of the above-mentioned Gal-3 inhibitors and their binding affinities, see Table 1.
| No | Name | Structure | Binding studies | Ref. |
|---|---|---|---|---|
| 44 | GR-MD-02 |
|
NMR KD = 8.0 μM (Gal-1) KD = 2.8 μM (Gal-3) | Chan et al.245 |
| 45 | DAVANAT GM-CT-01 |
|
NMR KD = 10.0 μM (Gal-1) KD = 2.9 μM (Gal-3) | Chan et al.245 |
| 46 |
|
ELISA IC50 = 4.4 μM | Sörme et al.116 | |
| 47 |
|
Competitive fluorescence polarization assay KD = 320 nM | Sörme et al.271 | |
| 48 |
|
Competitive fluorescence polarization assay KD = 370 μM (Gal-1) KD = 87 μM (Gal-3) | Öberg et al.272 | |
| 49 |
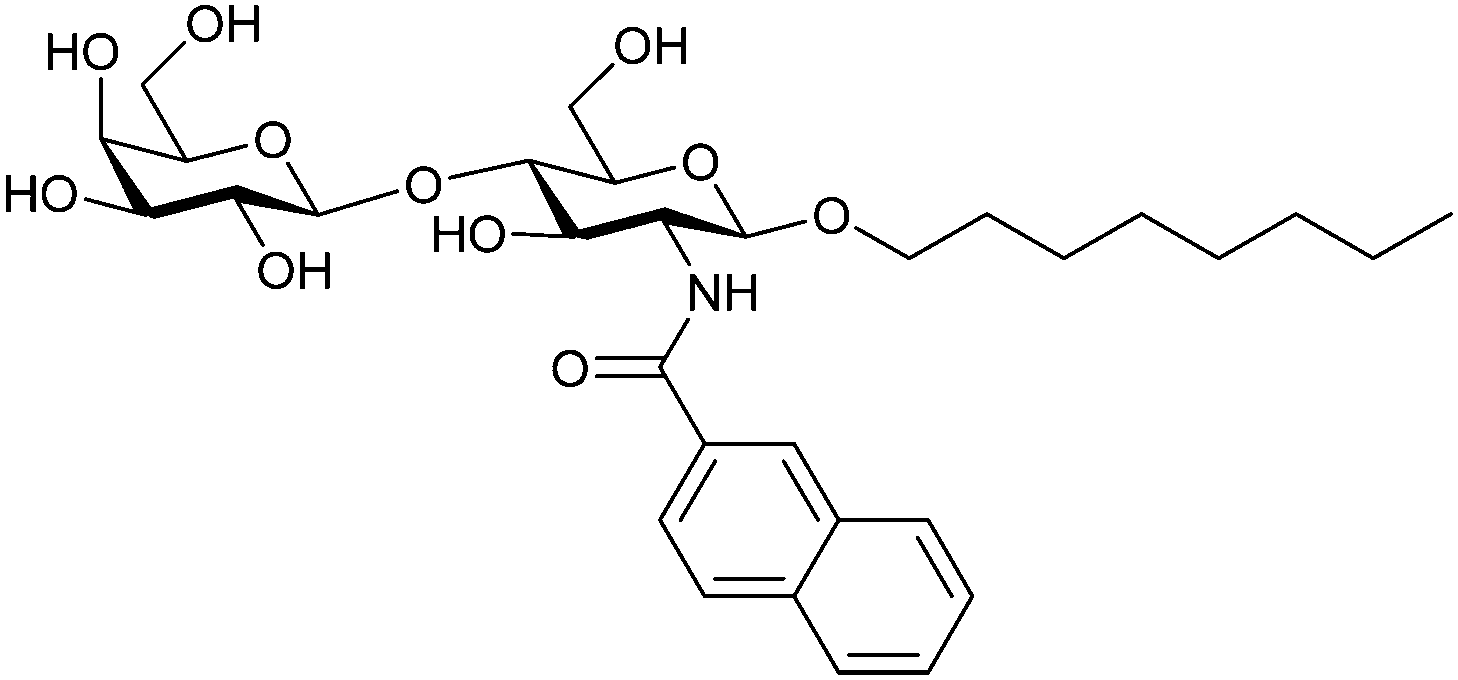
|
Frontal affinity chromatography/mass spectrometry KD = 10.6 μM | Hindsgaul et al.274 | |
| 50 |
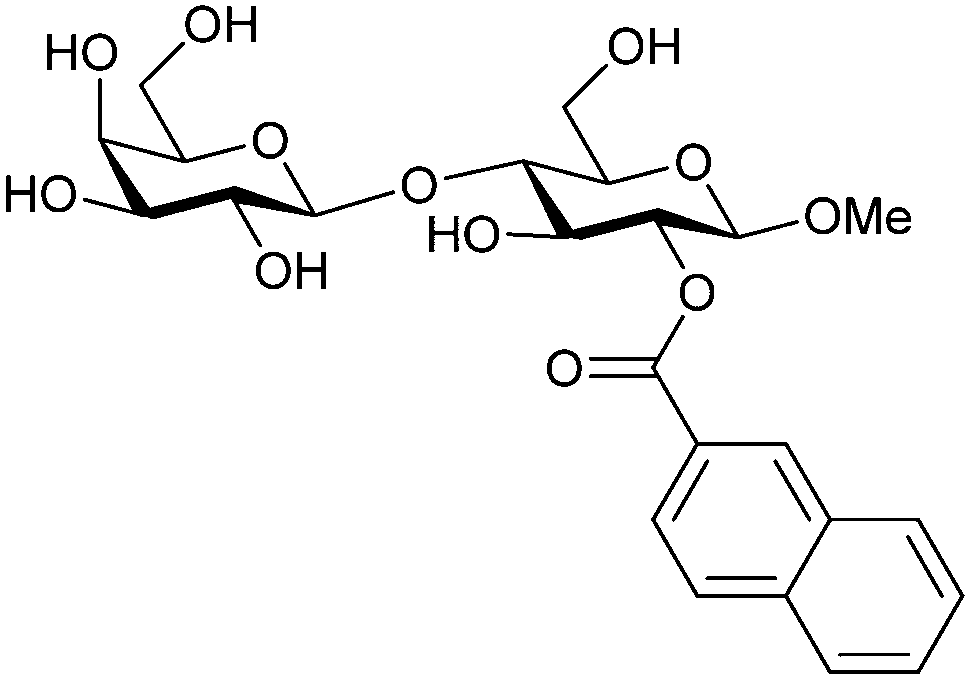
|
Competitive fluorescence polarization assay KD = 14 μM (Gal-1) KD = 2.5 μM (Gal-3) | Cumpstey et al.276 | |
| 51 |
|
Competitive fluorescence polarization assay KD = 280 μM (Gal-1) KD = 1.2 μM (Gal-3) | Pieters et al.275 | |
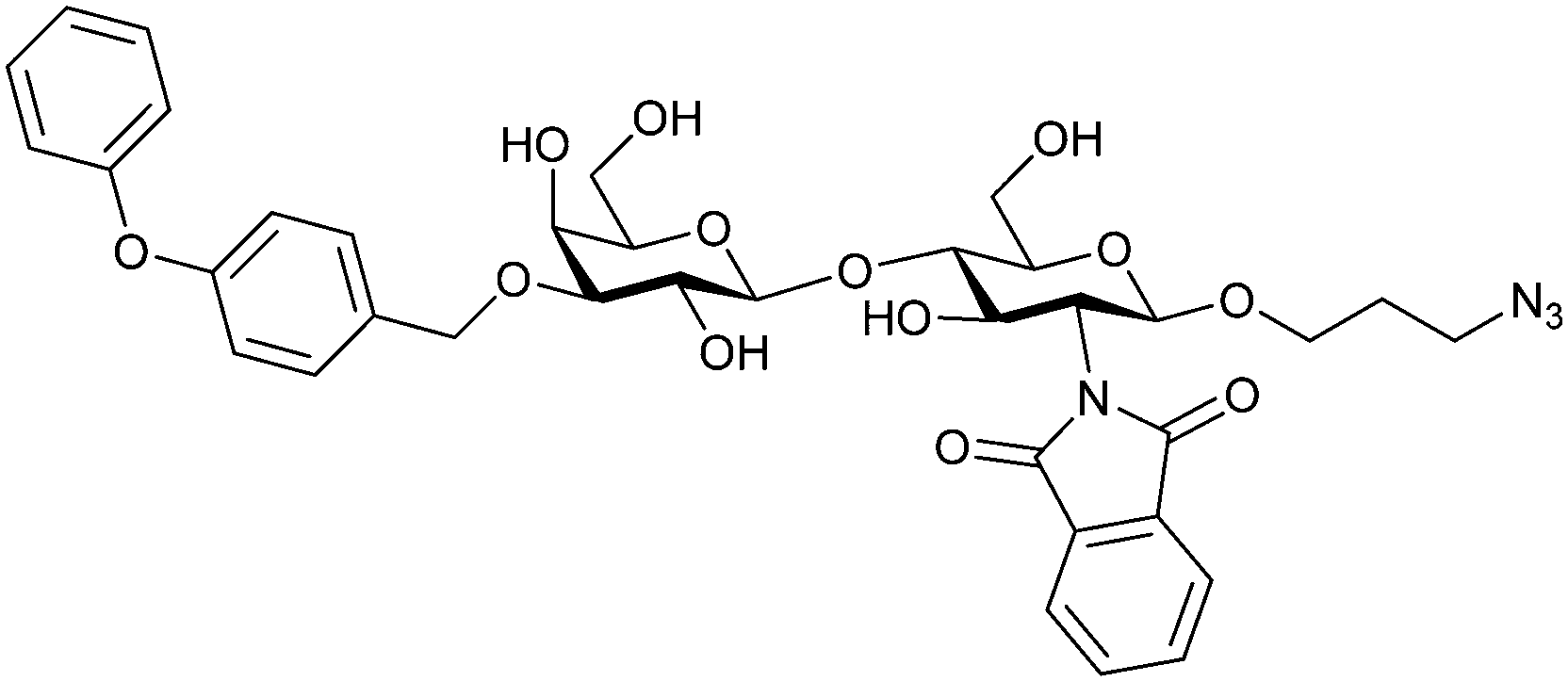
| 52 |
|
Competitive fluorescence polarization assay KD = 33 nM | Cumpstey et al.278 | |
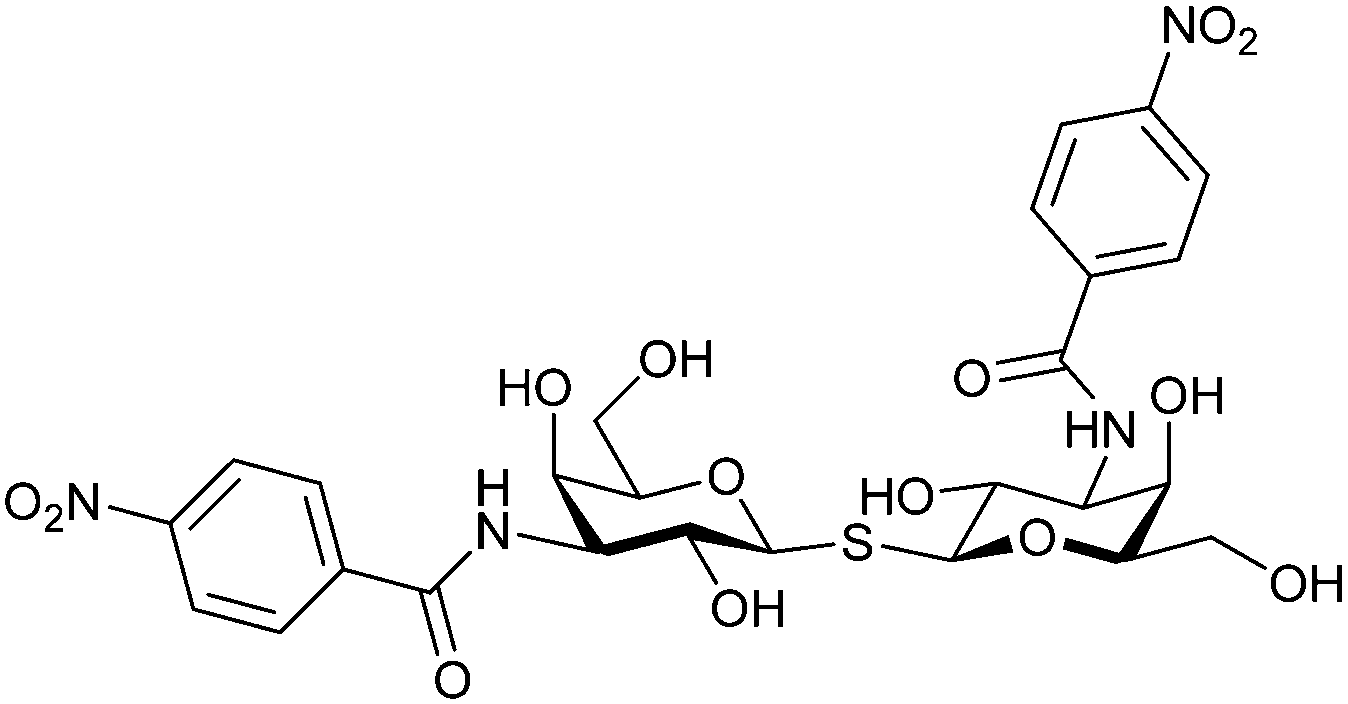
| 53 |
|
Competitive fluorescence polarization assay KD = 4.7 μM (Gal-1) KD = 46 nM (Gal-3) KD = 17 μM (Gal-7) KD high (Gal-8N) KD = 0.9 μM (Gal-9N) | Cumpstey et al.117 | |
| 54 |
|
Competitive fluorescence polarization assay KD = 29 nM (Gal-3) KD = 5.4 μM (Gal-7) KD = 58 μM (Gal-8N) KD = 1.1 μM (Gal-9N) | Salameh et al.279 | |
| 55 | TD139, GB0139, Olitigaltin |
|
Competitive fluorescence polarization assay KD = 12 nM (Gal-1) KD > 5 μM (Gal-2) KD = 14 nM (Gal-3) KD = 0.17 μM (Gal-4N) KD = 0.14 μM (Gal-4C) KD = 1.9 μM (Gal-7) KD = 86 μM (Gal-8N) KD = 0.68 μM (Gal-9N) KD = 0.12 μM (Gal-9C) | Delaine et al.292 |
| 56 |
|
Competitive fluorescence polarization assay KD = 84 μM (Gal-1) KD = 0.36 μM (Gal-3) | Pieters et al.275 | |
| 14 | GB1211 |
|
Competitive fluorescence polarization assay KD = 23 nM (Gal-3) | Brimert et al.288 |
| 57 |
|
Fluorescence binding assay IC50 = 6.9 nM (Gal-3) IC50 = 2660 nM (Gal-1) IC50 = 2500 nM (Gal-9) | Liu et al.291 |
3.1.2.2 Galectin-1. Gal-1 is a prototype galectin that exists as a dimer with two identical CRDs. It is involved in cell growth, differentiation, and signalling. Being highly expressed in the thymus, lymph nodes, as well as in immune cells such as T cells and activated macrophages, Gal-1 plays a key role in immune response regulation. Its expression is increased in tumour tissues as compared to normal healthy tissues.293 High expression of Gal-1 favours growth and progression of tumours and metastases by suppressing the immune response242 and by modulating cell migration, adhesion and angiogenesis.294–296 It also plays a key role in promoting escape from T cell-dependent immunity.295 Furthermore, Gal-1 can selectively induce apoptosis in activated Th1 and Th17 cells, thereby turning down the T-cell immunity, which is the basis of its anti-inflammatory activity.230,297 Additionally, HIV-1 exploits the host's Gal-1 to increase its attachment to host cells, thus increasing its overall infectivity in susceptible cells.229,298–300
Most compounds developed as Gal-3 inhibitors also show significant affinity for Gal-1 (see Table 1). Although selective inhibition of Gal-1 was not the prime focus of the above-mentioned studies, the reported data suggest that thiodigalactoside, modified pectins and galactomannans all inhibit Gal-1. Introducing an aromatic heterocycle to position 4 of C3-triazolo-thiogalactosides and triazolo-thiodigalactosides provided compounds with single-digit nM Gal-1 affinity and almost 10-fold Gal-1 selectivity over Gal-3 (compound 58, Table 2). X-Ray crystallography of the complex showed that the heterocycle is positioned deeper in a pocket between Ser29 and Asp123 of Gal-1 than the six-membered phenyl ring in the non-selective Gal-1/Gal-3 ligands.301 Various other carbohydrate-based scaffolds have been studied, out of which 3-deoxy-3-methyl-gulosides302 and aryltriazolylmethyl C-galactopyranosides303 act as selective Gal-1 inhibitors.
| No | Name | Structure | Binding studies | Ref. |
|---|---|---|---|---|
| 58 |
|
Competitive fluorescence polarisation assay KD = 6.1 nM (Gal-1) KD = 59 nM (Gal-3) | Peterson et al.301 | |
| 59 | Anginex |
|
Competitive fluorescence polarisation assay KD = 25 nM (Gal-1) | Griffionen et al.305 |
| 60 | 6DBF7 |
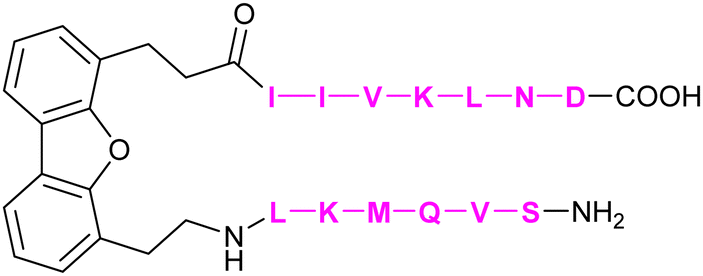
|
Mayo et al.308 | |
| 61 | DB16 |
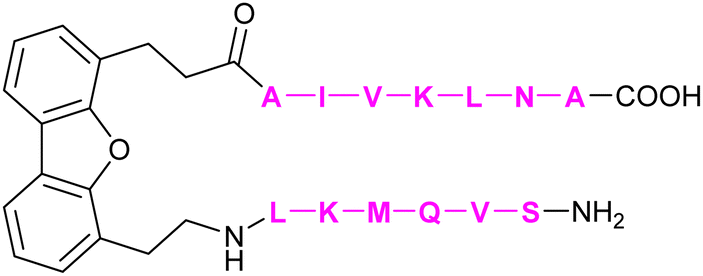
|
Dings et al.128 | |
| 62 | KM0118 OTX008 |
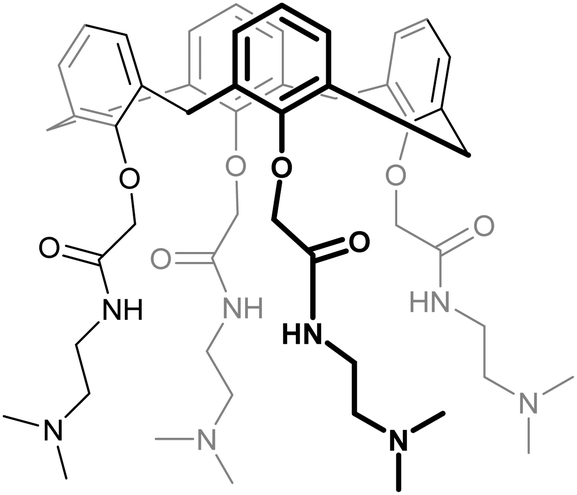
|
Dings et al.309 |
In addition to the carbohydrate-based glycomimetics mentioned above, peptides and peptidomimetics have been intensely studied as selective Gal-1 inhibitors to block angiogenesis. Anginex (59) is a 33-mer synthetic peptide that was originally designed to resemble the β-sheet structure of antiangiogenic proteins such as platelet factor 4 (PF4) and interleukin (IL)-8.304–306 Apart from Gal-1, 59 was reported to bind to some other galectins, namely Gal-2, -7, -8N, -9N, but not to Gal-3, -4, and -9C.307 Compounds 6DBF7 (60) and DB16 (61) are partial peptidomimetics of Anginex and have six amino acid residues at the N-terminus and seven at the C-terminus linked by a hydrophobic dibenzofuran (DBF) scaffold to achieve a β-sheet peptide conformation. When compared to Anginex, the compounds exhibit better angiostatic properties.308 However, Anginex and its partial peptidomimetics are peptides and prone to hydrolysis by proteases. In order to overcome this metabolic instability and maintain the hydrophobic and hydrophilic faces of the molecule, a non-peptidic topomimetic OTX008 (Calixarene 0118, PTX008, 62), was designed.3091H–15N HSQC NMR spectroscopy studies showed that the compound binds at a site different from the CBS and thus acts as a non-competitive allosteric inhibitor of Gal-1 (see Section 3.3.2).310 The compound downregulated cancer cell proliferation, invasion, and tumour angiogenesis in a variety of tumour cells.311 In addition, it has also shown synergistic effects with the tyrosine kinase inhibitor sunitinib in human ovarian carcinoma and glioblastoma.312 In May 2013, OTX008 was successfully evaluated in a Phase I clinical trial (OncoEthix, NCT01724320).313 In 2014, OncoEthix was acquired by Merck & Co. and since then, no further clinical studies have been performed according to clinicaltrials.gov. For an overview of the above-mentioned Gal-1 inhibitors and their binding affinities, see Table 2.
3.1.2.3 Galectin-9. The tandem-repeat galectin Gal-9 has N- and C-terminal carbohydrate-binding domains connected by a peptide link. Gal-9 interacts with programmed cell death protein 1 (PD-1, CD279) and T cell immunoglobulin and mucin-domain containing-3 (TIM-3). While PD-1 is an immune checkpoint that promotes apoptosis of antigen-specific T-cells in lymph nodes and reduces apoptosis in regulatory T cells, TIM-3 is a T cell checkpoint inhibitor. TIM-3 expression on T cells, together with other check-point molecules, in chronic infections and cancers can hinder productive immune responses. The interaction of Gal-9 with PD-1 and TIM-3 regulates T cell death, making these promising targets for cancer immunotherapy.314 The activity of the TIM-3/Gal-9 checkpoint can be modulated in two ways: (i) blockage of TIM-3 with monoclonal antibodies or small molecules and (ii) blockage of Gal-9.315 Three monoclonal antibodies to TIM-3 are already in clinical trials for the treatment of solid tumours. Each of the programmes is evaluating the safety and efficacy either as monotherapy or in combination with anti-PD-1 antibodies in patients with advanced solid tumours or hematologic malignancies.228 In 2016, the Nilsson group reported galactoside-N-sulfonyl amidines (e.g. compound 63) with good affinities and selectivity towards Gal-9N over other galectins (Fig. 12A).316 Later on, they showed an interesting epimer switch of Gal-9 domain selectivity: while 3-N-aryl galactosides 64 bind the C-terminal domain, the respective gulosides 65 bind the N-terminal domain (Fig. 12B).317
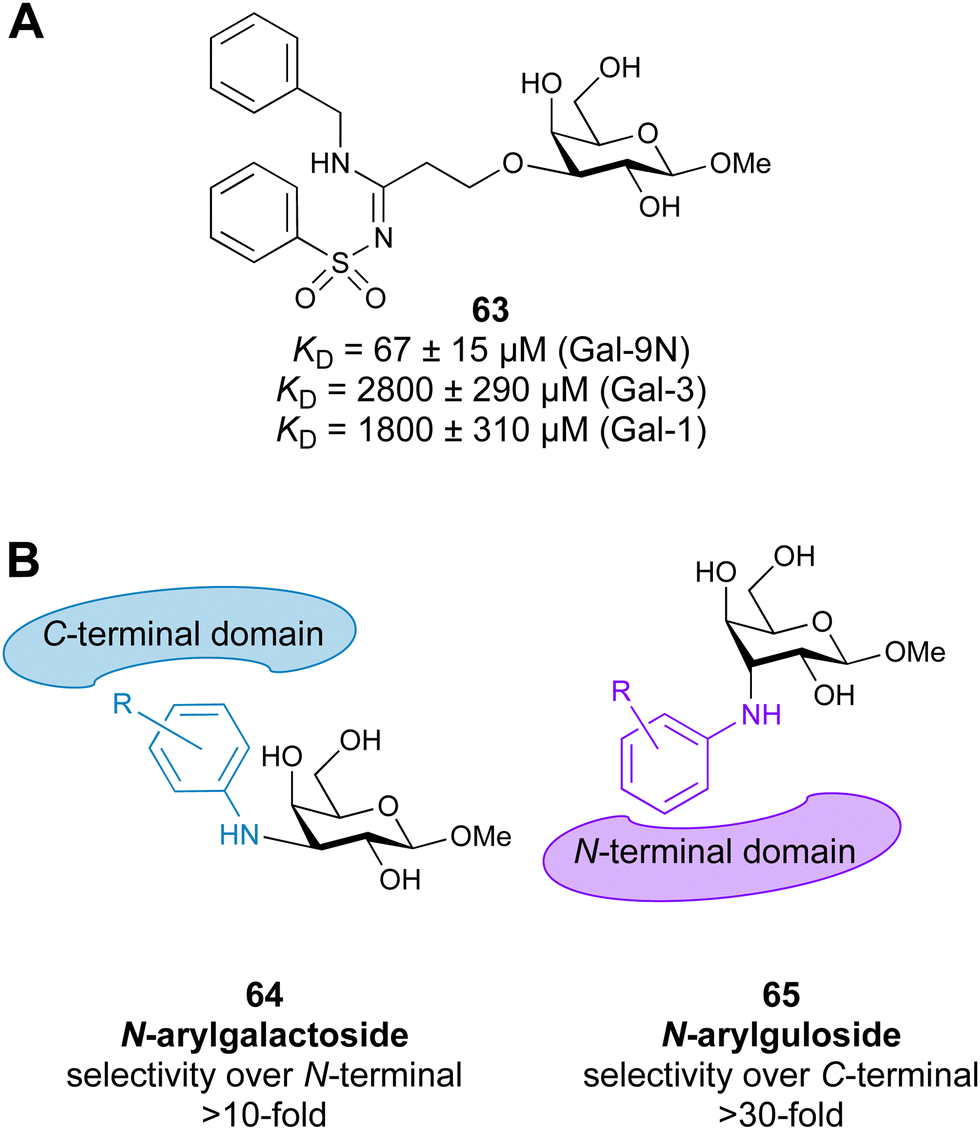 | ||
| Fig. 12 Gal-9 inhibitors. (A) Galactoside-N-sulfonyl amidine, (B) selectivity switch between 3-N-aryl galactosides and 3-N-aryl gulosides. | ||
3.1.2.4 Galectin-8. Gal-8 is a tandem repeat galectin consisting of two CRDs, one N-terminal and the other C-terminal. These two domains share 35% sequence identity and bind differently to the natural ligands. Recently, Gal-8 has emerged as a potential pharmacological target for the treatment of various diseases, including cancer318 and inflammation.319,320 Unique functions of Gal-8 have been linked to the specificity of its N-terminal CRD. Therefore, there is a high demand for selective and high-affinity Gal-8N ligands that could potentially have anti-tumour and anti-inflammatory properties. To date, several galactose-based Gal-8N inhibitors have been reported, but mostly suffer from poor selectivity.321–324 Recently, D-galactal derivatives (e.g. compounds 66 and 67, Fig. 13A) have shown not only high affinity, but also good selectivity over other galectins.325,326 Other recently developed antagonists include 3-lactoyl-α-D-thiogalactopyranosides (e.g. compound 68, Fig. 13B).327
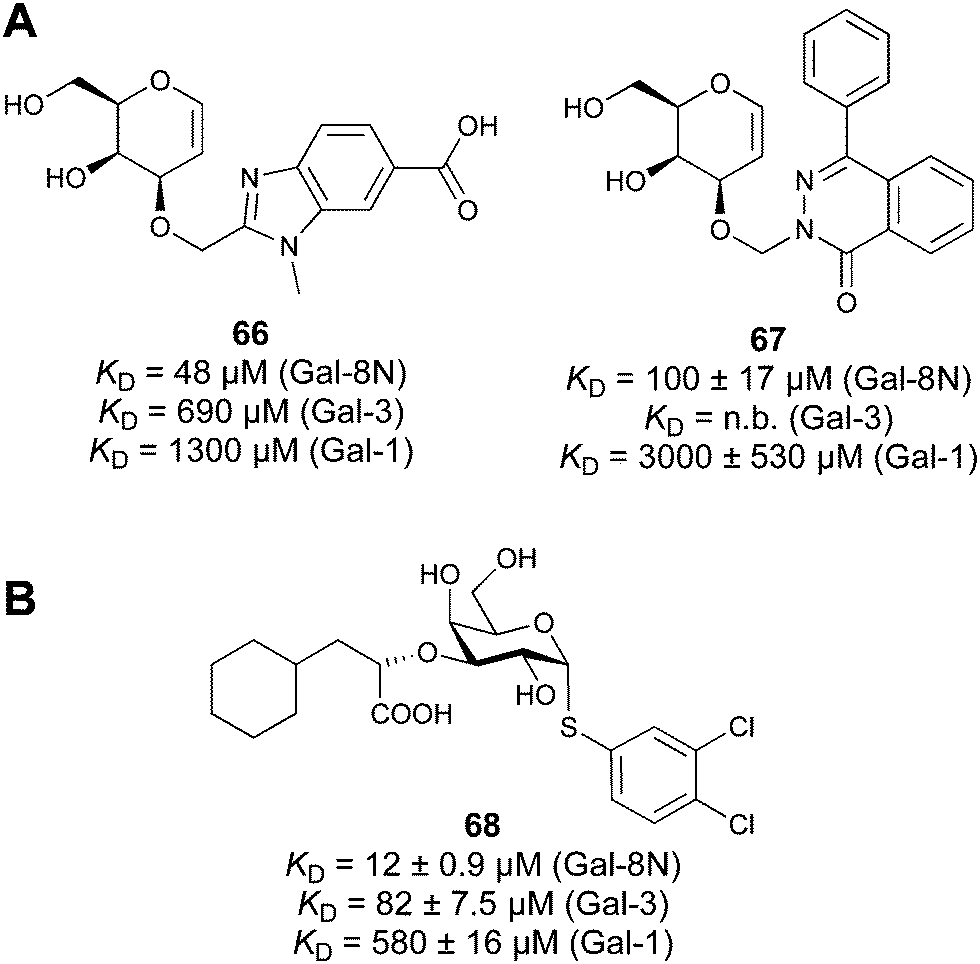 | ||
| Fig. 13 Gal-8N inhibitors and their binding affinities for Gal-8N, Gal-3 and Gal-1, highlighting the selectivity for Gal-8N. (A) D-Galactals, (B) 3-lactoyl-α-D-thiogalactopyranoside. n.b. = no binding. | ||
All Siglecs share a similar overall protein architecture. An N-terminal V-Ig like domain harbours the ligand binding site for sialic acid recognition. This domain is extended away from the cell surface by a series of one to sixteen C1 or C2-Ig like domains.332,333 A C-terminal intracellular domain is present in some Siglecs and contains an immunoreceptor tyrosine-based inhibition motif (ITIM) for cell signalling inhibition (i.e. CD22). Alternatively, positively charged amino acids located in the transmembrane region enable association with Dap12 carrying an immunoreceptor tyrosine-based activation motif (ITAM) for cell signalling activation.334 Overall, Siglecs can be divided into two groups with classic Siglecs being more conserved among species and CD33-related Siglecs having a higher sequence variability between homologues.329
Individual Siglecs have a restricted expression pattern on immune cells, rendering them suitable targets for cell-specific therapies. Additionally, many, if not all, are endocytic receptors allowing the delivery of various cargos to immune cells.329,335 This concept was successfully followed for several Siglecs: Siglec-8 is expressed on eosinophiles and mast cells and is explored for therapies of asthma and allergies,336 Siglec-15 is expressed on osteoclasts and is a target for the treatment of osteoporosis.329,337 For myelin associated glycoprotein (MAG/Siglec-4),333 glycomimetics have been developed for the enhancement of neurite outgrowth, whereas a completely orthogonal approached was reported recently in which PPSGG (PN-1007, Polyneuron Pharmaceuticals), an undisclosed glycomimetic, was claimed to prevent anti-MAG IgM autoantibodies to deplete MAG expressing cells (NCT04568174). CD22 (Siglec-2) is highly expressed on B cells and has been successfully used as a target for the treatment of B cell lymphoma. The anti-CD22 antibody drug conjugate (ADC) inotuzumab ozogamicin (Besponsa, Pfizer/UCB) is used for the treatment of acute lymphoblastic leukaemia.338 The anti-CD33 (Siglec-3) antibody gemtuzumab ozogamicin (Mylotarg, Pfizer) has been approved since 2000 for the treatment of acute myeloid leukaemia.339 Moreover, a CD33 glycomimetic might be an effective therapy against late-onset Alzheimer disease (AD), which increases the uptake of the toxic amyloid-β (Aβ) peptide into microglial cells and thus might promote clearance of the Aβ peptide causing AD progression.340 For a summary of current antibodies against Siglecs the reader is referred to a more in-depth review.341
With respect to the development of glycomimetic ligands for Siglecs, one has to take into account that the sialic acid family is a large and diverse group of sugars.342,343N-Acetyl neuraminic acid (Neu5Ac) is the most common nine-carbon representative of the family and can potentially carry a large variety of substitution patterns, such as sulfation and acetylation.328 Additionally, Neu5Ac can be further sialylated in the 3-, 6-, 8-, and 9-position. Consequently, the Siglec protein structure provides grounds for a high level of sialic acid specificity based on the linkage of the sialic acid or substitution pattern. For carbohydrate-based glycomimetics, this translates into the basis for many design approaches starting from extended carbohydrate epitopes, up to trisaccharides to reduce off-targets within the family. From this, a significant loss of drug-likeliness and less favourable pharmacological properties arise. To the best of our knowledge, there are no reports on replacing the core sialic acid with a drug-like scaffold.
The development of carbohydrate-based glycomimetics for Siglecs was pioneered by early work reporting on sialic acid derivatives substituted in position 9 to gain affinity for several targets.344 The general approach of expanding the core Neu5Ac using hydrophobic substituents in position 9 was later followed by attempts to grow this central element into C-2, -4, -5 direction and finds its justification in a number of distinct structural features of the Siglec architecture.345–347 Firstly, a central arginine is present in a conserved position of the antiparallel β-sheet making interactions with the carboxylate of the sialic acid (Fig. 14). Secondly, there is an interesting feature of the sialic acid recognition: the monosaccharide is essentially a peptidomimetic. It extends the anti-parallel β-sheet with its hydrogen-bond donors and acceptors mimicking the next antiparallel β-strand (Fig. 14B).348 Next, this GG′ strand to which the sialic acid aligns like a peptide has high structural variability amongst the Siglecs exactly when extending from position 9.332 The biological role of this variability is not fully described, but it can be speculated to be involved in generating specificity for substitution patterns of the 7, 8, and 9 position of the Neu5Ac core. For the carbohydrate-based glycomimetics, this implies high likelihood for gaining both affinity and specificity when extending from this position. Additionally, the same holds true for the CC′ loop which can extend towards the 9 position of the carbohydrate binding site (Fig. 14A). Interestingly, the CC′ loop can also dictate the linkage specificity as evidenced for CD22 and Siglec-7.332,349 Other positions that have been explored for extending the carbohydrate core such as the 5 position show lower structural variability compared to the 9 position. Overall, some Siglecs have preformed binding sites devoid of any conformational selection upon ligand binding, such as CD22 and Siglec-1, while others do have such as Siglec-4 and -7. Capitalizing on these structural features of the protein, and the advancement of sialic acid synthesis, a number of reports have shed light on carbohydrate-based glycomimetics for several Siglecs using focused library screening.335,345–347,350–356
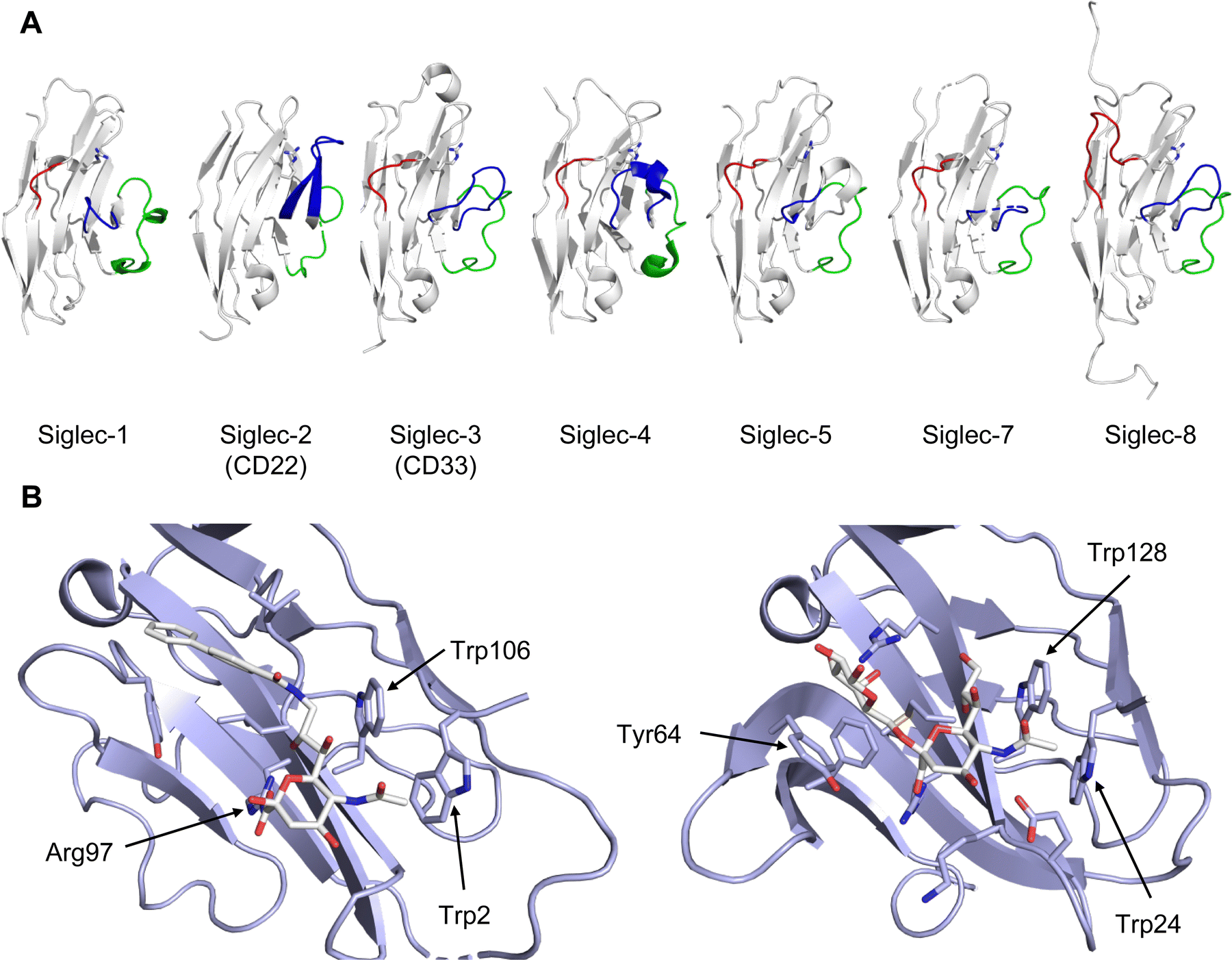 | ||
| Fig. 14 Siglec V-Ig domain fold. (A) Siglec-1, CD22 (Siglec-2), CD33 (Siglec-3), Siglec-4, Siglec-5, Siglec-7, Siglec-8, C’D loop region (green), G strand (red), CC′ loop (blue), essential arginine shown in stick (PDB codes 1QFP, 5VKM, 6D49, 5FLV, 2ZG2, 2G5R, 2N7B, respectively). (B) (left) Siglec-1 in complex with 69 showing 5’ site stacking of Trp2 and stabilization of the glycerol side chain of the Neu5Ac by Trp106.350 This pose leads to the extension of the anti-parallel β-sheet and is further stabilized by a salt bridge to the so-called “essential arginine” (here Arg97). The GG′ loop harbours Val109 in Siglec-1 providing affinity for the biphenyl substituent in 69 (PDB code 1ODA). (right) CD22 does not have a GG′ loop and is less restricted in this position. However, compared to Siglec-1, the role of the CC′ loop is much more pronounced. In CD22, Tyr64 located in this loop does a stacking interaction explaining the strong linkage preference of CD22 for α-2,6 linked sialic acid ligands (PDB code 5VKM). | ||
An additional challenge for the development of inhibitors for many Siglecs are cis-ligands. These are sialosides present on the same cell and although the protein structure extends the active site away from the cell surface, Siglecs are exposed to these sialic acids, which can block the active site. These cis-ligands are present in high local concentrations, which are estimated to be in the 100 mM range, imposing a massive competition for potential glycomimetics.329,357 These ligands can even be present on the same receptor, leading to the formation of homooligomeric receptor assemblies on the cell, e.g. CD22.358 Taken together, the vast majority of glycomimetics for Siglecs are carbohydrate-based extending of a limited set of substitution vectors benefiting from the local site variability to gain specificity and lipophilicity to gain affinity. However, none of these molecules has made it into the clinics or as monovalent chemical probe into in vivo preclinical development, likely because of the high affinity necessary. For a more detailed summary of small molecule inhibitors of Siglecs the reader is also referred to other excellent reviews.335,341,355
3.1.3.1 Siglec-1. Siglec-1 (sialoadhesin, CD169) is expressed on macrophages and is the largest member of the Siglec family, with 17 Ig repeats extending this protein far into the extracellular space. It was the first Siglec for which the X-ray structure was solved, teaching us many of the above-mentioned structural characteristics of the recognition process: the essential arginine, the extension of the anti-parallel beta sheet.359 Later, the complex between Siglec-1 and 9-N-BPC-Neu5Acα2OMe (69, Table 3) was solved by X-ray crystallography and the affinity gain through interaction with the GG′ loop became apparent (Fig. 14B).350 The biphenyl substituent results in a 13-fold affinity increase over the Neu5Ac monosaccharide.350,360 A clear specificity gain was seen when included in the trisaccharide structure 9-N-BPC-Neu5Acα(2,3)Galβ(1,4)GlcNAc (70) compared to the 9-N-BPC-Neu5Acα(2,6)Galβ(1,4)GlcNAc (75, Fig. 15) being a CD22 ligand provided by the α(2,3) over the α(2,6) glycosidic linkage.361 However, used in a multivalent display for targeted delivery on liposomes in vivo the compound failed because of insufficient specificity.362 Following up on this work, using rational design and virtual screening a focused library of substituents in the 9 position, a thienochromene substituent was identified (71, Table 3) leading to more than one order of magnitude affinity increase and importantly sufficient specificity to be applied in mice as part of a liposomal formation.353 This affinity and specificity increase can likely be rationalized by the higher shape complementarity of the thienochromene group extending into the GG′ region. This in vivo specificity was later used for the delivery of αGal ceramide loaded lipid nanoparticles to Siglec-1 expressing macrophages and activation of iNKT cells through CD1d.363 Furthermore, the model antigen ovalbumin (OVA) was delivered to Siglec-1+ macrophages.364
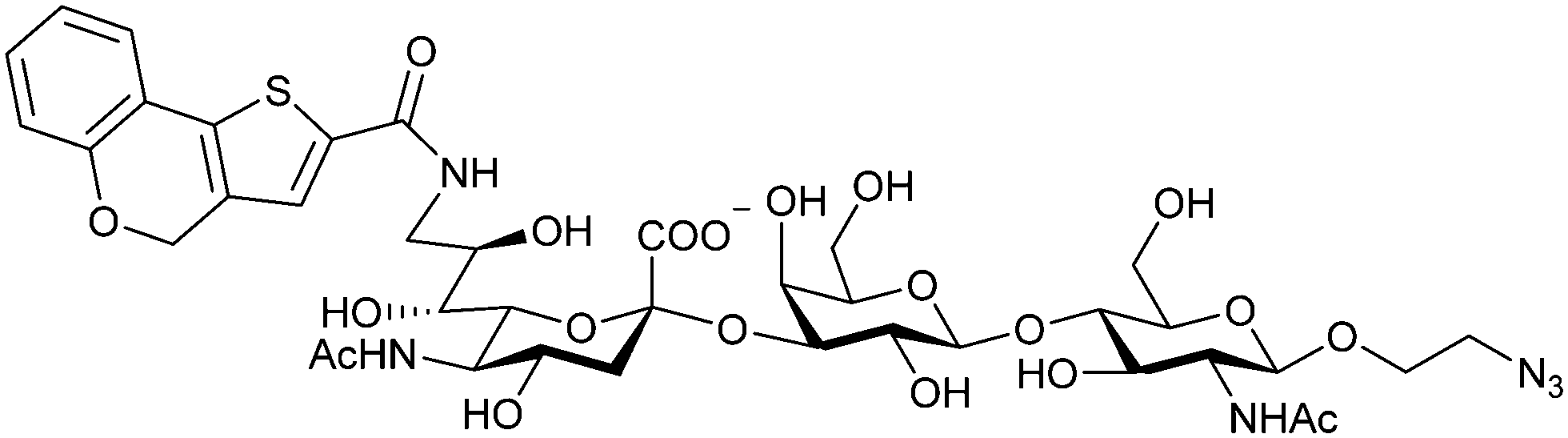
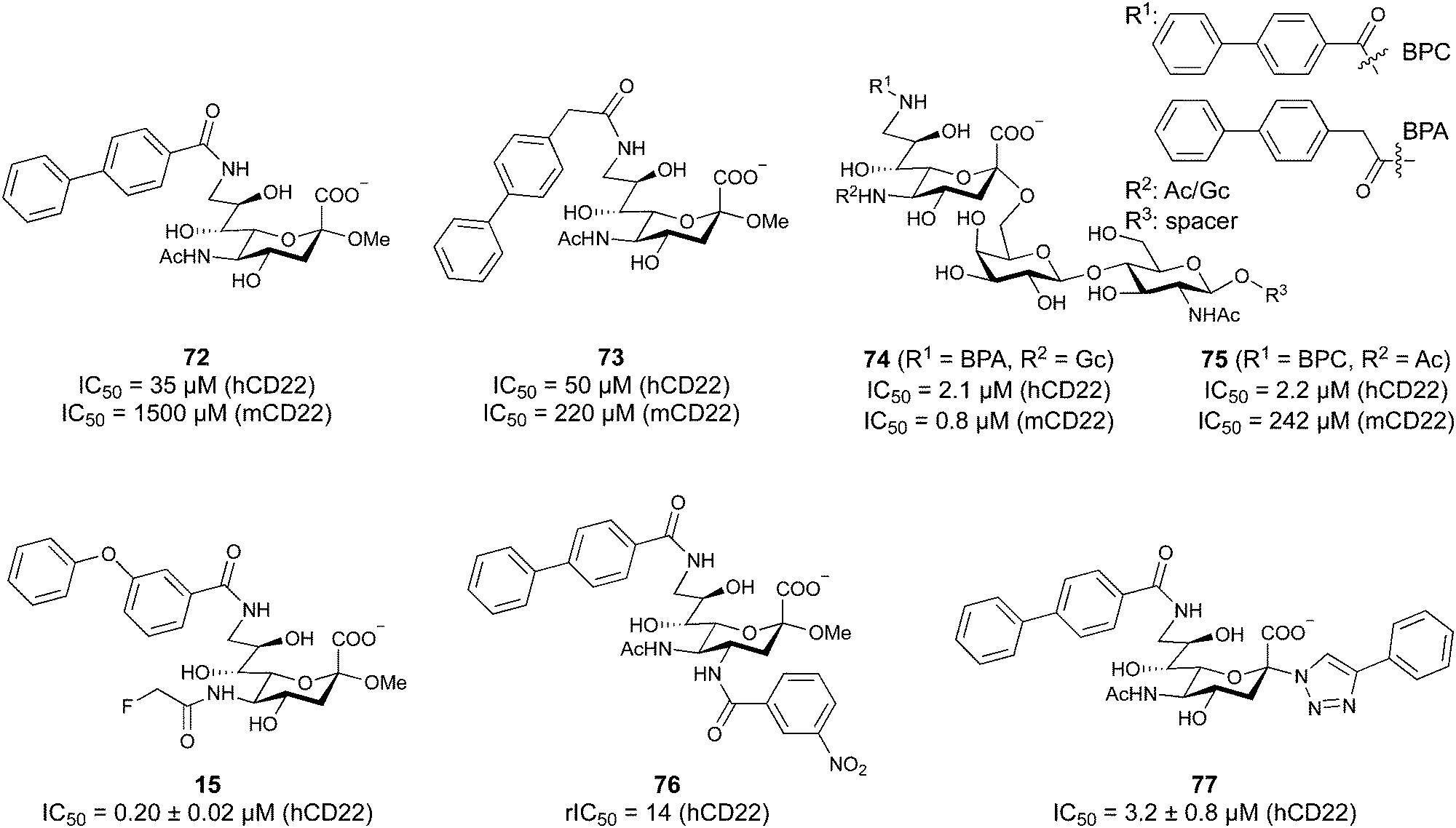 | ||
| Fig. 15 Murine and human CD22 antagonists. A selectivity gain towards human CD22 (hCD22) over murine CD22 (mCD22) is achieved by BPC substitution of the 9’ position and acetylation of position 5. C4 modification resulted in further improved affinity. 76 here is reported in relative IC50 compared to 72 (IC50 = 3 μM) in the corresponding assay setup with rIC50 = IC50 (72)/IC50 (76).345 | ||
3.1.3.2 Siglec-2/CD22. CD22 (Siglec-2) is a 140 kDa single transmembrane protein carrying six C Ig-like domains and one V-Ig domain that extends CD22 like a tilted rod into the extracellular space.332 This inhibitory immune cell receptor is highly expressed on B cells, with reported low expression on mast cells and conventional dendritic cells.365 This restricted expression pattern has advanced CD22 as an attractive target for immune therapy, early validated using anti-CD22 antibodies.338
Major advancement in the development of carbohydrate-based glycomimetics targeting CD22 came from early work of Kelm et al., and later was followed by Paulson and co-workers reporting a 224-fold increase in affinity for 9-BPC-Neu5Acα2Me (72) for the human CD22 and a 123-fold increase for 9-BPA-Neu5Acα2Me (73) for the murine homolog, compared to their unsubstituted parent monosaccharides, respectively (Fig. 15).344,345,366 Since the murine CD22 has a strong preference for N-glycolyl neuraminic acid (Neu5Gc), including this moiety in the design led to an over 250-fold potency gain when combined with this 9’ substitution in the framework of the trisaccharide 74 compared to 75.351 Compared to the α(2-6)-sialyllactose core scaffold, with an affinity of 281 ± 10 μM as assessed by isothermal titration calorimetry (ITC), this 9-BPA-NeuGcα(2-6)Galβ(1-4)GlcNAcβ-spacer (74) achieved more than three orders increase in affinity.332,351 Taken together, this advancement in the 5 and 9 positions provided sufficient affinity and specificity to target murine CD22 using multivalent display on nanoparticles in vivo.367,368 For the human homolog, a suitable ligand with sufficient affinity and specificity came from focused library screening of the 9 position, being 6‘MBP-5F-Neu5Ac (15).115 Although CD22 glycomimetic recognition is mediated by a largely preformed binding site, NMR analysis highlights that the role of the biphenyl substituent is more complex and provides room for future improvement.332,369
Besides advancement in the 5 and 9 position, further improvement came from a C-4 modified structure-based design of 9-BPC-4-mNPC-Neu5Acα2Me (76) with a sub-micromolar affinity or C-2 modifications (77).345,346
To the best of our knowledge the only non-carbohydrate CD22 ligand is the peptide PV3, which was derived from epratuzumab Fab and binds CD22 with 9 μM affinity.370 The peptide binding site is unrelated to the canonical carbohydrate binding site and does not have to overcome the cis-ligand challenge. Applied as a multivalent probe for targeted delivery on a liposome, it allows for in vivo delivery when high receptor density is available.370
One important pathogenicity factor containing lectin domains are bacterial toxins comprising Shiga toxin, present in haemorrhagic Enterobacteriaceae. In these AB5 toxins, the pentameric B-domain repeat is a lectin and responsible for carbohydrate-dependent binding to the cells, followed by internalisation to exert their intracellular toxicity. Due to their geometry, these AB5 toxins have been successfully inhibited with complementary pentavalent ligands, such as STARFISH or DAISY containing native oligosaccharide epitopes.33,374 However, their further development as antitoxins has stalled for unknown reasons.
Furthermore, in many bacteria, surface exposed lectins serve as adhesins for host colonization and persistence. Several lectins of the most problematic Gram-negative bacteria are therefore currently targeted with glycomimetics.
3.1.4.1 Escherichia coli fimbrial adhesin FimH. One intensively studied lectin is FimH, an adhesin at the tip of E. coli type 1 fimbriae that is important for tissue binding in urinary tract infections and inflammatory bowel disease. Thus, clinical candidates that inhibit FimH are being developed for both conditions.
With about 150 million cases annually, urinary tract infections (UTIs) are among the most frequent bacterial infections.375,376 Notably, up to 80% of all uncomplicated UTIs are caused by uropathogenic E. coli (UPEC).377,378 UPEC utilize type 1 fimbriae to adhere to urothelial cells which line the host bladder, a pivotal step to establish an infection.379 Host cell adhesion allows UPEC to avoid clearance during micturition and additionally invasion of host cells is triggered. Once inside of host cells, UPEC again utilize FimH to form biofilm-like structures, called intracellular bacterial communities (IBCs), to replicate and hide from the immune system or antibiotic treatment. In the last step of the bacterial infection cycle, the bacteria disperse from the IBCs and exfoliate to invade neighbouring cells.380 Expression of type 1 fimbriae was not only shown to be essential for cell adhesion, but also for formation of IBCs.381
Furthermore, the increased prevalence of adherent-invasive E. coli (AIEC) in patients suffering from specific inflammatory bowel diseases, such as Crohn's disease (CD), has attracted growing attention for development of new treatment strategies.382,383 Like UPEC, AIEC adhere to intestinal epithelial cells via FimH. The chronic inflammation elicited by the intestinal dysbiosis in CD leads to tissue damage and progressing organ degradation.
Therefore, the development of high affinity FimH ligands presents an opportunity to complement antibiotic use and improve the treatment of Crohn's disease and UTIs.
Type 1 fimbriae are 0.1–2 μm long filaments on the bacterial surface.384,385 They consist of approximately 1000 copies of the subunit FimA, forming a right-handed helical rod, which attaches to a single FimF and FimG subunit, and is capped at the tip by the carbohydrate-binding adhesin, FimH (Fig. 16A).384,385 Notably, the subunits engage in a donor-strand completion mechanism, in which the incomplete immunoglobulin-fold of each subunit is completed by an N-terminal extension of the next subunit.386,387 Structurally, the 30 kDa protein FimH consists of two domains: the amino-terminal carbohydrate-binding FimHL (aa 1–158) and the carboxyl-terminal FimHP (aa 159–279), which connects FimHL to the following subunit FimG and therefore to the pilus (Fig. 16B). Importantly, the domain interaction of FimHL and FimHP allows formation of so-called catch-bonds, characterised by an increased binding strength under shear stress, e.g. during micturation.388–390 Those catch bonds help UPEC to avoid clearance during urination, while preserving the ability to migrate and infect new cells in absence of a tractive force.391
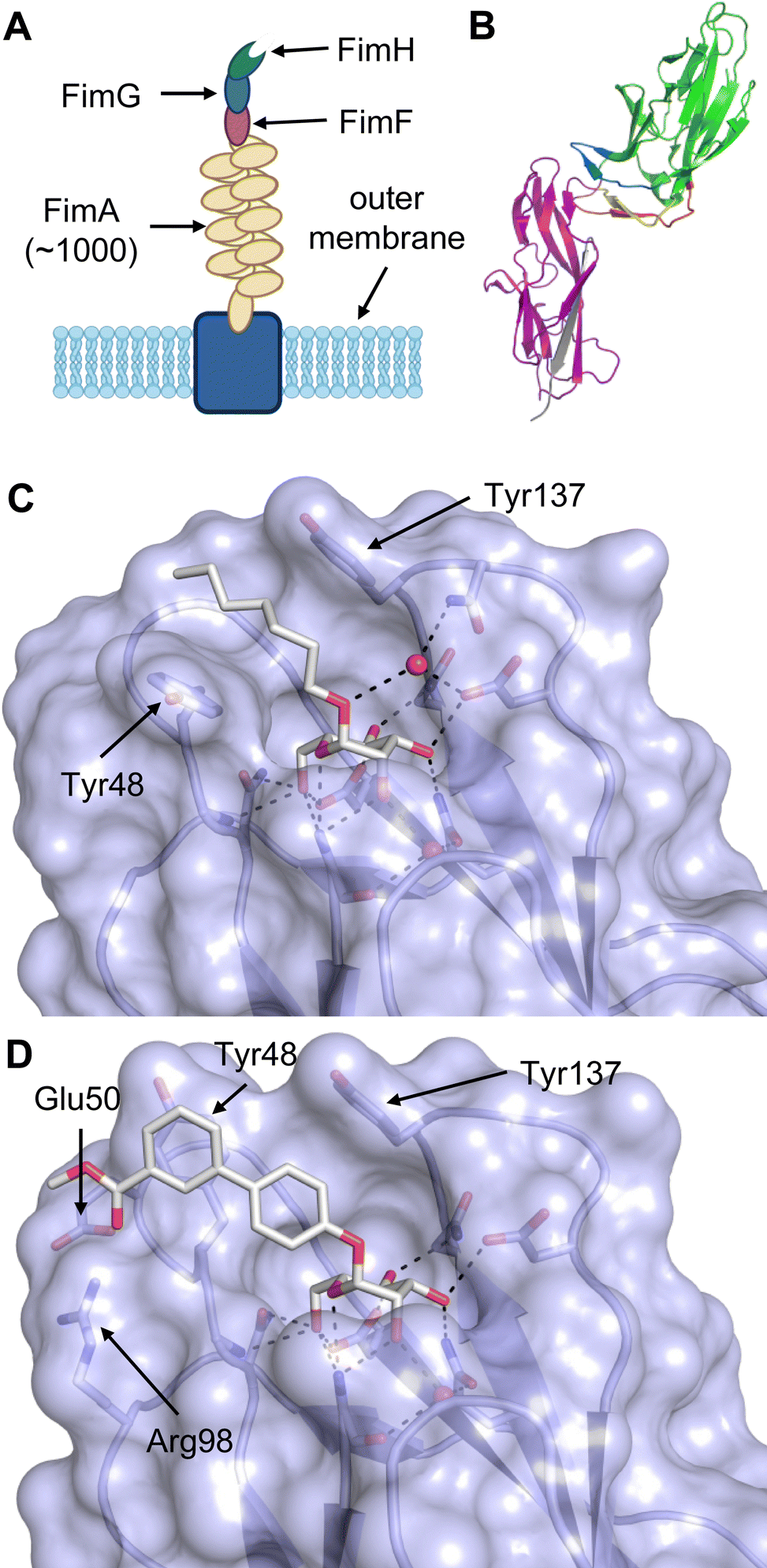 | ||
| Fig. 16 (A) Structure of a type 1 fimbrium: anchored to the membrane ∼1000 FimA subunits form a helical rod to which the mannose-binding subunit FimH is attached via the subunits FimF and FimG.408 (B) Two-domain fimbrium subunit FimH (PDB code 4XOE).389 The binding affinity of the carbohydrate-binding domain FimHL (green) is influenced by contacts of the swing loop (blue, aa 23–33), linker loop (yellow, aa 151–158) and insertion loop (red, aa 112–125) to the fimbrial domain FimHP (pink) enabling the formation of catch-bonds. Donor strand to complete the Ig-fold of FimHP shown in grey. (C) Ligand heptyl α-D-mannoside (78) bound to FimH (PDB code 4XOE).389 The mannose moiety engages in extensive hydrogen bonding in a deep binding pocket while the alkyl chain interacts hydrophobically with Tyr48 and Tyr137 of the tyrosine gate in the in-docking mode. Water shown in red. (D) Biphenyl ester 79 in complex with FimH (PDB code 3MCY).409 Hydrophobic interactions of the aglycon with the tyrosine gate as well as interaction with the salt-bridge of Glu50-Arg98 significantly contribute to high affinity binding. In contrast to heptyl α-D-mannoside, 79 interacts with FimH in the out-docking mode. | ||
The carbohydrate-binding adhesin domain of FimHL is located at the distal end of a β-sheet opposite of the FimHP domain. In absence of shear stress, three loops of FimHL contact FimHP, resulting in distortion of the β-sheets, affecting the CRD. Consequently, the binding pocket widens, leading to weakened ligand interactions and therefore reduced ligand affinity and quicker ligand dissociation. In contrast, tensile force leads to domain separation of FimHL and FimHP, enabling long-lived binding events with high affinity.389,392 This mechanism of allosteric regulation by domain interaction is underlined by the work of the Maier and Glockshuber groups.389 A 3300-fold higher affinity for n-heptyl α-D-mannoside (HM, 78) was reported for isolated FimHL (representing the high affinity conformation) compared to full length FimH in the low affinity conformation (KD = 3.0 ± 0.2 nM vs. 9900 ± 150 nM). Natural ligands of FimH are α-D-mannosides of N-glycans, such as the uroepithelial glycoprotein uroplakin 1a or CEACAM6 (CD66c) in the ileum.393,394 Importantly, the amino acids of the CRD of FimH are highly conserved among different E. coli isolates and many further Enterobacteriaceae, suggesting a reduced risk of resistance development.395–398 Studies with deletion mutants of fimH validated FimH as potential drug target. The fimH mutants lost the ability to bind to human and murine bladder cells and a lowered bacterial survival rate in murine kidney and bladder was reported.399,400 In addition, the therapeutic value of targeting FimH was further underlined by vaccination against FimH and use of antibodies directed against FimH, both resulting in significantly reduced colonization of murine bladder cells in vivo.400,401
The mannose ligand is bound by FimH in a deep, negatively charged pocket formed by Asn46, Asp47, Asp54, Gln133, Asn135, Asn138 and Asp140.395 An extensive network of direct and water-mediated hydrogen bonds, is formed between FimH and mannose (36), resulting in an unusual high affinity (KD = 2.3 μM, determined by SPR)402 for a monovalent lectin–carbohydrate interaction (Fig. 17).395 In this complex, every hydroxy group of mannose establishes direct hydrogen bonding except for hemiacetal O1, which engages in indirect H bonding via a water.402
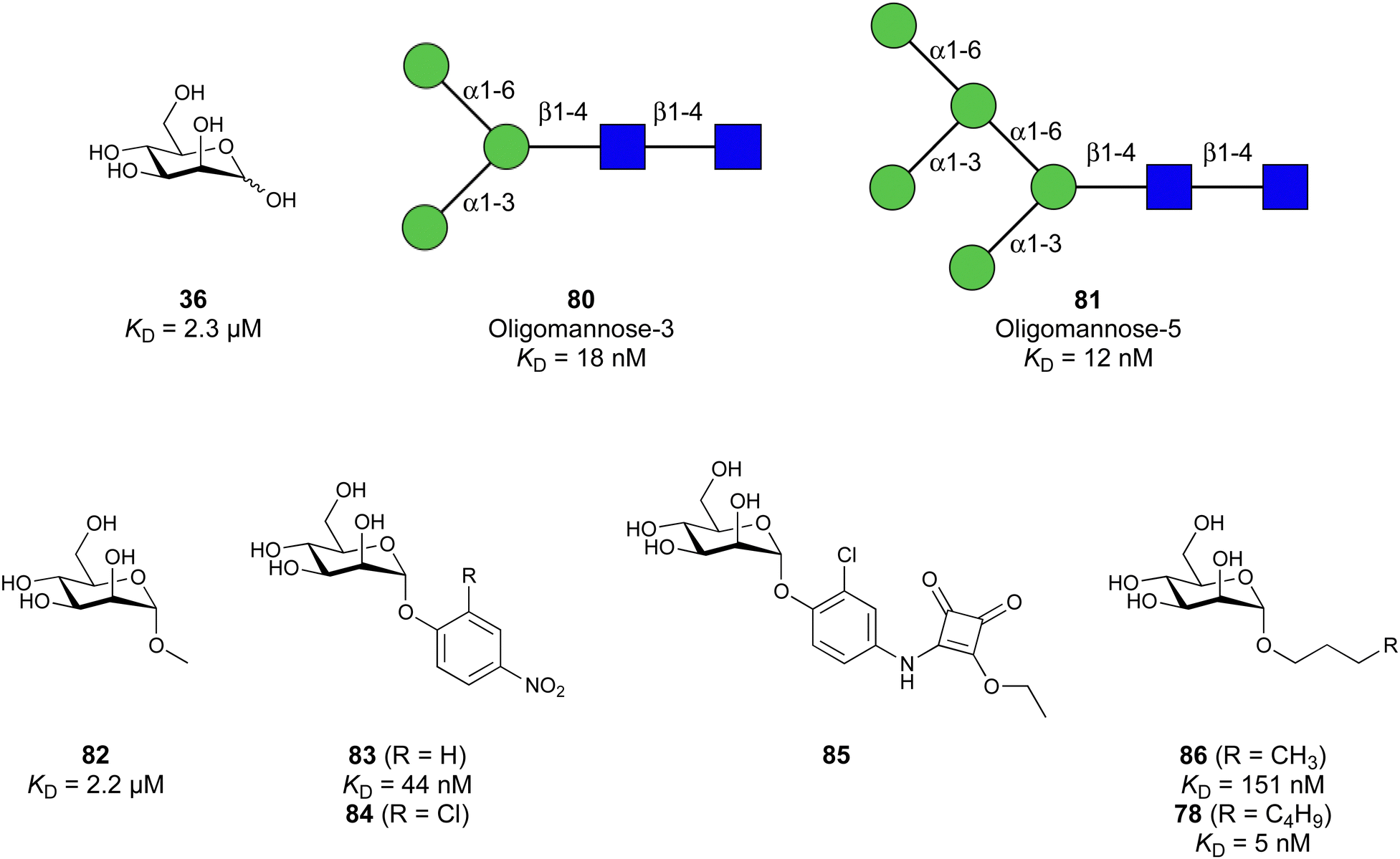 | ||
| Fig. 17 Natural occurring mannosides and early synthetic FimH ligands. Alkyl and aryl mannosides profit from additional hydrophobic interactions with the tyrosine gate surrounding the CRD compared to D-mannose (36). A small substituent in ortho position of the phenyl aglycon further improves affinity. | ||
α-Mannosides are bound with the aglycon pointing outwards of the binding pocket towards the protein surface, where a hydrophobic rim comprised of Phe1, Ile13 and Phe142 as well as the so-called ‘tyrosine gate’ of Tyr48, Ile52 and Tyr137 surround the entry to the deep, hydrophilic CRD.402 In particular, the tyrosine gate allows for differentiation between different mannose oligosaccharides and favourable interaction with the tyrosine gate is a key prerequisite for the development of high-affinity FimH inhibitors. The extraordinary affinities of the naturally occurring branched mannosides oligomannose-3 (80) and oligomannose-5 (81) (KD = 18 nM and 12 nM, determined by SPR) highlight the possibility to achieve nanomolar binding to FimH.403 Notably, Tyr48 of the tyrosine gate is conformationally flexible, resulting in an ‘in-docking’ pose (e.g. oligomannose-3, PDB code 2VCO404 or HM, PDB code 4XOE, Fig. 16C) and an ‘out-docking’ mode frequently reported for synthetic FimH inhibitors (e.g.3MCY,112Fig. 16D).405 Replacement of the mannose moiety by different sugars such as glucose leads to a significant loss of affinity, resulting in millimolar binding.402 Notably, some affinity is retained for binding of fructose (KD = 31 μM, determined by SPR).406 Further important features of the FimH binding pocket relevant for drug development are the Arg98–Glu50 salt bridge and a small hydrophobic pocket next to the CRD.407
Because of the extraordinary binding specificity of FimH for α-D-mannosides, the development of FimH antagonists mainly focused on the introduction of aglycons to mannose to improve binding by hydrophobic interactions. A notable exception to this is the report on septanoses to replace the mannose moiety, by Peczuh and Ernst et al. resulting in compounds with low micromolar IC50.410 Generally, the potency of compounds reported by different working groups cannot be directly compared due to a variety of different in vitro assays and in vivo models. An overview of the different in vitro and in vivo assays employed as well as a comprehensive summary of affinity data was published by Mydock-McGrane et al. in 2017.407
Early glycomimetics. Development of FimH inhibitors started in 1977, when methyl α-D-mannoside (MeαMan, 82) was reported as inhibitor of E. coli cell adhesion to human mucosal cells in vitro (Fig. 17).411 A 1979 study in mice revealed that MeαMan could prevent and reverse bacterial adhesion to bladder cells in vivo.412 Thereafter, MeαMan was used as reference for the potency of new FimH inhibitors. Sharon and Ofek, who conducted those studies, also postulated the existence of a hydrophobic region in close proximity to the CRD in 1983. In an aggregation assay of guinea pig erythrocytes with E. coli p-nitrophenyl α-mannoside (pNPαMan, 83) was found 125-fold more potent than MeαMan.413 Further investigation of aromatic α-mannosides by Sharon et al., led to the key discovery that phenyl ortho-substituents improve inhibitory potency. Inhibition of adhesion of E. coli O128 to guinea pig ileal epithelial cells by o-chloro-p-nitrophenyl α-mannoside (84) surpassed the inhibitory activity of MeαMan by a factor of 470, and importantly, the activity of pNPαMan (without ortho-substituent) almost 7-fold.414 Despite these early key observations in the structure–activity relationship of FimH inhibitors no significant progress in the development of aromatic α-mannosides was made until Lindhorst and colleagues published a novel scaffold in 2006, in which a squaric acid moiety was introduced to the para position of the phenyl aglycon.113 The resulting antagonist 85 showed further improved inhibitory activity due to enhanced interaction with the tyrosine gate. In an ELISA based assay a relative potency of 6900 (with reference to MeαMan) was achieved, an almost 35-fold increase compared to 84.
The affinity gain achieved by interacting with the hydrophobic tyrosine gate also led to the discovery of alkyl mannosides as FimH inhibitors. In crystallisation experiments by Knight and De Greve et al., butyl α-D-mannoside (BM, 86) was found in the binding pocket of FimH, although no sugar was added during crystallisation.402 The Luria-Bertani (LB) medium used for protein expression was hypothesized as origin of the high affinity ligand BM (KD = 151 nM, SPR), and after further investigation of simple alkyl mannosides, heptyl α-D-mannoside (HM, 78) was reported as potent binder of FimH (KD = 5 nM, SPR).402 Despite the high affinity for isolated FimH, a concentration of 1 mM HM was needed to completely prevent bacterial binding to bladder cells in vitro. Additionally, HM only showed a significant reduction of bacterial binding and cell invasion in a murine cystitis model when the uropathogenic E. coli strain UTI89 was incubated with a high concentration of 5 mM HM before inoculation.404
FimH inhibitors to treat and prevent UTI. A milestone in the development of FimH antagonists was the introduction of the biphenyl aglycon to mannosides. Following a structure-based drug design approach aided by docking experiments, the first compounds of this new class were reported by Janetka et al. and Ernst et al. in 2010.112,114 The highest affinities were reported for derivatives carrying the second phenyl ring (B ring) in para position of the first phenyl ring (A ring). Furthermore, a loss of affinity was reported for O-glycoside derivatives carrying one to three methylene units as a spacer between the mannose and aromatic aglycon, highlighting the close proximity of the CRD and the tyrosine gate.112,114
Exploration of the substitution pattern on the B ring by Janetka et al. revealed a preference of meta substituents over para and ortho, attributed to improved interaction with Tyr48 of the tyrosine gate and Arg90.112 To quantify the activity of the synthesized derivates, the Janetka group determined the 90% inhibition titers of guinea pig erythrocyte hemagglutination (HAI titer), of which biphenyl ester 79 and methyl amide 87 were potent inhibitors (HAI titers = 1 μM), an improvement of more than 1000-fold compared to MeαMan (HAI titer >1 mM), and 15-fold compared to HM (HAI titer = 15 μM) (Fig. 18). The co-crystal structure of FimH and ester 79 revealed the basis for the greatly improved activity. While the mannose residue was bound in the CBS as previously reported,402 the biphenyl moiety engaged in π-stacking with Tyr48 and hydrophobic interactions with Tyr137 of the tyrosine gate. Importantly, the methyl ester in meta position of the second ring (B ring) contributed to binding via an interaction with the salt bridge of Arg98 and Glu50 at the outer rim of the binding region (Fig. 16D).112
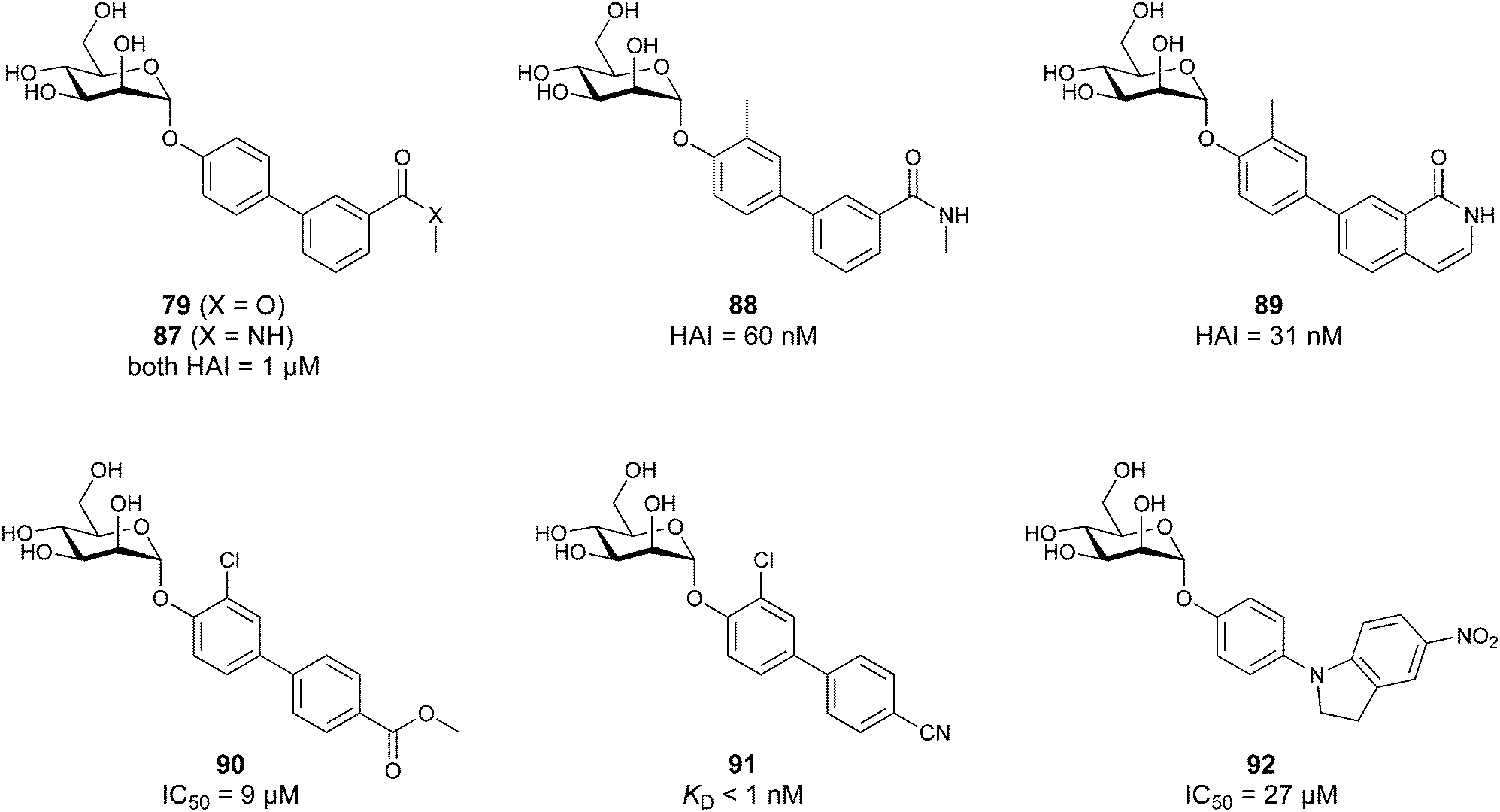 | ||
| Fig. 18 Aromatic FimH inhibitors developed by the Janetka (79, 87 to 89) and Ernst (90 to 92) groups. Biphenyl aglycons and their heterocyclic analogues engage in favourable interactions with the tyrosine gate of FimH, explaining the high affinity and efficacy of the respective mannosides. | ||
Methyl amide 87 was chosen as the preferred lead structure due to a higher metabolic stability than ester 79. Introduction of a methyl group as ortho-substituent on the A ring resulted in the orally bioavailable lead compound 88, with improved affinity (HAI titer 60 nM) (Fig. 18).415 Dosed at 50 mg kg−1 by peroral administration (p.o.), 88 was able to reduce the bacterial burden in a murine chronic UTI model by approximately 3 log10 units 6 h after infection.416,417 Furthermore, establishment of an UTI with multidrug-resistant E. coli ST131 was prevented by 88, an effect not achieved by preventive administration of the antibiotic combination trimethoprim/sulfamethoxazole.418 These excellent results in the chronic UTI model were further surpassed by a reduction of 4 log10 units by isoquinolone 89 (HAI titer 31 nM), a cyclic amide analog of 88.416 Notably, derivatives of isoquinolone 89 showed further improved in vitro activity, in the low nanomolar range, but lacked significant efficacy in vivo in mice (50 mg kg−1 p.o.).
From a series of para substituted biphenyl mannosides the Ernst group identified ester 90 as early lead compound. 90 reached an IC50 value of 9 μM in an E. coli disaggregation assay from guinea pig erythrocytes compared to the IC50 value of 77 μM reported for HM.114 Importantly, the carboxylate metabolite does not significantly lose activity (IC50 = 10 μM). After extensive in vitro studies on binding affinity and pharmacokinetic properties, as well as the first in vivo PK study, the efficacy of lead compound 90 was tested in a murine UTI model. Dosed at 50 mg kg−1 p.o. before transurethral infection with the UPEC strain UTI89, 90 led to a decrease of bacterial burden in urine by 2 log10 colony forming units (CFU) and in the bladder by 4 log10 CFU. Importantly, the same reduction was achieved by intravenous (i.v.) application of the carboxylate metabolite of ester 90 (50 mg kg−1) confirming oral bioavailability and potency of lead mannoside 90.114 Optimisation of the PK/PD profile, with special focus on the plasma half-life to prolong the effective duration resulted in the bioisostere nitrile 91, with steady renal excretion over 8 hours.419 Preventive administration of 91 (10 mg kg−1, p.o.) was more effective than administration of the antibiotic ciprofloxacin (8 mg kg−1, subcutaneous, s.c.) in reducing the bacterial count in the bladder (reduction of 2.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10vs. 2.4log10 CFU). Further, fluorination of the B ring led to sub-nanomolar binders to the isolated (high-affinity) FimH domain, but efficacy data were not reported.420
log10vs. 2.4log10 CFU). Further, fluorination of the B ring led to sub-nanomolar binders to the isolated (high-affinity) FimH domain, but efficacy data were not reported.420
To allow binding in the “in-docking” mode, Ernst et al. synthesized a series of phenyl triazole compounds with varying spacer length and modifications to the anomeric centre.421 No improvement of the binding affinity compared to HM or biphenyl aglycons was achieved and, furthermore, triazole mannosides are not predicted to be orally bioavailable due to limited membrane permeation.
Replacement of the B ring phenyl with heterocycles led to the discovery of 92 as highly potent FimH inhibitor (Fig. 18).422 Despite dosage limits due to low solubility, the concentration to prevent 90% of bacterial adhesion by UPEC (EC90) was exceeded for more than 8 hours after i.v. administration of 1 mg kg−1 in a PK mouse study. Efficacy of 92 was proven by preventive administration in a mouse model. Dosed at 1 mg kg−1 (i.v.), 92 achieved a 10000-fold reduction of CFU in the bladder of mice, an effect equally potent to the antibiotic ciprofloxacin (8 mg kg−1, s.c.) commonly used to treat UTIs. Despite higher in vitro potency of the analogue carrying an additional chloro substituent in ortho position of the proximal phenyl ring (ring A), the compound was not evaluated in vivo due to limited solubility.422
Due to the plethora of diverse mannose-binding proteins in humans (e.g. DC-SIGN, mannose binding protein, dectin-2, langerin), selectivity of glycomimetics for their intended target is of crucial importance. A selection of compounds, including HM and biphenyl mannosides, showed a 100000-fold higher affinity for FimH compared to the tested human mannose binding proteins, indicating binding selectivity is not a problem in the development of monovalent FimH inhibitors.423
A general problem of biphenyl mannosides is their low solubility, limiting the therapeutic dose, and their low bioavailability, at least partially due to the low stability of the O-glycosidic bond. Janetka and colleagues investigated acetate and phosphate esters, as well as a glycine derivative as prodrug strategies to improve oral bioavailability and prolong compound exposure, with positive results for tetraacetate (93) and 6-phosphate mannoside prodrugs (Fig. 19).424
 | ||
| Fig. 19 Strategies to improve oral bioavailability of aromatic FimH inhibitors include prodrugs (top) and modification of the O-glycosidic bond (bottom). | ||
In particular, the use of phosphate prodrugs is commonly applied to drugs suffering from limited solubility. The phosphate prodrug lacks membrane permeability and acts as soluble reservoir for the active agent, which is released by enzymatic hydrolysis prior to uptake. Thereby, the concentration of free, poorly soluble drug is limited, but a high dose can be administered. Following this strategy, the Ernst group synthesized a series of mannosides as phosphate esters (94 and 95) reaching excellent aqueous solubility (125-fold improvement for heterocyclic 92; 15-fold improvement for nitrile 91, >3000 μg mL−1vs. 192 μg mL−1) (Fig. 19).425
Depending on the position of the phosphate ester, the rate of hydrolysis varied. Compounds carrying the phosphate at O4 or O6 of mannose displayed a desired longer half-life (t1/2 > 40 min) compared to O2 or O3 derivatives (t1/2 < 15 min). In an in vivo PK study in mice, a doubling of the urine AUC0-24 was achieved by p.o. administration of the O4 phosphate ester prodrug (94) of lead nitrile 91 (Fig. 18 and 19).
Efforts to improve metabolic stability of the mannosides included replacement of the glycosidic oxygen by carbon, nitrogen, and sulfur (96–98).424,426,427 In a seminal work, Janetka and colleagues reported on C-mannosides with an (R)-hydroxy methylene unit linking the carbohydrate and its biphenyl aglycon.424 This replacement resulted in potent inhibitors of same or higher affinity, with significantly improved metabolic stability compared to the O-glycoside. In a murine chronic cystitis model, C-mannoside 99 dosed at 25 mg kg−1 (p.o.) reduced the CFU in the bladder by 3 log10 units after 12 hours, while O-glycoside analogue 88 did not result in a significant reduction of bacterial burden. Furthermore, introduction of the (R)-configured hydroxy methylene unit to inhibitors bearing an isoquinolone moiety (as 89) resulted in additional promising preclinical candidates. The pronounced stereochemical selectivity for the (R)-epimer is due to a water-mediated hydrogen bond to Asp140 and Asn135 formed by the (R)-hydroxy group, an interaction that is not possible for the (S)-epimer, as predicted by docking.
In 2012, Janetka and Hultgren co-founded Fimbrion Therapeutics which further pursues the development of FimH inhibitors, since 2016 in cooperation with GlaxoSmithKline. With financial support by CARB-X, the first clinical Phase I evaluation of the joint lead compound GSK3882347 (undisclosed structure) in healthy individuals was successfully completed in May 2021 (NCT04488770). A second Phase I study to investigate the pharmacokinetics and microbial response in women with uncomplicated acute UTI was started in May 2022 and is expected to end in December 2023 (NCT05138822).
FimH inhibitors to treat Crohn's disease. In the context of Crohn's disease (CD), the site of infection is the intestine, requiring alternative strategies for inhibitor design with regards to pharmacokinetics: while treating UTIs requires enteral absorbance and renal excretion of the parent drug or of an active metabolite, no systemic bioavailability is required to treat CD in the gut. Gouin and colleagues investigated thiazolylaminomannosides (TazMan) with heterocyclic aglycons to treat CD.428 Notably, lead 100 (relative inhibitory concentration compared to HM, 0.2) engages in more favourable fashion with Tyr48 and Tyr137 of the tyrosine gate, compared to the phenyl moiety in biphenyl aglycons (Fig. 20). Variation of the linker length and replacement of the anomeric nitrogen by carbon or sulfur improved stability but resulted in less potent compounds.429
 | ||
| Fig. 20 FimH inhibitors under investigation for the treatment of Crohn's disease. 100 is a representative of the thiazolylaminomannoside (TazMan) class of FimH inhibitors, which noticeably interact with the tyrosine gate in a more favourable way compared to biphenyl aglycons. Terminal modification of the heptyl chain in 101 significantly improved the in vivo efficacy compared to HM. Replacement of the anomeric oxygen improved metabolic stability (102). Divalent C-mannoside Sibofimloc has been advanced to Phase IIa clinical trials. | ||
Additionally, Gouin et al. investigated further alkyl mannosides. They hypothesized unfavourable pharmacokinetic properties caused by the unmodified alkyl chain in HM as cause for its poor activity in vivo. By terminal functionalization amide derivative 101 was obtained, which in contrast to HM showed promising results of reduced gut colonization and inflammation in vivo in a murine model of Crohn's disease (Fig. 20).430 Exchanging the anomeric oxygen for a methylene in 102 increased metabolic stability and simultaneously lowered systemic bioavailability from 25% to 16%, as desired for the treatment of Crohn's disease. Importantly, compound 102 (10 mg kg−1, p.o.) was able to eradicate AIEC from the ileum of transgenic mice. This observation serves as proof of concept that patients suffering from Crohn's disease with increased prevalence of AIEC can be treated orally with FimH antagonists.431
A major industrial player in the preclinical development of FimH inhibitors is Vertex Pharmaceuticals, who filed patents on a plethora of diverse mannose-based FimH inhibitors, including disaccharides and divalent ligands, with varying aglycons, intended for the treatment of UTIs and inflammatory bowel diseases.432–436 The portfolio was licensed to Enterome in 2016, where Sibofimloc (EB8018/TAK-018, 103), a divalent and gut-restricted FimH ligand, was identified as lead compound for the treatment of Crohn's disease (Fig. 20). In vitro adhesion of AIEC to primary ileal cells and T84 epithelial cells was blocked by 1 μM Sibofimloc, resulting in reduced inflammation and tissue damage.383 After successful completion of Phase I studies on safety and tolerability (NCT02998190) as well as pharmacokinetics (NCT03709628), a Phase IIa study to investigate the preventive effect of Sibofimloc on the postoperative recurrence of Crohn's disease was initiated in cooperation with Takeda Pharmaceutical in 2020 (NCT03943446). However, problems to recruit suitable participants led to termination of the study in August 2022.
3.1.4.2 E. coli F9 fimbrial adhesin FmlH. In addition to type 1 fimbriae and the FimH adhesin, E. coli often utilize the related UPEC F9 fimbriae with the adhesin FmlH to adhere to galactose and N-acetyl galactosamine conjugates, such as the Thomsen–Friedenreich (TF) antigen on kidney and bladder tissue in urinary tract infections.437 F9 fimbriae are homologous to type 1 fimbriae, and as such, FmlH is also related to FimH, although it displays a different carbohydrate-binding specificity.
Complementary to their FimH research, Janetka and Hultgren have therefore embarked on the synthesis of FmlH inhibitors.118 To this end, a number of GalNAc glycosides with varying aglycons have been synthesized and tested for inhibition of FmlH binding to immobilized TF-antigen, followed by infection studies in mice. The crystal structures of FmlH in complex with the TF-antigen or oNP-Gal (104) revealed a tight coordination of the galactose moiety in both ligands and demonstrated the possibility of varying the aglycon (Fig. 21). It was further demonstrated that substitution of galactose (KD = 694 μM) with GalNAc (KD = 189 μM) enhances binding affinity of FmlH inhibitors, as determined by bio-layer interferometry. An extensive synthesis campaign also revealed substituted biphenyl glycosides as potent binders of FmlH. The most potent ligand was the β-GalNAc biphenyl carboxylate 106 with a KD of 89 nM, and for its galactose analogue 105, a 50-fold lower binding of KD = 2.1 μM was determined. This selectivity of FmlH for GalNAc was rationalized by an attractive hydrogen-bonding interaction of the acetamide with a protein-bound water molecule in the co-crystal structure with the protein. In a murine cystitis model, it was demonstrated by transurethral instillation that the potent FmlH inhibitor 106 synergizes with a FimH inhibitor (mannoside 4Z269) in reducing colonization of the kidney, while in the bladder no synergy was observed and the FimH inhibitor dominated (Fig. 21).
 | ||
| Fig. 21 Inhibitors of the F9 fimbrial adhesin FmlH of uropathogenic E. coli. In contrast to its namesake FimH, the adhesin FmlH shows the highest affinity for ortho-biphenyl substituted galactosides and GalNAc glycosides. | ||
The team then further optimised these compounds and identified a methylsulfonamide as a bioisostere of the carboxylate, as well as a beneficial effect of an additional CF3 group in the proximal phenyl meta to the carbohydrate.119 The resulting compound 107 showed an IC50 of 34 nM in a competitive, ELISA based binding assay, which was 20-fold more potent than the initial lead 106 (Fig. 21). The co-crystal structure of 107 with FmlH revealed that both the additional CF3 group and the sulfonamide residue form attractive interactions with the protein, providing an explanation for the observed activity increase. Importantly, compound 107 displayed an increased metabolic stability and an extended plasma half-life, but unfortunately, also a low bioavailability of only 1% after p.o. administration and only a low renal clearance were observed, requiring further optimisation of its PK properties.
3.1.4.3 The soluble lectins LecA and LecB of Pseudomonas aeruginosa. The Gram-negative bacterium P. aeruginosa is currently the most critical bacterial pathogen as defined by the WHO priority pathogen list. This bacterium is difficult to treat due to excessive development of resistance to antibiotics and its abundant biofilm formation.438 The latter is a major resistance determinant of this pathogen since the biofilm shields embedded bacteria from chemotherapy and host defence. Therefore, several approaches to identify new anti-infectives against this bacterium aim to block biofilm formation.439
P. aeruginosa utilises the two lectins LecA (PA-IL) and LecB (PA-IIL) for initial adhesion to the host, for biofilm formation and as virulence factors.440–442 Both proteins have first been identified by Gilboa-Garber et al. and their carbohydrate binding specificity was determined.443 In particular, it was shown that both lectins inhibit ciliary beating,444,445 wound healing446,447 and impact on cell physiology448,449 and immunity.450,451 In addition, LecA was shown to promote host cell invasion by P. aeruginosa.452 In first-in-human studies, aerosols containing the monosaccharides D-galactose and L-fucose as ligands of LecA and LecB, respectively, have shown beneficial effects on patients with P. aeruginosa lung infections after inhalative administration.453,454 Further, adjunctive therapy in mice with the antibiotics ceftazidine or ciprofloxacin revealed synergistic effects when galactose, mannose and fucose were co-administered with the bacterial inoculum to the murine lung.455
LecA binds D-galactose (108, Fig. 22) and conjugates thereof and its native ligand is presumably the glycosphingolipid Gb3 that was identified from a glycan array screen.456 The structure of LecA in complex with galactose was solved by X-ray crystallography (Fig. 23A).457 This structure reveals the oligomerization of LecA into a homotetramer with the binding sites on the vertices of a rectangle and further demonstrates the calcium ion-mediated recognition of the carbohydrate ligand: the 3- and 4-hydroxy groups coordinate to the protein-bound calcium ion whereas the 6-hydroxy group is recognized in a small water-occupied pocket and involved in direct hydrogen bonding with His50 and Gln53, as well as with Gln53 and Pro51 via water-mediated hydrogen bonds. In addition, the 2-, 3-, and 4-hydroxy groups form hydrogen bonds with Asn107 and Asp100.
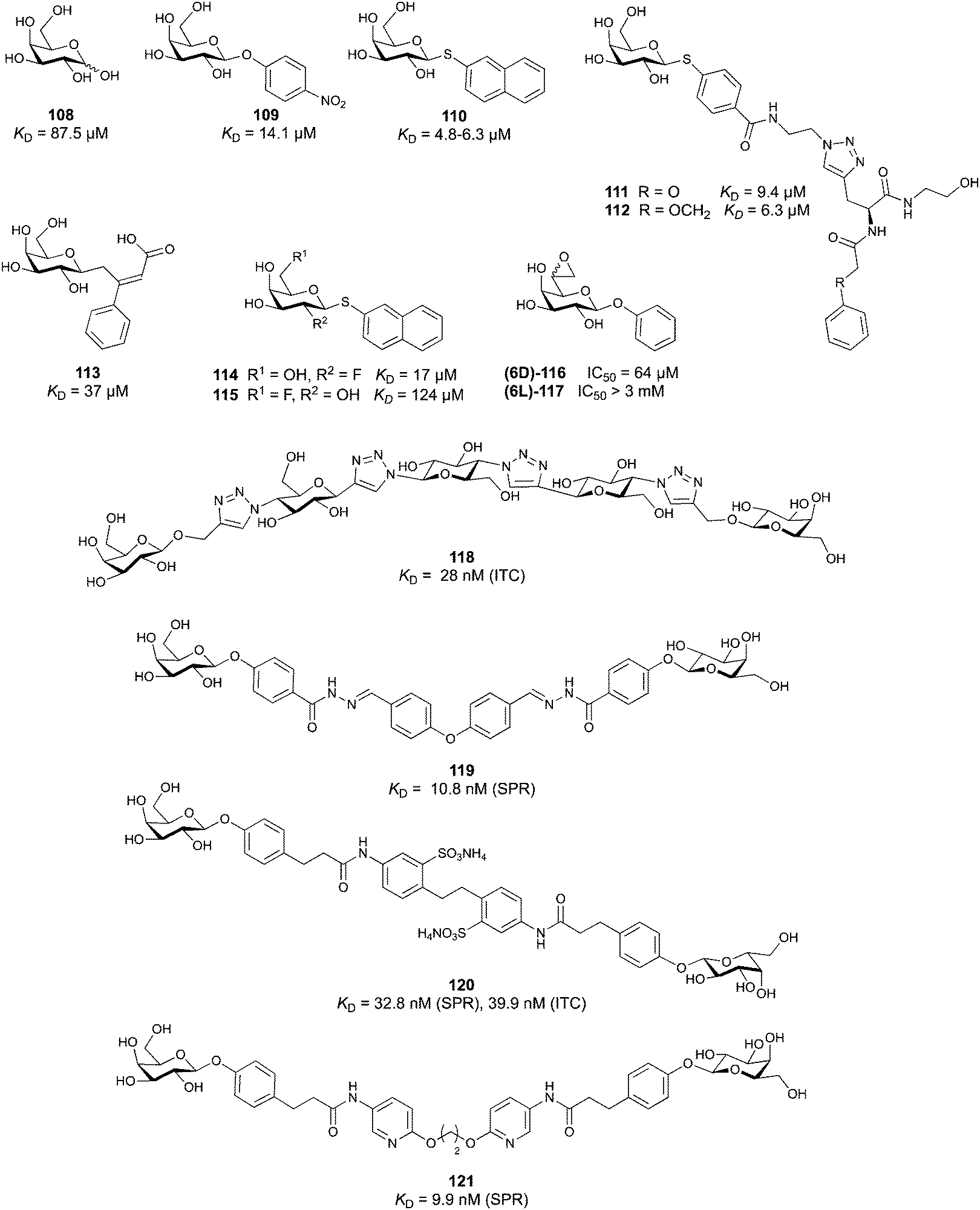 | ||
| Fig. 22 Glycomimetic inhibitors derived from galactose for the bacterial lectin LecA. Notably, (6D)-116 is an irreversible LecA inhibitor as nucleophilic attack of Cys62 to the epoxide warhead leads to covalent binding. | ||
 | ||
| Fig. 23 LecA forms a homotetramer with the carbohydrate binding sites arranged on the vertices of a rectangle (A, PDB code 1OKO). Binding sites occupied with various ligands are magnified: Gal-α(1,3)-Gal-β(1,4)-Glc (B, PDB code 2VXJ), 109 (C, PDB code 3ZYF), epoxide (6D)-116 (D, PDB code 5MIH), and central pocket targeting ligand 111 (E, PDB code 7FIO). The divalent ligand 118 bridges two adjacent binding sites in the LecA tetramer (F, PDB code 4YWA). His50 establishes CH–π contacts with aryl aglycons in (C–E) and forms a hydrogen bond with O6 in LecA ligands. In the crystal structure of LecA with (6D)-116 obtained at pH 4.6 (D), Cys62 does not form the covalent bond observed in solution at physiological pH and the epoxide remains intact. Calcium ions are depicted as green spheres, a tightly coordinated water molecule is shown as red sphere. | ||
The search for inhibitors of LecA surprisingly identified β-aryl galactosides (e.g. pNP-Gal 109) as more potent inhibitors for LecA compared to D-galactose or α-galactosides (Fig. 23B).458 In a crystallographic study, the molecular basis has been attributed to a CH–π interaction between the aryl aglycone and the imidazol side chain of His50 (Fig. 23C),459 and consequently the naphthyl galactoside 110 showed high binding to LecA (KD = 4.2 μM), while D-galactose has a KD of 87.5 μM. Various SAR studies on an aromatic aglycone have been reported and in general, substitution is tolerated on the phenyl ring, but major additional increase in affinity has not been achieved to date.459–461 Recently, the Titz group identified a further druggable pocket located between two LecA monomers and close to the carbohydrate binding site and adjacent to the His50-bound phenyl aglycon.462 To this end and inspired by a multivalent system of Winssinger et al.,463 they systematically elongated the aglycon with flexible spacers and attached aryl groups for their ligation to the central pocket as predicted by docking. The crystal structure of 111 (KD = 9.4 μM) in complex with LecA indicated the phenyl pharmacophore close to the entry of the central pocket (Fig. 23E). Further, a ligand with an increased chain length (112, KD = 6.3 μM) showed a high enthalpy of binding (−50 kJ mol−1) which was counterbalanced by high entropic costs (20.7 kJ mol−1) indicating a conformationally restricted bound state. However, further experiments are necessary to validate its binding inside the central pocket.
Replacing the glycosidic oxygen to increase metabolic stability by introducing thioglycosides (see 110) has been pursued and is well tolerated by LecA.458,460 Another work reports on the synthesis of β-C-galactosides with positioning of a phenyl ring further away from the galactose residue which resulted in significant loss of binding affinity with one of the derivatives reaching a KD of 37 μM (compound 113, Fig. 22).464
Giguère and co-workers reported on the systematic single and multiple exchange of all hydroxy groups with fluorine atoms in galactosides as tools to assess lectin binding.465 It was shown that the 2-deoxy-2-fluoro galactoside 114 resulted in an approximately 3-fold loss in binding affinity by ITC when compared to the parent galactoside (e.g. 17 μM vs. 4.8 μM for the thionaphthyl glycoside 110). The enthalpy of LecA-binding is strongly reduced in those fluoro-analogues by approx. 7–10 kJ mol−1. However, this work also demonstrates the lower entropic costs by 4–6 kJ mol−1 for binding of these fluoroderivatives compared to the galactosides as a result of different desolvation penalties which to some extent counterbalances the unfavourable enthalpy. For the 6-deoxy-6-fluoro analogue 115, the binding affinity was drastically reduced and 25-fold lower affinities were reported. A similar observation of an affinity reduction has been made when the 2-OH group in D-galactose was replaced by NHAc in GalNAc.458,466
Binding kinetics are often fast for carbohydrate–lectin interactions and consequently, the binding affinity of a given ligand is lowered when fast dissociation rates are in place (lit.467 and references therein). Furthermore, a sufficiently long residence time of a given inhibitor on a receptor has been identified as crucial for success in vivo.468 In some cases, the success of glycomimetic inhibitors resulted from their extended receptor-residence times, e.g. for the FimH antagonists467 or LecB inhibitors.469,470 However, for LecA even some of the best monovalent ligands with KDs <3 μM showed a very fast dissociation from the protein.471
Therefore, the covalent inhibition of a given target can provide an opportunity to overcome fast dissociation by irreversibly blocking its function. Covalent inhibition is an approach with increasing success in clinical use for inhibition of unrelated proteins, e.g. proteases or kinases.472 Inspection of the crystal structure of LecA in complex with a galactoside reveals the presence of a cysteine residue in close proximity to the carbohydrate. Cysteines are nucleophiles and therefore often targeted using electrophilic warheads, for example in covalent inhibitors of cysteine proteases. In the case of LecA, Cys62 is located in close proximity to the hydroxymethyl group of galactose, and therefore this position was selected for the introduction of an electrophile in LecA inhibitors.473 Since this part of galactose is accommodated in a rather small pocket in LecA, epoxides were chosen as sterically least demanding electrophiles. To this end, phenyl galactoside was extended to the diastereomeric heptose epoxides 116 and 117 (Fig. 22). Biophysical analysis of these molecules with LecA revealed a weak but selective inhibition of LecA by one diastereomer (116, IC50 = 64 μM), whereas the other diastereomer 117 was inactive. Protein mass spectrometry then unambiguously demonstrated the covalent bond formed between Cys62 and the epoxide in 116 under physiological conditions, providing the experimental proof for the first-in-class covalent lectin inhibitor. Surprisingly, crystallographic analysis of LecA in complex with 116 revealed its non-covalent ligation with the epoxide ring intact (Fig. 23D). This unexpected observation could be explained by the acidic conditions for LecA crystallisation which lower the cysteine's nucleophilicity.
In general, numerous attempts to improve binding affinity for LecA on the monovalent galactoside level have delivered an in-depth knowledge on the SAR. Unfortunately, all those studies resulted in only moderate affinity increases and the best compounds consist of β-galactosides carrying naphthyl (110) or coumaryl aglycones. These substituents may, however, encounter problems in further translation due to toxicity.
To overcome this rather moderate affinity of the small molecules, numerous approaches have exploited the quaternary structure of LecA as target rather than focussing on a single binding site. To this end, oligo- and multivalent galactosides presented on a large diversity of scaffolds were developed which are beyond the scope of this review (the interested reader is referred to references75,131,474 and references of this ChemSocRev issue). However, most of these multivalent ligands have been studied for lectin binding only. Only few molecules were tested further: galactosylated peptide dendrimers have shown good potency in antibiofilm experiments in vitro475 and galactosylated calixarenes showed in vivo efficacy to reduce lung damage in an acute murine lung infection model in a preventive treatment regime using co-administration of bacteria and inhibitor.476 Another approach worth mentioning here are divalent precision ligands containing two terminal galactosides tailored to match the simultaneous binding to two adjacent galactose-binding sites in LecA. This approach was pioneered by the Pieters lab and exploits the close proximity of these two sites.477–480 One molecule that consists of two galactosides spaced by a number of triazoles and glucose moities, 118, could even be cocrystallised with LecA and the chelating binding mode was proven (Fig. 23F). Non-carbohydrate spacers have been developed by Titz et al. to enable rapid synthetic access and implement drug-like properties, yielding a highly potent divalent LecA inhibitor 119 (KD = 10.8 nM) that showed a high selectivity over human Galectin-1 as potential off-target.471 Recently, these bisacylhydrazone spacers have been bioisosterically replaced with amides to overcome the drawbacks of this labile and potentially toxic linker function.446 The resulting molecules showed superior metabolic stability and much higher aqueous solubility (e.g. for 120 up to >1.5 mM vs. 1.6 μM for the bisacylhydrazones) and the best derivative 121 bound to LecA with a KD of 9.9 nM. More importantly, the increased solubility now enabled ITC analysis and evaluating the molecules’ antivirulence properties in cell culture and in vitro infection experiments.
In contrast, LecB binds to L-fucose 122 and D-mannose 36 and their conjugates (Fig. 24). Following a glycan array screening, the blood group antigen LewisA has been identified as ligand with the highest affinity.481,482 The crystal structure of LecB from P. aeruginosa PAO1 has been solved in complex with its ligands (Fig. 25)483,484 and a recent neutron diffraction structure of LecB in complex with L-fucose allowed the analysis of ligand and protein protonation states.485 As observed for galactophilic LecA and despite the unrelated primary protein sequences, LecB also forms homotetramers albeit with a different orientation of the binding sites. In LecB, the carbohydrate binding sites are located on the vertices of a tetrahedron and thus, spatially maximally apart from each other (Fig. 25A). Surprisingly, the crystal structure revealed that two calcium ions are located inside a single carbohydrate binding pocket and these ions mediate the binding of the carbohydrate ligand with the protein. The 2-, 3-, and 4-hydroxy groups of fucose are directly coordinated to the two calcium ions with the 3-hydroxy group simultaneously serving as a μ-bridging ligand to both metal ions. In addition, the ligand is also heavily involved in hydrogen bonding with the protein. The presence of the two metal ions as well as an additional lipophilic contact of the C6 methyl group of fucose served as an explanation for the unusual high affinity of a monosaccharide for a lectin (KD = 2.9 μM).482D-Mannose possesses the same relative orientation of its three ring hydroxy groups as those in L-fucose. D-Mannose can therefore also bind in a similar orientation to LecB, but with the absence of the lipophilic interaction of fucose, the affinity for D-mannose is reduced.
 | ||
| Fig. 24 Inhibitors of the P. aeruginosa lectin LecB are derived from L-fucose (122), D-mannose (36) or a hybrid of both (133). | ||
 | ||
| Fig. 25 LecB forms a homotetramer with the carbohydrate binding sites arranged on the vertices of a tetrahedron (A, PDB code 1GZT). Binding sites occupied with various ligands are magnified: LewisA (B, PDB code 5A6Z), C-fucosyl peptide 127 (C, PDB code 3DCQ), fucose–mannose hybrid 137 (D, PDB code 5MAZ), β-N-fucoside 139 (E, PDB code 8AIY), and α-N-fucoside 140 (F, PDB code 8AIJ). Calcium ions are depicted as green spheres. | ||
Importantly, despite the fact that P. aeruginosa is a highly variable organism at the genomic level, the genes encoding for LecA and LecB are part of the more conserved core genome.486 Nevertheless, the sequence of LecB varies among clinical and environmental isolates.487,488 Interestingly, the sequence of LecB has been established as correlative marker for the two major clades in P. aeruginosa phylogeny, one containing the clinical isolate PAO1 and the other containing the highly virulent clinical isolate PA14.487 It was demonstrated by glycan array analysis and biophysical characterization that the carbohydrate-specificity in LecB from PA14 is conserved, despite a high number of amino acid variations, some of which located in the carbohydrate binding site. Moreover, it was shown that the binding affinity of LecBPA14 for its ligands is even 3- to 7-fold higher than the one of LecBPAO1. As observed from mammalian glycan array binding data, LecB from both strains potently binds to a broad diversity of glycans carrying either fucose residues, or mannose residues or both, which suggests a decisive role of LecB for bacterial adhesion to the host.
Due to the high affinity of LecB for L-fucose, the search for LecB inhibitors focussed on this deoxyhexose as a starting point (Fig. 24).439,489 In eukaryotes, L-fucose is generally an α-linked terminal carbohydrate in glycoconjugates. Therefore, α-linked fucosides have initially been at the centre of attention and methyl α-L-fucoside 123 revealed a strong affinity for LecBPAO1 (KD = 0.43 μM).482 In fact, the anomeric isomer methyl β-L-fucoside 124 is an up to 500-fold weaker ligand for LecB.487 Since the trisaccharide LewisA (Fuc-α-1,4-(Gal-β-1,3)-GlcNAc, Fig. 25-B) showed the highest potency for LecBPAO1 (KD = 0.21 μM)482 and its interaction with LecB revealed no contact with the galactose residue, Fuc-α-1,4-GlcNAc truncates with triazoles at the reducing end of GlcNAc have been synthesized and their affinity (e.g.125, KD = 0.29–0.31 μM) remained nearly as potent as the one of LewisA.490 Along these lines, Bernardi, Imberty et al. reported α-linked fucosyl amides that were predicted by docking to establish an additional hydrogen bond between their carbonyl oxygen and Ser23.491 The best derivative 126 showed low micromolar affinity with a KD of 1.2 μM. α-C-fucosides have also been described to implement chemical/metabolic stability and allow conjugation to peptide dendrimers.492 The affinity of the tripeptide conjugate cFuc-Lys-Pro-Leu-NH2 (127) was equipotent to nitrophenyl α-L-fucoside (IC50 = 5.3–5.9 μM), which was twice better than the reducing L-fucose in the same assay (Fig. 25C).
Since terminal α-L-fucosides are abundant in humans and numerous endogenous lectins bind to fucosides,1 carefully addressing the selectivity of LecB inhibitors is highly relevant, especially when used in multivalent systems. To address this concern, the Titz group has chosen methyl mannoside 128 as a starting point for derivatization.493 The methylene-linked 6-OH group is an equatorial substituent in mannose and therefore allows contact to LecB, in contrast to the aglycones of α-fucosides that are axial substituents and point towards the solvent. The latter could serve as an explanation that no increase in affinity for α-fucosides has been achieved.
In the first set of molecules, the 6-hydroxy group was replaced by an azide that yielded a library of triazoles and after reduction amides, sulfonamides and amines. Sulfonamides have been identified as the most potent derivatives and the binding affinity was increased from 71 μM (KD for methyl α-D-mannoside) to a KD of 3.3 μM for sulfonamide 130 and cinnamide 129 also showed moderate affinity (KD = 18.5 μM). It was demonstrated that the increased binding affinity of sulfonamides and cinnamides results from an approx. 10–25-fold decreased dissociation rates of those derivatives from their complex with LecB (koff = 0.6–1.8 × 103 s−1) compared to MeαMan (koff = 15.5 × 103 s−1).469 In the next step, these mannosides were homologized to mannoheptoses with two aims: (i) those mannoheptoses have a free 6-OH that could engage in hydrogen bonding with Ser23 as observed for mannose in the crystal structure494 with LecB and (ii) the (sulfon-) amide substituent could be moved further towards the identified subpocket in this direction.495 Both diastereomers were synthesized and analysed for LecB inhibition: although a selectivity for the (6S)-heptose 131 over its (6R)-diastereomer 132 was demonstrated, IC50 values remained high (IC50 = 82–217 μM). In another work, they further analysed the function of this OH group, originating from Man-O6 which establishes a hydrogen bond to Ser23 in the crystal, to quantify its contribution to binding in solution.496 Furthermore, they set out to test the hypothesis of a possible synergistic effect of the Fuc methyl group and said Man-O6 hydroxy group in LecB inhibitors. To this end, a hybrid of L-fucose and D-mannose was synthesised, resulting in compound 133 and deoxygenated to 134. Both molecules were tested in a competitive binding assay and by ITC for LecB binding and surprisingly, they showed very similar thermodynamic profiles with comparable KDs of 16–17 μM. These data led to the conclusion that synergism for binding affinity is not obtained and that the hydrogen bond formed between mannose-6-OH and Ser23 in the solid state does not contribute to the binding affinity in solution.
However, these molecules are C-glycosides which increases their chemical and metabolic stability. Furthermore, their combination with the sulfonamide substituents likely also increases selectivity since the resulting molecules are neither terminal α-linked fucosides nor terminal mannosides, the common ligands for many endogenous lectins. Therefore, the Titz group designed and synthesized a series of these derivatives as sulfonamides and amides.470,497 As observed for the mannosides, especially the sulfonamide derivatives 135, 136 and 137 showed very potent binding of LecBPAO1 (KD = 0.83–1.27 μM) and LecBPA14 (KD = 290–320 nM). The crystal structure of those molecules in complex with LecB revealed that the aromatic substituents are ligated to a subpocket adjacent to the carbohydrate binding site and form extensive contacts with various hydrophobic amino acids, e.g. Val69, and methyl-substituted derivatives 135 and 137 revealed a deeper binding in the pocket (Fig. 25D). With regards to binding kinetics, the ligands’ off-rate could be further reduced to koff = 0.41 × 103 s−1 which results in receptor-residence times of 28 min, compared to 45 s for MeMan. Importantly, two derivatives were then assessed for selectivity over the human lectin langerin. While the thermodynamic affinity of 135 for LecB is 1.27 μM (from PAO1) or 310 nM (from PA14), the KI for the fucose-binding langerin was 2.2 mM, defining a selectivity window of a factor 1700–7000. In addition, 135 and other glycomimetics were also tested more globally on their effect on the immune response by quantifying the TNF-α response from primary murine spleen cells upon addition of various immune stimuli acting via diverse immune response pathways. No effect of the sulfonamide glycomimetics 130 or 135 (up to 1 mM) on the production of TNF-α was observed after stimulation, further supporting their selectivity.
Several glycomimetics were then tested in a biofilm inhibition assay using P. aeruginosa PA14 constitutively expressing mCherry and quantification of biofilm mass by confocal fluorescence microscopy. In 48 h biofilm formation experiments, a dense biofilm was observed in absence of LecB inhibitor, whereas in the presence of 100 μM glycomimetics biofilm formation was inhibited by 50–90%, and C-glycosidic sulfonamides 135 and 136 showed the highest potency of inhibition (80–90%). Importantly, in presence of MeαMan or even the very potent LecB ligand MeαFuc (KD = 202 nM for LecBPA14)487 biofilm mass was only non-significantly reduced under identical conditions, which impressively demonstrates the advantages of glycomimetics over native glycosides.
Because these glycomimetics showed nanomolar inhibition of LecBPA14, prevention of P. aeruginosa biofilm formation and excellent in vitro ADME/toxicity properties, 135 and 136 were then analysed in a murine pharmacokinetics study dosed at 10 mg kg−1 i.v. or p.o. Both molecules were orally bioavailable and the experiments revealed their renal excretion. However, large differences after intravenous administration in exposure (AUC 1.72 μg mL−1 h−1 and 7.40 μg mL−1 h−1 for 135 and 136, respectively) and half-lives (t1/2 = 0.28 h and 0.57 h for 135 and 136, respectively) have been detected with 136 exhibiting a superior pharmacokinetic profile, also after per oral administration. Consequently, these molecules are currently under investigation in murine models of P. aeruginosa infection.
In the search for new glycomimetics addressing the subpocket adjacent to the anomeric centre of the bound fucose, Titz et al. have recently reported N-linked β-fucosyl amides as potent LecB ligands.498 In contrast to the α-linked derivatives reported by Bernardi before (e.g.126), these β-anomers revealed an unexpected high potency for LecB of IC50s down to 85 nM and a KD of 195 nM for β-fucosyl benzamide 138 as determined by a competitive binding assay and ITC. Thus, these compounds have shown a >10-fold increase in binding affinity for LecB compared to the isomeric α-fucosyl benzamide 140, which has a KD of 2310 nM by ITC. The increased potency results from an increased entropy of binding of the β-anomer, while the enthalpy of binding was comparable for both. The crystal structure of β-amide 139 in complex with LecB revealed additional interactions of the aglycon with the protein when compared with the crystal structure of α-amide 140 in complex with LecB (Fig. 25E and F). Attractive interactions of the aglycon can only be established for the β-anomer, explaining at least in part the higher binding affinity while the increased binding entropy probably results from favourable release of water from the protein surface. ADME/Tox evaluation of these compounds revealed good metabolic stability in mouse and human plasma, as well as in presence of liver microsomes from both species. Also, plasma protein binding was generally low and all compounds have been further analysed for cytotoxicity against 3 cell lines in vitro. Interestingly, a differential picture of cytotoxicity for some derivatives was found with HepG2 cells (human liver), while the compounds were generally non-toxic against CHO (hamster ovaries) and A549 (human lung) cells.
As for LecA, most multivalent LecB-targeting ligands were analysed for lectin binding only. Some were tested further and the tetravalent C-fucoside FD2 by Reymond et al. presenting ligand 127 has shown good potency in antibiofilm experiments492 and restored antibiotic activity in vitro.499 The current development status of FD2 is unknown. In addition, calixarene-mounted fucosides by Vidal et al. have been tested in an acute lung infection model with P. aeruginosa in mice and showed efficacy in preventing lung colonisation and reducing lung injury, their current development status is also unknown.476
3.1.4.4 The lectins from Burkholderia cenocepacia. The Burkholderia cepacia complex comprises several species of human pathogenic bacteria which often cause lung infections, for example in cystic fibrosis (CF) patients.500 These bacteria express a number of lectins of which the β-propeller lectin BambL from Burkholderia ambifaria and two LecB-homologs, BC2L-A and BC2L-C, have been structurally characterized and inhibitors are being developed.
BambL is a six-bladed beta propeller lectin consisting of a trimeric repeat of its 87 amino acid polypeptide.501 The protein binds fucose with high affinity in two distinct binding sites per repeat, one within one protomer and one at the interface between two protomers (Fig. 26). In contrast to LecB, ligated fucose is deeply buried in BambL with an interaction of its lipophilic α-face with the side chain indole of Trp74, a recognition motif commonly observed in galectins. The KD for Me-α-L-Fuc (142) is 0.96 μM and the best known ligand of BambL is blood group A trisaccharide (143, KD = 0.46 μM) (Fig. 27). Richichi et al. developed the bicyclic fucose derivative 144 which showed very potent binding to BambL with a KD of 1.54 μM. This compound was also tested with LecB but no binding was observed. Further, the compound was also unable to inhibit DC-SIGN, suggesting a selectivity of this fucomimetic for BambL.502 Extension of the molecule at its carboxylic acid group to 3-hydroxy propylamide resulted in 141 with 240 nM affinity for BambL.503 Later, a virtual screening was performed with BambL and two ligands were experimentally confirmed.504 One of these was the glycomimetic 2-deoxy-2-fluoro fucose 145 with an IC50 of 19.9 μM (KD = 18.8 μM) and selectivity over LecB (IC50 > 1 mM).
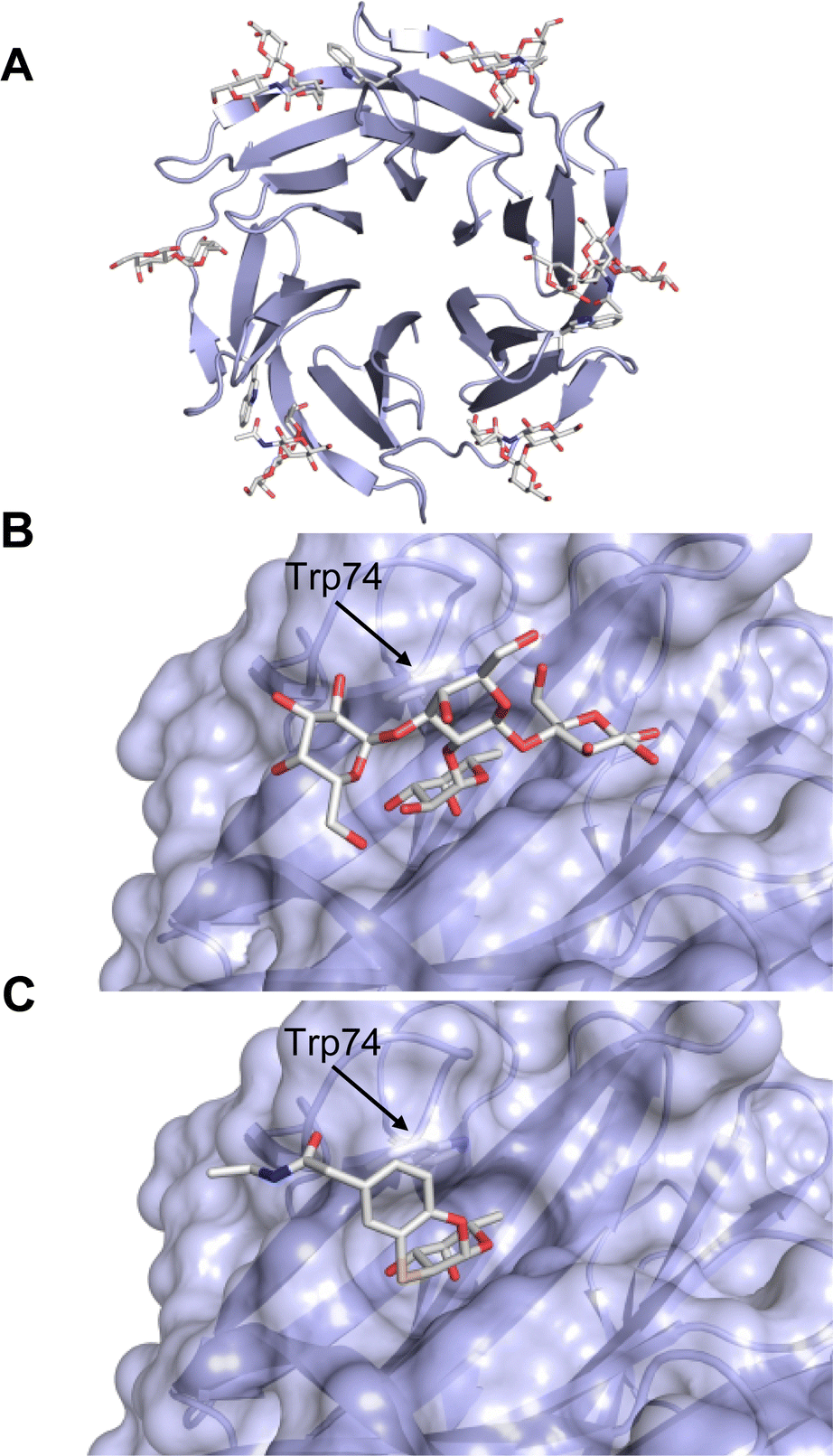 | ||
| Fig. 26 Quarternary structure and binding site of BambL. (A) BambL forms a homotrimer resulting in a β-propeller fold with two slightly different types of fucose binding sites per monomer, one site inside one monomer and one site between two monomers (PDB code 3ZW2). Magnified binding site within one monomer occupied by (B) blood group B antigen (PDB code 3ZWE) or (C) glycomimetic 141 (PDB code 6ZFC). | ||
 | ||
| Fig. 27 Inhibitors of the Burkholderia lectins BambL are derived from fucose, whereas BC2L-A inhibitors derive from mannose. BC2L-C consists of two domains, the N-terminus binds fucosides and the C-terminus is homologous to LecB and binds to mannosides. | ||
The bclBCA gene cluster encodes for three lectins BC2L-A, BC2L-B and BC2L-C. Their names were chosen due to their homology to LecB previously called PA-IIL. These three Burkholderia lectins are adhesins and play a role in biofilm development as reported by Eberl et al., who demonstrated that a knock-out of this gene cluster in the CF isolate B. cenocepacia H111 resulted in hollow biofilm microcolonies when grown under flow conditions.505 Furthermore, it was shown that reversion of the bclBCA-deficient strain's biofilm phenotype required complementation with all three genes, while bclA, bclB, or bclAC alone were insufficient.
The structure of BC2L-A has been solved as a dimer and it was shown that this protein preferably binds mannosylated glycans (Fig. 28).506,507 In contrast to its homolog LecB, the IC50 of Me-α-D-Man 146 with BC2L-A is 18.2 μM, while the corresponding value for Me-α-L-Fuc 142 is 20.9 mM, indicating a 1000-fold selectivity of this lectin for mannosides. These observations can be explained by the crystal structure of the complex with Me-α-D-Man: the 6-OH of mannose deeply enters the protein (Fig. 28B) and is fully hydrogen-bonded to Ala30, Glu31 and Asp110. Since fucosides have no analogous hydroxy group, the 6-OH group is the main driver of the selectivity of BC2L-A. In addition, the side chain of His112 could be responsible for a steric clash with the aglycon in fucosides, further contributing to selectivity. It was shown by Imberty, Silipio and co-workers that BC2L-A also binds with good potency to heptoses present in bacterial lipopolysaccharides.508 The L-glycero-D-manno-heptose 147 binds to BC2L-A with a KD of 137 μM (Fig. 28C) while the diastereomeric D-glycero-D-manno-heptose 148 is not recognized, demonstrating the impact of the stereochemistry of the 6-OH group. A series of synthetic mannose and mannoheptose analogues was analyzed for their BC2L-A inhibition.509 In this study, the essential role of this OH group was further confirmed by substitution with the halogens Cl, Br, or I and with an amino group, none of these derivatives inhibited BC2L-A. Also, when the ring hydroxy groups were exchanged with fluorine or altered by O-methylation, a loss of activity was observed. Interestingly, the glycomimetic (6S)-mannoheptoses with amide and especially sulfonamide substituents at C7 were superior inhibitors of BC2L-A (IC50 = 13.8–116 μM, e.g.149) while the (6R)-diastereomers, e.g.150, were inactive, consistent with the data for unmodified heptoses by Marchetti et al.
 | ||
| Fig. 28 Quarternary structure and binding site of BC2L-A. (A) BC2L-A forms a homodimer with the carbohydrate binding sites oriented in opposite directions (PDB code 2VNV). Magnified binding site occupied with (B) methyl mannoside 146 (PDB code 2VNV) or (C) mannoheptose 147 (PDB code 4AOC). Despite being a LecB ortholog, BC2L-A shows a pronounced selectivity for mannosides over fucosides explained by hydrogen bonding of the mannose 6 OH. Calcium ions are depicted as green spheres. | ||
BC2L-C was also crystallised510,511 and this protein consists of two domains: an N-terminal TNF-like lectin domain and a C-terminal LecB-domain, which then assemble into hexamers. Small angle X-ray scattering and transmission electron microscopy data allowed to postulate a model where the hexamer consists of two trimeric N-terminal domains (Fig. 29A). and three dimeric C-terminal domains. This complex heterobifunctional superlectin was shown to possess proinflammatory activity and to stimulate IL-8 expression in cell culture. The N-terminal domain of BC2L-C binds a diverse set of fucosylated oligosaccharides with LewisY and H-type 1 antigen as most potent binders (KDs of 53.9 and 77.2 μM, respectively) and the Lewis blood group antigens LeA, LeB, and LeX have KDs of 132, 213, and 196 μM. Its affinity for Me-α-L-Fuc is significantly lower (KD = 2.7 mM). In contrast, the C-terminal domain is homologous to BC2L-A and binds selectively (high)mannose, mannoheptose and conjugates thereof. Affinities were determined by ITC for mono-, di-, and trisaccharides and KDs ranged from 27.6 to 88.1 μM. Because BC2L-A was shown to bind to B. cenocepacia cells, BC2L-C was postulated to crosslink bacteria and host cells via C- and N-terminus, respectively.
 | ||
| Fig. 29 B. cenocepacia BC2L-C is a lectin composed of an N-terminal fucose-binding domain and a C-terminal LecB-orthologous mannose-binding domain. (A) The N-terminal domain of BC2L-C trimerizes and binds fucosides at the interface of two monomers (PDB code 2WQ4), (B) a bound fragment was identified in a second site of BC2L-C N-terminal domain adjacent to the CRD ligated with the oligosaccharide Globo H (PDB code 6ZZW), (C) β-fucosylamide glycomimetic 152 cocrystallized with BC2L-C N-terminal domain (PDB code 7OLW). Residues involved in binding to fucose are highlighted: Tyr48 forms CH–π interactions with Fuc-C6, Arg85 coordinates to Fuc-4-OH and -5-O, Arg111 binds Fuc-2-OH and -3-OH. The fragment in (B) and the aglycon in (C) is recognized in a hydrophobic cleft and forms attractive interaction with Tyr58. | ||
In the N-terminal domain of BC2L-C, a second site has been identified in close proximity to the fucose binding site.512 After a large virtual screening for this second site, shortlisted fragments have been analysed for binding to BC2L-C N-terminus by differential scanning fluorometry and STD-NMR. Finally, a co-crystal structure has been obtained of this domain in complex with the tetrasaccharide Globo H and one fragment residing in the second site (Fig. 29B). These results set the stage for the development of fucose-derived glycomimetics where fucose and fragments have been linked via different linker chemistries. Fucosylamide 151 with an attached indole carboxamide showed an affinity of 0.76 mM, which is a >3-fold improvement compared to MeFuc with 2.7 mM.513 Glycomimetic 152 could be cocrystallized with BC2L-C, demontrating the tight binding of the aglycone in the second site (Fig. 29C). Interestingly, these molecules are structurally similar to the very potent LecB inhibitors 138 and 139 and might therefore constitute a class of glycomimetics that simultaneously fight two pathogens which often co-infect patients.
Sialic acids are negatively charged carbohydrates based on 9-carbon atom α-ketoacids and N-acetyl neuraminic acid (Neu5Ac) is the most abundant sialic acid monosaccharide in humans that also exists with distinct chemical modifications, e.g. acetylation.343 These sialic acids are frequently used as primary receptors by many viruses for establishing infection, among them are influenza viruses, some coronaviruses and many others.515–517 Therefore, the design of inhibitors of viral agglutinins is a well-studied field, which is however relying almost exclusively on the multivalent display of native Neu5Ac to exploit the avidity of the trimerizing influenza hemagglutinin.518,519 In a recent example, a large multivalent display of Neu5Ac with additional neuraminidase inhibitors has been designed to wrap around the viral particle and hereby block infection.520 In influenza A, these hemagglutinins differ in their sequence, which results in numerous serotypes of the influenza virus.514 These differences also impact on the recognition of the Neu5Ac moiety in its natural glycoconjugates, as demonstrated by the selectivity for α-2,3-linked glycans in avian influenza and for α-2,6-linked Neu5Ac in human influenza.
An early study on modifying the Neu5Ac residue itself and probing its interaction with influenza hemagglutinin H3 was performed by Sauter et al.521 In this important study, it was shown for the methyl glycoside of Neu5Ac (153) that its non-interacting 7-OH could be removed in 154 without impact on binding to H3 and modifying 4-OH by esterification (155) or 9-OH by transforming into an amine (156) was possible (Fig. 30). Binding of these modified monosaccharides was in the low mM range and thus comparably potent to the parent methyl glycoside of Neu5Ac, 153. It was also observed that removal of the hydrogen bond donor in position 9 by acetylation entirely abolished binding (KD > 100 mM). Further, acetylation of 7-OH or exchange of the acetamide with an azide also abolished recognition by H3. Thus, this work provided an early structure–activity relationship for Neu5Ac recognition in one influenza hemagglutinin.
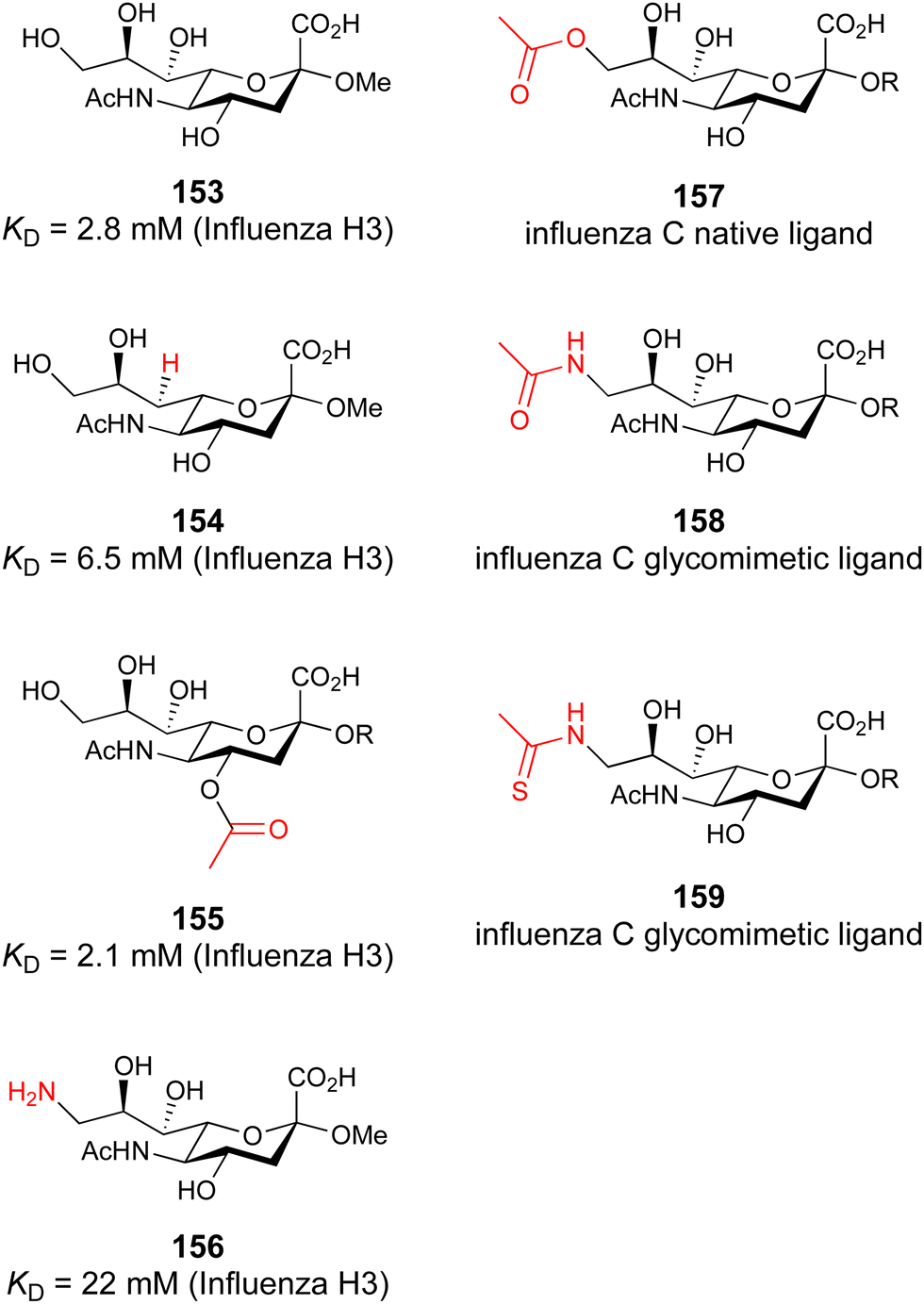 | ||
| Fig. 30 Glycomimetics of Neu5Ac as inhibitors of viral hemagglutinins from influenza A and hemagglutinin-esterase from influenza C. | ||
In contrast to influenza A hemagglutinin H3, the influenza C hemagglutinin–esterase fusion protein recognises 9-O-acetyl Neu5Ac conjugates 157 as its native ligands and substrates. Paulson and co-workers have reported the synthetic 9-acetamido 9-deoxy Neu5Ac derivative 158 that was enzymatically conjugated for display on erythrocytes. Cells carrying this glycomimetic conjugate then successfully agglutinated influenza C,522 while influenza A or B were not agglutinated. A small structure activity–relationship study showed that erythrocytes carrying Neu5Ac derivatives with position 9 modified as azide, amine or hexanoyl amide did not agglutinate influenza C. This work was further extended by demonstrating that the 9-deoxy 9-thioacetamido Neu5Ac 159 is indeed a glycomimetic for influenza C hemagglutinin function but prevents further processing by the esterase function of the fusion protein.523
The knowledge obtained from these SAR studies on glycomimetics of Neu5Ac for hemagglutinins will allow the generation of receptor/strain specificity of inhibitors. The mandatory high affinity for efficient viral trapping will be achieved after multivalent display of those glycomimetics.
3.2 Novel glycomimetic scaffolds for Ca2+-dependent lectins
In the early 2000s, the Kiessling group designed one of the first non-carbohydrate glycomimetics for DC-SIGN. The aim was to mimic the canonical carbohydrate recognition mediated by a central Ca2+ ion using a shikimic acid-derived scaffold.524–527 This core structure was supposed to provide a collection of diverse ligands for targeting a range of C-type lectin receptors. Interestingly, the shikimic acid-derived scaffold targeted the carbohydrate-binding site of DC-SIGN through the hydroxy groups at positions 3 and 4 similarly to mannose. Thus, it has been considered as a good mimetic of mannose for Ca2+-dependent lectins. Additionally, the carboxylic acid and thiols offer two potential points of diversification, which were exploited to introduce different substituents and to synthesize a library of 192 compounds. Their ability to compete with immobilized mannan for binding to the fluorophore-labelled extracellular domain of DC-SIGN was tested in a fluorescence-based competition assay. The best shikimic acid–based glycomimetic of the library showed only weak affinity for DC-SIGN (18, Table 4, IC50 = 3.2 mM525). However, the potency of the glycomimetic was later enhanced by three orders of magnitude following multivalent presentation (IC50 = 2.9 μM527).| No | Structure | Protein | Binding affinity | Ref. |
|---|---|---|---|---|
| 18 |
|
DC-SIGN | IC50 = 3.2 mM | Garber et al.525 |
| 161 |
|
P-selectin | IC50 = 0.57 μM | Kranich et al.528 |
| 16 |
|
LecA | K D = 1.11 ± 0.07 mM | Kuhaudomlarp et al.121 |
| 160 |
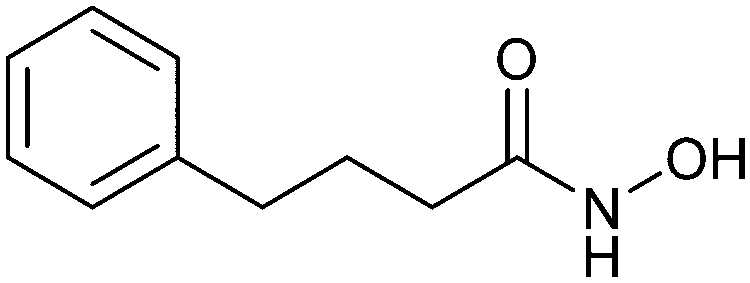
|
LecA | K D = 4.6 ± 0.9 mM | Shanina et al.122 |
| 162 |
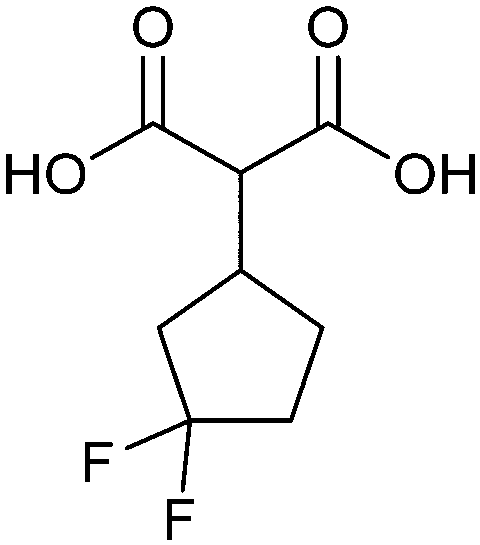
|
LecB | K D = 1.2 ± 0.4 mM | Shanina et al.122 |
| DC-SIGN | K D = 1.2 ± 0.5 mM |
Another class of glycomimetics was discovered for P-selectin by Revotar.528 A rational design approach led to the development of non-carbohydrate P-selectin antagonists mimicking sLeX. These glycomimetics contain the hydroxy groups in positions 4, 3 and 2 of an attached pyrogallol, intended to mimic the hydroxy groups of fucose. A comprehensive structure–activity relationship study provided the upper range nM inhibitor of P-selectin 161 (Table 4, IC50 = 0.57 μM), which showed high inhibition values up to 83% in the HL-60 cell attachment assay.
A further example of targeting a Ca2+ dependent lectin using non-carbohydrate small molecules has been recently reported for LecA from P. aeruginosa.121In silico screening of the National Cancer Institute (NCI) Diversity IV database and validation of hits by several biophysical assays identified catechols as ligands taking the canonical coordination of the central Ca2+ ion in this lectin. Notably, this compound class is also known as part of the pan-assay interference substances (PAINS529), which are prone to undergo unspecific interaction with proteins.530 Despite the fact that catechols can be oxidized to reactive quinones, Imberty, Titz and co-workers proved that electron-deficient catechols are stable under the conditions tested and in their interaction with LecA. Thus, catechols are not necessarily ‘bad actors’. A crystal structure in complex with LecA confirms the catechol 16 as a mimic of carbohydrates in Ca2+-binding. Similar to the LecA–galactose complex (Fig. 31A), the catechol coordinates the Ca2+ ion through two vicinal hydroxy groups, which mimic the 3 and 4 hydroxy groups of galactose (Fig. 31B). In particular, this catechol derivative forms H-bonds with Asn107 and Asp100 and the backbone oxygen of Tyr36. Even though this exemplifies that small molecules can coordinate the ions in the carbohydrate-binding site of a lectin, the Ca2+ coordination alone is not responsible for the binding. The nitrile group in catechol 16 plays a crucial role and mimics the hydroxy group of galactose in position 6. It uses a conserved water molecule (WAT1) to form a H-bond bridge between the nitrogen atom of the nitrile group in catechol and the oxygen atom of the carbonyl group of Pro51 in LecA. Its removal or shifting to the neighbouring carbon atom abrogated or decreased the binding efficiency measured by SPR and the inhibition in a fluorescence polarization (FP) assay, respectively. Despite the millimolar range binding affinities of catechols (16, Table 4), they bind tightly enough relative to their size (MW of 135–155 g mol−1) and number of heavy atoms (HA).531 Given the fact that catechols show ligand efficiency (LE) values of 0.4, they provide a good basis for future compound growing to increase their binding potency for LecA. Moreover, it was shown that catechols may target the carbohydrate-binding site of other Ca2+-dependent lectins, such as langerin. However, the mechanism of action remains to be confirmed for CTLs. Overall, electron poor catechols challenge the paradigm of ‘undruggable’ lectins as non-carbohydrate glycomimetics and show the potential of the structure-based design studies with the aim to improve their potency and drug-like properties as potential antimicrobial agents.
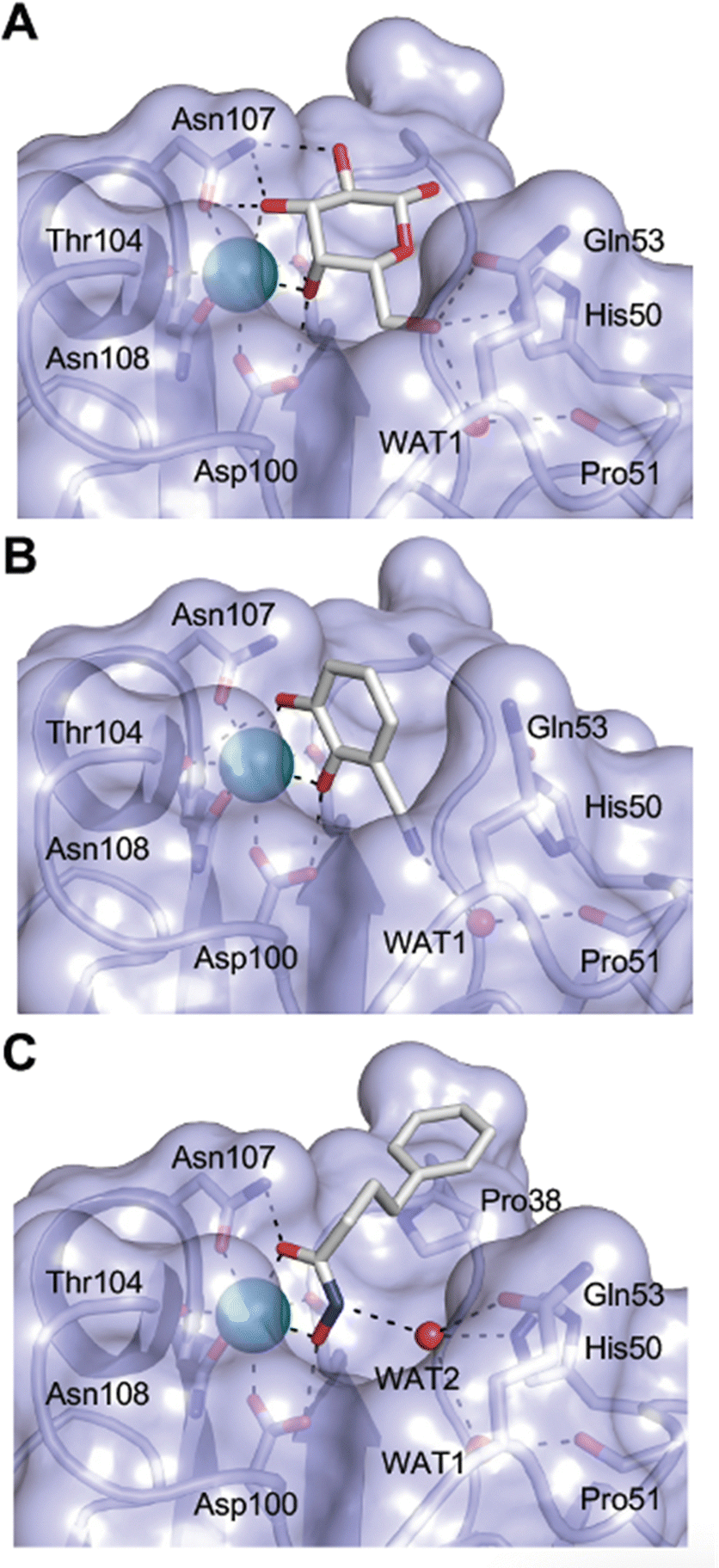 | ||
| Fig. 31 Non-carbohydrate glycomimetics for the Ca2+-dependent lectin LecA. Crystal structures reveal the interactions between LecA and (A) galactose (PDB code 1OKO), (B) catechol 16 (PDB code 6YO3), and (C) hydroxamic acid 160 (PDB code 7FJH). The non-carbohydrates 16 and 160 bind to LecA via calcium complexation in similar fashion to the native ligand galactose. Calcium ions depicted as green spheres and tightly coordinated water molecules as red sphere. | ||
In another attempt to identify non-carbohydrate pharmacophores for Ca2+-dependent lectins by Rademacher et al., an NMR-based screening of a chemically diverse library of 1000 fragments identified a compound with a hydroxamic acid moiety that coordinates the Ca2+ ion in the carbohydrate binding site of LecA (160, Table 4).532 The structure–activity relationship (SAR) study revealed a sterically optimal presentation of the hydroxamic acid group as demonstrated by a crystal structure of LecA in complex with 160 (Fig. 31C). Similar to galactose and catechols, the hydroxamic acid moiety coordinates to the Ca2+ ion. In particular, the nitrogen and the two oxygen atoms form H-bonds with His50, Gln53, Pro51 via a water molecule (WAT2) and Asn107, Asp100 mimicking the hydroxy groups of galactose in positions 6, 4 and 3, respectively. Notably, the phenyl moiety of the hydroxamic acid compound 160 forms CH–π interaction with Pro38 that is not present in catechols.
Inspired by the novel scaffolds, Rademacher and co-workers explored the application of a metal-binding pharmacophore (MBP) library aiming to improve the targeting of Ca2+-dependent lectins.122 A 19F NMR screening was performed to compare the hit rates of three fragment libraries against four Ca2+-dependent lectins (LecA, LecB, langerin and DC-SIGN). Notably, the MBP library showed superior hit rates compared to previous screening attempts against these targets. In particular, 1D and 2D NMR studies demonstrated the potential of a malonic acid scaffold in targeting the carbohydrate-binding sites of LecA, LecB and DC-SIGN (162, KD = 1.2 mM for LecB and DC-SIGN, Table 4). Even though the group has demonstrated the Ca2+-dependency of these interactions, the binding mechanism remains to be confirmed by crystallography studies.
Altogether, three non-carbohydrate glycomimetic scaffolds and an MBP library have been proposed to improve the targeting of the ‘undruggable’ site of the Ca2+-dependent lectins. Since these molecules are small (MW < 300 Da) and coordinate the Ca2+ ion, they are promising starting structures for the design of drug-like non-carbohydrate glycomimetics of lectins.
3.3 Allosteric modulation of lectins
Allosteric modulators have gained increasing interest in the development of selective and potent agonists and antagonists of protein function. While many drug targets share a common architecture of the primary active site with other members in their protein family, allosteric sites are less conserved between members of the same protein family and hence likely offer a better starting point for the reduction of off-target effects. Additionally, allosteric drugs can provide novel ways of action, such as changing protein levels, localization within the cell or even target activation.533–535 In this interplay between distal target sites, the mechanism by which orthosteric and allosteric site are coupled can be diverse ranging from larger protein rearrangements to alteration of vibrational modes of amino acid side chains that promote reciprocal interaction of the two sites.536 The concept of allostery in drug design has been widely and successfully explored for GPCRs and other intrinsically dynamic, allosteric drug targets and is more and more applied to other targets such as kinases, phosphatases and other protein classes with low selectivity.537,538 For carbohydrate-binding proteins, the concept of allostery in the design of non-carbohydrate glycomimetics has been investigated only sparsely, giving rise to new opportunities for the future. Allosteric sites can offer favourable properties for the development of functional glycomimetics, such as better physicochemical properties and the absence of competition with endogenous ligands, as highlighted for Siglecs (see Section 3.1.3). Here, we summarize information on allosteric mechanisms, how they were identified and how, if at all, they were put into the design process. | ||
| Fig. 32 FimH allostery. The close-up of the low affinity (light blue) and high affinity (light pink) conformation of FimH at the domain interface (A) reveals significant conformational differences which affect the carbohydrate binding site (B) via long-range effects. In detail, this results in alterations of amino acids sidechains, in particular Tyr48 and Tyr137 (shown as stick), in the carbohydrate binding site. Figure adopted from lit.546 | ||
Compared to FimH, allostery is less apparent and a less explored phenomenon for other bacterial lectins. Cholera toxin is an AB5 toxin with a lectin domain promoting cell tropism and uptake. This lectin domain is well studied and its recognition of the ganglioside GM1 has been described in detail. However, a second carbohydrate binding site has been discovered and this site is proposed to be allosterically coupled to the primary site.547 To the best of our knowledge this has not been followed for the design of glycomimetics. In contrast, coming from a fragment-based screening approach, several sub-millimolar inhibitors of BambL have been recently identified.548 BambL is a lectin of B. ambifaria, causing chronic infections.501 Evidence from biophysical analysis and single point mutations introduced into the protein suggest an allosteric mode of action. A binding site was identified distal to the orthosteric fucose recognition site (Fig. 33). In this study, 1H–15N HSQC data are presented showing that changes of the chemical environment in secondary sites induced either by weak small molecule binding or by mutation are transferred to remote sites in the protein.125 Higher affinity ligands are necessary to move these allosteric glycomimetics into further assessment.
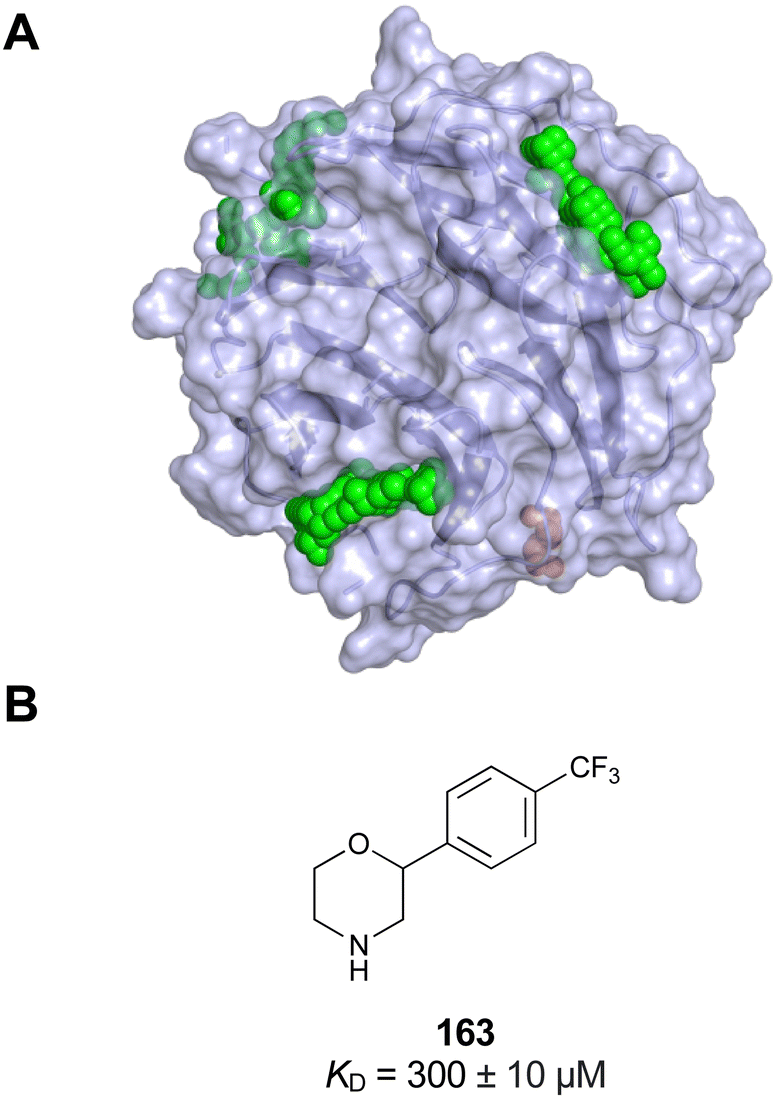
 | ||
| Fig. 34 Surface representation of the binding region of allosteric modulators for Gal-1. Residues undergoing chemical shift perturbation upon 62 binding are shown in red.127 Yellow indicates amino acids interacting with DB16 (61) at the edge of the monomer.128 Carbohydrate ligand indicated by green spheres. Structures of 61 and 62 are shown in Table 2. | ||
 | ||
| Fig. 35 LLS2 (164) is a proposed allosteric inhibitor of Gal-1 with a reported inhibitory constant from an in vitro cell killing assay using SKOV3 cells.550 | ||
The notion that galectins are amenable to allosteric modulation is further supported by two reported Gal-3 inhibitors, both analogues of tetrahydroisoquinoline natural products (Fig. 36).552 DX-52-1 (165) was suggested to covalently bind to Gal-3 and both compounds bind Gal-3 outside of its β-galactoside-binding site and inhibit cell migration. The exact binding site is not reported. In light of these potentially allosteric inhibitors for Gal-1 and -3, it is not surprising that also for Gal-7 allosteric communication was described. Longer-range communication of loops 1, 2 and 5 perturbing protein–protein interaction was suggested from protein engineering and biophysical characterization.553
 | ||
| Fig. 36 Allosteric Gal-3 inhibitors DX-52-1 (165) and HUK-921 (166) with reported inhibitory constants from epithelial cell migration assays.552,554 | ||
Selectins are type I membrane proteins that mediate cell adhesion, assist in leukocyte tethering and rolling, and exhibit a catch bond behaviour with a shear threshold feature.570–574 For P-selectin two states, having a bent or extended EGF orientation, are reported. These states undergo dynamic conformational exchange, sampled by forced induced allostery, initially thought to be limited to the interaction of the CRD and the adjacent EGF-like domains.575 Later, these insights were extended into long scale allostery over the entire protein structure, now including the consensus repeat (CR) domains.576 Knowing that such long-range interactions exist has expanded our view on potential functional sites for the development of allosteric glycomimetics. This opens doors for modulation of the carbohydrate recognition process with new chemical matter.
A good example of how protein dynamics and allostery are connected to the design of a carbohydrate-based glycomimetic is sLeX (26) recognition by E-selectin (Fig. 7). These data became available when the pan-selectin antagonist GMI-1070 (31) was already in Phase II trials.149 While it was known in the time of development that the first domain was sufficient to allow binding,577 affinity increase with increasing number of CR domains was also reported similar to P-selectin.578 The breakthrough how long-range force induced allostery can directly relate to carbohydrate-based glycomimetic recognition came from work employing an extended protein construct design and a co-crystallisation approach of 28 rather than soaking of a two-domain construct.149,167 This resulted in three regions of the lectin domain being affected upon ligand binding: (1) the long loop structure (residues 81 to 89) rearranging, (2) the so-called bridging region mediating the (3) transition motion originating from the pivot region connected to the EGF-like domain. For the design of the carbohydrate-based glycomimetic, the alteration of the long loop structure by almost 10 Å was most obvious (Fig. 7). While such a large-scale rearrangement is often observed for CTLs upon Ca2+ binding and release,150,555,560–562 the remarkable observation for E-selectin was that the central fucose got covered by the long loop.149 The fucose occupies the canonical carbohydrate site, coordinating the Ca2+ with its 3- and 4-hydroxy groups, now also interacting with the long loop, leading to formation of a hydrogen bond of the 2-hydroxy group in the extended conformation of E-selectin. This switch between the open and closed conformation of the loop is suggested to happen spontaneously after ligand recognition.579 Moreover, solution state, label-free small angle X-ray scattering (SAXS) data strongly suggest that the structural transition to an extended conformation can be initiated even by small molecule recognition.149 These data explained the previously reported, notorious importance of the fucose 2-hydroxy group for the activity of GMI-1070 (31) series and other glycomimetics.580,581 In analogy to P-selectin, these new insights into conformational transitions open the path for the development of novel allosteric glycomimetics.
A look at the long loop mobility in the CTL langerin complements the picture that arose from E-selectin. Langerin is a type II transmembrane receptor that forms homotrimers, promoted by the neck domain extending it 24 nm from the plasma membrane. Compared to the type I transmembrane selectins, there is no EGF-like domain, and instead the long helical neck domain of langerin renders E-selectin-like long-range conformational transitions unlikely. Moreover, interdomain interactions between the langerin CRDs can be excluded, as no difference for the carbohydrate affinity of the monomer and the trimer was detected.582 Such interdomain interactions have not been reported for other hetero- or homooligomeric CTLs. However, in analogy to E-selectin for which the mobility of the long loop structure was reported, micro- to millisecond dynamics was inferred from NMR studies.582 Furthermore, a short loop structure of langerin, called the bridging region for the selectins, is coupled to the long loop mobility.582,583 The cleft between the two loops harbours His294, the pH sensor of this CTL.582,583 Combining molecular dynamics simulations, NMR and mutational scanning of the CRD, a conserved allosteric network of communicating amino acids was identified for human langerin, which was further substantiated by a Ca2+-independent activation upon recognition of larger heparin fragments.158 Overall, a similar picture of the coupled loop mobilities emerges for langerin and E-selectin.
Taking the next step, a fragment-based design approach was conducted against murine langerin and several allosteric modulators were identified, some such as 20 with two-digit micromolar potency as measured in an inhibition assay in which lectin binding to a modified lipid surface was monitored (Fig. 37).161 In recent follow-up on this work, the binding site for this compound class was determined using NMR and computational methods.584 The allosteric site of langerin is located in the cleft between the long and short loop, the so-called bridging region (Fig. 38). Moreover, this study suggested long loop mobility upon carbohydrate recognition (Fig. 38A and B).584 Whether the inhibition mechanism of the thiazolopyrimidines is based on the interference with the long loop, similar to what has been observed for E-selectin, covering the central fucose, remains to be elucidated. Interestingly, this cleft between the long and the short loop was previously reported to harbour a short peptide, suggesting it is amenable to modulation via small molecule inhibitors.585 Additionally, the short loop itself is involved in formation of langerin/langerin interactions in the endosomal compartment, forming Birbeck granules, a langerin-specific organelle.586 Taken together, several lines of evidence support the notion that allosteric inhibition of langerin through intercalation in the bridging region is possible. The next step will be to develop this lead into higher affinity ligands to explore their utility in functional, cell-based assays and in vivo studies.
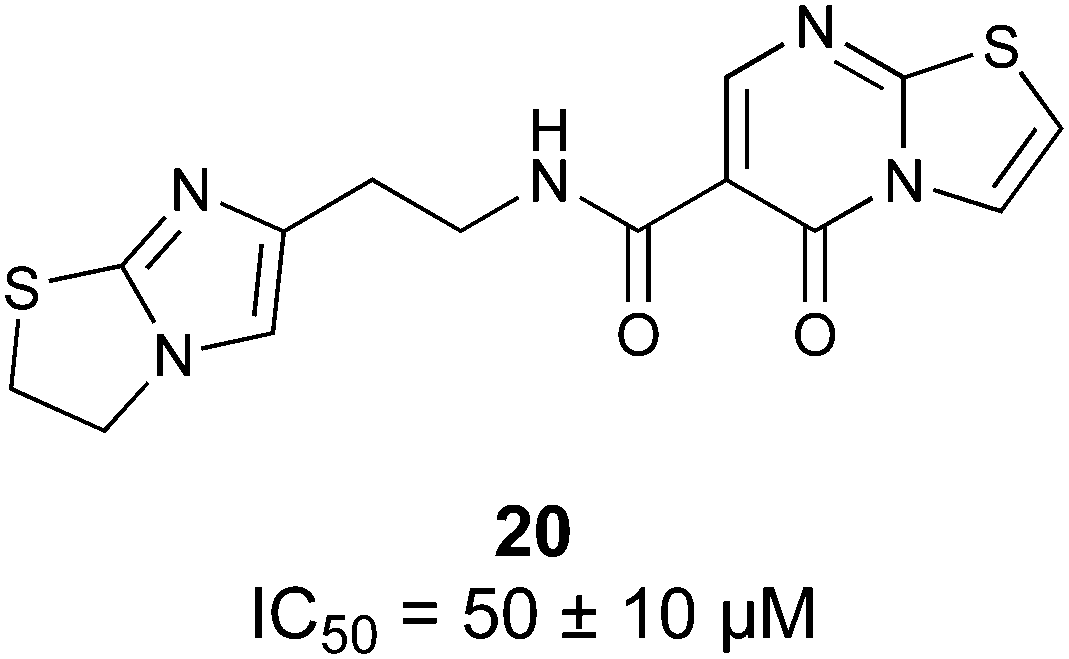 | ||
| Fig. 37 Allosteric inhibitor of murine langerin 20.126 | ||
 | ||
| Fig. 38 Long loop (red) and short loop (blue) structures in E-selectin, langerin and DC-SIGN. (A and B) Short loop, long loop of E-selectin with (A) open conformation (PBD code 1G1T) and (B) closed conformation (PBD code 4C16). (C) Langerin in complex with a peptide in the cleft (‘bridging region’) between the two loops (PBD code 3P7H).582 Amino acids involved in an allosteric network are highlighted with green spheres.582 (D) One allosteric site of DC-SIGN is indicated with green spheres (PBD code 1SL4).162 Calcium ions are shown as grey spheres. | ||
For DC-SIGN, an orthogonal approach to carbohydrate-based inhibitor design was pioneered by the Kiessling group reporting on a high throughput screening over 30000 small molecules against DC-SIGN.587 This led to the discovery of quinoxalinones and thiazoles (167) as drug-like inhibitors in the low micromolar range (Fig. 39). In a follow-up study by the same group, these ligands were optimized to overcome the liabilities of the previous scaffolds such as the oxidation of the quinoxalinones’ thioether functionality resulting in IC50 values of 0.3 μM for 168.587,588 As these compounds lacked functional groups to complex Ca2+ ions, a carbohydrate binding site-independent mechanism of interaction was suggested.44,138
 | ||
| Fig. 39 Non-carbohydrate-based glycomimetics for DC-SIGN. For 173, the anomeric biphenyl substituent was identified to bind to a secondary site without significant contributions from the original mannose scaffold.162 | ||
More insight into the recognition of non-carbohydrate ligands came from a series of studies describing fragment screening against DC-SIGN.160,568,589–591 One report identified several secondary sites from a fragment-based screening using ligand-based NMR, SPR, microarray and an ELLA-type plate-based assay (Fig. 40) and revealed compound 169.160 Additionally, a library coming from diversity-oriented synthesis was screened using 19F NMR and yielded high micromolar ligands (170) (Fig. 39).589 Orthogonal to that, cell-based fragment screening led to the identification of low micromolar inhibitors of DC-SIGN.590 Several compounds structurally related to the previously reported quinoxalinones were identified. IC50 values of 39 μM and 80 μM were reported for 171 and 172, respectively (Fig. 39).590 Overall, these fragments were of considerably lower molecular weights than the HTS hits and revealed several interesting starting scaffolds. These compounds have reasonably high affinities with respect to their overall molecular weight, the so-called ligand efficiency (LE).124 Moreover, this led to the discovery of additional binding sites for small molecules, distal to the primary carbohydrate site (Fig. 40).160 For a compound of 168 series a secondary site close to the primary site was suggested to inhibit carbohydrate binding by steric repulsion. However, inspired by the quinoxalinone scaffold, a series of quinolones such as 19 were explored as inhibitors for DC-SIGN and close inspection suggested an allosteric inhibition, opening the question for a mode of action for the quinoxalinones again.591 Moreover, one of these secondary sites was further supported by insights coming from crystal packaging of carbohydrate-based glycomimetics (24, Fig. 8).210 The carbohydrate-based glycomimetic 24 was found to cross-link two DC-SIGN monomers, one with the mannose, the other one with the hydroxymethylphenyl substituent as evidenced by X-ray crystallography. However, cross-linking of a cell surface receptor should not be excluded as a viable mechanism route to inhibition as evidenced by dimerization of the Siglec CD22 or the CTL LOX-1.592,593
 | ||
| Fig. 40 Potential secondary sites available for drug-like molecules to bind to DC-SIGN. The location of the carbohydrate-binding site is indicated by the orthosteric ligand 23 and the calcium ion (shown as green sphere).160 | ||
In a recent report, carbohydrate-based glycomimetic 173 was designed for langerin to benefit from a biphenyl extending of the anomeric position (Fig. 39). 173 showed high affinity for langerin (KD = 250 ± 70 μM); however, excellent binding to DC-SIGN expressing cells when 173 was presented on liposomal formulations was reported when analysing off-target recognition.162 Closer inspection revealed that this molecule has two functions: the primary Ca2+ site is engaged by the mannose scaffold and the aromatic substituent is bound to a previously proposed secondary site (Fig. 40).160 Binding of the biphenyl substituent to the secondary site led to the allosteric activation of the primary carbohydrate site. A site around Met270 binds the biphenyl agylcon of an initially envisioned carbohydrate-based glycomimetic and this site as well as the mode of action were supported by several biophysical assays, mutational analysis and molecular dynamics simulation (Fig. 38D).
Taken together, DC-SIGN is amenable to allosteric modulation and first ligands are explored to make use of it, albeit these compounds are still of low affinity and more experiments are necessary to elucidate the pathways allowing coupling of the orthosteric and allosteric sites.
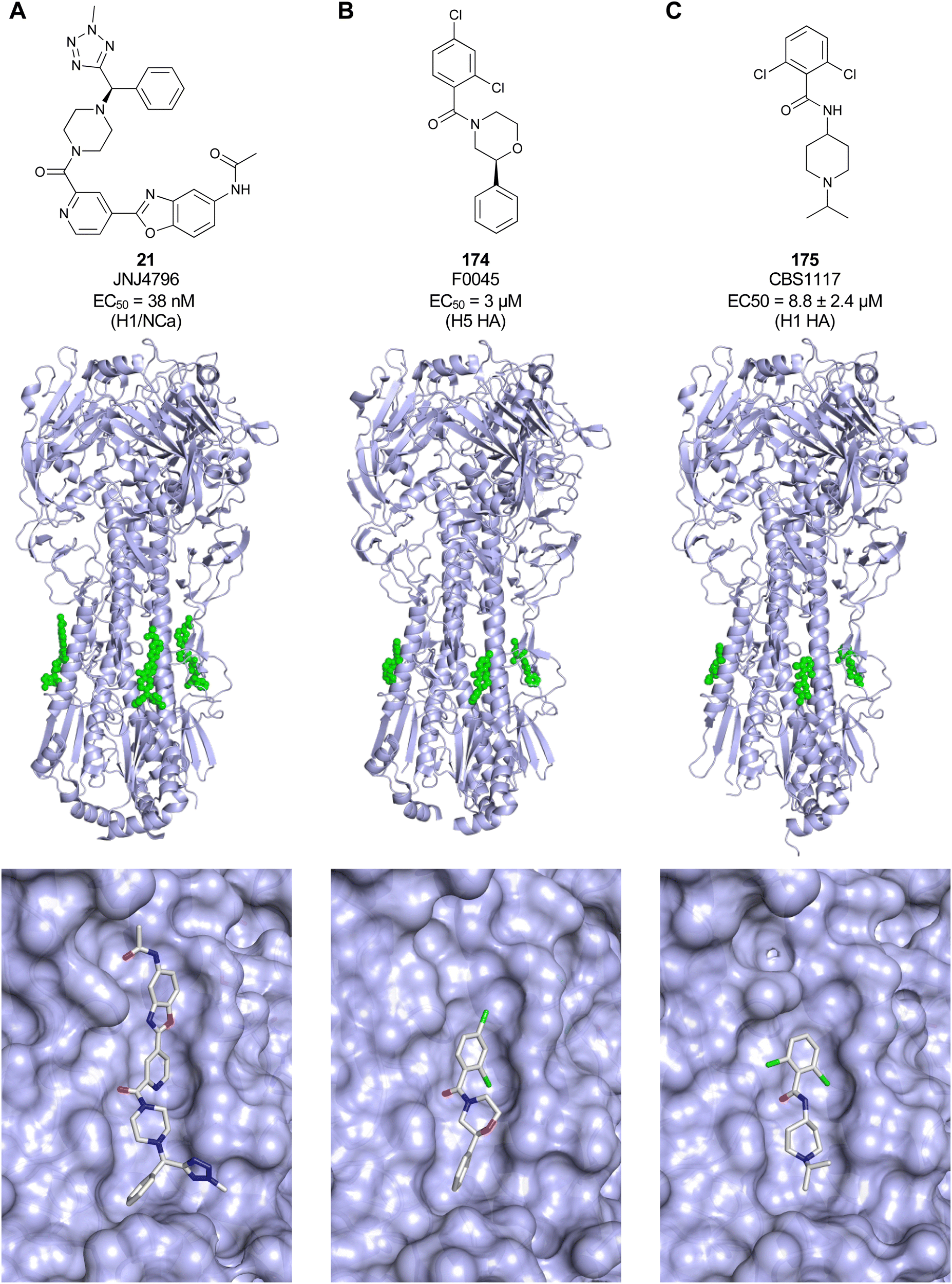 | ||
| Fig. 41 Allosteric inhibitors 21 (A, PDB code 6CFG), 174 (B, PDB code 6WCR) and 175 (C, PDB code 6VMZ) of influenza A virus hemagglutinin are shown (top) with their respective binding sites (bottom). Binding of these molecules in an allosteric site inhibits HA transition to the post-fusion state, thereby preventing endosomal escape of influenza A. | ||
Taken together, the development of functional glycomimetics following an allosteric drug design approach remains rarely pursued, yet the number of allosterically modulated lectins accumulates. This is not surprising as also for other drug target classes the use of allostery as a design principle emerged only within the last two decades and was often driven by serendipity.566,567 For galectins and CTLs, allosteric sites were identified during a mode of action analysis following a screening of a library of drug-like small molecules or peptides. For carbohydrate-binding proteins, an approach targeting a druggable allosteric site is very attractive compared to focusing on the orthosteric, hydrophilic, flat and solvent-exposed carbohydrate binding site.44,569,601 Finally, this opens the question for the biological relevance of such sites in lectins.
4. Novel applications of glycomimetics: receptor targeting
4.1 The asialoglycoprotein receptor (ASGPR) as target for drug delivery
The asialoglycoprotein receptor (ASGPR), also known as the Ashwell–Morell receptor, was the first mammalian lectin to be discovered.602 As a C-type lectin, it requires Ca2+ ions for the interaction with carbohydrate ligands. It recognizes galactose and N-acetylgalactosamine and clears desialylated glycoproteins with exposed Gal or GalNAc as non-reducing end groups. The mammalian ASGPR is composed of two homologous subunits, major subunit H1 and minor subunit H2, that are encoded by two distinct genes. In humans, the subunits form homo- and hetero-oligomers with different receptor configurations, among which a trimer with two H1 and one H2 is the most abundant.602–604 ASGPR is primarily expressed on hepatocytes and minimally on extra-hepatic cells. It belongs to the recycling receptors and is endocytosed and recycled constitutively every ca. 15 min, with or without the ligands.605,606 Thanks to its unique localization and high expression of up to 500000 ASGPRs per hepatocyte,607 it is an attractive target for receptor-mediated drug delivery to the liver.21,608,609 Diseases such as hepatocellular carcinoma,610 hepatitis B611 and C,612 and malaria613 are all associated with hepatocytes. Indeed, infection with HBV or HCV may lead to chronic hepatitis, which can cause cirrhosis and liver cancer. Therefore, targeting hepatocytes through the interaction with ASGPR is a viable strategy to treat these diseases.
Two approaches are currently being used: ligand-anchored nanocarriers and drug–ligand conjugates. In addition, RNA interference (RNAi) is becoming increasingly popular to treat a number of liver-related diseases. Four commercially available drugs are currently on the market: givosiran to treat acute hepatic porphyria, lumasiran for severe primary hyperoxaluria type 1, inclisiran for hypercholesterolemia, and vutrisiran for transthyretin-mediated amyloidosis. Targeting these drugs to hepatocytes exploits a trivalent monosaccharide ligand. This application paved the way for least 25 other drug candidates currently in clinical development, and most importantly, it opened the door for novel strategies based on monovalent ligands.
 | ||
| Fig. 42 Monovalent ASGPR ligands and their binding affinities. | ||
In contrast to monovalent ligands, multivalent ligands show a dramatically enhanced binding affinity for ASGPR. The affinity increases in the order mono- < bi- < triantennary ligand, with IC50 values of 1 mM, 1 μM and 1 nM, respectively.619 Only a minor improvement was observed when using more than three GalNAc units per ligand.619,620 A trivalent ligand is typically built on the molecule of tris(hydroxymethyl)aminomethane (Tris) to which carbohydrate or glycomimetic ligands are attached through amide, ester or ether-based linkers. The geometry of the molecule and the hydrophilic–hydrophobic balance have a great impact on ligand binding. It has been determined that the optimum distance between the three carbohydrate moieties to bind simultaneously to all three CBSs should be between 20 and 30 Å.621 The various structural and spatial aspects of multivalent ASGPR ligands have recently been reviewed by Huang et al.622
The glycoproteins asialofetuin (AF, 12 Gal and 3 GalNAc residues/mol(protein)) and asialoorosomucoid (ASOR, 20 Gal residues/mol(protein)) are endogenous glycoproteins known to bind strongly to ASGPR.21 Arabinogalactan and pullulan, a galactose- and glucose-based polymer, respectively, are polysaccharides extensively studied for ASGPR-mediated targeting.623,624 Pullulan relies on the ASGPR inability to discriminate between D-Gal and D-Glc.21 However, the uptake of pullulan is lower than that of arabinogalactan.625
PK2 (FCE28069) is a copolymer based on N-(2-hydroxypropyl)methacrylamide (HPMA) with N-linked galactosamine and doxorubicin (DOX). In a preclinical study, the copolymer displayed a 5-fold reduction in cardiotoxicity relative to free DOX.635 A Phase I clinical trial study in patients with primary or metastatic liver cancer proved that liver-specific doxorubicin delivery utilizing a galactosamine-modified polymer is achievable.636,637 A biodistribution study showed that 24 h after administration, 17% of the administered dose of doxorubicin targeted to the liver while a doxorubicin–polymer conjugate without galactosamine showed no targeting. Although the dosage for Phase II trials was recommended, to the best of our knowledge, there was no further development of PK2.
Targeted nanoparticles take advantage of receptor-mediated endocytosis to deliver a high payload of a drug to the liver. In addition, they protect the drug from degradation and enable the transport of both hydrophobic and hydrophilic drugs. Biodegradability of nanocarriers to non-toxic components is important to preclude toxicity. Hydrophobic nanoparticles are readily cleared off by reticuloendothelial system (RES), and therefore, hydrophilic or stealth particles are preferentially used, with PEG being the most popular stealth agent. While liposomes638 are the most widely tested type of nanoparticles, other types are tested, too, e.g. micelles,639,640 or dendrimers.641,642 Another strategy is the direct synthesis of covalent drug–ligand conjugates. This strategy is studied for example for the delivery of radiopharmaceuticals to the liver (for a review, see ref. 21).
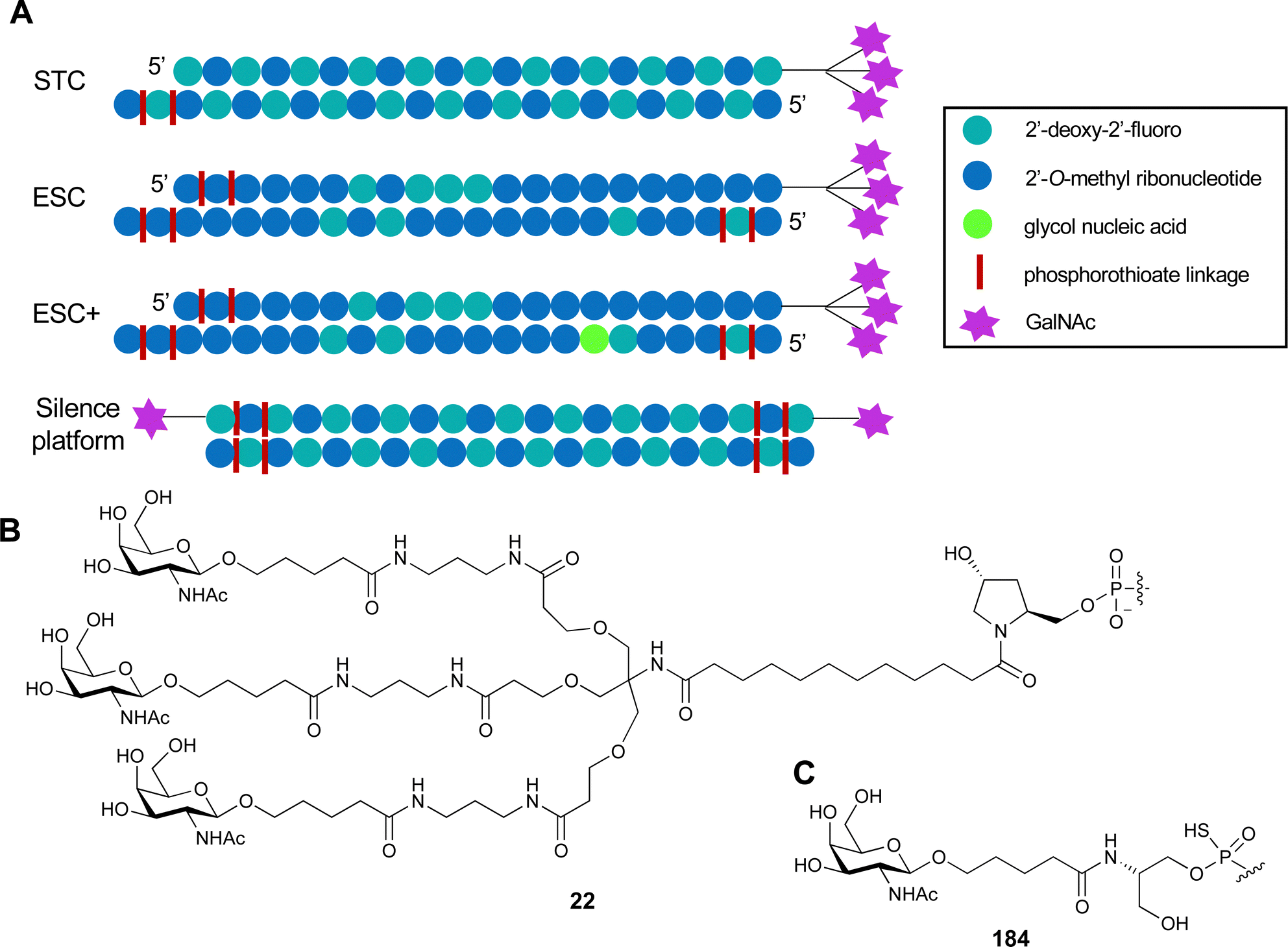 | ||
| Fig. 43 (A) Design of GalNAc–siRNA conjugates: STC-standard template chemistry, ESC-extended stabilization chemistry, ESC+-extended stabilization chemistry plus, Silence platform-single GalNAc positioned at opposite sides of the sense strand. The top strand is the sense strand and bottom the antisense strand. (B) Structure of triantennary GalNAc ligand 22 built on a Tris scaffold. (C) Structure of monovalent GalNAc ligand 184 built on a serinol scaffold. | ||
GalNAc–siRNA conjugates bind to the ASGPR and are taken up in endosomes where the conjugate dissociates from the receptor. Additionally, the sugar moieties and branches are very quickly lysed from the oligonucleotide652 and then the oligonucleotide translocates to the cytoplasm by endosomal escape (Fig. 44).650
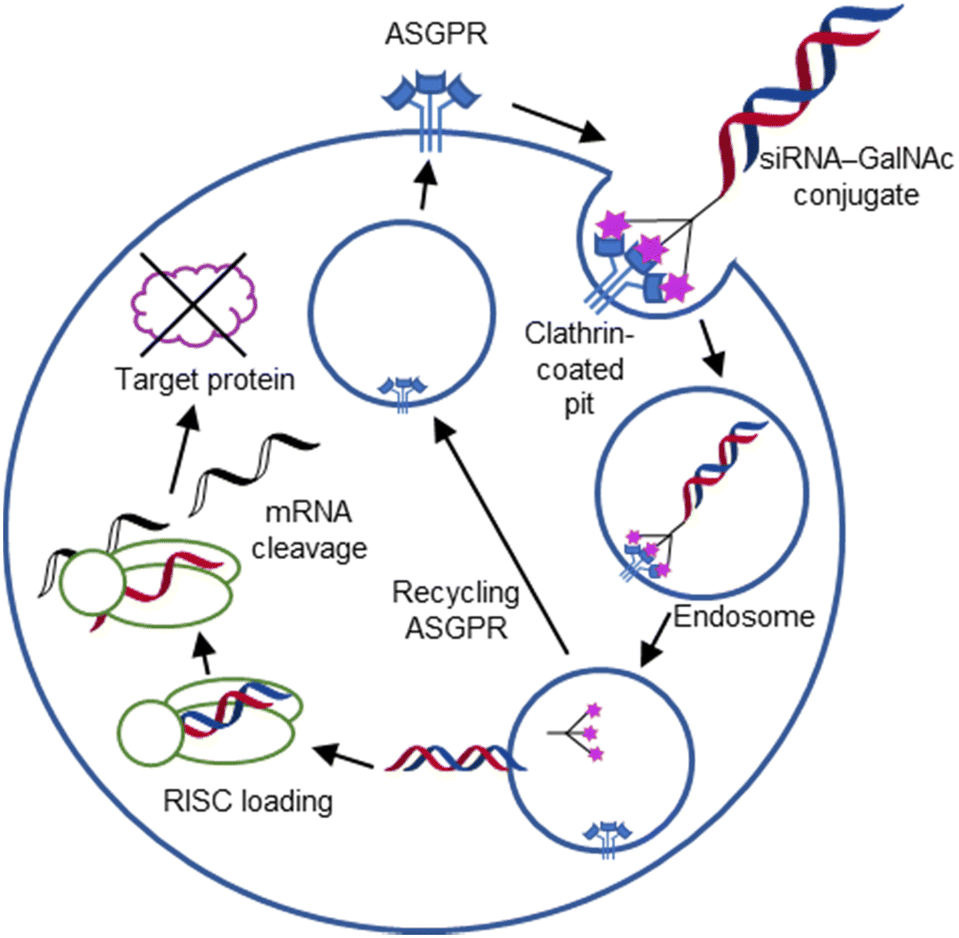 | ||
| Fig. 44 Mechanism of action of siRNA–GalNAc conjugates. After GalNAc-mediated asialoglycoprotein receptor (ASGPR) binding and uptake, the siRNA integrates into an RNA induced silencing complex (RISC) leading to degradation of the respective complementary mRNA. The mechanism for endosomal escape of the siRNA is still unknown.650 Reproduced from ref. 653 with permission from The American Society of Gene and Cell Therapy/Elsevier, copyright 2017. | ||
As of September 2022, four GalNAc–siRNA drugs are on the market (Table 5), five in the late stage of development (Phase III) and at least 20 in the early stages of development in Phase I or II (sources: clinicaltrials.gov and the pipeline data on the websites of the companies Alnylam, Dicerna, Novo Nordisk, Arrowhead Pharmaceuticals, Silence Therapeutics, and Arbutus Biopharma).654–658
| No | Name | Company | Indication | Target | Ref. |
|---|---|---|---|---|---|
| 185 | Givosiran (GIVLAARI), ALN-AS1 | Alnylam | Acute hepatic porphyria | 5-Aminolevulinic acid synthase 1 (ALAS1) | NCT03338816659–661 |
| 186 | Lumasiran (OXLUMO), ALN-GO1 | Alnylam | Primary hyperoxaluria type 1 (PH1) | Glycolate oxidase 1 (GO) | NCT03681184662–665 |
| 187 | Inclisiran (LEQVIO), AlN-PCSsc | Alnylam/Novartis | Hypercholesterolemia (heterozygous familial and non-familial) or mixed dyslipidaemia | Proprotein convertase subtilisin–kexin type 9 (PCSK9) | NCT03397121, NCT03399370, NCT03400800666–668 |
| 188 | Vutrisiran (AMVUTTRA), ALN-TTRsc02 | Alnylam | Transthyretin-mediated amyloidosis (ATTR) | Transthyretin (TTR) | NCT03759379, NCT04153149669 |
4.2 Targeted protein degradation
Targeted protein degradation (TPD) has opened many avenues for the treatment of various diseases and provided multiple routes to study fundamental biology.670,671 Proteolysis-targeting chimeras (PROTACs) are amongst the best understood applications of TPD, now on their way through clinical trials.672 Briefly, the approach is based on the application of a heterobifunctional small molecule comprising a ligand for an E3 ligase of the ubiquitin proteasome system and a ligand for the protein of interest, connected by a linker modality. Cross-linking the two proteins then induces covalent tagging of the protein of interest with ubiquitin and consequently leads to its degradation in the proteasome. One major advantage of this technology is the catalytic elimination of the target, compared to blocking the target occupancy. However, this approach is limited to cytosolic proteins while omitting about 40% of the proteome being extracellular or membrane-associated targets. Lysosome-targeting chimeras (LYTACs) make use of lysosome shuttling receptors taking over the E3 ligase function and set the protein of interest en route to the lysosome for degradation. It is not surprising to find carbohydrate-binding receptors to be the first to be explored for such an application: the 300 kDa multidomain cation-independent mannose-6-phosphate receptor (CI-M6PR, Fig. 45A and B)673,674 and the 100 kDa heterotrivalent C-type lectin ASGPR.675,676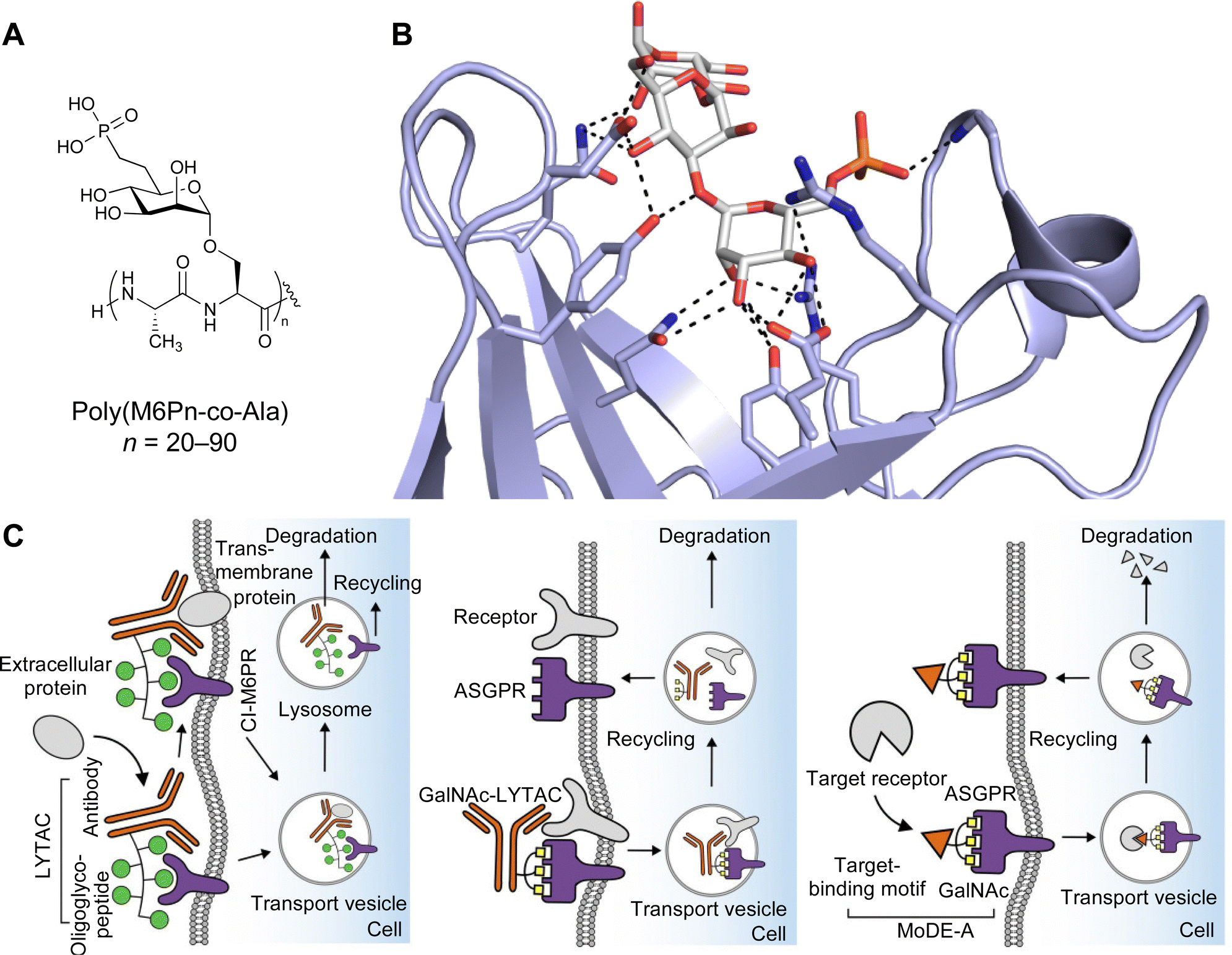 | ||
| Fig. 45 Lectin-mediated protein degradation. (A) Mannose-6-phosphonate peptide polymer used for CI-M6PR targeting.673 (B) Pentamannosyl phosphate in complex with bovine CI-M6PR (PDB code 1C39).684 (C) Targeted protein degradation through CI-M6PR using LYTAC (left),673 and the ASGPR based strategies of GalNAc-LYTAC (middle),676 and MoDE-A (right).680 While LYTACS rely on antibodies for binding of their extracellular degradation target, MoDE-As utilise small molecules. | ||
The first-generation LYTACs, developed by Bertozzi, targeted the CI-M6PR to exploit receptor-mediated endocytosis of extracellular proteins into the lysosome (Fig. 45C).673 The CI-M6PR is a P-type lectin that recognizes mannose-6-phosphate (M6P) residue caps on N-glycans of endogenous proteins with a micromolar affinity to transport them from the extracellular space into the lysosome, a system that has been used for treatment of lysosomal storage diseases for a long time.677,678 At lower pH in the endosomal compartment prior to the arrival in the lysosome, the cargo is released and CI-M6PR recycles back to the surface resembling an efficient system for cargo uptake. Mimicking the multivalent display of M6P on a glycoprotein cargo for the CI-M6PR, Bertozzi and colleagues used a known multivalent, biocompatible, phosphatase-inert mannose-6-phosphonate (M6Pn) to showcase efficient and constant uptake via CI-M6PR in absence of receptor degradation.673 These polymers were conjugated to antibodies specific for soluble components of the extracellular space as well as for membrane bound receptors and led to their targeted degradation. The CI-M6PR is amongst a number of potential lysosome trafficking receptors that could potentially be used for the LYTAC approach. Other receptors might provide tissue specificity.
In 2021, the research groups of Bertozzi, Spiegel and Tang independently reported chimeric molecules with triantennary GalNAc for targeted protein degradation in the liver via the ASGPR (Fig. 45C).675,676,679 This CTL is not ubiquitously expressed as the CI-M6PR and is found primarily on hepatocytes with minimal expression on other cells (see Section 4.1). As a proof of concept, Bertozzi introduced a second-generation LYTAC consisting of a 3.4 kDa peptide binder linked to a trivalent GalNAc ligand that degraded integrins and reduced cancer cell proliferation.676 Spiegel, in contrast, focused on small molecule-based lysosome-targeting degraders. He called the heterobifunctional molecules MoDE-As (molecular degraders of extracellular proteins through the asialoglycoprotein receptor, Fig. 45C) and proved the concept by inducing depletion of an antibody and a proinflammatory cytokine.680 Tang further confirmed that lysosomal degradation of protein targets through ASGPR is possible by both small molecule- and antibody-based lysosome-targeting degraders. In addition, he showed that molecular size plays an important role and that internalization through ASGPR is more efficient for smaller degrader–target protein complexes.679
Avilar Therapeutics focused on extracellular protein degradation and recently introduced the ATAC platform, designed to target diverse pathological proteins and exploit the natural ASGPR protein degradation pathway. ATACs (ASGPR-targeting chimeras) are bifunctional molecules composed of a ligand that binds to ASGPR, linked to a second ligand which binds to a disease-causing extracellular protein. The chemical nature of the ASGPR ligand used in ATACs has not been disclosed, and the company has synthesized hundreds of monosaccharide-based ASGPR ligands, out of which over 100 had a KD ≤ 1000 nM and around 40 had a KD ≤ 100 nM. In addition, they have over 20 X-ray structures of ASGPR–ligand complexes.681 The chosen ASGPR ligand has approximately 2000-fold higher affinity than GalNAc and >60-fold increase in affinity over bicyclic bridged 182. Compared to the previous generation of compounds682 which contained trivalent GalNAc ligands attached to an antibody, ATACs are based on bi- and mono-dentate ligands attached to a peptide or small molecule.683 For initial proof-of-concept studies, ATACs were designed to target two extracellular proteins with different concentration and kinetic properties: IgG (high plasma concentration and long half-life) and TNF-α (low plasma concentration and short half-life). The in vitro studies demonstrated ligand binding, ternary complex formation, cellular uptake, and degradation of the target proteins, IgG and TNF-α.
Taken together, glycomimetics with a proper design to take advantage of specific receptor recognition, but also excellent endosomal release properties to allow receptor recycling and cargo release, will certainly be opening the way for many other lysosome shuttling receptors to be explored.
4.3 CD22 targeting – Siglec-engaging tolerance-inducing antigenic liposomes (STALs)
The limited expression of CD22 on B cells and its role as an inhibitory receptor for B cell receptor (BCR) signalling made this Siglec an attractive target for the treatment of various B cell-associated diseases. The carbohydrate-based glycomimetics 6′-MBP-5F-Neu5Ac (15) and 9-BPA-NeuGcα(2-6)Galβ(1-4)GlcNAcβ-spacer (74) developed for human and murine CD22, respectively, were the door openers.115,685 As a first application, 9-BPA-NeuGcα(2-6)Galβ(1-4)GlcNAcβ-spacer (74) co-presented with nitrophenol as a model antigen on a polymer backbone was used to enforce colocalization of the BCR and CD22 leading not only to shutdown of BCR signalling and reduced anti-nitrophenol antibody secretion, but also to B cell apoptosis.685 In the next step, 9-BPC-NeuAcα(2-6)Galβ(1-4)GlcNAcβ-spacer (75), a ligand still lacking sufficient specificity for human CD22, was successfully applied to kill human B cell lymphoma cells in a mouse; however, still suffering from the unwanted Siglec-1 off-target effect in the animals.686 Conceptually, this work was key, since the delivery of doxorubicin encapsulated in liposomes allowed to draw important conclusions about the endocytic properties, alternative particles to overcome cis-ligands and the general principle of B cell killing through toxin delivery.These findings were brought to a new level by implementing Siglec-engaging tolerance-inducing antigenic liposomes (STALs): co-display of an antigen with the 9-BPA-NeuGcα(2-6)Galβ(1-4)GlcNAcβ-spacer (74) ligand for murine CD22 induced B cells apoptosis in mice (Fig. 46A).367 Using a haemophilia model in which autoantibody production against factor VII would lead to severe bleeding in mice, Paulson and co-workers showed significantly reduced phenotype by antigen specific B cell ablation.367 Similarly efficient, the application of STALs showed great utility of this approach to reduce citrulline-specific B cells in vivo. Anti-citrulline autoantibodies are a hallmark of rheumatoid arthritis, an autoimmune disease for which the glycomimetic-based STALs showed applicability.687
 | ||
| Fig. 46 Targeting of the Siglec CD22. (A) STALs reduce antigen specific B cell response by colocalisation of the BCR and the inhibitory CD22. (B) After CD22-mediated uptake of a CD22 ligand–siRNA conjugate the expression of a target protein is suppressed via siRNA-mediated gene silencing. (C) Exploiting the same principle as STALs CD22 ligand–antibody conjugates reduce signalling of specific receptors. CD22L = CD22 ligand. Adopted from lit.367,693,694 | ||
In a recent follow-up, Macauley and colleagues have shown that changing the carrier from a liposome to red blood cells by direct insertion of the glycomimetic lipid conjugate, similar effects on B cells could be induced. Inhibition of cellular activation, reduction of cytokine secretion and cellular proliferation were achieved.688 To even further enhance the peripheral tolerance induction, STALs were co-formulated with rapamycin and using the model antigen ovalbumin it was shown that the anti-OVA antibody production was even more reduced compared to the common STAL formulation.689 To expand the application to human disease, the glycomimetic 6′-MBP-5F-Neu5Ac (15) was applied in a human CD22 transgenic mouse in a peanut allergy oral sensitization model.690 Together with previous data supporting the coformulation of rapamycin,689 the co-administration of STALs with poly(lactic-co-glycolic acid)–rapamycin nanoparticles (PLGA-R) induced robust peripheral tolerance compared to PLGA-R particles alone in a model of spontaneous autoimmune arthritis to the self-antigen glucose-6-phosphate-isomerase (GPI).691
Overall, CD22 glycomimetics have been successfully applied in vivo in various systems to showcase utility and applicability for the delivery of various cargo to B cells, not limited to toxins,686,692 model antigens,685,689 immunosuppressants,689,691 antibodies,693 and RNA therapeutics (Fig. 46B and C).694 These molecules gave important insights into the role of B cells and the potential treatment of haemophilia,367 B cell lymphoma,686,688 rheumatoid arthritis,687 and allergies,368,690,691,695 lately also their application for cellular reengineering to allow natural killer cells to achieve tumour-specific CD22 targeting.696
4.4 Langerin targeting
Langerin (CD207) is a CTL expressed on Langerhans cells (LCs), the major antigen presenting cells in the epidermis of the human skin. LCs are sentinels in the skin, protecting us against incoming threats,697 and central for the induction of an appropriate immune response.698 As an innate immune cell receptor, langerin is uniquely expressed by LCs and is involved in pathogen recognition promoting the uptake of several viruses such as HIV,699 measles,700 and influenza virus701 as well as fungi702 and mycobacteria.703 Langerin is very efficient in pathogen uptake since it is a fast recycling receptor, similar to the ASGPR and hence holds promise to be of similar utility for cell-specific delivery of therapeutics.152,704 The utility of langerin-based delivery has been explored previously using antibodies and has revealed that langerin mediated cargo uptake leads to cross-presentation of exogenous antigens, important for anti-viral and anti-cancer therapeutics.705–710 In this respect, the recent development of the carbohydrate-based glycomimetic 189 for human langerin was reported.711 The N-tosylated glucosamine 189 makes use of the Ca2+-mediated canonical hydroxy group coordination of the 3- and 4-OH groups. It was suggested that the tosyl group is involved in a T-stack interaction with Phe315 in the langerin binding site, resulting in an overall affinity of 230 μM including additional effects coming from the linker at the anomeric position (Fig. 47).711 Overall, these interactions add up to an overall 100-fold affinity gain over the parent glucose (Ki = 21 ± 4 mM). Remarkable affinity gains of the N-tosylated glucosamine could already be achieved by bivalent display on a DNA/PNA backbone with optimized ligand spacing712 and multivalent display on liposomes or proteins leading to highly specific delivery to LCs.711,713,714 When a protein antigen was directly conjugated to multiple glycomimetic ligands, LC specific delivery could also be shown ex vivo in intact human skin samples, unlocking new potentials for therapeutical exploring LC targeting and modulating the immune system.715 | ||
| Fig. 47 Carbohydrate-based glycomimetic 189 is a ligand for human langerin. (A) Chemical structure of 189.711 (B) Computational modelling of the complex between 189 and human langerin was supported by NMR data and shows interaction of Lys313 with the sulfonamide linker and a T-stacking interaction between Phe315 and the tosyl group. Adopted from lit.711 | ||
4.5 Selectin-targeting
Following the success of glycomimetics in various indications, they have consequently also found use as delivery agents. Besides the clinically used ASGPR ligands, the use of carbohydrates as targeting ligands has been widely studied in numerous applications using nanoparticles, e.g. for the imaging of inflammation in the brain via sialyl LewisX-decorated MRI active nanoparticles716 or galactosides as mono- or oligosaccharides for the targeting of galectins.717 Using the above-mentioned advantages of glycomimetics over natural glycosides, this concept has also been implemented for the targeting of the selectins.Sialyl LewisX glycomimetics have been developed for the production of E-selectin targeting liposomes.718 To this end, 33 (Fig. 6) bearing an N-acetyl group in position 2 of the galactose residue was modified at its carboxylic acid and coupled to the lipid 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE). Then, liposomes have been produced and were analysed for their E-selectin binding and targeting efficacy in vitro and in vivo. Targeted liposomes were shown to bind to E-selectin in an SPR experiment while untargeted liposomes were devoid of binding. Similar results were observed when fluorescent liposomes were assessed by FACS for binding to TNF-α-activated HUVEC cells where specificity was obtained upon E-selectin expression, further demonstrating the targeting efficacy in a cellular context. Last, the imaging of subcutaneously implanted non small cell lung carcinoma tumours in mice was successfully achieved with those glycomimetic liposomes, suggesting future applications for the delivery of loaded drug cargo.
4.6 Delivery of antibiotics via lectin targeting for P. aeruginosa
Bacterial infections are increasingly difficult to treat due rising antimicrobial resistance of the infectious agents and their additional defense within biofilms, e.g. for P. aeruginosa.439 Simply increasing the antibiotic concentration to break resistance and achieve effective antibacterial affinity is, however, not a tractable approach in the patient due to the toxicity of the drugs. Thus, one way to overcome this drawback is to conjugate antibiotics to targeting moieties for the selective enrichment at the pathogen inside the patient, either via siderophore-mediated uptake into the bacteria719,720 or through binding to their surface via antibodies721 and thereby increase the drug's local concentration for efficient killing. As a proof-of-concept, it was demonstrated that the LecA-targeted glycomimetic 116 conjugated to a fluorescent probe can bind and image P. aeruginosa biofilms.473 To implement the targeting, glycomimetics developed for the inhibition of P. aeruginosa lectins have been exploited as targeting units in three approaches (Fig. 48). | ||
| Fig. 48 Targeting of P. aeruginosa via its lectins LecA and LecB. Glycomimetics 190 and 191 are uncleavable antibiotic conjugates, while 192–195 are their cleavable successors designed for P. aeruginosa triggered proteolytic release of the antibiotic. Liposomes containing targeting lipids 196 and 197 can be used for the delivery of diverse cargo towards P. aeruginosa. | ||
First, the conjugation of LecA- and LecB-targeting moieties to the clinically used antibiotic ciprofloxacin, that suffers from rare but severe side effects, was reported.722 In this work, the targeting units were covalently attached to ciprofloxacin through a triazole ring and varying linker lengths resulting in conjugates 190 and 191. While the lectin binding of these conjugates was effective in the low μM range, the antimicrobial efficacy of the conjugates was reduced probably due to a reduced uptake to reach their intrabacterial targets, topoisomerase and gyrase. However, an enrichment of the conjugates at the P. aeruginosa biofilm and a reduced cytotoxicity of the conjugates compared to the free drug was achieved.
In a follow-up report, the linker was varied and the tetrapeptide sequence Gly-Ala-Leu-Ala was implemented as a scission motif for pathogen-specific activation by the secreted bacterial protease LasB to liberate the free drug at the infection site.723 The resulting conjugates 192–195 have been analysed for their degradation kinetics in various matrices and their antimicrobial activity. While all conjugates were stable in human blood plasma in vitro, they were rapidly cleaved in the presence of a P. aeruginosa culture supernatant containing the activating enzyme LasB. The degradation products from initial LasB scission were the lectin targeting moiety, available for blocking the respective lectin to weaken the biofilm, and a dipeptidyl antibiotic, which was further processed by plasma peptidases to liberate the free drug. Interestingly, the ciprofloxacin conjugates 192 and 193 were not fully cleaved into the free drug as a result of the tertiary amide at its piperazine moiety, resulting in reduced antimicrobial activity of the cleavage product. To overcome this drawback, fluoroquinolone derivatives with primary amides were used instead and a full proteolytic processing of conjugates 194 and 195 was observed and active drugs were liberated.
In a third approach by Titz and co-workers,724 the same LecA- and LecB-targeting ligands were attached to diacyl glycerol lipids (196 and 197) for incorporation into liposomes aiming at the delivery of unmodified antibiotics loaded into the nanoparticles. In an artificial biofilm model where the lectins LecA or LecB have been immobilized on an abiotic surface, it was shown that the targeted liposomes are specifically retained under flow conditions in a carbohydrate-dependent manner. Importantly, the binding was very tight as a result of the liposomes' multivalency which was demonstrated by the facts that for the LecA-targeted liposomes, 100 mM galactose in the buffer was required for displacement from the LecA surface, while for the LecB-targeted liposomes, even 250 mM fucose only partially displaced the glycomimetic liposomes and required the addition of EDTA to remove the calcium ions from LecB for full dissociation of the liposome from the lectin surface. Furthermore, it was demonstrated by SPR that the LecA-targeted liposome binding affinity is so strong due to the multivalency within the liposome that the dissociation of the liposomes from immobilised lectin is virtually absent. Thus, these glycomimetic liposomes constitute a promising delivery device for the targeting of antibiotics, which requires further research.
5. Conclusions and outlook
The more we learn about the biology of lectins, the more this exciting target class will move into the focus of drug discovery. As of now, lectins have slowly emerged as targets for pharmaceutical treatment of a number of diseases, either validated by the use of therapeutic antibodies or the persistent work of the community to develop small-molecule mimetics of their natural carbohydrate substrates.Carbohydrates as natural ligands themselves have been challenging to promote into a pharmaceutical active ingredient. However, they are stereochemically rich starting structures with good ligand efficiencies, relating the low molecular weight of a monosaccharide and the affinity resulting from the high number of directed interactions. These often enthalpy-driven interactions provide specificity with respect to the many off-targets other than lectins themselves. Glycomimetics are not reported to have significant unwanted targets other than lectins. Still, within the lectins as a target group, specificity can be generated through the monosaccharide, by modification of the original scaffold only. Advancements in carbohydrate chemistry have made this compound class more accessible while still being challenging compared to other scaffolds. Additionally, while a growing number of glycomimetics has been developed into orally available drug candidates, several applications of glycomimetics are independent of oral application routes, similar to the treatment with therapeutic antibodies.
To overcome the limitations of maintaining the original carbohydrate scaffold, alternative approaches have emerged by replacing the sugar core by keeping only the most relevant pharmacophores to establish affinity and specificity leading to non-carbohydrate based, functional glycomimetics. Another approach that might lead to a modulation of lectins for therapeutic intervention are allosteric modulators as functional glycomimetics. These molecules might serve as antagonists or even agonists, via binding to allosteric sites, remote from the carbohydrate recognition site, rendering the necessity to resemble a carbohydrate obsolete. Our enthusiasm over this promising route to tackle lectins is fuelled by the advancement it brought to other challenging drug target classes such as kinases and phosphatases.
Infection processes are inherently linked to carbohydrate–lectin interactions. This fact therefore provides a valuable starting point for the development of new antiinfectives. To this end, the rich field of glycomimetics in the discovery phase has already led to numerous compounds in (pre-)clinical development, especially for bacterial infections by E. coli in urinary tract infections and inflammatory bowel disease.
Besides the intriguing biological functions of lectins in inflammation, cancer and infection, mammalian lectins are typically limited in their expression profile for defined cells, which makes them attractive targets for targeted delivery purposes. The ASGPR as an accessible hepatic receptor has spearheaded this development. The trivalent GalNAc ligand 22 has assisted to bring four drugs onto the market and led to numerous new therapeutics in clinical trials. Similarly, the CI-M6PR has been proposed as target protein for the development of LYTACs, based on simple mannose-6-phosphonate polymers that allow targeted degradation of extracellular proteins. For the future clinical application of this concept it will be helpful to identify more tissue specific, endocytic receptors with high expression profiles. These will be key towards the specific degradation of targeted membrane receptors.
Taken together, we anticipate a prosperous future for glycomimetics to modulate biological processes in chemical biology research but also as drugs in a large diversity of indications.
Conflicts of interest
C. R. is co-founder, advisor, and shareholder of Cutanos GmbH, a company that is working on the commercialization of a glycomimetic-based targeting of Langerhans cells. C. R. and A. T. are inventors on patents covering glycomimetics.Acknowledgements
C. R. thanks the funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 956314 ALLODD and the funding from European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement 614 no. 716024). This research was funded in part by the Austrian Science Fund (FWF) [I 5157-B] and the Deutsche Forschungsgemeinschaft (RA1944/7-1). P. M. thanks Max Planck Institute (partner group programme) and IOCB Tech for support. A. T. acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement No. 716311, Sweetbullets), DZIF (TTU 09.718) and Deutsche Forschungsgemeinschaft (grant no. Ti756/5-1). Some figures were created using biorender.com.References
- A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 4th edn, 2022 Search PubMed.
- R. A. Laine, Glycobiology, 1994, 4, 759–767 CrossRef CAS PubMed.
- A. Martínez-Palomo, Int. Rev. Cytol., 1970, 29, 29–75 Search PubMed.
- J. W. Costerton, R. T. Irvin and K.-J. Cheng, Annu. Rev. Microbiol., 1981, 35, 299–324 CrossRef CAS PubMed.
- P. A. Haynes, Glycobiology, 1998, 8, 1–5 CrossRef CAS PubMed.
- S. L. Crine and K. R. Acharya, FEBS J., 2022, 289(24), 7670–7687 CrossRef CAS PubMed.
- M. A. J. Ferguson and A. F. Williams, Annu. Rev. Biochem., 1988, 57, 285–320 CrossRef CAS PubMed.
- H. Ikezawa, Biol. Pharm. Bull., 2002, 25, 409–417 CrossRef CAS PubMed.
- F. Ge, L. Zhu, A. Aang, P. Song, W. Li, Y. Tao and G. Du, Biotechnol. Lett., 2018, 40, 847–854 CrossRef CAS PubMed.
- B. M. Harvey and R. S. Haltiwanger, in Molecular Mechanisms of Notch Signaling, ed. T. Borggrefe and B. D. Giaimo, Springer International Publishing, Cham, 2018, pp. 59–78 DOI:10.1007/978-3-319-89512-3_4.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef PubMed.
- A. Buffone, Jr. and V. M. Weaver, J. Cell Biol., 2019, 219, e201910070 CrossRef PubMed.
- G. S. Offeddu, C. Hajal, C. R. Foley, Z. Wan, L. Ibrahim, M. F. Coughlin and R. D. Kamm, Commun. Biol., 2021, 4, 255 CrossRef CAS PubMed.
- J. C. Lumibao, J. R. Tremblay, J. Hsu and D. D. Engle, J. Exp. Med., 2022, 219, e20211505 CrossRef CAS PubMed.
- A. Radhakrishnan, K. Park, I. Kwak, M. Jaabir and J. Sivakamavalli, in Lectins: Innate immune defense and Therapeutics, ed. P. Elumalai and S. Lakshmi, Springer Singapore, Singapore, 2021, pp. 51–72 DOI:10.1007/978-981-16-7462-4_3.
- A. Engering, T. B. Geijtenbeek, S. J. van Vliet, M. Wijers, E. van Liempt, N. Demaurex, A. Lanzavecchia, J. Fransen, C. G. Figdor, V. Piguet and Y. van Kooyk, J. Immunol., 2002, 168, 2118–2126 CrossRef CAS PubMed.
- J. den Dunnen, S. I. Gringhuis and T. B. Geijtenbeek, Cancer Immunol. Immunother., 2009, 58, 1149–1157 CrossRef CAS PubMed.
- N. Romani, B. E. Clausen and P. Stoitzner, Immunol. Rev., 2010, 234, 120–141 CrossRef CAS PubMed.
- R. P. McEver, Cardiovasc. Res., 2015, 107, 331–339 CrossRef CAS PubMed.
- T. Krause and G. A. Turner, Clin. Exp. Metastas., 1999, 17, 183–192 CrossRef CAS PubMed.
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139 CrossRef PubMed.
- R.-Y. Yang, G. A. Rabinovich and F.-T. Liu, Expert Rev. Mol. Med., 2008, 10, e17 CrossRef PubMed.
- T. B. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. van Vliet, G. C. F. van Duijnhoven, J. Middel, I. L. H. A. Cornelissen, H. S. L. M. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor and Y. van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS PubMed.
- P.-Y. Lozach, H. Lortat-Jacob, A. de Lacroix de Lavalette, I. Staropoli, S. Foung, A. Amara, C. Houles, F. Fieschi, O. Schwartz, J. L. Virelizier, F. Arenzana-Seisdedos and R. Altmeyer, J. Biol. Chem., 2003, 278, 20358–20366 CrossRef CAS PubMed.
- R. Amraei, W. Yin, M. A. Napoleon, E. L. Suder, J. Berrigan, Q. Zhao, J. Olejnik, K. B. Chandler, C. Xia, J. Feldman, B. M. Hauser, T. M. Caradonna, A. G. Schmidt, S. Gummuluru, E. Muhlberger, V. Chitalia, C. E. Costello and N. Rahimi, ACS Cent. Sci., 2021, 7, 1156–1165 CrossRef CAS PubMed.
- C. P. Alvarez, F. Lasala, J. Carrillo, O. Muniz, A. L. Corbi and R. Delgado, J. Virol., 2002, 76, 6841–6844 CrossRef CAS PubMed.
- A. L. Lewis, J. L. Kohler and M. Aebi, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, 2022, ch. 37 DOI:10.1101/glycobiology.4e.37.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- N. Sharon, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 527–537 CrossRef CAS PubMed.
- D. C. Smith, J. M. Lord, L. M. Roberts and L. Johannes, Semin. Cell Dev. Biol., 2004, 15, 397–408 CrossRef CAS PubMed.
- R. Pellizzari, O. Rossetto, G. Schiavo and C. Montecucco, Philos. Trans. R. Soc., B, 1999, 354, 259–268 CrossRef CAS PubMed.
- J. M. Lord, L. M. Roberts and J. D. Robertus, FASEB J., 1994, 8, 201–208 CrossRef CAS PubMed.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS PubMed.
- P. Neri, S. I. Nagano, S.-I. Yokoyama, H. Dohi, K. Kobayashi, T. Miura, T. Inazu, T. Sugiyama, Y. Nishida and H. Mori, Microbiol. Immunol., 2007, 51, 581–592 CrossRef CAS PubMed.
- A. A. Kulkarni, C. Fuller, H. Korman, A. A. Weiss and S. S. Iyer, Bioconjugate Chem., 2010, 21, 1486–1493 CrossRef CAS PubMed.
- H. O. Yosief, S. S. Iyer and A. A. Weiss, Infect. Immun., 2013, 81, 2753–2760 CrossRef CAS PubMed.
- C. P. Sager, D. Eriş, M. Smieško, R. Hevey and B. Ernst, Beilstein J. Org. Chem., 2017, 13, 2584–2595 CrossRef CAS PubMed.
- N. P. Mullin, P. G. Hitchen and M. E. Taylor, J. Biol. Chem., 1997, 272, 5668–5681 CrossRef CAS PubMed.
- F. P. Schwarz, K. D. Puri, R. G. Bhat and A. Surolia, J. Biol. Chem., 1993, 268, 7668–7677 CrossRef CAS PubMed.
- S. Cabani, P. Gianni, V. Mollica and L. Lepori, J. Solution Chem., 1981, 10, 563–595 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 49–76 Search PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, 1997 Search PubMed.
- K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152–15160 CrossRef CAS PubMed.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug. Discov., 2009, 8, 661–677 CrossRef CAS PubMed.
- R. A. Copeland, Expert Opin. Drug Dis., 2021, 16, 1441–1451 CrossRef PubMed.
- V. Georgi, F. Schiele, B.-T. Berger, A. Steffen, P. A. Marin Zapata, H. Briem, S. Menz, C. Preusse, J. D. Vasta, M. B. Robers, M. Brands, S. Knapp and A. Fernández-Montalván, J. Am. Chem. Soc., 2018, 140, 15774–15782 CrossRef CAS PubMed.
- C. A. Lipinski, J. Pharmacol. Toxicol., 2000, 44, 235–249 CrossRef CAS PubMed.
- N. J. Overeem, P. H. Hamming, M. Tieke, E. van der Vries and J. Huskens, ACS Nano, 2021, 15, 8525–8536 CrossRef CAS PubMed.
- G. V. Dubacheva, T. Curk, B. M. Mognetti, R. Auzély-Velty, D. Frenkel and R. P. Richter, J. Am. Chem. Soc., 2014, 136, 1722–1725 CrossRef CAS PubMed.
- G. V. Dubacheva, T. Curk, D. Frenkel and R. P. Richter, J. Am. Chem. Soc., 2019, 141, 2577–2588 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- A. Holla and A. Skerra, Protein Eng., Des. Sel., 2011, 24, 659–669 CrossRef CAS PubMed.
- B. M. Curtis, S. Scharnowske and A. J. Watson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8356–8360 CrossRef CAS PubMed.
- M. E. Taylor and K. Drickamer, Curr. Opin. Struc. Biol., 2014, 28, 14–22 CrossRef CAS PubMed.
- X. Cao, X. Du, H. Jiao, Q. An, R. Chen, P. Fang, J. Wang and B. Yu, Acta Pharm. Sin. B, 2022, 12, 3783–3821 CrossRef CAS PubMed.
- J. Patocka, E. Nepovimova, W. Wu and K. Kuca, Environ. Toxicol. Pharmacol., 2020, 79, 103400 CrossRef CAS PubMed.
- C. Schwarz, G. Taccetti, P.-R. Burgel and S. Mulrennan, Resp. Med., 2022, 195, 106778 CrossRef PubMed.
- V. Heinemann, L. W. Hertel, G. B. Grindey and W. Plunkett, Cancer Res., 1988, 48, 4024–4031 CAS.
- D. Siegel, H. C. Hui, E. Doerffler, M. O. Clarke, K. Chun, L. Zhang, S. Neville, E. Carra, W. Lew, B. Ross, Q. Wang, L. Wolfe, R. Jordan, V. Soloveva, J. Knox, J. Perry, M. Perron, K. M. Stray, O. Barauskas, J. Y. Feng, Y. Xu, G. Lee, A. L. Rheingold, A. S. Ray, R. Bannister, R. Strickley, S. Swaminathan, W. A. Lee, S. Bavari, T. Cihlar, M. K. Lo, T. K. Warren and R. L. Mackman, J. Med. Chem., 2017, 60, 1648–1661 CrossRef CAS PubMed.
- P. Som, H. L. Atkins, D. Bandoypadhyay, J. S. Fowler, R. R. MacGregor, K. Matsui, Z. H. Oster, D. F. Sacker, C. Y. Shiue, H. Turner, C. N. Wan, A. P. Wolf and S. V. Zabinski, J. Nucl. Med., 1980, 21, 670–675 CAS.
- I. Hillebrand, K. Boehme, G. Frank, H. Fink and P. Berchtold, Res. Exp. Med., 1979, 175, 81–86 CrossRef CAS PubMed.
- J. M. Woods, R. C. Bethell, J. A. Coates, N. Healy, S. A. Hiscox, B. A. Pearson, D. M. Ryan, J. Ticehurst, J. Tilling and S. M. Walcott, et al. , Antimicrob. Agents Chemother., 1993, 37, 1473–1479 CrossRef CAS PubMed.
- J. W. M. Cheng, Clin. Ther., 2002, 24, 1757–1769 CrossRef CAS PubMed.
- C. U. Kim, W. Lew, M. A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver and R. C. Stevens, J. Am. Chem. Soc., 1997, 119, 681–690 CrossRef CAS PubMed.
- B. E. Maryanoff, S. O. Nortey, J. F. Gardocki, R. P. Shank and S. P. Dodgson, J. Med. Chem., 1987, 30, 880–887 CrossRef CAS PubMed.
- B. Lembcke, U. R. Fölsch and W. Creutzfeldt, Digestion, 1985, 31, 120–127 CAS.
- W. Meng, B. A. Ellsworth, A. A. Nirschl, P. J. McCann, M. Patel, R. N. Girotra, G. Wu, P. M. Sher, E. P. Morrison, S. A. Biller, R. Zahler, P. P. Deshpande, A. Pullockaran, D. L. Hagan, N. Morgan, J. R. Taylor, M. T. Obermeier, W. G. Humphreys, A. Khanna, L. Discenza, J. G. Robertson, A. Wang, S. Han, J. R. Wetterau, E. B. Janovitz, O. P. Flint, J. M. Whaley and W. N. Washburn, J. Med. Chem., 2008, 51, 1145–1149 CrossRef CAS PubMed.
- H. C. Kolb and B. Ernst, Chem. – Eur. J., 1997, 3, 1571–1578 CrossRef CAS.
- G. Thoma, J. L. Magnani, J. T. Patton, B. Ernst and W. Jahnke, Angew. Chem., Int. Ed., 2001, 40, 1941–1945 CrossRef CAS PubMed.
- A. Tamburrini, C. Colombo and A. Bernardi, Med. Res. Rew., 2020, 40, 495–531 CrossRef CAS PubMed.
- S. Sattin and A. Bernardi, Carbohydrate Chemistry, The Royal Society of Chemistry, 2016, vol. 41, pp. 1–25 Search PubMed.
- V. C. Damalanka, A. R. Maddirala and J. W. Janetka, Expert Opin. Drug Disc., 2021, 16, 513–536 CrossRef CAS PubMed.
- R. Hevey, Pharmaceuticals, 2019, 12, 55 CrossRef CAS PubMed.
- R. Hevey, Chem. – Eur. J., 2021, 27, 2240–2253 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- C. Colombo and A. Bernardi, Eur. J. Org. Chem., 2011, 3911–3919 CrossRef CAS.
- W. Wang, P. Rattananakin and P. G. Goekjian, J. Carbohydr. Chem., 2003, 22, 743–751 CrossRef CAS.
- K. M. Driller, S. Libnow, M. Hein, M. Harms, K. Wende, M. Lalk, D. Michalik, H. Reinke and P. Langer, Org. Biomol. Chem., 2008, 6, 4218–4223 RSC.
- I. Cumpstey, S. Agrawal, K. E. Borbas and B. Martín-Matute, Chem. Commun., 2011, 47, 7827–7829 RSC.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS PubMed.
- K. Bijian, Z. Zhang, B. Xu, S. Jie, B. Chen, S. Wan, J. Wu, T. Jiang and M. A. Alaoui-Jamali, Eur. J. Med. Chem., 2012, 48, 143–152 CrossRef CAS PubMed.
- K. Sidoryk, L. Rárová, J. Oklešťková, Z. Pakulski, M. Strnad, P. Cmoch and R. Luboradzki, Org. Biomol. Chem., 2016, 14, 10238–10248 RSC.
- A. W. McDonagh, M. F. Mahon and P. V. Murphy, Org. Lett., 2016, 18, 552–555 CrossRef CAS PubMed.
- T. Suzuki, H. Makyio, H. Ando, N. Komura, M. Menjo, Y. Yamada, A. Imamura, H. Ishida, S. Wakatsuki and R. Kato, Bioorg. Med. Chem., 2014, 22, 2090–2101 CrossRef CAS PubMed.
- I. Pérez-Victoria, O. Boutureira, T. D. Claridge and B. G. Davis, Chem. Commun., 2015, 51, 12208–12211 RSC.
- S. André, K. E. Kövér, H.-J. Gabius and L. Szilágyi, Bioorg. Med. Chem. Lett., 2015, 25, 931–935 CrossRef PubMed.
- R. Sommer, O. N. Makshakova, T. Wohlschlager, S. Hutin, M. Marsh, A. Titz, M. Künzler and A. Varrot, Structure, 2018, 26, 391–402 CrossRef CAS PubMed.
- N. Kostlánová, E. P. Mitchell, H. Lortat-Jacob, S. Oscarson, M. Lahmann, N. Gilboa-Garber, G. Chambat, M. Wimmerová and A. Imberty, J. Biol. Chem., 2005, 280, 27839–27849 CrossRef PubMed.
- A. V. Demchenko, Handbook of chemical glycosylation: advances in stereoselectivity and therapeutic relevance, John Wiley & Sons, 2008 Search PubMed.
- M. Spell, X. Wang, A. E. Wahba, E. Conner and J. Ragains, Carbohydr. Res., 2013, 369, 42–47 CrossRef CAS PubMed.
- T. Furuta, K. Takeuchi and M. Iwamura, Chem. Commun., 1996, 157–158 RSC.
- G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS PubMed.
- R. Lahiri, A. A. Ansari and Y. D. Vankar, Chem. Soc. Rev., 2013, 42, 5102–5118 RSC.
- A. E. Stütz and T. M. Wrodnigg, Adv. Carbohydr. Chem. Biochem., 2011, 66, 187–298 CrossRef PubMed.
- X. Gu, V. Gupta, Y. Yang, J. Y. Zhu, E. J. Carlson, C. Kingsley, J. S. Tash, E. Schönbrunn, J. Hawkinson and G. I. Georg, ChemMedChem, 2017, 12, 1977–1984 CrossRef CAS PubMed.
- H. Paulsen, Angew. Chem., Int. Ed. Engl., 1966, 5, 495–510 CrossRef CAS.
- R. A. Dwek, T. D. Butters, F. M. Platt and N. Zitzmann, Nat. Rev. Drug Discov., 2002, 1, 65–75 CrossRef CAS PubMed.
- O. Arjona, A. M. Gomez, J. C. Lopez and J. Plumet, Chem. Rev., 2007, 107, 1919–2036 CrossRef CAS PubMed.
- S. Roscales and J. Plumet, Int. J. Carbohydr. Chem., 2016, 2016, 760548 Search PubMed.
- B. López-Méndez, C. Jia, Y. Zhang, L. H. Zhang, P. Sinay, J. Jiménez-Barbero and M. Sollogoub, Chem. – Asian J., 2008, 3, 51–58 CrossRef PubMed.
- I. Robina, P. Vogel and Z. J. Witczak, Curr. Org. Chem., 2001, 5, 1177–1214 CrossRef CAS.
- Z. J. Witczak, Curr. Med. Chem., 1999, 6, 165–178 CrossRef CAS PubMed.
- X. Liao, V. c Větvička and D. Crich, J. Org. Chem., 2018, 83, 14894–14904 CrossRef CAS PubMed.
- S. Ito, M. Yamashita, T. Niimi, M. Fujie, V. K. Reddy, H. Totsuka, B. Haritha, K. Maddali, S. Nakamura and K. Asai, Heterocycl. Commun., 2009, 15, 23–30 CAS.
- B. Dayde, C. Pierra, G. Gosselin, D. Surleraux, A. T. Ilagouma, C. Laborde, J. N. Volle, D. Virieux and J. L. Pirat, Eur. J. Org. Chem., 2014, 1333–1337 CrossRef CAS.
- B. Xu, L. Unione, J. Sardinha, S. Wu, M. Ethève-Quelquejeu, A. Pilar Rauter, Y. Blériot, Y. Zhang, S. Martín-Santamaría and D. Díaz, Angew. Chem., Int. Ed., 2014, 126, 9751–9756 CrossRef.
- B. Linclau, S. Golten, M. Light, M. Sebban and H. Oulyadi, Carbohydr. Res., 2011, 346, 1129–1139 CrossRef CAS PubMed.
- R. Hevey, Biomimetics, 2019, 4, 53 CrossRef CAS PubMed.
- J. C. Biffinger, H. W. Kim and S. G. DiMagno, ChemBioChem, 2004, 5, 622–627 CrossRef CAS PubMed.
- S. G. Withers, D. J. MacLennan and I. P. Street, Carbohydr. Res., 1986, 154, 127–144 CrossRef CAS.
- T. Wohlschlager, A. Butschi, P. Grassi, G. Sutov, R. Gauss, D. Hauck, S. S. Schmieder, M. Knobel, A. Titz, A. Dell, S. M. Haslam, M. O. Hengartner, M. Aebi and M. Künzler, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2787–E2796 CrossRef CAS PubMed.
- Z. Han, J. S. Pinkner, B. Ford, R. Obermann, W. Nolan, S. A. Wildman, D. Hobbs, T. Ellenberger, C. K. Cusumano and S. J. Hultgren, J. Med. Chem., 2010, 53, 4779–4792 CrossRef CAS PubMed.
- O. Sperling, A. Fuchs and T. K. Lindhorst, Org. Biomol. Chem., 2006, 4, 3913–3922 RSC.
- T. Klein, D. Abgottspon, M. Wittwer, S. Rabbani, J. Herold, X. Jiang, S. Kleeb, C. Lüthi, M. Scharenberg, J. Bezençon, E. Gubler, L. Pang, M. Smiesko, B. Cutting, O. Schwardt and B. Ernst, J. Med. Chem., 2010, 53, 8627–8641 CrossRef CAS PubMed.
- C. D. Rillahan, M. S. Macauley, E. Schwartz, Y. He, R. McBride, B. M. Arlian, J. Rangarajan, V. V. Fokin and J. C. Paulson, Chem. Sci., 2014, 5, 2398–2406 RSC.
- P. Sörme, Y. Qian, P. G. Nyholm, H. Leffler and U. J. Nilsson, ChemBioChem, 2002, 3, 183–189 CrossRef.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, Chem. – Eur. J., 2008, 14, 4233–4245 CrossRef CAS PubMed.
- V. Kalas, M. E. Hibbing, A. R. Maddirala, R. Chugani, J. S. Pinkner, L. K. Mydock-McGrane, M. S. Conover, J. W. Janetka and S. J. Hultgren, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2819–E2828 CrossRef CAS PubMed.
- A. R. Maddirala, R. Klein, J. S. Pinkner, V. Kalas, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2019, 62, 467–479 CrossRef CAS PubMed.
- D. Schwizer, H. Gäthje, S. Kelm, M. Porro, O. Schwardt and B. Ernst, Bioorg. Med. Chem., 2006, 14, 4944–4957 CrossRef CAS PubMed.
- S. Kuhaudomlarp, E. Siebs, E. Shanina, J. Topin, I. Joachim, P. da Silva Figueiredo Celestino Gomes, A. Varrot, D. Rognan, C. Rademacher, A. Imberty and A. Titz, Angew. Chem., Int. Ed., 2021, 60, 8104–8114 CrossRef CAS PubMed.
- E. Shanina, S. Kuhaudomlarp, E. Siebs, F. F. Fuchsberger, M. Denis, P. da Silva Figueiredo Celestino Gomes, M. H. Clausen, P. H. Seeberger, D. Rognan, A. Titz, A. Imberty and C. Rademacher, Commun. Chem., 2022, 5, 64 CrossRef CAS PubMed.
- K. C. A. Garber, K. Wangkanont, E. E. Carlson and L. L. Kiessling, Chem. Commun., 2010, 46, 6747–6749 RSC.
- J. Aretz, E.-C. Wamhoff, J. Hanske, D. Heymann and C. Rademacher, Front. Immunol., 2014, 5, 323 Search PubMed.
- E. Shanina, S. Kuhaudomlarp, K. Lal, P. H. Seeberger, A. Imberty and C. Rademacher, Angew. Chem., Int. Ed., 2022, 61, e202109339 CrossRef CAS PubMed.
- J. Aretz, U. R. Anumala, F. F. Fuchsberger, N. Molavi, N. Ziebart, H. Zhang, M. Nazaré and C. Rademacher, J. Am. Chem. Soc., 2018, 140, 14915–14925 CrossRef CAS PubMed.
- R. P. Dings, E. S. Van Laar, J. Webber, Y. Zhang, R. J. Griffin, S. J. Waters, J. R. MacDonald and K. H. Mayo, Cancer Lett., 2008, 265, 270–280 CrossRef CAS PubMed.
- R. P. Dings, N. Kumar, M. C. Miller, M. Loren, H. Rangwala, T. R. Hoye and K. H. Mayo, J. Pharmacol. Exp. Ther., 2013, 344, 589–599 CrossRef CAS PubMed.
- H. Zhang, O. Daněk, D. Makarov, S. Rádl, D. Kim, J. Ledvinka, K. Vychodilová, J. Hlaváč, J. Lefèbre, M. Denis, C. Rademacher and P. Ménová, ACS Med. Chem. Lett., 2022, 13, 935–942 CrossRef CAS PubMed.
- Z. Chen, Q. Cui, M. Caffrey, L. Rong and R. Du, Pharmaceuticals, 2021, 14, 587 CrossRef CAS PubMed.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013–2025 RSC.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- R. D. Cummings, E. Chiffoleau, Y. van Kyook and R. P. McEver, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, 2022, ch. 34 DOI:10.1101/glycobiology.4e.34.
- G. D. Brown, J. A. Willment and L. Whitehead, Nat. Rev. Immunol., 2018, 18, 374–389 CrossRef CAS PubMed.
- K. Drickamer and M. E. Taylor, Curr. Opin. Struct. Biol., 2015, 34, 26–34 CrossRef CAS PubMed.
- S. Mayer, M. K. Raulf and B. Lepenies, Histochem. Cell Biol., 2017, 147, 223–237 CrossRef CAS PubMed.
- J. Cramer, RSC Med. Chem., 2021, 12, 1985–2000 RSC.
- P. Valverde, J. D. Martinez, F. J. Canada, A. Arda and J. Jimenez-Barbero, ChemBioChem, 2020, 21, 2999–3025 CrossRef CAS PubMed.
- W. I. Weis, K. Drickamer and W. A. Hendrickson, Nature, 1992, 360, 127–134 CrossRef CAS PubMed.
- W. I. Weis, R. Kahn, R. Fourme, K. Drickamer and W. A. Hendrickson, Science, 1991, 254, 1608–1615 CrossRef CAS PubMed.
- A. N. Zelensky and J. E. Gready, FEBS J., 2005, 272, 6179–6217 CrossRef CAS PubMed.
- M. E. Taylor and K. Drickamer, J. Biol. Chem., 1993, 268, 399–404 CrossRef CAS PubMed.
- S. A. McMahon, J. L. Miller, J. A. Lawton, D. E. Kerkow, A. Hodes, M. A. Marti-Renom, S. Doulatov, E. Narayanan, A. Sali, J. F. Miller and P. Ghosh, Nat. Struct. Mol. Biol., 2005, 12, 886–892 CrossRef CAS PubMed.
- K. Drickamer, Nature, 1992, 360, 183–186 CrossRef CAS PubMed.
- W. I. Weis, M. E. Taylor and K. Drickamer, Immunol. Rev., 1998, 163, 19–34 CrossRef CAS PubMed.
- M. E. Taylor and K. Drickamer, Curr. Opin. Struct. Biol., 2014, 28, 14–22 CrossRef CAS PubMed.
- C. P. Sager, D. Eris, M. Smiesko, R. Hevey and B. Ernst, Beilstein J. Org. Chem., 2017, 13, 2584–2595 CrossRef CAS PubMed.
- R. C. Preston, R. P. Jakob, F. P. Binder, C. P. Sager, B. Ernst and T. Maier, J. Mol. Cell Biol., 2016, 8, 62–72 CrossRef CAS PubMed.
- K. K. Ng, S. Park-Snyder and W. I. Weis, Biochemistry, 1998, 37, 17965–17976 CrossRef CAS PubMed.
- T. Kawasaki and G. Ashwell, J. Biol. Chem., 1977, 252, 6536–6543 CrossRef CAS PubMed.
- R. Mc Dermott, U. Ziylan, D. Spehner, H. Bausinger, D. Lipsker, M. Mommaas, J. P. Cazenave, G. Raposo, B. Goud, H. de la Salle, J. Salamero and D. Hanau, Mol. Biol. Cell, 2002, 13, 317–335 CrossRef CAS PubMed.
- A. L. Schwartz, H. J. Geuze and H. F. Lodish, Philos. Trans. R. Soc., B, 1982, 300, 229–235 CAS.
- J. Brown, C. A. O'Callaghan, A. S. Marshall, R. J. Gilbert, C. Siebold, S. Gordon, G. D. Brown and E. Y. Jones, Protein Sci., 2007, 16, 1042–1052 CrossRef CAS PubMed.
- M. Nagae, K. Morita-Matsumoto, M. Kato, M. K. Kaneko, Y. Kato and Y. Yamaguchi, Structure, 2014, 22, 1711–1721 CrossRef CAS PubMed.
- N. Silva-Martin, S. G. Bartual, E. Ramirez-Aportela, P. Chacon, C. G. Park and J. A. Hermoso, Structure, 2014, 22, 1595–1606 CrossRef CAS PubMed.
- J. C. Munoz-Garcia, E. Chabrol, R. R. Vives, A. Thomas, J. L. de Paz, J. Rojo, A. Imberty, F. Fieschi, P. M. Nieto and J. Angulo, J. Am. Chem. Soc., 2015, 137, 4100–4110 CrossRef CAS PubMed.
- J. Hanske, R. Wawrzinek, A. Geissner, E. C. Wamhoff, K. Sellrie, H. Schmidt, P. H. Seeberger and C. Rademacher, ChemBioChem, 2017, 18, 1183–1187 CrossRef CAS PubMed.
- J. Egger, C. Weckerle, B. Cutting, O. Schwardt, S. Rabbani, K. Lemme and B. Ernst, J. Am. Chem. Soc., 2013, 135, 9820–9828 CrossRef CAS PubMed.
- J. Aretz, H. Baukmann, E. Shanina, J. Hanske, R. Wawrzinek, V. A. Zapol'skii, P. H. Seeberger, D. E. Kaufmann and C. Rademacher, Angew. Chem., Int. Ed., 2017, 56, 7292–7296 CrossRef CAS PubMed.
- J. Aretz, U. R. Anumala, F. F. Fuchsberger, N. Molavi, N. Ziebart, H. Zhang, M. Nazare and C. Rademacher, J. Am. Chem. Soc., 2018, 140, 14915–14925 CrossRef CAS PubMed.
- R. Wawrzinek, E. C. Wamhoff, J. Lefebre, M. Rentzsch, G. Bachem, G. Domeniconi, J. Schulze, F. F. Fuchsberger, H. Zhang, C. Modenutti, L. Schnirch, M. A. Marti, O. Schwardt, M. Brautigam, M. Guberman, D. Hauck, P. H. Seeberger, O. Seitz, A. Titz, B. Ernst and C. Rademacher, J. Am. Chem. Soc., 2021, 143, 18977–18988 CrossRef CAS PubMed.
- C. Huysamen, J. A. Willment, K. M. Dennehy and G. D. Brown, J. Biol. Chem., 2008, 283, 16693–16701 CrossRef CAS PubMed.
- R. E. Shrimpton, M. Butler, A.-S. Morel, E. Eren, S. S. Hue and M. A. Ritter, Mol. Immunol., 2009, 46, 1229–1239 CrossRef CAS PubMed.
- P. L. Kavanagh, T. A. Fasipe and T. Wun, JAMA, 2022, 328, 57–68 CrossRef CAS PubMed.
- H. Laubli and L. Borsig, Semin. Cancer Biol., 2010, 20, 169–177 CrossRef PubMed.
- W. S. Somers, J. Tang, G. D. Shaw and R. T. Camphausen, Cell, 2000, 103, 467–479 CrossRef CAS PubMed.
- F. P. Binder, K. Lemme, R. C. Preston and B. Ernst, Angew. Chem., Int. Ed., 2012, 51, 7327–7331 CrossRef CAS PubMed.
- P. W. Bedard and N. Kaila, Expert Opin. Ther. Pat., 2010, 20, 781–793 CrossRef CAS PubMed.
- N. Kaila and B. E. t Thomas, Med. Res. Rev., 2002, 22, 566–601 CrossRef CAS PubMed.
- K. Scheffler, J. R. Brisson, R. Weisemann, J. L. Magnani, W. T. Wong, B. Ernst and T. Peters, J. Biomol. NMR, 1997, 9, 423–436 CrossRef CAS PubMed.
- K. E. Norman, G. P. Anderson, H. C. Kolb, K. Ley and B. Ernst, Blood, 1998, 91, 475–483 CrossRef CAS PubMed.
- D. Schwizer, J. T. Patton, B. Cutting, M. Smiesko, B. Wagner, A. Kato, C. Weckerle, F. P. Binder, S. Rabbani, O. Schwardt, J. L. Magnani and B. Ernst, Chemistry, 2012, 18, 1342–1351 CrossRef CAS PubMed.
- J. Chang, J. T. Patton, A. Sarkar, B. Ernst, J. L. Magnani and P. S. Frenette, Blood, 2010, 116, 1779–1786 CrossRef PubMed.
- M. J. Telen, T. Wun, T. L. McCavit, L. M. De Castro, L. Krishnamurti, S. Lanzkron, L. L. Hsu, W. R. Smith, S. Rhee, J. L. Magnani and H. Thackray, Blood, 2015, 125, 2656–2664 CrossRef CAS PubMed.
- J. L. Magnani, Presented in part at the ACS Spring Meeting 2021, 2021.
- P. Dätwyler, X. Jiang, B. Wagner, N. Varga, T. Mühlethaler, K. Hostettler, S. Rabbani, O. Schwardt and B. Ernst, ChemMedChem, 2022, 17, e202100634 CrossRef PubMed.
- J. Peterson, M.-G. Baek, S. Locatelli-Hoops, J.-W. Lee, L. Deng, D. A. Stewart, T. A. Smith, D. D. Myers, W. E. Fogler and J. L. Magnani, Blood, 2018, 132, 4678 CrossRef.
- GlycoMimetics: GMI-1687, https://glycomimetics.com/pipeline/programs/gmi-1687/).
- M. M. Steele, P. Radhakrishnan, J. L. Magnani and M. A. Hollingsworth, Cancer Res., 2014, 74, 4503 CrossRef.
- B. Muz, A. Abdelghafer, M. Markovic, J. Yavner, A. Melam, N. N. Salama and A. K. Azab, Cancers, 2021, 13(2), 335 CrossRef CAS PubMed.
- V. Barbier, J. Erbani, C. Fiveash, J. M. Davies, J. Tay, M. R. Tallack, J. Lowe, J. L. Magnani, D. R. Pattabiraman, A. C. Perkins, J. Lisle, J. E. J. Rasko, J.-P. Levesque and I. G. Winkler, Nat. Commun., 2020, 11, 2042 CrossRef CAS PubMed.
- J. L. Magnani and W. E. Fogler, WO2019108750A1, 2019.
- L. O. Gerlach, R. T. Skerlj, G. J. Bridger and T. W. Schwartz, J. Biol. Chem., 2001, 276, 14153–14160 CrossRef CAS PubMed.
- G. L. Gravina, A. Mancini, A. Colapietro, S. D. Monache, A. Angelucci, A. Calgani, W. E. Fogler, J. L. Magnani and C. Festuccia, Cancer Res., 2015, 75, 428 CrossRef.
- M. M. Steele, W. E. Fogler, J. L. Magnani and M. A. Hollingsworth, Cancer Res., 2015, 75, 425 CrossRef.
- W. Zhang, N. Patel, W. E. Fogler, J. L. Magnani and M. Andreeff, Blood, 2015, 126, 3790 CrossRef.
- T. P. Kogan, B. Dupre, H. Bui, K. L. McAbee, J. M. Kassir, I. L. Scott, X. Hu, P. Vanderslice, P. J. Beck and R. A. Dixon, J. Med. Chem., 1998, 41, 1099–1111 CrossRef CAS PubMed.
- A. E. Hicks, K. B. Abbitt, P. Dodd, V. C. Ridger, P. G. Hellewell and K. E. Norman, J. Leukoc Biol., 2005, 77, 59–66 CrossRef CAS PubMed.
- K. M. Beeh, J. Beier, M. Meyer, R. Buhl, R. Zahlten and G. Wolff, Pulm. Pharmacol. Ther., 2006, 19, 233–241 CrossRef CAS PubMed.
- H. Watz, D. Bock, M. Meyer, K. Schierhorn, K. Vollhardt, C. Woischwill, F. Pedersen, A. Kirsten, K. M. Beeh, W. Meyer-Sabellek, H. Magnussen and J. Beier, Pulm. Pharmacol. Ther., 2013, 26, 265–270 CrossRef CAS PubMed.
- A. Kirsten, H. Watz, G. Kretschmar, F. Pedersen, D. Bock, W. Meyer-Sabellek and H. Magnussen, Pulm. Pharmacol. Ther., 2011, 24, 555–558 CrossRef CAS PubMed.
- N. Kaila, K. Janz, A. Huang, A. Moretto, S. DeBernardo, P. W. Bedard, S. Tam, V. Clerin, J. C. Keith, D. H. H. Tsao, N. Sushkova, G. D. Shaw, R. T. Camphausen, R. G. Schaub and Q. Wang, J. Med. Chem., 2007, 50, 40–64 CrossRef CAS PubMed.
- P. W. Bedard, V. Clerin, N. Sushkova, B. Tchernychev, T. Antrilli, C. Resmini, J. C. Keith, J. K. Hennan, N. Kaila and S. DeBernardo, J. Pharmacol. Exp. Ther., 2007, 324, 497–506 CrossRef PubMed.
- A. G. Japp, R. Chelliah, L. Tattersall, N. N. Lang, X. Meng, K. Weisel, A. Katz, D. Burt, K. A. A. Fox, G. Z. Feuerstein, T. M. Connolly and D. E. Newby, J. Am. Heart Assoc., 2013, 2, e006007 CrossRef PubMed.
- T. B. Geijtenbeek, R. Torensma, S. J. van Vliet, G. C. van Duijnhoven, G. J. Adema, Y. van Kooyk and C. G. Figdor, Cell, 2000, 100, 575–585 CrossRef CAS PubMed.
- B. M. Curtis, S. Scharnowske and A. J. Watson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8356–8360 CrossRef CAS PubMed.
- C. Chaipan, E. J. Soilleux, P. Simpson, H. Hofmann, T. Gramberg, A. Marzi, M. Geier, E. A. Stewart, J. Eisemann, A. Steinkasserer, K. Suzuki-Inoue, G. L. Fuller, A. C. Pearce, S. P. Watson, J. A. Hoxie, F. Baribaud and S. Pohlmann, J. Virol., 2006, 80, 8951–8960 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- T. B. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. van Vliet, G. C. van Duijnhoven, J. Middel, I. L. Cornelissen, H. S. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor and Y. van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- T. B. Geijtenbeek, S. J. Van Vliet, E. A. Koppel, M. Sanchez-Hernandez, C. M. Vandenbroucke-Grauls, B. Appelmelk and Y. Van Kooyk, J. Exp. Med., 2003, 197, 7–17 CrossRef CAS PubMed.
- M. Colmenares, A. Puig-Kroger, O. M. Pello, A. L. Corbi and L. Rivas, J. Biol. Chem., 2002, 277, 36766–36769 CrossRef CAS PubMed.
- A. Cambi, K. Gijzen, J. de Vries, R. Torensma, B. Joosten, G. J. Adema, M. G. Netea, B. J. Kullberg, L. Romani and C. G. Figdor, Eur. J. Immunol., 2003, 33, 532–538 CrossRef CAS PubMed.
- Q. Lu, J. Liu, S. Zhao, M. F. Gomez Castro, M. Laurent-Rolle, J. Dong, X. Ran, P. Damani-Yokota, H. Tang, T. Karakousi, J. Son, M. E. Kaczmarek, Z. Zhang, S. T. Yeung, B. T. McCune, R. E. Chen, F. Tang, X. Ren, X. Chen, J. C. C. Hsu, M. Teplova, B. Huang, H. Deng, Z. Long, T. Mudianto, S. Jin, P. Lin, J. Du, R. Zang, T. T. Su, A. Herrera, M. Zhou, R. Yan, J. Cui, J. Zhu, Q. Zhou, T. Wang, J. Ma, S. B. Koralov, Z. Zhang, I. Aifantis, L. N. Segal, M. S. Diamond, K. M. Khanna, K. A. Stapleford, P. Cresswell, Y. Liu, S. Ding, Q. Xie and J. Wang, Immunity, 2021, 54, 1304–1319.e1309 CrossRef CAS PubMed.
- K. C. Garber, K. Wangkanont, E. E. Carlson and L. L. Kiessling, Chem. Commun., 2010, 46, 6747–6749 RSC.
- S. Mari, D. Serrano-Gomez, F. J. Canada, A. L. Corbi and J. Jimenez-Barbero, Angew. Chem., 2004, 44, 296–298 CrossRef PubMed.
- M. Thepaut, C. Guzzi, I. Sutkeviciute, S. Sattin, R. Ribeiro-Viana, N. Varga, E. Chabrol, J. Rojo, A. Bernardi, J. Angulo, P. M. Nieto and F. Fieschi, J. Am. Chem. Soc., 2013, 135, 2518–2529 CrossRef CAS PubMed.
- T. Tomasic, D. Hajsek, U. Svajger, J. Luzar, N. Obermajer, I. Petit-Haertlein, F. Fieschi and M. Anderluh, Eur. J. Med. Chem., 2014, 75, 308–326 CrossRef CAS PubMed.
- L. Medve, S. Achilli, J. Guzman-Caldentey, M. Thepaut, L. Senaldi, A. Le Roy, S. Sattin, C. Ebel, C. Vives, S. Martin-Santamaria, A. Bernardi and F. Fieschi, Chemistry, 2019, 25, 14659–14668 CrossRef CAS PubMed.
- D. A. Mitchell, N. A. Jones, S. J. Hunter, J. M. D. Cook, S. F. Jenkinson, M. R. Wormald, R. A. Dwek and G. W. J. Fleet, Tetrahedron-Asymmetr, 2007, 18, 1502–1510 CrossRef CAS.
- A. Bernardi, D. Arosio, L. Manzoni, F. Micheli, A. Pasquarello and P. Seneci, J. Org. Chem., 2001, 66, 6209–6216 CrossRef CAS PubMed.
- I. Sutkeviciute, M. Thepaut, S. Sattin, A. Berzi, J. McGeagh, S. Grudinin, J. Weiser, A. Le Roy, J. J. Reina, J. Rojo, M. Clerici, A. Bernardi, C. Ebel and F. Fieschi, ACS Chem. Biol., 2014, 9, 1377–1385 CrossRef CAS PubMed.
- A. Holla and A. Skerra, Protein Eng. Des. Sel., 2011, 24, 659–669 CrossRef CAS PubMed.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Nat. Struct. Mol. Biol., 2004, 11, 591–598 CrossRef CAS PubMed.
- M. Andreini, D. Doknic, I. Sutkeviciute, J. J. Reina, J. Duan, E. Chabrol, M. Thepaut, E. Moroni, F. Doro, L. Belvisi, J. Weiser, J. Rojo, F. Fieschi and A. Bernardi, Org. Biomol. Chem., 2011, 9, 5778–5786 RSC.
- J. J. Reina, S. Sattin, D. Invernizzi, S. Mari, L. Martínez-Prats, G. Tabarani, F. Fieschi, R. Delgado, P. M. Nieto, J. Rojo and A. Bernardi, ChemMedChem, 2007, 2, 1030–1036 CrossRef CAS PubMed.
- N. Varga, I. Sutkeviciute, C. Guzzi, J. McGeagh, I. Petit-Haertlein, S. Gugliotta, J. Weiser, J. Angulo, F. Fieschi and A. Bernardi, Chem. – Eur. J., 2013, 19, 4786–4797 CrossRef CAS PubMed.
- J. Cramer, A. Lakkaichi, B. Aliu, R. P. Jakob, S. Klein, I. Cattaneo, X. Jiang, S. Rabbani, O. Schwardt, G. Zimmer, M. Ciancaglini, T. Abreu Mota, T. Maier and B. Ernst, J. Am. Chem. Soc., 2021, 143, 17465–17478 CrossRef CAS PubMed.
- S. Ng, E. Lin, P. I. Kitov, K. F. Tjhung, O. O. Gerlits, L. Deng, B. Kasper, A. Sood, B. M. Paschal, P. Zhang, C. C. Ling, J. S. Klassen, C. J. Noren, L. K. Mahal, R. J. Woods, L. Coates and R. Derda, J. Am. Chem. Soc., 2015, 137, 5248–5251 CrossRef CAS PubMed.
- M. Thepaut, J. Luczkowiak, C. Vives, N. Labiod, I. Bally, F. Lasala, Y. Grimoire, D. Fenel, S. Sattin, N. Thielens, G. Schoehn, A. Bernardi, R. Delgado and F. Fieschi, PLoS Pathog, 2021, 17, e1009576 CrossRef CAS PubMed.
- G. Timpano, G. Tabarani, M. Anderluh, D. Invernizzi, F. Vasile, D. Potenza, P. M. Nieto, J. Rojo, F. Fieschi and A. Bernardi, ChemBioChem, 2008, 9, 1921–1930 CrossRef CAS PubMed.
- B. Bertolotti, B. Oroszová, I. Sutkeviciute, L. Kniežo, F. Fieschi, K. Parkan, Z. Lovyová, M. Kašáková and J. Moravcová, Carbohydr. Res., 2016, 435, 7–18 CrossRef CAS PubMed.
- K. Henrick, S. Bawumia, E. A. Barboni, B. Mehul and R. C. Hughes, Glycobiology, 1998, 8, 45–57 CrossRef CAS PubMed.
- S. H. Barondes, V. Castronovo, D. N. W. Cooper, R. D. Cummings, K. Drickamer, T. Felzi, M. A. Gitt, J. Hirabayashi, C. Hughes, K.-I. Kasai, H. Leffler, F.-T. Liu, R. Lotan, A. M. Mercurio, M. Monsigny, S. Pillai, F. Poirer, A. Raz, P. W. J. Rigby, J. M. Rini and J. L. Wang, Cell, 1994, 76, 597–598 CrossRef CAS PubMed.
- C. P. Modenutti, J. I. B. Capurro, S. Di Lella and M. A. Martí, Front. Chem., 2019, 7, 823 CrossRef CAS PubMed.
- D. Ayona, P.-E. Fournier, B. Henrissat and B. Desnues, Front. Immunol., 2020, 11, 1877 CrossRef CAS PubMed.
- C. M. Arthur, L. C. Rodrigues, M. D. Baruffi, H. C. Sullivan, J. Heimburg-Molinaro, D. F. Smith, R. D. Cummings and S. R. Stowell, Galectins, Springer, 2015, pp. 115–131 Search PubMed.
- M. Le Mercier, V. Mathieu, B. Haibe-Kains, G. Bontempi, T. Mijatovic, C. Decaestecker, R. Kiss and F. Lefranc, J. Neuropathol. Exp. Neurol., 2008, 67, 456–469 CrossRef CAS PubMed.
- M. C. Kiefer, M. J. Brauer, V. C. Powers, J. J. Wu, S. R. Umansky, L. D. Tomei and P. J. Barr, Nature, 1995, 374, 736–739 CrossRef CAS PubMed.
- S. Nakahara, N. Oka and A. Raz, Apoptosis, 2005, 10, 267–275 CrossRef CAS PubMed.
- R. D. Cummings, F.-T. Liu, G. A. Rabinovich, S. R. Stowell and G. R. Vasta, Essentials of glycobiology, Cold Spring Harbor Laboratory Press, 4th edn, 2022, ch. 36 DOI:10.1101/glycobiology.4e.36.
- F. T. Liu and G. A. Rabinovich, Ann. NY Acad. Sci., 2010, 1183, 158–182 CrossRef CAS PubMed.
- S. Thiemann and L. G. Baum, Annu. Rev. Immunol., 2016, 34, 243–264 CrossRef CAS PubMed.
- J. Ilarregui, G. Bianco, M. Toscano and G. Rabinovich, Ann. Rheum. Dis., 2005, 64, iv96–iv103 CrossRef CAS PubMed.
- H. Lahm, S. André, A. Hoeflich, H. Kaltner, H.-C. Siebert, B. Sordat, C.-W. von der Lieth, E. Wolf and H.-J. Gabius, Glycoconjugate J., 2003, 20, 227–238 CrossRef PubMed.
- M. R. Girotti, M. Salatino, T. Dalotto-Moreno and G. A. Rabinovich, J. Exp. Med., 2020, 217, e20182041 CrossRef PubMed.
- Y. Takenaka, T. Fukumori and A. Raz, Glycoconj. J., 2002, 19, 543–549 CrossRef CAS PubMed.
- F.-T. Liu and G. A. Rabinovich, Nat. Rev. Cancer, 2005, 5, 29–41 CrossRef CAS PubMed.
- I. Camby, N. Belot, F. Lefranc, N. Sadeghi, Y. de Launoit, H. Kaltner, S. Musette, F. Darro, A. Danguy, I. Salmon, H.-J. Gabius and R. Kiss, J. Neuropathol. Exp. Neurol., 2002, 61, 585–596 CrossRef CAS PubMed.
- A. Hittelet, H. Legendre, N. Nagy, Y. Bronckart, J.-C. Pector, I. Salmon, P. Yeaton, H.-J. Gabius, R. Kiss and I. Camby, Int. J. Cancer, 2003, 103, 370–379 CrossRef CAS PubMed.
- A. J. Cagnoni, J. M. Perez Saez, G. A. Rabinovich and K. V. Mariño, Front. Oncol., 2016, 6, 109 Search PubMed.
- H. Leffler, S. Carlsson, M. Hedlund, Y. Qian and F. Poirier, Glycoconjugate J., 2002, 19, 433–440 CrossRef CAS PubMed.
- T.-J. Hsieh, H.-Y. Lin, Z. Tu, B.-S. Huang, S.-C. Wu and C.-H. Lin, PLoS One, 2015, 10, e0125946 CrossRef PubMed.
- Y.-C. Chan, H.-Y. Lin, Z. Tu, Y.-H. Kuo, S.-T. D. Hsu and C.-H. Lin, Int. J. Mol. Sci., 2018, 19, 392 CrossRef PubMed.
- C. T. Oberg, H. Leffler and U. J. Nilsson, Chimia (Aarau), 2011, 65, 18–23 CrossRef PubMed.
- H. Blanchard, X. Yu, P. M. Collins and K. Bum-Erdene, Expert Opin. Ther. Pat., 2014, 24, 1053–1065 CrossRef CAS PubMed.
- H. Blanchard, K. Bum-Erdene, M. H. Bohari and X. Yu, Expert Opin. Ther. Pat., 2016, 26, 537–554 CrossRef CAS PubMed.
- A. Girard and J. L. Magnani, Trends Glycosci. Gly., 2018, 30, SE211–SE220 CrossRef.
- P. Argüeso and N. Panjwani, Exp. Eye Res., 2011, 92, 2–3 CrossRef PubMed.
- N. C. Henderson, A. C. Mackinnon, S. L. Farnworth, F. Poirier, F. P. Russo, J. P. Iredale, C. Haslett, K. J. Simpson and T. Sethi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5060–5065 CrossRef CAS PubMed.
- N. C. Henderson, A. C. Mackinnon, S. L. Farnworth, T. Kipari, C. Haslett, J. P. Iredale, F. T. Liu, J. Hughes and T. Sethi, Am. J. Pathol., 2008, 172, 288–298 CrossRef CAS PubMed.
- Y. Nishi, H. Sano, T. Kawashima, T. Okada, T. Kuroda, K. Kikkawa, S. Kawashima, M. Tanabe, T. Goto, Y. Matsuzawa, R. Matsumura, H. Tomioka, F.-T. Liu and K. Shirai, Allergol. Int., 2007, 56, 57–65 CrossRef CAS PubMed.
- A. C. MacKinnon, M. A. Gibbons, S. L. Farnworth, H. Leffler, U. J. Nilsson, T. Delaine, A. J. Simpson, S. J. Forbes, N. Hirani, J. Gauldie and T. Sethi, Am. J. Resp. Crit. Care Med., 2012, 185, 537–546 CrossRef CAS PubMed.
- J. Zou, V. V. Glinsky, L. A. Landon, L. Matthews and S. L. Deutscher, Carcinogenesis, 2005, 26, 309–318 CrossRef CAS PubMed.
- J. R. Newton-Northup, M. T. Dickerson, L. Ma, C. L. Besch-Williford and S. L. Deutscher, Clin. Exp. Metastasis, 2013, 30, 119–132 CrossRef CAS PubMed.
- Y. Yang, Z. Zhou, S. He, T. Fan, Y. Jin, X. Zhu, C. Chen, Z.-R. Zhang and Y. Huang, Biomaterials, 2012, 33, 2260–2271 CrossRef CAS PubMed.
- W. Sun, L. Li, Q. Yang, W. Shan, Z. Zhang and Y. Huang, Mol. Pharm., 2015, 12, 4124–4136 CrossRef CAS PubMed.
- W. Sun, L. Li, L.-J. Li, Q.-Q. Yang, Z.-R. Zhang and Y. Huang, Acta Pharmacol. Sin., 2017, 38, 806–822 CrossRef CAS PubMed.
- W. Zhang, P. Xu and H. Zhang, Trends Food Sci. Technol., 2015, 44, 258–271 CrossRef CAS.
- D. Chauhan, G. Li, K. Podar, T. Hideshima, P. Neri, D. He, N. Mitsiades, P. Richardson, Y. Chang, J. Schindler, B. Carver and K. C. Anderson, Cancer Res., 2005, 65, 8350–8358 CrossRef CAS PubMed.
- J. J. Grous, C. H. Redfern, D. Mahadevan and J. Schindler, J. Clin. Oncol., 2006, 24, 13023 Search PubMed.
- F. Cotter, D. A. Smith, T. E. Boyd, D. A. Richards, C. Alemany, D. Loesch, G. Salogub, G. F. Tidmarsh, G. M. Gammon and J. Gribben, J. Clin. Oncol., 2009, 27, 7006 Search PubMed.
- L. Jolla, La Jolla Pharmaceutical Company Reports Positive, Top-Line Results from Phase 2 Clinical Trial of GCS-100 in Chronic Kidney Disease, https://www.sec.gov/Archives/edgar/data/920465/000092046514000012/pressreleasedatamar10.htm).
- P. G. Traber and E. Zomer, PLoS One, 2013, 8, e83481 CrossRef PubMed.
- P. G. Traber, H. Chou, E. Zomer, F. Hong, A. Klyosov, M.-I. Fiel and S. L. Friedman, PLoS One, 2013, 8, e75361 CrossRef CAS PubMed.
- N. Chalasani, M. F. Abdelmalek, G. Garcia-Tsao, R. Vuppalanchi, N. Alkhouri, M. Rinella, M. Noureddin, M. Pyko, M. Shiffman, A. Sanyal, A. Allgood, H. Shlevin, R. Horton, E. Zomer, W. Irish, Z. Goodman, S. A. Harrison, P. G. Traber, M. Abdelmalek, L. Balart, B. Borg, N. Chalasani, M. Charlton, H. Conjeevaram, M. Fuchs, R. Ghalib, P. Gholam, D. Halegoua-De Marzio, S. Harrison, C. Jue, N. Kemmer, K. Kowdley, M. Lai, E. Lawitz, R. Loomba, M. Noureddin, A. Paredes, M. Rinella, D. Rockey, M. Rodriguez, R. Rubin, M. Ryan, A. Sanyal, A. Scanga, T. Sepe, M. Shiffman, M. Shiffman, B. Tetri, P. Thuluvath, D. Torres, J. Vierling, J. Wattacheril, A. Weiland and D. Zogg, Gastroenterology, 2020, 158, 1334–1345 CrossRef CAS PubMed.
- B. D. Curti, Y. Koguchi, R. S. Leidner, A. S. Rolig, E. R. Sturgill, Z. Sun, Y. Wu, V. Rajamanickam, B. Bernard and I. Hilgart-Martiszus, J. Immunother. Cancer, 2021, 9, e002371 CrossRef PubMed.
- A. Klyosov, E. Zomer and D. Platt, Glycobiology and Drug Design, American Chemical Society, 2012, vol. 1102, ch. 4, pp. 89–130 Search PubMed.
- P. Sörme, Y. Qian, P.-G. Nyholm, H. Leffler and U. J. Nilsson, ChemBioChem, 2002, 3, 183–189 CrossRef.
- P. Sörme, P. Arnoux, B. Kahl-Knutsson, H. Leffler, J. M. Rini and U. J. Nilsson, J. Am. Chem. Soc., 2005, 127, 1737–1743 CrossRef PubMed.
- C. T. Öberg, H. Leffler and U. J. Nilsson, J. Med. Chem., 2008, 51, 2297–2301 CrossRef PubMed.
- C. T. Öberg, A.-L. Noresson, H. Leffler and U. J. Nilsson, Chem. – Eur. J., 2011, 17, 8139–8144 CrossRef PubMed.
- S. Fort, H.-S. Kim and O. Hindsgaul, J. Org. Chem., 2006, 71, 7146–7154 CrossRef CAS PubMed.
- H. van Hattum, H. M. Branderhorst, E. E. Moret, U. J. Nilsson, H. Leffler and R. J. Pieters, J. Med. Chem., 2013, 56, 1350–1354 CrossRef CAS PubMed.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, ChemBioChem, 2007, 8, 1389–1398 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, J. Am. Chem. Soc., 2000, 122, 870–874 CrossRef CAS.
- I. Cumpstey, A. Sundin, H. Leffler and U. J. Nilsson, Angew. Chem., Int. Ed., 2005, 44, 5110–5112 CrossRef CAS PubMed.
- B. A. Salameh, I. Cumpstey, A. Sundin, H. Leffler and U. J. Nilsson, Bioorg. Med. Chem., 2010, 18, 5367–5378 CrossRef CAS PubMed.
- N. Henderson, T. Sethi, A. Mackinnon, H. Leffler and U. Nilsson, Canadian Pat., CA2794066C, 2017 Search PubMed.
- N. Hirani, L. Nicol, A. C. MacKinnon, P. Ford, H. Schambye, Pedersen, U. Nilsson, H. Leffler, T. Thomas, O. Knott, M. Gibbons, J. Simpson and T. Maher, QJM: Int. J. Med., 2016, 109, S16–S16 Search PubMed.
- N. Hirani, A. C. MacKinnon, L. Nicol, P. Ford, H. Schambye, A. Pedersen, U. J. Nilsson, H. Leffler, T. Sethi, S. Tantawi, L. Gravelle, R. J. Slack, R. Mills, U. Karmakar, D. Humphries, F. Zetterberg, L. Keeling, L. Paul, P. L. Molyneaux, F. Li, W. Funston, I. A. Forrest, A. J. Simpson, M. A. Gibbons and T. M. Maher, Eur. Respir. J., 2021, 57(5), 2002559 CrossRef CAS PubMed.
- E. Gaughan, T. Quinn, A. Bruce, J. Antonelli, V. Young, J. Mair, A. Akram, N. Hirani, O. Koch, C. Mackintosh, J. Norrie, J. W. Dear and K. Dhaliwal, BMJ Open, 2021, 11, e054442 CrossRef PubMed.
- V. K. Rajput, A. MacKinnon, S. Mandal, P. Collins, H. Blanchard, H. Leffler, T. Sethi, H. Schambye, B. Mukhopadhyay and U. J. Nilsson, J. Med. Chem., 2016, 59, 8141–8147 CrossRef CAS PubMed.
- S. Bertuzzi, J. I. Quintana, A. Ardá, A. Gimeno and J. Jiménez-Barbero, Front. Chem., 2020, 8, 593 CrossRef CAS PubMed.
- F. R. Zetterberg, K. Peterson, R. E. Johnsson, T. Brimert, M. Håkansson, D. T. Logan, H. Leffler and U. J. Nilsson, ChemMedChem, 2018, 13, 133–137 CrossRef CAS PubMed.
- F. R. Zetterberg, A. MacKinnon, T. Brimert, L. Gravelle, R. E. Johnsson, B. Kahl-Knutson, H. Leffler, U. J. Nilsson, A. Pedersen, K. Peterson, J. A. Roper, H. Schambye, R. J. Slack and S. Tantawi, J. Med. Chem., 2022, 65, 12626–12638 CrossRef CAS PubMed.
- T. Brimert, R. Johnsson, H. Leffler, U. Nilsson and F. Zetterberg, WO2016120403, 2016.
- F. Zetterberg, U. Nilsson and H. Leffler, WO2018011094, 2018.
- Galecto Announces First Patient Treated in Phase 2 Trial of Oral Galectin-3 Inhibitor GB1211 in Liver CirrhosisGalecto now has three ongoing Phase 2 clinical trials with three different drug candidates in three high value indications, https://www.biospace.com/article/releases/galecto-announces-first-patient-treated-in-phase-2-trial-of-oral-galectin-3-inhibitor-gb1211-in-liver-cirrhosisgalecto-now-has-three-ongoing-phase-2-clinical-trials-with-three-different-drug-candidates-in-three-high-value-indications/.
- C. Liu, P. R. Jalagam, J. Feng, W. Wang, T. Raja, M. R. Sura, R. K. V. L. P. Manepalli, B. R. Aliphedi, S. Medavarapu, S. K. Nair, V. Muthalagu, R. Natesan, A. Gupta, B. Beno, M. Panda, K. Ghosh, J. K. Shukla, H. Sale, P. Haldar, N. Kalidindi, D. Shah, D. Patel, A. Mathur, B. A. Ellsworth, D. Cheng and A. Regueiro-Ren, J. Med. Chem., 2022, 65, 11084–11099 CrossRef CAS PubMed.
- T. Delaine, P. Collins, A. MacKinnon, G. Sharma, J. Stegmayr, V. K. Rajput, S. Mandal, I. Cumpstey, A. Larumbe, B. A. Salameh, B. Kahl-Knutsson, H. van Hattum, M. van Scherpenzeel, R. J. Pieters, T. Sethi, H. Schambye, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, ChemBioChem, 2016, 17, 1759–1770 CrossRef CAS PubMed.
- V. L. Thijssen, R. Heusschen, J. Caers and A. W. Griffioen, Biochim. Biophys. Acta, Rev. Cancer, 2015, 1855, 235–247 CrossRef CAS PubMed.
- N. L. Perillo, M. E. Marcus and L. G. Baum, J. Mol. Med., 1998, 76, 402–412 CrossRef CAS PubMed.
- N. Rubinstein, M. Alvarez, N. W. Zwirner, M. A. Toscano, J. M. Ilarregui, A. Bravo, J. Mordoh, L. Fainboim, O. L. Podhajcer and G. A. Rabinovich, Cancer Cell, 2004, 5, 241–251 CrossRef CAS PubMed.
- N. D'Haene, S. Sauvage, C. Maris, I. Adanja, M. Le Mercier, C. Decaestecker, L. Baum and I. Salmon, PLoS One, 2013, 8, e67029 CrossRef PubMed.
- M. A. Toscano, G. A. Bianco, J. M. Ilarregui, D. O. Croci, J. Correale, J. D. Hernandez, N. W. Zwirner, F. Poirier, E. M. Riley, L. G. Baum and G. A. Rabinovich, Nat. Immunol., 2007, 8, 825–834 CrossRef CAS PubMed.
- M. Ouellet, S. Mercier, I. Pelletier, S. Bounou, J. Roy, J. Hirabayashi, S. Sato and M. J. Tremblay, J. Immunol., 2005, 174, 4120–4126 CrossRef CAS PubMed.
- C. St-Pierre, H. Manya, M. Ouellet, G. F. Clark, T. Endo, M. J. Tremblay and S. Sato, J. Virol., 2011, 85, 11742–11751 CrossRef PubMed.
- C. St-Pierre, M. Ouellet, D. Giguère, R. Ohtake, R. Roy, S. Sato and M. J. Tremblay, Antimicrob. Agents Chemother., 2012, 56, 154–162 CrossRef CAS PubMed.
- K. Peterson, P. M. Collins, X. Huang, B. Kahl-Knutsson, S. Essén, F. R. Zetterberg, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, RSC Adv., 2018, 8, 24913–24922 RSC.
- K. B. Pal, M. Mahanti, H. Leffler and U. J. Nilsson, Int. J. Mol. Sci., 2019, 20, 3786 CrossRef CAS PubMed.
- A. Dahlqvist, A. Furevi, N. Warlin, H. Leffler and U. J. Nilsson, Beilstein J. Org. Chem., 2019, 15, 1046–1060 CrossRef CAS PubMed.
- J. B. Wang, M. D. Wang, E. X. Li and D. F. Dong, Peptides, 2012, 38, 457–462 CrossRef CAS PubMed.
- A. W. Griffioen, D. W. J. van der Schaft, A. F. Barendsz-Janson, A. Cox, H. A. J. S. Boudier, H. F. P. Hillen and K. H. Mayo, Biochem. J., 2001, 354, 233–242 CrossRef CAS PubMed.
- R. J. M. G. E. Brandwijk, R. P. M. Dings, E. van der Linden, K. H. Mayo, V. L. J. L. Thijssen and A. W. Griffioen, Biochem. Biophys. Res. Commun., 2006, 349, 1073–1078 CrossRef CAS PubMed.
- E. Salomonsson, V. L. Thijssen, A. W. Griffioen, U. J. Nilsson and H. Leffler, J. Biol. Chem., 2011, 286, 13801–13804 CrossRef CAS PubMed.
- K. H. Mayo, R. P. M. Dings, C. Flader, I. Nesmelova, B. Hargittai, D. W. J. van der Schaft, L. I. van Eijk, D. Walek, J. Haseman, T. R. Hoye and A. W. Griffioen, J. Biol. Chem., 2003, 278, 45746–45752 CrossRef CAS PubMed.
- R. P. M. Dings, X. Chen, D. M. E. I. Hellebrekers, L. I. van Eijk, Y. Zhang, T. R. Hoye, A. W. Griffioen and K. H. Mayo, JNCI: J. Natl. Cancer I., 2006, 98, 932–936 CrossRef CAS PubMed.
- R. P. M. Dings, M. C. Miller, I. Nesmelova, L. Astorgues-Xerri, N. Kumar, M. Serova, X. Chen, E. Raymond, T. R. Hoye and K. H. Mayo, J. Med. Chem., 2012, 55, 5121–5129 CrossRef CAS PubMed.
- L. Astorgues-Xerri, M. E. Riveiro, A. Tijeras-Raballand, M. Serova, G. A. Rabinovich, I. Bieche, M. Vidaud, A. de Gramont, M. Martinet, E. Cvitkovic, S. Faivre and E. Raymond, Eur. J. Cancer, 2014, 50, 2463–2477 CrossRef CAS PubMed.
- M. Zucchetti, K. Bonezzi, R. Frapolli, F. Sala, P. Borsotti, M. Zangarini, E. Cvitkovic, K. Noel, P. Ubezio, R. Giavazzi, M. D’Incalci and G. Taraboletti, Cancer Chemother. Pharmacol., 2013, 72, 879–887 CrossRef CAS PubMed.
- K. Rezai, S. Durand, N. Lachaux, E. Raymond, P. Herait and F. Lokiec, Cancer Res., 2013, 73, 33 CrossRef.
- R. Yang, L. Sun, C.-F. Li, Y.-H. Wang, J. Yao, H. Li, M. Yan, W.-C. Chang, J.-M. Hsu, J.-H. Cha, J. L. Hsu, C.-W. Chou, X. Sun, Y. Deng, C.-K. Chou, D. Yu and M.-C. Hung, Nat. Commun., 2021, 12, 832 CrossRef CAS PubMed.
- C. Bailly, X. Thuru and B. Quesnel, Cancers, 2021, 13, 6365 CrossRef CAS PubMed.
- S. Mandal, V. K. Rajput, A. P. Sundin, H. Leffler, B. Mukhopadhyay and U. J. Nilsson, Can. J. Chem., 2016, 94, 936–939 CrossRef CAS.
- M. Mahanti, K. B. Pal, A. P. Sundin, H. Leffler and U. J. Nilsson, ACS Med. Chem. Lett., 2020, 11, 34–39 CrossRef CAS PubMed.
- W.-S. Chen, Z. Cao, S. Sugaya, M. J. Lopez, V. G. Sendra, N. Laver, H. Leffler, U. J. Nilsson, J. Fu, J. Song, L. Xia, P. Hamrah and N. Panjwani, Nat. Commun., 2016, 7, 11302 CrossRef CAS PubMed.
- M. V. Tribulatti, J. Carabelli, C. A. Prato and O. Campetella, Glycobiology, 2019, 30, 134–142 CrossRef PubMed.
- J. F. Sampson, A. Suryawanshi, W.-S. Chen, G. A. Rabinovich and N. Panjwani, Immunol. Cell Biol., 2016, 94, 213–219 CrossRef CAS PubMed.
- M. H. Bohari, X. Yu, C. Kishor, B. Patel, R. M. Go, H. A. Eslampanah Seyedi, Y. Vinik, I. D. Grice, Y. Zick and H. Blanchard, ChemMedChem, 2018, 13, 1664–1672 CrossRef CAS PubMed.
- B. Patel, C. Kishor, T. A. Houston, H. Shatz-Azoulay, Y. Zick, Y. Vinik and H. Blanchard, J. Med. Chem., 2020, 63, 11573–11584 CrossRef CAS PubMed.
- C. Wu, C. Yong, Q. Zhong, Z. Wang, U. J. Nilsson and Y. Zhang, RSC Adv., 2020, 10, 19636–19642 RSC.
- M. Hassan, S. van Klaveren, M. Håkansson, C. Diehl, R. Kovačič, F. Baussière, A. P. Sundin, J. Dernovšek, B. Walse, F. Zetterberg, H. Leffler, M. Anderluh, T. Tomašič, Ž. Jakopin and U. J. Nilsson, Eur. J. Med. Chem., 2021, 223, 113664 CrossRef CAS PubMed.
- M. Hassan, F. Baussière, S. Guzelj, A. P. Sundin, M. Håkansson, R. Kovačič, H. Leffler, T. Tomašič, M. Anderluh, Ž. Jakopin and U. J. Nilsson, ACS Med. Chem. Lett., 2021, 12, 1745–1752 CrossRef CAS PubMed.
- S. van Klaveren, A. P. Sundin, Ž. Jakopin, M. Anderluh, H. Leffler, U. J. Nilsson and T. Tomašič, ChemMedChem, 2022, 17, e202100575 CAS.
- B. Girardi, M. Manna, S. Van Klaveren, T. Tomašič, Ž. Jakopin, H. Leffler, U. J. Nilsson, D. Ricklin, J. Mravljak, O. Schwardt and M. Anderluh, ChemMedChem, 2022, 17, e202100514 CAS.
- A. Varki, Glycobiology, 1992, 2, 25–40 CrossRef CAS PubMed.
- M. S. Macauley, P. R. Crocker and J. C. Paulson, Nat. Rev. Immunol., 2014, 14, 653–666 CrossRef CAS PubMed.
- S. Duan and J. C. Paulson, Annu. Rev. Immunol., 2020, 38, 365–395 CrossRef CAS PubMed.
- Y. Zhu, S. Yao and L. Chen, Immunity, 2011, 34, 466–478 CrossRef CAS PubMed.
- J. Ereno-Orbea, T. Sicard, H. Cui, M. T. Mazhab-Jafari, S. Benlekbir, A. Guarne, J. L. Rubinstein and J. P. Julien, Nat. Commun., 2017, 8, 764 CrossRef PubMed.
- M. F. Pronker, S. Lemstra, J. Snijder, A. J. R. Heck, D. M. E. Thies-Weesie, R. J. Pasterkamp and B. J. C. Janssen, Nat. Commun., 2016, 7, 13584 CrossRef PubMed.
- T. Angata, Adv. Exp. Med. Biol., 2020, 1204, 215–230 CrossRef CAS PubMed.
- T. Angata, C. M. Nycholat and M. S. Macauley, Trends Pharmacol. Sci., 2015, 36, 645–660 CrossRef CAS PubMed.
- T. Kiwamoto, N. Kawasaki, J. C. Paulson and B. S. Bochner, Pharmacol. Ther., 2012, 135, 327–336 CrossRef CAS PubMed.
- J. Wang, J. Sun, L. N. Liu, D. B. Flies, X. Nie, M. Toki, J. Zhang, C. Song, M. Zarr, X. Zhou, X. Han, K. A. Archer, T. O'Neill, R. S. Herbst, A. N. Boto, M. F. Sanmamed, S. Langermann, D. L. Rimm and L. Chen, Nat. Med., 2019, 25, 656–666 CrossRef CAS PubMed.
- J. F. DiJoseph, D. C. Armellino, E. R. Boghaert, K. Khandke, M. M. Dougher, L. Sridharan, A. Kunz, P. R. Hamann, B. Gorovits, C. Udata, J. K. Moran, A. G. Popplewell, S. Stephens, P. Frost and N. K. Damle, Blood, 2004, 103, 1807–1814 CrossRef CAS PubMed.
- V. H. van Der Velden, J. G. te Marvelde, P. G. Hoogeveen, I. D. Bernstein, A. B. Houtsmuller, M. S. Berger and J. J. van Dongen, Blood, 2001, 97, 3197–3204 CrossRef CAS PubMed.
- L. A. Miles, S. J. Hermans, G. A. N. Crespi, J. H. Gooi, L. Doughty, T. L. Nero, J. Markulić, A. Ebneth, B. Wroblowski, D. Oehlrich, A. A. Trabanco, M. L. Rives, I. Royaux, N. C. Hancock and M. W. Parker, iScience, 2019, 19, 110–118 CrossRef CAS PubMed.
- M. P. Lenza, U. Atxabal, I. Oyenarte, J. Jimenez-Barbero and J. Ereno-Orbea, Cells, 2020, 9(12), 2691 CrossRef CAS PubMed.
- T. Angata and A. Varki, Chem. Rev., 2002, 102, 439–470 CrossRef CAS PubMed.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS PubMed.
- S. Kelm, R. Brossmer, R. Isecke, H. J. Gross, K. Strenge and R. Schauer, Eur. J. Biochem., 1998, 255, 663–672 CrossRef CAS PubMed.
- S. Kelm, P. Madge, T. Islam, R. Bennett, H. Koliwer-Brandl, M. Waespy, M. von Itzstein and T. Haselhorst, Angew. Chem., Int. Ed., 2013, 52, 3616–3620 CrossRef CAS PubMed.
- H. Prescher, A. Schweizer, E. Kuhfeldt, L. Nitschke and R. Brossmer, ChemBioChem, 2017, 18, 1216–1225 CrossRef CAS PubMed.
- C. D. Rillahan, E. Schwartz, C. Rademacher, R. McBride, J. Rangarajan, V. V. Fokin and J. C. Paulson, ACS Chem. Biol., 2013, 8, 1417–1422 CrossRef CAS PubMed.
- M. A. Zhuravleva, K. Trandem and P. D. Sun, J. Mol. Biol., 2008, 375, 437–447 CrossRef CAS PubMed.
- H. Attrill, A. Imamura, R. S. Sharma, M. Kiso, P. R. Crocker and D. M. Van Aalten, J. Biol. Chem., 2006, 281, 32774–32783 CrossRef CAS PubMed.
- N. R. Zaccai, K. Maenaka, T. Maenaka, P. R. Crocker, R. Brossmer, S. Kelm and E. Y. Jones, Structure, 2003, 11, 557–567 CrossRef CAS PubMed.
- B. E. Collins, O. Blixt, S. Han, B. Duong, H. Li, J. K. Nathan, N. Bovin and J. C. Paulson, J. Immunol., 2006, 177, 2994–3003 CrossRef CAS PubMed.
- Y. Zeng, C. Rademacher, C. M. Nycholat, S. Futakawa, K. Lemme, B. Ernst and J. C. Paulson, Bioorg. Med. Chem. Lett., 2011, 21, 5045–5049 CrossRef CAS PubMed.
- C. M. Nycholat, C. Rademacher, N. Kawasaki and J. C. Paulson, J. Am. Chem. Soc., 2012, 134, 15696–15699 CrossRef CAS PubMed.
- A. J. Cagnoni, J. M. Perez Saez, G. A. Rabinovich and K. V. Marino, Front. Oncol., 2016, 6, 109 Search PubMed.
- G. j Liu, L. y Jia and G. w Xing, Asian J. Org. Chem., 2019, 9, 42–52 CrossRef.
- C. M. Nycholat, S. Duan, E. Knuplez, C. Worth, M. Elich, A. Yao, J. O’Sullivan, R. McBride, Y. Wei, S. M. Fernandes, Z. Zhu, R. L. Schnaar, B. S. Bochner and J. C. Paulson, J. Am. Chem. Soc., 2019, 141, 14032–14037 CrossRef CAS PubMed.
- B. E. Collins, O. Blixt, A. R. DeSieno, N. Bovin, J. D. Marth and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6104–6109 CrossRef CAS PubMed.
- S. Han, B. E. Collins, P. Bengtson and J. C. Paulson, Nat. Chem. Biol., 2005, 1, 93–97 CrossRef CAS PubMed.
- A. P. May, R. C. Robinson, M. Vinson, P. R. Crocker and E. Y. Jones, Mol. Cell, 1998, 1, 719–728 CrossRef CAS PubMed.
- S. Kelm, J. Gerlach, R. Brossmer, C. P. Danzer and L. Nitschke, J. Exp. Med., 2002, 195, 1207–1213 CrossRef CAS PubMed.
- O. Blixt, S. Han, L. Liao, Y. Zeng, J. Hoffmann, S. Futakawa and J. C. Paulson, J. Am. Chem. Soc., 2008, 130, 6680–6681 CrossRef CAS PubMed.
- W. C. Chen, N. Kawasaki, C. M. Nycholat, S. Han, J. Pilotte, P. R. Crocker and J. C. Paulson, PLoS One, 2012, 7, e39039 CrossRef CAS PubMed.
- N. Kawasaki, J. L. Vela, C. M. Nycholat, C. Rademacher, A. Khurana, N. van Rooijen, P. R. Crocker, M. Kronenberg and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7826–7831 CrossRef CAS PubMed.
- L. J. Edgar, N. Kawasaki, C. M. Nycholat and J. C. Paulson, Cell Chem. Biol., 2019, 26, 131–136 CrossRef CAS PubMed.
- S. Duan, B. M. Arlian, C. M. Nycholat, Y. Wei, H. Tateno, S. A. Smith, M. S. Macauley, Z. Zhu, B. S. Bochner and J. C. Paulson, J. Immunol., 2021, 206, 2290–2300 CrossRef CAS PubMed.
- B. E. Collins, O. Blixt, S. Han, B. Duong, H. Li, J. K. Nathan, N. Bovin and J. C. Paulson, J. Immunol., 2006, 177, 2994–3003 CrossRef CAS PubMed.
- M. S. Macauley, F. Pfrengle, C. Rademacher, C. M. Nycholat, A. J. Gale, A. von Drygalski and J. C. Paulson, J. Clin. Invest., 2013, 123, 3074–3083 CrossRef CAS PubMed.
- L. C. Hardy, J. Smeekens, D. Raghuwanshi, S. Sarkar, G. C. Daskhan, S. Rogers, C. Nycholat, S. Maleki, A. W. Burks, J. C. Paulson, M. S. Macauley and M. D. Kulis, J. Allergy Clin. Immunol., 2022, 150(6), 1476–1485.e4 CrossRef CAS PubMed.
- R. E. Forgione, F. F. Nieto, C. Di Carluccio, F. Milanesi, M. Fruscella, F. Papi, C. Nativi, A. Molinaro, P. Palladino, S. Scarano, M. Minunni, M. Montefiori, M. Civera, S. Sattin, O. Francesconi, R. Marchetti and A. Silipo, Chembiochem, 2022, 23, e202200076 CrossRef CAS PubMed.
- B. Kim, J. Shin, T. Kiziltepe and B. Bilgicer, Nanoscale, 2020, 12, 11672–11683 RSC.
- M. Miethke, M. Pieroni, T. Weber, M. Brönstrup, P. Hammann, L. Halby, P. B. Arimondo, P. Glaser, B. Aigle, H. B. Bode, R. Moreira, Y. Li, A. Luzhetskyy, M. H. Medema, J. L. Pernodet, M. Stadler, J. R. Tormo, O. Genilloud, A. W. Truman, K. J. Weissman, E. Takano, S. Sabatini, E. Stegmann, H. Brötz-Oesterhelt, W. Wohlleben, M. Seemann, M. Empting, A. K. H. Hirsch, B. Loretz, C. M. Lehr, A. Titz, J. Herrmann, T. Jaeger, S. Alt, T. Hesterkamp, M. Winterhalter, A. Schiefer, K. Pfarr, A. Hoerauf, H. Graz, M. Graz, M. Lindvall, S. Ramurthy, A. Karlén, M. van Dongen, H. Petkovic, A. Keller, F. Peyrane, S. Donadio, L. Fraisse, L. J. V. Piddock, I. H. Gilbert, H. E. Moser and R. Müller, Nat. Rev. Chem., 2021, 5, 726–749 CrossRef CAS PubMed.
- M. B. Calvert, V. R. Jumde and A. Titz, Beilstein J. Org. Chem., 2018, 14, 2607–2617 CrossRef CAS PubMed.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS PubMed.
- G. L. Mulvey, P. Marcato, P. I. Kitov, J. Sadowska, D. R. Bundle and G. D. Armstrong, J. Infect. Dis., 2003, 187, 640–649 CrossRef CAS PubMed.
- L. E. Nicolle, Clin. Microbiol. Newsl., 2002, 24, 135–140 CrossRef.
- G. K. Harding and A. R. Ronald, Int. J. Antimicrob. Agents, 1994, 4, 83–88 CrossRef CAS PubMed.
- A. L. Flores-Mireles, J. N. Walker, M. Caparon and S. J. Hultgren, Nat. Rev. Microbiol., 2015, 13, 269–284 CrossRef CAS PubMed.
- A. R. Ronald, Am. J. Med., 2002, 113, 14–19 CrossRef PubMed.
- D. S. Eto, T. A. Jones, J. L. Sundsbak and M. A. Mulvey, PLoS Pathog., 2007, 3, e100 CrossRef PubMed.
- D. Abgottspon and B. Ernst, Chimia (Aarau), 2012, 66, 166–169 CrossRef CAS PubMed.
- K. J. Wright, P. C. Seed and S. J. Hultgren, Cell. Microbiol., 2007, 9, 2230–2241 CrossRef CAS PubMed.
- A. Sivignon, J. Bouckaert, J. Bernard, S. G. Gouin and N. Barnich, Expert Opin. Ther. Tar., 2017, 21, 837–847 CrossRef CAS PubMed.
- G. Chevalier, A. Laveissière, G. Desachy, N. Barnich, A. Sivignon, M. Maresca, C. Nicoletti, E. Di Pasquale, M. Martinez-Medina, K. W. Simpson, V. Yajnik, H. Sokol, M. s. investigators, J. Plassais, F. Strozzi, A. Cervino, R. Morra and C. Bonny, Microbiome, 2021, 9, 176 CrossRef CAS PubMed.
- E. Hahn, P. Wild, U. Hermanns, P. Sebbel, R. Glockshuber, M. Häner, N. Taschner, P. Burkhard, U. Aebi and S. A. Müller, J. Mol. Biol., 2002, 323, 845–857 CrossRef CAS PubMed.
- G. Waksman and S. J. Hultgren, Nat. Rev. Microbiol., 2009, 7, 765–774 CrossRef CAS PubMed.
- D. Choudhury, A. Thompson, V. Stojanoff, S. Langermann, J. Pinkner, S. J. Hultgren and S. D. Knight, Science, 1999, 285, 1061–1066 CrossRef CAS PubMed.
- H. Remaut, R. J. Rose, T. J. Hannan, S. J. Hultgren, S. E. Radford, A. E. Ashcroft and G. Waksman, Mol. Cell, 2006, 22, 831–842 CrossRef CAS PubMed.
- P. Aprikian, V. Tchesnokova, B. Kidd, O. Yakovenko, V. Yarov-Yarovoy, E. Trinchina, V. Vogel, W. Thomas and E. Sokurenko, J. Biol. Chem., 2007, 282, 23437–23446 CrossRef CAS PubMed.
- M. M. Sauer, R. P. Jakob, J. Eras, S. Baday, D. Eriş, G. Navarra, S. Bernèche, B. Ernst, T. Maier and R. Glockshuber, Nat. Commun., 2016, 7, 10738 CrossRef CAS PubMed.
- M. M. Sauer, R. P. Jakob, T. Luber, F. Canonica, G. Navarra, B. Ernst, C. Unverzagt, T. Maier and R. Glockshuber, J. Am. Chem. Soc., 2019, 141, 936–944 CrossRef CAS PubMed.
- W. E. Thomas, L. M. Nilsson, M. Forero, E. V. Sokurenko and V. Vogel, Mol. Microbiol., 2004, 53, 1545–1557 CrossRef CAS PubMed.
- I. Le Trong, P. Aprikian, B. A. Kidd, M. Forero-Shelton, V. Tchesnokova, P. Rajagopal, V. Rodriguez, G. Interlandi, R. Klevit, V. Vogel, R. E. Stenkamp, E. V. Sokurenko and W. E. Thomas, Cell, 2010, 141, 645–655 CrossRef CAS PubMed.
- G. Zhou, W.-J. Mo, P. Sebbel, G. Min, T. A. Neubert, R. Glockshuber, X.-R. Wu, T.-T. Sun and X.-P. Kong, J. Cell Sci., 2001, 114, 4095–4103 CrossRef CAS PubMed.
- N. Barnich, F. A. Carvalho, A.-L. Glasser, C. Darcha, P. Jantscheff, M. Allez, H. Peeters, G. Bommelaer, P. Desreumaux, J. F. Colombel and A. Darfeuille-Michaud, J. Clin. Invest., 2007, 117, 1566–1574 CrossRef CAS PubMed.
- C. S. Hung, J. Bouckaert, D. Hung, J. Pinkner, C. Widberg, A. DeFusco, C. G. Auguste, R. Strouse, S. Langermann, G. Waksman and S. J. Hultgren, Mol. Microbiol., 2002, 44, 903–915 CrossRef CAS PubMed.
- F. Vandemaele, D. Vandekerchove, M. Vereecken, J. Derijcke, M. Dho-Moulin and B. M. Goddeeris, Vet. Res., 2003, 34, 153–163 CrossRef CAS PubMed.
- F. J. Vandemaele, S. M. Hensen and B. M. Goddeeris, Vet. Microbiol., 2004, 101, 147–152 CrossRef CAS PubMed.
- S. N. Abraham, D. Sun, J. B. Dale and E. H. Beachey, Nature, 1988, 336, 682–684 CrossRef CAS PubMed.
- I. Connell, W. Agace, P. Klemm, M. Schembri, S. Mårild and C. Svanborg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9827–9832 CrossRef CAS PubMed.
- S. Langermann, S. Palaszynski, M. Barnhart, G. Auguste, J. S. Pinkner, J. Burlein, P. Barren, S. Koenig, S. Leath, C. H. Jones and S. J. Hultgren, Science, 1997, 276, 607–611 CrossRef CAS PubMed.
- K. Thankavel, B. Madison, T. Ikeda, R. Malaviya, A. H. Shah, P. M. Arumugam and S. N. Abraham, J. Clin. Invest., 1997, 100, 1123–1136 CrossRef CAS PubMed.
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C. S. Hung, J. Pinkner, R. Slättegård, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441–455 CrossRef CAS PubMed.
- J. Bouckaert, J. Mackenzie, J. L. de Paz, B. Chipwaza, D. Choudhury, A. Zavialov, K. Mannerstedt, J. Anderson, D. Piérard, L. Wyns, P. H. Seeberger, S. Oscarson, H. De Greve and S. D. Knight, Mol. Microbiol., 2006, 61, 1556–1568 CrossRef CAS PubMed.
- A. Wellens, C. Garofalo, H. Nguyen, N. Van Gerven, R. Slättegård, J. P. Hernalsteens, L. Wyns, S. Oscarson, H. De Greve, S. Hultgren and J. Bouckaert, PLOS One, 2008, 3, e2040 CrossRef PubMed.
- A. Wellens, M. Lahmann, M. Touaibia, J. Vaucher, S. Oscarson, R. Roy, H. Remaut and J. Bouckaert, Biochemistry, 2012, 51, 4790–4799 CrossRef CAS PubMed.
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C. S. Hung, J. Pinkner, R. Slättegård, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441–455 CrossRef CAS PubMed.
- L. K. Mydock-McGrane, T. J. Hannan and J. W. Janetka, Expert Opin. Drug Dis., 2017, 12, 711–731 CrossRef CAS PubMed.
- H. Remaut, C. Tang, N. S. Henderson, J. S. Pinkner, T. Wang, S. J. Hultgren, D. G. Thanassi, G. Waksman and H. Li, Cell, 2008, 133, 640–652 CrossRef CAS PubMed.
- Z. Han, J. S. Pinkner, B. Ford, R. Obermann, W. Nolan, S. A. Wildman, D. Hobbs, T. Ellenberger, C. K. Cusumano, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2010, 53, 4779–4792 CrossRef CAS PubMed.
- C. P. Sager, B. Fiege, P. Zihlmann, R. Vannam, S. Rabbani, R. P. Jakob, R. C. Preston, A. Zalewski, T. Maier, M. W. Peczuh and B. Ernst, Chem. Sci., 2018, 9, 646–654 RSC.
- I. Ofek, D. Mirelman and N. Sharon, Nature, 1977, 265, 623–625 CrossRef CAS PubMed.
- M. Aronson, O. Medalia, L. Schori, D. Mirelman, N. Sharon and I. Ofek, J. Infect. Dis., 1979, 139, 329–332 CrossRef CAS PubMed.
- N. Firon, I. Ofek and N. Sharon, Carbohyd. Res., 1983, 120, 235–249 CrossRef CAS PubMed.
- N. Firon, S. Ashkenazi, D. Mirelman, I. Ofek and N. Sharon, Infect. Immun., 1987, 55, 472–476 CrossRef CAS PubMed.
- Z. Han, J. S. Pinkner, B. Ford, E. Chorell, J. M. Crowley, C. K. Cusumano, S. Campbell, J. P. Henderson, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2012, 55, 3945–3959 CrossRef CAS PubMed.
- C. Jarvis, Z. Han, V. Kalas, R. Klein, J. S. Pinkner, B. Ford, J. Binkley, C. K. Cusumano, Z. Cusumano, L. Mydock-McGrane, S. J. Hultgren and J. W. Janetka, ChemMedChem, 2016, 11, 367–373 CrossRef CAS PubMed.
- C. K. Cusumano, J. S. Pinkner, Z. Han, S. E. Greene, B. A. Ford, J. R. Crowley, J. P. Henderson, J. W. Janetka and S. J. Hultgren, Sci. Transl. Med., 2011, 3, 109–115 Search PubMed.
- M. Totsika, M. Kostakioti, T. J. Hannan, M. Upton, S. A. Beatson, J. W. Janetka, S. J. Hultgren and M. A. Schembri, J. Infect. Dis, 2013, 208, 921–928 CrossRef CAS PubMed.
- S. Kleeb, L. Pang, K. Mayer, D. Eris, A. Sigl, R. C. Preston, P. Zihlmann, T. Sharpe, R. P. Jakob, D. Abgottspon, A. S. Hutter, M. Scharenberg, X. Jiang, G. Navarra, S. Rabbani, M. Smiesko, N. Lüdin, J. Bezençon, O. Schwardt, T. Maier and B. Ernst, J. Med. Chem., 2015, 58, 2221–2239 CrossRef CAS PubMed.
- W. Schönemann, J. Cramer, T. Mühlethaler, B. Fiege, M. Silbermann, S. Rabbani, P. Dätwyler, P. Zihlmann, R. P. Jakob, C. P. Sager, M. Smieško, O. Schwardt, T. Maier and B. Ernst, ChemMedChem, 2019, 14, 749–757 CrossRef PubMed.
- O. Schwardt, S. Rabbani, M. Hartmann, D. Abgottspon, M. Wittwer, S. Kleeb, A. Zalewski, M. Smieško, B. Cutting and B. Ernst, Bioorg. Med. Chem., 2011, 19, 6454–6473 CrossRef CAS PubMed.
- X. Jiang, D. Abgottspon, S. Kleeb, S. Rabbani, M. Scharenberg, M. Wittwer, M. Haug, O. Schwardt and B. Ernst, J. Med. Chem., 2012, 55, 4700–4713 CrossRef CAS PubMed.
- M. Scharenberg, O. Schwardt, S. Rabbani and B. Ernst, J. Med. Chem., 2012, 55, 9810–9816 CrossRef CAS PubMed.
- L. Mydock-McGrane, Z. Cusumano, Z. Han, J. Binkley, M. Kostakioti, T. Hannan, J. S. Pinkner, R. Klein, V. Kalas, J. Crowley, N. P. Rath, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2016, 59, 9390–9408 CrossRef CAS PubMed.
- S. Kleeb, X. Jiang, P. Frei, A. Sigl, J. Bezençon, K. Bamberger, O. Schwardt and B. Ernst, J. Med. Chem., 2016, 59, 3163–3182 CrossRef CAS PubMed.
- J. A. Sattigeri, M. Garg, P. Bhateja, A. Soni, A. R. A. Rauf, M. Gupta, M. S. Deshmukh, T. Jain, N. Alekar, T. K. Barman, P. Jha, T. Chaira, R. B. Bambal, D. J. Upadhyay and T. Nishi, Bioorg. Med. Chem. Lett., 2018, 28, 2993–2997 CrossRef CAS PubMed.
- L. Mousavifar, M. Touaibia and R. Roy, Acc. Chem. Res., 2018, 51, 2937–2948 CrossRef CAS PubMed.
- S. Brument, A. Sivignon, T. I. Dumych, N. Moreau, G. Roos, Y. Guérardel, T. Chalopin, D. Deniaud, R. O. Bilyy, A. Darfeuille-Michaud, J. Bouckaert and S. G. Gouin, J. Med. Chem., 2013, 56, 5395–5406 CrossRef CAS PubMed.
- T. Chalopin, D. Alvarez Dorta, A. Sivignon, M. Caudan, T. I. Dumych, R. O. Bilyy, D. Deniaud, N. Barnich, J. Bouckaert and S. G. Gouin, Org. Biomol. Chem., 2016, 14, 3913–3925 RSC.
- A. Sivignon, X. Yan, D. Alvarez Dorta, R. Bonnet, J. Bouckaert, E. Fleury, J. Bernard, S. G. Gouin, A. Darfeuille-Michaud and N. Barnich, mBio, 2015, 6, e01298–01215 CrossRef CAS PubMed.
- D. Alvarez Dorta, A. Sivignon, T. Chalopin, T. I. Dumych, G. Roos, R. O. Bilyy, D. Deniaud, E.-M. Krammer, J. de Ruyck, M. F. Lensink, J. Bouckaert, N. Barnich and S. G. Gouin, ChemBioChem, 2016, 17, 936–952 CrossRef CAS PubMed.
- Y. L. Bennani and B. Liu, WO2014055474, 2014.
- Y. L. Bennani, C. Cadilhac, S. K. Das, E. Dietrich, J. Gallant, B. Liu, O. Z. Pereira, Y. K. Ramtohul, T. J. Reddy, L. Vaillancourt, C. Yannopoulos and F. Vallee, WO2013134415, 2013.
- E. Dietrich, C. Poisson, M. Gallant, S. Lessard, B. Liu, S. K. Das, Y. Ramtohul, T. J. Reddy, J. Martel, F. Vallee and J.-F. Levesque, WO2014165107, 2014.
- Y. Ramtohul, P. K. Das, C. Cadilhac, T. J. Reddy, M. Gallant, B. Liu, E. Dietrich, F. Vallee, J. Martel and C. Poisson, WO2014100158A1, 2014.
- M. Gallant, J.-F. Truchon, T. J. Reddy, E. Dietrich, L. Vaillancourt and F. Vallee, WO2016199105, 2016.
- R. D. Klein and S. J. Hultgren, Nat. Rev. Microbiol., 2020, 18, 211–226 CrossRef CAS PubMed.
- K. Poole, Fron. Microbiol., 2011, 2, 65 CAS.
- S. Wagner, R. Sommer, S. Hinsberger, C. Lu, R. W. Hartmann, M. Empting and A. Titz, J. Med. Chem., 2016, 59, 5929–5969 CrossRef CAS PubMed.
- C. Chemani, A. Imberty, S. de Bentzmann, M. Pierre, M. Wimmerová, B. P. Guery and K. Faure, Infect. Immun., 2009, 77, 2065–2075 CrossRef CAS PubMed.
- D. Tielker, S. Hacker, R. Loris, M. Strathmann, J. Wingender, S. Wilhelm, F. Rosenau and K. E. Jaeger, Microbiology, 2005, 151, 1313–1323 CrossRef CAS PubMed.
- S. P. Diggle, R. E. Stacey, C. Dodd, M. Cámara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8, 1095–1104 CrossRef CAS PubMed.
- N. Gilboa-Garber, Methods in Enzymology, Academic Press, 1982, vol. 83, pp. 378–385 Search PubMed.
- E. C. Adam, B. S. Mitchell, D. U. Schumacher, G. Grant and U. Schumacher, Am. J. Respir. Crit. Care Med., 1997, 155, 2102–2104 CrossRef CAS PubMed.
- O. Bajolet-Laudinat, S. Girod-de Bentzmann, J. M. Tournier, C. Madoulet, M. C. Plotkowski, C. Chippaux and E. Puchelle, Infect. Immun., 1994, 62, 4481–4487 CrossRef CAS PubMed.
- E. Zahorska, F. Rosato, K. Stober, S. Kuhaudomlarp, J. Meiers, D. Hauck, D. Reith, E. Gillon, K. Rox, A. Imberty, W. Römer and A. Titz, Angew. Chem., Int. Ed., 2023, 62, e202215535 CrossRef CAS PubMed.
- R. Thuenauer, A. Landi, A. Trefzer, S. Altmann, S. Wehrum, T. Eierhoff, B. Diedrich, J. Dengjel, A. Nyström, A. Imberty and W. Römer, mBio, 2020, 11, e03260–03219 CrossRef PubMed.
- S. Zheng, T. Eierhoff, S. Aigal, A. Brandel, R. Thuenauer, S. de Bentzmann, A. Imberty and W. Römer, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864, 1236–1245 CrossRef CAS PubMed.
- C. Cott, R. Thuenauer, A. Landi, K. Kühn, S. Juillot, A. Imberty, J. Madl, T. Eierhoff and W. Römer, Biochim. Biophys. Acta, 2016, 1863, 1106–1118 CrossRef CAS PubMed.
- I. Wilhelm, E. Levit-Zerdoun, J. Jakob, S. Villringer, M. Frensch, R. Übelhart, A. Landi, P. Müller, A. Imberty, R. Thuenauer, J. Claudinon, H. Jumaa, M. Reth, H. Eibel, E. Hobeika and W. Römer, Sci. Signal., 2019, 12, aao7194 CrossRef PubMed.
- J. Sponsel, Y. Guo, L. Hamzam, A. C. Lavanant, A. Pérez-Riverón, E. Partiot, Q. Muller, J. Rottura, R. Gaudin, D. Hauck, A. Titz, V. Flacher, W. Römer and C. G. Mueller, EMBO Rep., 2023, e55971, DOI:10.15252/embr.202255971.
- T. Eierhoff, B. Bastian, R. Thuenauer, J. Madl, A. Audfray, S. Aigal, S. Juillot, G. E. Rydell, S. Müller, S. de Bentzmann, A. Imberty, C. Fleck and W. Römer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12895–12900 CrossRef CAS PubMed.
- P. von Bismarck, R. Schneppenheim and U. Schumacher, Klin. Padiatr., 2001, 213, 285–287 CrossRef CAS PubMed.
- H. P. Hauber, M. Schulz, A. Pforte, D. Mack, P. Zabel and U. Schumacher, Int. J. Med. Sci., 2008, 5, 371–376 CrossRef CAS PubMed.
- I. Bucior, J. Abbott, Y. Song, M. A. Matthay and J. N. Engel, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2013, 305, L352–L363 CrossRef CAS PubMed.
- B. Blanchard, A. Nurisso, E. Hollville, C. Tétaud, J. Wiels, M. Pokorná, M. Wimmerová, A. Varrot and A. Imberty, J. Mol. Biol., 2008, 383, 837–853 CrossRef CAS PubMed.
- G. Cioci, E. P. Mitchell, C. Gautier, M. Wimmerová, D. Sudakevitz, S. Pérez, N. Gilboa-Garber and A. Imberty, FEBS Lett., 2003, 555, 297–301 CrossRef CAS PubMed.
- N. Garber, U. Guempel, A. Belz, N. Gilboa-Garber and R. J. Doyle, Biochim. Biophys. Acta, 1992, 1116, 331–333 CrossRef CAS PubMed.
- R. U. Kadam, D. Garg, J. Schwartz, R. Visini, M. Sattler, A. Stocker, T. Darbre and J. L. Reymond, ACS Chem. Biol., 2013, 8, 1925–1930 CrossRef CAS PubMed.
- J. Rodrigue, G. Ganne, B. Blanchard, C. Saucier, D. Giguère, T. C. Shiao, A. Varrot, A. Imberty and R. Roy, Org. Biomol. Chem., 2013, 11, 6906–6918 RSC.
- I. Joachim, S. Rikker, D. Hauck, D. Ponader, S. Boden, R. Sommer, L. Hartmann and A. Titz, Org. Biomol. Chem., 2016, 14, 7933–7948 RSC.
- E. Siebs, E. Shanina, S. Kuhaudomlarp, P. da Silva Figueiredo Celestino Gomes, C. Fortin, P. H. Seeberger, D. Rognan, C. Rademacher, A. Imberty and A. Titz, ChemBioChem, 2022, 23, e202100563 CrossRef CAS PubMed.
- A. Novoa, T. Eierhoff, J. Topin, A. Varrot, S. Barluenga, A. Imberty, W. Römer and N. Winssinger, Angew. Chem., Int. Ed., 2014, 53, 8885–8889 CrossRef CAS PubMed.
- Y. M. Chabre, D. Giguère, B. Blanchard, J. Rodrigue, S. Rocheleau, M. Neault, S. Rauthu, A. Papadopoulos, A. A. Arnold, A. Imberty and R. Roy, Chemistry, 2011, 17, 6545–6562 CrossRef CAS PubMed.
- V. Denavit, D. Lainé, C. Bouzriba, E. Shanina, É. Gillon, S. Fortin, C. Rademacher, A. Imberty and D. Giguère, Chemistry, 2019, 25, 4478–4490 CrossRef CAS PubMed.
- E. Shanina, E. Siebs, H. Zhang, D. Varón Silva, I. Joachim, A. Titz and C. Rademacher, Glycobiology, 2021, 31, 159–165 CrossRef CAS PubMed.
- M. Scharenberg, X. Jiang, L. Pang, G. Navarra, S. Rabbani, F. Binder, O. Schwardt and B. Ernst, ChemMedChem, 2014, 9, 78–83 CrossRef CAS PubMed.
- R. A. Copeland, D. L. Pompliano and T. D. Meek, Nat. Rev. Drug Discov., 2006, 5, 730–739 CrossRef CAS PubMed.
- R. Sommer, D. Hauck, A. Varrot, S. Wagner, A. Audfray, A. Prestel, H. M. Möller, A. Imberty and A. Titz, ChemistryOpen, 2015, 4, 756–767 CrossRef CAS PubMed.
- R. Sommer, S. Wagner, K. Rox, A. Varrot, D. Hauck, E. C. Wamhoff, J. Schreiber, T. Ryckmans, T. Brunner, C. Rademacher, R. W. Hartmann, M. Brönstrup, A. Imberty and A. Titz, J. Am. Chem. Soc., 2018, 140, 2537–2545 CrossRef CAS PubMed.
- E. Zahorska, S. Kuhaudomlarp, S. Minervini, S. Yousaf, M. Lepsik, T. Kinsinger, A. K. H. Hirsch, A. Imberty and A. Titz, Chem. Commun., 2020, 56, 8822–8825 RSC.
- R. A. Bauer, Drug Discov. Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrot and A. Titz, Angew. Chem., Int. Ed., 2017, 56, 16559–16564 CrossRef CAS PubMed.
- J. L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Cámara, A. Stocker, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2011, 50, 10631–10635 CrossRef CAS PubMed.
- A. M. Boukerb, A. Rousset, N. Galanos, J. B. Méar, M. Thépaut, T. Grandjean, E. Gillon, S. Cecioni, C. Abderrahmen, K. Faure, D. Redelberger, E. Kipnis, R. Dessein, S. Havet, B. Darblade, S. E. Matthews, S. de Bentzmann, B. Guéry, B. Cournoyer, A. Imberty and S. Vidal, J. Med. Chem., 2014, 57, 10275–10289 CrossRef CAS PubMed.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48, 4008–4010 RSC.
- F. Pertici, N. J. de Mol, J. Kemmink and R. J. Pieters, Chemistry, 2013, 19, 16923–169237 CrossRef CAS PubMed.
- R. Visini, X. Jin, M. Bergmann, G. Michaud, F. Pertici, O. Fu, A. Pukin, T. R. Branson, D. M. Thies-Weesie, J. Kemmink, E. Gillon, A. Imberty, A. Stocker, T. Darbre, R. J. Pieters and J. L. Reymond, ACS Chem. Biol., 2015, 10, 2455–2462 CrossRef CAS PubMed.
- G. Yu, A. C. Vicini and R. J. Pieters, J. Org. Chem., 2019, 84, 2470–2488 CrossRef CAS PubMed.
- A. Imberty, M. Wimmerová, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- S. Perret, C. Sabin, C. Dumon, M. Pokorná, C. Gautier, O. Galanina, S. Ilia, N. Bovin, M. Nicaise, M. Desmadril, N. Gilboa-Garber, M. Wimmerová, E. P. Mitchell and A. Imberty, Biochem. J., 2005, 389, 325–332 CrossRef CAS PubMed.
- E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Pérez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Biol., 2002, 9, 918–921 CrossRef CAS PubMed.
- E. P. Mitchell, C. Sabin, L. Snajdrová, M. Pokorná, S. Perret, C. Gautier, C. Hofr, N. Gilboa-Garber, J. Koca, M. Wimmerová and A. Imberty, Proteins, 2005, 58, 735–746 CrossRef CAS PubMed.
- L. Gajdos, M. P. Blakeley, M. Haertlein, V. T. Forsyth, J. M. Devos and A. Imberty, Nat. Commun., 2022, 13, 194 CrossRef CAS PubMed.
- J. Klockgether, N. Cramer, L. Wiehlmann, C. F. Davenport and B. Tümmler, Front. Microbiol., 2011, 2, 150 CAS.
- R. Sommer, S. Wagner, A. Varrot, C. M. Nycholat, A. Khaledi, S. Häussler, J. C. Paulson, A. Imberty and A. Titz, Chem. Sci., 2016, 7, 4990–5001 RSC.
- A. M. Boukerb, A. Decor, S. Ribun, R. Tabaroni, A. Rousset, L. Commin, S. Buff, A. Doléans-Jordheim, S. Vidal, A. Varrot, A. Imberty and B. Cournoyer, Front. Microbiol., 2016, 7, 811 RSC.
- J. Meiers, E. Siebs, E. Zahorska and A. Titz, Curr. Opin. Chem. Biol., 2019, 53, 51–67 CrossRef CAS PubMed.
- K. Marotte, C. Sabin, C. Préville, M. Moumé-Pymbock, M. Wimmerová, E. P. Mitchell, A. Imberty and R. Roy, ChemMedChem, 2007, 2, 1328–1338 CrossRef CAS PubMed.
- M. Andreini, M. Anderluh, A. Audfray, A. Bernardi and A. Imberty, Carbohydr. Res., 2010, 345, 1400–1407 CrossRef CAS PubMed.
- E. M. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K. M. Bartels, S. P. Diggle, M. Cámara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K. E. Jaeger, T. Darbre and J. L. Reymond, Chem. Biol., 2008, 15, 1249–1257 CrossRef CAS PubMed.
- D. Hauck, I. Joachim, B. Frommeyer, A. Varrot, B. Philipp, H. M. Möller, A. Imberty, T. E. Exner and A. Titz, ACS Chem. Biol., 2013, 8, 1775–1784 CrossRef CAS PubMed.
- R. Loris, D. Tielker, K. E. Jaeger and L. Wyns, J. Mol. Biol., 2003, 331, 861–870 CrossRef CAS PubMed.
- A. Hofmann, R. Sommer, D. Hauck, J. Stifel, I. Göttker-Schnetmann and A. Titz, Carbohydr. Res., 2015, 412, 34–42 CrossRef CAS PubMed.
- R. Sommer, T. E. Exner and A. Titz, PLoS One, 2014, 9, e112822 CrossRef PubMed.
- R. Sommer, K. Rox, S. Wagner, D. Hauck, S. S. Henrikus, S. Newsad, T. Arnold, T. Ryckmans, M. Brönstrup, A. Imberty, A. Varrot, R. W. Hartmann and A. Titz, J. Med. Chem., 2019, 62, 9201–9216 CrossRef CAS PubMed.
- P. Mała, E. Siebs, J. Meiers, K. Rox, A. Varrot, A. Imberty and A. Titz, J. Med. Chem., 2022, 65, 14180–14200 CrossRef PubMed.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J. L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- E. Mahenthiralingam, A. Baldwin and C. G. Dowson, J. Appl. Microbiol., 2008, 104, 1539–1551 CrossRef CAS PubMed.
- A. Audfray, J. Claudinon, S. Abounit, N. Ruvoën-Clouet, G. Larson, D. F. Smith, M. Wimmerová, J. Le Pendu, W. Römer, A. Varrot and A. Imberty, J. Biol. Chem., 2012, 287, 4335–4347 CrossRef CAS PubMed.
- B. Richichi, A. Imberty, E. Gillon, R. Bosco, I. Sutkeviciute, F. Fieschi and C. Nativi, Org. Biomol. Chem., 2013, 11, 4086–4094 RSC.
- S. Kuhaudomlarp, L. Cerofolini, S. Santarsia, E. Gillon, S. Fallarini, G. Lombardi, M. Denis, S. Giuntini, C. Valori, M. Fragai, A. Imberty, A. Dondoni and C. Nativi, Chem. Sci., 2020, 11, 12662–12670 RSC.
- T. Dingjan, É. Gillon, A. Imberty, S. Pérez, A. Titz, P. A. Ramsland and E. Yuriev, J. Chem. Inf. Model., 2018, 58, 1976–1989 CrossRef CAS PubMed.
- S. Inhülsen, C. Aguilar, N. Schmid, A. Suppiger, K. Riedel and L. Eberl, MicrobiologyOpen, 2012, 1, 225–242 CrossRef PubMed.
- E. Lameignere, L. Malinovská, M. Sláviková, E. Duchaud, E. P. Mitchell, A. Varrot, O. Sedo, A. Imberty and M. Wimmerová, Biochem. J., 2008, 411, 307–318 CrossRef CAS PubMed.
- E. Lameignere, T. C. Shiao, R. Roy, M. Wimmerova, F. Dubreuil, A. Varrot and A. Imberty, Glycobiology, 2010, 20, 87–98 CrossRef CAS PubMed.
- R. Marchetti, L. Malinovska, E. Lameignère, L. Adamova, C. de Castro, G. Cioci, C. Stanetty, P. Kosma, A. Molinaro, M. Wimmerova, A. Imberty and A. Silipo, Glycobiology, 2012, 22, 1387–1398 CrossRef CAS PubMed.
- G. Beshr, R. Sommer, D. Hauck, D. C. B. Siebert, A. Hofmann, A. Imberty and A. Titz, MedChemComm, 2016, 7, 519–530 RSC.
- O. Sulák, G. Cioci, M. Delia, M. Lahmann, A. Varrot, A. Imberty and M. Wimmerová, Structure, 2010, 18, 59–72 CrossRef PubMed.
- O. Sulák, G. Cioci, E. Lameignère, V. Balloy, A. Round, I. Gutsche, L. Malinovská, M. Chignard, P. Kosma, D. F. Aubert, C. L. Marolda, M. A. Valvano, M. Wimmerová and A. Imberty, PLoS Pathog., 2011, 7, e1002238 CrossRef PubMed.
- K. Lal, R. Bermeo, J. Cramer, F. Vasile, B. Ernst, A. Imberty, A. Bernardi, A. Varrot and L. Belvisi, Chemistry, 2021, 27, 10341–10348 CrossRef CAS PubMed.
- R. Bermeo, K. Lal, D. Ruggeri, D. Lanaro, S. Mazzotta, F. Vasile, A. Imberty, L. Belvisi, A. Varrot and A. Bernardi, ACS Chem. Biol., 2022, 17, 2899–2910 CrossRef CAS PubMed.
- A. J. Thompson, R. P. de Vries and J. C. Paulson, Curr. Opin. Virol., 2019, 34, 117–129 CrossRef CAS PubMed.
- J. S. Long, B. Mistry, S. M. Haslam and W. S. Barclay, Nat. Rev. Microbiol., 2019, 17, 67–81 CrossRef CAS PubMed.
- B. S. Blaum and T. Stehle, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. C. Baker, Academic Press, 2019, vol. 76, pp. 65–111 Search PubMed.
- J. E. Stencel-Baerenwald, K. Reiss, D. M. Reiter, T. Stehle and T. S. Dermody, Nat. Rev. Microbiol., 2014, 12, 739–749 CrossRef CAS PubMed.
- I. Papp, C. Sieben, K. Ludwig, M. Roskamp, C. Böttcher, S. Schlecht, A. Herrmann and R. Haag, Small, 2010, 6, 2900–2906 CrossRef CAS PubMed.
- W. Lu, W. Du, V. J. Somovilla, G. Yu, D. Haksar, E. de Vries, G.-J. Boons, R. P. de Vries, C. A. M. de Haan and R. J. Pieters, J. Med. Chem., 2019, 62, 6398–6404 CrossRef CAS PubMed.
- C. Nie, M. Stadtmüller, B. Parshad, M. Wallert, V. Ahmadi, Y. Kerkhoff, S. Bhatia, S. Block, C. Cheng and T. Wolff, Sci. Adv., 2021, 7, eabd3803 CrossRef CAS PubMed.
- N. K. Sauter, J. E. Hanson, G. D. Glick, J. H. Brown, R. L. Crowther, S. J. Park, J. J. Skehel and D. C. Wiley, Biochemistry, 1992, 31, 9609–9621 CrossRef CAS PubMed.
- G. Herrler, H. J. Gross, A. Imhof, R. Brossmer, G. Milks and J. C. Paulson, J. Biol. Chem., 1992, 267, 12501–12505 CrossRef CAS PubMed.
- R. Brossmer, R. Isecke and G. Herrler, FEBS Lett., 1993, 323, 96–98 CrossRef CAS PubMed.
- M. C. Schuster, D. A. Mann, T. J. Buchholz, K. M. Johnson, W. D. Thomas and L. L. Kiessling, Org. Lett., 2003, 5, 1407–1410 CrossRef CAS PubMed.
- K. C. A. Garber, K. Wangkanont, E. E. Carlson and L. L. Kiessling, Chem. Commun., 2010, 46, 6747–6749 RSC.
- J. C. Grim, K. C. A. Garber and L. L. Kiessling, Org. Lett., 2011, 13, 3790–3793 CrossRef CAS PubMed.
- L. R. Prost, J. C. Grim, M. Tonelli and L. L. Kiessling, ACS Chem. Biol., 2012, 7, 1603–1608 CrossRef CAS PubMed.
- R. Kranich, A. S. Busemann, D. Bock, S. Schroeter-Maas, D. Beyer, B. Heinemann, M. Meyer, K. Schierhorn, R. Zahlten, G. Wolff and E. M. Aydt, J. Med. Chem., 2007, 50, 1101–1115 CrossRef CAS PubMed.
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- A. L. Hopkins, C. R. Groom and A. Alex, Drug Discov. Today, 2004, 9, 430–431 CrossRef PubMed.
- E. Shanina, S. Kuhaudomlarp, E. Siebs, F. F. Fuchsberger, M. Denis, P. da Silva Figueiredo Celestino Gomes, M. H. Clausen, P. H. Seeberger, D. Rognan, A. Titz, A. Imberty and C. Rademacher, Commun. Chem., 2022, 5, 64 CrossRef CAS PubMed.
- J. C. Milne, P. D. Lambert, S. Schenk, D. P. Carney, J. J. Smith, D. J. Gagne, L. Jin, O. Boss, R. B. Perni, C. B. Vu, J. E. Bemis, R. Xie, J. S. Disch, P. Y. Ng, J. J. Nunes, A. V. Lynch, H. Yang, H. Galonek, K. Israelian, W. Choy, A. Iffland, S. Lavu, O. Medvedik, D. A. Sinclair, J. M. Olefsky, M. R. Jirousek, P. J. Elliott and C. H. Westphal, Nature, 2007, 450, 712–716 CrossRef CAS PubMed.
- S. F. Generoso, M. Giustiniano, G. La Regina, S. Bottone, S. Passacantilli, S. Di Maro, H. Cassese, A. Bruno, M. Mallardo, M. Dentice, R. Silvestri, L. Marinelli, D. Sarnataro, S. Bonatti, E. Novellino and M. Stornaiuolo, Nat. Chem. Biol., 2015, 11, 280–286 CrossRef CAS PubMed.
- S. K. Bagal, K. Omoto, D. C. Blakemore, P. J. Bungay, J. G. Bilsland, P. J. Clarke, M. S. Corbett, C. N. Cronin, J. J. Cui, R. Dias, N. J. Flanagan, S. E. Greasley, R. Grimley, E. Johnson, D. Fengas, L. Kitching, M. L. Kraus, I. McAlpine, A. Nagata, G. J. Waldron and J. S. Warmus, J. Med. Chem., 2019, 62, 247–265 CrossRef CAS PubMed.
- R. Nussinov and C. J. Tsai, Cell, 2013, 153, 293–305 CrossRef CAS PubMed.
- X. Lu, J. B. Smaill and K. Ding, Angew. Chem., Int. Ed., 2020, 59, 13764–13776 CrossRef CAS PubMed.
- S. Lu, Q. Shen and J. Zhang, Acc. Chem. Res., 2019, 52, 492–500 CrossRef CAS PubMed.
- V. Kalas, J. S. Pinkner, T. J. Hannan, M. E. Hibbing, K. W. Dodson, A. S. Holehouse, H. Zhang, N. H. Tolia, M. L. Gross, R. V. Pappu, J. Janetka and S. J. Hultgren, Sci. Adv., 2017, 3, e1601944 CrossRef PubMed.
- D. I. Kisiela, P. Magala, G. Interlandi, L. A. Carlucci, A. Ramos, V. Tchesnokova, B. Basanta, V. Yarov-Yarovoy, H. Avagyan, A. Hovhannisyan, W. E. Thomas, R. E. Stenkamp, R. E. Klevit and E. V. Sokurenko, PLoS Pathog., 2021, 17, e1009440 CrossRef CAS PubMed.
- V. B. Rodriguez, B. A. Kidd, G. Interlandi, V. Tchesnokova, E. V. Sokurenko and W. E. Thomas, J. Biol. Chem., 2013, 288, 24128–24139 CrossRef CAS PubMed.
- V. Tchesnokova, P. Aprikian, O. Yakovenko, C. Larock, B. Kidd, V. Vogel, W. Thomas and E. Sokurenko, J. Biol. Chem., 2008, 283, 7823–7833 CrossRef CAS PubMed.
- V. Tchesnokova, P. Aprikian, D. Kisiela, S. Gowey, N. Korotkova, W. Thomas and E. Sokurenko, Infect. Immun., 2011, 79, 3895–3904 CrossRef CAS PubMed.
- S. Rabbani, B. Fiege, D. Eris, M. Silbermann, R. P. Jakob, G. Navarra, T. Maier and B. Ernst, J. Biol. Chem., 2018, 293, 1835–1849 CrossRef CAS PubMed.
- E. M. Krammer, J. de Ruyck, G. Roos, J. Bouckaert and M. F. Lensink, Molecules, 2018, 23(7), 1641 CrossRef PubMed.
- M. M. Sauer, R. P. Jakob, J. Eras, S. Baday, D. Eriş, G. Navarra, S. Bernèche, B. Ernst, T. Maier and R. Glockshuber, Nat. Commun., 2016, 7, 10738 CrossRef CAS PubMed.
- J. B. Heim, V. Hodnik, J. E. Heggelund, G. Anderluh and U. Krengel, Sci. Rep., 2019, 9, 12243 CrossRef PubMed.
- E. Shanina, S. Kuhaudomlarp, K. Lal, P. H. Seeberger, A. Imberty and C. Rademacher, Angew. Chem., Int. Ed., 2022, 61, e202109339 CrossRef CAS PubMed.
- R. P. Dings, M. C. Miller, I. Nesmelova, L. Astorgues-Xerri, N. Kumar, M. Serova, X. Chen, E. Raymond, T. R. Hoye and K. H. Mayo, J. Med. Chem., 2012, 55, 5121–5129 CrossRef CAS PubMed.
- T. C. Shih, R. Liu, G. Fung, G. Bhardwaj, P. M. Ghosh and K. S. Lam, Mol. Cancer Ther., 2017, 16, 1212–1223 CrossRef CAS PubMed.
- T. C. Shih, Y. Fan, S. Kiss, X. Li, X. N. Deng, R. Liu, X. J. Chen, R. Carney, A. Chen, P. M. Ghosh and K. S. Lam, Neuro Oncol., 2019, 21, 1389–1400 CrossRef CAS PubMed.
- A. W. Kahsai, J. Cui, H. Ü. Kaniskan, P. P. Garner and G. Fenteany, J. Biol. Chem., 2008, 283, 24534–24545 CrossRef CAS PubMed.
- N. T. H. Pham, M. Letourneau, M. Fortier, G. Begin, M. S. Al-Abdul-Wahid, F. Pucci, B. Folch, M. Rooman, D. Chatenet, Y. St-Pierre, P. Lague, C. Calmettes and N. Doucet, J. Biol. Chem., 2021, 297, 101308 CrossRef CAS PubMed.
- A. W. Kahsai, S. Zhu, D. J. Wardrop, W. S. Lane and G. Fenteany, Chem. Biol., 2006, 13, 973–983 CrossRef CAS PubMed.
- T. Onizuka, H. Shimizu, Y. Moriwaki, T. Nakano, S. Kanai, I. Shimada and H. Takahashi, FEBS J., 2012, 279, 2645–2656 CrossRef CAS PubMed.
- S. Wragg and K. Drickamer, J. Biol. Chem., 1999, 274, 35400–35406 CrossRef CAS PubMed.
- H. Feinberg, D. Torgersen, K. Drickamer and W. I. Weis, J. Biol. Chem., 2000, 275, 35176–35184 CrossRef CAS PubMed.
- T. Gramberg, E. Soilleux, T. Fisch, P. F. Lalor, H. Hofmann, S. Wheeldon, A. Cotterill, A. Wegele, T. Winkler, D. H. Adams and S. Pohlmann, Virology, 2008, 373, 189–201 CrossRef CAS PubMed.
- F. Probert, D. A. Mitchell and A. M. Dixon, FEBS J., 2014, 281, 3739–3750 CrossRef CAS PubMed.
- K. K. Ng and W. I. Weis, Biochemistry, 1998, 37, 17977–17989 CrossRef CAS PubMed.
- S. F. Poget, S. M. Freund, M. J. Howard and M. Bycroft, Biochemistry, 2001, 40, 10966–10972 CrossRef CAS PubMed.
- S. Nielbo, J. K. Thomsen, J. H. Graversen, P. H. Jensen, M. Etzerodt, F. M. Poulsen and H. C. Thogersen, Biochemistry, 2004, 43, 8636–8643 CrossRef CAS PubMed.
- F. Marcelo, N. Supekar, F. Corzana, J. C. van der Horst, I. M. Vuist, D. Live, G. P. H. Boons, D. F. Smith and S. J. van Vliet, J. Biol. Chem., 2019, 294, 1300–1311 CrossRef CAS PubMed.
- S. A. Jegouzo, H. Feinberg, T. Dungarwalla, K. Drickamer, W. I. Weis and M. E. Taylor, J. Biol. Chem., 2015, 290, 16759–16771 CrossRef CAS PubMed.
- M. Nagae, K. Yamanaka, S. Hanashima, A. Ikeda, K. Morita-Matsumoto, T. Satoh, N. Matsumoto, K. Yamamoto and Y. Yamaguchi, J. Biol. Chem., 2013, 288, 33598–33610 CrossRef CAS PubMed.
- J. A. Hardy and J. A. Wells, Curr. Opin. Struct. Biol., 2004, 14, 706–715 CrossRef CAS PubMed.
- S. Lu, M. Ji, D. Ni and J. Zhang, Drug Discov. Today, 2018, 23, 359–365 CrossRef CAS PubMed.
- J. Aretz, E. C. Wamhoff, J. Hanske, D. Heymann and C. Rademacher, Front. Immunol., 2014, 5, 323 Search PubMed.
- B. G. Keller and C. Rademacher, Curr. Opin. Struct. Biol., 2020, 62, 31–38 CrossRef CAS PubMed.
- E. B. Finger, K. D. Puri, R. Alon, M. B. Lawrence, U. H. von Andrian and T. A. Springer, Nature, 1996, 379, 266–269 CrossRef CAS PubMed.
- M. B. Lawrence, G. S. Kansas, E. J. Kunkel and K. Ley, J. Cell Biol., 1997, 136, 717–727 CrossRef CAS PubMed.
- T. T. Waldron and T. A. Springer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 85–90 CrossRef CAS PubMed.
- B. T. Marshall, M. Long, J. W. Piper, T. Yago, R. P. McEver and C. Zhu, Nature, 2003, 423, 190–193 CrossRef CAS PubMed.
- K. K. Sarangapani, T. Yago, A. G. Klopocki, M. B. Lawrence, C. B. Fieger, S. D. Rosen, R. P. McEver and C. Zhu, J. Biol. Chem., 2004, 279, 2291–2298 CrossRef CAS PubMed.
- S. Lu, Y. Zhang and M. Long, PLoS One, 2010, 5, e15417 CrossRef PubMed.
- S. Lu, S. Chen, D. Mao, Y. Zhang and M. Long, PLoS One, 2015, 10, e0118083 CrossRef PubMed.
- D. V. Erbe, B. A. Wolitzky, L. G. Presta, C. R. Norton, R. J. Ramos, D. K. Burns, J. M. Rumberger, B. N. Rao, C. Foxall and B. K. Brandley, et al. , J. Cell Biol., 1992, 119, 215–227 CrossRef CAS PubMed.
- F. A. Aleisa, K. Sakashita, J. M. Lee, D. B. AbuSamra, B. Al Alwan, S. Nozue, M. Tehseen, S. M. Hamdan, S. Habuchi, T. Kusakabe and J. S. Merzaban, J. Biol. Chem., 2020, 295, 3719–3733 CrossRef CAS PubMed.
- X. Wang, L. Bie, J. Fei and J. Gao, J. Chem. Inf. Model., 2020, 60, 5153–5161 CrossRef CAS PubMed.
- B. K. Brandley, M. Kiso, S. Abbas, P. Nikrad, O. Srivasatava, C. Foxall, Y. Oda and A. Hasegawa, Glycobiology, 1993, 3, 633–641 CrossRef CAS PubMed.
- J. Y. Ramphal, Z. L. Zheng, C. Perez, L. E. Walker, S. A. DeFrees and F. C. Gaeta, J. Med. Chem., 1994, 37, 3459–3463 CrossRef CAS PubMed.
- J. Hanske, S. Aleksić, M. Ballaschk, M. Jurk, E. Shanina, M. Beerbaum, P. Schmieder, B. G. Keller and C. Rademacher, J. Am. Chem. Soc., 2016, 138, 12176–12186 CrossRef CAS PubMed.
- J. O. Joswig, J. Anders, H. Zhang, C. Rademacher and B. G. Keller, J. Biol. Chem., 2021, 296, 100718 CrossRef CAS PubMed.
- H. Zhang, C. Modenutti, Y. P. K. Nekkanti, M. Denis, I. A. Bermejo, J. Lefèbre, K. Che, D. Kim, M. Kagelmacher, D. Kurzbach, M. Nazaré and C. Rademacher, ACS Chem. Biol., 2022, 17(10), 2728–2733 CrossRef CAS PubMed.
- L. Chatwell, A. Holla, B. B. Kaufer and A. Skerra, Mol. Immunol., 2008, 45, 1981–1994 CrossRef CAS PubMed.
- T. Oda, H. Yanagisawa, H. Shinmori, Y. Ogawa and T. Kawamura, eLife, 2022, 11, e79990 CrossRef CAS PubMed.
- M. J. Borrok and L. L. Kiessling, J. Am. Chem. Soc., 2007, 129, 12780–12785 CrossRef CAS PubMed.
- S. L. Mangold, L. R. Prost and L. L. Kiessling, Chem. Sci., 2012, 3, 772–777 RSC.
- N. S. Troelsen, E. Shanina, D. Gonzalez-Romero, D. Dankova, I. S. A. Jensen, K. J. Sniady, F. Nami, H. Zhang, C. Rademacher, A. Cuenda, C. H. Gotfredsen and M. H. Clausen, Angew. Chem., Int. Ed., 2020, 59, 2204–2210 CrossRef CAS PubMed.
- J. Schulze, H. Baukmann, R. Wawrzinek, F. F. Fuchsberger, E. Specker, J. Aretz, M. Nazare and C. Rademacher, ACS Chem. Biol., 2018, 13, 3229–3235 CrossRef CAS PubMed.
- H. Zhang, O. Danek, D. Makarov, S. Radl, D. Kim, J. Ledvinka, K. Vychodilova, J. Hlavac, J. Lefebre, M. Denis, C. Rademacher and P. Menova, ACS Med. Chem. Lett., 2022, 13, 935–942 CrossRef CAS PubMed.
- H. Prescher, A. Schweizer, M. Frank, E. Kuhfeldt, J. Ring and L. Nitschke, J. Med. Chem., 2022, 65, 10588–10610 CrossRef CAS PubMed.
- G. Schnapp, H. Neubauer, F. H. Büttner, S. Handschuh, I. Lingard, R. Heilker, K. Klinder, J. Prestle, R. Walter, M. Wolff, M. Zeeb, F. Debaene, H. Nar and D. Fiegen, Commun. Chem., 2020, 3, 75 CrossRef CAS PubMed.
- N. Yamakawa, Y. Yasuda, A. Yoshimura, A. Goshima, P. R. Crocker, G. Vergoten, Y. Nishiura, T. Takahashi, S. Hanashima, K. Matsumoto, Y. Yamaguchi, H. Tanaka, K. Kitajima and C. Sato, Sci. Rep., 2020, 10, 8647 CrossRef CAS PubMed.
- M. J. P. van Dongen, R. U. Kadam, J. Juraszek, E. Lawson, B. Brandenburg, F. Schmitz, W. B. G. Schepens, B. Stoops, H. A. van Diepen, M. Jongeneelen, C. Tang, J. Vermond, A. van Eijgen-Obregoso Real, S. Blokland, D. Garg, W. Yu, W. Goutier, E. Lanckacker, J. M. Klap, D. C. G. Peeters, J. Wu, C. Buyck, T. H. M. Jonckers, D. Roymans, P. Roevens, R. Vogels, W. Koudstaal, R. H. E. Friesen, P. Raboisson, D. Dhanak, J. Goudsmit and I. A. Wilson, Science, 2019, 363, eaar6221 CrossRef CAS PubMed.
- G. Tang, X. Lin, Z. Qiu, W. Li, L. Zhu, L. Wang, S. Li, H. Li, W. Lin, M. Yang, T. Guo, L. Chen, D. Lee, J. Z. Wu and W. Yang, ACS Med. Chem. Lett., 2011, 2, 603–607 CrossRef CAS PubMed.
- K. M. White, P. De Jesus, Z. Chen, P. Abreu, Jr., E. Barile, P. A. Mak, P. Anderson, Q. T. Nguyen, A. Inoue, S. Stertz, R. Koenig, M. Pellecchia, P. Palese, K. Kuhen, A. García-Sastre, S. K. Chanda and M. L. Shaw, ACS Infect. Dis., 2015, 1, 98–109 CrossRef CAS PubMed.
- Y. Yao, R. U. Kadam, C. D. Lee, J. L. Woehl, N. C. Wu, X. Zhu, S. Kitamura, I. A. Wilson and D. W. Wolan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18431–18438 CrossRef CAS PubMed.
- A. Antanasijevic, M. A. Durst, H. Cheng, I. N. Gaisina, J. T. Perez, B. Manicassamy, L. Rong, A. Lavie and M. Caffrey, Life Sci. Alliance, 2020, 3, e202000724 CrossRef PubMed.
- H. Y. Liu and P. L. Yang, Annu. Rev. Virol., 2021, 8, 459–489 CrossRef PubMed.
- M. Civera, E. Moroni, L. Sorrentino, F. Vasile and S. Sattin, Eur. J. Org. Chem., 2021, 4245–4259 CrossRef CAS.
- P. K. Grewal, in Methods in Enzymology, ed. M. Fukuda, Academic Press, 2010, vol. 479, pp. 223–241 Search PubMed.
- Y. I. Henis, Z. Katzir, M. A. Shia and H. F. Lodish, J. Cell Biol., 1990, 111, 1409–1418 CrossRef CAS PubMed.
- J. Bischoff and H. F. Lodish, J. Biol. Chem., 1987, 262, 11825–11832 CrossRef CAS PubMed.
- M. Spiess, Biochemistry, 1990, 29, 10009–10018 CrossRef CAS PubMed.
- A. L. Schwartz, S. E. Fridovich and H. F. Lodish, J. Biol. Chem., 1982, 257, 4230–4237 CrossRef CAS PubMed.
- P. H. Weigel and J. A. Oka, J. Biol. Chem., 1983, 258, 5095–5102 CrossRef CAS PubMed.
- Y. A. Ivanenkov, S. Y. Maklakova, E. K. Beloglazkina, N. V. Zyk, A. G. Nazarenko, A. G. Tonevitsky, V. E. M. Kotelianski and A. G. Majouga, Russ. Chem. Rev., 2017, 86, 750–776 CrossRef CAS.
- D. E. Large, J. R. Soucy, J. Hebert and D. T. Auguste, Adv. Ther., 2019, 2, 1800091 CrossRef.
- R. Dutta and R. I. Mahato, Pharmacol. Therapeut., 2017, 173, 106–117 CrossRef CAS PubMed.
- L. Singh, S. Indermun, M. Govender, P. Kumar, L. C. Du Toit, Y. E. Choonara and V. Pillay, Viruses, 2018, 10, 267 CrossRef PubMed.
- M. H. Elberry, N. H. E. Darwish and S. A. Mousa, Virol. J., 2017, 14, 88 CrossRef PubMed.
- S. Borrmann and K. Matuschewski, Trends Mol. Med., 2011, 17, 527–536 CrossRef CAS PubMed.
- S. K. Mamidyala, S. Dutta, B. A. Chrunyk, C. Préville, H. Wang, J. M. Withka, A. McColl, T. A. Subashi, S. J. Hawrylik, M. C. Griffor, S. Kim, J. A. Pfefferkorn, D. A. Price, E. Menhaji-Klotz, V. Mascitti and M. G. Finn, J. Am. Chem. Soc., 2012, 134, 1978–1981 CrossRef CAS PubMed.
- J. Wu, M. H. Nantz and M. A. Zern, Front. Biosci., 2002, 7, 717–725 Search PubMed.
- N. I. Ruiz and K. Drickamer, Glycobiology, 1996, 6, 551–559 CrossRef CAS PubMed.
- D. T. Connolly, R. R. Townsend, K. Kawaguchi, W. R. Bell and Y. C. Lee, J. Biol. Chem., 1982, 257, 939–945 CrossRef CAS PubMed.
- A. R. Kolatkar, A. K. Leung, R. Isecke, R. Brossmer, K. Drickamer and W. I. Weis, J. Biol. Chem., 1998, 273, 19502–19508 CrossRef CAS PubMed.
- Y. C. Lee, R. R. Townsend, M. R. Hardy, J. Lönngren, J. Arnarp, M. Haraldsson and H. Lönn, J. Biol. Chem., 1983, 258, 199–202 CrossRef CAS PubMed.
- U. Westerlind, J. Westman, E. Törnquist, C. I. E. Smith, S. Oscarson, M. Lahmann and T. Norberg, Glycoconj. J., 2004, 21, 227–241 CrossRef CAS PubMed.
- E. A. L. Biessen, D. M. Beuting, H. C. P. F. Roelen, G. A. van de Marel, J. H. Van Boom and T. J. C. Van Berkel, J. Med. Chem., 1995, 38, 1538–1546 CrossRef CAS PubMed.
- X. Huang, J.-C. Leroux and B. Castagner, Bioconjugate Chem., 2017, 28, 283–295 CrossRef CAS PubMed.
- D. U. Warrier, A. K. Dhanabalan, G. Krishnasamy, H. Kolge, V. Ghormade, C. R. Gupta, P. K. Ambre and U. A. Shinde, Int. J. Biol. Macromol., 2022, 207, 683–699 CrossRef CAS PubMed.
- T. Tanaka, Y. Fujishima, S. Hamano and Y. Kaneo, Eur. J. Pharm. Sci., 2004, 22, 435–444 CrossRef CAS PubMed.
- Y. Kaneo, T. Tanaka, T. Nakano and Y. Yamaguchi, J. Controlled Release, 2001, 70, 365–373 CrossRef CAS PubMed.
- Y. C. Lee, FASEB J., 1992, 6, 3193–3200 CrossRef CAS PubMed.
- S. Liras, V. Mascitti and B. Thuma, US20160207953A1, 2016.
- C. A. Sanhueza, M. M. Baksh, B. Thuma, M. D. Roy, S. Dutta, C. Préville, B. A. Chrunyk, K. Beaumont, R. Dullea, M. Ammirati, S. Liu, D. Gebhard, J. E. Finley, C. T. Salatto, A. King-Ahmad, I. Stock, K. Atkinson, B. Reidich, W. Lin, R. Kumar, M. Tu, E. Menhaji-Klotz, D. A. Price, S. Liras, M. G. Finn and V. Mascitti, J. Am. Chem. Soc., 2017, 139, 3528–3536 CrossRef CAS PubMed.
- S. Liras, V. Mascitti and B. Thuma, US9340553B2, 2016.
- M. Meier, M. D. Bider, V. N. Malashkevich, M. Spiess and P. Burkhard, J. Mol. Biol., 2000, 300, 857–865 CrossRef CAS PubMed.
- M. Gonçalves, S. Mignani, J. Rodrigues and H. Tomás, J. Controlled Release, 2020, 317, 347–374 CrossRef PubMed.
- M. Hashida, M. Nishikawa, F. Yamashita and Y. Takakura, Adv. Drug Deliv. Rev., 2001, 52, 187–196 CrossRef CAS PubMed.
- K. Akamatsu, M. Imai, Y. Yamasaki, M. Nishikawa, Y. Takakura and M. Hashida, J. Drug Target., 1998, 6, 229–239 CrossRef CAS PubMed.
- S. Wang, L. Cheng, F. Yu, W. Pan and J. Zhang, Int. J. Pharm., 2006, 311, 82–88 CrossRef CAS PubMed.
- J. W. Hopewell, R. Duncan, D. Wilding and K. Chakrabarti, Hum. Exp. Toxicol., 2001, 20, 461–470 CrossRef CAS PubMed.
- P. J. Julyan, L. W. Seymour, D. R. Ferry, S. Daryani, C. M. Boivin, J. Doran, M. David, D. Anderson, C. Christodoulou, A. M. Young, S. Hesslewood and D. J. Kerr, J. Controlled Release, 1999, 57, 281–290 CrossRef CAS PubMed.
- L. W. Seymour, D. R. Ferry, D. Anderson, S. Hesslewood, P. J. Julyan, R. Poyner, J. Doran, A. M. Young, S. Burtles and D. J. Kerr, J. Clin. Oncol., 2002, 20, 1668–1676 CrossRef CAS PubMed.
- R. Böttger, G. Pauli, P.-H. Chao, N. Al Fayez, L. Hohenwarter and S.-D. Li, Adv. Drug Delivery Rev., 2020, 154–155, 79–101 CrossRef PubMed.
- E. F. Craparo, D. Triolo, G. Pitarresi, G. Giammona and G. Cavallaro, Biomacromolecules, 2013, 14, 1838–1849 CrossRef CAS PubMed.
- Y. Zou, Y. Song, W. Yang, F. Meng, H. Liu and Z. Zhong, J. Controlled Release, 2014, 193, 154–161 CrossRef CAS PubMed.
- S. H. Medina, V. Tekumalla, M. V. Chevliakov, D. S. Shewach, W. D. Ensminger and M. E. H. El-Sayed, Biomaterials, 2011, 32, 4118–4129 CrossRef CAS PubMed.
- Y. Hayashi, T. Higashi, K. Motoyama, Y. Mori, H. Jono, Y. Ando and H. Arima, J. Drug Target., 2013, 21, 487–496 CrossRef CAS PubMed.
- H. Cui, X. Zhu, S. Li, P. Wang and J. Fang, ACS Omega, 2021, 6, 16259–16265 CrossRef CAS PubMed.
- J. K. Nair, J. L. S. Willoughby, A. Chan, K. Charisse, M. R. Alam, Q. Wang, M. Hoekstra, P. Kandasamy, A. V. Kel’in, S. Milstein, N. Taneja, J. O’Shea, S. Shaikh, L. Zhang, R. J. van der Sluis, M. E. Jung, A. Akinc, R. Hutabarat, S. Kuchimanchi, K. Fitzgerald, T. Zimmermann, T. J. C. van Berkel, M. A. Maier, K. G. Rajeev and M. Manoharan, J. Am. Chem. Soc., 2014, 136, 16958–16961 CrossRef CAS PubMed.
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef CAS PubMed.
- J. K. Nair, H. Attarwala, A. Sehgal, Q. Wang, K. Aluri, X. Zhang, M. Gao, J. Liu, R. Indrakanti, S. Schofield, P. Kretschmer, C. R. Brown, S. Gupta, J. L. S. Willoughby, J. A. Boshar, V. Jadhav, K. Charisse, T. Zimmermann, K. Fitzgerald, M. Manoharan, K. G. Rajeev, A. Akinc, R. Hutabarat and M. A. Maier, Nucleic Acids Res., 2017, 45, 10969–10977 CrossRef CAS PubMed.
- M. R. Hassler, A. A. Turanov, J. F. Alterman, R. A. Haraszti, A. H. Coles, M. F. Osborn, D. Echeverria, M. Nikan, W. E. Salomon, L. Roux, B. Godinho, S. M. Davis, D. V. Morrissey, P. D. Zamore, S. A. Karumanchi, M. J. Moore, N. Aronin and A. Khvorova, Nucleic Acids Res., 2018, 46, 2185–2196 CrossRef CAS PubMed.
- M. K. Schlegel, D. J. Foster, A. V. Kel’in, I. Zlatev, A. Bisbe, M. Jayaraman, J. G. Lackey, K. G. Rajeev, K. Charissé, J. Harp, P. S. Pallan, M. A. Maier, M. Egli and M. Manoharan, J. Am. Chem. Soc., 2017, 139, 8537–8546 CrossRef CAS PubMed.
- M. M. Janas, M. K. Schlegel, C. E. Harbison, V. O. Yilmaz, Y. Jiang, R. Parmar, I. Zlatev, A. Castoreno, H. Xu, S. Shulga-Morskaya, K. G. Rajeev, M. Manoharan, N. D. Keirstead, M. A. Maier and V. Jadhav, Nat. Commun., 2018, 9, 723 CrossRef PubMed.
- A. J. Debacker, J. Voutila, M. Catley, D. Blakey and N. Habib, Mol. Ther., 2020, 28, 1759–1771 CrossRef CAS PubMed.
- A. Weingärtner, L. Bethge, L. Weiss, M. Sternberger and M. W. Lindholm, Mol. Ther. Nucleic Acids, 2020, 21, 242–250 CrossRef PubMed.
- G. Gregoriadis, A. G. Morell, I. Sternlieb and I. H. Scheinberg, J. Biol. Chem., 1970, 245, 5833–5837 CrossRef CAS PubMed.
- T. S. Zimmermann, V. Karsten, A. Chan, J. Chiesa, M. Boyce, B. R. Bettencourt, R. Hutabarat, S. Nochur, A. Vaishnaw and J. Gollob, Mol. Ther., 2017, 25, 71–78 CrossRef CAS PubMed.
- Alnylam-Our pipeline, https://www.alnylam.com/alnylam-rnai-pipeline, (accessed September 2022, 2022).
- Dicerna Pipeline, https://dicerna.com/pipeline/, (accessed September 2022, 2022).
- Novo Nordisk R & D Pipeline, https://www.novonordisk.com/science-and-technology/r-d-pipeline.html, (accessed September 2022, 2022).
- Silence Therapeutics-Our pipeline, https://silence-therapeutics.com/our-pipeline/default.aspx, (accessed September 2022, 2022).
- Arbutus Biopharma-Pipeline, https://www.arbutusbio.com/pipeline/, (accessed September 2022, 2022).
- M. Balwani, E. Sardh, P. Ventura, P. A. Peiró, D. C. Rees, U. Stölzel, D. M. Bissell, H. L. Bonkovsky, J. Windyga, K. E. Anderson, C. Parker, S. M. Silver, S. B. Keel, J.-D. Wang, P. E. Stein, P. Harper, D. Vassiliou, B. Wang, J. Phillips, A. Ivanova, J. G. Langendonk, R. Kauppinen, E. Minder, Y. Horie, C. Penz, J. Chen, S. Liu, J. J. Ko, M. T. Sweetser, P. Garg, A. Vaishnaw, J. B. Kim, A. R. Simon and L. Gouya, New Engl. J. Med., 2020, 382, 2289–2301 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2021, 81, 841–848 CrossRef CAS PubMed.
- R. Marchi, L. Duarte, A. Ke, B. Hl and E. Al, Hematol. Transfus. Cell Ther., 2020, 42, 17–18 CrossRef.
- S. F. Garrelfs, Y. Frishberg, S. A. Hulton, M. J. Koren, W. D. O’Riordan, P. Cochat, G. Deschênes, H. Shasha-Lavsky, J. M. Saland, W. G. van’t Hoff, D. G. Fuster, D. Magen, S. H. Moochhala, G. Schalk, E. Simkova, J. W. Groothoff, D. J. Sas, K. A. Meliambro, J. Lu, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. M. Gansner, T. L. McGregor and J. C. Lieske, New Engl. J. Med., 2021, 384, 1216–1226 CrossRef CAS PubMed.
- Y. Frishberg, G. Deschênes, J. W. Groothoff, S.-A. Hulton, D. Magen, J. Harambat, W. G. van’t Hoff, U. Lorch, D. S. Milliner, J. C. Lieske, P. Haslett, P. P. Garg, A. K. Vaishnaw, S. Talamudupula, J. Lu, B. A. Habtemariam, D. V. Erbe, T. L. McGregor and P. Cochat, Clin. J. Am. Soc. Nephrol., 2021, 16, 1025–1036 CrossRef CAS PubMed.
- F. Erger and B. B. Beck, Nat. Rev. Nephrol., 2021, 17, 573–574 CrossRef CAS PubMed.
- Z. A. Massy and T. B. Drueke, Kidney Int., 2022, 101, 208–211 CrossRef CAS PubMed.
- F. J. Raal, D. Kallend, K. K. Ray, T. Turner, W. Koenig, R. S. Wright, P. L. J. Wijngaard, D. Curcio, M. J. Jaros, L. A. Leiter and J. J. P. Kastelein, N. Engl. J. Med., 2020, 382, 1520–1530 CrossRef CAS PubMed.
- K. K. Ray, R. S. Wright, D. Kallend, W. Koenig, L. A. Leiter, F. J. Raal, J. A. Bisch, T. Richardson, M. Jaros, P. L. J. Wijngaard and J. J. P. Kastelein, N. Engl. J. Med., 2020, 382, 1507–1519 CrossRef CAS PubMed.
- Y. N. Lamb, Drugs, 2021, 81, 389–395 CrossRef CAS PubMed.
- B. A. Habtemariam, V. Karsten, H. Attarwala, V. Goel, M. Melch, V. A. Clausen, P. Garg, A. K. Vaishnaw, M. T. Sweetser, G. J. Robbie and J. Vest, Clin. Pharmacol. Ther., 2021, 109, 372–382 CrossRef CAS PubMed.
- M. Bekes, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discov., 2022, 21, 181–200 CrossRef CAS PubMed.
- Y. Ding, D. Xing, Y. Fei and B. Lu, Chem. Soc. Rev., 2022, 51, 8832–8876 RSC.
- D. A. Nalawansha and C. M. Crews, Cell Chem. Biol., 2020, 27, 998–1014 CrossRef CAS PubMed.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed.
- X. Zhang, H. Liu, J. He, C. Ou, T. C. Donahue, M. M. Muthana, L. Su and L. X. Wang, ACS Chem. Biol., 2022, 17(11), 3013–3023 CrossRef CAS PubMed.
- D. F. Caianiello, M. Zhang, J. D. Ray, R. A. Howell, J. C. Swartzel, E. M. J. Branham, E. Chirkin, V. R. Sabbasani, A. Z. Gong, D. M. McDonald, V. Muthusamy and D. A. Spiegel, Nat. Chem. Biol., 2021, 17, 947–953 CrossRef CAS PubMed.
- G. Ahn, S. M. Banik, C. L. Miller, N. M. Riley, J. R. Cochran and C. R. Bertozzi, Nat. Chem. Biol., 2021, 17, 937–946 CrossRef CAS PubMed.
- M. Gary-Bobo, P. Nirde, A. Jeanjean, A. Morere and M. Garcia, Curr. Med. Chem., 2007, 14, 2945–2953 CrossRef CAS PubMed.
- P. Y. Tong, W. Gregory and S. Kornfeld, J. Biol. Chem., 1989, 264, 7962–7969 CrossRef CAS PubMed.
- Y. Zhou, P. Teng, N. T. Montgomery, X. Li and W. Tang, ACS Cent. Sci., 2021, 7, 499–506 CrossRef CAS PubMed.
- D. F. Caianiello, M. Zhang, J. D. Ray, R. A. Howell, J. C. Swartzel, E. M. J. Branham, E. Chirkin, V. R. Sabbasani, A. Z. Gong, D. M. McDonald, V. Muthusamy and D. A. Spiegel, Nat. Chem. Biol., 2021, 17, 947–953 CrossRef CAS PubMed.
- S. Liras, V. Mascitti, B. A. Thuma and R. Rouet, WO2021155317A1, 2021.
- M. G. Saulnier, J. J. Chen, S. Karra, K. T. Sprott, J. A. Wiles and S. Ray, WO2021155317, 2021.
- E. Tozzo, Presented in part at the 9th Drug Discovery Strategic Summit (DDSS), 2022.
- L. J. Olson, J. Zhang, Y. C. Lee, N. M. Dahms and J. J. Kim, J. Biol. Chem., 1999, 274, 29889–29896 CrossRef CAS PubMed.
- B. H. Duong, H. Tian, T. Ota, G. Completo, S. Han, J. L. Vela, M. Ota, M. Kubitz, N. Bovin, J. C. Paulson and D. Nemazee, J. Exp. Med., 2010, 207, 173–187 CrossRef CAS PubMed.
- W. C. Chen, G. C. Completo, D. S. Sigal, P. R. Crocker, A. Saven and J. C. Paulson, Blood, 2010, 115, 4778–4786 CrossRef CAS PubMed.
- K. J. Bednar, C. M. Nycholat, T. S. Rao, J. C. Paulson, W. P. Fung-Leung and M. S. Macauley, ACS Chem. Biol., 2019, 14, 644–654 CrossRef CAS PubMed.
- F. Spiller, C. M. Nycholat, C. Kikuchi, J. C. Paulson and M. S. Macauley, J. Immunol., 2018, 200, 949–956 CrossRef CAS PubMed.
- L. Pang, M. S. Macauley, B. M. Arlian, C. M. Nycholat and J. C. Paulson, ChemBioChem, 2017, 18, 1226–1233 CrossRef CAS PubMed.
- K. J. Bednar, E. Shanina, R. Ballet, E. P. Connors, S. Duan, J. Juan, B. M. Arlian, M. D. Kulis, E. C. Butcher, W. P. Fung-Leung, T. S. Rao, J. C. Paulson and M. S. Macauley, J. Immunol., 2017, 199, 3116–3128 CrossRef CAS PubMed.
- A. Srivastava, B. M. Arlian, L. Pang, T. K. Kishimoto and J. C. Paulson, ACS Chem. Biol., 2021, 16, 1985–1993 CrossRef CAS PubMed.
- W. Peng and J. C. Paulson, J. Am. Chem. Soc., 2017, 139, 12450–12458 CrossRef CAS PubMed.
- M. Islam, B. M. Arlian, F. Pfrengle, S. Duan, S. A. Smith and J. C. Paulson, J. Am. Chem. Soc., 2022, 144, 9302–9311 CrossRef CAS PubMed.
- T. Harumoto, H. Iwai, M. Tanigawa, T. Kubo, T. Atsumi, K. Tsutsumi, M. Takashima, G. Destito, R. Soloff, K. Tomizuka, C. Nycholat, J. Paulson and K. Uehara, ACS Chem. Biol., 2022, 17, 292–298 CrossRef CAS PubMed.
- K. A. Orgel, S. Duan, B. L. Wright, S. J. Maleki, J. C. Wolf, B. P. Vickery, A. W. Burks, J. C. Paulson, M. D. Kulis and M. S. Macauley, J. Allergy Clin. Immunol., 2017, 139, 366–369 CrossRef CAS PubMed.
- S. Hong, C. Yu, P. Wang, Y. Shi, W. Cao, B. Cheng, D. G. Chapla, Y. Ma, J. Li, E. Rodrigues, Y. Narimatsu, J. R. Yates, 3rd, X. Chen, H. Clausen, K. W. Moremen, M. S. Macauley, J. C. Paulson and P. Wu, Angew. Chem., Int. Ed., 2021, 60, 3603–3610 CrossRef CAS PubMed.
- B. Malissen, S. Tamoutounour and S. Henri, Nat. Rev. Immunol., 2014, 14, 417–428 CrossRef CAS PubMed.
- E. Wong, B. Montoya, C. Stotesbury, M. Ferez, R. H. Xu and L. J. Sigal, Cell Rep., 2019, 29, 3047–3059 CrossRef CAS PubMed.
- L. de Witte, A. Nabatov, M. Pion, D. Fluitsma, M. A. de Jong, T. de Gruijl, V. Piguet, Y. van Kooyk and T. B. Geijtenbeek, Nat. Med., 2007, 13, 367–371 CrossRef CAS PubMed.
- M. van der Vlist, L. de Witte, R. D. de Vries, M. Litjens, M. A. de Jong, D. Fluitsma, R. L. de Swart and T. B. Geijtenbeek, Eur. J. Immunol., 2011, 41, 2619–2631 CrossRef CAS PubMed.
- W. C. Ng, S. L. Londrigan, N. Nasr, A. L. Cunningham, S. Turville, A. G. Brooks and P. C. Reading, J. Virol., 2015, 90, 206–221 CrossRef PubMed.
- M. A. de Jong, L. E. Vriend, B. Theelen, M. E. Taylor, D. Fluitsma, T. Boekhout and T. B. Geijtenbeek, Mol. Immunol., 2010, 47, 1216–1225 CrossRef CAS PubMed.
- R. E. Hunger, P. A. Sieling, M. T. Ochoa, M. Sugaya, A. E. Burdick, T. H. Rea, P. J. Brennan, J. T. Belisle, A. Blauvelt, S. A. Porcelli and R. L. Modlin, J. Clin. Invest., 2004, 113, 701–708 CrossRef CAS PubMed.
- R. McDermott, H. Bausinger, D. Fricker, D. Spehner, F. Proamer, D. Lipsker, J. P. Cazenave, B. Goud, H. De La Salle, J. Salamero and D. Hanau, J. Invest. Dermatol., 2004, 123, 72–77 CrossRef CAS PubMed.
- J. Idoyaga, A. Lubkin, C. Fiorese, M. H. Lahoud, I. Caminschi, Y. Huang, A. Rodriguez, B. E. Clausen, C. G. Park, C. Trumpfheller and R. M. Steinman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2384–2389 CrossRef CAS PubMed.
- J. Idoyaga, N. Suda, K. Suda, C. G. Park and R. M. Steinman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1524–1529 CrossRef CAS PubMed.
- P. Stoitzner, C. H. Tripp, A. Eberhart, K. M. Price, J. Y. Jung, L. Bursch, F. Ronchese and N. Romani, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7783–7788 CrossRef CAS PubMed.
- J. Kervevan, A. Bouteau, J. S. Lanza, A. Hammoudi, S. Zurawski, M. Surenaud, L. Dieudonne, M. Bonnet, C. Lefebvre, H. Hocini, R. Marlin, A. Guguin, B. Hersant, O. Hermeziu, E. Menu, C. Lacabaratz, J. D. Lelievre, G. Zurawski, V. Godot, S. Henri, B. Z. Igyarto, Y. Levy and S. Cardinaud, PLoS Pathog, 2021, 17, e1009749 CrossRef CAS PubMed.
- C. H. Tripp, H. Voit, A. An, M. Seidl-Philipp, J. Krapf, S. Sigl, N. Romani, B. Del Frari and P. Stoitzner, Exp. Dermatol., 2021, 30, 1279–1289 CrossRef CAS PubMed.
- E. Klechevsky, R. Morita, M. Liu, Y. Cao, S. Coquery, L. Thompson-Snipes, F. Briere, D. Chaussabel, G. Zurawski, A. K. Palucka, Y. Reiter, J. Banchereau and H. Ueno, Immunity, 2008, 29, 497–510 CrossRef CAS PubMed.
- E. C. Wamhoff, J. Schulze, L. Bellmann, M. Rentzsch, G. Bachem, F. F. Fuchsberger, J. Rademacher, M. Hermann, B. Del Frari, R. van Dalen, D. Hartmann, N. M. van Sorge, O. Seitz, P. Stoitzner and C. Rademacher, ACS Cent. Sci., 2019, 5, 808–820 CrossRef CAS PubMed.
- G. Bachem, E. C. Wamhoff, K. Silberreis, D. Kim, H. Baukmann, F. Fuchsberger, J. Dernedde, C. Rademacher and O. Seitz, Angew. Chem., Int. Ed., 2020, 59, 21016–21022 CrossRef CAS PubMed.
- J. Schulze, M. Rentzsch, D. Kim, L. Bellmann, P. Stoitzner and C. Rademacher, Biochemistry, 2019, 58, 2576–2580 CrossRef CAS PubMed.
- L. Bellmann, C. Zelle-Rieser, P. Milne, A. Resteu, C. H. Tripp, N. Hermann-Kleiter, V. Zaderer, D. Wilflingseder, P. Hortnagl, M. Theochari, J. Schulze, M. Rentzsch, B. Del Frari, M. Collin, C. Rademacher, N. Romani and P. Stoitzner, J. Invest. Dermatol., 2021, 141, 84–94 CrossRef CAS PubMed.
- M. Rentzsch, R. Wawrzinek, C. Zelle-Rieser, H. Strandt, L. Bellmann, F. F. Fuchsberger, J. Schulze, J. Busmann, J. Rademacher, S. Sigl, B. Del Frari, P. Stoitzner and C. Rademacher, Front. Immunol., 2021, 12, 732298 CrossRef CAS PubMed.
- S. I. van Kasteren, S. J. Campbell, S. Serres, D. C. Anthony, N. R. Sibson and B. G. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18–23 CrossRef CAS PubMed.
- E. S. Shchegravina, A. A. Sachkova, S. D. Usova, A. V. Nyuchev, Y. A. Gracheva and A. Y. Fedorov, Russ. J. Bioorg. Chem., 2021, 47, 71–98 CrossRef.
- J. L. Magnani, J. M. Peterson, W. E. Fogler and M.-G. Baek, WO2022061168A1, 2022.
- M. G. P. Page, Ann. NY Acad. Sci., 2013, 1277, 115–126 CrossRef CAS PubMed.
- P. Klahn and M. Brönstrup, Nat. Prod. Rep., 2017, 34, 832–885 RSC.
- S. M. Lehar, T. Pillow, M. Xu, L. Staben, K. K. Kajihara, R. Vandlen, L. DePalatis, H. Raab, W. L. Hazenbos, J. Hiroshi Morisaki, J. Kim, S. Park, M. Darwish, B.-C. Lee, H. Hernandez, K. M. Loyet, P. Lupardus, R. Fong, D. Yan, C. Chalouni, E. Luis, Y. Khalfin, E. Plise, J. Cheong, J. P. Lyssikatos, M. Strandh, K. Koefoed, P. S. Andersen, J. A. Flygare, M. Wah Tan, E. J. Brown and S. Mariathasan, Nature, 2015, 527, 323–328 CrossRef CAS PubMed.
- J. Meiers, E. Zahorska, T. Röhrig, D. Hauck, S. Wagner and A. Titz, J. Med. Chem., 2020, 63, 11707–11724 CrossRef CAS PubMed.
- J. Meiers, K. Rox and A. Titz, J. Med. Chem., 2022, 65, 13988–14014 CrossRef CAS PubMed.
- O. Metelkina, B. Huck, J. S. O'Connor, M. Koch, A. Manz, C. M. Lehr and A. Titz, J. Mater. Chem. B, 2022, 10, 537–548 RSC.
| This journal is © The Royal Society of Chemistry 2023 |